

Abstract
In the mammalian embryo, epiblast cells must exit the naïve state and acquire formative pluripotency. This cell state transition is recapitulated by mouse embryonic stem cells (ESCs), which undergo pluripotency progression in defined conditions in vitro. However, our understanding of the molecular cascades and gene networks involved in the exit from naïve pluripotency remains fragmentary. Here, we employed a combination of genetic screens in haploid ESCs, CRISPR/Cas9 gene disruption, large‐scale transcriptomics and computational systems biology to delineate the regulatory circuits governing naïve state exit. Transcriptome profiles for 73 ESC lines deficient for regulators of the exit from naïve pluripotency predominantly manifest delays on the trajectory from naïve to formative epiblast. We find that gene networks operative in ESCs are also active during transition from pre‐ to post‐implantation epiblast in utero. We identified 496 naïve state‐associated genes tightly connected to the in vivo epiblast state transition and largely conserved in primate embryos. Integrated analysis of mutant transcriptomes revealed funnelling of multiple gene activities into discrete regulatory modules. Finally, we delineate how intersections with signalling pathways direct this pivotal mammalian cell state transition.
Keywords: exit from naïve pluripotency, haploid ES cells, naïve to formative transition, signalling, systems biology
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Methods & Resources; Regenerative Medicine
An extensive mutagenesis screen identifies regulatory gene modules governing exit from naïve mammalian pluripotency.

Introduction
Mouse embryonic stem cells (ESCs) can self‐renew in defined conditions in a state of naïve pluripotency (Smith, 2017). The ESC exit from naïve pluripotency provides an amenable experimental system for dissection of a cell fate decision paradigm (Buecker et al, 2014; Kalkan et al, 2017). Naïve pluripotency is under control of a gene regulatory network (GRN) containing the core pluripotency transcription factors (TFs) Pou5f1, Sox2 and naïve‐specific TFs such as Nanog, Esrrb, Klf4 and others (Chen et al, 2008; Dunn et al, 2014; Niwa 2018). In defined cell culture conditions that include inhibitors against Mek1/2 (PD0325901) and Gsk3 (CHIR990201, CH; collectively termed “2i”), ESCs can be homogenously maintained in the naïve state (Ying et al, 2008). Within 24–36 h after withdrawal of 2i, ESCs transit into formative pluripotency, entirely losing naïve identity (Kalkan et al, 2017). During this transition, the naïve GRN is extinguished and expression of formative factors such as Otx2, Pou3f1, Dnmt3a/b and Fgf5 is initiated. A similar transition is evident during peri‐implantation development, where the TF network maintaining naïve pluripotency dissolves between embryonic day (E) 4.5 and E5.5 (Boroviak et al, 2014; Acampora et al, 2016; Mohammed et al, 2017). The speed of the naïve to formative GRN transition is notable because (i) the cell cycle is around 12 h long; (ii) all factors that are required to establish and maintain naïve pluripotency are expressed robustly in naïve cells; and (iii) the naïve pluripotency network is recursively self‐reinforcing. The rapid dissolution of naïve pluripotency implies the existence of circuit‐breaking mechanisms. In recent years, we and others have identified various factors promoting ESC differentiation using screens in haploid and diploid ES cells (Guo et al, 2011; Leeb & Wutz, 2011; Betschinger et al, 2013; Leeb et al, 2014; Li et al, 2018).
Robust assays employing ESCs expressing a Rex1 promoter‐driven destabilised GFP reporter (Rex1::GFPd2) enable the dissection of the exit from naïve pluripotency in high resolution (Kalkan et al, 2017; Mulas et al, 2017). Rex1‐GFP downregulation is initiated within 24h after 2i withdrawal (N24) and completed after 48h (N48). Nevertheless, the exact nature, mechanistic underpinnings and sequence of events during exit from naïve pluripotency remain only partially understood. In particular, we lack insight into how the different molecular components of the system co‐operate to elicit proper cell fate transition. Here, we have driven a Rex1‐GFP reporter screen to saturation, thus providing an extensive list of genes and pathways involved in the exit from naïve pluripotency. We utilised this information in a systems biology approach to explore regulatory principles of the exit from naïve pluripotency. To evaluate dependencies and causal relationships within the pluripotency and differentiation circuitries, we probed the response of the differentiation programme to a comprehensive series of exit factor gene knockouts. Through computational integration of molecular profiling data with regulatory networks and in vivo GRN trajectories, we expose the regulatory foundations of a cell fate choice paradigm at a pivotal junction in early mammalian development.
Results
Haploid ES cell saturation screen
Haploid ES cells are an efficient platform for insertional mutagenesis‐based screens (Elling et al, 2011; Leeb & Wutz, 2011; Kokubu & Takeda, 2014). We previously reported a medium‐scale screen comprising approximately 5 × 104 mutagenic events to identify factors regulating the exit from naïve pluripotency (Leeb et al, 2014). We have now driven this approach to saturation by assaying approximately 1.2 million mutations in receptive genomic regions that cause delays in Rex1 downregulation in two independent Rex1‐reporter cell lines (Fig 1A and B), utilising three different mutagenic transposon vectors in 35 independent screens (Fig 1C and D).
