

Abstract
Antigen-specific lung-resident memory T cells (TRMs) constitute the first line of defense that mediates rapid protection against respiratory pathogens and inspires novel vaccine designs against infectious pandemic threats, yet effective means of inducing TRMs, particularly via non-viral vectors, remain challenging. Here, we demonstrate safe and potent induction of lung-resident TRMs using a biodegradable polymeric nanoshell that co-encapsulates antigenic peptides and TLR9 agonist CpG-oligodeoxynucleotide (CpG-ODN) in a virus-mimicking structure. Through subcutaneous priming and intranasal boosting, the combinatorial nanoshell vaccine elicits prominent lung-resident CD4+ and CD8+ T cells that surprisingly show better durability than live viral infections. In particular, nanoshells containing CpG-ODN and a pair of conserved class I and II major histocompatibility complex-restricted influenza nucleoprotein-derived antigenic peptides are demonstrated to induce near-sterilizing immunity against lethal infections with influenza A viruses of different strains and subtypes in mice, resulting in rapid elimination of replicating viruses. We further examine the pulmonary transport dynamic and optimal composition of the nanoshell vaccine conducive for robust TRM induction as well as the benefit of subcutaneous priming on TRM replenishment. The study presents a practical vaccination strategy for inducing protective TRM-mediated immunity, offering a compelling platform and critical insights in the ongoing quest toward a broadly protective vaccine against universal influenza as well as other respiratory pathogens.
Keywords: resident memory T cell, nanoshell, T cell vaccine, peptide vaccine, influenza virus, universal influenza vaccine, respiratory infection
Graphical Abstract
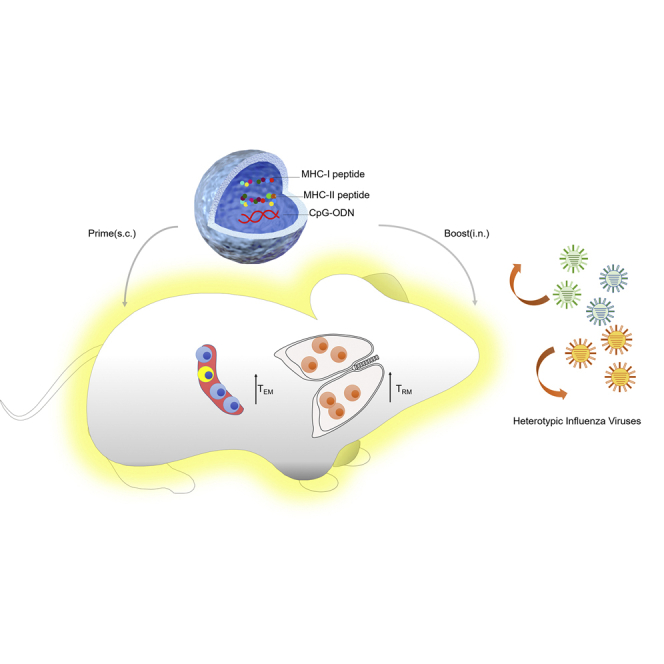
Nanoshell vaccines encapsulating CpG and both MHC-I and MHC-II-restricted conserved peptides derived from influenza virus proteomes can elicit robust CD4+ and CD8+ lung-resident memory T cells (TRMs) and circulatory memory T cells through the subcutaneous priming and intranasal boosting strategy to protect against influenza viruses of different strains and subtypes.
Introduction
Distinct from their circulatory central memory (TCM) and effector memory T cell (TEM) counterparts, resident memory T cells (TRMs) have been recently identified as a specialized population of non-circulatory memory T cells that are poised to immediately respond to invading pathogens.1,2 TRMs trigger rapid innate and adaptive immune responses by secretion of cytokines and provide first-line immune protection.3 In the lungs, pulmonary TRMs possess unique homing molecules and surface markers such as CD69 and CD103 that facilitate durable residence in lung mucosa, and these cells have been proven critical for cross-protection against multiple respiratory diseases including influenza,4, 5, 6 tuberculosis,7 respiratory syncytial virus,8 and coronaviruses.9 While many studies conducted by infection with viral vectors have shown compelling evidence of pulmonary TRM-mediated antiviral protection,10,11 non-viral strategies at inducing pulmonary TRMs remain largely inadequate at achieving the sterilizing immunity attainable via live infections.12, 13, 14 As non-viral vectors, particularly those containing defined T cell peptide antigens, offer the allure of improved safety, logistics, and defined and controllable TRM induction, their development can help answer essential questions in TRM biology and provide a viable platform for TRM vaccine preparation against pandemic threats.
Among respiratory infections, influenza has remained one of the most primary public health concerns that emerge seasonally, and this infectious disease has also drawn much TRM-related vaccine research, as current influenza vaccines that induce antibodies are ineffective in coping with frequently mutated viral surface proteins, namely hemagglutinin (HA) and neuraminidase (NA), and fail to protect against distantly related strains or different subtypes. Annual reformulation of influenza vaccines is thus required to keep pace with the continuous viral evolution.15 In contrast to neutralizing antibody responses that are often strain specific, T cells recognizing the epitopes derived from the conserved internal viral proteins, like nucleoprotein (NP) and matrix protein 1 (M1), bear the potential to cross-protect against different influenza strains and subtypes, including emerging viral strains.16, 17, 18 Identification of highly conserved CD8+ T cell epitopes across all three influenza virus types A, B, and C19,20 as well as the higher fitness costs for viral mutation from these T cell epitopes21,22 further offer glimpses of hope that a universal T cell-based influenza vaccine may be attainable upon effective TRM priming. As recent reports highlight the continuing challenge in antibody-based vaccination strategy against mutation-prone viral strains,23,24 strategies to induce protective T cell immunity warrant major considerations in the quest for a universal influenza vaccine.25,26
In order to effectively stimulate T cell responses to peptide antigens and minimize the reactogenic effect of immunologic adjuvants, synthetic nanocarriers have emerged as a compelling non-viral approach for stimulating immune potentiation with minimal safety concerns.27, 28, 29, 30, 31 Recent advances in emulsion protocols and formulation control have made possible the preparation of biodegradable polymeric nanoshells with structural semblance to virus capsids,32, 33, 34 allowing for efficient partitioning of water-soluble cargoes in the aqueous core and opening new opportunities for formulation designs. With the aim of examining the prospect of optimizing T cell-based influenza vaccines, we here adopted the thin-shell hollow polymeric nanoparticle, called nanoshell, to formulate influenza virus-derived T cell epitope peptides. Combinatorial nanoshells co-encapsulating CpG-oligodeoxynucleotide (CpG-ODN) and major histocompatibility complex (MHC) class I- and MHC class II-restricted antigenic peptides of influenza A virus (IAV) were prepared, and their physicochemical properties, immunogenicity, reactogenicity, and antiviral protectivity were systematically characterized.
Amid growing interest in the role of TRM cells in protection against respiratory virus infection as well as increasing reports highlighting the implication of T cell immunity in antiviral protection in the current COVID-19 pandemic,35, 36, 37 the present study further explores the influence of vaccine composition and vaccination route on the establishment of pulmonary TRMs. Of great translational interest, we observe that robust induction of TRM cells in mice vaccinated with a rationally designed vaccination formulation and scheme confers full resistance to lethal IAV infections of four different strains and subtypes. The study demonstrates insights and a viable non-viral vaccination strategy toward eliciting robust and protective lung-resident T cell immunity against lethal respiratory viral infections. The finding offers new opportunities for universal influenza vaccine development and provides a versatile strategy against mutation-prone pandemic threats.
Results
Preparation and characterization of combinatorial nanoshell vaccines co-encapsulating peptides and CpG-ODN
To assess T cell-mediated antiviral effect and to examine T cell dynamics via lineage tracking, we prepared vaccines containing two different pairs of class I and II MHC-restricted antigenic peptides, either NP366–374 (NPI peptide)/NP311–325 (NPII peptide) or OVA257–264 (OVAI)/OVA323–339 (OVAII). NPI/NPII peptides were derived from the authentic influenza viral NP protein; OVAI/OVAII peptides from the model antigen ovalbumin (OVA) were adopted to monitor antigen-specific T cells in vivo in conjunction with clonotypic OT-I and OT-II T cells after adoptive T cell transfer. Building on our previous report that integrates CpG-ODN adjuvant (CpG) for boosting antiviral T cells,13 we prepared combinatorial nanoshell vaccines co-encapsulating CpG with either pair of antigenic peptides (Figure 1A). The desired encapsulant mixtures were first prepared in 200 mM sodium bicarbonate, which was subsequently used as the inner aqueous phase for nanoshell preparation. Using an optimized double emulsion protocol previously shown to readily encapsulate aqueous cargoes dissolved in the inner aqueous phase,32, 33, 34 hollow nanoparticles possessing a shell ~10 nm in thickness and a unimodal size distribution with large aqueous inner cores were consistently prepared (Figures 1B–1D). High-efficiency peptide and CpG encapsulation was confirmed with high-performance liquid chromatography (HPLC) (Figures S1A–S1C; Table S1), which showed that the solubilized peptides and CpG were readily partitioned into the virus-sized hollow nanoparticles at ~50% efficiency. Successful cargo encapsulation was also visually affirmed under cryoelectronic microscopy (cryoEM), which revealed a prominent grainy texture indicative of concentrated biologic content inside an NPI/NPII/CpG nanoshell (NS(NPI/II + CpG)) (Figure 1C) or an OVAI/OVAII/CpG nanoshell (NS(OVAI/II + CpG)) (Figure S1D). The physicochemical properties of the nanoshells were largely similar for the different combinatorial encapsulants used in the present study (Table S1). Immediately after the nanoshell preparation, the nanoformulations were stored at −20°C and thawed right before use. One milligram of the nanoparticles contains ~10 μg of OVAI/NPI peptides, 10 μg of OVAII/NPII peptides, and/or 5 μg of CpG, which corresponds to ~9,000 peptides and/or 450 CpG per nanoshell based on particle concentration measurement by nanoparticle tracking analysis (Figure S1E).
