

Abstract
Break‐induced replication (BIR) is a specialized homologous‐recombination pathway for DNA double‐strand break (DSB) repair, which often induces genome instability. In this study, we establish EGFP‐based recombination reporters to systematically study BIR in mammalian cells and demonstrate an important role of human PIF1 helicase in promoting BIR. We show that at endonuclease cleavage sites, PIF1‐dependent BIR is used for homology‐initiated recombination requiring long track DNA synthesis, but not short track gene conversion (STGC). We also show that structure formation‐prone AT‐rich DNA sequences derived from common fragile sites (CFS‐ATs) induce BIR upon replication stress and oncogenic stress, and PCNA‐dependent loading of PIF1 onto collapsed/broken forks is critical for BIR activation. At broken replication forks, even STGC‐mediated repair of double‐ended DSBs depends on POLD3 and PIF1, revealing an unexpected mechanism of BIR activation upon replication stress that differs from the conventional BIR activation model requiring DSB end sensing at endonuclease‐generated breaks. Furthermore, loss of PIF1 is synthetically lethal with loss of FANCM, which is involved in protecting CFS‐ATs. The breast cancer‐associated PIF1 mutant L319P is defective in BIR, suggesting a direct link of BIR to oncogenic processes.
Keywords: break‐induced replication, long track gene conversion, PIF1, replication stress, short track gene conversion
Subject Categories: DNA Replication, Repair & Recombination
BIR‐mediated homologous recombination allows mammalian cells to cope with replication stress and is differently controlled at fork‐breakage‐induced vs endonucleases‐generated DSBs.
Introduction
DNA double‐strand breaks (DSBs) are a major cause inducing chromosome rearrangements, a hallmark of cancer cells (Aguilera & Gomez‐Gonzalez, 2008; Negrini et al, 2010). Oncogene expression often results in replication stress, leading to fork collapse and DSB formation (Bartkova et al, 2006; Di Micco et al, 2006), which is one driving force behind genome instability. While DSBs can be repaired by different pathways, homologous recombination (HR) is believed to be the most conserved mechanism to repair DSBs (Paques & Haber, 1999; Jasin & Rothstein, 2013).
One principal mechanism of HR is gene conversion (GC), which is utilized when both sides of a DSB are homologous to the donor (Appendix Fig S1A, left). In mitotic cells, GC is initiated by 5′–3′ end resection, followed by invading the 3′ single‐strand DNA (ssDNA) end into the homologous template to form a displacement loop (D‐loop), after which the 3′ end of the invading strand is used as a primer for new DNA synthesis. In mitotic cells, GC occurs mainly by synthesis‐dependent strand annealing (SDSA), wherein the other resected end anneals to the newly synthesized strand once displaced from its template. GC can also occur through the double‐strand break repair pathway (DSBR, also called the double Holliday junction [dHJ] pathway), in which the other resected end anneals to the displaced strand of D‐loop to form dHJ. Resolution of dHJ produces non‐crossover (NCO) and crossover (CO) products. GC tracks are usually short, typically < 100 bps in mammalian cells and 50–300 bps in yeast (Sweetser et al, 1994; Taghian & Nickoloff, 1997; Elliott et al, 1998; Nickoloff et al, 1999; Palmer et al, 2003).
Break‐induced replication (BIR) is another type of HR mechanism that is used when homology is detected at only one DSB end to the donor sequence (Appendix Fig S1A, right) (Llorente et al, 2008; Anand et al, 2013; Malkova & Ira, 2013). Based on the study from yeast, BIR is initiated by strand invasion to form a D‐loop and progresses via D‐loop migration. The key difference between GC and BIR is that BIR, but not GC, relies on Pol32, a non‐essential subunit of Polδ in yeast, and helicase Pif1 (Lydeard et al, 2007; Deem et al, 2011; Donnianni & Symington, 2013; Saini et al, 2013; Wilson et al, 2013; Sakofsky et al, 2014). Once established, BIR often proceeds for a long distance in yeast and can copy hundreds of kilobases of DNA to the end of a chromosome (Davis & Symington, 2004; Malkova et al, 2005). During BIR, replisomes for repair synthesis often disassociate from the templates, resulting in frequent template switching (Smith et al, 2007). Besides single‐ended DSBs (seDSBs), BIR is also activated at double‐ended DSBs (deDSBs) when only one DSB end contains homology to the donor or when the distance of the homologies in the donor to the two DSB ends is more than 1–2 kb apart (Appendix Fig S1B, bottom, (Jain et al, 2009; Mehta et al, 2017)). A “recombination execution checkpoint” (REC) was proposed by the Haber group to sense the DSB ends and make the choice of GC or BIR: if the homologies to the two DSB ends are close to each other, GC is activated, but if homology can only be detected at one DSB end, BIR is activated (Jain et al, 2009). However, it is still not clear how REC detects the engagement of DSBs to the donor to make the selection of GC or BIR. Consistent with the role of BIR in repairing seDSBs, the Ira group showed in yeast that BIR is involved in repair of replication fork breakage, although it does not appear to be a primary pathway as adjacent converging forks often complete replication at collapsed forks and suppress BIR (Mayle et al, 2015).
In mammalian cells, it has been described that oncogene‐induced DNA replication, replication stress‐induced DNA repair synthesis in mitosis (mitotic DNA synthesis, MiDAS) and alternative lengthening of telomeres (ALT) exhibit BIR characteristics (Costantino et al, 2014; Minocherhomji et al, 2015; Dilley et al, 2016; Roumelioti et al, 2016; Sotiriou et al, 2016). These important discoveries highlighted the significant roles of BIR in DSB repair especially under replication stress. However, it remains unclear how BIR is activated and operated in mammalian cells and what determines the repair pathway selection between BIR and GC.
In this study, we established EGFP‐based reporters to monitor GC and BIR in mammalian cells. We demonstrated that at DSBs induced by endonucleases, BIR is used for homology‐initiated recombination requiring long track DNA synthesis, but not for short track gene conversion (STGC). However, different from BIR in yeast, which can proceed for more than 100 kbs (Davis & Symington, 2004; Malkova et al, 2005), BIR track length at endonuclease‐generated DSBs in mammalian cells rarely exceeds 4 kb, and BIR can be completed by either SDSA or end joining. We also found that POLD3‐dependent BIR is activated when forks are broken upon encountering DNA nicks or at common fragile site (CFS)‐derived AT‐rich sequences (CFS‐ATs) upon replication and oncogenic stress. Unexpectedly, at broken forks, POLD3‐dependent BIR is established even for STGC involving two DSB ends. PCNA and RFC1 are required for BIR activation, and PIF1 recruitment to collapsed/broken forks is dependent on PCNA. We propose that BIR activation mechanism at collapsed/broken forks is different from that at DSBs generated by endonucleases, and does not require sensing DSB ends for activation. Furthermore, we showed that PIF1 and its helicase activity are important for BIR in mammalian cells, and the breast cancer‐associated PIF1 mutant L319P is defective in BIR. PIF1 exhibits a synthetic lethal interaction with FANCM that is important for protecting CFS‐ATs.
Results
Establish an EGFP‐based reporter to monitor BIR in mammalian cells
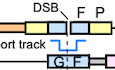
In yeast, when the distance between the two homologies in the donor template to the two DSB ends in the recipient is 1–2 kb or longer, BIR is preferentially used over GC to repair these DSBs (Appendix Fig S1B, (Jain et al, 2009; Mehta et al, 2017)). We thus established an EGFP‐based reporter (EGFP‐BIR‐5085, Fig 1A) in mammalian cells to analyze homology‐initiated recombination that requires long track of DNA synthesis (0.9–3.8 kb). In this reporter, the EGFP open reading frame is split into the N‐ and C‐terminal fragments (EG and FP) which are linked to one of the two intron sequences (intron 1 and 2) derived from insulin‐like growth factor 2 mRNA‐binding protein 1 (IGF2BP1) (Wang et al, 2004), respectively. The intron sequences are fused with a 1.3 kb Cypridina luciferase fragment (Luc). To avoid single‐strand annealing (SSA), the Luc‐FP cassette is placed in front of the EG‐Luc cassette that contains two inverted I‐SceI sites inserted at the end of the Luc sequence. The reporter was introduced into U2OS cells, and clones with a single copy of the reporter stably integrated into the genome were obtained after Southern blot analysis.
Figure 1. PIF1 is important for BIR at endonuclease‐generated DSBs.
- Schematic drawing of the EGFP‐BIR‐5085 reporter and the BIR repair product by SDSA (BIR‐SDSA) or end joining (BIR‐EJ) which results in EGFP expression after splicing.
- U2OS (EGFP‐BIR‐5085) reporter cell line was infected by lentiviruses encoding endonuclease I‐SceI or empty vector, followed by puromycin selection (2 µg/ml, 2 days) and assayed for the percentage of EGFP‐positive cells by FACS analysis 4 days post‐infection.
- BIR track length was determined in the single EGFP‐positive clones derived from U2OS (EGFP‐BIR‐5085) WT and PIF1 KO reporter cell lines after I‐SceI (left) or Cas9/sgRNA (right) cleavage by sequencing the PCR products of repair junctions using genomic DNA. Group means are shown and error bars represent ± SD. Dashed lines (3.8 and 0.9 kb) indicate the upper and lower limits of track length that can be scored by this reporter.
- U2OS (EGFP‐BIR‐5085) cells expressing shRNAs for RAD51, POLD3, or shRNA vector (Ctrl) were infected by lentiviruses encoding endonuclease I‐SceI. The percentage of EGFP‐positive cells was assayed by FACS analysis 4 days later (left). Expression of RAD51 and POLD3 is shown by Western blot analysis (right).
- U2OS (EGFP‐BIR‐5085) cells carrying Tet‐On Cas9/sgRNA‐5085 (Appendix Table S1) and expressing indicated shRNA were treated by Nocodozale (0.3 μM) and 40 h later, Doxycycline (Dox, 5 μg/ml) was added. The percentage of EGFP‐positive cells was quantified by FACS analysis 48 h after induction (left). Cell cycles before and after Nocodazole treatment were analyzed by FACS following propidium iodide (PI) staining (middle). Expression of RAD51, POLD3, and RAD52 is shown by Western blot analysis (right).
- U2OS (EGFP‐BIR‐5085) cells expressing PIF1 shRNA or shRNA vector (Ctrl; left) or expressing PIF1‐WT or E307Q mutant with endogenous PIF1 depleted by shRNA (right) were infected by lentiviruses expressing I‐SceI. The percentage of EGFP‐positive cells was assayed by FACS analysis 4 days post‐infection. PIF1 expression level was determined by qPCR (Appendix Fig S3A), and the expression of PIF1‐WT or E307Q mutant is shown in Appendix Fig S3C.
- U2OS (EGFP‐BIR‐5085) cells and two PIF1 knocked‐out (KO) clones derived from the U2OS (EGFP‐BIR‐5085) cell line and generated by CRISPR KO were assayed for the percentage of EGFP‐positive cells by FACS 4 days after I‐SceI lentiviral infection.
- Schematic drawing of the EGFP‐STGC‐1731 reporter and the repair product generated by STGC is shown (top). iEGFP: internal EGFP. U2OS (EGFP‐STGC‐1731) cells expressing RAD51, POLD3, PIF1 shRNA, or shRNA vector (Ctrl) were assayed for EGFP‐positive repair events by FACS analysis 4 days post‐infection of I‐SceI lentiviruses (middle). Expression RAD51 and POLD3 is shown by Western blot analysis (bottom).
Data information: Error bars represent the standard deviation (SD) of at least three independent experiments. Significance of the differences was assayed by two‐tailed non‐paired parameters were applied in Student's t‐test. The P value is indicated as **P < 0.01, ****P < 0.0001 and n.s. (not significant) P > 0.05.
Source data are available online for this figure.
Upon I‐SceI cleavage in the EG‐Luc cassette, the repair is initiated by invading the homologous Luc sequence (1.3 kb long homology) to the Luc‐FP cassette on its sister chromatid (Fig 1A). The GC track needs to proceed for 3.8 kb to reach the second homologous sequence outside of the reporter (right‐side homology, shown in pink) to complete GC. In mammalian cells, GC with short track length is called STGC, but when the GC track length is longer than 1–2 kb, GC is termed as long track gene conversion (LTGC) (Johnson & Jasin, 2000; Puget et al, 2005). Thus, in our reporter, if the invading strand reaches the second homology locating 3.8 kb away, the repair is completed by LTGC via SDSA. As a result of LTGC, the recipient chromatid would contain a new fused EG‐Luc and Luc‐FP cassette, which will produce a functional EGFP after splicing (Fig 1A, repair products, BIR‐SDSA [LTGC]). However, if the replicating strand is prematurely disassociated from its template before reaching the second homology 3.8 kb away, the newly synthesized DNA end may be ligated to the other end of the original DSB via end joining. In this scenario, if the invading strand has completed the replication of the intron‐FP fragment (0.9 kb), green cells can also be produced (Fig 1A, repair products, BIR‐EJ [end joining]). As expected, I‐SceI expression induced green cell formation in the U2OS (EGFP‐BIR‐5085) reporter cell line (Fig 1B). We performed PCR and sequencing analysis of the resulted single green clones. Surprisingly, only 1 out of 30 green clones (3.3%) completed replication of 3.8 kb and finished LTGC by using the second end homology (right‐side homology, Fig 1A and Appendix Fig S14). For the rest 29 clones (96.7%), replication is aborted before reaching the second end homology, and the disassociated replicating strand is ligated to the second DSB end by end joining. The replication track length of each event is determined (Fig 1C, left), and Southern blot analysis was performed to verify the observation (Appendix Fig S2A). Characterization of the end joining junctions revealed that more than 60% of the end joining events contain 1–5 bp microhomology and more than 10% of the events have 1–8 bp insertions at the repair breakpoints (Appendix Fig S2B), suggesting that microhomology‐mediated end joining (MMEJ) is involved in ligating the broken ends when BIR is aborted. We further showed that recombination scored by the EGFP‐BIR‐5085 reporter is dependent on RAD51 and POLD3 (Fig 1D), consistent with a requirement for RAD51‐dependent strand invasion and BIR‐specific DNA synthesize of 0.9 to 3.8 kb or longer. Short BIR track length and using end joining to complete BIR in mammalian cells were also observed by the Halazonetis group (Costantino et al, 2014). These studies suggest that BIR replication in mammalian cells is not very processive and usually cannot exceed 4 kb, which is quite different from BIR replication in yeast which can proceed for hundreds of kbs (Davis & Symington, 2004; Malkova et al, 2005). In order to distinguish the two mechanisms for completing BIR in mammalian cells, we term the BIR events that are finished by strand annealing to the second homology end as BIR/SDSA and by end joining as BIR/EJ (Fig 1A).