Figure 1. Establishment of exit factor deficient KO ESC lines informed by a haploid ESC saturation screen.

- Illustration of the Rex1‐GFPd2 reporter cell line and its exit from naïve pluripotency. Rex1‐GFPd2 (in short Rex1‐GFP) expression is tightly linked to naïve pluripotency. Shutdown of GFP expression indicates commitment to differentiation.
- FACS analysis of Rex1‐GFP reporter levels throughout a 72h differentiation time course after 2i withdrawal.
- Scheme of the screening strategy to identify candidate genes involved in the exit from naïve pluripotency. After random insertional mutagenesis using piggyBac transposon‐based gene‐trap vectors, haploid Rex1‐GFP ESCs were released into differentiation. Cells maintaining GFP expression after exposure to differentiation conditions were isolated and the gene‐trap insertion sites mapped.
- The cumulative number of hits (red) and the cumulative number of novel hits (blue) in 35 independent insertional mutagenesis screens in haploid ESCs are shown.
- Representative Rex1‐GFP FACS plots showing the differentiation delays 24 h after 2i withdrawal of Tcf7l1, Rbpj, Trim71, Smg5 and Pten KOs. A Myc KO served as a negative control. Blue indicates the Rex1‐GFP FACS profiles for KO, and dashed lines indicate WT.
- Differential expression of genes at N24 versus 2i in WT RC9 cells. Black dots show significance (FDR ≤ 0.05, H0: |log2FC| < log2(1.5)). Pluripotency genes are red dots, formative genes are orange dots, haploid screen hits are blue dots, and the 73 KO genes are green dots.
- t‐SNE projection of the 73 KOs in 2i, based on expression of the 3068 differentially expressed genes between N24 and 2i in WT. The strength of differentiation delay observed at N24 in the respective KOs are indicated by a colour gradient and measured as average naïve marker log2 fold change (log2 FC, based on expression levels of Esrrb, Nanog, Tfcp2l1, Tbx3, Prdm14 and Klf4, Zfp42) in the respective KO at N24. Red: delayed differentiation; blue: accelerated differentiation.
- Similar to (G) for KOs at N24.
Stringent filtering resulted in a candidate list comprising 489 genes (Dataset EV1). These screens generated a candidate inventory of the machinery that mediates exit from naïve pluripotency. Reassuringly, the known exit from naïve pluripotency regulators Tcf7l1, Fgfr2, Jarid2 and Mapk1 (Erk2) (Kalkan & Smith, 2014) were among the highest ranked genes (Appendix Fig S1A). The candidate hit genes were enriched for processes involved in transcription regulation, epigenetics and signalling‐related functions (Appendix Fig S1B and C; Dataset EV1), as well as RNA‐binding functions in‐line with emerging evidence of RNA regulatory mechanisms in cell fate control (Ye & Blelloch, 2014).
Many identified genes are not specific to pluripotency and have functions in common pathways and processes, implying that the exit from naïve pluripotency utilises widely expressed cellular machinery. Therefore, mechanisms mediating ESC transition might also be utilised in other differentiation processes.
Establishment of a mutant ESC library for systematic transcriptional profiling
To characterise deficiencies in naïve exit in molecular detail, we generated KO ESC lines deficient for 73 selected genes, comprising top ranked genes from the mutagenic screen. We also included components from pathways and protein complexes for which multiple members were recovered, even if just below the cut‐off threshold (e.g. the Paf complex member Leo1, the mTORC1 regulator Tsc2 and the NMD component Smg6), and Mbd3, Zfp281 and L3mbtl3 as known players in the exit from naïve pluripotency (Betschinger et al, 2013). Three control genes were included that are either not expressed in ES cells (Nestin), expected to be neutral (Hprt) or whose ablation was expected to accelerate differentiation (c‐Myc). Paired gRNAs were used to disrupt target genes in a diploid biparental Rex1::GFPd2 reporter ES cell line carrying a Cas9 transgene (henceforth termed RC9 cells) (Fig 1A) (Li et al, 2018). Following an efficient parallelised approach, we established passage matched and isogenic homozygous KO cell lines, thus maximising comparability (Appendix Fig S1D and E). All KOs were validated by genomic PCR, followed by Sanger sequencing when required.
Full protein deficiency was validated for 14 KOs (Eed, Suz12, Jarid2, Kdm6a, Smg5, Smg6, Smg7, Tsc2, Pten, Raf1, Tcf7l1, Leo1, Nmt1 and Csnk1a1) (Appendix Fig S2A). Only heterozygote clones could be generated for Mapk1 (Erk2), resulting in reduced protein levels. Notably, increased levels of Erk1 in Erk2 heterozygous mutant cells failed to rescue the strong differentiation delay in the Mapk1het KOs. For five further genes (Alg13, Dido1, Msi2, Etv5, Jmjd1c), we confirmed the absence of the corresponding transcripts or specific out of frame deletion of an exon by RT–qPCR or Sanger sequencing of RT–PCR products. Successful rescue experiments using 3xflag‐tagged transgenes for six genes (Rbpj, Etv5, Fgfr1, Jarid2, Mbd3 and Tcf7l1) established causality between the observed genotype and phenotype (Appendix Fig S2B and C). Thus, all tested knockouts showed the expected impact on RNA or protein expression. However, we cannot exclude the possibility of hypomorphic phenotypes in some cases.
