

Abstract
Cells release diverse types of extracellular vesicles (EVs), which transfer complex signals to surrounding cells. Specific markers to distinguish different EVs (e.g. exosomes, ectosomes, enveloped viruses like HIV) are still lacking. We have developed a proteomic profiling approach for characterizing EV subtype composition and applied it to human Jurkat T cells. We generated an interactive database to define groups of proteins with similar profiles, suggesting release in similar EVs. Biochemical validation confirmed the presence of preferred partners of commonly used exosome markers in EVs: CD81/ADAM10/ITGB1, and CD63/syntenin. We then compared EVs from control and HIV‐1‐infected cells. HIV infection altered EV profiles of several cellular proteins, including MOV10 and SPN, which became incorporated into HIV virions, and SERINC3, which was re‐routed to non‐viral EVs in a Nef‐dependent manner. Furthermore, we found that SERINC3 controls the surface composition of EVs. Our workflow provides an unbiased approach for identifying candidate markers and potential regulators of EV subtypes. It can be widely applied to in vitro experimental systems for investigating physiological or pathological modifications of EV release.
Keywords: exosomes, extracellular vesicles, HIV, proteomics, T cells
Subject Categories: Membrane & Intracellular Transport; Microbiology, Virology & Host Pathogen Interaction;
A new proteomic approach allows comprehensive characterisation of exosome heterogeneity in human T cells.
Introduction
Extracellular vesicles (EVs) play important roles in cell‐to‐cell communication in both normal physiology and pathological conditions. EVs are lipid bilayered structures released by all cells and can contain transmembrane proteins, membrane‐associated proteins, cytosolic proteins and nucleic acids (van Niel et al, 2018). Following release, EVs may interact with neighbouring or distant cells and modulate their function by initiating signalling via surface contact or by transferring molecules present on or within the EV. The role of EVs, and in particular, exosomes, has received increasing attention in recent years (Mathieu et al, 2019). Exosomes are EVs of small size (50–150 nm) that form inside intracellular multivesicular endosomal compartments (MVE). However, small EVs (sEVs) of similar size as exosomes can also be released by shedding from the plasma membrane. Defining exhaustively the heterogeneity of EVs is crucial for development of accurate therapies or biomarker applications, since it has been shown that different subtypes of EVs can exert some common but also different functions (Tkach et al, 2017). Today, it is clear that classical exosome markers used widely in the literature, such as the tetraspanins CD81 or CD9, are also included in other subtypes of EVs (Kowal et al, 2016). Unambiguous definition of EV subtypes requires simultaneous detection of multiple markers: for instance, co‐expression of CD63 and other tetraspanins has been proposed as specific for exosomes (Kowal et al, 2016; Jeppesen et al, 2019). Therefore, identifying markers that are invariably present on the same EV is important to classify EV subtypes reliably.
In the specific case of cells infected with HIV‐1, an extra level of complexity is added to the heterogeneity of EVs. Membrane‐enveloped replication‐competent viruses, virus‐like particles with incomplete viral contents, as well as other EVs modified by the infection are released together with subtypes of EVs that may not be altered by infection (Nolte‐'t Hoen et al, 2016). In T cells, virus assembly occurs at the plasma membrane, where recruitment of the HIV‐1 Gag protein is necessary for efficient production of the virions (Freed, 2015). Gag interaction with components of the ESCRT machinery allows sequential recruitment of other ESCRT members and accessory proteins required during viral egress (Votteler & Sundquist, 2013). The nascent viral particle is similar to other sEVs in size, apparent composition and physical features. Consequently, separation of host sEVs from HIV virions is extremely challenging (Nolte‐'t Hoen et al, 2016). While numerous studies have shown that HIV infection alters the functionality of EVs produced by HIV‐infected cells, and that these EVs play an important role in HIV replication and pathogenesis (Pérez et al, 2019), separation of EVs and HIV is suboptimal in most cases. Thus, it is crucial to better define changes induced by the virus on EVs to allow accurate characterization of their function.
The great heterogeneity in EV subtypes contrasts with the sparseness of markers that have been used for the definition of EVs. This fact is even more evident in the case of EVs produced by HIV‐infected cells, where single markers have been proposed to differentiate EVs from virions and to study their function and composition. For example, CD45 and acetylcholinesterase (AChE) have been reported to be absent from viral particles (Esser et al, 2001; Cantin et al, 2008) and thus have been widely used as general markers of EVs, although it is unclear to what extent these markers are found in different types of EVs.
Here, we develop an unbiased proteomic profiling approach for characterizing the composition of sEV subtypes released by a human T lymphoma cell line, allowing us to evaluate known and novel EV marker proteins. Our data are accessible via an online tool that offers many interactive display and analysis features (http://evprofiler.institut‐curie.org). Our approach differs conceptually from previous studies, which have mostly relied on protein identification from a fraction enriched in a particular EV subtype. In contrast, we separate all EV subtypes partially by centrifugation, and profile protein abundances across the subfractions. Cluster analysis then indicates groups of proteins likely to be associated with the same EV subtypes, which can be further analysed by orthogonal biochemical methods. Furthermore, our approach can be used comparatively, to detect changes in EV composition under different conditions. As a proof of concept, we assess changes in EVs released upon infection with HIV‐1 and thereby identify previously unknown viral and host EV‐specific components.
Results
Biochemical and ultrastructural characterization of EVs released by HIV‐infected and control Jurkat cells
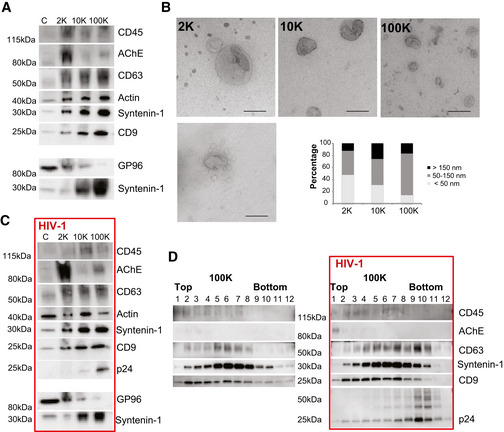
We used the Jurkat T‐cell line as an experimental model to investigate the diversity of EVs released under different physiological conditions. First, we separated EVs from cell‐conditioned medium (CCM) of Jurkat cells. After differential ultracentrifugation, we analysed EVs pelleted at low speed centrifugation (2,000 g, leading to a 2K pellet), EVs pelleted at 10,000 g (10K pellet) and EVs recovered at 100,000 g ultracentrifugation (100K pellet), as previously described by our group (Kowal et al, 2016). As recommended by the MISEV2018 guidelines (Théry et al, 2018), the 2K, 10K and 100K pellets were analysed for the presence of transmembrane proteins (CD9 and CD63 tetraspanins) and cytosolic proteins (syntenin‐1 or actin): this demonstrates the presence of EVs, while the endoplasmic reticulum protein endoplasmin (also called GP96, gene HSP90B1) is not expected in exosomes but possibly in other EVs. In addition, we analysed the distribution of the markers classically used to distinguish sEVs from HIV: the transmembrane protein CD45 (gene PTPRC) (Ott, 2009) and the secreted or GPI‐anchored protein AChE (gene ACHE) (Cantin et al, 2008). As shown in Fig 1A, GP96 was mainly found in the 2K pellet, whereas CD63, actin and CD9 were present in the majority of EVs, as previously described (Kowal et al, 2016). The small EVs (sEVs) marker syntenin‐1 (gene SDCBP) was enriched in the 100K pellets but also found in the 10K and 2K pellets from Jurkat CCM. Interestingly, CD45 and AChE were more abundant in the 2K and 10K pellets than in the 100K pellet (for AChE, as we described before: Liao et al, 2019). Transmission electron microscopy analysis (TEM; Fig 1B) of materials recovered in the 100K pellet showed a majority of small‐sized EVs (50–150 nm diameter). Some sEVs are also found in the 10K and 2K pellets, often aggregated with medium EVs (Fig 1B, lower left panel). EVs bigger than 200 nm were particularly abundant in the 10K pellet. In contrast, a wide range of particle sizes was observed in the 2K pellet, with a high proportion of electrodense nanoparticles smaller than 50 nm. This analysis supports that sEVs are highly enriched in the 100K pellet.
Figure 1. Characterization of EVs produced by non‐infected and HIV‐infected Jurkat cells.
-
A–DEVs were isolated from CCM of uninfected (A, B, D) or HIV‐1‐infected (C, D) Jurkat cells by differential ultracentrifugation (A–C), and sEVs contained in the 100K pellet were further separated by velocity iodixanol gradient (D). (A, C) The successive pellets (2K, 10K, 100K) recovered from the CCM of 20 × 106 cells were analysed by Western blot for the indicated proteins side‐by‐side with the lysate of 0.4 × 106 producing cells: (C) Images from two different representative experiments are shown. Amount of proteins recovered in 2K, 10K and 100K pellets analysed in stain‐free gel images was similar between pellets: AU (protein amount in a given pellet/total protein amount in 2K + 10K + 100K) for 2K, 10K and 100K pellets were 1.13 ± 0.28, 0.96 ± 0.10 and 0.91 ± 0.33, respectively, in control cells (A), and 1.22 ± 0.29, 0,84 ± 0.35 and 0.94 ± 0.30, respectively, in infected cells (C) (mean ± SD). (B) Whole‐mount EM analysis showing representative images and size distribution of particles in 2K, 10K and 100K pellets (Scale bar, 200 nm). The diameter (nm) of particles was determined with ImageJ for five different experiments. 387, 363 and 385 particles were counted for 2K, 10K and 100K pellets, respectively. (D) 12 fractions were recovered from iodixanol gradients of the 100K pellets obtained from CCM of non‐infected and infected Jurkat cells and analysed by Western blot for the indicated proteins.
Next, we infected Jurkat cells with HIV‐1 (NL4‐3 EGFP‐Nef+ virus). After infection, Jurkat cells were washed extensively and cultured in EV‐depleted medium. EVs (including newly formed virus) were isolated from CCM, and their protein content was analysed with the same markers as above, plus Gag p24 protein as a marker for the virus (Fig 1C). The distribution of marker proteins among the different fractions was comparable to that of the uninfected situation, and the viral p24 protein was mainly present in 100K pellets. Importantly, during infection, as in control cells (Fig 1A), CD45 and AChE were more abundant in large/medium EV fractions (2K and 10K pellets) than in sEVs (100K pellet).
We further separated the sEVs contained within the 100K pellet by velocity iodixanol gradient (Fig 1D) as previously described (Dettenhofer & Yu, 1999; Cantin et al, 2008). We found at least two different types of sEVs in the 100K pellet, both containing the general EV marker CD9. Fractions recovered at the top of the gradient (fractions 1–4) contained some CD45 but little CD63 (i.e. probably corresponding mainly to non‐exosomal sEVs), whereas intermediate fractions (fractions 5–8) contained more CD63, and were hence enriched in exosomes. In addition, in bottom fractions from infected cells (fractions 9–12), we detected enrichment of the viral p24 protein and novel presence of CD63, but not of CD9 and CD45 (Fig 1D). Importantly, AChE, when detected, was only in the very top fractions; thus, not only was it found in fractions different from those enriched in p24 but also different from those enriched in the other sEVs markers. Collectively, these observations suggest that CD45+ and AChE+ EVs represent, respectively, mainly medium/large EVs, or other pelletable material contaminating the sEV pellets, rather than non‐viral sEVs, as we recently described for AChE (Liao et al, 2019). Therefore, although they are actually excluded from virus‐containing fractions, neither AChE nor CD45 are useful markers to distinguish HIV from sEVs. Moreover, changes in distribution across fractions of velocity gradients (for instance, for CD63) suggest that some proteins may be incorporated into HIV particles and/or that the pattern of released EVs is modified upon HIV‐1 infection. A detailed characterization of sEV subtypes is needed to explore these two possibilities.
Unbiased quantitative proteomic profiling identifies clusters of EV‐associated proteins
Since marker proteins for different EV subtypes show overlapping profiles during density gradient centrifugation, proteomic analysis of peak fractions is insufficient to define EV compositions unambiguously. A more powerful approach is to profile the distribution of proteins across the entire gradient and to cluster proteins based on profile similarity (reviewed in (Borner, 2020)). However, the analysis of 12 fractions, across multiple replicates and biological conditions, requires considerable resources and is fraught with technical variability; the limited reproducibility of such experiments renders comparative applications challenging (Gatto et al, 2014). A simpler alternative is to devise a short series of differential centrifugation steps to separate EV subtypes partially and to quantify protein distributions with very high accuracy against a SILAC reference fraction. This approach, initially developed for the analysis of intracellular vesicles (Borner et al, 2014), is highly reproducible and can resolve intracellular structures ranging from large organelles down to protein complexes (Itzhak et al, 2016). Here, we have adapted this approach to the analysis of medium and small EVs contained in the CCM (10K and 100K). Importantly, the differential ultracentrifugation parameters (speed and time) had to be optimized to allow recovery of similar amounts of protein at each centrifugation step (Appendix Fig S1A and B). CCM from SILAC medium‐labelled Jurkat cultures was depleted of cells and heavy EVs by a 2K spin, before being subjected to three successive centrifugations at increasing speeds (10K, 30K and 100K) (please note that this scheme is different from the standard 10K and 100K spins commonly used to investigate EVs). Pellets were recovered to obtain CCM subfractions F1, F2 and F3. In parallel, a “reference” fraction from SILAC heavy‐labelled cells was obtained by a single 100K spin (equivalent to all subfractions F1‐F3 combined; Appendix Fig S1C). Three biological replicates (A, B and C) were performed, and the subfractions were mixed with their cognate reference fractions prior to mass spectrometric analysis and quantification. Over 3,300 proteins were quantified in all three replicates (Dataset EV1). Mass spectrometry analysis was also performed on the corresponding releasing cells, to generate a quantitative whole proteome (Dataset EV1).
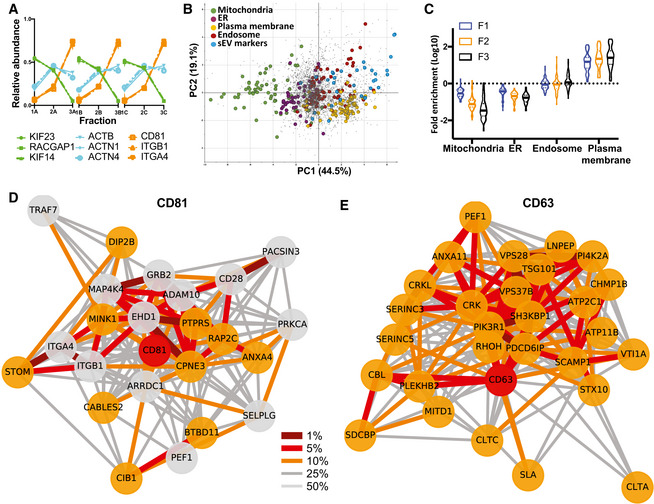
Protein abundance profiles across subfractions F1–F3 allows definition of groups of proteins that have similar fractionation behaviour, i.e. that are present in extracellular structures or EVs that have identical pelleting (i.e. size/density combination) properties. For example, the F‐actin‐associated proteins ACTB, ACTN1 and ACTN4 presented closely linked profiles with high relative abundance in the F2 and F3 subfractions (Fig 2A). In contrast, proteins involved in cytokinesis/midbody formation, such as KIF23, RACGAP1 and KIF14, had extremely similar profiles and were most abundant in the first fraction F1. CD81 displayed a near‐identical profile to integrin alpha 4 (ITGA4) and integrin beta 1 (ITGB1), known partners of an integrin heterodimeric complex, all peaking in the F3 fraction. Owing to the high accuracy of the SILAC quantification, even very subtle differences in profiles can be resolved (Borner et al, 2014). Thus, while both CD63 and CD81 peaked in F3, their profiles were consistently different across replicates, suggesting incorporation into at least some different EV subtypes (Fig EV1A). Next, we performed a principal component analysis (PCA) of all profiled proteins for visualization of the data and superposed the scores plot with known organellar markers to generate an “organellar map” (Itzhak et al, 2016). This revealed that the profiling resolved subtypes of EVs based on their subcellular origin; for instance, mitochondrial proteins were clearly separated from plasma membrane or endosomal proteins (Fig 2B). We then superposed a list of marker proteins present in sEVs expressing either CD63 or CD9 or CD81 tetraspanins (this list was selected from our previous proteomic analysis of human dendritic cell‐derived EV subtypes (Kowal et al, 2016)) (Appendix Table S1, Fig 2B). We observed that sEV markers localized in an intermediate position between markers of plasma membrane and of endosomes and were clearly separated from proteins with intracellular location in the endoplasmic reticulum or mitochondria (Fig 2B). Furthermore, we calculated the median enrichment of organellar markers detected in each of the Jurkat CCM subfractions relative to their abundance in Jurkat full proteomes (Fig 2C; Dataset EV1). This revealed strong depletion of mitochondrial (ca. 30‐fold in F3) and ER proteins (5‐fold in F3), but strong enrichment of plasma membrane proteins (ca. 20‐fold enrichment in F2 and F3). These observations indicate that the Jurkat CCM has a composition substantially different from whole Jurkat cells. While it contains components from different intracellular origins, it is highly enriched in plasma membrane‐derived components.