Figure 1.

Induction of robust antigen-specific T cell responses with minimal systemic adverse effects by combinatorial nanoshell vaccines
(A) Schematic representation of a combinatorial nanoshell vaccine containing MHC class I and class II- restricted peptide antigens and CpG. (B and C) CryoEM visualizations of empty PLGA nanoshells (B) and peptide- and CpG-loaded nanoshells (C). (D) Dynamic light scattering measurement of nanoparticles loaded with OVAI/II peptides and CpG (NS(OVAI/II + CpG)). (E) IFN-γ production of CD8+ and CD4+ cells in mice receiving different immunization formulas. WT (Thy1.2) mice were s.c. immunized with PBS control, empty nanoshell control, different doses of simple mixture of NPI/II (L: 5 μg/5 μg), NPI/II (H: 10 μg/10 μg), NPI/II + CpG (L: 5 μg/5 μg + 2.5 μg), and NPI/II + CpG (H: 10 μg/10 μg + 50 μg), and indicated doses of NS (NPI/II + CpG). At day 7 post immunization, mice were sacrificed for analysis of IFN-γ production of CD8+ and CD4+ cells in the spleen and lymph node (n ≥ 5 mice per group compiled from 2 independent experiments). ∗∗∗∗p ≤ 0.0001 (one-way ANOVA). (F) The proportional body weight changes of recipient WT mice were monitored at the indicated days post immunization. ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001 (Student’s t test). (G and H) At day 7 post-harvested spleens were measured for organ weight (G) and were also photographed (H). Data were pooled from 2~3 independent experiments. Individual organ weight of immunized mice for spleens with means plus SE (n ≥ 4 mice per group). ∗∗∗p ≤ 0.001 (one-way ANOVA).
Combinatorial nanoshell peptide vaccines with CpG induce robust antigen-specific T cell responses without systemic inflammatory effects
To evaluate the antigen-specific CD4+ and CD8+ T cell responses induced by the combinatorial nanoshell peptide vaccines, we subcutaneously (s.c.) immunized C57BL/6 mice with titrated doses of nanoshells, ranging from 20 μg to 500 μg, that co-encapsulated NPI/NPII peptides and CpG (NS(NPI/NPII + CpG)). For comparison purposes, mice were immunized with NPI/NPII peptide nanoshell vaccine without CpG NS(NPI/NPII) or with the simple mixture (no nanoshell carrier) of NPI/NPII peptides and CpG (NPI/NPII + CpG) either in a lower dose (5 μg/5 μg NPI/NPII peptides and 2.5 μg CpG) equivalent to that in 500 μg of NS(NPI/NPII + CpG) or in a higher dose (10 μg/10 μg NPI/NPII peptides and 50 μg CpG) that was a minimal peptide/adjuvant content required for T cell stimulation as assessed in our prior study.13 On day 7 after immunization, analysis of cells in the spleens and inguinal draining lymph nodes (dLNs) of vaccinated mice by flow cytometry showed that NS(NPI/NPII + CpG) induced robust interferon (IFN)-γ production in a dose-dependent manner. Moreover, 500 μg of NS(NPI/NPII + CpG) induced significantly higher IFN-γ production than 500 μg of NS(NPI/NPII) and the simple mixture NPI/NPII + CpG in both doses, highlighting the enhanced immunogenicity by CpG and nanoshell formulation (Figure 1E). The immunogenicity of NS(OVAI/II + CpG) was assessed in Thy1.2+/+ mice adoptively transferred with carboxyfluorescein succinimidyl ester (CFSE)-stained Thy1.1+/+ OT-I and Thy1.1/Thy1.2 OT-II T cells (Figure S2A). Similarly, we observed that NS(OVAI/II + CpG) induced prominent levels of proliferating and IFN-γ-producing CD4+ OT-II T cells in both spleens and dLNs and IFN-γ-producing CD8+ OT-I T cells in spleens (Figures S2B–S2E). Notably, no obvious weight loss was observed in all nanoshell-vaccinated mice, whereas significant weight loss was observed in mice administered the simple mixture OVAI/II + CpG (Figure 1F). Furthermore, nanoshell encapsulation was also shown to minimize the effect of spleen enlargement by CpG (Figures 1G and 1H). These results demonstrate that the nanoshell peptide vaccine with CpG exhibits excellent immunogenicity and allows for significant adjuvant dose sparing and reduction in undesirable reactogenicity for antiviral T cell priming.
Combinatorial nanoshell vaccines elicit near-sterilizing cross-protective T cell immunity against lethal IAV infection
Next, we assessed the antiviral protectivity of the combinatorial nanoshell peptide vaccines against influenza virus infection. We chose 500 μg of combinatorial nanoshell vaccines for the ensuing experiments because it exhibited the highest immunogenicity and negligible systemic reactogenicity. With the aim of inducing TRMs for effective antiviral protection, we implemented the peripheral-prime-local-boost (s.c./intranasal [i.n.]) strategy with a 28-day interval between priming and boosting (Figure 2A). Our preliminary result also showed that i.n. priming by nanoshell vaccine with combined MHC-I and MHC-II epitope peptides NS(NPI/II + CpG) induced significantly higher protective T cell immunity against influenza virus infection than priming by either MHC-I epitope NS(NPI + CpG) or MHC-II epitope NS(NPII + CpG) alone (Figure S3). Thus, through this s.c./i.n. prime-boost strategy, we compared the protectivity of NS(NPI/II + CpG) against a lethal dose of influenza PR8 strain with other control vaccine formulations, including empty nanoshell, NPI/II peptides alone, simple mixture of NPI/II plus CpG, and NS(NPI/II). In a stark contrast to control formulations and to the subdued protective effect typically exhibited by T cell peptide vaccines against influenza,38,39 we observed that immunization with s.c./i.n. NS(NPI/II + CpG) fully protected mice against the PR8 infection, with all the mice surviving and recovering with minimal body weight loss (Figures 2B and 2C). None of the control formulations, on the other hand, showed improvement in morbidity and mortality in the challenged mice. Very strikingly, the mice immunized with NS(NPI/II + CpG) exhibited undetectable viral loads on day 7 post infection, whereas all other groups showed high viral loads in lungs (>104 plaque-forming units [PFU] per lung) (Figure 2D). Viral kinetics analysis revealed rapid and profound suppression of replicating viruses in the lungs of mice receiving s.c./i.n. NS(NPI/II + CpG), but the lung viral loads of the control mice vaccinated by empty nanoshells remained largely steady through day 7 post infection (Figure 2E). We also found that immunization with s.c./i.n. NS(NPI/II + CpG) resulted in the highest IFN-γ-producing NPI/II-specific CD8+ and CD4+ T cells in lungs (Figure 2F) and in dLNs and spleens as well at 7 days post PR8 infection (Figure S4).
Figure 2.

The immunogenicity and protectivity of combinatorial nanoshells with NP366–374/NP311–325
(A) Schematic illustration of vaccination strategy with NS(NPI/II + CpG) to induce protective T cell immunity against heterosubtypic IAV infection. Mice were immunized by s.c. priming (500 μg/mouse) and i.n. boosting (300 μg/mouse) with the indicated vaccine formulas and then infected by different strains of influenza virus (PR8, WSN, HKx31, and VNHA,NA (H5N1)). (B and C) The body weight (B) and survival rates (C) of PR8-infected (110 PFU) mice immunized by indicated vaccine formulas. A body weight reduction of 25% or more was defined as the experimental end point, and mice were then euthanized at day 7 post infection. (D) Lung viral loads of mice immunized by different vaccine formulas were analyzed at day 7 post PR8 infection (n ≥ 4 mice per group compiled from 2 independent experiments). (E and F) The viral kinetics (E) and T cell immunity (F) of nanoshell vaccine-immunized mice after challenge by lethal PR8 infection. (F) Individual percentages with means plus SE of NPI-specific and NPII-specific IFN-γ-producing CD8+ and CD4+ T cells, respectively, in lungs at day 7 post-PR8 infection (n ≥ 5 mice per group compiled from 2 independent experiments). (G–I) Mice were immunized by s.c. priming and i.n. boosting with the either empty nanoshells or NS(NPI/II + CpG) and then infected by lethal doses of WSN (1 × 104 PFU/mouse) (G), HKx31 (1 × 105 PFU/mouse) (H), or VNHA,NA (H5N1) (400 PFU/mouse) (I) following the experimental protocol shown in (A). Survival rates (top), body weight loss (middle), and lung viral loads (bottom) were analyzed at day 7 post infection of WSN (G), HKx31 (H), and VNHA,NA (H5N1) (I) (n ≥ 5 mice per group compiled from 2 independent experiments). ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001 (Fisher’s exact test for survival rate, Student’s t test for viral kinetics, and one-way ANOVA for lung viral load and percentages of IFN-γ production).