ESC differentiation behaviour is highly dependent on cell density and timing of medium changes. To enable robust comparison of the differentiation of multiple KO ESC lines, we performed parallel differentiations batch wise in duplicates, always including WT ESCs and negative controls across seven experiments. At N24, we assayed the differentiation status by FACS analysis (Fig 1E) and extracted RNA for transcriptome analysis.
The exit machinery is already poised in 2i
Batch‐corrected RNA‐seq data (Appendix Fig S3A and B) comprising 14 replicates of WT ES cells identified 3068 differentially expressed genes (DEGs) between 2i and N24 (H0: |log2FC| < log2(1.5), FDR ≤ 0.05; Fig 1F, Appendix Fig S4A–C and Dataset EV1). Interestingly, most of the 489 genes identified in the haploid screens including the 73 genes (Dataset EV6) selected for KO did not change significantly in expression between 2i and N24 and were not present in the list of DEGs (Fig 1F and Appendix Fig S4C), with only 21% of screen hits showing differential expression at N24 (6% up‐, 15% downregulated). This implies that the exit machinery is already embedded in the ground state and ESCs are poised for rapid decommissioning of naïve identity and entry into differentiation (Kalkan & Smith, 2014).
To facilitate interrogation of the KO gene expression datasets, we developed an interactive online tool (GENEES—Genetic Network Explorer for Embryonic Stem Cells—http://shiny.cecad.uni‐koeln.de:3838/stemcell_network/).
Using t‐Distributed Stochastic Neighbour Embedding (t‐SNE), we visualised similarities between KOs based on expression of the DEG in 2i and at N24 (Fig 1G and H). We observed clustering of members of the same complex or pathway: Eed‐Suz12 (PRC2), Ptpn11‐Raf1‐Fgfr1‐Etv5 (Fgf/ERK), Smg5‐Smg6 (NMD; nonsense mediated decay), Mta3‐Mbd3 (NuRD) and Pten‐Tsc2 (mTORC1 signalling) (Fig 1G and H). The transcription profiles of the KO ESCs clustered by genotype, but mainly by culture condition (2i and N24) (Appendix Fig S4A). This is consistent with the observation that despite manifesting differentiation delays at N24, all of the KO ESCs ultimately departed from the naïve state during longer differentiation time courses, as measured by loss of Rex1‐GFP. Furthermore, even KOs that showed extensive Rex1‐GFP downregulation delays at N24 displayed transcription profiles that were globally adjusted towards differentiation. Therefore, the knockout of a single gene is not sufficient to permanently block exit from naïve pluripotency in culture, in accord with the finding that ternary depletion of Tcf7l1, Etv5 and Rbpj is required for sustained self‐renewal in the absence of 2i or LIF (Kalkan et al, 2019).
The exceptional role of Csnk1a1 and the involvement of compensatory mechanisms
At N24, the Csnk1a1 KO clustered with 2i samples (Appendix Fig S4A), indicating a special behaviour for this mutant. However, at N48 most Rex1‐GFP expression was downregulated, illustrating a strong delay but not a block in differentiation (Appendix Fig S5A). siRNA treatment as well as treatment with Epiblastin A, a chemical inhibitor of Csnk1a1 (Ursu et al, 2016), delayed the exit from naïve pluripotency without apparently affecting proliferation within the duration of the assay (Appendix Fig S5B–D). Csnk1a1 is a serine threonine kinase and a component of the beta‐catenin destruction complex. Although KO of another destruction complex member Apc, or of the downstream repressor Tcf7l1 resulted in the upregulation of similar gene‐sets (Appendix Fig S5E), we observed stronger differentiation defects and larger amplitude of gene deregulation in two independently derived Csnk1a1 mutants. However, these mutants also exhibited markedly impaired cell cycle profiles in N2B27‐based media (Appendix Fig S5F). Upon continuous culture (~5 passages), proliferation was restored in Csnk1a1 KO cells and differentiation potential was regained, suggesting upregulation of compensatory mechanisms and a likely effect of proliferation rate on differentiation kinetics, complicating mechanistic characterisation. A second case of phenotype adaptation was observed in Pum1 mutants. Pum1 KOs showed pronounced differentiation delays during early passages (Appendix Fig S5G), as also seen for acute Pum1 depletion by siRNA and in previously generated CRISPR KO ESCs (Leeb et al, 2014). However, the phenotype was lost in later passages, and Pum1 KO cells showed WT‐like Rex1‐GFP expression levels at N24 (Fig 2A and Appendix Fig S5G).
Figure 2. Systematic transcriptional profiling of a mutant ESC library.