Figure 2. Unbiased proteomic profiling analysis of EV subtypes released by Jurkat cells.
-
AProteomic profiles of different proteins, showing the relative abundance distribution across the 3 × 3 subfractions obtained from Jurkat cells. Proteins with very similar profiles (represented by the same colours) are likely part of the same EV subtypes. Each profile consists of three independent data triplets (F1‐F2‐F3A, F1‐F2‐F3B and F1‐F2‐F3C).
-
BAbundance profiles of over 3,000 proteins in EVs recovered after 10K (F1), 30K (F2) and 100K (F3) centrifugations (see Appendix Fig S1), from Jurkat cells were subjected to principal component analysis (PCA). Each scatter point represents one protein; proximity indicates similar profiles and hence similar distributions across EVs. The PCA scores plot was annotated with known marker proteins of intracellular organelles, and with markers identified previously (Kowal et al, 2016) as specific for CD9‐ or CD63‐ or CD81‐bearing sEVs in human dendritic cells (Appendix Table S1), as indicated in the legend. Non‐marker proteins are shown as grey dots. EVs of different subcellular origin are clearly separated by the profiling analysis. PC1 and PC2 account for 44.5 and 19.1% of the variability in the data, respectively.
-
CViolin plots showing enrichment of the protein markers of intracellular organelles across the F1‐F2‐F3 subfractions as compared to expression in the total cell proteome. Solid horizontal lines indicate medians, and dashed line indicates quartiles (n = 3). Mitochondria and ER markers are de‐enriched, and plasma membrane markers are enriched in EV fractions, with progressively pronounced effects from F1 to F3.
-
D, ENeighbourhood Network plots (top 25% quantile edges) for single queries CD63 (E, **network members, cut‐off for replicates = 3) and CD81 (D, *network members, cut‐off for replicates = 2), show several ESCRT components in the CD63 network, and ARRDC1 and integrins ITGA4/B1 in the CD81 network. A multi‐query network for these two proteins and CD3G, shown in Fig EV1, shows that all three networks are separated, indicating presence of the markers in different EV subtypes. Nodes: red = query, orange = close neighbour in all three replicates, grey = close neighbour in two out of three replicates. Edges: percentile within the local distance distribution (thicker edge and darker shade = smaller distance, i.e. closer neighbour); see Materials and Methods for details.
Figure EV1. Identification by unbiased proteomic analysis of groups of proteins likely released in the same EV subtypes by Jurkat cells.
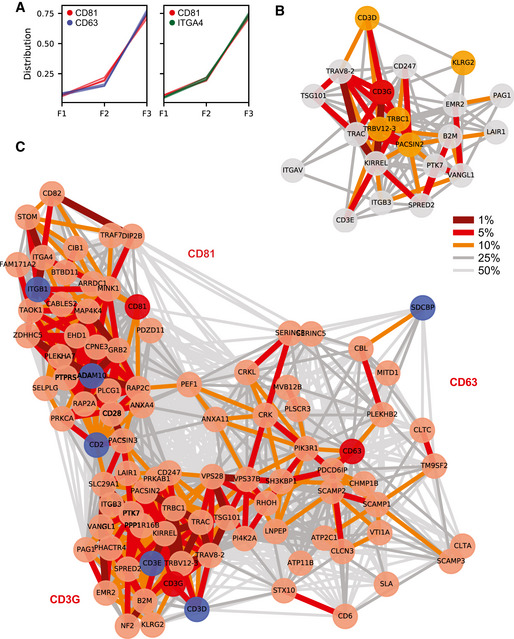
- Overlay of proteomic profiles of CD63 and CD81 (left) versus CD81 and ITGB4 (right) showing the relative abundance distribution across the 3 × 3 subfractions obtained from untreated Jurkat cells. Although all three proteins are strongly enriched in F3 fractions, the profiles of CD63 and CD81 are reproducibly different, whereas profiles of CD81 and ITGB4 are extremely similar.
- Neighbourhood Network plot of CD3G as a single query (replicate tolerance = 50, **network members, cut‐off for replicates = 2, 25% distance percentile for edges). Nodes: red = query, orange = close neighbour in all three replicates, grey = close neighbour in two out of three replicates. Edges: percentile within the local distance distribution (thicker edge and darker shade = smaller distance, i.e. closer neighbour); see Materials and Methods for details.
- Multiple query Neighbourhood Network plot for CD63, CD81 and CD3G. The top 30 close neighbours of each query were jointly used for the network layout (B ranking network members, 50% distance percentile of edges). Nodes: red = query, light red = close neighbours in 2 or 3 replicates, blue = neighbours validated by Immunoisolation, Immuno‐EM or MacsPlex Exo in figure 3. Edges: percentile within the local distance distribution (thicker edge and darker shade = smaller distance, i.e. closer neighbour); see Materials and Methods for details. The three networks are remotely connected, but clearly separate.
To identify groups of proteins with similar profiles, we then built a “neighbourhood network predictor” (NNP; see Table EV1, for the data with basic analysis functionality, and http://evprofiler.institut‐curie.org for the complete interactive database with network generation features). This interactive tool can be used to obtain a classified list of proteins with similar profiles against a query protein. By modelling the local distance distributions of proteins close to a query, and taking into account the reproducibility across biological replicates, we derived variable distance cut‐offs to define the query's “neighbourhood”. Furthermore, by scoring profile similarity among members of the query's neighbourhood, the NNP generates network plots that reveal clusters of tightly co‐profiling proteins. Since some proteins are shared between different types of EVs, it cannot predict hard cluster boundaries or pure population compositions. Nevertheless, the NNP allows discernment of groups of proteins with similar profiles at steady state, suggesting preferential incorporation into the same type(s) of EVs or extracellular structures.
First, we tested the ability of the NNP to identify stable protein complexes, which are expected to have closely linked profiles. Indeed, known multiprotein complexes displayed dense networks of the different subunits. For instance, querying the NNP with the gamma chain of the CD3 complex (CD3G) revealed a tight network including CD3D, CD3E and CD3Z (CD247), as well as the T‐cell receptor alpha and beta chains (TRAC, TRAV, TRBC1, TRBV12‐3) (Table EV1, Fig EV1B); both integrin alpha 4 (ITGA4) and integrin alpha v (ITGAV) had their respective known partners (ITGB1 and ITGB3) as their closest neighbours (Table EV1). These data support that our approach can identify functionally linked groups of proteins.
We next queried the NNP with frequently used EV and/or exosome marker proteins, to uncover groups of proteins likely to be released in similar EVs by Jurkat cells. We selected stringent cut‐offs to focus on the most immediate neighbourhood networks. CD81 and CD63 had dense neighbourhoods, which were clearly distinct (Fig 2D and E). The CD81 neighbourhood included ARRDC1, ADAM10 and also ITGA4/ITGB1, suggesting that under these conditions CD81 is predominantly found in plasma membrane‐derived EVs. In contrast, close neighbours of CD63 were mostly endosomal, including SCAMP1, Alix (PDCD6IP), the SNARE VTI1A and several ESCRT proteins, suggesting that this corresponds to EVs formed in an endosomal compartment or endosome‐like domains of the plasma membrane (Booth et al, 2006). To gain a more extensive perspective of how the different EV clusters relate to each other, we then queried the NNP with three simultaneous inputs: CD81, CD63 and CD3G (Fig EV1C). The resulting multi‐network plot supports the conclusion that the three proteins appear to mark largely non‐overlapping clusters. Furthermore, this more extended network reveals that the CD81 cluster also includes CD82, and that the CD63 cluster includes Syntenin‐1 (SDCBP). ESCRT‐I proteins (TSG101, VPS28, VPS37B) have strong links to the CD63 cluster and the TCR/CD3 cluster, suggesting a potential association with both.
The NNP's output does not provide EV proteomes; instead, the clusters of close neighbours indicate groups of proteins that have similar fractionation behaviour, consistent with a similar distribution across one or more EV types. We predict, for example, that CD81 and ADAM10 are found on the same EV subtype(s), i.e. that any EV that contains CD81 should also contain ADAM10 and vice versa. The same applies to CD63 and Syntenin‐1 (SDCBP). The NNP is thus a useful tool to define multiple markers for the same EV subtype. Conversely, if two proteins are in separate NNP clusters, they may either (i) be mutually exclusive markers for separate EV types, (ii) one might mark a subpopulation of EVs all carrying the other marker, or (iii) there are three populations on which they occur either separate or jointly. For example, the separate clusters for CD63 and CD81 indicate that there are at least two out of three EV subtypes: CD81+/CD63−, CD81−/CD63+ and CD81+/CD63+, but the NNP alone cannot predict which ones exist.
In summary, our analysis suggests that Jurkat cells release multiple types of EVs, with specific proteins predominantly associated with each type, and highlight the power of our unbiased approach for defining EV marker proteins. The NNP can be queried with any protein profiled in the Jurkat CCM.
In‐depth biochemical analysis of EV markers identified in separate networks
We next used orthogonal biochemical approaches to validate and refine the predictions inferred from the NNP. We focussed on CD63, CD81 and CD3 which, according to the multi‐network analysis shown in Fig EV1C, are in well‐separated clusters.
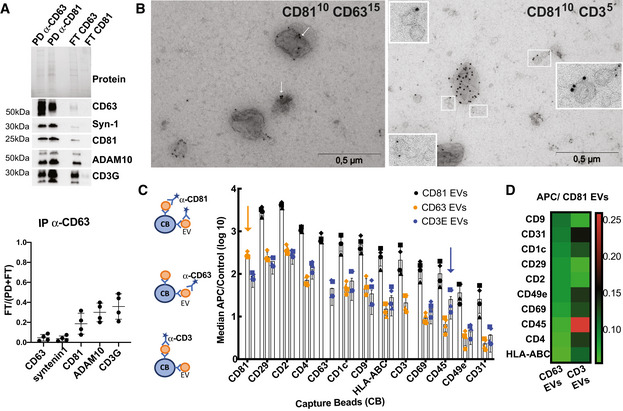
First, EVs were immuno‐isolated from Jurkat CCM depleted from cells and large EVs (2K pellet), using beads coated with antibodies targeting either CD63 or CD81. We compared the pulled‐down (PD) EVs with the EVs remaining in the flow‐through (FT) by Western blot analysis (Fig 3A). We analysed the presence of the two tetraspanins, CD3G and two proteins included in the clusters of CD81 and CD63 (ADAM10 and Syntenin‐1, respectively). Analysis of the PD and FT performed with anti‐CD81 beads showed that all EVs bearing CD63, Syntenin‐1 and ADAM10 were captured with CD81 antibody, but that a small amount of CD3G‐bearing EVs were detected in the FT. Therefore, CD81+ EVs represent a major population of EVs in Jurkat and both CD81+ and CD81−CD3G+ EVs co‐exists, consistent with our NNP analysis. After capture by anti‐CD63 beads, which isolated more than 90% of CD63 and syntenin‐containing EVs, over 30% of ADAM10, CD81 and CD3G signals remained in the FT. These results suggest that CD63‐bearing sEVs contain Syntenin‐1 and represent a subpopulation of CD81‐EVs (Fig 3A and quantification), which again agrees with our NNP network analysis. Importantly, similar results were observed in EVs isolated from CD4+ primary T cells (Fig EV2A), with CD63+ EVs representing a subpopulation of CD81+ EVs and with quite abundant CD81+CD3G+ and CD81−CD3G+ EVs. In order to analyse presence of these markers on single EVs, we used immuno‐EM (Fig 3B). We thus confirmed the presence of CD81+ EVs and of CD81+/CD63+ EVs and likewise observed CD3G+ EVs that are CD81−.
Figure 3. Biochemical analysis of the composition of EVs released by Jurkat cells.
EVs were purified by SEC from supernatants of Jurkat cells.
- EVs were subjected to immunoisolation with beads coupled to antibodies against CD81 or CD63. Bead‐associated (Pull‐down: PD) vesicles and those left behind (Flow‐Through: FT) were loaded on a gel for Western blot analysis with antibodies specific for CD63, Syntenin‐1, CD81, ADAM10 and CD3G. A representative blot (top panel) and quantification (mean ± SD) of the proportion of signal in FT as compared with total (PD + FT) from four independent experiments (bottom panel) are shown.
- Representative images of double‐immunogold labelling of EVs purified by SEC from Jurkat cells. Left panel, CD81/CD63; right panel, CD81/CD3E. Arrows show CD63 staining in CD81+CD63+ EVs.
- Multiplex bead‐based flow cytometry assay for detection of EV surface markers. Antibody‐coated capture beads were incubated with 2 × 109 particles. Captured EVs were detected with either APC‐labelled anti‐CD81, anti‐CD63 or anti‐CD3E. (C) Median APC fluorescence values for the different bead populations is shown as the ratio to the median APC fluorescence of control beads (log10 scale). Mean ± SD for four independent experiments is shown.
- Heat‐map representation of the median APC fluorescence values for the different bead populations detected with anti‐CD63 or anti‐CD3E antibodies relative to the values detected with anti‐CD81. Mean of four independent experiments.
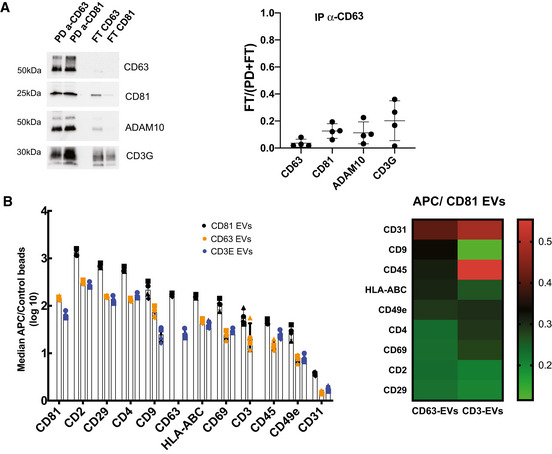
Figure EV2. Biochemical analysis of the composition of EVs released by primary CD4+ T cells.
EVs were purified by SEC from supernatant of activated CD4+T cells.
- EVs were subjected to immunoisolation with beads coupled to antibodies against CD81 or CD63. Bead‐associated (Pull‐down: PD) vesicles and those left behind (Flow‐Through: FT) were loaded on a gel for Western blot analysis with antibodies specific for CD63, CD81, ADAM10 and CD3G. A representative image and quantification (mean ± SD) of the proportion of signal in FT as compared with total (PD + FT) in samples obtained from four independent donors are shown.
- Multiplex bead‐based flow cytometry assay for detection of EV surface markers. Antibody‐coated capture beads were incubated with 2 × 109 particles. Captured EVs were detected with either APC‐labelled anti‐CD81, anti‐CD63 or anti‐CD3E. Left: Median APC fluorescence values for the different bead populations are shown as a ratio to the median APC fluorescence of control beads (log10 scale). Mean ± SD for four independent experiments is shown. Right: Heat‐map representation of the median APC fluorescence values for the different bead populations detected with anti‐CD63 or anti‐CD3E antibodies relative to the values detected with anti‐CD81 (mean value of 4 independent donors).
Last, we analysed the surface of EVs by a commercial multiplexed flow cytometry assay (Koliha et al, 2016) which allows detection of co‐occurrence of two proteins on EVs (Fig 3C). Beads coated with different monoclonal antibodies against surface markers were used to capture EVs, which were then labelled using APC‐conjugated antibodies against CD81, CD63 or CD3E. For all capture beads, APC signal obtained with anti‐CD81 antibody was higher compared to signal obtained with anti‐CD63 and anti‐CD3E, confirming that CD81 is abundantly found in EVs released from Jurkat cells (Fig 3B). Moreover, EVs captured by beads coated with anti‐CD2 and CD29 (= ITGB1) were strongly positive for CD81, confirming that these two markers are found in CD81+ EVs, as suggested by NNP analysis (Fig EV1C). Compared with EVs detected with anti‐CD81 antibody, CD63+ EVs presented lower APC mean values (Fig 3C). However, the ratio of CD63/CD81 signal was consistently between 0.08 and 0.11 for all detected markers (Fig 3D), further supporting that CD63+ EVs are a subpopulation of CD81+ EVs. Although the signal detected for CD3E+ EVs was also lower than the signal for CD81, some markers displayed relatively higher signal (CD45) or conversely lower signal (CD81) when compared with CD63‐EVs (Fig 3C and D). Interestingly, when searching the NNP for these 4 proteins, CD45 (= PTPRC) was closer to the CD3E than to the CD63 networks, in agreement with the biochemical validation. The same analysis performed on EVs from primary CD4+ T cells (Fig EV2B) revealed similar results with CD3E signal, as compared to CD63 signal, lower on CD81+ EVs and higher on CD45+ EVs.
In conclusion, the biochemical analysis validates several predictions of the NNP analysis and further refines their interpretation. Remarkably, similar results were obtained in primary CD4+ T cells, suggesting that our Jurkat‐based NNP tool has predictive value for these cells, too.
Changes in sEV protein composition upon HIV‐1 infection of Jurkat cells
We next used our approach to understand changes in the release of EVs from Jurkat cells during HIV‐1 infection. This pathological situation involves the partial usurpation of the cellular machinery of EV biogenesis by the virus.