Given the promising protectivity of NS(NPI/II + CpG) against PR8, we further examined the vaccine’s protective effect against different viral strains and subtypes. The antigenic peptides NPI and NPII are conserved among a number of IAV strains, including PR8 (H1N1), WSN (H1N1), HKx31 (H3N2), and VNHA,NA (H5N1). Of note, both HKx31 and VNHA,NA are recombinant influenza viruses with identical internal genes derived from the PR8 but with surface HA and NA proteins from A/Aichi/2/68 (H3N2) and A/Vietnam/1203/04 (H5N1), respectively.40 Mice receiving s.c./i.n. NS(NPI/II + CpG) vaccination also exhibited 100% survival after lethal challenges with WSN, HKx31, or VNHA,NA and had undetectable viral loads on day 7 post infection (Figures 2G–2I), whereas all mice immunized by empty nanoshells died by day 8. These results highlight the feasibility of T cell-based antiviral peptide vaccine with broadly neutralizing potential using the nanoshell platform.
Efficient uptake and transport of combinatorial nanoshell vaccines by CD11c-positive dendritic cells (DCs)
Intrigued by the prominent T cell activation and antiviral protectivity by the nanoshell vaccines, we further explored their cellular uptake and pulmonary transport in vivo by incorporating a water-soluble tracking dye, Alexa Fluor 555 (AF555). For ease of T cell tracking by lineage marker in subsequent studies, nanoshell vaccines incorporating OT-I and OT-II peptides (OVAI/II) were adopted from this point onward. Subcutaneously administered nanoshells readily drained to the dLNs, as is the case typically observed with delivery carriers of similar dimensions (Figure S5).34 As the pulmonary transport of nanocarriers is less commonly studied, we further explored the fate of the nanoshells upon i.n. administration, after which the cellular uptake of nanoshells was determined by analysis of fluorescent cells isolated from the lungs and dLNs at 12, 24, and 48 h post immunization. We found that in the lungs nanoshells were taken by a significant portion of SSChighCD11c+F4/80+ macrophages and SSClowCD11c+ conventional DCs (cDCs), including CD103+CD11b− and CD103−CD11b+ cDCs (Figures S6A and S6B). Uptake of nanoshells by macrophages and DCs peaked at 24 h post immunization, and, notably, CpG adjuvant significantly increased the uptake of nanoshells by CD103+CD11b− cDCs (29.0% versus 46.4%, p < 0.01) and CD103−CD11b+ cDCs (51.0% versus 65.3%, p < 0.05) but not by macrophages (78.3% versus 82.8%, p > 0.05) (Figure 3A). We then measured the expression of CD86, an activation and maturation marker of DC, in nanoshell-loaded DCs (AF555+ DCs). We found that CpG adjuvant enhanced the expression of CD86 in CD103+CD11b− cDCs at 24 h post immunization and in CD103−CD11b+ cDCs at 24 and 48 h post immunization (Figures 3B and 3C). Upon tracking the nanoshell-loaded (AF555+) cells in the dLNs, only AF555+CD11c+ DCs could be observed and no AF555+F4/80+ macrophages were found. In addition, CD11c+ DCs of dLNs in mice immunized by NS(OVAI/II + CpG + AF555) had significantly higher green fluorescence intensity of AF555 than those in mice immunized by NS(OVAI/II + AF555), whereas CD11c− cells exhibited low green fluorescence intensity (Figures 3D and 3E). Collectively, the data show that a high proportion of macrophages and DCs effectively took nanoshells in the lungs, but only CD11c+ DCs migrated to the dLNs. Furthermore, CpG enhanced the uptake of nanoshells and maturation of DCs.
Figure 3.

Uptake and tracking of nanoshell vaccines in lungs and dLNs
(A) The percentages of macrophages (SSCHighCD11c+MHC-IILowF4/80+) and DCs (SSCLowCD11c+MHC-IIHighCD103+ and SSCLowCD11c+MHC-IIHighCD11b+) in lungs at 12 h, 24 h, and 48 h after i.n. administration of nanoparticles (n = 4 mice per group compiled from 2 independent experiments). (B) t-distributed stochastic neighbor embedding (t-SNE) map of different subset of DCs (AF555+) colored by FlowSOM metaclusters in lungs at 24 h. Data are downsampled to 1 × 104 cells/mouse (from 3 mice/group), and representative heatmap statistic is 1 mouse per group. The right and middle t-SNE maps were gated by AF555+ cells. The color bar represents the expression levels of CD86 in NS-positive (AF555+) cells. (C) The percentages of CD86+ cells in NS-positive (AF555+) CD11c+CD103+ and CD11c+CD11b+ DCs of lungs at 12 h, 24 h, and 48 h after i.n. administration of nanoparticles (n ≥ 3 mice per group compiled from 2 independent experiments). (D) t-SNE map of different subset of DCs (AF555+) colored by FlowSOM metaclusters for dLNs. Data are downsampled to 1 × 104 cells/mouse (from 3 mice/group, contour), and representative heatmap statistic is 1 mouse per group. Color bar means the proportion of NS-positive cells. (E) The mean fluorescent (AF555+) intensity (MFI) of NS-positive (AF555+) CD11c− and CD11c+ cells of dLNs at 12 h, 24 h, and 48 h after i.n. administration of nanoparticles (n ≥ 3 mice per group compiled from 2 independent experiments). ∗p ≤ 0.05; ∗∗p ≤ 0.01 (Student’s t test).
CD11c+ DCs are critical for activation of T cells by combinatorial nanoshell vaccines
To validate the role of CD11c+ DCs in stimulating antigen-specific T cells by the combinatorial nanoshell vaccines in lungs, we utilized CD11c-DTR mice, in which CD11c+ cells, primarily DCs and some macrophages, can be specifically depleted by addition of diphtheria toxin (DT). Naive Thy1.2+/+ CD11c-DTR mice were treated with DT once via intraperitoneal (i.p.) administration and then adoptively transferred with CFSE-stained Thy1.1+/+ OT-I and Thy1.1/Thy1.2 OT-II T cells on the next day. Subsequently, mice were i.n. instilled with NS(OVAI/II + CpG) and sacrificed for analysis 3 days later (Figure 4A). We observed that DT treatment resulted in significant reduction of CD11c+ cells in the lungs and the dLNs (Figure 4B; Figure S7A). Depletion of CD11c+ cells dramatically attenuated the proliferation of antigen-specific CD4+ OT-II and CD8+ OT-I T cells (Figure 4C; Figure S6B) and caused a 2-log decrease of their cell numbers (Figure 4D). Taken together, the results indicate that CD11c+ DCs were the main antigen-presenting cells (APCs) responsible for activating antigen-specific T cells by the combinatorial nanoshell vaccines and echo recent efforts in vaccination modification strategies specifically tailored to facilitate CD11c+ DC-mediated T cell induction.41,42
Figure 4.

T cell proliferation stimulated by combinatorial nanoshell vaccine after depletion of CD11c-positive DCs
(A) Schematic representation of the experimental protocol. CD11c-DTR mice received PBS or DT depletion once. On the next day mice were co-transferred with CFSE-stained Thy1.1+/+ CD8+OT-I and Thy1.1/Thy1.2 CD4+OT-II cells and then i.n. immunized with NS (OVAI/II + CpG). (B) The fold change of CD11c+ cells in immunized mice at day 5 post PBS or DT treatment. (C and D) Individual percentages (C) and cell numbers (D) of proliferating CD4+OT-II and CD8+OT-I cells in dLNs in immunized mice at day 5 post PBS or DT treatment. (n ≥ 6 mice per group compiled from 3 experiments). ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001 (Student’s t test).