- Naïve (top) and formative (bottom) marker gene expression changes at N24 compared with WT in all 73 KO ESCs. Clustering based on naïve marker gene expression shows a wide distribution of differentiation delay phenotypes.
- Rex1‐GFP FACS analysis of Jarid2 and Tcf7l1 KO ESCs after transfection with negative control or Klf2‐specific siRNAs.
- Plot showing a comparison of the log2 fold change (KO versus WT) of mean naïve marker gene expression at N24 to the extent of change of the global transcriptome between 2i and N24 (defined as the correlation of log2FCs in WT differentiation and log2FCs in KOs at N24). Each dot corresponds to one of the 73 KOs. Pearson’s correlation is shown in the plot.
- Comparison of the number of genes deregulated in 2i and at N24 in all KOs (FDR ≤ 0.05, H0: |log2FC| < log2(1.5)). Differentiation phenotypes are colour coded according to average naïve marker log2FCs in KOs at N24.
- Rex1‐GFP FACS analysis of WT, Jmjd1c and Tcf7l1 KO cells at N48 cultured with and without the Gsk3 inhibitor CH.
Robust feedback wiring in the naïve TF network
Interestingly, the transcriptome data revealed that exit factors do not, in general, reduce naïve transcription factor (TF) expression in the ground state. However, Rbpj KO resulted in moderate but significant increases in Klf4, while the aforementioned Csnk1a1 KO ESCs showed limited upregulation of both Klf4 and Tbx3 in 2i (Fig EV1A). Ctbp2 KO significantly upregulated Nr0b1 in the ground state. Other KOs had no significant effects on factors of the naïve network in 2i. These data are consistent with robust feedback wiring in the naïve TF network (Dunn et al, 2014; Niwa, 2018) and neutralisation of most differentiation factors in 2i in culture (Martello & Smith, 2014). In contrast, we observed a more extensive impact on formative markers. Several KO cell lines showed lower baseline expression in 2i of Otx2, Fgf5, Dnmt3a/b and Pou3f1 (Oct6) (Fig EV1B). In‐line with recent results, we noted that depletion of several Fgf/ERK components resulted in reduced Dnmt3a/b, Pou3f1 and Fgf5 expression in 2i (Kalkan et al, 2019). Although Fgf/ERK signalling is effectively inhibited in 2i (Ying et al, 2008), our data suggest that either residual pathway activity or potential moonlighting functions of pathway components mediate poised expression of the formative pluripotency programme in 2i.
Figure EV1. Systematic transcriptional profiling of a mutant ESC library.

- RNA‐seq‐derived fold changes relative to WT of indicated naïve marker genes in indicated KOs in 2i (colour scale shows FDR, only genes with FDR ≤ 0.05 are shown).
- RNA‐seq‐derived fold changes relative to WT of indicated formative marker genes in indicated KOs in 2i (colour scale shows FDR, only genes with FDR ≤ 0.05 are shown).
- Rex1‐GFP FACS analysis of WT cells in 2i, at N24 and at N30, transfected with negative control or Klf2‐specific siRNAs.
- Comparison of the number of differentially expressed genes at N24 (FDR ≤ 0.05, H0: |log2FC| < log2(1.5)) to the average naïve marker log2FC at N24 (phenotype strength).
- Correlation of Tcf7l1 KOs to Jmjd1c KOs regarding log2FCs at N24 between knockout and WT control. Each dot corresponds to one gene. Only genes showing significance (FDR ≤ 0.05, H0: |log2FC| < log2(1.5)) in either one of the two KOs are plotted. Red line: total least square regression; regression coefficients are shown.
Clustering based on the expression of ten naïve pluripotency marker genes showed that the downregulation of the naïve pluripotency TFs during formative differentiation is defective across multiple KOs (Fig 2A). Although overall expression of the naïve TFs was highly correlated, Klf2 appeared to be an exception. Klf2 downregulation was notably impaired in Tcf7l1 KO ESCs, whereas it was unaffected by several KOs, including Jarid2, despite a comparable extent of deregulation of most other naïve marker genes. This indicates that Klf2 expression can be uncoupled from the core naïve network. Forced Klf2 expression stabilises self‐renewal (Hall et al, 2009; Qiu et al, 2015). Klf2 depletion destabilises mouse ESC identity (Yeo et al, 2014) and increases the speed of Rex1‐GFP downregulation upon 2i withdrawal (Fig EV1C). The differentiation delay in Tcf7l1 KO ES cells is partially dependent on Klf2 (Fig 2B), consistent with direct regulation (Martello et al, 2012). In contrast, Klf2 depletion in Jarid2 KOs did not restore differentiation timing, indicating that separable gene networks contribute to dismantling naïve pluripotency.