Two biological replicates for control and two for NL4‐3 EGFP‐Nef+ HIV‐infected Jurkat cells were processed in parallel, as for the previous analysis (Appendix Fig S1). By mass spectrometry, we profiled over 3,000 proteins in the CCMs from control and HIV‐1‐infected samples, with 2,805 proteins common to all four sets (Fig 4A, Dataset EV2). In CCMs from infected cells, HIV‐1 proteins show very closely matched profiles with high abundance in the last (F3) fraction, but these were clearly distinguishable from the profiles of host EV proteins CD81, ITGA4 and ITGB1 (Fig 4B). PCA of the abundance profiles of all proteins revealed that the majority of HIV‐1 proteins form a well‐separated cluster in the same quadrant as most sEV proteins (Appendix Fig S2A).
Figure 4. Identification of candidate proteins whose secretion is modified upon HIV infection.
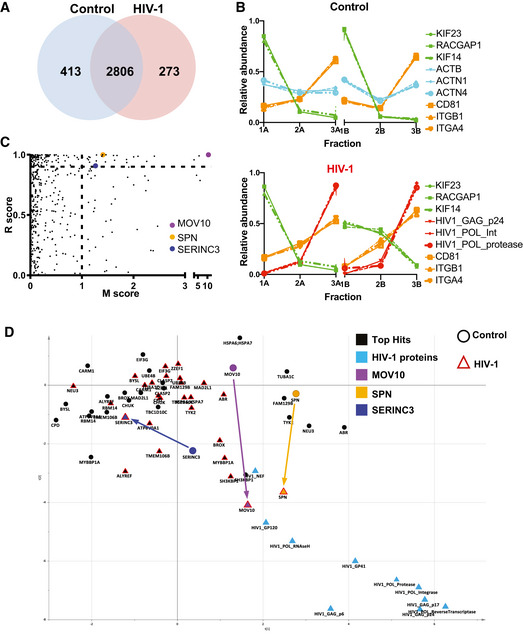
- Number of proteins identified after analysis by mass spectrometry in the six fractions of EVs from control and HIV‐infected cells. The majority of proteins were quantified in both samples.
- Proteomic profiles of different proteins (as in Fig 2), showing the relative abundance distribution across the six subfractions. Each profile consists of two independent data triplets (F1‐F2‐F3A and F1‐F2‐F3B). Abundance profiles of the same groups of proteins as in Fig 2A showed the same clustering behaviour, although in this second dataset, the proportion of proteins recovered in the F2 was more variable. These differences, however, did not affect the results of the NNP or movement analysis.
- Unbiased identification of significant translocation events triggered by HIV‐1 infection. Each protein is scored for magnitude of translocation (M score, x‐axis) and reproducibility of translocation direction (R score, y‐axis) across the two replicates. MR plot analysis reveals significant translocations in the top right quadrant. Proteins with M > 1 and R > 0.9 are candidate hits for changing localization (estimated FDR = 8%).
- Movement of the 26 proteins identified as top hits for significant and reproducible translocation upon HIV‐1 infection (from Fig 2C and Table 2). Superimposition of two PCA plots with top hits in black and key hits (MOV10, SPN, SERINC3) highlighted with different colours. The position of each protein is shown both in non‐infected (circle) and HIV‐1 infected (red triangle) conditions. Arrows indicate movements of candidate proteins upon infection.
We first asked if any host proteins profiled close to the HIV cluster. We constructed a nearest neighbour predictor tool as above (Table EV2). Querying the data with HIV p24, we retrieved all detected HIV‐1 proteins, except Nef, as top hits, followed by the host proteins SPN and MOV10 (Table 1), suggesting that these cellular components could be contained in the viral particle. Querying all HIV‐1 proteins separately identified SPN and MOV10 in the list of closest top hits in each case, with the exception of HIV‐Nef, whose close neighbours included TSG101, CD63 and VPS37b, but no other viral proteins. These observations suggest that HIV‐infected cells release the complete virus, but also other mainly endogenous EVs that contain the viral Nef protein.
Table 1.
Classified list of proteins with similar profiles to HIV‐1 Gag p24 using parameter cut‐offs Min ratio count: 1, Min correlation of replicates: 0, Min F3 enrichment: −1.
| Gene names | Min Ratio Count HIV | Abs Dist HIV | Relative proximity |
|---|---|---|---|
| HIV1_GAG_p24 | 124 | 0.00 | Query |
| HIV1_POL_ReverseTranscriptase | 60 | 1.24 | *** |
| HIV1_GAG_p17 | 47 | 1.60 | *** |
| HIV1_POL_Integrase | 28 | 2.15 | *** |
| HIV1_GP41 | 10 | 4.44 | *** |
| HIV1_GAG_p6 | 8 | 5.06 | *** |
| HIV1_POL_Protease | 11 | 5.25 | *** |
| HIV1_GP120 | 7 | 6.60 | *** |
| HIV1_POL_RNAseH | 4 | 7.23 | *** |
| SPN | 17 | 8.00 | *** |
| MOV10 | 50 | 8.59 | *** |
| AEBP1 | 31 | 8.65 | *** |
| CD82 | 28 | 8.79 | *** |
| UBASH3A | 52 | 9.07 | *** |
| C16orf54 | 6 | 9.10 | *** |
| IGSF8 | 43 | 9.15 | *** |
| CLTC | 253 | 9.23 | *** |
| HIV1_NEF | 5 | 9.25 | *** |
| SNRPD1 | 7 | 9.25 | *** |
| SH3GL1 | 8 | 9.50 | *** |
| CD53 | 2 | 9.55 | ** |
| SLC38A2 | 9 | 9.60 | ** |
| CBL | 59 | 9.60 | ** |
| SLA2 | 2 | 9.73 | ** |
| DIP2B | 83 | 9.82 | ** |
| SDCBP | 37 | 10.04 | * |
| GYPC | 6 | 10.22 | * |
| MVB12B | 18 | 10.29 | * |
| RPS2 | 20 | 10.34 | * |
| TNFAIP3 | 25 | 10.38 | * |
*, fairly close neighbour; **, close neighbour; ***, very close neighbour.
We then compared infected and uninfected conditions to identify host proteins that changed distribution between the subfractions upon infection, and, consequently, position in the PCA map. We performed an outlier test quantifying the movement (M) and reproducibility (R) of the profile shifts across biological replicates (Itzhak et al, 2016). We thereby identified 26 candidate proteins (high stringency filtering, FDR = 8%) that undergo significant movements (Table 2, Fig 4C, Dataset EV2 [including the extended list of hits, medium stringency, FDR = 20%]). Consistent with MR analysis results, the profile of MOV10 (the top hit, with M = 10.55) substantially changed upon HIV‐1 infection towards an increase of abundance in the F3 fraction, whereas, for instance, CD55 profiles (M = 0.01) did not change significantly between control and HIV‐1 infected situations (Appendix Fig S2B). Changes in the fractionation patterns of the 26 candidates suggested either a change in their association with different EV subtypes, or a change of the size, shape and/or density of the EVs themselves (although in this case multiple proteins should undergo the exact same shift). We then evaluated the nature of the profile changes by comparing the position of each candidate protein in the PCA maps before and after HIV infection (Fig 4D; see Table 2 for a detailed analysis). The two proteins identified as close neighbours of HIV proteins by the neighbourhood predictor (MOV10, SPN) moved strongly towards the HIV cluster upon infection. By contrast, SERINC3 (a member of the CD63 network in the previous analysis: Figs 2E and EV1C) moved strongly away from the HIV cluster. By neighbourhood analysis, SERINC3 was never found in proximity to HIV‐1 proteins, suggesting that this protein is predominantly contained in non‐virion EVs. Based on these results, we performed a more detailed analysis of SERINC3, as a candidate cargo of non‐viral EVs that moves away from the HIV cluster, and MOV10 and SPN, as candidates for virion inclusion according to movement towards HIV proteins and the nearest neighbour predictor.
Table 2.
Proteins with strong modification of EV‐associated secretion (M score > 1) with high reproducibility (R > 0.9) upon HIV infection of Jurkat cells (top right quadrant of Fig 4C). Estimated FDR = 8%.
| Gene name | Major subcellular localization | Known functions | Movement towards (IN) or away from (OUT) HIV cluster (M/R scores) |
|---|---|---|---|
| ABR | Cytosolic | Regulates GTPases RAC1, RAC2, CDC42 | OUT (3.05/0.958) |
| ALYREF | Nuclear/cytosolic | Nuclear export of mRNA | IN (5.67/0.991) |
| ATP6V0A1 | Multipass transmembrane, intracellular acidic compartment (?) | H+ translocation for acidification of vacuole | IN (1.14/0.983) |
| BROX | Cytosolic membrane associated? (CAAX motif) | IN (1.40/1) | |
| BYSL | Cytosol and nucleolus | rRNA processing | Lateral (1.46/0.940) |
| CARM1 | Nucleus (cytoplasm during mitosis) | Histone methylation | IN (1.13/0.958) |
| CHUK | Cytosol and nucleus | Part of the IKK complex that inhibits NFkB | Lateral (1.01/0.98) |
| CLASP2 | Cytoskeleton, plasma membrane, Golgi | Stabilizes dynamic microtubules | Lateral (1.47/1) |
| CPD | Single‐pass transmembrane, Plasma membrane | Releases C‐term Arg and Lys from polypeptides | Lateral (1.11/0.968) |
| EIF3G | Nucleus, cytoplasm | Required for initiation of protein synthesis by associating with 40S ribosome | Lateral (1.61/0.994) |
| FAM129B = NIBAN2 | Cytosol | OUT (1.39/0.987) | |
| HSPA6/HSPA7 | Cytoplasm and extracellular EVs | Chaperone | Lateral (1.87/0.960) |
| MAD2L1 | Nucleus and mitotic spindle during mitosis | Part of the complex that prevents anaphase until all chromosomes are aligned during mitosis | Lateral (2.26/0944) |
| MOV10 | Nucleus and cytoplasmic granules (P‐Body...) | RNA helicase, required for miRNA‐mediated gene silencing | IN (10.55/1) |
| MYBBP1A | Nucleus and cytoplasm | May activate or repress transcription | IN (2.01/0.982) |
| NEU3 | Peripheral membrane (no signal peptide: cytosolic?) | Modulates ganglioside content at the lipid bilayer | OUT (1.87/0.96) |
| RBM14 | Nucleus, cytosol | Transcriptional activator or repressor (2 isoforms), participate in innate immune response via cGAS‐STING | Lateral (1.28/0.949) |
| SERINC3 | Multipass transmembrane, plasma membrane and Golgi | HIV restriction factor, inhibits early step of viral infection | OUT (1.26/ 0.908) |
| SH3KBP1 | Cytosol, endocytic vesicles | Regulation of endocytosis and receptor signalling, interacts with PDCD6IP | IN (1.63/0.954) |
| SPN | Single‐Pass Type I transmembrane, plasma membrane | Regulates T‐cell functions | IN (1.41/1) |
| TBC1D10C | Cytosol, plasma membrane filopodia | Inhibits Ras signalling and calcineurin | Lateral (2.09/0.987) |
| TMEM106B | Single‐pass type II transmembrane, Lysosomes, endosomes | Regulates lysosomal trafficking via interaction with MAP6 | IN (1.68/0.994) |
| TUBA1 | Cytoskeleton | Constituent of microtubules | OUT (2.41/0.988) |
| TYK2 | Cytosol and extracellular vesicles | Phosphorylates IFNa/b‐Receptor | OUT(1.28/0.998) |
| UBE4B | Nucleus, cytoplasm | Ubiquitin‐protein ligase (E3 ligase) | Lateral (1.16/0.978) |
| ZZEF1 | Unknown (no signal peptide) | Lateral (1.15/0.938) |
Bold formatting indicates proteins that were followed up in detail in this study.
SERINC3 is found in host EVs while MOV10 and SPN are included in the virion
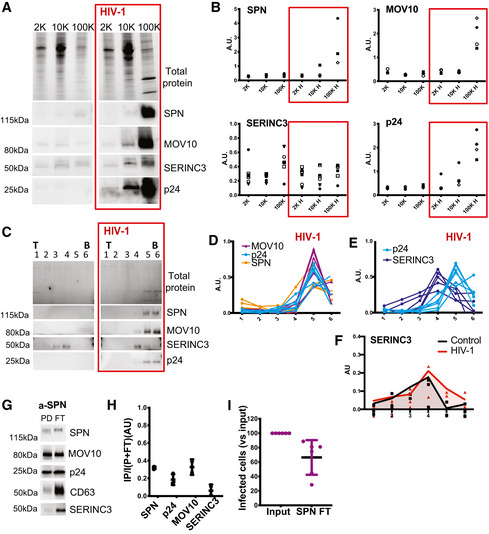
To validate the candidate proteins as specific host sEVs or virion components, we first enriched viral particles and EVs from the conditioned medium from both control and infected cells by differential centrifugation. Following classical serial centrifugation (2K, 10K, 100K), the presence of SPN, MOV10, SERINC3 and the viral marker p24 in the different pellets was assessed using commercial antibodies. As shown in Fig 5A, we detected SPN and MOV10 mainly in the 100K pellet from Jurkat infected cells together with p24. SERINC3 was detected in the 100K pellet from Jurkat infected cells but also in the different pellets from control and infected CCM. Quantification of the signal for each protein related to the total protein content showed that SERINC3 is not specifically enriched in any of the pellets from either control or infected CCM from five independent experiments (Fig 5B). In contrast, SPN and MOV10 were strongly enriched in the 100K pellet from infected CCM, in a similar way as viral p24.
Figure 5. MOV10 and SPN are included in the viral particle, whereas SERINC3 is excluded from the virus and present in virus‐modified EVs.
-
A, B(A) Analysis of the three candidate markers in EV pellets isolated from CCM of control and infected (red boxes) Jurkat cells. The successive pellets (2K, 10K, 100K) were analysed by Western blot, using antibodies against SPN, MOV10, SERINC3 and p24 (HIV‐1 protein). The 2K, 10K and 100K pellets obtained from 20 × 106 cells were loaded on the gel. Representative images (A) and quantifications in three to five independent experiments (B) are shown. For each pellet, arbitrary units (AU) represent the ratio of signal intensity for the analysed protein in the given pellet to the total amount of proteins in the same pellet. In cell lysates from the same experiments, AU (given protein/total amount of protein) for SPN and MOV10 were 1.29 ± 0.15 and 0.84 ± 0.33 times the levels in control cells, respectively (Mean ± SD).
-
CWB showing the distribution of the same proteins in six fractions recovered after velocity gradient separation of vesicles contained in the 100K pellets of control or HIV‐1 infected (red boxes) cells. Viral p24 protein was recovered in the bottom fractions of infected samples together with MOV10 and SPN. SERINC3 was recovered in the middle fractions. (T = top; B = bottom).
-
D, EQuantification of MOV10, SPN, SERINC3 and p24 signals in 4 to 7 independent Western blots from HIV‐1 infected samples. AU = band intensity in a given pellet / sum (band intensity in all six pellets).
-
FQuantification of SERINC3 signals in the different fractions from control and infected samples. AU = band intensity in a given pellet/sum (band intensity in all 12 pellets). Lines represent the medians of three independent experiments.
-
G, HEVs from supernatant of HIV‐infected Jurkat cells were subjected to immunoisolation with beads coupled to antibody against SPN. Bead‐associated (Pull‐down: PD) vesicles and those left behind (Flow‐Through: FT) were loaded on a gel for Western blot analysis with antibodies specific for SPN, MOV10, CD63, SERINC3 and viral p24. A representative image (G) and quantification (H) Mean ± SD of the proportion of signal in PD as compared with total (PD + FT) in three independent experiments are shown.
-
IEVs remaining in the FT or EVs secreted by the same number of cells but not subjected to immunoisolation (input) were used to infect a reporter cell line. Mean ± SD of six independent experiments is shown.
To better understand if these proteins were specific for different subtypes of EVs/virions within the 100K pellet, we analysed the same markers after iodixanol gradient separation of this pellet. Within the 100K pellet from infected CCM, viral protein p24 was found mainly in the bottom fractions of the gradient. The same distribution was observed for SPN and MOV10 (Fig 5C). Quantification of the relative abundance of MOV10, SPN and p24 within the different fractions obtained after separation of CCM from infected cells showed that these proteins are likely contained in the same particles (Fig 5D). In contrast, SERINC3‐enriched fractions were close to but different from those containing p24 (Fig 5C and E), pointing to the presence of this protein in EVs different from viral particles. Moreover, when comparing control and infected samples, we observed that the distribution of SERINC3 changed upon infection (Fig 5C). In infected samples, SERINC3‐containing vesicles showed a small shift towards more dense fractions (Fig 5F). The changes in behaviour of SERINC3 detected within the different fractions upon infection, together with the MR analysis results suggest that this protein is contained in host EVs that are modified by HIV‐1 infection.