We also showed that like RAD51, inactivation of CtIP, BRCA1, or BRCA2 significantly reduces BIR efficiency scored by the EGFP‐BIR‐5085 reporter (Appendix Fig S2C, Cas9), consistent with the requirement for end resection and homology‐dependent strand invasion in BIR. However, MiDAS, which has been shown to use BIR, is RAD51 independent but requires RAD52 (Bhowmick et al, 2016). We thus examined BIR in mitotic cells using the EGFP‐BIR‐5085 reporter. Interestingly, BIR in mitotic arrested cells, using 1.3 kb homology for strand invasion, is independent of RAD51 but dependent on RAD52 (Fig 1E), and this is likely due to a suppression of RAD51‐mediated HR in mitotic cells (Esashi et al, 2005; Ayoub et al, 2009) (see Discussion).
PIF1 is important for BIR in mammalian cells
PIF1 is important for promoting BIR in yeast (Wilson et al, 2013). To examine whether PIF1 in mammalian cells is also important for BIR, we silenced human PIF1 by shRNAs in U2OS (EGFP‐BIR‐5085) cells and showed that depleting PIF1 significantly reduces BIR frequency (Fig 1F, left and Appendix Fig S3A, left), suggesting that PIF1 is also important for BIR in mammalian cells. We also generated PIF1‐KO U2OS (EGFP‐BIR‐5085) cells by CRISPR/Cas9 and showed that BIR frequency is reduced in PIF1‐KO cells (Fig 1G and Appendix Fig S3B). In contrast, STGC, as scored using the EGFP‐STGC‐1731 reporter, which contains ~ 0.3 kb homology on either side of the I‐SceI cleavage site, is not reduced after I‐SceI cut when PIF1 or POLD3 is depleted by shRNAs (Fig 1H). To determine what are the remaining BIR events in PIF1‐KO cells, we analyzed single green clones derived from U2OS (EGFP‐BIR‐5085) PIF1‐KO cells after I‐SceI or Cas9 cleavage (Fig 1C). The track length of the remaining BIR events in PIF1‐KO cells is significantly reduced with the average track length of 1.5 kb (I‐SceI) or 1.4 kb (Cas9) vs 2.2 kb (I‐SceI) or 1.9 kb (Cas9) in wild‐type (WT) cells. Collectively, these data suggest that PIF1 has a conserved function in BIR to promote long track DNA synthesis in mammalian cells. To examine whether the helicase activity of PIF1 is needed for BIR, we expressed PIF1‐WT and the helicase mutant PIF1‐E307Q in U2OS (EGFP‐BIR‐5085) cells and inactivated endogenous PIF1 by shRNAs. We showed that BIR is significantly reduced in the PIF1‐E307Q mutant, indicating that the helicase activity of PIF1 is required for its function in BIR (Fig 1F, right and Appendix Fig S3C).
PIF1 and RAD51 are required for BIR at Flex1, an AT‐rich and structure‐prone sequence from CFS, upon replication and oncogenic stress
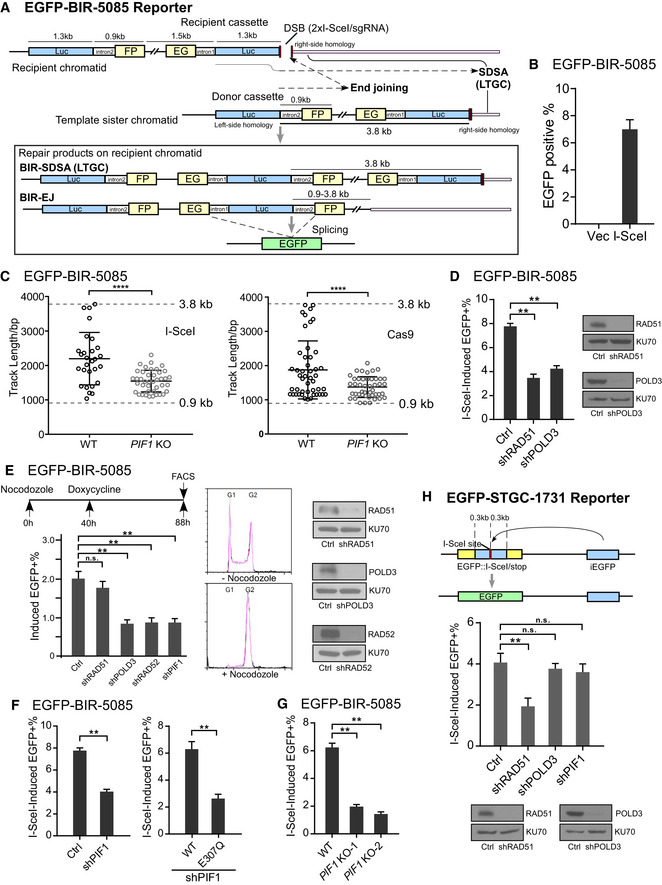
BIR is implicated in replication restart at seDSBs upon fork collapse. Consistent with a role of PIF1 in BIR, we showed that PIF1 depletion significantly increases cell sensitivity to hydroxyurea (HU) and aphidicolin (APH; Fig 2A). We also showed that PIF1‐deficient cells are sensitive to other DNA damaging agents which disturb DNA replication, such that ATR inhibitor AZD6738, topisomerase inhibitor camptothecin (CPT), DNA alkylating agent methyl methanesulfonate (MMS), and PARP inhibitor Olaparib (Appendix Fig S4). Single molecule DNA combing analysis further demonstrated that PIF1‐KO cells are defective in replication restart after HU treatment (Fig 2B).
Figure 2. PIF1 is required for BIR to repair DSBs at broken forks to promote replication restart.
- U2OS WT or PIF1‐KO cells were treated with the indicated concentrations of HU (left) or APH (right) for 72 h, and the cell viability assay was performed.
- U2OS WT or PIF1‐KO cells were labeled with CldU for 30 min followed by incubation with 2 mM HU for 2 h and then IdU for another 30 min. Labeled cells were processed for DNA fiber analysis. Representative images of stalled or restarted forks and forks with new origin firing were shown (left). The percentage of restarted forks was quantified by analyzing of 110–130 fibers for each experiment (right). Experiments were repeated four times for each sample.
- Schematic drawing of the EGFP‐BIR‐5085 reporter and the repair steps leading to the repair product expressing EGFP after Cas9n/sgRNA‐5085 expression.
- U2OS (EGFP‐BIR‐5085) cell lines carrying Dox‐inducible Cas9/sgRNA‐5085 (Dox‐Cas9) or Cas9n/sgRNA‐5085 (Dox‐Cas9n) were incubated with or without Dox (5 µg/ml) and assayed by FACS analysis 2 days later (left). U2OS (EGFP‐BIR‐5085, Dox‐Cas9 or Dox‐Cas9n) cells expressing shRNAs RAD51, POLD3, and PIF1 or shRNA vector (Ctrl) were incubated with 5 µg/ml Dox, and FACS analysis was performed after 2 days (right).
- Track length of single EGFP‐positive clones derived from U2OS (EGFP‐BIR‐5085) cells after Cas9/sgRNA‐5085 (n = 47) or Cas9n/sgRNA‐5085 (n = 39) expression was analyzed by sequencing of the PCR products from genomic DNA covering the repair junctions.
Data information: Error bars represent the standard deviation (SD) of at least three independent experiments. Significance of the differences was assayed by two‐tailed non‐paired parameters were applied in Student's t‐test. The P value is indicated as **P < 0.01, ***P < 0.001.
To investigate BIR mechanisms in repair of broken replication forks, we used Cas9 nickase (Cas9n: Cas9‐D10A) to generate nicks on DNA (Jinek et al, 2012) in the EGFP‐BIR‐5085 reporter, and when replication encounters the nicks, forks would break, leading to DSB formation (Fig 2C). Both Cas9‐ and Cas9n‐induced HR scored by EGFP‐BIR‐5085 reporter is RAD51, POLD3, and PIF1 dependent (Fig 2D), consistent with the usage of BIR mechanism. We also showed that Cas9n‐induced BIR depends on CtIP, BRCA1, and BRCA2 (Appendix Fig S2C, Cas9n). Interestingly, however, repair track length is much longer at DNA breaks caused by nicks (Cas9n) on replication forks with 27 out of 39 events (69.3%) reached the second homology ends (3.8 kb track length), whereas when DSBs were generated directly by Cas9 cleavage, only two out of 47 events (4.3%) completed 3.8 kb of DNA synthesis and the rest events used BIR/EJ (Fig 2E and Appendix Fig S14).
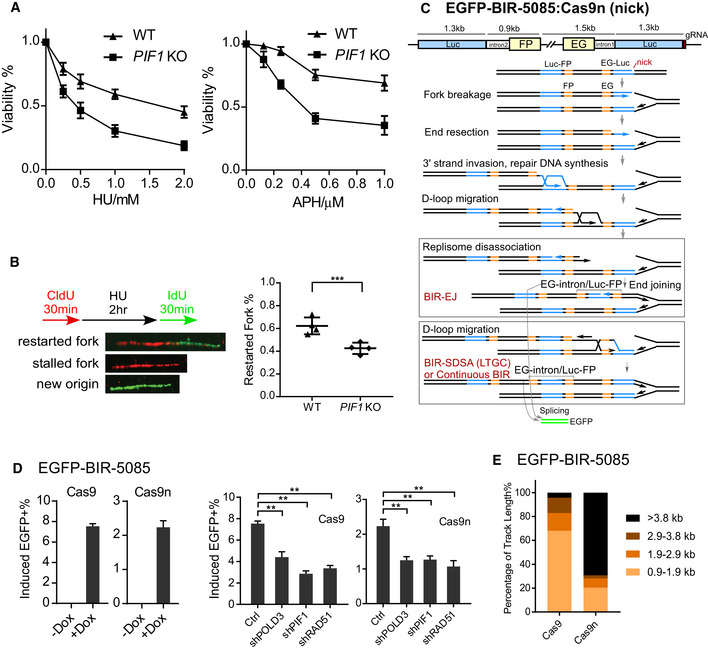
Upon replication stress, replication forks are often collapsed and broken at the genomic loci containing DNA secondary structures. To test whether BIR would be induced upon fork breakage at DNA secondary structures, we engineered a new BIR reporter, EGFP‐Flex1‐BIR‐5086, by replacing the I‐SceI site in the EGFP‐BIR‐5085 reporter with an AT‐rich sequence, Flex1, derived from CFS FRA16D (Fig 3A and Appendix Fig S5). Flex1 forms DNA secondary structures at replication forks which cause replication fork stalling and fork collapse, especially under replication stress (Zhang & Freudenreich, 2007; Wang et al, 2014; Wang et al, 2018). Indeed, green cells are accumulated upon HU and aphidicolin (APH) treatment in the U2OS (EGFP‐Flex1‐BIR‐5086) reporter cell line (Fig 3B and Appendix Fig S2D), and depletion of POLD3, PIF1, or RAD51 reduced HU‐induced green cell accumulation (Fig 3C and Appendix Fig S2D). CtIP, BRCA1, and BRCA2 are also needed for BIR‐mediated repair of DSBs caused by replication stress upon APH or HU treatment (Appendix Fig S2D). Thus, BIR plays an important role in repairing DSBs when forks are broken at structure‐prone DNA sequences. We also showed that depleting MUS81 reduces γH2AX accumulation at Flex1 after HU treatment (Appendix Fig S6A) and causes a reduction of Flex1‐induced BIR (Appendix Fig S6B), suggesting that MUS81 is involved in cleaving stalled forks at Flex1 and resulted DSBs are repaired by BIR. We analyzed the BIR track length in the EGFP‐Flex1‐BIR‐5086 reporter after APH treatment and found that all 38 green clones (100%) have completed 3.8 kb of DNA synthesis (Appendix Fig S14). We do not know the exact BIR track length beyond 3.8 kb, as it is the maximal BIR track length that can be determined in this reporter. Nevertheless, this study suggests that DNA synthesis track length during BIR is much longer at broken replication forks than that at DSBs generated directly by endonucleases.
Figure 3. PIF1 is required for BIR at Flex1 upon replication and oncogenic stress.
- Schematic drawing of the EGFP‐Flex1‐BIR‐5086 reporter. Flex1: 0.3 kb Flex1(AT)34 derived from CFS FRA16D.
- U2OS (EGFP‐Flex1‐BIR‐5086) cells were treated with or without 2 mM HU for 24 h, and the percentage of EGFP‐positive cells was quantified by FACS analysis 3 days after HU removal.
- U2OS (EGFP‐Flex1‐BIR‐5086) cells expressing shRNAs for RAD51, POLD3, and PIF1 or shRNA vector (Ctrl) were treated with 2 mM HU for 24 h. The percentage of EGFP‐positive cells by HU induction was quantified by FACS analysis 3 days after HU removal.
- U2OS (EGFP‐Flex1‐BIR‐5086) cells were infected by retrovirus expressing H‐RAS‐V12‐FLAG (RAS) or Cyclin E‐HA (Cyc E), and the percentage of EGFP‐positive cells was quantified by FACS analysis 4 days after retrovirus infection (top). The expression of RAS or Cyclin E is shown by Western blot analysis (bottom).
- U2OS (EGFP‐Flex1‐BIR‐5086) cells expressing shRNAs for RAD51, POLD3, and PIF1 or shRNA vector (Ctrl) were infected by retroviruses expressing RAS, and the percentage of EGFP‐positive cells was quantified by FACS analysis 4 days after retrovirus infection (top). Expression level of RAD51 and POLD3 is shown by Western blot analysis (bottom).
- U2OS WT or PIF1‐KO cells were infected by retrovirus expressing RAS or empty vector. Cell profiling was plotted by counting cell numbers every 24 h, and normalized to the cell number on the first day.
Data information: Error bars represent the standard deviation (SD) of at least three independent experiments. Significance of the differences was assayed by two‐tailed non‐paired parameters were applied in Student's t‐test. The P value is indicated as **P < 0.01.
Source data are available online for this figure.
Since oncogene expression causes replication stress (Costantino et al, 2014; Sotiriou et al, 2016), we examined whether oncogene expression would induce BIR. Overexpression of H‐RAS‐V12 (RAS) or Cyclin E significantly induces BIR at Flex1 as revealed by the EGFP‐Flex1‐BIR‐5086 reporter (Fig 3D). This is consistent with the previous observation that DSBs at Flex1 are accumulated upon oncogene expression (Wang et al, 2018). We further showed that oncogene‐induced BIR at Flex1 is significantly reduced when POLD3, PIF1, or RAD51 is depleted by shRNAs (Fig 3E). These data support the idea that BIR would be commonly used during the oncogenic process to repair DSBs at collapsed forks. We further showed that overexpression of RAS significantly inhibits growth of PIF1‐KO cells but not WT cells (Fig 3F), consistent with a previous study finding that RAS‐transformed fibroblasts grow more slowly upon depletion of PIF1 (Gagou et al, 2014). We propose that oncogenic stress causes a reliance on PIF1 for survival due to the critical role of PIF1 in BIR to promote replication restart at collapsed forks upon oncogenic stress.
BIR is also used for STGC involving two ends of a DSB at broken forks
When replication forks are stalled at DNA secondary structures or other replication blockers, forks can be broken around the blockers at different positions. This could lead to generation of seDSBs, but can also produce deDSBs (Fig 4A). For example, when replication forks are stalled at DNA secondary structures such as Flex1, breaks can be generated at the replication fork junctions to create seDSBs or at a distance from the fork junctions to generate deDSBs. In addition, when adjacent forks converge, seDSBs can also be converted to deDSBs.