Effects of exit gene depletion on global gene regulation
We used two measures to gauge differentiation delays in KOs compared with WT cells: first, the average naïve marker change defined throughout this manuscript as the log2 fold change of a set of seven naïve TFs, Esrrb, Nanog, Tfcp2l1, Tbx3, Prdm14, Klf4 and Zfp42 at N24 at N24 compared with WT; second, the extent of global transcriptome adjustments between 2i and N24 in each KO compared with changes occurring in WT. Generally, the delayed extinction of the naïve TF network in KOs at N24 was accompanied by reduced gene expression change between 2i and N24 (R = −0.91; Fig 2C). Trim71 KOs formed an exception. They exhibited a relatively strong impact on naïve TF expression levels, while displaying only modest global transcription changes. Thus, the RNA‐binding protein Trim71 appears to be focused on the regulation of naïve pluripotency TF genes, although the ensuing exit‐delay in Trim71 KO cells is modest.
A general correlation was observed between the numbers of genes deregulated in 2i and at N24 (Fig 2D). Mutants with the strongest impact on the transcriptome (e.g. Csnk1a1, Eed, Suz12, Zfp281 and Smg5) showed deregulation of several thousand genes both in 2i and at N24. Fgfr1 and Tcf7l1 KOs were two exceptions to this correlation with relatively few genes deregulated in 2i but several hundred at N24. We surmise that the effect of these two genes in 2i is largely masked because they are in the same pathways as the inhibitor targets Mek1/2 (PD0325901) and Gsk3 (Chiron 990201) (Ying et al, 2008; Martello & Smith, 2014).
Defective differentiation could simply equate with the extent of overall gene deregulation. We therefore mapped the average deregulation of naïve marker genes onto Fig 2D. Most KOs showing the strongest increase in naïve marker gene expression correlated with large‐scale gene deregulation (both in 2i and at N24). However, there were several exceptions: for example, KO of Eed, Suz12, Smg7, Leo1 and Ctbp2 affected a substantial number of genes, but caused naïve marker deregulation that was weaker than expected assuming a direct correlation between the number of deregulated genes and the differentiation phenotype (Figs 2D and EV1D). Vice versa, KO of Mbd3, Cdk8 or Pten affect naïve marker downregulation, despite a relatively mild impact on the number of DEGs in the respective KOs.
Transcriptome analysis reveals a genetic interaction between Jmjd1c and Tcf7l1
The putative histone 3 lysine 9 (H3K9) demethylase Jmjd1c (Kdm3c) KO is a case of particular note. No genes were significantly deregulated at N24 (Fig 2D), and a mild global neutralisation of differentiation‐induced transcriptome changes was observed (Fig 2C). Neither the role of Jmjd1c in pluripotency regulation nor its mode of action is known. We used multiple regression analysis to determine the similarity of the Jmjd1c KO to all other KO RNA‐seq profiles. At N24, the Jmjd1c KO transcriptome was most similar to the Tcf7l1 KO profiles (Fig EV1E), suggesting a potential functional connection between Jmjd1c and Tcf7l1. Indeed, Jmjd1c has recently been reported as a high‐confidence protein interactor of Tcf7l1 (preprint: Moreira et al, 2018). The 2i component Chiron 99021 is a specific inhibitor of Gsk3 and phenocopies deletion of Tcf7l1, which is the downstream repressor in ESCs (Wray et al, 2011; Shy et al, 2013). Accordingly, Chiron 99021 delays the differentiation of WT ESCs, but had little additional effect on Tcf7l1 KOs, without affecting growth or survival of treated cells (Fig 2E and Appendix Fig S5H). Jmjd1c‐deficient ESCs did not show a discernible phenotype at N48, in‐line with minimal deregulation of the naïve TF network. Addition of Chiron 99021 had a stronger than expected effect on Jmjd1c KOs and resulted in a synthetic enhanced delay phenotype (Fig 2E), suggesting a cooperative activity of Jmjd1c and the Wnt/Tcf7l1 axis in the exit from naïve pluripotency.
Relative quantification of differentiation delays in vitro
To quantify differentiation delays, we obtained RNA‐seq data from a 2h‐resolved WT ESC differentiation time course (Fig 3A and Appendix Fig S6A). We then compared the expression patterns of a set of naïve markers in a given KO at N24 with the expression of the same gene‐set along the WT 2i to N32 trajectory. This enabled us to “position” each KO along the trajectory and thus quantify the differentiation delay with a precision of about 4h (Fig 3B, Dataset EV2). In a complementary approach, we used all 3068 DEGs between 2i and N24 as an alternative reference set. This yielded similar, but on average slightly less pronounced differentiation delays (Fig 3C and Appendix Fig S6B), supporting the hypothesis that the naïve TF network is regulated in concordance with but partially independently from the rest of the transcriptome during exit from naïve pluripotency.
Figure 3. Relative quantification of differentiation delays in vitro .

- Scheme illustrating comparisons of the 73 KO gene expression profiles at N24 with an in vitro differentiation time course of WT ESCs with a 2‐h resolution.
- Heatmap showing differentiation delays of the 73 KO lines quantified by average naïve marker gene expression. Red bars indicate the closest correlation to that specific time point. Positive values indicate delayed differentiation. Negative values indicate more rapid differentiation. Each line corresponds to one KO. Selected KOs are indicated. See Dataset EV2 for the full hierarchy of the 73 KOs.