Elicitation of robust lung-resident and circulatory memory T cells by the peripheral-prime-local-boost vaccination strategy
TRM cells in lungs are essential for the first-line defense against respiratory viral infections, and vaccine administration through the i.n. route is considered critical to generation of tissue-resident memory T cells. However, several studies have reported that TRM replenishment is mediated by TEMs, which is most effectively stimulated via parenteral vaccination.43 Intrigued by the influence of and with the aim of optimizing vaccination routes for TRM induction, we set out to enumerate epitope-specific TRM cells in vivo, following different vaccination schemes with the nanoshell vaccine. We first compared the protective efficacy of NS(OVAI/II + CpG) vaccine primed and boosted via s.c./i.n., i.n./i.n., or s.c./s.c. vaccination routes in wild-type (WT) mice in the absence of adoptive transfer. Four weeks after boosting, mice were challenged by i.n. instillation with a lethal dose of HKx31-OVAI/II (Figure 5A). Mice that were immunized with NS(OVAI/II + CpG) by s.c. priming and i.n. boosting exhibited less weight loss (Figure 5B) and the best survival outcome, with no observed mortality (Figure 5C), and mice receiving i.n./i.n. priming and boosting with NS(OVAI/II + CpG) had the second-best survival outcome. Notably, despite the high immunogenicity of the NS(OVAI/II + CpG), s.c./s.c. immunization conferred very limited antiviral protectivity. Consistent with their improved mortality and morbidity, the s.c./i.n. and i.n./i.n. NS(OVAI/II + CpG) groups had the lowest lung viral loads upon examination at day 5 post infection (Figure 5D). In addition, mice in the s.c./i.n. NS(OVAI/II + CpG) group showed the strongest CD8+ and CD4+ T cell responses in both spleens and mediastinal dLNs at day 5 post infection (Figure 5E). Collectively, the results demonstrate that the local (i.n.) boosting strategy is critical for the induction of protective T cell immunity against IAV infection.
Figure 5.

The immunogenicity and protectivity of combinatorial nanoshell vaccines by different vaccination strategies
(A) Schematic illustration of the experimental protocol. C57BL/6 mice received primary and secondary immunization by the indicated vaccine formulations and routes in a 28-day interval. The doses used are as follows: s.c. (500 μg of nanoshells) and i.n. (300 μg of nanoshells). (B and C) Mice were then infected with 5 × 105 PFU of HKx31-HA-OVAI/II at day 56 post primary immunization (28 days after secondary immunization) and monitored for the body weight loss (B) and survival rate (C). (D) Lung viral loads were analyzed at day 5 post-HKx31-HA-OVAI/II infection. Data are individual viral loads with means plus SE (n ≥ 5 mice per group compiled from 3 independent experiments). (E) Individual percentages of virus-specific IFN-γ-producing CD4+ and CD8+ T cells in spleens and dLNs with means plus SE at day 5 post-HKx31-HA-OVAI/II infection (n ≥ 5 mice per group compiled from 3 independent experiments). ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001 (log-rank test for the survival rate and one-way ANOVA for lung viral loads and percentages of IFN-γ production).
As both s.c./i.n. and i.n./i.n. immunizations with combinatorial nanoshell vaccines conferred more effective antiviral immunity compared with the s.c./s.c. vaccination strategy, we further investigated how peripheral-prime-local-boost strategy may influence the circulatory and lung-resident T cell populations. To measure the antigen-specific memory T cells, naive Thy1.1+OT-I CD8+ T cells isolated from splenocytes were adoptively transferred to WT Thy1.2+ C57BL/6 mice, which were subsequently immunized by i.n./i.n. NS(OVAI/II + CpG) or s.c./i.n. NS(OVAI/II + CpG). One month after boosting, subpopulations of memory T cells, including TCMs, TEMs, and TRMs, were analyzed by flow cytometry (Figure 6A). Based on the expression of KLRG and CD62L, TCM cells were defined as KLRGlowCD62high and TEM cells were KLRGhighCD62Llow (Figure S8A), and TRM cells were determined by in vivo staining through the expression of CD69 or CD103 (Figure S8B).44 Our analysis showed that although the two groups had similar numbers of OT-I TRM cells in lungs, mice that were immunized via the s.c./i.n. route generated significantly more TEM and TCM cells in the spleens than mice with i.n./i.n. vaccination (Figure 6B). We further examined the influence of elevated circulatory T cell response on TRM dynamics by depleting lung-resident CD8+ T cells via intratracheal (i.t.) administration of anti-CD8 antibody, based on a protocol that can solely deplete lung-resident CD8+ T cells without affecting circulatory CD8+ T cells.45 Our pilot experiment showed that this protocol could deplete >90% lung-resident CD8+ T cells effectively without significantly affecting the circulatory CD8+ T cells (data not shown). Seven days post CD8+ TRM depletion on day 28 after vaccine boosting, mice immunized by the s.c./i.n. route exhibited significantly more TRM cells in lungs compared with mice immunized by the i.n./i.n. route (Figure 6C). Interestingly, despite having higher levels of TEMs prior to TRM depletion, mice receiving s.c./i.n. immunization showed similar numbers of TEM cells in spleens compared with those receiving i.n./i.n. immunization following TRM depletion (Figure 6C). The more efficient TRM recovery as well as the larger TEM differential observed for the s.c./i.n. group in the depletion study strengthens the correlation between TEM levels and their conversion to TRM cells. Collectively, although i.n./i.n. and s.c./i.n. immunizations induced similar levels of TRM cells in the lungs 1 month after booster vaccination, the enhanced antiviral protectivity, elevated circulatory TEM cell population, and better TRM replenishment in lungs support s.c./i.n. prime-boost as a superior vaccination strategy.
Figure 6.

Elicitation of memory T cell populations by nanoshell vaccines with different vaccination strategies
(A) WT (Thy1.2) mice were transferred with naive Thy1.1+CD8+OT-I cells 1 day before immunization and immunized by the indicated individual protocols. Memory T cells were analyzed at 28 days post secondary immunization. (B) Lung samples were gated on Thy1.1+CD3e−CD8+CD62L−KLRG-1− cells and analyzed for the total number of CD69+, CD103+, and CD69+CD103+ cells (n ≥ 7 mice per group compiled from 3 independent experiments). Spleen samples were analyzed for the total number of TEM (CD62L−KLRG-1+) and TCM (CD62L+KLRG-1−) cells (n ≥ 7 mice per group compiled from 3 independent experiments). (C) TRM cells in lungs and TEM cells in spleens were measured after depletion of lung-resident CD8+ T cells by intratracheal administration with anti-CD8 antibody (n ≥ 5 mice per group compiled from 2 independent experiments) ∗p ≤ 0.05; ∗∗p ≤ 0.01 (Student’s t test).
Discussion
Non-viral vector peptide vaccines intended to elicit T cell immunity against viral infections have generally generated disappointing levels of protection,25,26 and, to the best of our knowledge, an effective T cell peptide vaccine capable of conferring potent cross-strain influenza protection has not been previously reported. In this study, we demonstrated that a combinatorial nanoshell vaccine co-encapsulating a single pair of class I and class II MHC-restricted epitope peptides and CpG, when given by peripheral s.c. priming and local i.n. boosting, was able to induce near-sterilizing antigen-specific lung-resident and circulatory T cell immunity, which in turn conferred cross-protection against lethal IAVs of different strains (PR8, WSN, HKx31, and VNHA,NA) and subtypes (H1N1, H3N2, and H5N1). Mice immunized by nanoshells with NP366–374/NP311–325 suppressed viral replication by ~1,000-fold at day 5 and reduced lung viral loads to undetectable levels by day 7 post infection (>100,000-fold less). The vaccination strategy that elicits protective T cell immunity against IAV infection is summarized and depicted in Figure 7, and both magnitude and location of antigen-specific T cells were found to be critical to antiviral protectivity. The study demonstrates the viability of peptide-based T cell vaccines for cross-reactive influenza protection, highlighting key insights in formulation design, delivery strategy, vaccination routes, and mechanistic basis for optimizing T cell immunity against IAV infections.
Figure 7.

Schematic summary of the cross-protective T cell immunity against IAVs elicited by the combinatorial nanoshell vaccine
After peripheral subcutaneous priming, intranasal boosting with the combinatorial nanoshell vaccine containing two class I and class II MHC-restricted epitope peptides (i.e., NPI/II) and CpG-ODN led to efficient uptake and maturation of pulmonary CD11c+ DCs. Mature CD11c+ DCs subsequently migrate to draining lymph nodes, where they present epitope peptides and stimulate cognate T cells recruited from the circulatory memory T cell population. Finally, lung-resident memory T cells and circulatory memory T cells, including TCMs and TEMs, are established and act synergistically to provide near-sterilizing cross-protection against IAVs of different strains and subtypes.