Figure 4. BIR is used at replication‐associated DSBs even for STGC.
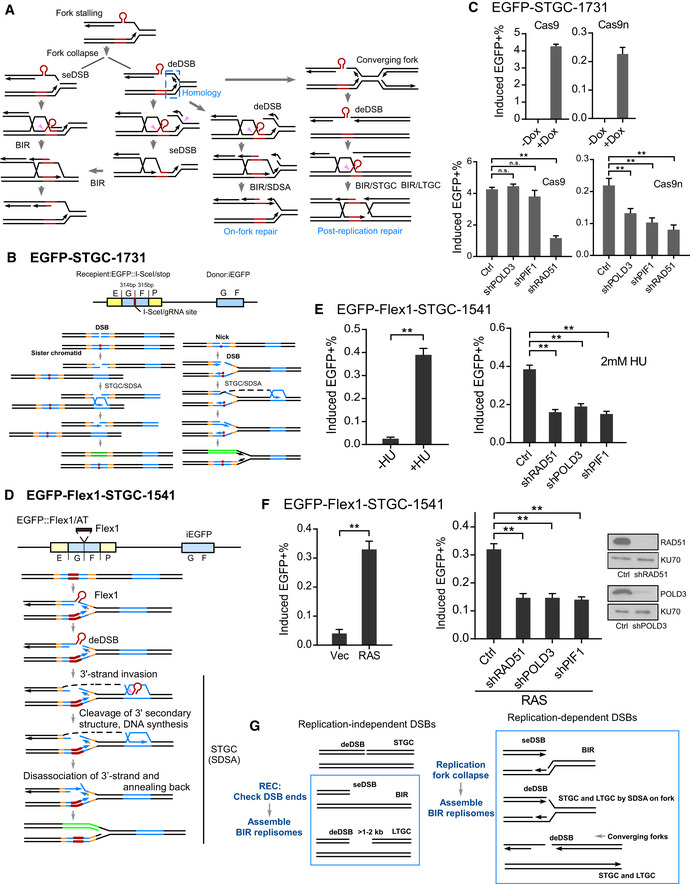
- Proposed models for repair of DSBs generated at broken replication forks (see text for details). seDSB: single‐ended DSB; deDSB: double‐ended DSB. Pink arrows: endonucleases to remove Flex1 or other secondary structures at DSB ends or DNA tails at the fork junctions.
- Schematic drawing of the EGFP‐STGC‐1731 reporter and proposed pathways to repair DSBs generated by endonuclease cleavage (I‐SceI or Cas9, left) or converted from nicks (Cas9n, right).
- U2OS (EGFP‐STGC‐1731) cell lines carrying Dox‐inducible Cas9/sgRNA‐1731 (Dox‐Cas9) or Cas9n/sgRNA (Dox‐Cas9n) were incubated with or without Dox (5 µg/ml), and the percentage of EGFP‐positive cells was quantified by FACS analysis 2 days later (top). U2OS (EGFP‐STGC‐1731, Dox‐Cas9 or Dox‐Cas9n) cells expressing shRNAs for RAD51, POLD3, and PIF1 or shRNA control (Ctrl) were incubated with Dox (5 µg/ml), and the percentage of EGFP‐positive cells was quantified by FACS analysis after 2 days (bottom).
- Schematic drawing of the EGFP‐Flex1‐STGC‐1541 reporter and the proposed mechanism to repair DSBs at Flex1 generated upon fork breakage by SDSA which involves two DSB ends.
- U2OS (EGFP‐Flex1‐STGC‐1541) cells were treated with or without 2 mM HU for 24 h, and the percentage of EGFP‐positive cells by HU induction was quantified by FACS analysis 4 days after removal of HU (left). U2OS (EGFP‐Flex1‐STGC‐1541) cells expressing shRNAs for RAD51, POLD3, and PIF1 or shRNA vector (Ctrl) were treated with 2 mM HU for 24 h, and the percentage of EGFP‐positive cells was quantified by FACS analysis 4 days later (right).
- U2OS (EGFP‐Flex1‐STGC‐1541) cells were infected by retroviruses expressing RAS or empty vector, and the percentage of EGFP‐positive cells was assayed by FACS 5 days following infection (left). U2OS (EGFP‐Flex1‐STGC‐1541) cells expressing shRNAs for RAD51, POLD3, and PIF1 shRNAs or shRNA vector were infected by retroviruses expressing RAS, and the percentage of EGFP‐positive cells was assayed by FACS 5 days after infection (middle). Expression level of RAD51 and POLD3 is shown by Western blot analysis (right).
- Proposed models for activating BIR at replication‐dependent and replication‐independent DSBs (see text for details).
Data information: Error bars represent the standard deviation (SD) of at least three independent experiments. Significance of the differences was assayed by two‐tailed non‐paired parameters were applied in Student's t‐test. The P value is indicated as **P < 0.01 and n.s. (not significant) P > 0.05.
Source data are available online for this figure.
We showed that when replication forks are broken (EGFP‐BIR‐5085, Cas9n; EGFP‐Flex1‐BIR‐5086, HU), BIR involving long track of DNA synthesis can be used to repair DSBs on forks (Figs 2D and 3C and Appendix Fig S14). To test whether STGC involving two DSB ends can also occur upon fork breakage, we expressed Cas9n in U2OS (EGFP‐STGC‐1731) cells to score STGC at collapsed forks. The donor template in the EGFP‐STGC‐1731 reporter has homologies to both sides of the DSB but contains only the internal part of EGFP (iEGFP: GF fragment). Since the C‐terminal end of EGFP in the iEGFP donor is missing, the invading strand from the recipient EGFP cassette has to disassociate from the donor template after short track DNA synthesis and anneal back to the recipient in order to obtain the C terminus of EGFP for restoring the EGFP open reading frame (Fig 4B, right and Appendix Fig S7B). Thus, this EGFP‐STGC‐1731 reporter scores only STGC that requires the involvement of two DSB ends. We showed that like Cas9, Cas9n also induces green cell accumulation in the EGFP‐STGC‐1731 reporter (Fig 4C, top), suggesting that STGC involving two DSB ends is also used at broken replication forks. Furthermore, we showed that while Cas9‐induced STGC (at endonuclease‐generated DSBs) scored by the EGFP‐STGC‐1731 reporter is independent of POLD3 and PIF1, Cas9n‐induced STGC (at fork‐associated DSBs) depends on both POLD3 and PIF1 activities (Fig 4C, bottom), suggesting that BIR mechanism is used even for STGC‐mediated repair of DSBs at broken replication forks.
We also replaced the I‐SceI/Cas9 cleavage sites in the EGFP‐STGC‐1731 reporter (Fig 4B) with Flex1, to generate the reporter EGFP‐Flex1‐STGC‐1541 (Fig 4D), where DSBs can be induced upon replication stress (Wang et al, 2014; Wang et al, 2018). Indeed, upon HU treatment, green cells accumulate in U2OS (EGFP‐Flex1‐STGC‐1541) cells (Fig 4E, left). Like the EGFP‐STGC‐1731 reporter, the C‐terminal end of EGFP is missing in the donor (iEGFP) and thus the EGFP‐Flex1‐STGC‐1541 reporter also scores only STGC that involves two DSB ends (Fig 4D and Appendix Fig S8). We showed that when STGC is induced by HU at Flex1 in the EGFP‐Flex1‐STGC‐1541 reporter, RAD51, POLD3, and PIF1 are all needed (Fig 4E, right). RAS‐induced STGC at Flex1 is also RAD51‐, POLD3‐, and PIF1‐dependent (Fig 4F, right). These data suggest that BIR is used for STGC upon fork breakage at CFS‐ATs.
To confirm that STGC is indeed used in the EGFP‐STGC‐1731 reporter induced by nicks (Cas9n) and in the EGFP‐Flex1‐STGC‐1541 reporter at Flex1 after APH treatment, we sequenced the repair products of green single clones and found that all events (20 events for each reporter) are completed by STGC using two DSB ends as described in the model (Fig 4B and D).
Collectively, we showed that following fork breakage induced either by DNA nicks (Fig 4B) or by DNA secondary structure formation (Fig 4D), BIR is activated, which can be used to repair not only seDSBs, but also deDSBs even for STGC at broken replication forks. We propose that establishment of BIR is differently regulated at endonuclease‐generated DSBs (replication‐independent) and at broken replication forks (replication‐dependent). Upon DSB formation induced by endonucleases such as I‐SceI and Cas9, REC checks the DSB ends and determines whether BIR needs to be used. BIR is activated when only one DSB end has homology to the donor (e.g., seDSBs) or when the homologies in the donor to the two DSB ends are too far apart (e.g., a gap between the homologies, Fig 4G, left). However, upon fork breakage, BIR is immediately established irrespectively of the configuration of the DSB ends (Fig 4G, right). BIR is used at both seDSBs and deDSBs, and mediates both STGC and LTGC on broken forks. Fork collapse/breakage may serve as a signal to activate BIR.
BIR with long track DNA synthesis is promoted upon replication fork breakage
We have shown that in the EGFP‐BIR‐5085 reporter (Figs 1A and 2C), repair track length is much longer at nick‐induced (Cas9n) DSBs compared with endonuclease (Cas9)‐induced DSBs (Fig 2E and Appendix Fig S14). In addition, all repair events scored by the EGFP‐Flex1‐BIR‐5086 reporter at Flex1 site upon replication stress have completed 3.8 kb DNA synthesis (Appendix Fig S14). Thus, DNA synthesis track becomes much longer at DSBs on broken forks than that generated directly by endonucleases.
To better understand the repair mechanisms at DSBs, we designed a new EGFP/STGC‐mCherry/LTGC‐5034 reporter which allows us to monitor the competition of STGC and LTGC (Fig 5A). In this reporter, a full‐length copy of EGFP under the CMV promoter is inactivated by inserting three stop codons along with an I‐SceI/sgRNA site in the middle and used as the cleavage recipient cassette. The EGFP internal fragment (iEGFP: GF fragment) identical to the donor sequence used in the EGFP‐STGC‐1731 reporter is fused to a mCherry cassette by using a self‐cleavage peptide T2A (Szymczak et al, 2004), and the fused iEGFP/T2A/mCherry cassette is placed upstream of the cleavage recipient cassette. Repair of the DSB by STGC via annealing the invading strand back to the FP homology in the EGFP recipient cassette would restore the EGFP open reading frame and produce green cells (Fig 5A, STGC). If replication of the invading strand proceeds through iEGFP (GF fragment) and mCherry, and reaches the FP homology (2.2 kb away from the cleavage site) in the recipient cassette (Fig 5A, LTGC/BIR, right‐side homology), LTGC via SDSA can be used, resulting in red but not green cells (the c‐terminal EGFP is missing, Fig 5A, LTGC/BIR). However, red cells can also be produced when DNA synthesis of the invading strand goes beyond the end of mCherry open reading frame (1.1 kb) but aborts before reaching the FP homology (2.2 kb away), and BIR is completed by end joining.
Figure 5. BIR with long repair track is promoted at DSBs on broken forks, and PCNA and RFC1 are required for activating BIR.
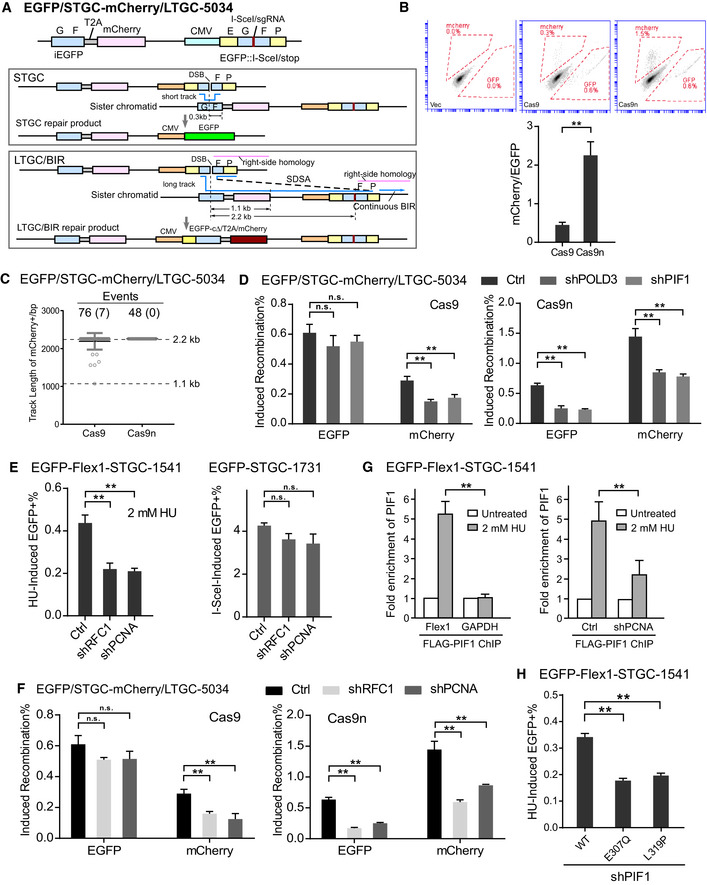
- Schematic drawing of EGFP/STGC‐mCherry/LTGC‐5034 reporter and the repair products generated by the STGC or LTGC/BIR mechanisms.
- U2OS (EGFP/STGC‐mCherry/LTGC‐5034) with Dox‐inducible Cas9/sgRNA‐1731 (Cas9) or Cas9n/sgRNA‐1731 (Cas9n) or empty vector (Vec) were assayed for recombination by FACS analysis 2 days after adding Dox (5 µg/ml). Representative FACS data (top), and the ratios of mCherry to EGFP in the indicated cell lines after Dox induction were shown (bottom).
- Track length of single mCherry‐positive clones derived from U2OS (EGFP/STGC‐mCherry/LTGC‐5034) cells after Cas9/sgRNA‐1731 or Cas9n/sgRNA‐1731 cleavage was analyzed by sequencing PCR products from genomic DNA at repair junctions. The numbers of the total events analyzed after Cas9 and Cas9n cleavage are shown on the top with the numbers of events using BIR‐EJ indicated in brackets. Group means are shown. Error bars represent ± SD. Dashed lines (2.2 and 1.1 kb) indicate the upper and lower limits of track length that can be scored by this reporter.
- U2OS (EGFP/STGC‐mCherry/LTGC‐5034, Dox‐Cas9 [left] or Dox‐Cas9n [right]) cells expressing shRNAs for POLD3 and PIF1 or shRNA vector (Ctrl) were incubated with Dox (5 µg/ml). The percentage of EGFP‐ or mCherry‐positive cells after induction was quantified by FACS analysis 2 days later to determine the percentage of EGFP‐ or mCherry‐positive cells.