- Plot showing a comparison of the differentiation delays calculated using naïve markers as in (B) to differentiation delays calculated using all 3068 genes differentially expressed in WT differentiation (N24 vs. 2i) (see Appendix Fig 6B).
The regulatory programme for the in vivo pre‐ to post‐implantation epiblast transition is preserved in most mutants
To explore the extent to which our in vitro data captures in vivo regulation, we first compared global transcriptomes of WT and KO ESCs with single‐cell transcriptome data from the in vivo pre‐ (E4.5) and post‐implantation (E5.5 and E6.5) mouse epiblast (Mohammed et al, 2017) (Fig 4A and Dataset EV3). As expected (Boroviak et al, 2014), WT cells in 2i showed greater similarity (fraction of identity, FOI) to the E4.5 than to the E5.5 epiblast (Gong & Szustakowski, 2013). At N24, in contrast, higher FOI to the E5.5 epiblast was observed. We could not detect a clear difference between FOIs of cells in 2i or at N24 when compared to the E6.5 epiblast. This underscores that 24 h of 2i withdrawal models the E4.5 to E5.5 epiblast transition (Kalkan et al, 2017).
Figure 4. Comparing KO expression profiles to the in vivo pre‐ to post‐implantation epiblast transition.

- Fractions of identity (FOI) between 2i and N24 expression data and mouse E4.5, E5.5 and E6.5 epiblast, computed using all expressed genes (log2FPKM > 0) for all KOs. Each dot represents one KO. Average naïve marker log2FC in KOs at N24 is indicated by a colour gradient.
- FOIs of WT and KO cells in 2i compared with the E4.5 and E5.5 mouse epiblast, computed using all expressed genes (log2FPKM > 0); average naïve marker log2FCs in KOs at N24 are indicated by a colour gradient.
- FOIs of WT and KO cells at N24 compared with the E4.5 and E5.5 mouse epiblast, computed using all expressed genes (log2FPKM > 0); average naïve marker log2FCs in KOs at N24 are indicated by a colour gradient.
- FOIs of WT and KO cells in 2i (green) and at N24 (grey) compared with E4.5 and E5.5 mouse epiblast, computed using all expressed genes (log2FPKM > 0).
- PCA showing all KOs in 2i (triangles) and at N24 (diamonds) and embryo‐derived data sets (ICM, primitive endoderm [PrE], E4.5, E5.5 E6.5 epiblast). Colour gradient indicates naïve marker log2FC at N24 of the respective KO.
Interestingly, some KO ESCs showed expression features in 2i that indicated increased similarity to the in vivo epiblast (Fig 4B). Among those were KOs showing strong in vitro differentiation defects, such as Zfp281, Rbpj, Fgfr1 and Etv5, but also Trim71, Fbxw7 and Smg7, which showed only modest differentiation delays in culture. PRC2 mutants (Eed, Suz12 and Jarid2 KOs) showed an opposing pattern and lost similarity to the E4.5 epiblast in 2i.
At N24 several KOs, such as Csnk1a1, Zfp281, Fgfr1, Etv5, Rbpj and Smg5, retained strong similarity to the E4.5 epiblast (Fig 4C). Strikingly, Zfp281 and Csnk1a1 KO profiles at N24 showed similarity to the E4.5 epiblast on par with WT cells cultured in 2i (Fig 4D). The strong Zfp281 phenotype is consistent with an overt differentiation delay phenotype (Mayer et al, 2020). Overall, there was a good correlation between the in vitro differentiation delay and the similarity with pre‐implantation epiblast cells, and both the 2i and N24 KO transcriptomes that were more similar to the E4.5 epiblast exhibited stronger exit‐delay phenotypes in vitro (Figs 4C and D, and EV2A). Together, this indicates that similar transcriptional networks are regulated by similar mechanisms during in vivo and in vitro transitions to formative pluripotency, with the exception of the Jarid2 KO, which at N24 showed a similarity to the E4.5 epiblast that was smaller than expected based on its strong in vitro differentiation defect (Figs 4B and C, and EV2A).
Figure EV2. Comparing KO expression profiles to the in vivo pre‐ to post‐implantation epiblast transition.