SPN is a transmembrane protein and thus has domains exposed on the surface of EVs. Hence, we next performed immunoisolation using beads coated with an antibody against SPN, to ascertain if SPN is incorporated into HIV virions. Analysis by Western blot of the immuno‐isolated particles (PD) and those remaining behind (FT; Fig 5G) showed that we captured about 30% of SPN‐containing EVs (70% of SPN‐EVs remained in the FT [Fig 5H]). Importantly, anti‐SPN also captured about 30% of MOV10 and 25% of p24, but < 1% of SERINC3. Furthermore, the removal of 30% of SPN+ EVs from the sample led to an equivalent reduction in the percentage of infected reporter cells in an HIV infectivity assay (Fig 5I). These results demonstrate that SPN‐bearing EVs represent HIV particles that also contain MOV10, but no or very little SERINC3, as predicted by our MR analysis.
SERINC3 down‐regulation modulates the number and surface composition of EVs
SERINC3 has been described as a viral restriction factor counteracted by Nef, which prevents its incorporation into the HIV‐1 viral particle (Rosa et al, 2015; Usami et al, 2015). Indeed, when we separated EVs from cells infected with Nef‐deficient HIV‐1 in an Iodixanol gradient, SERINC3 was found in the same fractions as viral p24 (Fig EV3A). In contrast, upon infection with Nef‐containing virus, SERINC3 was absent from the viral particle and incorporated into different types of EVs produced by uninfected and infected cells (Fig 5C). To analyse the possible role of SERINC3 in the biogenesis of EVs, we down‐regulated its expression in Jurkat cells (Fig EV3B). SERINC3 KD cells were left uninfected or were infected with HIV‐1 with or without Nef. As expected from the literature, upon infection with Nef‐deficient virus, the supernatants from SERINC3 KD cells were more infectious than the supernatants produced by control cells (Fig EV3C). However, in the presence of Nef, the down‐regulation of SERINC3 did not have a significant impact on their infectious potential when compared with control cells (Figs 6A and EV3C), although it increased the number of particles in the supernatant 48 h after infection (Figs 6B and EV3D) in cells expressing two of the three SERINC3 shRNA sequences. Iodixanol gradient separation of the sEVs produced upon infection showed that the amount of p24 and its distribution among the fractions of the gradient were not consistently affected in SERINC3 KD cells compared with control cells (Fig 6C and D, KD cells obtained by inhibition with the pooled 3 shRNA). Thus, the increase in number of particles released from SERINC3 KD cells can be attributed to an increase in the release of EVs different from viral particles.
Figure EV3. Lentivirus‐mediated silencing of SERINC3.
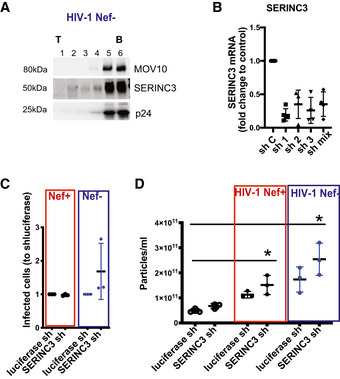
-
A100K pellets from Jurkat cells infected with Nef‐defective HIV‐1 were subjected to iodixanol velocity gradient separation. Six fractions were recovered and analysed by Western blot for the presence of SERINC3 and p24. (T = top; B = bottom).
-
BJurkat cells were transduced with lentiviruses encoding different shRNA sequences specific for SERINC3, or a control shRNA (Sh C). SERINC3 mRNA levels were determined by qPCR. Mean ± SD of three‐four different experiments is shown.
-
C, DControl and SERINC3 KD Jurkat cells were left uninfected or infected with NL4‐3 EGFP HIV‐1 containing or not containing Nef. (C) Numbers of GFP+ Ghost X4R5 cells obtained after treatment with supernatant from infected SERINC3 KD cells. Results are expressed as a ratio to the number of GFP+ cells after treatment with supernatant from infected control cells. Mean ± SD for three independent experiments is shown This experiment confirms that SERINC3 restricts HIV‐1 infectivity only for Nef‐defective virus. (D) SERINC3 KD cells were infected or not infected, and numbers of secreted particles were quantified by NTA. Results are expressed as the number of secreted particles/ml of conditioned medium. Mean ± SD for three independent experiments is shown. *P < 0.05 (Friedman test followed by Dunn's post‐test).
Figure 6. SERINC3 down‐regulation modifies the EVs released by Jurkat cells.
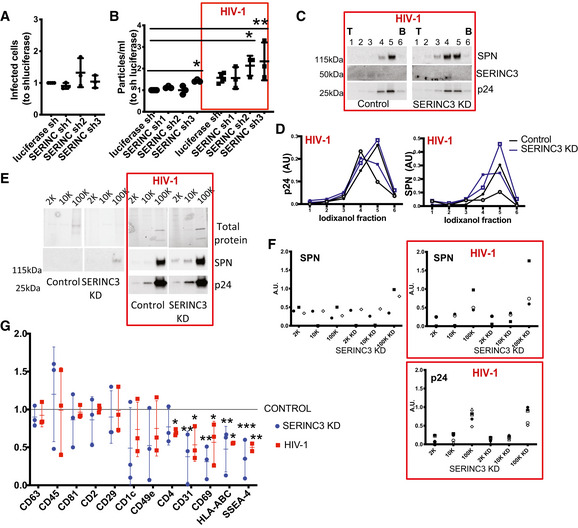
Jurkat cells were infected with lentivirus expressing control luciferase shRNA or shRNAs targeting SERINC3, followed or not by infection with NL4‐3 EGFP‐Nef+ HIV‐1 (red boxes).
-
AGFP+ Ghost X4R5 cells obtained after treatment with supernatant from infected SERINC3 KD cells. Results are expressed as a ratio to the number of GFP+ cells after treatment with supernatant from infected control cells. Mean ± SD of three independent experiments is shown.
-
BNumber of particles secreted were quantified by NTA. Results are expressed as a ratio to the number of particles secreted by uninfected luciferase shRNA cells. *P < 0.05, **P < 0.01 (one‐way ANOVA followed by Dunnett's post‐test). Mean ± SD of three independent experiments is shown.
-
C100K pellets from infected cells were subjected to iodixanol velocity gradient separation. Six fractions were recovered and analysed by Western blot for the presence of SPN, SERINC3 and p24. (T = top; B = bottom).
-
DQuantification of SPN and p24 signals in the different fractions of two independent iodixanol fractionations (exp 1: open symbols, exp 2: closed symbols). AU = band intensity in a given pellet/sum (band intensity in all 12 pellets).
-
E, F2K, 10K, 100K obtained from CCM of control cells and SERINC3 KD cells either uninfected of infected with NL4‐3 EGFP‐Nef+ were analysed by Western blot with antibodies against SPN and p24. The 2K, 10K and 100K pellets obtained from 20 × 106 cells were loaded on the gel. Representative images (E) and quantification of SPN and p24 signals (F) are shown. For each pellet, arbitrary units (AU) represent the ratio of signal intensity in the given pellet to the total amount of protein in the same given pellet and relative to the total amount in 2K + 10K + 100K from control cells.
-
GMultiplex bead‐based flow cytometry assay for detection of EV surface markers. Antibody‐coated capture beads were incubated with EV samples. Captured EVs were detected with a mixture of APC‐labelled anti‐CD9, anti‐CD63 and anti‐CD81. Quantification of the median APC fluorescence values for all bead populations after background and isotype antibody correction. Results were relativized to the values obtained with control sample for each experiment. Dotted line represents values obtained for EVs isolated from control cell CM. Mean ± SD of three independent experiments is shown. *P < 0.05, **P < 0.01, ***P < 0.001 (one‐way ANOVA followed by Dunnett's post‐test).
Although the amount of p24 is not affected by SERINC3 down‐regulation, we detected an increase in the amount of SPN incorporation into sEVs/virus after HIV‐1 infection (Fig 6C and D). Analysis of 2K, 10K, 100K pellets obtained from CCM showed that the increase in SPN occurs also in EVs released by uninfected SERINC KD cells (Fig 6E and F). Since SERINC3 down‐regulation changed SPN expression on the surface of EVs/virus, we decided to analyse other EV surface proteins by the multiplexed flow cytometry assay (Koliha et al, 2016). After capture on beads coated with the different monoclonal antibodies, EVs were detected by a combination of three APC‐labelled antibodies against CD9, CD81 and CD63 tetraspanins (TSP). EVs from SERINC3 KD cells differed in surface composition compared with EVs from control cells. TSP+‐EV surface composition in samples from SERINC3 KD cells phenocopied the surface signature found in EVs from HIV‐1 infected cells. Compared with EVs isolated from control cells, TSP+‐EVs from both SERINC3 KD and HIV‐1‐infected cells presented decreased levels of CD31, CD69, HLA‐ABC, SSEA‐4 and, to a lesser extent, CD4, CD1c, CD49e (ITGA5; Fig 6G). Thus, down‐regulation of SERINC3 modifies the surface of EVs.
Discussion
Here we present a novel pipeline for identification of components of EV subtypes, which will be suitable for a wide range of experimental systems. Following an unbiased quantitative proteomic profiling analysis, a neighbourhood network predictor (NNP) interactive tool is generated (provided here for Jurkat cells: http://evprofiler.institut‐curie.org) to identify putative partners of a protein of interest released in the same EVs, or at least in EVs endowed with similar biophysical (size/density) pelleting properties. The analysis can be extended to a comparison of EVs released under two or more different patho/physiological conditions, using the movement‐reproducibility (MR) analysis framework (provided here for control vs HIV‐1 infected cells in Dataset EV2), and to reveal modifications of the protein composition of released EVs. Applied to the specific case of Jurkat T cells infected with HIV‐1, the NNP and MR tools allowed us to identify several candidate host proteins specifically secreted in the viral particle or, conversely in non‐viral EVs. We validated these predictions for two putative viral proteins (MOV10 and SPN) and one protein exclusively found in the sEVs (SERINC3). Our results also open new avenues for further biogenetic and functional studies, since we demonstrate that SERINC3 can control the surface composition of EVs.
Traditionally, EVs have been classified according to their biogenesis origin into three major groups: (i) EVs budding directly from the plasma membrane (ectosomes, microvesicles or microparticles); (ii) EVs formed in intracellular MVEs (exosomes); (iii) EVs generated from cells undergoing apoptosis (apoptotic bodies). However, heterogeneity of EVs, even within these major subtypes, has become evident in recent years. To date, the majority of proteomic studies have provided crude lists of proteins found in bulk preparations of EVs containing various mixtures of subtypes. The development of approaches to discover potential new components that associate with EV subtypes will be essential to improve our knowledge of the respective functions of distinct EVs (Tkach et al, 2018). Our study provides such a tool and constitutes a major advance towards understanding molecular heterogeneity of EVs.
Previous studies have also used proteomics of different EV subtypes to begin to evaluate the specificity of protein markers classically used to define exosomes and other EV subtypes (Keerthikumar et al, 2015; Minciacchi et al, 2015; Kowal et al, 2016; Jeppesen et al, 2019). However, the generated lists of EV‐assigned proteins could only be analysed manually a posteriori by other researchers, to search for associations of proteins with the subtypes of EVs analysed in each study. Here, instead, we generated an interactive tool that covers all EVs secreted from Jurkat cells, and that can be flexibly queried to define associations of EV proteins. For instance, we interrogated our NNP with proteins found in a recent proteomic analysis of plasma membrane‐derived ectosomes released by T cells (Saliba et al, 2019). Although the other study showed increased secretion of both CD81 and CD6 in ectosomes upon cognate engagement at the immune synapse, the two proteins are distant from each other in our NNP, suggesting that they may be released in two or more distinct types of EVs, at least in Jurkat cells.
Our approach is unbiased and effectively separates EVs containing components of different subcellular origin (e.g. plasma membrane and endosome‐derived markers have very different profiles from markers of the ER or mitochondria). However, it is important to keep in mind its conceptual limitations. First, EVs with very similar fractionation properties under the chosen separation conditions may not be resolved. Second, for proteins associated with multiple types of EVs (a common occurrence), a mixed profile is obtained, weighted by how much of the protein is associated with each EV type. Such a mixed profile may not be directly interpretable, as the underlying “pure” EV profiles and their weights are not known. Nevertheless, any profile still carries significant information, especially when compared with other profiles: if two proteins have the same distribution across multiple types of EVs, they will also have similar profiles. Therefore, the NNP's output of close neighbours should best be seen as a group of proteins that have similar fractionation behaviour, consistent with a similar distribution across one or more EV types. This in turn may also suggest a functional association of these proteins. Conversely, if two proteins have consistently different profiles, they are unlikely to have the same distribution across EVs. It is important to note though that this does not mean that they are never present in the same type of EV; rather, it suggests different majority steady state distributions across EVs. This is illustrated by our own findings regarding CD81 and CD63. The NNP places them in different clusters, suggesting that they are present in at least two types of EVs; indeed, our biochemical validation showed evidence of CD81+/CD63‐ as well as CD81+/CD63+ EVs. Finally, the NNP does not provide hard cut‐offs or boundaries between EV subtypes, but rather reveals proteins with linked EV profiles.
Another important feature of this study is the comparative profile analysis, which identifies modulation of EV cargoes upon cell perturbation. Here too, our approach has some limitations: first, absence of apparent change does not mean that no change occurs; for instance, the protein could move to another type of EV with similar pelleting properties. More importantly though, the MR analysis detects only robust changes between biological replicates. As with any experimental setup, biological and technical noise can mask real changes, and we stringently prefiltered our profiling data to exclude poorly reproducible shifts (see Materials and Methods). For example, we did not include CD63 in the comparative +/− HIV analysis, as the data were insufficient in this case, due to the low number of CD63 peptides quantified in the comparative dataset. So while our gradient analysis in Fig 1 suggests that CD63 shifts upon HIV infection, we could not address this point by profiling. Nevertheless, the MR analysis identified numerous proteins whose secretory behaviour is strongly affected by HIV infection, of which we selected three for validation. Other proteins with identified MR shifts, or with more subtle changes that were not highlighted, may also be important. For such candidates, the dedicated interactive tool (Table EV2) may be used manually to evaluate the reproducibility of results.
An important result of our profiling concerns the HIV‐1 protein Nef. The literature provides contradictory observations on the presence of Nef in EVs versus virions. Nef had been observed in clusters of microvesicles (Muratori et al, 2009) or recovered exclusively in CD45+ EVs (Raymond et al, 2011), or in both virus‐ and non‐virus‐containing fractions of a velocity gradient (Lenassi et al, 2010). Here, when querying our NNP for the HIV1‐Nef protein, we found as nearest neighbours mainly host proteins, including several proteins of the ESCRT pathway and CD63, but no other HIV‐1‐encoded proteins. Therefore, this observation suggests that the majority of Nef present in the CCM is included in a subtype of possibly endosome‐derived vesicles, rather than in the virus itself. This result thus opens new ways to isolate Nef‐containing EVs free of virus (for example by using Nef's neighbours as baits), for an accurate evaluation of their role in HIV‐mediated pathologies.
Concerning the host proteins, our re‐evaluation of previously suggested markers of non‐viral EVs such as CD45 (Arakelyan et al, 2017) and AChE (Cantin et al, 2008) underscores their limited usefulness (see Fig 1 and our previous study, (Liao et al, 2019)). Our proteomic analysis of EVs released by Jurkat cells did not detect any SILAC‐labelled peptides of AChE, confirming our previous finding that the majority of AChE in EV preparations comes from serum used to culture the cells (Liao et al, 2019). For CD45, we found that this protein is excluded from virus‐containing fractions (Fig 1D) and is more abundant in 2K and 10K pellets than in 100K pellets, where the majority of sEVs and virus are found. On the other hand, we identified two host proteins, MOV10 and SPN, which are specifically included in virions. MOV10 is a RNA helicase that has been previously detected in HIV‐1 viral preparations (Abudu et al, 2012), supporting our findings. SPN is a highly abundant surface protein of T lymphocytes and regulates T‐cell functions including activation, proliferation and migration (Clark & Baum, 2012). In polarized T cells, SPN is specifically associated with PM microdomains where Gag is recruited (Llewellyn et al, 2013). Consistent with this, we found specific incorporation of SPN into the viral particle.
Furthermore, we describe here that SERINC3 is included specifically in host sEVs with altered pelleting properties after infection with Nef‐containing HIV (Fig 5C). SERINC3 was described together with SERINC5 as a restriction factor when incorporated into the viral particle (Usami et al, 2015; Rosa et al, 2015). This incorporation is counteracted by Nef (Usami et al, 2015; Rosa et al, 2015), which reduces the surface levels of SERINC3 and SERINC5 (Matheson et al, 2015) and relocalizes SERINC5 from the cell surface to perinuclear vesicles (Usami et al, 2015). Our observations on SERINC3 suggest another (or a complementary) way for the virus to eliminate this restriction factor: by releasing it in EVs, instead of routing it to a degradation pathway.
Nef also downregulates CD4 and MHCI by modifying their trafficking within the late secretory pathway (Pereira & daSilva, 2016). Here, we describe that infection of Jurkat T cells with Nef‐containing HIV‐1 changes the composition of released EVs (Fig 6E and G). Since we find reduced levels of CD4 and HLA in tetraspanin + EVs, these changes could, at least in part, reflect alterations in the PM such as those elegantly measured in a previous proteomic analysis of the T‐cell PM (Matheson et al, 2015). Importantly, we found that SERINC3 down‐regulation leads to similar changes in EVs as does infection with HIV. Knockdown of SERINC3 increased release of EVs and changed their surface protein composition, allowing incorporation of SPN. Thus, Nef‐mediated SERINC3 re‐routing could explain some changes in protein trafficking upon infection that lead to compositional changes of both host EVs and virus. To what extent down‐regulation of SERINC3 accounts for Nef‐mediated changes in PM or protein trafficking remains to be determined.