- U2OS (EGFP‐Flex1‐STGC‐1541) cells expressing shRNAs for RFC1 and PCNA or vector (Ctrl) were treated with 2 mM HU for 24 h. The percentage of EGFP‐positive cells after HU treatment was quantified by FACS analysis 3 days after HU removal (left). U2OS (EGFP‐STGC‐1731) cells expressing shRNAs for RFC1 and PCNA or vector (Ctrl) were infected by lentivirus expressing I‐SceI. The percentage of EGFP‐positive cells by I‐SceI induction was quantified by FACS analysis 4 days later (right).
- U2OS (EGFP/STGC‐mCherry/LTGC‐5034, Dox‐Cas9 [left] or Dox‐Cas9n [right]) cells expressing shRNAs for RFC1 and PCNA or vector (Ctrl) were incubated with Dox (5 µg/ml). The percentage of EGFP‐ or mCherry‐positive cells after induction was quantified by FACS analysis 2 days later.
- FLAG‐PIF1 was stably expressed in U2OS (EGFP‐Flex1‐STGC‐1541) cells by lentiviral infection. Enrichment of FLAG‐PIF1 at Flex1 site or GAPDH site with or without HU (2 mM, 10 h) treatment was calculated by anti‐FLAG ChIP (left). When PCNA was depleted by shRNA using vector as a control (Ctrl), enrichment of FLAG‐PIF1 at Flex1 site was calculated by anti‐FLAG ChIP (right). ChIP value in cells without HU treatment is set up as 1 for normalization.
- U2OS (EGFP‐Flex1‐STGC‐1541) cells expressing FLAG‐PIF1 WT or mutants with endogenous PIF1 depleted by shRNA were treated with 2 mM HU for 24 h. The percentage of EGFP‐positive cells after HU treatment was quantified by FACS analysis 3 days after HU removal.
Data information: Error bars represent the standard deviation (SD) of at least three independent experiments. Significance of the differences was assayed by two‐tailed non‐paired parameters were applied in Student's t‐test. The P value is indicated as **P < 0.01 and n.s. (not significant) P > 0.05.
We analyzed repair products of the single green or red clones. As expected, all single green cell clones, analyzed after either Cas9 or Cas9n cleavage (Cas9: 30 clones and Cas9n: 28 clones), completed STGC by annealing the F homology in iEGFP donor and the FP homology in the recipient cassette via SDSA. For obtained red clones after Cas9 cleavage (Fig 5C), only seven out of 76 (9.2%) aborted replication before 2.2 kb and used end joining to complete the repair, while the rest (90.8%, ≥ 2.2 kb track length) used LTGC via SDSA at the FP homology present in both donor and the recipient. Upon Cas9n cleavage, all red clones (48 clones analyzed) completed 2.2 kb replication and used LTGC/BIR (100%, ≥ 2.2 kb track length). Since majority (> 90% after Cas9) or all (after Cas9n) of red cells have completed LTGC (≥ 2.2 kb track length), green and red cells scored by this EGFP/STGC‐mCherry/LTGC‐5034 reporter reflect the competitive use of STGC and LTGC.
We further showed that in the EGFP/STGC‐mCherry/LTGC‐5034 reporter, accumulated green cells after Cas9 cleavage are twofold more than red cells, suggesting that STGC is used more frequently than LTGC (Fig 5B, Cas9). Green cell accumulation after Cas9 cleavage is independent of POLD3 and PIF1, whereas red cell accumulation is significantly reduced when POLD3 or PIF1 is depleted by shRNAs, supporting the idea that BIR is used for LTGC and not for STGC at endonuclease‐generated DSBs (Fig 5D, left). Interestingly, when Cas9n was used to cleave the EGFP/STGC‐mCherry/LTGC‐5034 reporter, the resulting accumulation of red cells was more than double the number of green cells (Fig 5B, Cas9n), suggesting that repair with long track DNA synthesis becomes dominant over short track at broken replication forks. Importantly, under this condition when DSBs are generated upon fork breakage, the accumulation of both red and green cells depends on POLD3 and PIF1 (Fig 5D, right). We also showed that both STGC and LTGC are decreased upon depletion of RAD51, CtIP, BRCA1, or BRCA2 after Cas9 or Cas9n cleavage (Appendix Fig S9A), consistent with that both end resection and strand invasion are required for STGC and LTGC. We also generated a new reporter EGFP/STGC‐mCherry/LTGC‐Flex1‐5304 by inserting the Flex1 sequence in the EGFP/STGC‐mCherry/LTGC‐5034 reporter (Appendix Fig S10A) and showed that Flex1 also induces accumulation of more red cells than green cells after HU treatment (Appendix Fig S10B), in a manner similar to that after Cas9n cleavage of the EGFP/STGC‐mCherry/LTGC‐5034 reporter (Fig 5B). This study suggests that LTGC is used more frequently than STGC on broken forks, which is opposite to the more frequent use of STGC at endonuclease‐generated DSBs that are independent of replication. This also supports the notion that BIR with relative long track length is actively used at broken forks.
PCNA and RFC1 are required for activating BIR
Since BIR is activated at broken replication forks, we asked whether certain key replication factors are important for BIR activation. We showed that STGC at Flex1 induced by HU is POLD3‐ and PIF1‐dependent, using the BIR mechanism (Fig 4E). When we silenced the expression of PCNA, or RFC1 by shRNAs, HU‐induced STGC at Flex1 as assayed by the EGFP‐Flex1‐STGC‐1541 reporter was significantly reduced (Fig 5E, left and Appendix Fig S11A–C). In contrast, STGC after I‐SceI cleavage in the EGFP‐STGC‐1731 reporter, which does not use the BIR mechanism (Fig 1G), is not significantly affected by the depletion of PCNA, and RFC1 (Fig 5E, right). Furthermore, in the STGC and LTGC competition reporter EGFP/STGC‐mCherry/LTGC‐5034, after Cas9 cleavage, the accumulation of red cells, but not green cells, is significantly dependent on PCNA and RFC1, whereas after Cas9n cleavage, both green and red cell accumulation requires PCNA and RFC1 (Fig 5F). Collectively, we observed a nice correlation between using the BIR mechanism and the involvement of PCNA and RFC1. By ChIP analysis, we further demonstrated that PIF1 is accumulated at Flex1 after HU and APH treatment and this HU‐ and APH‐induced PIF1 recruitment is significantly reduced when PCNA is depleted by shRNAs (Fig 5G and Appendix Fig S11D). We also showed that γH2AX is increased at Flex1 after HU treatment when PCNA or RFC1 is depleted in a manner similar to PIF1 depletion (Appendix Fig S11G). These data suggest that the replication factors PCNA and RFC1 are important for BIR activation at broken forks, and recruitment of PIF1 to the collapsed/broken replication forks is dependent on PCNA. We propose that upon fork collapse/breakage, BIR replisomes which has PIF1 as a component can be established immediately, largely due to the presence of replication factors such as PCNA and RFC1 in the vicinity. However, at replication‐independent DSBs, BIR replisomes are not assembled unless REC is activated after sensing that only one DSB end can be engaged.
PIF1 exhibits synthetic lethal interaction with FANCM
We showed previously that FANCM is important for protecting DNA secondary structures on forks and that in its absence, more DSBs are accumulated at Flex1 and at other structure‐prone DNA sequences on replication forks (Wang et al, 2018). We further demonstrated that inactivation of FANCM significantly induces BIR at Flex1 as scored by the EGFP‐Flex1‐BIR‐5086 reporter (Fig 6A, left and Appendix Fig S12A), and this increase depends on RAD51, POLD3, and PIF1 (Fig 6A, right). In addition, consistent with elevated fork collapse at Flex1 in FANCM‐deficient cells, STGC induced by depletion of FANCM also depends on POLD3 and PIF1 (Fig 6B and Appendix Fig S12B). γH2AX is increased at Flex1 when FANCM is depleted, suggesting that DSBs are accumulated at Flex1 in FANCM‐deficient cells (Appendix Fig S12C). These data support the model that when the FANCM‐mediated secondary structure removal mechanism is impaired, replication forks are collapsed to generate DSBs at Flex1, and BIR is used to repair these fork collapse‐associated DSBs (Fig 6C). To determine the biological importance of BIR in repair of DSBs caused by fork collapse due to FANCM deficiency, we examined cell viability of FANCM‐KO cells when BIR pathway is inactivated. We observed that depletion of POLD3 or PIF1 in FANCM‐KO causes more cell death compared with that in WT cells (Fig 6D). These data suggest that BIR is critical for repairing DSBs accumulated at DNA secondary structures on replication forks in FANCM‐deficient cells, and PIF1 exhibits synthetic lethality interaction with FANCM.
Figure 6. PIF1‐ and POLD3‐mediated BIR is important for repairing DSBs at Flex1 caused by FANCM deficiency.
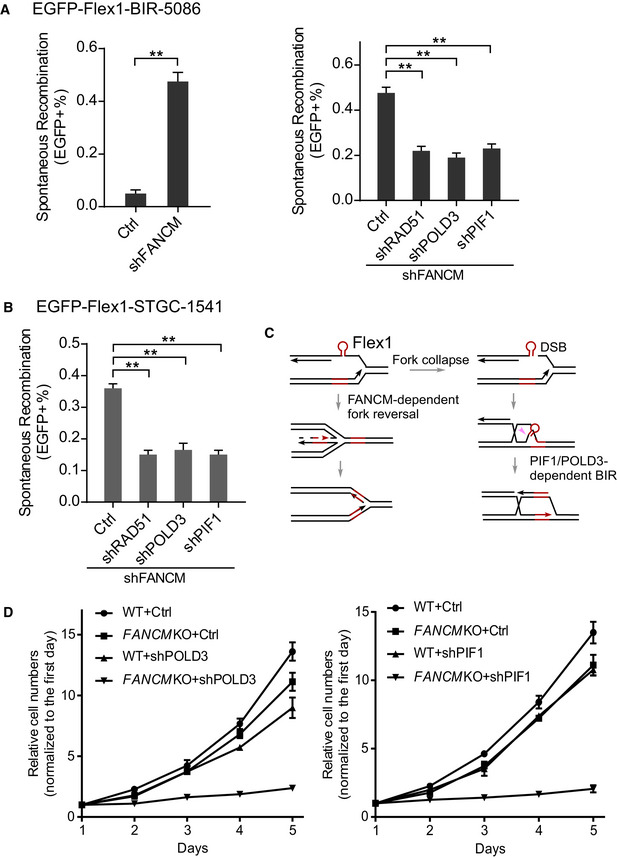
- U2OS (EGFP‐Flex1‐BIR‐5086) cells were infected by lentiviruses expressing FANCM shRNA or shRNA vector (Ctrl; left) or cells from the same reporter cell line expressing shRNAs for RAD51, POLD3, and PIF1 or shRNA vector (Ctrl) were infected by FANCM shRNA lentiviral viruses (right). The percentage of EGFP‐positive cells after spontaneous recombination was quantified by FACS analysis 5 days after lentiviral infection of FANCM shRNA. Knockdown of FANCM by shRNA is shown by qPCR and Western blot in Appendix Fig S12A.
- U2OS (EGFP‐Flex1‐STGC‐1541) cells expressing shRNAs for RAD51, POLD3, and PIF1 or shRNA vector were infected by lentivirus expressing FANCM shRNA. The percentage of EGFP‐positive cells was quantified by FACS analysis 5 days after FANCM shRNA lentiviral infection. Knockdown of FANCM by shRNA is shown by qPCR and Western blot in Appendix Fig S12B.
- Proposed model for concerted roles of FANCM‐dependent fork reversal and PIF1/POLD3‐dependent BIR in protection of Flex1 stability. Pink arrow: endonuclease cleavage to remove Flex1.
- Growth curves of WT or FANCM‐KO HCT116 cells were plotted after expressing POLD3 (left) or PIF1 (right) shRNA or shRNA vector (Ctrl). Cell number was normalized to that on day one.
Data information: Error bars represent the standard deviation (SD) of at least three independent experiments. Significance of the differences was assayed by two‐tailed non‐paired parameters were applied in Student's t‐test. The P value is indicated as **P < 0.01.
PIF1 is recruited to CFSs and plays an important role in preventing chromosomal breakage
Replication stress activates MiDAS, which requires POLD3, suggesting an involvement of the BIR mechanism (Minocherhomji et al, 2015; Bhowmick et al, 2016). We further demonstrated that MiDAS is significantly reduced when PIF1 is knocked out (Fig 7A). This suggests that PIF1 is required for MiDAS upon replication stress and further supports the notion that BIR is the mechanism underlying MiDAS.
Figure 7. PIF1 is recruited to CFSs and important for preventing chromosomal breakage.
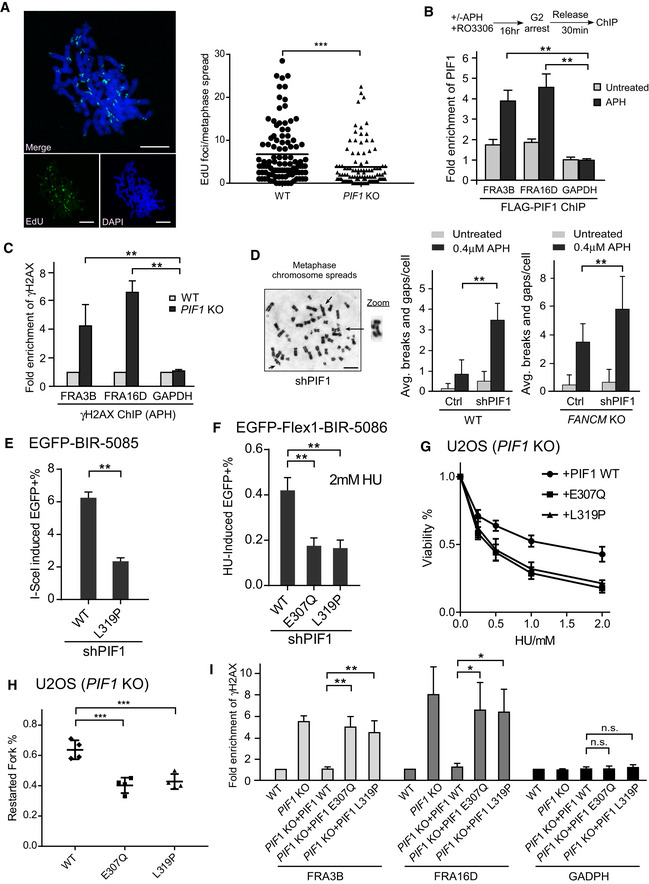
- MiDAS analysis was performed in U2OS WT and PIF1‐KO cells. Cells were treated with APH (0.4 µM) and RO‐3306 (7 µM), and then released into fresh medium containing EdU (20 µM) and Colcemid (0.1 µg/ml) as described in Materials and Methods. The image of EdU incorporation on metaphase chromosomes is shown (left) and EdU foci formed on each metaphase spread were quantified (right). ~100 metaphase spreads were analyzed in each sample. Scale bars, 10 μm.
- U2OS (HR‐Flex1‐STGC‐1541) cells expressing FLAG‐PIF1 or vector were treated with or without APH (0.4 μM, 22 h) in the presence of RO3306 (7 μM), and anti‐FLAG ChIP was performed 30 min after release from drug treatment and analyzed by qPCR. Enrichment of FLAG‐PIF1 at FRA3B, FRA16D, and GAPDH loci was calculated using ChIP value in cells expressing vector as 1 for normalization.