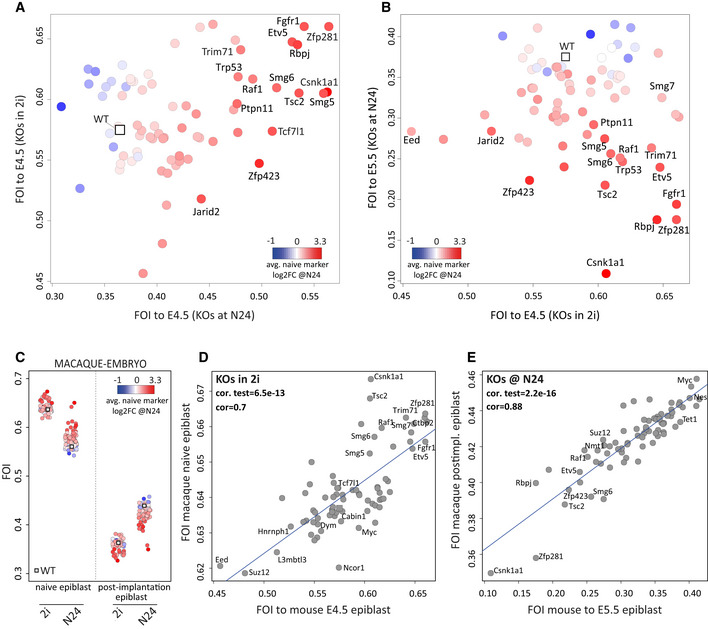
- FOIs to the E4.5 epiblast of all KOs in 2i compared with FOI to the E4.5 epiblast of all KOs at N24. The average naïve marker log2FC in KOs at N24 is indicated as colour gradient (strength of differentiation phenotype).
- FOIs of 2i samples to E4.5 epiblast (x‐axis) and FOIs of N24 samples to E5.5 epiblast (y‐axis). The average naïve marker log2FC in KOs at N24 is indicated as colour gradient.
- FOIs between 2i and N24 expression data and macaque naïve and post‐implantation epiblast, computed using all expressed genes (log2FPKM > 0) for all KOs. Each dot represents one KO. Average naïve marker log2FC in KOs at N24 is indicated by a colour gradient.
- Correlation plot of FOIs between 2i profiles and mouse naïve (E4.5) epiblast and FOIs between 2i profiles and macaque naïve epiblast (correlation significance is indicated in the plot).
- Correlation plot of FOIs between N24 profiles and mouse E5.5 epiblast and FOIs between N24 profiles and macaque post‐implantation epiblast (significance of correlation is indicated in the plot).
We then performed principal component analysis (PCA) for genes variably expressed in embryo development, using averaged values for inner cell mass (ICM), primitive endoderm (PrE) and E4.5, 5.5 and 6.5 epiblast (Mohammed et al, 2017). The PCA separated the 2i and N24 transcriptomes into two clusters with proximity to PrE/E4.5 and E5.5 epiblast, respectively (Fig 4E). Notably, in vitro differentiation defects were reflected by closer proximity of mutant N24 profiles to the E4.5 epiblast in the PCA.
We found that overall KOs showing strong in vitro differentiation phenotypes also exhibited increased similarity at N24 to the E4.5 epiblast and a concomitant reduction in the similarity to the E5.5 epiblast (Fig EV2B). This indicates a delay along the normal naïve to formative differentiation axis. Such a behaviour was most prominently detectable in KOs for Zfp281, Etv5, Fgfr1, Rbpj and Trim71. However, KOs for Csnk1a1, Zfp423 and Jarid2 deviated from this pattern, indicating that they might have disengaged from the normal embryonic developmental trajectory.
Correspondence to primate embryogenesis
Despite morphological and timing differences between rodent and primate peri‐implantation development, embryos of both orders appear to transit through similar pluripotency states (Smith, 2017; Rossant & Tam, 2018). To examine a potential role for the set of 73 exit factors in primate naïve to formative epiblast transition, we compared transcriptional profiles of our KO series and cells of the macaque in vivo naïve and post‐implantation epiblast (Nakamura et al, 2016). In general, ESCs in 2i were more similar than cells at N24 to the pre‐implantation macaque epiblast (Fig EV2C). Correspondingly, N24 cells were closer to the macaque post‐implantation epiblast. Levels of similarity of KOs in 2i to the pre‐implantation epiblast and at N24 to the post‐implantation stage were well correlated between mouse and macaque (Fig EV2D and E). These observations suggest that the overall GRN redeployment and the activity and role of exit factors during the naïve to formative transition are conserved between rodents and primates.
Identification of an extended naïve pluripotency network
ESC differentiation requires fine‐tuned coordination between extinction of the naïve and initiation of the formative transcription networks. To date, only incomplete inventories of the genes that functionally define these two states have been made. These genes cannot be defined based simply on differential expression between 2i and N24, because not all DEGs will be functionally linked to the naïve GRN. To identify those genes that show specific linkage to the core naïve pluripotency network, we trained regression models to predict gene expression changes across all KOs at N24 as a function of naïve marker log fold changes. The 496 genes whose expression was thereby identified to be tightly associated (R 2 ≥ 0.65) with one or several of the seven core pluripotency markers (Nanog, Esrrb, Tbx3, Tfcp2l1, Klf4, Prdm14 and Zfp42) were defined as “Naïve‐Associated Genes” (NAGs; Dataset EV3). Identification as NAG can be achieved by positive or negative association with naïve TF expression. Thus, we identified 278 NAGs that were downregulated (downNAGs—similar to e.g. Nanog) and 218 NAGs that were upregulated (upNAGs—similar to Pou3f1, Figs 5A and EV3A) during WT differentiation. Importantly, NAGs largely differ in identity from the top differentially expressed genes in WT differentiation (Fig 5A). Naïve pluripotency TFs not represented in the defining TF set were correctly identified as NAGs, including Klf5, Nr0b1 and Nr5a2. Klf2 is not one of the NAGs being only weakly associated (R 2 = 0.49) with the naïve network, in‐line with our earlier observation that Klf2 expression can be uncoupled from the naïve TF network. Of further note, Klf2 expression is barely detectable in marmoset or human pre‐implantation epiblast cells (Fig EV3B).