In conclusion, this work provides a powerful tool to unravel the diversity of EVs released by a particular cell type and to understand changes in release under different physiological and pathological conditions. We propose to use the same experimental design to detect molecular changes of EV composition in the context of other patho/physiological perturbations, for instance in tumour cells undergoing exposure to hypoxia, mimicking the poorly vascularized core of tumours. The unbiased identification of the cargoes of different EV subtypes that our approach allows, in combination with the analysis of single EVs through emerging technologies, will allow major developments for refining therapeutic applications of EVs.
Materials and Methods
Reagents and Tools table
| Reagent/Resource | Reference or Source | Identifier or Catalog Number |
|---|---|---|
| Experimental Models | ||
| Jurkat cells | This study | J77 clone 20 (short tandem repeat profiling) |
| 293‐LTV cells | Cell Biolabs |
LTV‐100 |
| GHOST X4R5 | from NIH AIDS Reagent Program | 3943 |
| Recombinant DNA | ||
| pBR‐NL4‐3 EGFP‐Nef+ (HIV‐1) | Schindler et al (2005) | N/A |
| pCMV‐VSV‐G | Adgene | Cat#8454 |
| pPAX2 | Adgene | Cat#12260 |
| X4GFP (HIV‐1) | Silvin et al (2017) | N/A |
| pLKO.1 sh luciferase | Sigma‐Aldrich | Mission shRNA SHC007 |
| pLKO.1 sh SERINC3_1 (Homo sapiens) | Sigma‐Aldrich | Mission shRNA, TRCN0000115948 |
| pLKO.1 sh SERINC3_2(H. sapiens) | Sigma‐Aldrich | Mission shRNA, TRCN0000115949 |
| pLKO.1 sh SERINC3_3(H. sapiens) | Sigma‐Aldrich | Mission shRNA, TRCN0000293864 |
| Antibodies | ||
| Mouse anti‐human CD63 (clone H5C6) | BD Bioscience | Cat#557305 |
| Mouse anti‐human CD9 (clone MM2/57) | Millipore | Cat#cbl162 |
| Mouse anti‐human CD45 (clone HI30) | BD Bioscience | Cat#557748 |
| Rat anti‐GP96 (clone 9G10) | Stressgen | Cat# ADI‐SPA‐850‐D |
| Goat anti‐human AChE | Abcam | Cat# ab31276 |
| Mouse anti‐human actin (clone C4) | Millipore | Cat# MAB1501 |
| Rabbit anti‐human syntenin‐1 (clone C2C3) | GeneTex | Cat# GTX10847 |
| Mouse anti‐human CD81 (clone 5A6) | Santa Cruz | Cat# sc‐23692 |
| Rabbit anti‐human SERINC3 | Abcam | Cat#ab153748 |
| Mouse anti‐human SPN (clone MEM‐59) | Abcam | Cat#ab9088 |
| Rabbit anti‐ MOV10 (clone EPR14478) | Abcam | Cat# ab189919 |
| Mouse anti‐human ADAM10 (clone 163003) | R&D Systems | Cat# MAB1427 |
| Rabbit anti‐CD3G (clone EPR4517) | Abcam | Cat# ab134096 |
| Mouse anti‐HIV‐1 p24 Monoclonal (183‐H12‐5C) | NIH AIDS reagent program | Cat#1513 |
| HRP‐conjugated goat anti‐rabbit IgG (H + L) | Jackson | Cat#111‐035‐144 |
| HRP conjugated goat anti‐mouse IgG (H + L) | Jackson | Cat#111‐035‐146 |
| HRP‐conjugated donkey anti‐goat IgG (H + L) | Jackson | Cat#705‐035‐147 |
| Rabbit anti‐human CD81 (clone EPR21916) | Abcam | Cat# ab233692 |
| Mouse anti‐human CD63 (clone TS63) | Diaclone | Cat# 857.770.000 |
| Rabbit anti‐mouse | Sigma | Cat#SAB3701080 |
| APC‐conjugated anti‐human CD3 (clone REA 613) | Miltenyi | Cat#130‐113‐697 |
| Oligonucleotides and other sequence‐based reagents | ||
| PCR primers GAPDH forward | This study | 5′ATGTTCGTCATGGGTGTGAA3′ |
| PCR primers GAPDH reverse | This study | 5′ATGTTCGTCATGGGTGTGAA3′ |
| PCR primers SERINC3 forward | This study | 5′ATTCTAGCATCCGCACTTCC3′ |
| PCR primers SERINC3 reverse | This study | 5′CGAGGCTGTCCATCTTCTTC3′ |
| Chemicals, enzymes and other reagents | ||
| RPMI‐1640‐GlutamaxTM medium | Gibco | Cat # 11554516 |
| Penicillin‐Streptomycin | Gibco | Cat#11548876 |
| Fetal bovine serum | Gibco | Batch#42F2567K |
| DMEM‐GlutamaxTM | Gibco | Cat#11594446 |
| GeneticinTM | Gibco | Cat#11558616 |
| PBS | Gibco | Cat# 11530546 |
| Hygromycin B | Invitrogen | Cat#10687010 |
| LymphoPrepTM tubes | Axis Shield | Cat#11548535 |
| Dynabeads™ Human T‐Activator CD3/CD28 for T Cell Expansion and Activation | Gibco | Cat#111.61D |
| Hepes | Gibco | Cat#12509079 |
| Non‐essential aminoacids | Gibco | Cat#11140050 |
| Sodium pyruvate | Gibco | Cat#11360070 |
| IL2 | R&D Systems | Cat#202‐IL‐010 |
| Puromycin | Invitrogen | Cat#A1113803 |
| TransIT‐293 reagent | Mirus Bio | Cat#MIR27906 |
| Fixable viability dye efluor 780 | eBioscience | Cat#65‐0865‐14 |
| OptiprepTM, | Sigma‐Aldrich | Cat#D1556 |
| 4x Laemmli Sample buffer | Biorad | Cat#1610747 |
| 4–15% Mini‐Protean® TGX Stain‐Free™ gels | Bio‐Rad | Cat#4568083 |
| 4–15% Mini‐Protean® TGX Stain‐Free™ gels | Bio‐Rad | Cat#4568086 |
| Immun‐Blot PVDF | Bio‐Rad | Cat#170‐4272 |
| Clarity western ECL substrate | Bio‐Rad | Cat#1705061 |
| Formvar | Agar | Cat#AGR1202 |
| Copper/palladium grids | Agar | Cat#AGG7262PD |
| Uranyl/acetate | LFG | Cat#6159‐44‐0 |
| Methyl‐cellulose, viscosity:25cP | Sigma | Cat#M6385 |
| Protein A‐gold | CMC, UMC Utrecht, Netherlands | |
| l‐arginine‐13C6 | Thermo Scientific | Cat#88210 |
| l‐lysine‐4,4,5,5‐D4 | Thermo Scientific | Cat#88437 |
| l‐arginine‐13C6 15N4 | Thermo Scientific | Cat#89990 |
| l‐lysine‐13C6 15N2 | Thermo Scientific | Cat#88209 |
| Dialyzed Fetal Bovine Serum | Thermo Scientific | Cat#A3382001 |
| SDS | Roth | CN30.3 |
| Tris‐HCl pH 8.0 | Sigma | #T6666 |
| Acetone | Fischer Chemical | #A/0600/17 |
| Acetonitrile | Merck | Cat#1.00029.1000 |
| Urea | Sigma | Cat#U5378 |
| Dithiothreitol (DTT) | Euromedex | Cat#EU0006‐B |
| Idodoacetamide | Sigma | Cat#I6125 |
| LysC | Wako | #129‐02541 |
| Trypsin | Promega | Cat#V5111 |
| Trifluoroacetic acid (TFA), | Sigma | Cat#73645 |
| SDB‐RPS solid phase extraction material | VWR | #66886‐U |
| 50‐cm column with 75‐µm inner diameter, packed in‐house with 1.8‐µm C18 particles | Dr. Maisch GmbH | |
| C18 column (75 μm inner diameter × 2 cm; nanoViper Acclaim PepMapTM 100, | Thermo Scientific | Cat#164535 |
| 50 cm × 75 μm C18 column (nanoViper Acclaim PepMapTM RSLC, 2 μm, 100 Å,) | Thermo Scientific | Cat#164942 |
| Centrifugal Filter (MWCO = 100 kDa;) | Sartorius | Cat#VS2061 |
| qEV size‐exclusion columns | Izon | Cat#SP1 |
| Protein A Magnetic Beads | Pierce | Cat#88846 |
| BS3 | Thermo Scientific | Cat#A39266 |
| SuperScript II Reverse Transciptase | Thermo Scientific | Cat#18064022 |
| SYBRgreen | ThermoScientific | Cat#A25742 |
| Software | ||
| Image Lab v5.2.1 | Biorad | |
| Primer3Plus | http://www.bioinformatics.nl/cgi‐bin/primer3plus/primer3plus.cgi | |
| FlowJo software v10 | FlowJo LLC | |
| MaxQuant V 1.6.1.13 | Cox and Mann (2008) | |
| iTEM, Olympus Soft Imaging Solutions GmbH 5.2 | ||
| Fiji/ImageJ v2.0.0‐rc‐69/1.52p | https://imagej.net/ImageJ | |
| Python v3.5+ plus packages (holoviz, panel, pandas, network) | https:/www.github.com/JuliaS92/EVProfiler | |
| Other | ||
| CD4+ T Cell Isolation Kit | Miltenyi | Cat#130‐096‐533 |
| MACSPlex Exosome Kit, human | Miltenyi | Cat#130‐108‐813 |
| Exosome Isolation Kit CD63, human | Miltenyi | Cat#130‐110‐918 |
| Exosome Isolation Kit CD81, human | Miltenyi | Cat#130‐111‐575 |
| LightCycler®480 Instrument II | LifeScience | |
| RSLCnano system (Ultimate 3000) | Thermo Scientific | Cat#ULTIM3000RSLCNANO |
| Orbitrap Fusion Tribrid mass spectrometer | Thermo Scientific | Cat#IQLAAEGAAPFADBMBCX |
| Q Exactive HF Hybrid Quadrupole‐Orbitrap mass spectromete | Thermo Scientific | |
| EASY‐nLC 1000 | Thermo Scientific | |
| MACSQuant Analyzer 10 | Milteny | |
| Cytek Aurora analyzer | ||
| Ultracentrifugue LE80K | Beckman | |
| Ultracentri Optima ‐ L80XP | Beckman | |
| Ultracentrifugue OPTIMA MAX XP | Beckman | |
| Type 45 Ti rotor | Beckman | |
| SW32 Ti rotor | Beckman | |
| TLA‐45 rotor | Beckman | |
| ZetaView PMX‐120 v8.04.02 | Particle Metrix | |
Methods and Protocols
Cell lines
Jurkat cell line identity was verified by short tandem repeat profiling as similar to J77 clone 20. Jurkat cells were cultured in RPMI‐1640‐ GlutaMAXTM medium (Gibco) with Penicillin‐Streptomycin (Gibco) and 10% of foetal bovine serum (FBS, Gibco). 293‐LTV cells were purchased from Cell Biolabs and cultured in DMEM‐GlutaMAXTM supplemented with 10% FBS (Gibco) and Penicillin‐Streptomycin (Gibco). GHOST X4R5 cells were obtained from NIH AIDS Reagent Program and cultured in DMEM‐GlutaMAXTM, 10% FBS and supplemented with Penicillin‐Streptomycin (Gibco), 500 μg/ml GeneticinTM (IGibco), 100 μg/ml hygromycin B (Invitrogen) and 1 μg/ml puromycin (Invitrogen).
CD4+ T cells were obtained from buffy coats from healthy human donors as previously described (Tkach et al, 2017). Briefly, peripheral blood mononuclear cells (PBMCs) were purified by density gradient centrifugation (LymphoPrep, Axis Shield). Total CD4+ T cells were isolated from PBMCs by negative selection using magnetic beads (CD4+ T‐Cell Isolation Kit, Miltenyi) following the manufacturer indications. This study was conducted according to the Helsinki Declaration, with informed consent obtained from the blood donors, as requested by our Institutional Review Board. Total CD4+ T cells were activated with Dynabeads™ Human T‐Activator CD3/CD28 for T‐cell expansion and activation (Gibco) and cultured in RPMI‐1640‐GlutaMAXTM medium (Gibco) with Penicillin‐Streptomycin (Gibco), HEPES (Gibco), non‐essential amino acids (Gibco), sodium pyruvate (Gibco), 10 U/ml IL‐2 (R&D systems) and 10% of foetal bovine serum (FBS, Gibco).
Preparation of viral stocks and infections
Jurkat cells were infected transiently with VSV‐G pseudotyped NL4‐3 HIV‐1 virus. VSV‐G‐pseudotyped viral particles were produced by transfection of 293T‐LTV cells with a mixture of pBR‐NL4‐3 EGFP‐Nef+ (NL4‐3 EGFP, encoding full‐length HIV‐1 in the pBR322 backbone under the control of viral long terminal repeat promoter, generated by Dr. F. Kirchhoff (Schindler et al, 2005)) and pCMV‐VSV‐G using TransIT‐293 reagent (Mirus Bio), following the manufacturer's recommendations. Virus‐containing cell supernatants were recovered after 48–72 h, filtered at 0.45 μM, aliquoted and stored at −80°C. Jurkat cells were infected with VSV‐G‐pseudotyped NL4‐3 Nef+ virus (20–50 ng p24/ml) for 2 h. For experiments using NL4‐3 EGFP‐Nef‐HIV virus, the vector pBR‐NL4‐3 EGFP‐Nef+ was replaced by X4GFP plasmid, previously described in (Silvin et al, 2017). X4GFP consist in a NL4‐3 virus encoding EGFP instead of Nef.
Lentiviral particles were produced by transfecting 293‐LTV cells with pPAX2, pCMV‐VSV‐G and pLKO.1 plasmid encoding specific shRNA sequences. sh luciferase (Mission shRNA SHC007; Sigma‐Aldrich), SERINC3‐specific shRNA, sh1 (Mission shRNA, TRCN0000115948; Sigma‐Aldrich), sh2 (Mission shRNA, TRCN0000115949; Sigma‐Aldrich) and sh3 (Mission shRNA, TRCN0000293864; Sigma‐Aldrich) were used. Lentivirus were recovered in supernatant after 48 h. Supernatant was filtered and concentrated 40× by ultracentrifugation at 120,000 g during 1 h 30 min in a SW32 Ti rotor. 5 × 106 Jurkat T cells were infected and selected in puromycin (2 µg/ml; Invivogen). Cells were amplified in the presence of puromycin for a maximum of 5 weeks.
Serum EV‐depleted medium
Serum EV‐depleted medium was obtained by overnight ultracentrifugation at 100,000 g in a Type 45 Ti rotor (Beckman Coulter, k‐factor 1042.2) of RPMI‐1640‐GlutaMAXTM medium (Gibco) supplemented with 20% FBS (Gibco). After centrifugation, EV‐depleted supernatant was carefully pipetted from the top and leaving 5 ml in the bottom of each tube to avoid disturbing bottom layers or the pellet. Supernatants were filtered through a 0.22 μm bottle filter (Millipore), and additional RPMI medium and antibiotics were added to prepare complete medium (10% EV‐depleted FBS final).
EV isolation by differential centrifugation
To obtain CCM, cells were cultured at an initial density of 1 × 106 cells/ml in EV‐depleted medium for 48‐h. Jurkat cells were left uninfected or were infected with VSV‐G‐pseudotyped NL4‐3 EGFP‐Nef+ virus for 2 h before being washed three times with PBS and incubated with medium depleted from FCS‐derived EVs. CCM was harvested after 48‐h culture and EVs isolated by a variation of a previously described protocol (Thery et al, 2006). Briefly, cells were pelleted at 300 g for 20 min at 4°C. Supernatant was centrifuged at 2,000 g for 20 min at 4°C to obtain the 2K pellet. Supernatant was transferred to new tubes and centrifuged in a SW32 Ti rotor (Beckman) for 30 min at 10,000 g (10K pellet), and finally for 90 min at 100,000 g (100K pellet). All pellets were washed in 37 ml of PBS and recentrifuged at the corresponding speed before being resuspended in sterile PBS. Cells recovered from the 300 g pellet were stained with a live/dead dye (Fixable Viability Dye eFluor 780, eBioscience). Cells were then fixed and analysed on MACSQuant instrument (Miltenyi) to assess viability and percentage of infection (GFP expression).
We have submitted all relevant data of our experiments to the EV‐TRACK knowledgebase (EV‐TRACK ID: EV190107) (Van Deun et al, 2017).