- Anti‐γH2AX ChIP analysis at FRA3B, FRA16D, and GAPDH genomic loci was performed in U2OS WT and PIF1‐KO cells before and after APH treatment (0.4 μM, 24 h). Enrichment of γH2AX at FRA3B, FRA16D, and GAPDH loci in WT and PIF1 KO cells was calculated using ChIP value in WT cells as 1 for normalization.
- Metaphase spread of HCT116 WT and FANCM‐KO cells was performed with or without expressing PIF1 shRNA or shRNA vector (Ctrl) before and after APH treatment (0.4 μM, 24 h). Representative images of metaphase spread are shown (left), and overall chromosome breaks and gaps per cell are quantified (right). Scale bars, 5 μm.
- U2OS (EGFP‐BIR‐5085) cells expressing PIF1 WT or L319P mutant with endogenous PIF1 silenced by shRNA were infected by lentiviruses to express I‐SceI. The percentage of EGFP‐positive cells was quantified by FACS analysis 5 days after I‐SceI infection.
- U2OS (EGFP‐Flex1‐BIR‐5086) cells expressing PIF1 WT or E307Q, L319P mutants with endogenous PIF1 silenced by shRNA were treated with 2 mM HU for 24 h. The percentage of EGFP‐positive cells by HU treatment was quantified by FACS analysis 3 days after HU removal.
- U2OS PIF1‐KO cells expressing PIF1 WT or E307Q, L319P mutants were treated with the indicated concentrations of HU for 72 h, and cell viability assays were performed.
- U2OS PIF1‐KO cells expressing PIF1 WT or E307Q, L319P mutants were labeled with CldU for 30 min, followed by 2 mM HU treatment for 2 h and IdU labeling for 30 min, and processed for DNA fiber analysis. The percentage of replication restarted DNA fibers is calculated.
- Anti‐γH2AX ChIP analysis at FRA3B, FRA16D, and GAPDH genomic loci was performed in U2OS WT and PIF1‐KO cells, or PIF1‐KO cells expressing PIF1‐WT, and E307Q and L319P mutants before and after APH treatment (0.4 μM, 24 h). Enrichment of γH2AX at FRA3B, FRA16D, and GAPDH loci was calculated using ChIP value in WT cells as 1 for normalization.
Data information: Error bars represent the standard deviation (SD) of at least three independent experiments. Significance of the differences was assayed by two‐tailed non‐paired parameters were applied in Student's t‐test. The P value is indicated as *P < 0.05, **P < 0.01, ***P < 0.001 and n.s. (not significant) P > 0.05.
Source data are available online for this figure.
Since PIF1 is important for promoting BIR upon fork collapse at the CFS‐derived Flex1 site and is also required for MiDAS which mainly occurs at CFSs (Minocherhomji et al, 2015), we asked whether PIF1 is recruited to CFSs and is important for preventing chromosomal breakage upon replication stress. By performing ChIP analysis, we found that PIF1 is significantly enriched at CFS FRA3B and FRA16D after treatment of a low dose of APH (0.4 μM; Fig 7B), and that depletion of PIF1 under this condition leads to a significant increase in γH2AX accumulation at FRA3B and FRA16D as compared to the GAPDH locus (Fig 7C). We also showed that recruitment of PIF1 to CFS FRA3B after APH treatment depends on PCNA (Appendix Fig S13). In addition, the number of chromatid breaks and gaps on prometaphase chromosome spreads is also significantly increased when PIF1 is depleted by shRNAs after treating cells with low dose of APH (0.4 μM), the condition to induce CFS expression, and depleting FANCM by shRNA further increases break and gap formation in PIF1‐deficient cells (Fig 7D). These data suggest that PIF1 is recruited to CFSs and plays an important role in preventing chromosomal breakage under replication stress.
Breast cancer‐associated PIF1‐mutant L319P is defective in BIR
Mutations of PIF1 are associated with predisposition to breast cancer (Chisholm et al, 2012). The PIF1‐L319P variant contains a mutation in the putative PIF1 family signature motif (Bochman et al, 2010), but it remains unclear how this mutant affects the disease. By expressing PIF1‐WT and PIF1‐L319P with endogenous PIF1 depleted by shRNAs, we found that BIR‐mediated LTGC (Fig 7E, EGFP‐BIR‐5085, I‐SceI) and STGC (Fig 5H, EGFP‐Flex1‐STGC‐1541, HU) are both significantly reduced in PIF1‐L319P mutant compared with PIF1‐WT cells. We also examined Flex1‐induced BIR in U2OS (EGFP‐Flex1‐BIR‐5086) cells and showed that PIF1‐L319P and the helicase mutant PIF1‐E307Q are compromised in BIR induced by HU treatment (Fig 7F). Similar results were obtained when PIF1‐L319P and PIF1‐E307Q were expressed in the PIF1‐KO U2OS cells carrying the EGFP‐BIR‐5085 or EGFP‐Flex1‐BIR‐5086 reporter (Appendix Fig S3D and E). These data suggest that breast cancer‐associated PIF1‐L319P mutant is defective in BIR. Consistently, like the helicase PIF1‐E307Q, PIF1‐L319P is sensitive to HU (Fig 7G) and is defective in replication restart (Fig 7H). ChIP analysis indicated that γH2AX accumulation at CFS FRA3B and FRA16D is significantly elevated in PIF1‐E307Q helicase mutant and PIF1‐L319P breast cancer‐associated mutant cells (Fig 7I). These data suggest that the breast cancer‐associated PIF1‐L319P mutant exhibits defects in BIR, leading to increased chromosomal breakage.
Discussion
By using defined EGFP‐based repair reporters, we demonstrated that the control of BIR activation is quite different at DSBs generated by endonucleases compared with those caused by replication fork breakage. PIF1 has a conserved role in promoting processive DNA synthesis during BIR in mammalian cells, and its recruitment to broken forks is dependent on PCNA. The role of PIF1 in BIR likely contributes to the prevention of chromosomal breakage and suppression of breast cancer.
BIR activation at endonuclease‐induced DSBs
Based on the study in yeast, BIR is used when DSBs are single ended or when only one DSB end can find its homology (Llorente et al, 2008; Anand et al, 2013; Malkova & Ira, 2013). Increasing the distance between the two DSB end homologies in the donor template also activates BIR (Jain et al, 2009; Mehta et al, 2017), possibly because only one homology at a DSB end can be sensed or engaged since the other homology is too far away. The initial homology search and strand invasion in GC and BIR are almost identical in efficiency and kinetics, but initiation of DNA synthesis from the 3′ invading strand in BIR is much delayed compared with that in GC (Malkova et al, 2005; Jain et al, 2009; Donnianni & Symington, 2013). Thus, modulating DNA replication initiation by REC is proposed to be the key step for the activation of GC or BIR in yeast (Jain et al, 2009).
Long track gene conversion, defined in mammalian cells with GC track length longer than 1–2 kb (Johnson & Jasin, 2000; Puget et al, 2005), is similar to gap repair with relatively large gap size described in yeast (Paques & Haber, 1999). It has been shown that BIR mechanism is used in yeast for gap repair when the gap size is more than 1–2 kb (Jain et al, 2009; Mehta et al, 2017). We showed that after Cas9 cleavage in the EGFP/STGC‐mCherry/LTGC‐5034 reporter, LTGC (red event), but not STGC (green events), is dependent on POLD3 and PIF1 (Fig 5D, left). Thus, like gap repair with relative large gap size in yeast, LTGC in mammalian cells is also mediated by BIR. These data support the notion that the mechanism to activate BIR at endonuclease‐generated DSBs is conserved from yeast to mammalian cells, and the presence of a single DSB end or two far‐apart DSB ends are likely the signals for REC to activate BIR.
In yeast, BIR replication can proceed for hundreds of kbs to the end of a chromosome (Davis & Symington, 2004; Malkova et al, 2005). However, almost all BIR events (> 95%), scored in the EGFP‐BIR‐5085 reporter after I‐SceI or Cas9 cleavage, fail to complete 3.8 kb of DNA synthesis to reach the second homology, and are completed by end joining (Fig 1C and Appendix Fig S14). The relatively short BIR replication track length and frequent use of end joining to finish BIR in mammalian cells were also observed in the previous study from the Halazonetis laboratory (Costantino et al, 2014). As proposed in yeast, BIR proceeds by a series of strand invasion, DNA synthesis, and strand disassociation steps after one DSB end invades to the homologous template, which causes template switch (Smith et al, 2007). In mammalian cells, such strand invasion, DNA synthesis, and disassociation cycles may also occur during BIR. If the newly synthesized invading strands are disassociated from the template and find the homology to the second end of the break, BIR can be completed by SDSA. However, if the disassociated strand ends cannot find the homology, reinvasion may occur to continue BIR, or end joining is used to terminate the repair. The short BIR track length in mammalian cells could be due to less processive replisomes used for BIR in mammalian cells compared with yeast, or more robust end joining activity in mammalian cells which increases the chance for the disassociated ends to be ligated to the second break end when homology is not there. It will be interesting to determine in mammalian cells whether strand disassociation and reinvasion occur as frequently as in yeast, and whether completing BIR by end joining would prevent template switching caused by BIR‐associated strand reinvasion.
BIR is activated at broken forks without checking DBS ends
Break‐induced replication is implicated in rescuing broken replication forks where seDSBs are generated (Llorente et al, 2008; Anand et al, 2013; Malkova & Ira, 2013). This is supported by the critical role of BIR in replication restart in bacteriophage T4 and Escherichia coli, which have a single replication origin (Formosa & Alberts, 1986; Kogoma, 1997; Mosig, 1998; Marians, 2000). In yeast, BIR is launched upon fork collapse but quickly terminated by merging replication forks or by MUS81‐mediated cleavage of D‐loops (Mayle et al, 2015). BIR is also shown to be used to promote expansion of triple nucleotides that would cause replication fork collapse in yeast (Kim et al, 2017). By using the EGFP‐Flex1‐BIR‐5086 repair reporter, we showed that structure‐prone CFS‐AT sequences such as Flex1 induce BIR at broken forks, especially upon replication and oncogenic stress. Consistent with the role of BIR to repair DSBs upon fork breakage, PIF1‐KO cells are sensitive to HU and APH, and replication restart is impaired in PIF1‐deficient cells.
Presumably, deDSBs can form at collapsed forks if breaks occur with some distance to the fork junctions, and they can also form if adjacent forks converge from the other side (Fig 4A). By using the EGFP‐STGC‐1731 reporter after Cas9n cleavage (Fig 4C, Cas9n) and the EGFP‐Flex1‐STGC‐1541 reporter after HU treatment (Fig 4E), we showed that Flex1‐ and nick‐induced STGC at broken forks is POLD3‐ and PIF1‐dependent, suggesting that at fork‐associated DSBs, the BIR mechanism can be used even for SDSA involving two DSB ends. This is in sharp contrast to STGC used at endonuclease‐generated DSBs which is POLD3‐ and PIF1‐independent (Figs 1H and 4C, Cas9). These data suggest that activation of BIR may be differently regulated at DSBs generated directly by endonucleases or upon fork breakage, and support the model that BIR is activated by replication‐associated DSBs without checking the DSB ends.
By using the EGFP/STGC‐mCherry/LTGC‐5034 reporter, we showed that LTGC is dominant over STGC at broken forks whereas STGC is used more frequently at endonuclease‐generated DSBs (Fig 5B), suggesting a more robust BIR activity with longer repair track present at broken forks. Furthermore, we also found that in the EGFP‐BIR‐5085 reporter, after I‐SceI or Cas9 cleavage, only 3% of BIR events have completed 3.8 kb of DNA synthesis, whereas about 70% of BIR events after Cas9n cleavage show 3.8 kb or longer BIR track length (Fig 2E and Appendix Fig S14). More significantly, all BIR events (100%) at Flex1‐induced BIR after HU treatment have a 3.8 kb or longer track length (Appendix Fig S14). Thus, the BIR track length at broken forks appears to be much longer than that at endonuclease‐generated DSBs, which could be due to more processive DNA synthesis associated with BIR at broken forks. It remains to be determined the exact extent of BIR track length (e.g., beyond 3.8 kb in our reporter) on broken forks and whether the BIR migrating D‐loops could eventually convert to replication forks after MUS81 cleavage as described in yeast (Mayle et al, 2015).
Models for BIR activation at endonuclease‐induced DSBs and fork‐associated DSBs
We propose that at replication‐independent DSBs, such as those created by site‐specific endonucleases, REC is required to detect the need for BIR and assemble BIR replisomes to launch BIR (Fig 4G, left). However, upon fork breakage, BIR is activated immediately without a requirement to check the DSB ends by REC, and BIR is used not only for seDBSs but also for STGC at DSBs with two ends (Fig 4G, right).
The key step to launch BIR is probably to assemble BIR replisomes. At replication‐independent DSBs, selection of GC or BIR is controlled by REC based on the detection of seDSBs or deDSBs. The GC replisomes are available at DSBs, but it may take some time for REC to decide that BIR is the choice and then start to assemble BIR replisomes, which would cause a delay to initiate BIR DNA synthesis as proposed in yeast (Jain et al, 2009). Alternatively, activation of BIR may happen after the initiation of DNA synthesis; there is no evidence yet that the initiation of DNA synthesis for BIR is also delayed in mammalian cells. In this scenario, following strand invasion, GC replisomes may be used first to synthesize DNA, but if newly synthesized strands are disassociated from the template but cannot find the homology at the second break end, the free ends of the newly synthesized strands may be detected by REC to activate BIR. It is also possible that multiple repeated strand invasion and disassociation cycles can be sensed by REC to launch BIR. Currently, the composition of replisomes to mediate GC (STGC) or BIR/LTGC in mammalian cell has not been well studied yet. One possibility is that additional components such as PIF1 are loaded onto the existing GC replisomes and convert GC replisomes to BIR replisomes. Alternatively, BIR replisomes may be assembled de novo upon REC activation to replace GC replisomes. Besides PIF1, BIR replisomes may include other helicases and replication factors that are not present in the GC replisomes. POLD3 may be a component in both GC replisomes and BIR replisomes, but its activity becomes essential only when BIR is used.
It remains an outstanding question how BIR activation is achieved differently at fork‐associated DSBs and at the replication‐independent DSBs. It is plausible that certain replication factors required for the assembly of BIR replisomes are already present on replication forks or are recruited or modified upon replication stress, and thus, BIR replisomes can be assembled immediately once forks are broken. Along this line, we showed that PCNA and RFC1 are not only required for BIR but also for STGC at broken forks (Fig 5E left and Fig 5F right), whereas these factors are only needed for BIR but not for STGC at I‐SceI‐generated DSBs (Fig 5E right), suggesting that PCNA and RFC are the critical factors to activate the BIR pathway. This is consistent with the previous finding that PCNA and RFC1 act as the initial sensors of telomere damage to activate ALT, which uses the BIR mechanism (Dilley et al, 2016). Importantly, we also found that PIF1 recruitment to Flex1 after HU treatment is dependent on PCNA. We speculate that once DSBs are generated at forks, PCNA immediately recruits PIF1 to assemble BIR replisomes, of which PIF1 is a critical component. In contrast to fork‐associated DSBs, at replication‐independent DSBs, PCNA may need to be recruited or activated first after REC detects a need for BIR, and then assemble BIR replisomes by recruiting factors such as PIF1 to activate BIR. Furthermore, since BIR track length at broken forks is much longer than that at endonuclease‐induced DSBs (Fig 2E and Appendix Fig S14), additional accessory factors such as helicases may be specifically added onto BIR replisomes at broken forks to make BIR replication more processive.