Poly(dl-lactide-co-glycolide) (PLGA) is a commonly used biomaterial for vaccine development owing to its biocompatibility and synthetic flexibility.46, 47, 48, 49 To maximize antiviral T cell immunity, T cell epitope peptides and adjuvants were co-encapsulated in PLGA-based hollow nanostructures with an average diameter of 160 nm, yielding vaccine particles with close structural semblance to natural virions. The particle formulation allows for modular antigen and adjuvant incorporation with consistent encapsulation efficiencies, and the ability to adjust nanoshell composition enables interrogation of the TRM induction mechanism. We showed that both antigen and adjuvant were conducive to TRM establishment during i.n. boosting, and through tracking with fluorescent dye-loaded nanoshells, their pulmonary transport dynamic was further elucidated. The nanoshells were efficiently taken up by CD11c+ macrophages and DCs, and nanoshell-associated CD11c+CD103+ and CD11c+CD11b+ DCs were readily identified in the dLNs. This transport pattern is strikingly similar to that of viral particles following pulmonary infections and informs design considerations for TRM-inducing delivery vehicles.50,51 We also observed that specific depletion of CD11c+ cells by DT dramatically reduced the proliferation of T cells stimulated by nanoshell vaccines, supporting that CD11c+ DCs were responsible for the priming activity of the nanoshell vaccines in lungs.52, 53, 54 It is noteworthy to point out that even though empty nanoshells and CpG-loaded nanoshells share similar physicochemical properties, incorporation of CpG was observed to increase DC uptake. With incorporation of DC-targeting ligands frequently employed to enhance vaccine potency,41,42 this finding highlights that the inherent immunogenicity of nanoparticles can be tuned to favor their cellular distributions to DCs.
With the quantity and persistence of pulmonary TRM cells being critical factors that can influence antiviral protectivity,2,5,6 the vaccination route is among the many important factors toward optimizing TRM establishment.55, 56, 57 We assessed several immunization strategies and demonstrated that local boosting was critical for the establishment of robust TRM and protectivity against IAV. By comparing i.n./i.n. and s.c./i.n. prime-boost vaccinations, we observed a correlation between the heightened TEM levels in the s.c./i.n. group and improved pulmonary TRM replenishment and antiviral protectivity. This finding is critical for development of vaccination regimens for reliable antiviral protectivity,4 and it also echoes reports that highlight circulatory TEM cells and parenteral immunization as major factors promoting pulmonary TRM durability and replenishment.43,58 As the dynamics and factors for TRM establishment remain ongoing topics of investigation, future studies with additional nanoshell compositions are warranted.
The prominent antiviral efficacy against lethal influenza challenge observed in the present work is highly encouraging and opens up the exciting prospect of nanoshell-based TRM vaccine for broadly neutralizing influenza vaccination as well as for other respiratory infections. Antigenic peptide also holds several advantages over recombinant protein for its vastly easier synthesis process and convenient scalability to effectively cope with emerging and re-emerging infectious diseases. To realize the vision of clinical translation, identification of influenza epitope peptides presented by common human leukocyte antigen alleles is required to achieve wide population coverage. Such efforts can be facilitated by the advances in modern technology for T cell epitope identification, including the use of epitope prediction algorithms and mass spectroscopy immunopeptidomics.59,60 Encouragingly, highly conserved CD8+ T cell epitope peptides across influenza A, B, and C viruses have been identified,19 lending plausibility to T cell-based universal influenza vaccination. Of note, a high level of cross-reactivity between severe acute respiratory syndrome (SARS)-NP induced T cells and SARS coronavirus 2-NP has also been reported,37 suggesting the feasibility of a pan-coronavirus vaccine based on TRM induction against conserved T cell epitopes. The combinatorial polymeric nanoshell provides a versatile platform that can be iteratively optimized to maximize epitope coverage and immune stimulation. It is also anticipated to bridge the gap between the exciting frontier of TRM research and its practical utility in disease prevention.
Materials and methods
Mice
All mouse experiment protocols were approved by the Laboratory Animal Committee of National Taiwan University College of Medicine. C57BL/6 (B6) WT mice came from the National Laboratory Animal Center in Taiwan. The breeder pairs of Foxp3gfp were a gift from Dr. Alexander Rudensky (New York, NY, USA). CD11c-DTR mice were a gift from Dr. Ping-Ning Hsu (National Taiwan University) and were maintained in the National Laboratory Animal Center in Taiwan. Thy1.1 × OT-I and Thy1.1/Thy1.2 × Foxp3gfp × OT-II mice were generated by cross-breeding the indicated mouse lines in a B6 background by ourselves and were maintained in the Animal Center of National Taiwan University College of Medicine.
Viruses, infection, and virus quantification
The plasmids hemagglutinin (HA) and neuraminidase (NA) of HKx31 cloned in pHW2000 were a gift from Dr. Paul Thomas (Memphis, TN, USA). WSN was a gift from Chi-Ju Chen (National Yang-Ming University, Taiwan). The VNHA,NA virus was kindly provided by Richard Webby, with its multibasic HA cleavage site replaced by that of a low-pathogenicity avian influenza virus.61 The HKx31-HA-OVAI/II was generated by a strategy described previously.13 Briefly, we first modified the OVA OT-I/OT-II peptide (OVA257–264/OVA323–339) by adding SR (XbaI site), GT (KpnI site), and GGGGS linker to generate SR-SIINFEKL/ISQAVHAAHAEINEAGRGT-GGGS and then inserted the sequence into HKx31-HA after the glycine at residue 17. All viruses, including HKx31-HA-OVAI/II, PR8, HKx31, and WSN, were produced by the 8-plasmid reverse genetics system and propagated in Madin-Darby canine kidney (MDCK) cell monolayers, and the titers were determined by the plaque assay. Six- to eight-week-old mice were fully anesthetized by i.p. injection of Rompun (xylazine) and Zoletil (tiletamine hydrochloride and zolazepam hydrochloride) and then infected with 20 μL of viral suspension with indicated doses of viruses via the i.n. route. For measuring the survival rate of mice infected by influenza virus, mice were humanely euthanized when they reached 25%–30% loss of their initial body weight. For measurement of lung viral titer of infected mice, lungs were isolated and homogenized in 1 mL of infection medium consisting of DMEM with non-essential amino acids solution (NEAA), sodium pyruvate, and bovine serum albumin. Lung viral loads were determined by inoculation of MDCK cells with serial 10-fold dilutions of the lung suspension with the plaque assay.
Preparation of combinatorial nanoparticle vaccines
Combinatorial nanoparticle vaccines were prepared by a water-in-oil-in-water double emulsion process adapted from a previously reported method.32 A polymer solution was prepared by dissolving 50 mg/mL of carboxy-terminated, 50:50 PLGA (0.15–0.25 dL/g, corresponding to ~10,000 Da; LACTEL Absorbable Polymers) in dichloromethane. The inner aqueous phase was prepared by dissolving the desired peptide mixtures, CpG-ODN 1826 (InvivoGen), and/or Alexa Fluor 555 Cadaverine dye (Sigma-Aldrich) in 200 mM sodium bicarbonate solution to enhance peptide and CpG-ODN solubility. For the different combinatorial nanoparticles, the inner aqueous phases contained 10 mg/mL of each designated peptide (i.e., OVA257–264 [OVAI peptide] [SIINFEKL], OVA323–339 [OVAII peptide] [ISQAVHAAHAEINEAGR], NP366–374 [NPI peptide] [ASNENMETM], and NP311–325 [NPII peptide] [QVYSLIRPNENPAHK]), 5 mg/mL of CpG-ODN, and/or 0.1 mg/mL of AF555. No precipitate was observed in any of the combinatorial formulations prior to nanoparticle preparation. For a typical nanoparticle preparation, 50 μL of aqueous solution containing the desired encapsulants was emulsified in 500 μL of polymer solution in ice with an Ultrasonic Probe Sonicator in the pulse mode with 40% amplitude and on-off durations of 1 and 2 s for 1 min. The first emulsion was subsequently added to 5 mL of 1 mM phosphate buffer (pH 7), which was then probe sonicated at 30% amplitude with on-off durations of 1 and 2 s for 2 min. The emulsion was subsequently poured into 8 mL of water and heated at 40°C under gentle stirring in a fume hood for solvent evaporation. After 1 h of solvent evaporation, the nanoparticles were collected using 100 kDa molecular weight cut off (MWCO) Amicon filters (Sigma-Aldrich) to remove unencapsulated materials. The collected nanoparticles were evaluated with dynamic light scattering, nanoparticle tracking analysis, cryoEM, and HPLC for physicochemical properties, particle concentration, particle morphology, and encapsulation efficiency, respectively. The nanoparticles were then suspended in 10 mM disodium phosphate and 10% sucrose prior to storage at −20°C. Prior to each immunization study, the nanoparticles were thawed in a 37°C water bath and diluted to desired concentrations with PBS.