Iodixanol gradient separation
100K pellets obtained by ultracentrifugation of conditioned medium from 60–120 million cells (Jurkat NL4‐3 Nef+‐infected or uninfected) were resuspended in 1 ml of PBS and layered on top of iodixanol (OptiprepTM, Sigma‐Aldrich #D1556) gradients. Gradients were prepared in PBS in 4% increments ranging from 6 to 18% and centrifuged for 1 h at 4°C at 200,000 g (35,000 rpm) in a SW 41 Ti rotor (Beckman Coulter), stopping without brake. After centrifugation, six (or 12) fractions of 2 (or 1) ml were collected from the top of the tube. Fractions were then diluted to 37 ml with PBS to be washed by ultracentrifugation for 40 min at 100,000 g (25,000 rpm) in a SW 32 Ti rotor (Beckman). The fractions were resuspended in 15 μl of PBS and used for WB analysis.
Western blotting
Vesicles recovered from conditioned medium from 20 × 106 cells (2K, 10K and 100K pellets) or the iodixanol gradient fractions from 60–120 × 106 cells (6–12 fractions, respectively) were resuspended in in 4x Laemmli Sample buffer (Bio‐Rad) diluted in PBS and loaded on 4–15% Mini‐Protean® TGX Stain‐Free™ gels (Bio‐Rad), under non‐reducing conditions. Transferred membranes (Immuno‐Blot PVDF Bio‐Rad) were developed using Clarity western ECL substrate (Bio‐Rad) and the ChemiDoc Touch imager (Bio‐Rad). Intensity of the bands was quantified using ImageLab Software (Bio‐Rad). Antibodies for WB were anti‐human: CD63 (clone H5C6, BD Bioscience 557305), CD9 (clone MM2/57, Millipore cbl162), CD45 ( clone HI30, BD Bioscience 557748, GP96 (clone 9G10, Stressgen ADI‐SPA‐850‐D), AChE (Abcam ab31276), actin (clone C4, Millipore MAB1501), syntenin‐1 (clone C2C3, GeneTex GTX10847), CD81 (clone 5A6, Santa Cruz sc‐23692), SERINC3 (Abcam 153748), SPN (clone MEM‐59, Abcam 9088) and MOV10 (clone EPR14478, Abcam ab189919), ADAM10 (clone 163003, R&D Systems MAB1427), CD3 (clone EPR4517, Abcam ab134096) and anti‐HIV‐1 p24 monoclonal (183‐H12‐5C, NIH AIDS reagent program 1513). Secondary antibodies included HRP‐conjugated goat anti‐rabbit IgG (H + L) (Jackson 111‐035‐144), HRP‐conjugated goat anti‐mouse IgG (H + L) (Jackson 111‐035‐146) and HRP‐conjugated donkey anti‐goat IgG (H + L) (Jackson 705‐035‐147). Images were obtained with the Chemidoc Imaging System (Bio‐Rad), and quantifications were done using Image Lab software (Bio‐Rad).
EV isolation by size‐exclusion chromatography
Jurkat cell lines were left uninfected or infected during 2 h before being cultured in EV‐depleted medium for 48 h. CCM was harvested by pelleting cells at 300 g for 20 min at 4°C. Supernatant was centrifuged at 2,000 g for 20 min at 4°C to discard 2K pellet and then concentrated on an Sartorius Centrifugal Filter (MWCO = 100 kDa; VS2061). Medium was concentrated to 500 μl and overlaid on 70 nm qEV size‐exclusion columns (Izon, SP1). EVs were recovered in fractions 7–11, following manufacturers' instructions. These fractions were further subjected to immunoisolation, bead‐based multiplex flow cytometry and electron microscopy analysis.
Immunoisolation
Immunoisolation of CD81 or CD63 EVs (Figs 3A and EV2A) was performed with 6–10 × 1010 particles purified by size‐exclusion chromatography (SEC) from Jurkat CCM using Exosome Isolation Kit CD81 or Exosome Isolation Kit CD63 (Miltenyi) and following manufacturer's instructions. FT was subjected to ultracentrifugation for 30 min at 100,000 g in a TLA‐45 rotor (Beckman).
For immunoisolation of SPN‐EVs (Fig 5G), 100 μl of Protein A Magnetic Beads were coupled to anti‐SPN antibody (Abcam 9088) by overnight incubation at 4°C. Beads were then washed three times with 500 μl of PBS, and the antibody was crosslinked by incubation with 500 μl of BS3 solution in PBS (Thermo Scientific) for 1 h at 4°C. After crosslinking, beads were washed with 500 μl of PBS‐Tween 0.001%. EVs from 20 million Jurkat cells isolated by SEC were added to the beads and incubated under rotation for 2 h at RT. Bead‐bound EVs were washed three times in 500 μl of PBS. Nonbead‐bound EVs were concentrated on a Sartorius Centrifugal Filter (MWCO = 100 kDa; VS0142) to generate FT. FT was used to infect GHOST cells. FT and bead‐bound EV samples were resuspended in 20 μl of loading buffer 1× (Bio‐Rad) to be loaded on gel.
Bead‐based multiplex flow cytometry assay
EVs isolated from Jurkat CCM (Fig 3B) and primary CD4+ T CCM (Fig EV2B) by SEC were subjected to bead‐based multiplex analysis by flow cytometry (MACSPlex Exosome Kit, human, Miltenyi). Samples were processed according to manufacturer's instructions, with 3 detection antibodies used separately. Particle counts quantified by NTA were used to estimate input EV amounts. 2 × 109 EVs were diluted with MACSPlex buffer to a final volume of 120, and 10 µl of MACSPlex Exosome Capture Beads was added. Samples were incubated on an orbital shaker overnight at room temperature protected from light. After washing, detection antibodies (APC‐conjugated anti‐CD81 or anti‐CD63 [included in the kit] or 5 μl of anti‐CD3E [Miltenyi, 130‐113‐697]) were incubated for 1 h at RT. Flow cytometric analysis was performed with Aurora analyser (Cytek) and data analysed with FlowJo software (v10, FlowJo LLC). The 39 single bead populations were gated to allow determination of the APC signal intensity on the respective bead population and median fluorescence intensity (MFI) for each capture bead was measured. Out of the 37 markers analysed, only those detected in the proteome of Jurkat are shown.
100K pellets from control cells, SERINC3 KD cells and control cells after NL4‐3 EGFP‐Nef+ infection (Fig 6G) were analysed by the MACSPlex Exosome Kit as above, but with detection by a mix of APC‐conjugated anti‐CD9/CD81/CD63 antibodies provided by the manufacturer, and including a fixation step with 4% PFA for 1 h to inactivate the virus. 3–5 × 108 fixed EVs and 15 µl of MACSPlex Exosome Capture Beads were used. Flow cytometric analysis was performed with a MACSQuant Analyzer 10 with the corresponding software (Miltenyi Biotec) following the acquisition recommendations for the MACSPlex Exosome kit (Miltenyi Biotec). FlowJo software (v10, FlowJo LLC) was used to analyse flow cytometric data. For each capture bead, background was corrected by subtracting respective MFI values from matched non‐EV controls that were treated exactly like EV‐containing samples, and values of the corresponding isotype control were further subtracted. Due to low amount of sample and fixation, some markers, such as CD3 and CD9, were not detected.
Electron microscopy
Electron microscopy was performed on pellets resuspended in PBS and stored at − 80°C that had never been thawed and re‐frozen. EV suspension in PBS was deposited on formvar/carbon‐coated copper/palladium grids and adsorbed for 20 min before uranyl/acetate contrasting and methyl cellulose embedding for whole‐mount analysis as described previously (Thery et al, 2006). Cryosections were prepared and single‐ or double‐labelled with different antibodies according to the Protein A‐gold method (Slot & Geuze, 2007). CD81/CD63 double immunostaining was performed by successively incubating with rabbit anti‐CD81 (Abcam ab233692 1:50) for 30 min, 10 nm protein A‐gold (CMC, Utrecht, The Netherlands) for 20 min, fixed for 5 min with 1% glutaraldehyde (Electron Microscopy Sciences), followed by mouse anti‐CD63 (TS63 Diaclone 857.770.000 1:50) in PBS‐BSA 1% for 30 min, rabbit anti‐mouse (Sigma SAB3701080 1:100) for 30 min, 15 nm protein A‐gold (CMC, Utrecht, The Netherlands) for 20 min and fixed for 5 min with 1% (w/v) glutaraldehyde in PBS. Subsequently, after a wash on 10 droplets of distilled water, grids were transferred to droplets of 0.4% (w/v) uranyl acetate (UA) staining and 1.8% (w/v) methyl cellulose embedding solution. After 10 min of incubation, grids were picked up in a wire loop. Most of the excess of the viscous embedding solution was drained away with filter paper after which the grids with sections were air‐dried forming a thin layer of embedding solution. A similar procedure was followed for CD3/CD81 double labelling, using the antibodies in this order: rabbit anti‐CD3 (Abcam, ab134096 1:50), 5 nm protein A‐gold (CMC, Utrecht, The Netherlands), fixed for 5 min with 1% glutaraldehyde (Electron Microscopy Sciences, for 5 min), followed by with a rabbit anti‐CD81 (Abcam ab233692 1:50) antibody in PBS‐BSA 1% for 30 min, 10 nm protein A‐gold (CMC, Utrecht, The Netherlands) and fixed for 5 min with 1% (w/v) glutaraldehyde in PBS. Images were acquired with a digital camera Quemesa (EMSIS GmbH, Münster, Germany) mounted on a Tecnai Spirit transmission electron microscope (FEI Company) operated at 80kV. Vesicle size was assessed with ImageJ software.
Sample preparation for proteomics
Establish conditions for sample subfractionation to obtain similar amount of proteins in each fraction.
Establish optimal conditions for HIV‐1 infection to obtain at least 20% of infected cells with < 20% cell death.
Prepare RPMI for SILAC (Thermo Scientific) supplemented with 10% dialysed foetal bovine serum (Thermo Scientific) and “medium” amino acids l‐arginine‐13C6 and l‐lysine‐4,4,5,5‐D4 (medium‐SILAC) or “heavy” amino acids l‐arginine‐13C6 15N4 and l‐lysine‐13C6 15N2 (heavy‐SILAC)(Thermo Scientific).
Culture Jurkat cells for seven passages in medium‐SILAC RPMI or heavy‐SILAC RPMI to achieve metabolic labelling.
Determine the amino acid incorporation efficiency by liquid chromatography–tandem mass spectrometry (LC‐MS/MS).
-
Obtain a reference pellet (Ref) from cells grown in “heavy”‐labelled SILAC medium.
-
o
Incubate cells in FBS‐EV‐depleted heavy‐SILAC medium for 48 h.
-
o
Collect the supernatant and measure the cell viability. Cells used to obtain the reference pellet (Ref) from control sample and from infected sample (HIV‐Ref) presented 91.45 and 89.2% viability.
-
o
Centrifuge supernatants at 300 g (for 20 min) and 2,000 g (for 20 min) to obtain a clear supernatant.
-
o
Centrifuge the cleared supernatants at 100,000 g for 90 min in a SW32 Ti rotor to obtain the reference pellet (Ref).
-
o
-
Obtain subfractions from SILAC “medium”‐labelled cells.
-
o
Incubate cells in FBS‐EV‐depleted medium‐SILAC medium for 48 h.
-
o
Collect the supernatant and measure the cell viability. Three biological replicates of subfractions were generated independently from control Jurkat cells presenting more than 90% viability. Two biological replicates (A, B) of subfractions were obtained in parallel from uninfected and from NL4‐3 EGFP‐Nef+ infected cells. Subfractions (F1), (F2) and (F3) from replicate A and B were obtained from cells with 91 and 94% viability, respectively. Infected cells used to obtain subfractions (HIV‐F1), (HIV‐F2) and (HIV‐F3) from replicate A and B were 89.4 and 93% viable and presented 20.08 and 37% of infection, respectively.
-
o
Centrifuge supernatants at 300 g (for 20 min) and 2,000 g (for 20 min) to obtain a clear supernatant.
Centrifuge the cleared supernatants at 10,000 g for 20 min in a SW32 Ti rotor to obtain F1.
Centrifuge the cleared supernatants at 30,000 g for 20 min in a SW32 Ti rotor to obtain F2.
Centrifuge the cleared supernatants at 100,000 g for 90 min in a SW32 Ti rotor to obtain F3.
Resuspend pellets in (2.5% (w/v) SDS, Tris–HCl pH 8.0 and heated to 90°C for 5 min.
Mix subfraction with the reference fraction.
For the three preparations from untreated Jurkat cells, a protein assay (BCA, Thermo Fisher) was performed, and equal quantities of subfractions F1‐F3 and the reference fraction obtained from heavy‐SILAC‐labelled cells were mixed. For the HIV infection experiment (untreated vs HIV‐infected), each subfraction (F1), (F2) and (F3) from uninfected cells was mixed with one third of the reference fraction from untreated cells. The subfractions from infected cells (HIV‐F1), (HIV‐F2) and (HIV‐F3) were mixed with one third of the reference fraction from infected cells (HIV‐Ref).
Use a small aliquot of cells for each fractionation to independently lyse in SDS buffer and processed as described below for analysis of full proteomes.
MS and proteomic analysis
Untreated Jurkat cells (“NNP set”)
The online Neighbour Network Predictor tool can be accessed at: http://evprofiler.institut‐curie.org. For a detailed step by step instruction for the following protocol, please refer to (Itzhak et al, 2019).
Sample processing
The three sets of SILAC mixed subfractions (F1–3 plus heavy reference, in triplicate), as well as 30 µg SILAC medium‐labelled full proteome sample from each batch of uninfected Jurkat cells, were precipitated in parallel in 5× volume ice‐cold acetone at −20°C over night, and each was resuspended in 30 µl 50 mM Tris pH 8.1 containing 8 M urea and 1 mM dithiothreitol (DTT) at room temperature. The proteins were then alkylated with 5 mM iodoacetamide. In‐solution digestion was performed first with LysC overnight at room temperature, and for a further 3 h with additional trypsin upon dilution to 2 M Urea. The enzyme‐to‐protein ratio was 1:50 (w/w), but at least 500 ng of each enzyme was used. Peptides were then acidified to 1% (v/v) trifluoroacetic acid (TFA), and up to 20 µg peptides were loaded onto SDB‐RPS stage tips for peptide clean‐up and triple fractionation as previously described (Kulak et al, 2014). Peptides were vacuum‐concentrated to dryness, reconstituted in buffer A* (0.1% TFA, 2% acetonitrile [CAN]) and adjusted to 100–250 ng/µl.
LC‐MS/MS analysis
500 ng of peptides was loaded on a 50‐cm column with 75‐µm inner diameter, packed in‐house with 1.8‐µm C18 particles (Dr Maisch GmbH, Germany). Peptide separation by reverse phase chromatography was performed using an EASY‐nLC 1000 (Thermo Fisher Scientific), running a linear gradient from buffer A (0.1% formic acid) containing 5% buffer B (80% acetonitrile in 0.1% formic acid) to 30% buffer B over 95 min at 300 nl/min flow rate and 55°C. Runs were separated by 5 min wash‐outs with 95% buffer B and re‐equilibration. The LC was coupled to a Q Exactive HF‐X Hybrid Quadrupole‐Orbitrap Mass Spectrometer via a nanoelectrospray source (Thermo Fisher Scientific). MS data were acquired using a data‐dependent top 15 method, dynamically excluding precursors picked during the last 30 s. MS1 survey scans were acquired at a resolution of 60,000 in a 300–1,650 Th range, with a maximum injection time of 20 ms and 3e6 target ions, as determined with predictive automatic gain control. Sequencing was performed via higher energy collisional dissociation fragmentation of ions isolated from a 1.4 Th window, with a maximum injection time of 28 ms and 1e5 target ions. MS2 fragment scans were acquired at a resolution of 15,000 in a 200–2,000 Th range. The minimum predicted ion count to be reached per injection was 2.9e3. Raw data have been deposited to the ProteomeXchange Consortium via the PRIDE (Vizcaíno et al, 2016) partner repository with the dataset identifier PXD019074.
Processing of proteomic data
Raw files were analysed with MaxQuant Version 1.6.10.43 (Cox & Mann, 2008) with default settings, except for the following. SILAC mixed samples and full proteome samples were separated into two parameter groups for relative SILAC quantification and label‐free quantification using the MaxLFQ algorithm (Cox et al, 2014), respectively. For SILAC samples, the multiplicity was set to 2, with Arg6 and Lys4 as light labels, and Arg10 and Lys8 as heavy labels; re‐quantification was enabled. For full proteomes, only the light SILAC labels were specified. The minimum ratio count was set to 1 for both SILAC and LFQ, and matching between runs was enabled. Only identifications from the same cellular fraction and adjacent SDB‐RPS peptide fractions were matched across replicates. The human SwissProt protein reference database was downloaded from UniProt (2019_11_29, canonical and isoform data).
Comparison of control and HIV‐infected Jurkat cells (the “comparative set”)
Sample processing
SILAC protein mixed samples were separated by polyacrylamide gel electrophoresis (PAGE), and separated proteins were recovered in 7 bands and in‐gel digested as described in standard protocols. Briefly, following the SDS–PAGE and washing of the excised gel slices, proteins were reduced by adding 10 mM dithiothreitol (DTT) (Sigma‐Aldrich) prior to alkylation with 55 mM iodoacetamide (Sigma‐Aldrich). After washing and dehydrating the gel pieces with 100% acetonitrile, trypsin (Sequencing Grade, Promega) was added and proteins were digested overnight in 25 mM ammonium bicarbonate at 30°C. Peptides were extracted (70/35/5, MeCN/H2O/formic acid) from each gel slice, vacuum concentrated to dryness and reconstituted in injection buffer (2% MeCN, 0.3% TFA) before LC‐MS/MS analysis.