Utilizing different BIR activation mechanisms to repair DSBs that are associated with and without replication can ensure the most efficient BIR usage when it is in need but limits its involvement if unnecessary since BIR is highly mutagenic (Llorente et al, 2008; Malkova & Haber, 2012; Malkova & Ira, 2013). When replication‐independent DSBs are generated, sister chromatids are expected to be used as templates and STGC should be sufficient. BIR with long track DNA synthesis should be inhibited unless under the unusual conditions that DSBs are single‐ended or only one DSB end can be engaged to the template. Thus, at replication‐independent DSBs, BIR is only activated when REC senses a necessity for BIR; this mechanism restricts BIR and prevents BIR‐associated mutagenic effects. However, upon fork breakage, substantial DSBs could be single‐ended and BIR is required for replication restart from these seDSBs. Immediate activation of BIR at collapsed replication forks would allow a quick response in order to rescue replication forks, which is vital for cells. Once BIR‐specific replisomes are assembled upon fork collapse, they can also be used for STGC and LTGC at deDSBs that are formed directly at collapsed forks or after convergence of adjacent forks. By performing DNA combing experiments, we demonstrated that PIF1 is required for efficient replication restart upon replication stress, which supports the idea that the PIF1‐ and POLD3‐dependent BIR mechanism is probably used directly at broken forks rather than post‐replication after fork convergence. Thus, despite that human cells contain abundant replication origins and collapsed forks presumably can be rescued by converging forks, BIR is still used as an important mechanism to repair broken forks in mammalian cells.
Defining BIR in mammalian cells
Based on the study in yeast, BIR is defined as a specialized HR mechanism that is used at seDSBs or when only one DSB end can find its homology (Llorente et al, 2008; Anand et al, 2013; Malkova & Ira, 2013). BIR has a unique dependence on a non‐essential Polδ subunit Pol32, and this is used as a criterion to distinguish BIR from gene conversion. In mammalian cells, POLD3 dependence is also used as a feature to define BIR. MiDAS and ALT, which exhibit POLD3‐dependent DNA synthesis, are thought to utilize BIR (Minocherhomji et al, 2015). In yeast, BIR replication track can be as long as hundreds of kbs (Davis & Symington, 2004; Malkova et al, 2005). However, in mammalian cells, BIR track length is short at endonuclease‐generated DSBs, rarely exceeding 3.8 kb, and BIR can be completed by SDSA or by end joining (Fig 1C and Appendix Fig S14). Thus, continuous replication for extremely long tracks cannot be used as a key feature to judge of whether BIR is used in mammalian cells. Additionally, we found that BIR at broken forks is immediately activated in mammalian cells regardless of the configuration of DSB ends and is even used for STGC involving two DSB ends. Therefore, the model of BIR activation by seDSBs may not be applied to fork‐associated DSBs. It remains to be determined whether the same mechanism found in mammalian cells is used in yeast for BIR activation at fork‐associated DSBs.
Taking all into account, in mammalian cells, BIR is initiated by one end homology search and proceeds by POLD3‐dependent DNA synthesis, which can be terminated by SDSA (BIR/SDSA) or end joining (BIR‐EJ). Additionally, during the repair of broken forks, BIR D‐loops may be converted to replication forks after MUS81 cleavage. PIF1 has a conserved role in BIR in mammalian cells and is likely an important component of the BIR replisomes. It is worth to note that BIR/EJ that we have described is initiated with homology invasion and probably completed by MMEJ (Appendix Fig S2B), and this is different from previously described microhomology‐mediated BIR (MMBIR), which is launched by microhomology search (Hastings et al, 2009; Malkova & Ira, 2013).
We also tested the role of other HR players in BIR. As scored by the EGFP‐BIR‐5085 and EGFP‐Flex1‐BIR‐5086 reporters, which carry 1.3 kb homology for strand invasion, BIR at both endonuclease‐generated DSBs (Cas9 and I‐SceI) and at broken forks (Cas9n and Flex1), shows RAD51 dependence, even for the BIR/EJ events, and this is consistent with the previous observation by Halazonetis' laboratory (Costantino et al, 2014). Depletion of BRCA1, BRCA2 or CtIP also results in reduced BIR, which is in line with the requirement of RAD51 and end resection for BIR (Appendix Figs S2C and D, and S9A). It was shown previously that when BRCA1, CtIP, RAD51, or other RAD51 paralogs are deficient, STGC and LTGC are both reduced after I‐SceI cleavage in mouse embryonic stem (ES) cells, with a more significant reduction of STGC than LTGC, leading to an increased ratio of LTGC/STGC and a bias toward LTGC (Nagaraju et al, 2006; Nagaraju et al, 2009; Chandramouly et al, 2013). However, using the EGFP/STGC‐mCherry/LTGC‐5034 competition reporter in both U2OS cells (Appendix Fig S9A) and HCT116 cells (Appendix Fig S9B), we did not observe a significant increase in the ratio of LTGC/STGC when BRCA1, BRCA2, RAD51, or CtIP is depleted. This difference could be the result of using different types of cells; in mouse ES cells, HR is super active and is the dominant DSB repair pathway, whereas in human somatic cells, HR activity is relatively low and non‐homologous end joining is used more frequently. It is possible that the BRCA1 pathway has a unique activity in suppression of LTGC when HR is elevated in ES cells.
PIF1‐dependent BIR is important for protecting structure‐prone DNA sequences
It has been shown in yeast that Pif1 is important for BIR through establishing migrating D‐loops and facilitating extensive DNA synthesis (Saini et al, 2013; Wilson et al, 2013). In this study, we demonstrated that human PIF1 has a conserved function in BIR and is required for BIR not only at DSBs generated by endonucleases but also upon fork breakage. In this aspect, we found that PIF1 is required for repairing DSBs formed at CFS‐ATs, such as Flex1. Although multiple characteristics of CFSs contribute to their fragility, it has been shown that replication often stalls at AT‐rich sequences in CFSs, causing DSB formation (Zhang & Freudenreich, 2007; Ozeri‐Galai et al, 2011; Wang et al, 2014; Wang et al, 2018), and CFS‐ATs is considered as one of the important elements contributing to CFS instability (Irony‐Tur Sinai & Kerem, 2019; Irony‐Tur Sinai et al, 2019). Oncogene overexpression stimulates BIR at CFS‐ATs (Fig 3D), suggesting a critical role of BIR in protecting structure‐prone DNA sequences in our genome including CFSs during oncogenesis.
In our previous study, we showed that FANCM is important for removing DNA secondary structures at CFS‐ATs and preventing DSB formation there (Wang et al, 2018). In this study, we further demonstrated that inactivation of FANCM leads to increased BIR at CFS‐ATs, which depends on PIF1 and POLD3. In accordance with the notion that BIR is important for repairing fork‐associated DSBs, simultaneous inactivation of FANCM and PIF1 causes synthetic lethality. DNA secondary structures are abundantly present in the human genome; for instance, more than 700,000 sequences are predicted to have the potential to form G quadruplexes (G4s) (Rhodes & Lipps, 2015). The synthetic lethal interactions between FANCM and PIF1 suggest that the concerted roles of preventing DSB formation by FANCM and promoting BIR for repair‐coupled replication restart by PIF1 (Fig 6C) are important for protecting the integrity of genomic sites with DNA secondary structures.
MiDAS often occurs at CFSs upon replication stress, and its dependence on POLD3 suggests that BIR is involved (Minocherhomji et al, 2015; Bhowmick et al, 2016). However, the DNA lesion structures that induce MiDAS and the exact mechanism of MiDAS to repair DNA lesions are not completely understood. One possibility is that CFSs often replicate late, and a portion of DSBs generated upon replication stress are carried over to early mitosis for repair. Alternatively, replication at CFSs is often disturbed upon replication stress, causing incomplete DNA replication when cells enter mitosis (Glover et al, 2017). MUS81 cleaves under‐replicated DNA, resulting in DNA gaps that involve MiDAS for repair (Naim et al, 2013; Ying et al, 2013; Minocherhomji et al, 2015). We showed that MiDAS is dependent on PIF1, further supporting the notion that MiDAS is mediated by BIR. However, in contrast to RAD51‐dependent BIR observed in synchronized cells, MiDAS is RAD51 independent. This difference is likely due to compromised HR activity in mitosis. It has been shown that while DSBs are still sensed and γH2AX, MRE11, and MDC1 foci are localized to mitotic DSBs (Kato et al, 2008; Giunta et al, 2010; Gomez‐Godinez et al, 2010; van Vugt et al, 2010; Britton et al, 2013), the damage signaling downstream of MDC1 for recruitment of RNF8, RNF168, and BRCA1 is prohibited in mitosis (Nelson et al, 2009; Giunta et al, 2010; van Vugt et al, 2010). Extensive end resection, CHK1 activation, and RAD51 filament formation are abrogated, resulting in suppression of HR in mitotic cells (Esashi et al, 2005; Freire et al, 2006; Ayoub et al, 2009; Peterson et al, 2011; Krajewska et al, 2013). Along this line, using the same EGFP‐BIR‐5085 reporter carrying 1.3 kb homology for strand invasion, we showed that while BIR is RAD51 dependent in asynchronized cells, it becomes RAD51 independent in mitotic cells (Fig 1D and E). Consistent with the role of PIF1 in MiDAS, we found that upon replication stress, PIF1 is recruited to CFSs (Fig 7B) and is important for preventing chromosomal breakage upon replication stress (Fig 7D).
PIF1‐dependent BIR is important for tumor suppression and PIF1 can serve as an anti‐cancer target
We showed that human PIF1 is important for replication restart upon replication stress and for preventing chromosomal breakage, suggesting a critical role of PIF1 in the maintenance of genome stability. Along these lines, we found that a breast cancer predisposition mutant PIF1‐L319P (Chisholm et al, 2012) is defective in BIR/LTGC and replication restart and causes chromosomal breakage. L319P is present in the conserved PIF1 signature motif, and the corresponding mutant pfh1‐L430P in Schizosaccharomyces pombe is inviable (Chisholm et al, 2012), suggesting that this specific site is critical for PIF1 function. The connection of breast cancer predisposition to impaired function of PIF1 implies that BIR may be linked to tumor suppression.
While loss of PIF1 induces genome instability, contributing to tumorigenesis, BIR is also stimulated by oncogenic replication stress. We also showed that loss of PIF1 causes cell death upon oncogene expression, suggesting that tumor cells are addicted to, or rely on, PIF1‐dependent BIR for survival. This is consistent with our observation that BIR plays an essential role in repairing DSBs generated upon fork collapse. Since cancer cells are often under replicative stress (Lecona & Fernandez‐Capetillo, 2014), it is conceivable that targeting the BIR pathway could be exploited as an effective therapeutic approach to specifically kill tumor cells and enhance the efficacy of genotoxic agents that cause replication stress. ATR has been shown to have two sides of tumor suppression and tumor maintenance function, and its inhibitors are being used as antitumor agents (Murga et al, 2011; Campaner & Amati, 2012; Schoppy et al, 2012; Karnitz & Zou, 2015). Our study suggests that PIF1 is another player acting as a double‐edged sword during tumor initiation and tumor maintenance; genome instability caused by PIF1 deficiency promotes tumorigenesis, while PIF1 is required for tumor maintenance after tumor establishment. Since PIF1 is a non‐essential gene in normal cells, it could serve as a good target for cancer treatment with low toxicity. The synthetic lethal interaction between PIF1 and FANCM also lays a foundation for targeted treatment of FANCM‐deficient tumors through inactivating PIF1, which is especially relevant breast cancers since FANCM deficiency is often associated with breast cancer especially hard‐to‐treat triple negative breast tumors (Kiiski et al, 2014; Kiiski et al, 2017; Neidhardt et al, 2017).
Materials and Methods
Cell cultures
U2OS (from ATCC), Lenti‐X 293T (from Clontech), HCT116 WT, and FANCM‐KO (Wang et al, 2013) cells were cultured in DMEM with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37°C with 5% CO2.
Plasmids construction and generation of reporter cell lines
A FLAG tag was placed to the N terminus of human PIF1 cDNA, kindly provided by Cyril Sanders (Sanders, 2010), and FLAG‐PIF1 was inserted into the pCDH‐CMV‐MCS‐EF1‐NEO lentiviral vector, which was derived from pCDH‐CMV‐MCS‐EF1‐PURO (System Biosciences). Doxycycline‐inducible Cas9 plasmid pCW‐Cas9 was obtained from Addgene (#50661). PIF1‐E307Q and L319P mutants and Cas9 D10A mutant were generated by site‐directed mutagenesis. sgRNAs (listed in the Appendix Table S1) for generating DSBs or nicks at the sites close to the I‐SceI recognition site in various repair reporters were cloned into the lentiguide‐puro vector (Addgene #52963). H‐RAS‐V12‐FLAG and CyclinE‐HA were cloned into the pBabe‐puro vector (Addgene #1764). I‐SceI endonuclease was cloned into pCDH‐CMV‐MCS‐EF1‐PURO lentiviral vector (System Biosciences).
The EGFP‐STGC‐1731 and EGFP‐Flex1‐STGC‐1541 reporters were described previously (Wang et al, 2014). To generate the EGFP‐BIR‐5085 reporter, EGFP was split into the EG and FP two parts. As shown in Fig 1A, the N‐terminal EGFP fragment EG, including the CMV promoter, was fused to the intron1‐Luc‐I‐SceI fragment (Luc: 1.3 kb Cypridina luciferase fragment). The C‐terminal EGFP fragment FP, including the 3′ SV40 poly(A) tail, was fused to the Luc‐intron2‐FP fragment and placed in front of the EG‐intron1‐Luc‐I‐SceI cassette. Intron1 and intron2 are derived from insulin‐like growth factor 2 mRNA‐binding protein 1 (IGF2BP1)(Wang et al, 2004). To obtain the EGFP‐Flex1‐BIR‐5086 reporter (as shown in Fig 3A), a 0.3 kb Flex1(AT)34 fragment (Zhang & Freudenreich, 2007; Wang et al, 2014) was inserted at the I‐SceI site in the EGFP‐BIR‐5085 reporter. The STGC and LTGC competition reporter, EGFP/STGC‐mCherry/LTGC‐5034 (Fig 5A), was generated by fusing the EGFP internal fragment (iEGFP) present in the EGFP‐STGC‐1731 reporter (Fig 1H) in frame with mCherry cDNA through the T2A sequence(Szymczak et al, 2004), and the resulting iEGFP‐T2A‐mCherry cassette was placed in front of the CMV‐EGFP::I‐Sce1/stop cassette used in the EGFP‐STGC‐1731 reporter. The Flex1‐EGFP/STGC‐mCherry/LTGC‐5304 reporter was generated by inserting Flex1(AT)34 to the I‐SceI site in the EGFP/STGC‐mCherry/LTGC‐5034 reporter. The backbone of all the reporters is pUC19 with hygromycin selection marker.