Quantification of peptide and CpG-ODN loading by HPLC analysis
Trifluoroacetic acid (TFA) was purchased from Alfa Aesar (Heysham, UK). Acetonitrile was purchased from J.T. Baker (Avantor Performance Materials, Center Valley, PA, USA). Glacial acetic acid was purchased from Macron Fine Chemicals (Avantor Performance Materials, Center Valley, PA, USA). Triethylamine was purchased from Sigma-Aldrich (St. Louis, MO, USA). All reagents and chemicals were analytical grade. The HPLC analysis was carried out in an Agilent Technologies Series 1100 apparatus (Waldbronn, Germany). The analytical column was an Ascentis Express C18 reverse-phase column (Supelco). The column temperature was kept at 25°C during the quantification. For peptide antigen quantification, the mobile phase consisted of phase A (0.1% TFA in acetonitrile) and phase B (0.1% TFA in water). The samples were run with a linear gradient elution from 10% to 95% of phase A over 15 min and then kept constant at 95% phase A for 5 min. Then, the eluent was reversed to the initial composition within 5 min. The wavelength of detection was set at 220 nm for OVAI and OVAII peptides, and the flow rate was set at 1.0 mL/min. NPI and NPII peptides were measured at 215 nm and with a flow rate of 0.75 mL/min. For CpG-ODN quantification, the mobile phase A consisted of 5.70 mL of glacial acetic acid and 13.90 mL of triethylamine mixed with Milli-Q water, which yielded a 0.1 M triethylammonium acetate buffer. The pH was adjusted to 7. The mobile phase B was acetonitrile. The mobile phases A and B were run at a 50:50 volume ratio during elution. CpG-ODN was measured at 260 nm and with a flow rate of 0.7 mL/min.
Reagents, cell staining, and flow cytometry analysis
Cells were washed twice with staining buffer (PBS containing 2% fetal bovine serum [FBS]) and stained with fluorescence-conjugated antibodies against surface markers wherever appropriate, including anti-Thy1.1 BV510 clone OX-7 (BioLegend), anti-CD4 PerCP-Cy5.5 clone RM4-5 (eBioscience), anti-CD8α PE-Cy7 clone 53-6.7 (eBioscience), anti-CD44 BV650 clone IM7 (BioLegend), anti-CD69 PE clone H1.2F3 (eBioscience), anti-CD103 BV421 clone 2E7 (BioLegend), anti-CD62L BUV737 clone MEL-14 (BD Biosciences), anti-KLRG-1 BUV395 clone 2F1 (BD Biosciences), anti-CD11c BB515 clone N418 (BD Biosciences), anti-CD11b BV711 clone M1/70 (BD Biosciences), anti-MHC-II, anti-CD86, anti-CD103, and anti-F4/80 for 30 min. For intracellular IFN-γ staining, cells were fixed and permeabilized (Foxp3/Transcription Factor Fixation/Permeabilization Concentrate and Diluent, eBioscience) after surface staining and then stained intracellularly with anti-IFN-γ-APC (BD Biosciences). Cells were analyzed with a fluorescence-activated cell sorting (FACS) LSRFortessa flow cytometer (BD Biosciences).
Adoptive transfer of naive OT-I and OT-II T cells
Splenocytes from indicated donor mice were prepared as a single-cell suspension and FACS sorted for CD8+ OT-I cells and CD4+OT-II+CD25−GFP− cells, which were adjusted to an appropriate final volume with sterile serum-free RPMI 1640 (Gibco). Each mouse received 0.3 mL of cell suspension with the OT-I cell number of 0.8–1 × 106 cells and OT-II cell number of 3 × 106 cells. Donor cells were transferred into recipient mice by intravenous (i.v.) injection through the tail veins.
Isolation of DCs from the lung and lymph node
NS (OVAI/II + AF555)- or NS (OVAI/II + CpG + AF555)-treated mice were sacrificed and received systemic PBS perfusion to exclude red blood cells (RBCs), and lung or dLN samples were homogenized by scissors into 1 mm3 sections. Samples were then treated with digestion buffer (0.5 mg/mL collagenase type IV, 25 U/mL type IV DNase I in RPMI 1640 supplemented with 1% glutamine-penicillin-streptomycin) at 37°C for 30 min. Afterward, the reaction was stopped by addition of RPMI 1640 supplemented with 10% FBS and 1% glutamine-penicillin-streptomycin. Further, lung and LN samples were treated to single cell by 18G needle and 1 mL pipette tips, respectively. Cells were treated with RBC lysis buffer (eBioscience) and washed twice with staining buffer (PBS containing 2% FBS) for further staining.
Depletion of CD11c cells and measurement of T cell stimulation in CD11c-DTR mice
CD11c-DTR mice were treated once with i.p. DT (4 ng/g) (Calbiochem) to deplete CD11c+ cells and on the next day were transferred with 1 × 106 CFSE+ OT-I cells and 3 × 106 CFSE+ OT-II cells isolated from Thy1.1 × OT-I and Thy1.1/Thy1.2 × Foxp3gfp × OT-II mice, respectively. One day later, mice were immunized with NS(OVAI/II + CpG) through the i.n. route and analyzed for CD11c+ cell population and the proliferation of donor OT-I and OT-II cells in spleens, lymph nodes, or lungs at day 3 post immunization.
Measurement of antigen-specific T cells
To analyze the level of OVAI/II- or NPI/II-specific T cells after influenza virus infection, 2–5 × 106 primary cells from spleens, lymph nodes, or lungs were resuspended in 200 μL of T cell culture medium containing OVA257–264 (20 μg/mL)/OVA323–339 (40 μg/mL) peptide or NP366-374 (10 μg/mL)/NP311-325 (40 μg/mL) peptide, interleukin (IL)-2 (2 ng/mL), and GolgiPlug (BD Biosciences). Cells were incubated at 37°C for 6 h and analyzed by intracellular IFN-γ staining.
In vivo staining of lung-resident T cells
Mice were injected intravenously with 3 μg of APC-conjugated anti-CD3 Ab clone 145-2C11 (eBioscience). After 10 min, mice were sacrificed and received systemic PBS perfusion to exclude RBCs, and lung samples were digested by collagenase. Afterward, cells were treated with RBC lysis buffer (eBioscience) and washed twice with staining buffer (PBS containing 2% FBS) for further staining.
In vivo lung depletion of CD8 T cells
Mice were given two shots of 60 μg of anti-CD8 Ab (53-6.7, Bio X Cell) per mouse by i.t. injection.
Statistical analyses
Data are expressed as mean ± standard error (SE) values. The survival rates were analyzed by the log-rank test and Fisher’s exact test. Continuous variables, including lung viral titers and percentage and numbers of antigen-specific T cells, DCs, and macrophages, were analyzed by the Student’s t test or one-way ANOVA test followed by Tukey’s multiple comparison test. A p value <0.05 was considered statistically significant.
Data and materials availability
All data associated with this study are present in the paper or the Supplemental information.
Acknowledgments
We acknowledge the service provided by the Flow Cytometric Analyzing and Sorting Core of the First Core Laboratory and the Third Core Laboratory, National Taiwan University College of Medicine. This work was supported by grants from the National Taiwan University Hospital (UN106-057 and 109C101-5 to H.-C.Y.) and Ministry of Science and Technology, Taiwan (MOST105-2628-B-002-027-MY3 to H.-C.Y., MOST107-2321-B-002-024 to H.-C.Y. and C.-M.J.H., and MOST107-2119-M-001-042 to C.-M.J.H.).
Author contributions
P.-H.L., H.-W.C., P.-J.C., C.-M.J.H., and H.-C.Y. formed the original concepts and designs of the experiments; B.-Y.Y. and C.-M.J.H. generated polymeric nanoshells; P.-H.L. and C.-Y.L. performed the experiments and C.-F.P., Y.-H.L., and Y.-S.H. helped with tissue collection; Y.-H.L. performed the HPLC experiments; L.-L.W helped with t-SNE data analysis; P.-H.L. and C.-Y.L. acquired and interpreted data; P.-J.C., C.-M.J.H., and H.-C.Y. reviewed and helped interpret the data; P.-H.L., C.-M.J.H., and H.-C.Y. drafted and finalized the submitted manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.03.010.
Contributor Information
Che-Ming Jack Hu, Email: chu@ibms.sinica.edu.tw.
Hung-Chih Yang, Email: hcyang88@ntu.edu.tw.