LC‐MS/MS analysis
Liquid chromatography (LC) was performed with an RSLCnano system (Ultimate 3000, Thermo Scientific) coupled online to an Orbitrap Fusion Tribrid Mass Spectrometer (MS, Thermo Scientific). Peptides were trapped on a C18 column (75 μm inner diameter × 2 cm; nanoViper Acclaim PepMapTM 100, Thermo Scientific) with buffer A (2/98 MeCN/H2O in 0.1% formic acid) at a flow rate of 2.5 µl/min over 4 min. Separation was performed on a 50 cm × 75 μm C18 column (nanoViper Acclaim PepMapTM RSLC, 2 μm, 100 Å, Thermo Scientific) regulated to a temperature of 55°C at a flow rate of 300 nl/min by using four‐step linear gradients of 161 min: 2 min (from 5 to 6% (v/v)), 18 min (from 6 to 9% (v/v)), 132 min (from 9 to 30% (v/v)) and 9 min (from 30 to 40% (v/v)) of buffer B (100% MeCN in 0.1% formic acid). Full‐scan MS was acquired in the Orbitrap analyser with a resolution set to 120,000, a mass range of m/z 400–1,500 and a 4 × 105 ion count target. Tandem MS was performed by isolation at 1.6 Th with the quadrupole, HCD fragmentation with normalized collision energy of 28 (%) and rapid scan MS analysis in the ion trap. The MS2 ion count target was set to 1 × 104, and only those precursors with charge state from 2 to 7 were sampled for MS2 acquisition. The instrument was run in top speed mode with 3 s cycles. Raw data have been deposited to the ProteomeXchange Consortium via the PRIDE (Vizcaíno et al, 2016) partner repository with the dataset identifier PXD016626.
Processing of proteomic data
Raw files were analysed with MaxQuant Version 1.6.1.13 (Cox & Mann, 2008), with default settings, except for the following. Multiplicity was set to 2, with Arg6 and Lys4 as light labels, and Arg10 and Lys8 as heavy labels. Requantify, match between runs and iBAQ were enabled. The minimum ratio count for SILAC quantification was set to 1. For the experimental design, equivalent slices from the SDS–PAGE gels were labelled with consecutive numbers (1–7 for control fractionation profiling samples, 11–17 for HIV‐1 infected fractionation profiling samples, and 101–107 for whole proteome samples (1, 11 and 101 correspond to the top slices of the gels). Hence, only identifications from equivalent and directly adjacent slices were matched. The human SwissProt protein reference database was downloaded from UniProt (2019_07_29, canonical and isoform data), augmented with second a custom‐made HIV protein Fasta file. The latter was based on the sequence information for the HIV‐1 strain used in this study (HIV‐1 vector pNL4‐3, GenBank: AF324493.2). The file contained individual HIV proteins as would be obtained post‐processing of the viral multiprotein precursors (Dataset EV2), as suggested in Nemeth et al (2017) and UniProt.
Bioinformatic analysis of proteomic data
The protein groups output files from MaxQuant were first processed in Excel (separate analyses for the “NNP dataset” from untreated Jurkat cells, and the “comparative set” from untreated and HIV‐infected cells):
Remove protein groups marked as reverse hit/potential contaminant/only identified by site.
Normalize each sample for channel loading: Divide SILAC ratios by median ratio of the sample.
Normalize intensities (full proteome samples, and SILAC medium intensities from F1–F3) for equal loading: Divide all intensities by the summed intensity of the sample and multiply with largest sum.
This master dataset was then further subdivided for different types of analyses. The SILAC ratio data were grouped into three‐data point profiles, i.e. for each protein, the SILAC ratios in the F1, F2 and F3 fractions of a given replicate and biological condition. For each protein, there were up to three profiles in the NNP dataset, with up to nine SILAC ratios in total, and up to four profiles in the comparative set (two from control, and two from HIV‐infected samples), with up to 12 SILAC ratios in total.
Analysis of the untreated Jurkat NNP dataset
Remove all proteins with less than nine SILAC ratios (only full coverage data).
Annotate proteins with some of the organellar subcellular localization markers from (Itzhak et al, 2016), and with EV markers (see Appendix Table S1).
-
Visualize the data structure by performing PCA using SIMCA 15 (Umetrics), with log SILAC ratios scaled to unit variance.
-
o
Since PCA is sensitive to leverage by extreme values, which can render the scores plot less informative, 11 proteins were excluded (Dataset EV1). These proteins were however included in the nearest neighbour and network analysis described below.
-
o
-
Calculate relative protein recovery profiles:
-
o
Invert SILAC ratios.
-
o
Weight ratios by relative fraction yields (F1:F2:F3 = 0.19:0.34:0.47).
-
o
Normalize each three‐point profile to sum = 1.
-
o
-
Calculate profile reproducibility:
-
o
For all pairwise combinations of replicate profiles calculate the Pearson correlation.
-
o
For maximum stringency work with the minimal correlation from here.
-
o
-
Calculate enrichment of EV fractions F1–F3 versus full proteome.
-
o
Calculate median normalized protein intensities across replicates.
-
o
For each protein, divide the median intensities of fractions F1–F3 by the normalized full proteome intensity, yielding an enrichment factor Fraction X/Full proteome.
-
o
To give equal weight to enrichment and depletion, ratios are either log10‐transformed or transformed as follows: Ratios equal or greater than one are not transformed. Ratios smaller than one are inverted and multiplied by −1. Hence, a ratio of 0.1 is converted to −10, to indicate 10‐fold depletion (Dataset EV1).
-
o
All following analysis steps are calculated on the fly for one or several query proteins and can be reproduced either by following the analysis steps in the interactive Excel sheet or using the python code provided on github (https://www.github.com/JuliaS92/EVProfiler).
Nearest neighbour profile analysis
Nearest neighbour profile analysis across the EV fractions was performed largely as described in (Borner et al, 2014), who originally reported the approach for the analysis of intracellular vesicle populations. Briefly, proteins with similar fractionation profiles as a given query protein are retrieved. Profile similarity between proteins is evaluated by calculating the pairwise Manhattan distance (absolute summed difference at every point of the two profiles). The query itself is retrieved as top hit (with a distance of 0), and other proteins are listed in order of increasing distance to the query. Different quality filters can be applied (eg minimum number of SILAC quantification events, minimum cosine correlation of profiles across repeats, enrichment level in the F3 extracellular vesicle fraction), to adjust the stringency and sensitivity of the analysis. As a guide to define the close protein neighbourhood of a query, a simple estimate of the local profile density and profile distribution was performed. From the nearest 250 proteins, the median distance to the query (termed MDQ) was calculated. Next, for every one of these 250 proteins the absolute distance to the MDQ was determined. The median of these values corresponds to the MAD (median absolute distance to the MDQ). Each positive distance to the MDQ (ie the right tail of the distribution) was then transformed to a pseudo Z score, using a robust estimate of the standard deviation: pZ = ((distance to MDQ) − MDQ)/(1.483 × MAD). Profiles were than categorized by these pZ scores: very close neighbours (Z > 3.09), close (Z > 2.33), fairly close (Z > 1.64), borderline (Z > 1.28). Please note that these categories are meant to provide useful guidance to evaluate relative profile proximity in the context of other mapped proteins nearby. They neither reflect exact boundaries for local clusters, nor exact probabilities of association with the query protein. Please also note that while the distances between two proteins are identical regardless of which of the two proteins is submitted as query, the proximity guide classifier may be different. So for example, an isolated protein that is near the edge of a dense cluster of profiles will retrieve some of these proteins as relatively close neighbours; however, these proteins themselves may have many neighbours within the cluster that are more proximal than the first query protein. This asymmetry is apparent in the PCA scores plots of the profiling data (Fig 2B). Furthermore, depending on the local density of profiles, the absolute distances corresponding to the different classifiers will vary. Again, the PCA scores plot helps to conceptualize this: in sparsely populated areas, distances between proteins are generally larger, so proteins with a relatively large absolute distance can still be classified as relatively close neighbours. Conversely, in a densely populated area of profile space, proteins must have very short distances to be considered as relatively close neighbours. It may therefore be informative to consider also the absolute distances of proteins to the query, as indicated in the predictor output.
The predictor also allows setting a minimum enrichment level of proteins in the F3 extracellular vesicle fraction relative to whole cells. The rationale is that proteins that are specifically sorted into small EVs should show enrichment in this fraction compared with their cellular pools. This is true for many classical markers of EVs such as CD63 and CD81. While it is not a necessary criterion, it can boost the stringency of the clustering analysis for sEV proteins. The percentrank function in Excel was used to calculate the abundance percentile for each protein from intensities from whole cell lysates (median from three replicates). For the extracellular fractions, the same percentrank calculation was performed for the SILAC medium intensities of the F3 fractions. Thus, a relative abundance between 0 (least abundant) and 1 (most abundant) was obtained for proteins in the whole cells and in the F3 fractions. For proteins not detected in whole cell lysates, the abundance percentile was set to 0. Next, the abundance percentile in the whole cell was subtracted from the abundance percentile in the F3 fraction. Thus, for every protein, a relative abundance shift (F3 vs cells), ranging between −1 and 1, was obtained. Positive values indicate enrichment in the 100K fraction compared with whole cell lysates. Proteins detected only in the F3 fraction had positive shifts by default. A shift of 0 indicates neither enrichment nor depletion. Please note that protein abundances are log‐normally distributed, and that an abundance percentile shift is not a linear measure of absolute abundance change.
Nearest neighbour network generation (This function is only available via the online tool at http://evprofiler.institut‐curie.org; the Excel Predictor contains the underlying data, but cannot display network graphs)
To visualize the complex structure of local neighbourhoods, we utilized network analysis. This can reveal for example if the closest neighbours of a protein form a very tight network among themselves, indicating a functional cluster; if the neighbourhood contains one or more tight subclusters, which may correspond to protein complexes; or if the local neighbourhood is very evenly distributed. These insights cannot be judged from a PCA plot alone, as the dimensional flattening invariably loses information contained in the original nine‐dimensional dataset. The network analysis was carried out in python using the networkx library (Hagberg et al, 2008). Network nodes are selected based on the distance ranking and pseudo z‐scoring as described above. Per default, nodes were selected from the overall nearest 50 proteins that showed a positive profile correlation across replicates, were found in the nearest 75 neighbours in at least two replicates and were at least fairly close (pZ > 1.64) to the query. To achieve an unidirectional assessment of all connections within this network, all pairwise Manhattan distances between the query's 250 closest neighbours were calculated. The distances between the network nodes were then categorized by the quantile they occupy in this distribution; all distances beyond the 50th or 25th percentile (as indicated) were discarded from the network. All remaining distances were inverted and used as edge weights for a force‐directed layouting algorithm ((Fruchterman & Reingold, 1991), as implemented in networkx), which pulls proteins with short distances closer together. Finally, edges were coloured by their distance percentile to visually reveal tight clusters. To visualize the segregation/overlap of adjacent neighbourhoods, networks were also constructed from more than one query protein. For each of the individual queries, nodes were selected as above, but with slightly adjusted parameters. Only the 30 closest neighbours were considered, to clearly separate distant queries, but borderline (pZ > 1.28) distances were included, to allow for more connections between adjacent neighbourhoods. The quantile boundaries were calculated for each individual set of 250 neighbours and then averaged across the queries. Thus, if two queries from areas of dataspace with different density are selected, this will become apparent in the number and colour of the edges.
This network based analysis, as well as the underlying scores and linear neighbourhoods, as provided in the supplemented Excel files, was compiled into an interactive dashboard, which is available online (http://evprofiler.institut‐curie.org). This was implemented in python using the holoviz framework and its bindings to several other python libraries (https://holoviz.org). All genes in the NNP profiling dataset can be queried, and all parameters described above can be adjusted as needed.
Analysis of the comparative dataset (untreated vs HIV‐infected)
For global PCA maps and nearest neighbour analysis, control and HIV fractionation profiles were analysed separately. Only proteins with two complete profiles in control and/or two complete profiles in HIV‐1 infected conditions were retained. SILAC ratios were the log‐transformed. For profile analysis and MR plot profile shift analysis, only proteins with four complete profiles (ie 12 SILAC ratios) were included. (Of note, HIV proteins themselves were not part of this analysis, as they were absent from the control fractions.) Ratios were inverted, summed within each profile and then each inverted ratio was divided by the corresponding profile sum. This yielded 0–1 normalized profiles, which show the relative proportion of a protein in each of the three fractions of a profile, summing to 1 (see also (Itzhak et al, 2016, 2017)).
For PCA, proteins were annotated with some of the organellar subcellular localization markers from (Itzhak et al, 2016), and diverse extracellular vesicle markers (see Appendix Table S1). HIV‐1 proteins were also annotated. PCA was then performed using SIMCA 15 (Umetrics). Log ratio data were UV scaled. Of note, each protein entered into the analysis with one or two sets of six SILAC ratios (ProteinX_con (profiles replicates A and B), and/or Protein X_HIV (profiles replicates A and B)), to obtain one set of joint loadings for both conditions, and hence directly comparable scores plots (see (Itzhak et al, 2017)). Since PCA is sensitive to leverage by extreme values, which can render the scores plot less informative, 62 proteins were excluded from the analysis (Dataset EV2). These proteins were however included in the nearest neighbour prediction and MR plot analysis described below.
Nearest neighbour profile analysis across the extracellular vesicle fractions was performed largely as described above for the NNP set, but separately for untreated and HIV‐infected datasets. Slightly adjusted cut‐offs were chosen for the nearest neighbour classification (very close neighbours (Z > 2.88), close (Z > 2.33), fairly close (Z > 1.64), borderline (Z > 1.15).
For MR plot profile shift analysis, the analysis reported step by step in (Itzhak et al, 2016, 2017, 2019) was followed, with minor variations to accommodate the different data structure. Briefly, proteins with significantly different profiles are likely to shift EV association. Since experimental and biological variation will introduce differences between replicate extracellular vesicle preparations, statistical analysis is required to distinguish background noise from significant changes. For each protein, the 0–1 normalized profiles from control and HIV‐1 infected preparations were subtracted, to yield two Delta profiles (from replicates A and B). Since most proteins are not expected to change profiles between conditions, the delta profiles largely reflect experimental noise. Using the robust Mahalanobis distance calculation implemented in Perseus 1.6. software (Itzhak et al, 2016; Tyanova et al, 2016), for each delta profile a P‐value of likelihood of deviation from the population mean was obtained. A very small P‐value indicates a potential outlier, i.e. a protein undergoing a genuine profile shift. For each protein, two independent P‐values were thus obtained, from the two replicates. For a stringent analysis, the higher (ie less significant) of the two P‐values was chosen and squared (as the second replicate had an equal or lower P‐value). This was then corrected for multiple hypothesis testing using the Benjamini‐Hochberg approach. The corrected P‐value was then negLog10‐transformed to yield a Movement (M) score. Next, the similarity of the direction of the profile shifts was ascertained, using Pearson correlation of the delta profiles from the replicates, to obtain the reproducibility (R) score. To enhance the sensitivity of the analysis, M scores were calculated only for proteins with a minimum SILAC ratio count of 2 in each fraction, and a positive R score (1,342 proteins). M and R scores were then combined into a MR plot analysis. Proteins that passed both M and R score cut‐offs were considered as candidates whose extracellular vesicle distribution is changed in response to HIV‐1 infection. To identify stringent cut‐offs for M and R scores and control the false discovery rate (FDR), a mock MR analysis from a scrambled experimental design not expected to yield any true hits was performed. Mock delta profiles were calculated by subtracting the profiles from the two control replicates from each other, and the profiles from the two replicates from HIV‐1 infected preparations. R scores were then calculated as Pearson correlation coefficients of these two mock delta profiles. M scores were then calculated for all proteins with positive R scores and a minimum ratio count of two (720 proteins in total), as above. To calculate the FDR, the number of hits at a given set of cut‐offs for M and R scores in the mock experiment was divided by the number of hits obtained in the actual comparisons (control vs HIV‐1 infected), scaled by a factor (1,342/720 = 1.86) to account for the different number of analysed proteins. Two levels of stringency were chosen to define significant hits: high stringency (M score > 1, R score > 0.9), FDR estimated at 8%, and medium stringency (M scores > 1, R score > 0.75), FDR estimated at 20%.