The EGFP‐STGC‐1731 (originally called EGFP‐HR), EGFP‐Flex1‐STGC‐1541 (originally called EGFP‐HR‐Flex) reporter cell lines were constructed previously (Wang et al, 2014; Wang et al, 2018). The EGFP‐BIR‐5085, EGFP‐Flex1‐BIR‐5086, EGFP/STGC‐mCherry/LTGC‐5034, and Flex1‐EGFP/STGC‐mCherry/LTGC‐5304 reporter cell lines were generated by transfection of the reporter plasmids, followed hygromycin B (17 μg/ml, 5 days) selection. Clones with stable integration of single reporter cassette in the genome were confirmed by Southern blot analysis.
Generation of PIF1 KO cell line by CRISPR
Two sgRNAs targeting different sites in human PIF1 Exon5, which flank the critical catalytic site E307, were individually subcloned into the pSpCas9(BB)‐2A‐Puro (PX459) V2.0 vector (Addgene #62988). To knockout (KO) PIF1, these two plasmids each containing one sgRNA were transfected together to the target cell lines by Lipofectamine 2000. Transfected cells were selected with puromycin (2 μg/ml, 2 days), followed by single clone isolation. PIF1‐KO clones were confirmed by PCR of genomic DNA using primers (5′‐CTGGGTGACAGAAAGAGACCTT and 5′‐TTGTTCTGCTGCCGGACAGCTCTG) followed by sequencing. The obtained PIF1‐KO clones contain a deletion of 100 bp in Exon5 between the two sgRNA sites, which have deleted the PIF1 catalytic site and caused open reading frame shift (Appendix Fig S3B).
shRNA interference
Silencing of indicated endogenous genes was performed by lentiviral infection using pLKO.1‐blast vector (Addgene #26655) or pLKO.1‐TRC cloning vector (Addgene #10878) to express corresponding shRNAs followed by blasticidin (10 μg/ml, 2 days) selection or puromycin (2 μg/ml, 2 days) selection. The shRNA sequences targeting different genes are listed in Appendix Table S2. The knockdown efficiency of indicated genes by shRNA was verified by Western blot of whole‐cell lysates or RT–qPCR using primers listed in Appendix Table S3.
RT–qPCR
Total RNA was extracted from cell lines using the RNeasyMini Kit (Qiagen). cDNA was synthesized through reverse transcription using the iScript cDNA synthesis kit (Bio‐Rad). The iTaq Universal SYBR Green Supermix (Bio‐Rad) was used for qPCR on C1000 Thermal Cycler (Bio‐Rad).
Immunoblotting
Cells from a confluent 6‐cm plate were lysed in NETN buffer (20 mM Tris–HCl, pH 8.0, 100 mM NaCl, 0.5 mM EDTA, 0.5% NP‐40). After adding 2X loading buffer (100 mM Tris–Cl, pH 6.8, 4% SDS, 0.2% bromophenol blue, 20% glycerol, 200 mM DTT), the samples were heated at 95°C for 5 min and separated on 6–15% SDS–PAGE. Antibodies used are: BRCA1 (sc‐6954, Santa Cruz Biotechnology), BRCA2 (sc‐293185, Santa Cruz Biotechnology), CTIP (A300‐487A, Bethyl), RAD51 (sc‐398587, Santa Cruz Biotechnology), RAD52 (sc‐365341, Santa Cruz Biotechnology), POLD3 (ab182564, Abcam), PCNA (NA03, Millipore), FLAG (F1804, Sigma‐Aldrich), HA (MMS‐101P, Covance), RFC1 (A300‐320A, Bethyl), MUS81 (sc‐376661, Santa Cruz Biotechnology), KU70 (sc‐17789, Santa Cruz Biotechnology), Peroxidase AffiniPure Goat Anti‐Mouse IgG(H + L; #115‐035‐146, Jackson Immuno Research Labs), and Peroxidase AffiniPure Goat Anti‐Rabbit IgG (H + L; #111‐035‐144, Jackson Immuno Research Labs). Antibodies against FANCM were kindly provided by Dr. Weidong Wang (NIH).
Recombination assay using EGFP‐based reporters
To measure the repair of I‐SceI‐generated DSBs, the reporter cell lines were infected with lentiviruses expressing I‐SceI from the pCDH‐CMV‐I‐SceI‐EF1‐PURO vector, followed by puromycin selection (2 μg/ml, 2 days). Three days after the drug selection, the percentage of EGFP‐positive cells was analyzed by FACS on BD Accuri C6 flow cytometer. To assay HU‐ or APH‐induced recombination, EGFP‐Flex1‐STGC‐1541, EGFP‐Flex1‐BIR‐5086, and Flex1‐EGFP/STGC‐mCherry/LTGC‐5304 cell lines were treated by HU 2 mM or APH 0.4 μM as indicated. FACS was performed 3 days after drug removal.
To obtain Tet‐On Cas9 (or Cas9n)/sgRNA cell lines, U2OS EGFP‐STGC‐1731, EGFP‐BIR‐5085, or EGFP/STGC‐mCherry/LTGC‐5034 reporter cell lines were infected with lentiviruses expressing Cas9 (or Cas9 D10A) and sgRNA from the lentiguide‐puro vector (Addgene #52963). Single clones were selected for 2 days in puromycin (2 μg/ml) and picked up 10 days later. Single clones were screened by FACS to determine the percentage of EGFP‐ or mCherry‐positive cells before and 2 days after Dox (5 μg/ml) induction. Clones with the lowest background and high levels of recombination after Dox induction were retained. To measure the repair of Cas9 (or Cas9n)/sgRNA generated DSBs or nick‐derived DSBs, Doxycycline (Dox, 5 μg/ml) was added to induce the expression of Cas9 (or Cas9n), and FACS analysis was performed 2 days after Dox induction.
To analyze EGFP‐BIR‐5085 in mitotic cells, U2OS (EGFP‐BIR‐5085) cells carrying Tet‐On Cas9/sgRNA‐5085 were synchronized by treatment with Nocodazole (0.3 μM) for 40 h followed by the addition of Dox (5 μg/ml). FACS analysis was performed 48 h later to determine the percentage of EGFP cells. Cell cycle profiles were analyzed by FACS following propidium iodide (PI) staining.
To analyze the repair junction sequences or the track length of each repair events, EGFP‐ or mCherry‐positive cells were collected by FACS sorting, and allowed to grow for 3–4 weeks to form single clones. Genomic DNA extracted from the single clones was used for Southern blot analysis or as PCR template for sequencing repair junctions. The repair track length was determined by aligning the PCR sequencing results with the predicted BIR/SDSA repair product.
Growth curve and drug sensitivity assay
To monitor cell growth, cells plated in 10‐cm plates were trypsinized and counted every 24 h and growth curves were plotted after cell number was normalized to that of the first day. To determine drug sensitivity, cells were plated in 96‐well plates (5,000 cells/well) and treated with indicated drug at various concentrations for 72 h. Cell viability was determined by MTS‐based (Promega, G3582) readout at 490 nm on Spectramax M2 (Molecular Devices).
DNA fiber assay
Cells were incubated with 25 µM CldU for 30 min, followed by treatment with 2 mM HU for 2 h, and then incubated with 250 µM IdU for 30 min. DNA fiber was prepared using DNA Extraction Kit (Genomic Vision). Briefly, labeled cells were washed with PBS and resuspended in low melting point agarose. After solidification, agarose plugs were treated with Proteinase K (1 mg/ml, overnight) followed by agarose digestion (agarase, three units, overnight). DNA released from agarose plugs was stretched on silanized coverslips slowly. To detect CldU and IdU on DNA fibers, coverslips were incubated with rat anti‐BrdU (ab6326, Abcam) and mouse anti‐BrdU (347580, BD Biosciences) primary antibodies for 2 h, followed by incubation with Alexa 594 anti‐rat (A11007, Invitrogen) and Alexa 488 anti‐mouse (A11029, Invitrogen) secondary antibodies for 1 h at room temperature. Coverslips were washed with PBS with 0.1% Tween 20 and mounted in Prolong Gold antifade reagent (Invitrogen). Images were acquired with a LSM 780 confocal laser scanning microscope and analyzed using ImageJ software (NIH, USA).
MiDAS
MiDAS was performed as described (Garribba et al, 2018). U2OS cells were treated with 0.4 µM APH for 8 h, then synchronized to late G2 phase by addition of 7 µM RO‐3306 and incubation for another 8 h in the presence of APH. Cells were washed three times with cold PBS within 5 min and then released into the medium with 20 µM EdU and 0.1 µg/ml Colcemid at 37°C for 60 min. Cells were collected and resuspended in 5 ml 75 mM KCl for 20 min at 37°C. Swollen mitotic cells were collected and fixed in 5 ml fixative solution (methanol/acetic acid: 3/1) at room temperature for 30 min. Fixed cells were collected and resuspended in 100 µl fixative solution and dropped onto pre‐hydrated slides. After aging at room temperature overnight, EdU was detected using Click‐IT Plus EdU Alexa Fluor 488 Imaging Kit. Images were acquired using a LSM 780 confocal laser scanning microscope.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed as described (Wang et al, 2014). Briefly, cells were cross‐linked with 1% formaldehyde for 10 min at room temperature and glycine was added to the concentration of 0.125 M to stop the reaction. After washing twice with cold PBS, collected cells were resuspended in the lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris–HCl, pH 8.1) supplemented with the protease inhibitor cocktail (“PIC”, cOmplete, Roche) and subject to sonication to break chromatin. After centrifugation, the supernatant was collected and pre‐cleared with Protein A/G Sepharose beads. IP was performed overnight at 4°C with specific antibodies followed by washing with TSE I (0.1% SDS, 1% Triton X‐100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.1, 150 mM NaCl), TSE II (0.1% SDS, 1% Triton X‐100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.1, 500 mM NaCl), buffer III (0.25 M LiCl, 1% NP‐40, 1% Sodium deoxycholate, 1 mM EDTA, 10 mM Tris–HCl, pH 8.1), and TE. The protein–DNA complex was then eluted from beads by 120 μl elution buffer (1% SDS, 0.1 M NaHCO3). Cross‐linking was reversed by adding 4 μl of 5 M NaCl and incubating at 65°C for 6 h, followed by adding 2 μl of 20 mg/ml proteinase K for 2‐h digestion at 42°C. DNA was extracted by QIAquick kit (QIAGEN). Recovered DNA was analyzed by qPCR. GAPDH locus was used as a control to show the specificity of protein binding to the specific locus. The primers used for ChIP: Flex1 (Flex1‐F 5′GGCAGTACATCAATGGGCGTG, and Flex1‐R 5′CCTTTAGTGAGGGTTAATTGCGCG); FRA3B (3B‐F 5′ CACTTCCTAACAGGCCCAAA and 3B‐R 5′ CCTCCACTTCTCCTCCCTCT); FRA16D (16D‐F5′ TCCTGTGGAAGGGATATTTA and 16D‐R 5′ CCCCTCATATTCTGCTTCTA); GAPDH (GAPDH‐ F 5′CCCTCTGGTGGTGGCCCCTT and GAPDH‐R 5′GGCGCCCAGACACCCAATCC). Anti‐FLAG antibody (F3165, Sigma‐Aldrich) and Anti‐phospho‐H2AX (Ser139) antibody (07‐164, EMD Millipore) were used for ChIP of FLAG‐PIF1 and γH2AX, respectively.
Metaphase chromosome spread
Metaphase chromosome analysis was performed according to standard protocols. Briefly, cells were exposed to 0.4 μM APH for 18 h, followed by treatment of 0.1 µg/ml colcemid at 37°C for 45 min. Collected cells were resuspended in 75 mM KCl hypotonic solution and incubated at 37°C for 30 min, followed by several changes of fixative solution (methanol/acetic acid: 3/1). Cells were dropped onto slides and incubated for 2 h at 60°C prior to Giemsa staining. Breaks and gaps were quantified on Giemsa‐stained metaphases. Fifty metaphases per sample were scored for the number of overall chromosome gaps and breaks.
Statistical analysis
Excel was used for the statistical analyses. Two‐tailed non‐paired parameters were applied in Student's t‐test to analyze the significance of the differences between samples. In all experiments, error bars represent standard deviation (SD) of at least three independent experiments. The P value is indicated as *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and n.s. (not significant) P > 0.05.
Author contributions
SL, HW, SJ and XW designed and performed experiments, and analyzed the data; and ZiW, JL and SL performed some experiments; LT and ZeW participated in the initial design of the parental constructs of the reporters; TC contributed to student supervision. XW is responsible for the overall project's planning and experimental designing. XW, SL and HW wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Appendix
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Appendix
Source Data for Figure 7
Acknowledgements
We would like to thank Dr. Cyril Sanders (University of Sheffield, UK) for providing cDNA of human PIF1. We would like to thank Dr. Lei Li (University of Texas, MD Anderson Cancer Center) for providing HCT116 FANCM‐KO cell lines. We thank Dr. Weidong Wang for providing anti‐FANCM antibody. Plasmids pLKO.1‐blast vector (#26655), pLKO.1‐TRC cloning vector (#10878), pCW‐Cas9 (#50661), pSpCas9(BB)‐2A‐Puro (PX459) V2.0 (#62988), pBabe‐puro (Addgene #1764), and lentiguide‐puro (#52963) are from Addgene. This study is funded by NIH grants CA244912, CA187052, CA197995, and GM080677 to X.W.
The EMBO Journal (2021) 40: e104509.
Data availability
This study includes no data deposited in external repositories. Uncropped Western blots and microscope images were supplied as Source Data.