Supplemental information
References
- 1.Schenkel J.M., Masopust D. Tissue-resident memory T cells. Immunity. 2014;41:886–897. doi: 10.1016/j.immuni.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mueller S.N., Mackay L.K. Tissue-resident memory T cells: local specialists in immune defence. Nat. Rev. Immunol. 2016;16:79–89. doi: 10.1038/nri.2015.3. [DOI] [PubMed] [Google Scholar]
- 3.Schenkel J.M., Fraser K.A., Beura L.K., Pauken K.E., Vezys V., Masopust D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science. 2014;346:98–101. doi: 10.1126/science.1254536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu T., Hu Y., Lee Y.T., Bouchard K.R., Benechet A., Khanna K., Cauley L.S. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J. Leukoc. Biol. 2014;95:215–224. doi: 10.1189/jlb.0313180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zens K.D., Chen J.K., Farber D.L. Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight. 2016;1:85832. doi: 10.1172/jci.insight.85832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pizzolla A., Nguyen T.H.O., Smith J.M., Brooks A.G., Kedzieska K., Heath W.R., Reading P.C., Wakim L.M. Resident memory CD8+ T cells in the upper respiratory tract prevent pulmonary influenza virus infection. Sci. Immunol. 2017;2:eaam6970. doi: 10.1126/sciimmunol.aam6970. [DOI] [PubMed] [Google Scholar]
- 7.Beverley P.C., Sridhar S., Lalvani A., Tchilian E.Z. Harnessing local and systemic immunity for vaccines against tuberculosis. Mucosal Immunol. 2014;7:20–26. doi: 10.1038/mi.2013.99. [DOI] [PubMed] [Google Scholar]
- 8.Kinnear E., Lambert L., McDonald J.U., Cheeseman H.M., Caproni L.J., Tregoning J.S. Airway T cells protect against RSV infection in the absence of antibody. Mucosal Immunol. 2018;11:249–256. doi: 10.1038/mi.2017.46. [DOI] [PubMed] [Google Scholar]
- 9.Lipsitch M., Grad Y.H., Sette A., Crotty S. Cross-reactive memory T cells and herd immunity to SARS-CoV-2. Nat. Rev. Immunol. 2020;20:709–713. doi: 10.1038/s41577-020-00460-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Vries R.D., Rimmelzwaan G.F. Viral vector-based influenza vaccines. Hum. Vaccin. Immunother. 2016;12:2881–2901. doi: 10.1080/21645515.2016.1210729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng X., Oduro J.D., Boehme J.D., Borkner L., Ebensen T., Heise U., Gereke M., Pils M.C., Krmpotic A., Guzmán C.A. Mucosal CD8+ T cell responses induced by an MCMV based vaccine vector confer protection against influenza challenge. PLoS Pathog. 2019;15:e1008036. doi: 10.1371/journal.ppat.1008036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malonis R.J., Lai J.R., Vergnolle O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chem. Rev. 2020;120:3210–3229. doi: 10.1021/acs.chemrev.9b00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin P.H., Wong W.I., Wang Y.L., Hsieh M.P., Lu C.W., Liang C.Y., Jui S.H., Wu F.Y., Chen P.J., Yang H.C. Vaccine-induced antigen-specific regulatory T cells attenuate the antiviral immunity against acute influenza virus infection. Mucosal Immunol. 2018;11:1239–1253. doi: 10.1038/s41385-018-0004-9. [DOI] [PubMed] [Google Scholar]
- 14.Knight F.C., Gilchuk P., Kumar A., Becker K.W., Sevimli S., Jacobson M.E., Suryadevara N., Wang-Bishop L., Boyd K.L., Crowe J.E., Jr. Mucosal Immunization with a pH-Responsive Nanoparticle Vaccine Induces Protective CD8+ Lung-Resident Memory T Cells. ACS Nano. 2019;13:10939–10960. doi: 10.1021/acsnano.9b00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krammer F., Smith G.J.D., Fouchier R.A.M., Peiris M., Kedzierska K., Doherty P.C., Palese P., Shaw M.L., Treanor J., Webster R.G., García-Sastre A. Influenza. Nat. Rev. Dis. Primers. 2018;4:3. doi: 10.1038/s41572-018-0002-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilkinson T.M., Li C.K., Chui C.S., Huang A.K., Perkins M., Liebner J.C., Lambkin-Williams R., Gilbert A., Oxford J., Nicholas B. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat. Med. 2012;18:274–280. doi: 10.1038/nm.2612. [DOI] [PubMed] [Google Scholar]
- 17.Sridhar S., Begom S., Bermingham A., Hoschler K., Adamson W., Carman W., Bean T., Barclay W., Deeks J.J., Lalvani A. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat. Med. 2013;19:1305–1312. doi: 10.1038/nm.3350. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z., Wan Y., Qiu C., Quiñones-Parra S., Zhu Z., Loh L., Tian D., Ren Y., Hu Y., Zhang X. Recovery from severe H7N9 disease is associated with diverse response mechanisms dominated by CD8+ T cells. Nat. Commun. 2015;6:6833. doi: 10.1038/ncomms7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koutsakos M., Illing P.T., Nguyen T.H.O., Mifsud N.A., Crawford J.C., Rizzetto S., Eltahla A.A., Clemens E.B., Sant S., Chua B.Y. Human CD8+ T cell cross-reactivity across influenza A, B and C viruses. Nat. Immunol. 2019;20:613–625. doi: 10.1038/s41590-019-0320-6. [DOI] [PubMed] [Google Scholar]
- 20.Eickhoff C.S., Terry F.E., Peng L., Meza K.A., Sakala I.G., Van Aartsen D., Moise L., Martin W.D., Schriewer J., Buller R.M. Highly conserved influenza T cell epitopes induce broadly protective immunity. Vaccine. 2019;37:5371–5381. doi: 10.1016/j.vaccine.2019.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Z.T., Zarnitsyna V.I., Lowen A.C., Weissman D., Koelle K., Kohlmeier J.E., Antia R. Why Are CD8 T Cell Epitopes of Human Influenza A Virus Conserved? J. Virol. 2019;93:e01534-18. doi: 10.1128/JVI.01534-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berkhoff E.G., de Wit E., Geelhoed-Mieras M.M., Boon A.C., Symons J., Fouchier R.A., Osterhaus A.D., Rimmelzwaan G.F. Functional constraints of influenza A virus epitopes limit escape from cytotoxic T lymphocytes. J. Virol. 2005;79:11239–11246. doi: 10.1128/JVI.79.17.11239-11246.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu N.C., Thompson A.J., Lee J.M., Su W., Arlian B.M., Xie J., Lerner R.A., Yen H.-L., Bloom J.D., Wilson I.A. Different genetic barriers for resistance to HA stem antibodies in influenza H3 and H1 viruses. Science. 2020;368:1335–1340. doi: 10.1126/science.aaz5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gostic K.M., Ambrose M., Worobey M., Lloyd-Smith J.O. Potent protection against H5N1 and H7N9 influenza via childhood hemagglutinin imprinting. Science. 2016;354:722–726. doi: 10.1126/science.aag1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sridhar S. Heterosubtypic T-Cell Immunity to Influenza in Humans: Challenges for Universal T-Cell Influenza Vaccines. Front. Immunol. 2016;7:195. doi: 10.3389/fimmu.2016.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clemens E.B., van de Sandt C., Wong S.S., Wakim L.M., Valkenburg S.A. Harnessing the Power of T Cells: The Promising Hope for a Universal Influenza Vaccine. Vaccines (Basel) 2018;6:E18. doi: 10.3390/vaccines6020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chattopadhyay S., Chen J.Y., Chen H.W., Hu C.J. Nanoparticle Vaccines Adopting Virus-like Features for Enhanced Immune Potentiation. Nanotheranostics. 2017;1:244–260. doi: 10.7150/ntno.19796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Irvine D.J., Swartz M.A., Szeto G.L. Engineering synthetic vaccines using cues from natural immunity. Nat. Mater. 2013;12:978–990. doi: 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang R.H., Zhang L. Nanoparticle-Based Modulation of the Immune System. Annu. Rev. Chem. Biomol. Eng. 2016;7:305–326. doi: 10.1146/annurev-chembioeng-080615-034446. [DOI] [PubMed] [Google Scholar]
- 30.Deng L., Chang T.Z., Wang Y., Li S., Wang S., Matsuyama S., Yu G., Compans R.W., Li J.D., Prausnitz M.R. Heterosubtypic influenza protection elicited by double-layered polypeptide nanoparticles in mice. Proc. Natl. Acad. Sci. USA. 2018;115:E7758–E7767. doi: 10.1073/pnas.1805713115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng L., Mohan T., Chang T.Z., Gonzalez G.X., Wang Y., Kwon Y.M., Kang S.M., Compans R.W., Champion J.A., Wang B.Z. Double-layered protein nanoparticles induce broad protection against divergent influenza A viruses. Nat. Commun. 2018;9:359. doi: 10.1038/s41467-017-02725-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin L.C.-W., Huang C.-Y., Yao B.-Y., Lin J.-C., Agrawal A., Algaissi A., Peng B.-H., Liu Y.-H., Huang P.-H., Juang R.-H. Viromimetic STING agonist-loaded hollow polymeric nanoparticles for safe and effective vaccination against Middle East respiratory syndrome coronavirus. Adv. Funct. Mater. 2019;29:1807616. doi: 10.1002/adfm.201807616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin S.Y., Yao B.Y., Hu C.J., Chen H.W. Induction of Robust Immune Responses by CpG-ODN-Loaded Hollow Polymeric Nanoparticles for Antiviral and Vaccine Applications in Chickens. Int. J. Nanomedicine. 2020;15:3303–3318. doi: 10.2147/IJN.S241492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chattopadhyay S., Liu Y.H., Fang Z.S., Lin C.L., Lin J.C., Yao B.Y., Hu C.J. Synthetic Immunogenic Cell Death Mediated by Intracellular Delivery of STING Agonist Nanoshells Enhances Anticancer Chemo-immunotherapy. Nano Lett. 2020;20:2246–2256. doi: 10.1021/acs.nanolett.9b04094. [DOI] [PubMed] [Google Scholar]