To classify the movement of proteins relative to HIV, the profiles of candidates in control and HIV‐1 infected conditions were subtracted from the average profile of all detected HIV‐1 proteins, to yield two Manhattan distances (control and HIV‐1 infected). These were then divided, and log10‐transformed to get a shift score. Proteins whose profiles were more similar to HIV after infection (shift scores > 0.10) were deemed to move towards HIV‐1; proteins with the opposite trend (negative shift scores < −0.10) were considered to move away from HIV‐1. Proteins with very small changes in delta profiles (between −0.10 and 0.10) were classified as moving “laterally” to HIV‐1 (Table 2).
qPCR
RNAs were isolated from 1 × 106 JURKAT cells with the Qiagen RNeasy Mini Kit, and 1,000 ng was reverse transcribed with SuperScript II Reverse Transcriptase (ThermoFisher). 1/20th of the resulting cDNA was used for each PCR reaction, performed with detection by SYBR Green (Thermo Scientific) on a LightCycler®480 Instrument II (LifeScience). Primer sequences for each gene were designed using Primer3Plus (http://www.bioinformatics.nl/cgi‐bin/primer3plus/primer3plus.cgi). Cycle of quantitation thresholds (Cq) were normalized to Cq of GAPDH and fold enrichments were calculated as compared with the control shRNA‐transduced cells values. Primers for human GAPDH were the forward primer 5′‐ATGTTCGTCATGGGTGTGAA‐3′ and the reverse primer 5′‐ATGTTCGTCATGGGTGTGAA‐3′. For human SERINC3 we used the forward primer 5′‐ATTCTAGCATCCGCACTTCC‐3′ and reverse primer 5′‐CGAGGCTGTCCATCTTCTTC‐3′.
Infection of GHOST cells
Jurkat cell lines were left uninfected or were infected for 2 h before being cultured in EV‐depleted medium for 48 h. CCM was harvested by pelleting cells at 300 g for 20 min at 4°C. Supernatant was centrifuged at 2,000 g for 20 min at 4°C to discard 2K pellet. The resulting supernatant was concentrated with a Sartorius Centrifugal Filter (MWCO = 100 kDa; VS2061). Infectivity of concentrated CCM from infected Jurkat cells was measured by incubation of 50 μl of concentrated CCM with 15,000 GHOST cells for 48 h. Percentage of GFP‐positive GHOST cells was determined by FACS analysis.
Nanoparticle tracking analysis
NTA was performed with CCM using ZetaView PMX‐120 (Particle Metrix) with software version 8.04.02. The instrument settings were 22°C, sensitivity of 70 and shutter of 75. Measurements were done using two different dilutions, at 11 different positions (five cycles per position) and frame rate of 30 frames per second. Note that NTA quantifies particles in general, whether or not they are covered by a membrane (i.e. qualify as vesicles or virions).
Statistical analyses
Bioinformatic analysis of proteomic data, generation of the network neighbour predictor and analysis of the ctrl/HIV comparative dataset: see details in section “Bioinformatics analysis of proteomic data”.
Other statistical analyses: Statistical analyses of particle concentration and multiplex bead‐based flow cytometry data were performed with GraphPad Prism version 8.0.2 (GraphPad software, California USA), by one‐way ANOVA followed by Dunnett's post‐test (Fig 6B and G) or by Friedman test followed by Dunn's post‐test (Fig EV3D).
Author contributions
LM‐J prepared the samples and JPS performed the proteomics for the untreated Jurkat cells and CCMs, used to generate the online neighbourhood network predictor. JPS and GHHB analysed the proteomics data. JPS and GHHB designed the neighbourhood network predictor. JPS implemented the online tool. LM‐J, JPS, GHHB and CT interpreted the proteomics data. LM‐J prepared the samples and FD performed the proteomics for the Ctrl/HIV Jurkat cells and CCMs. GHHB and DL analysed the proteomics data. LM‐J, DL, GHHB and CT interpreted the proteomics data. For all other experiments: LM‐J, NN, MT and MJ performed the experiments. LM‐J, NN, MT, MJ and CT analysed the data. LM‐J, KWW, MO and CT interpreted the data. LM‐J, KWW, MO and CT designed the experiments. LM‐J, GHHB and CT wrote the paper. All authors read the manuscript and provided comments.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
Dataset EV2
Acknowledgements
Clotilde Théry and her team acknowledge the following INSERM U932 members for providing tools and helpful discussions: Dr Nicolas Manel, Dr Philippe Benaroch, Dr Claire Hivroz, Dr Sebastian Amigorena, Dr Nicolas Ruffin, Dr Andrès Zucchetti, Stéphanie Dogniaux. The authors acknowledge the Cytometry platform at Institut Curie's Research Centre, and the assistance of its team to perform Cytometry. The work was funded by INSERM and Institut Curie and grants from NIDA (DA040385, to KWW, CT and MO, and DA047807, to KWW); ANRS (2015‐1 CT, LMJ); SIDACTION (17‐1‐AAE‐1138, CT); INCa (INCA‐11548, CT), Fondation ARC (PGA1 RF20180206962, CT); French National Research Agency (ANR‐10‐IDEX‐0001‐02 PSL*, ANR‐11‐LABX‐0043 and ANR‐18‐CE13‐0017‐03 to CT); and by the NIH Common Fund through the Office of Strategic Coordination/Office of the NIH Director (UG3CA241694, to KWW). The Proteomic platform of Institut Curie is supported by “Région Ile‐de‐France” and Fondation pour la Recherche Médicale grants (to D.L.). Electron microscopy imaging was enabled by the French National Research Agency through the "Investments for the Future" program (France‐BioImaging, ANR‐10‐INSB‐04), and PICT‐IBiSA, member of the France‐BioImaging national research infrastructure, supported by the CelTisPhyBio Labex (N° ANR‐10‐LBX‐0038) part of the IDEX PSL (No. ANR‐10‐IDEX‐0001‐02 PSL). Georg H. H. Borner and Julia Schessner were funded by the Max Planck Society for the Advancement of Science, and by the German Research Foundation (DFG/Gottfried Wilhelm Leibniz Prize MA 1764/2‐1); they wish to thank Matthias Mann for his support.
The EMBO Journal (2021) 40: e105492.
Contributor Information
Georg H H Borner, Email: borner@biochem.mpg.de.
Clotilde Théry, Email: clotilde.thery@curie.fr.
Data availability
The nearest neighbour network generation online tool is available at http://evprofiler.institut‐curie.org. The datasets and python code produced in this study are available in the following databases:
Raw data: PRIDE PXD019074 (http://www.ebi.ac.uk/pride/archive/projects/PXD019074) for Untreated Jurkat (NNP dataset); PXD016626 (http://www.ebi.ac.uk/pride/archive/projects/PXD016626) for Control and HIV‐infected Jurkat (comparative dataset).
code: github: https://github.com/JuliaS92/EVProfiler.
References
- Abudu A, Wang X, Dang Y, Zhou T, Xiang S‐H, Zheng Y‐H (2012) Identification of molecular determinants from moloney leukemia virus 10 homolog (MOV10) protein for virion packaging and anti‐HIV‐1 activity. J Biol Chem 287: 1220–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakelyan A, Fitzgerald W, Zicari S, Vanpouille C, Margolis L (2017) Extracellular vesicles carry HIV Env and facilitate Hiv infection of human lymphoid tissue. Sci Rep 7: 1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth AM, Fang Y, Fallon JK, Yang J‐M, Hildreth JEK, Gould SJ (2006) Exosomes and HIV Gag bud from endosome‐like domains of the T cell plasma membrane. J Cell Biol 172: 923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borner GHH, Hein MY, Hirst J, Edgar JR, Mann M, Robinson MS (2014) Fractionation profiling: a fast and versatile approach for mapping vesicle proteomes and protein‐protein interactions. Mol Biol Cell 25: 3178–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borner GHH (2020) Organellar maps through proteomic profiling – a conceptual guide. Mol Cell Proteomics 19: 1076–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantin R, Diou J, Bélanger D, Tremblay AM, Gilbert C (2008) Discrimination between exosomes and HIV‐1: purification of both vesicles from cell‐free supernatants. J Immunol Methods 338: 21–30 [DOI] [PubMed] [Google Scholar]
- Clark MC, Baum LG (2012) T cells modulate glycans on CD43 and CD45 during development and activation, signal regulation, and survival. Ann N Y Acad Sci 1253: 58–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettenhofer M, Yu XF (1999) Highly purified human immunodeficiency virus type 1 reveals a virtual absence of Vif in virions. J Virol 73: 1460–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser MT, Graham DR, Coren LV, Trubey CM, Bess JW Jr, Arthur LO, Ott DE, Lifson JD (2001) Differential incorporation of CD45, CD80 (B7–1), CD86 (B7–2), and major histocompatibility complex class I and II molecules into human immunodeficiency virus type 1 virions and microvesicles: implications for viral pathogenesis and immune regulation. J Virol 75: 6173–6182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO (2015) HIV‐1 assembly, release and maturation. Nat Rev Microbiol 13: 484–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruchterman TMJ, Reingold EM (1991) Graph drawing by force‐directed placement. Softw Pract Exp 21: 1129–1164 [Google Scholar]
- Gatto L, Breckels LM, Burger T, Nightingale DJH, Groen AJ, Campbell C, Nikolovski N, Mulvey CM, Christoforou A, Ferro M et al (2014) A foundation for reliable spatial proteomics data analysis. Mol Cell Proteomics 13: 1937–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg AA, Schult DA, Swart PJ (2008) Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science Conference, Varoquaux G, Vaught T, Millman J (eds), pp 11–15. Pasadena, CA USA. https://networkx.org/documentation/stable//citing.html [Google Scholar]
- Itzhak DN, Tyanova S, Cox J, Borner GH (2016) Global, quantitative and dynamic mapping of protein subcellular localization. Elife 5: e16950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhak DN, Davies C, Tyanova S, Mishra A, Williamson J, Antrobus R, Cox J, Weekes MP, Borner GHH (2017) A Mass spectrometry‐based approach for mapping protein subcellular localization reveals the spatial proteome of mouse primary neurons. Cell Rep 20: 2706–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhak DN, Schessner JP, Borner GHH (2019) Dynamic organellar maps for spatial proteomics. Curr Protoc Cell Biol 83: e81 [DOI] [PubMed] [Google Scholar]
- Jeppesen DK, Fenix AM, Franklin JL, Higginbotham JN, Zhang Q, Zimmerman LJ, Liebler DC, Ping J, Liu Q, Evans R et al (2019) Reassessment of exosome composition. Cell 177: 428–445.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keerthikumar S, Gangoda L, Liem M, Fonseka P, Atukorala I, Ozcitti C, Mechler A, Adda CG, Ang CS, Mathivanan S (2015) Proteogenomic analysis reveals exosomes are more oncogenic than ectosomes. Oncotarget 6: 15375–15396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koliha N, Wiencek Y, Heider U, Jungst C, Kladt N, Krauthauser S, Johnston IC, Bosio A, Schauss A, Wild S (2016) A novel multiplex bead‐based platform highlights the diversity of extracellular vesicles. J Extracell Vesicles 5: 29975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal‐Bengtson B, Dingli F, Loew D, Tkach M, Théry C (2016) Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA 113: E968–E977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M (2014) Minimal, encapsulated proteomic‐sample processing applied to copy‐number estimation in eukaryotic cells. Nat Methods 11: 319–324 [DOI] [PubMed] [Google Scholar]
- Lenassi M, Cagney G, Liao M, Vaupotic T, Bartholomeeusen K, Cheng Y, Krogan NJ, Plemenitas A, Peterlin BM (2010) HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4 T cells. Traffic 11: 110–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Z, Martin Jaular L, Soueidi E, Jouve M, Muth DC, Schøyen TH, Seale T, Haughey NJ, Ostrowski M, Théry C et al (2019) Acetylcholinesterase is not a generic marker of extracellular vesicles. J Extracell Vesicles 8: 1628592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llewellyn GN, Grover JR, Olety B, Ono A (2013) HIV‐1 Gag associates with specific uropod‐directed microdomains in a manner dependent on its MA highly basic region. J Virol 87: 6441–6454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheson NJ, Sumner J, Wals K, Rapiteanu R, Weekes MP, Vigan R, Weinelt J, Schindler M, Antrobus R, Costa ASH et al (2015) Cell surface proteomic map of HIV infection reveals antagonism of amino acid metabolism by Vpu and Nef. Cell Host Microbe 18: 409–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu M, Martin‐Jaular L, Lavieu G, Théry C (2019) Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell‐to‐cell communication. Nat Cell Biol 21: 9–17 [DOI] [PubMed] [Google Scholar]
- Minciacchi VR, You S, Spinelli C, Morley S, Zandian M, Aspuria PJ, Cavallini L, Ciardiello C, Reis Sobreiro M, Morello M et al (2015) Large oncosomes contain distinct protein cargo and represent a separate functional class of tumor‐derived extracellular vesicles. Oncotarget 6: 11327–11341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratori C, Cavallin LE, Kratzel K, Tinari A, De Milito A, Fais S, D'Aloja P, Federico M, Vullo V, Fomina A et al (2009) Massive secretion by T cells is caused by HIV Nef in infected cells and by Nef transfer to bystander cells. Cell Host Microbe 6: 218–230 [DOI] [PubMed] [Google Scholar]
- Nemeth J, Vongrad V, Metzner KJ, Strouvelle VP, Weber R, Pedrioli P, Aebersold R, Günthard HF, Collins BC (2017) In vivo and in vitro proteome analysis of human immunodeficiency virus (HIV)‐1‐infected, human CD4 + T cells. Mol Cell Proteomics 16: S108–S123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Niel G, D'Angelo G, Raposo G (2018) Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 19: 213–228 [DOI] [PubMed] [Google Scholar]
- Nolte‐‘t Hoen E, Cremer T, Gallo RC, Margolis LB (2016) Extracellular vesicles and viruses: are they close relatives? Proc Natl Acad Sci USA 113: 9155–9161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott DE (2009) Purification of HIV‐1 virions by subtilisin digestion or CD45 immunoaffinity depletion for biochemical studies. Methods Mol Biol 485: 15–25 [DOI] [PubMed] [Google Scholar]
- Pereira EA, daSilva LLP (2016) HIV‐1 Nef: taking control of protein trafficking. Traffic 17: 976–996 [DOI] [PubMed] [Google Scholar]
- Pérez PS, Romaniuk MA, Duette GA, Zhao Z, Huang Y, Martin‐Jaular L, Witwer KW, Théry C, Ostrowski M (2019) Extracellular vesicles and chronic inflammation during HIV infection. J Extracell vesicles 8: 1687275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond AD, Campbell‐Sims TC, Khan M, Lang M, Huang MB, Bond VC, Powell MD (2011) HIV Type 1 Nef Is released from infected cells in CD45 + microvesicles and is present in the plasma of HIV‐infected individuals. AIDS Res Hum Retroviruses 27: 167–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa A, Chande A, Ziglio S, De Sanctis V, Bertorelli R, Goh SL, McCauley SM, Nowosielska A, Antonarakis SE, Luban J et al (2015) HIV‐1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 526: 212–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba DG, Céspedes‐Donoso PF, Bálint Š, Compeer EB, Korobchevskaya K, Valvo S, Mayya V, Kvalvaag A, Peng Y, Dong T et al (2019) Composition and structure of synaptic ectosomes exporting antigen receptor linked to functional CD40 ligand from helper T cells. Elife 8: e47528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler M, Münch J, Kirchhoff F (2005) Human immunodeficiency virus type 1 inhibits DNA damage‐triggered apoptosis by a Nef‐independent mechanism. J Virol 79: 5489–5498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvin A, Yu CI, Lahaye X, Imperatore F, Brault J‐B, Cardinaud S, Becker C, Kwan W‐H, Conrad C, Maurin M et al (2017) Constitutive resistance to viral infection in human CD141 + dendritic cells. Sci Immunol 2: eaai8071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot JW, Geuze HJ (2007) Cryosectioning and immunolabeling. Nat Protoc 2: 2480–2491 [DOI] [PubMed] [Google Scholar]
- Thery C, Amigorena S, Raposo G, Clayton A (2006) Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol Chapter 3: Unit 3.22 [DOI] [PubMed] [Google Scholar]
- Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F, Atkin‐Smith GK et al (2018) Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 7: 1535750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach M, Kowal J, Zucchetti AE, Enserink L, Jouve M, Lankar D, Saitakis M, Martin‐Jaular L, Théry C (2017) Qualitative differences in T‐cell activation by dendritic cell‐derived extracellular vesicle subtypes. EMBO J 36: 3012–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach M, Kowal J, Théry C (2018) Why the need and how to approach the functional diversity of extracellular vesicles. Philos Trans R Soc B Biol Sci 373: 20160479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- Usami Y, Wu Y, Göttlinger HG (2015) SERINC3 and SERINC5 restrict HIV‐1 infectivity and are counteracted by Nef. Nature 526: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deun J, Mestdagh P, Agostinis P, Akay O, Anand S, Anckaert J, Martinez ZA, Baetens T, Beghein E, Bertier L et al (2017) EV‐TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods 14: 228–232 [DOI] [PubMed] [Google Scholar]
- Vizcaíno JA, Csordas A, del‐Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez‐Riverol Y, Reisinger F, Ternent T et al (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44: D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votteler J, Sundquist WI (2013) Virus budding and the ESCRT pathway. Cell Host Microbe 14: 232–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
Dataset EV2
Data Availability Statement
The nearest neighbour network generation online tool is available at http://evprofiler.institut‐curie.org. The datasets and python code produced in this study are available in the following databases:
Raw data: PRIDE PXD019074 (http://www.ebi.ac.uk/pride/archive/projects/PXD019074) for Untreated Jurkat (NNP dataset); PXD016626 (http://www.ebi.ac.uk/pride/archive/projects/PXD016626) for Control and HIV‐infected Jurkat (comparative dataset).
code: github: https://github.com/JuliaS92/EVProfiler.