References
- Aguilera A, Gomez‐Gonzalez B (2008) Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet 9: 204–217 [DOI] [PubMed] [Google Scholar]
- Anand RP, Lovett ST, Haber JE (2013) Break‐induced DNA replication. Cold Spring Harb Perspect Biol 5: a010397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub N, Rajendra E, Su X, Jeyasekharan AD, Mahen R, Venkitaraman AR (2009) The carboxyl terminus of Brca2 links the disassembly of Rad51 complexes to mitotic entry. Curr Biol 19: 1075–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC et al (2006) Oncogene‐induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444: 633–637 [DOI] [PubMed] [Google Scholar]
- Bhowmick R, Minocherhomji S, Hickson ID (2016) RAD52 facilitates mitotic DNA synthesis following replication stress. Mol Cell 64: 1117–1126 [DOI] [PubMed] [Google Scholar]
- Bochman ML, Sabouri N, Zakian VA (2010) Unwinding the functions of the Pif1 family helicases. DNA Repair (Amst) 9: 237–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton S, Coates J, Jackson SP (2013) A new method for high‐resolution imaging of Ku foci to decipher mechanisms of DNA double‐strand break repair. J Cell Biol 202: 579–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campaner S, Amati B (2012) Two sides of the Myc‐induced DNA damage response: from tumor suppression to tumor maintenance. Cell Div 7: 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramouly G, Kwok A, Huang B, Willis NA, Xie A, Scully R (2013) BRCA1 and CtIP suppress long‐tract gene conversion between sister chromatids. Nat Commun 4: 2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm KM, Aubert SD, Freese KP, Zakian VA, King MC, Welcsh PL (2012) A genomewide screen for suppressors of Alu‐mediated rearrangements reveals a role for PIF1. PLoS One 7: e30748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, Halazonetis TD (2014) Break‐induced replication repair of damaged forks induces genomic duplications in human cells. Science 343: 88–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AP, Symington LS (2004) RAD51‐dependent break‐induced replication in yeast. Mol Cell Biol 24: 2344–2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, Malkova A (2011) Break‐induced replication is highly inaccurate. PLoS Biol 9: e1000594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A et al (2006) Oncogene‐induced senescence is a DNA damage response triggered by DNA hyper‐replication. Nature 444: 638–642 [DOI] [PubMed] [Google Scholar]
- Dilley RL, Verma P, Cho NW, Winters HD, Wondisford AR, Greenberg RA (2016) Break‐induced telomere synthesis underlies alternative telomere maintenance. Nature 539: 54–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnianni RA, Symington LS (2013) Break‐induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci USA 110: 13475–13480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott B, Richardson C, Winderbaum J, Nickoloff JA, Jasin M (1998) Gene conversion tracts from double‐strand break repair in mammalian cells. Mol Cell Biol 18: 93–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC (2005) CDK‐dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 434: 598–604 [DOI] [PubMed] [Google Scholar]
- Formosa T, Alberts BM (1986) DNA synthesis dependent on genetic recombination: characterization of a reaction catalyzed by purified bacteriophage T4 proteins. Cell 47: 793–806 [DOI] [PubMed] [Google Scholar]
- Freire R, van Vugt MA, Mamely I, Medema RH (2006) Claspin: timing the cell cycle arrest when the genome is damaged. Cell Cycle 5: 2831–2834 [DOI] [PubMed] [Google Scholar]
- Gagou ME, Ganesh A, Phear G, Robinson D, Petermann E, Cox A, Meuth M (2014) Human PIF1 helicase supports DNA replication and cell growth under oncogenic‐stress. Oncotarget 5: 11381–11398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garribba L, Wu W, Ozer O, Bhowmick R, Hickson ID, Liu Y (2018) Inducing and detecting mitotic DNA synthesis at difficult‐to‐replicate loci. Method Enzymol 601: 45–58. [DOI] [PubMed] [Google Scholar]
- Giunta S, Belotserkovskaya R, Jackson SP (2010) DNA damage signaling in response to double‐strand breaks during mitosis. J Cell Biol 190: 197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover TW, Wilson TE, Arlt MF (2017) Fragile sites in cancer: more than meets the eye. Nat Rev Cancer 17: 489–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Godinez V, Wu T, Sherman AJ, Lee CS, Liaw LH, Zhongsheng Y, Yokomori K, Berns MW (2010) Analysis of DNA double‐strand break response and chromatin structure in mitosis using laser microirradiation. Nucleic Acids Res 38: e202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR (2009) A microhomology‐mediated break‐induced replication model for the origin of human copy number variation. PLoS Genet 5: e1000327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irony‐Tur Sinai M, Kerem B (2019) Genomic instability in fragile sites‐still adding the pieces. Genes Chromosomes Cancer 58: 295–304 [DOI] [PubMed] [Google Scholar]
- Irony‐Tur Sinai M, Salamon A, Stanleigh N, Goldberg T, Weiss A, Wang YH, Kerem B (2019) AT‐dinucleotide rich sequences drive fragile site formation. Nucleic Acids Res 47: 9685–9695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Sugawara N, Lydeard J, Vaze M, Tanguy Le Gac N, Haber JE (2009) A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double‐strand break repair. Genes Dev 23: 291–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M, Rothstein R (2013) Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 5: a012740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RD, Jasin M (2000) Sister chromatid gene conversion is a prominent double‐strand break repair pathway in mammalian cells. EMBO J 19: 3398–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnitz LM, Zou L (2015) Molecular pathways: targeting ATR in cancer therapy. Clin Cancer Res 21: 4780–4785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato TA, Okayasu R, Bedford JS (2008) Comparison of the induction and disappearance of DNA double strand breaks and gamma‐H2AX foci after irradiation of chromosomes in G1‐phase or in condensed metaphase cells. Mutat Res 639: 108–112 [DOI] [PubMed] [Google Scholar]
- Kiiski JI, Pelttari LM, Khan S, Freysteinsdottir ES, Reynisdottir I, Hart SN, Shimelis H, Vilske S, Kallioniemi A, Schleutker J et al (2014) Exome sequencing identifies FANCM as a susceptibility gene for triple‐negative breast cancer. Proc Natl Acad Sci USA 111: 15172–15177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiiski JI, Tervasmaki A, Pelttari LM, Khan S, Mantere T, Pylkas K, Mannermaa A, Tengstrom M, Kvist A, Borg A et al (2017) FANCM mutation c.5791C>T is a risk factor for triple‐negative breast cancer in the Finnish population. Breast Cancer Res Treat 166: 217–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JC, Harris ST, Dinter T, Shah KA, Mirkin SM (2017) The role of break‐induced replication in large‐scale expansions of (CAG)n/(CTG)n repeats. Nat Struct Mol Biol 24: 55–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogoma T (1997) Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol Mol Biol Rev 61: 212–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewska M, Heijink AM, Bisselink YJ, Seinstra RI, Sillje HH, de Vries EG, van Vugt MA (2013) Forced activation of Cdk1 via wee1 inhibition impairs homologous recombination. Oncogene 32: 3001–3008 [DOI] [PubMed] [Google Scholar]
- Lecona E, Fernandez‐Capetillo O (2014) Replication stress and cancer: it takes two to tango. Exp Cell Res 329: 26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente B, Smith CE, Symington LS (2008) Break‐induced replication: what is it and what is it for? Cell Cycle 7: 859–864 [DOI] [PubMed] [Google Scholar]
- Lydeard JR, Jain S, Yamaguchi M, Haber JE (2007) Break‐induced replication and telomerase‐independent telomere maintenance require Pol32. Nature 448: 820–823 [DOI] [PubMed] [Google Scholar]
- Malkova A, Naylor ML, Yamaguchi M, Ira G, Haber JE (2005) RAD51‐dependent break‐induced replication differs in kinetics and checkpoint responses from RAD51‐mediated gene conversion. Mol Cell Biol 25: 933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova A, Haber JE (2012) Mutations arising during repair of chromosome breaks. Annu Rev Genet 46: 455–473 [DOI] [PubMed] [Google Scholar]
- Malkova A, Ira G (2013) Break‐induced replication: functions and molecular mechanism. Curr Opin Genet Dev 23: 271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marians KJ (2000) PriA‐directed replication fork restart in Escherichia coli . Trends Biochem Sci 25: 185–189 [DOI] [PubMed] [Google Scholar]
- Mayle R, Campbell IM, Beck CR, Yu Y, Wilson M, Shaw CA, Bjergbaek L, Lupski JR, Ira G (2015) DNA REPAIR. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science 349: 742–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Beach A, Haber JE (2017) Homology requirements and competition between gene conversion and break‐induced replication during double‐strand break repair. Mol Cell 65: 515–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, Hickson ID (2015) Replication stress activates DNA repair synthesis in mitosis. Nature 528: 286–290 [DOI] [PubMed] [Google Scholar]
- Mosig G (1998) Recombination and recombination‐dependent DNA replication in bacteriophage T4. Annu Rev Genet 32: 379–413 [DOI] [PubMed] [Google Scholar]
- Murga M, Campaner S, Lopez‐Contreras AJ, Toledo LI, Soria R, Montana MF, D'Artista L, Schleker T, Guerra C, Garcia E et al (2011) Exploiting oncogene‐induced replicative stress for the selective killing of Myc‐driven tumors. Nat Struct Mol Biol 18: 1331–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraju G, Odate S, Xie A, Scully R (2006) Differential regulation of short‐ and long‐tract gene conversion between sister chromatids by Rad51C. Mol Cell Biol 26: 8075–8086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraju G, Hartlerode A, Kwok A, Chandramouly G, Scully R (2009) XRCC2 and XRCC3 regulate the balance between short‐ and long‐tract gene conversions between sister chromatids. Mol Cell Biol 29: 4283–4294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naim V, Wilhelm T, Debatisse M, Rosselli F (2013) ERCC1 and MUS81‐EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat Cell Biol 15: 1008–1015 [DOI] [PubMed] [Google Scholar]
- Negrini S, Gorgoulis VG, Halazonetis TD (2010) Genomic instability–an evolving hallmark of cancer. Nat Rev Mol Cell Biol 11: 220–228 [DOI] [PubMed] [Google Scholar]
- Neidhardt G, Hauke J, Ramser J, Gross E, Gehrig A, Muller CR, Kahlert AK, Hackmann K, Honisch E, Niederacher D et al (2017) Association between loss‐of‐function mutations within the FANCM gene and early‐onset familial breast cancer. JAMA Oncol 3: 1245–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson G, Buhmann M, von Zglinicki T (2009) DNA damage foci in mitosis are devoid of 53BP1. Cell Cycle 8: 3379–3383 [DOI] [PubMed] [Google Scholar]
- Nickoloff JA, Sweetser DB, Clikeman JA, Khalsa GJ, Wheeler SL (1999) Multiple heterologies increase mitotic double‐strand break‐induced allelic gene conversion tract lengths in yeast. Genetics 153: 665–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozeri‐Galai E, Lebofsky R, Rahat A, Bester AC, Bensimon A, Kerem B (2011) Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol Cell 43: 122–131 [DOI] [PubMed] [Google Scholar]
- Palmer S, Schildkraut E, Lazarin R, Nguyen J, Nickoloff JA (2003) Gene conversion tracts in Saccharomyces cerevisiae can be extremely short and highly directional. Nucleic Acids Res 31: 1164–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F, Haber JE (1999) Multiple pathways of recombination induced by double‐strand breaks in Saccharomyces cerevisiae . Microbiol Mol Biol Rev 63: 349–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SE, Li Y, Chait BT, Gottesman ME, Baer R, Gautier J (2011) Cdk1 uncouples CtIP‐dependent resection and Rad51 filament formation during M‐phase double‐strand break repair. J Cell Biol 194: 705–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puget N, Knowlton M, Scully R (2005) Molecular analysis of sister chromatid recombination in mammalian cells. DNA Repair (Amst) 4: 149–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes D, Lipps HJ (2015) G‐quadruplexes and their regulatory roles in biology. Nucleic Acids Res 43: 8627–8637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumelioti FM, Sotiriou SK, Katsini V, Chiourea M, Halazonetis TD, Gagos S (2016) Alternative lengthening of human telomeres is a conservative DNA replication process with features of break‐induced replication. EMBO Rep 17: 1731–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini N, Ramakrishnan S, Elango R, Ayyar S, Zhang Y, Deem A, Ira G, Haber JE, Lobachev KS, Malkova A (2013) Migrating bubble during break‐induced replication drives conservative DNA synthesis. Nature 502: 389–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky CJ, Roberts SA, Malc E, Mieczkowski PA, Resnick MA, Gordenin DA, Malkova A (2014) Break‐induced replication is a source of mutation clusters underlying kataegis. Cell Rep 7: 1640–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders CM (2010) Human Pif1 helicase is a G‐quadruplex DNA‐binding protein with G‐quadruplex DNA‐unwinding activity. Biochem J 430: 119–128 [DOI] [PubMed] [Google Scholar]
- Schoppy DW, Ragland RL, Gilad O, Shastri N, Peters AA, Murga M, Fernandez‐Capetillo O, Diehl JA, Brown EJ (2012) Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J Clin Invest 122: 241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CE, Llorente B, Symington LS (2007) Template switching during break‐induced replication. Nature 447: 102–105 [DOI] [PubMed] [Google Scholar]
- Sotiriou SK, Kamileri I, Lugli N, Evangelou K, Da‐Re C, Huber F, Padayachy L, Tardy S, Nicati NL, Barriot S et al (2016) Mammalian RAD52 functions in break‐induced replication repair of collapsed DNA replication forks. Mol Cell 64: 1127–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweetser DB, Hough H, Whelden JF, Arbuckle M, Nickoloff JA (1994) Fine‐resolution mapping of spontaneous and double‐strand break‐induced gene conversion tracts in Saccharomyces cerevisiae reveals reversible mitotic conversion polarity. Mol Cell Biol 14: 3863–3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DAA (2004) Correction of multi‐gene deficiency in vivo using a single 'self‐cleaving' 2A peptide‐based retroviral vector. Nat Biotechnol 22: 589–594 [DOI] [PubMed] [Google Scholar]
- Taghian DG, Nickoloff JA (1997) Chromosomal double‐strand breaks induce gene conversion at high frequency in mammalian cells. Mol Cell Biol 17: 6386–6393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vugt MA, Gardino AK, Linding R, Ostheimer GJ, Reinhardt HC, Ong SE, Tan CS, Miao H, Keezer SM, Li J et al (2010) A mitotic phosphorylation feedback network connects Cdk1, Plk 1, 53BP1, and Chk2 to inactivate the G(2)/M DNA damage checkpoint. PLoS Biol 8: e1000287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB (2004) Systematic identification and analysis of exonic splicing silencers. Cell 119: 831–845 [DOI] [PubMed] [Google Scholar]
- Wang YC, Leung JW, Jiang YJ, Lowery MG, Do H, Vasquez KM, Chen JJ, Wang WD, Li L (2013) FANCM and FAAP24 maintain genome stability via cooperative as well as unique functions. Mol Cell 49: 997–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Li Y, Truong LN, Shi LZ, Hwang PY, He J, Do J, Cho MJ, Li H, Negrete A et al (2014) CtIP maintains stability at common fragile sites and inverted repeats by end resection‐independent endonuclease activity. Mol Cell 54: 1012–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Li S, Oaks J, Ren J, Li L, Wu X (2018) The concerted roles of FANCM and Rad52 in the protection of common fragile sites. Nat Commun 9: 2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, Kwon Y, Xu Y, Chung WH, Chi P, Niu H, Mayle R, Chen X, Malkova A, Sung P et al (2013) Pif1 helicase and Poldelta promote recombination‐coupled DNA synthesis via bubble migration. Nature 502: 393–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Minocherhomji S, Chan KL, Palmai‐Pallag T, Chu WK, Wass T, Mankouri HW, Liu Y, Hickson ID (2013) MUS81 promotes common fragile site expression. Nat Cell Biol 15: 1001–1007 [DOI] [PubMed] [Google Scholar]
- Zhang H, Freudenreich CH (2007) An AT‐rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae . Mol Cell 27: 367–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Appendix
Source Data for Figure 7
Data Availability Statement
This study includes no data deposited in external repositories. Uncropped Western blots and microscope images were supplied as Source Data.