- 35.Chen Z., John Wherry E. T cell responses in patients with COVID-19. Nat. Rev. Immunol. 2020;20:529–536. doi: 10.1038/s41577-020-0402-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Braun J., Loyal L., Frentsch M., Wendisch D., Georg P., Kurth F., Hippenstiel S., Dingeldey M., Kruse B., Fauchere F. SARS-CoV-2-reactive T cells in healthy donors and patients with COVID-19. Nature. 2020;587:270–274. doi: 10.1038/s41586-020-2598-9. [DOI] [PubMed] [Google Scholar]
- 37.Le Bert N., Tan A.T., Kunasegaran K., Tham C.Y.L., Hafezi M., Chia A., Chng M.H.Y., Lin M., Tan N., Linster M. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. 2020;584:457–462. doi: 10.1038/s41586-020-2550-z. [DOI] [PubMed] [Google Scholar]
- 38.Rosendahl Huber S., van Beek J., de Jonge J., Luytjes W., van Baarle D. T cell responses to viral infections - opportunities for Peptide vaccination. Front. Immunol. 2014;5:171. doi: 10.3389/fimmu.2014.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosendahl Huber S.K., Camps M.G., Jacobi R.H., Mouthaan J., van Dijken H., van Beek J., Ossendorp F., de Jonge J. Synthetic Long Peptide Influenza Vaccine Containing Conserved T and B Cell Epitopes Reduces Viral Load in Lungs of Mice and Ferrets. PLoS ONE. 2015;10:e0127969. doi: 10.1371/journal.pone.0127969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas P.G., Keating R., Hulse-Post D.J., Doherty P.C. Cell-mediated protection in influenza infection. Emerg. Infect. Dis. 2006;12:48–54. doi: 10.3201/eid1201.051237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson D.S., Hirosue S., Raczy M.M., Bonilla-Ramirez L., Jeanbart L., Wang R., Kwissa M., Franetich J.F., Broggi M.A.S., Diaceri G. Antigens reversibly conjugated to a polymeric glyco-adjuvant induce protective humoral and cellular immunity. Nat. Mater. 2019;18:175–185. doi: 10.1038/s41563-018-0256-5. [DOI] [PubMed] [Google Scholar]
- 42.Conniot J., Scomparin A., Peres C., Yeini E., Pozzi S., Matos A.I., Kleiner R., Moura L.I.F., Zupančič E., Viana A.S. Immunization with mannosylated nanovaccines and inhibition of the immune-suppressing microenvironment sensitizes melanoma to immune checkpoint modulators. Nat. Nanotechnol. 2019;14:891–901. doi: 10.1038/s41565-019-0512-0. [DOI] [PubMed] [Google Scholar]
- 43.Slütter B., Van Braeckel-Budimir N., Abboud G., Varga S.M., Salek-Ardakani S., Harty J.T. Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci. Immunol. 2017;2:eaag2031. doi: 10.1126/sciimmunol.aag2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anderson K.G., Sung H., Skon C.N., Lefrancois L., Deisinger A., Vezys V., Masopust D. Cutting edge: intravascular staining redefines lung CD8 T cell responses. J. Immunol. 2012;189:2702–2706. doi: 10.4049/jimmunol.1201682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee Y.N., Lee Y.T., Kim M.C., Gewirtz A.T., Kang S.M. A Novel Vaccination Strategy Mediating the Induction of Lung-Resident Memory CD8 T Cells Confers Heterosubtypic Immunity against Future Pandemic Influenza Virus. J. Immunol. 2016;196:2637–2645. doi: 10.4049/jimmunol.1501637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson E.A., Ols S., Miura K., Rausch K., Narum D.L., Spangberg M., Juraska M., Wille-Reece U., Weiner A., Howard R.F. TLR-adjuvanted nanoparticle vaccines differentially influence the quality and longevity of responses to malaria antigen Pfs25. JCI Insight. 2018;3:e120692. doi: 10.1172/jci.insight.120692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ilyinskii P.O., Kovalev G.I., O’Neil C.P., Roy C.J., Michaud A.M., Drefs N.M., Pechenkin M.A., Fu F.N., Johnston L.P.M., Ovchinnikov D.A. Synthetic vaccine particles for durable cytolytic T lymphocyte responses and anti-tumor immunotherapy. PLoS One. 2018;13:e0197694. doi: 10.1371/journal.pone.0197694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Metz S.W., Thomas A., Brackbill A., Yi X.W., Stone M., Horvath K., Miley M.J., Luft C., DeSimone J.M., Tian S.M. Nanoparticle Delivery of a Tetravalent E Protein Subunit Vaccine Induces Balanced, Type-Specific Neutralizing Antibodies to Each Dengue Virus Serotype. PLoS Neglect. Trop. Dis. 2018;12:e0006793. doi: 10.1371/journal.pntd.0006793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McHugh K.J., Nguyen T.D., Linehan A.R., Yang D., Behrens A.M., Rose S., Tochka Z.L., Tzeng S.Y., Norman J.J., Anselmo A.C. Fabrication of fillable microparticles and other complex 3D microstructures. Science. 2017;357:1138–1142. doi: 10.1126/science.aaf7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.GeurtsvanKessel C.H., Willart M.A.M., van Rijt L.S., Muskens F., Kool M., Baas C., Thielemans K., Bennett C., Clausen B.E., Hoogsteden H.C. Clearance of influenza virus from the lung depends on migratory langerin+CD11b- but not plasmacytoid dendritic cells. J. Exp. Med. 2008;205:1621–1634. doi: 10.1084/jem.20071365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ballesteros-Tato A., León B., Lund F.E., Randall T.D. Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8(+) T cell responses to influenza. Nat. Immunol. 2010;11:216–224. doi: 10.1038/ni.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wakim L.M., Gupta N., Mintern J.D., Villadangos J.A. Enhanced survival of lung tissue-resident memory CD8+ T cells during infection with influenza virus due to selective expression of IFITM3. Nat. Immunol. 2013;14:238–245. doi: 10.1038/ni.2525. [DOI] [PubMed] [Google Scholar]
- 53.Takamura S., Yagi H., Hakata Y., Motozono C., McMaster S.R., Masumoto T., Fujisawa M., Chikaishi T., Komeda J., Itoh J. Specific niches for lung-resident memory CD8+ T cells at the site of tissue regeneration enable CD69-independent maintenance. J. Exp. Med. 2016;213:3057–3073. doi: 10.1084/jem.20160938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wakim L.M., Smith J., Caminschi I., Lahoud M.H., Villadangos J.A. Antibody-targeted vaccination to lung dendritic cells generates tissue-resident memory CD8 T cells that are highly protective against influenza virus infection. Mucosal Immunol. 2015;8:1060–1071. doi: 10.1038/mi.2014.133. [DOI] [PubMed] [Google Scholar]
- 55.Mullins D.W., Sheasley S.L., Ream R.M., Bullock T.N., Fu Y.X., Engelhard V.H. Route of immunization with peptide-pulsed dendritic cells controls the distribution of memory and effector T cells in lymphoid tissues and determines the pattern of regional tumor control. J. Exp. Med. 2003;198:1023–1034. doi: 10.1084/jem.20021348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li A.V., Moon J.J., Abraham W., Suh H.Y., Elkhader J., Seidman M.A., Yen M.M., Im E.-J.J., Foley M.H., Barouch D.H. Generation of Effector Memory T Cell-Based Mucosal and Systemic Immunity with Pulmonary Nanoparticle Vaccination. Sci. Transl. Med. 2013;5:204ra130. doi: 10.1126/scitranslmed.3006516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stary G., Olive A., Radovic-Moreno A.F., Gondek D., Alvarez D., Basto P.A., Perro M., Vrbanac V.D., Tager A.M., Shi J. A mucosal vaccine against Chlamydia trachomatis generates two waves of protective memory T cells. Science. 2015;348:aaa8205. doi: 10.1126/science.aaa8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uddback I.E., Pedersen L.M., Pedersen S.R., Steffensen M.A., Holst P.J., Thomsen A.R., Christensen J.P. Combined local and systemic immunization is essential for durable T-cell mediated heterosubtypic immunity against influenza A virus. Sci. Rep. 2016;6:20137. doi: 10.1038/srep20137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Purcell A.W., Ramarathinam S.H., Ternette N. Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat. Protoc. 2019;14:1687–1707. doi: 10.1038/s41596-019-0133-y. [DOI] [PubMed] [Google Scholar]
- 60.Vita R., Mahajan S., Overton J.A., Dhanda S.K., Martini S., Cantrell J.R., Wheeler D.K., Sette A., Peters B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019;47(D1):D339–D343. doi: 10.1093/nar/gky1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li S., Liu C., Klimov A., Subbarao K., Perdue M.L., Mo D., Ji Y., Woods L., Hietala S., Bryant M. Recombinant influenza A virus vaccines for the pathogenic human A/Hong Kong/97 (H5N1) viruses. J. Infect. Dis. 1999;179:1132–1138. doi: 10.1086/314713. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.