Figure 5. Identification of an extended naïve pluripotency network.

- Venn diagrams showing the overlap between upNAGs and top‐upregulated genes (left) and downNAGs and top‐downregulated genes (right).
- The x‐axis displays all 3rd dimension (Dim3) genes, separating mouse naïve from post‐implantation epiblast (the inset shows the full PCA plot, see also Fig EV3D) in order of extent of contribution to Dim3. The contribution of the 496 NAGs (218 upNAGs, 278 downNAGs; orange line) to Dim3 and a size‐matched set of top differentially expressed genes (218 top‐upregulated, 278 top‐downregulated based on log2FC between 2i and N24; green) are plotted in a cumulative manner.
- Log2FC in mouse ESC differentiation (N24 versus 2i) compared with log2FC in mouse in vivo transition from pre‐ to post‐implantation epiblast (E5.5 versus E4.5). Selected gene groups are plotted from left to right: i) NAGs, ii) all 3068 differentially expressed genes between 2i and N24 and iii) top differentially expressed genes in mESC differentiation (218 top‐upregulated, 278 top‐downregulated). Rho values indicate the level of correlation between in vivo and in vitro differentiation of given gene groups. Alpha indicates the angle between the x‐axes and the orthogonal regression. Genes with |log2FC| > 0.1 in both the x‐ and y‐axes are highlighted in orange and blue, respectively.
- Log2FC in mouse ESC differentiation (N24 versus 2i) compared with log2FC of orthologues in macaque in vivo transition from pre‐ to post‐implantation epiblast. Gene groups from left to right are identical to (C).
- Frequency of co‐regulation of NAGs or the matching set of top differentially expressed genes and differentially expressed genes in mouse and macaque in vivo pre‐ to post‐implantation epiblast differentiation. Co‐regulation was defined as showing a |log2FC| > 0.1 in both conditions. Two‐tailed chi‐square test (CI 95%) was used to compute significance levels between the expected and the observed number of modulated genes.
- Heatmap derived from one‐way hierarchical clustering of log2FCs of NAGs (left) and top differentially expressed genes (right) in the in vitro exit from naïve pluripotency model (N24 versus 2i), mouse pre‐ to post‐implantation transition and macaque pre‐ to post‐implantation transition.
Figure EV3. Identification of an extended naïve pluripotency network.

- Comparison of the strength of the linkage to the naïve network (naïve association, R2 values; x‐axis) to the log2FC of all genes during the naïve to formative transition in WT (y‐axis). upNAGs are indicated in red, downNAGs in blue.
- Klf2 expression at the indicated stages. Expression levels (FPKM) in compacted morula (cMOR), expanded ICM (eICM), E4.5 epiblast (EPI) are shown derived from single‐cell RNA‐seq data sets from human, marmoset and mouse pre‐implantation development. Box‐plots were generated utilising the GRAPPA web app (Boroviak et al, 2018) and indicate the first and third quartile of expression levels. The midline identifies the median. Each dot corresponds to an individual cell.
- Plots showing expression of Pdgfa (top) and its cognate receptor Pdgfra (bottom) during mouse pre‐implantation embryo development in a white (low) to orange (high) gradient.
- Principal component analysis of published single‐cell RNA‐seq data sets showing that Dimension 3 (Dim 3) separates pre‐ from post‐implantation epiblast in vivo (magnification of plot in Fig 5A).
The NAG showing the strongest association with the naïve core network is Pdgfa, which is relatively highly expressed in ES cells (FPKM ~80). Pdgfa has no known role in ESC self‐renewal, but functions in segregation of the primitive endoderm (Artus et al, 2013). To examine whether the link between Pdgfa and the naïve transcription factor network is maintained in vivo, we utilised GRAPPA, a tool to visualise single‐cell expression data from pre‐implantation development (Boroviak et al, 2018). Indeed, Pdgfa is uniformly expressed in the E4.5 epiblast (Fig EV3C). Its cognate receptor Pdgfra, in contrast, is neither expressed in ES cells nor the naïve epiblast (FPKM in WT ESCs < 0.5), but specifically marks the neighbouring primitive endoderm at E4.5 (Plusa et al, 2008).
We surveyed the expression of NAGs at the transition from naïve to post‐implantation pluripotency in the single‐cell RNA‐seq data sets from E4.5, E5.5 and E6.5 epiblast cells (Mohammed et al, 2017). We detected a clear enrichment of NAGs within genes that separate the pre‐ from the post‐implantation epiblast in the differentiation state resolving dimension of a principal component analysis (PCA) (Figs 5B and EV3D), highlighting that NAGs are indicators for the naïve epiblast state also in vivo. Strikingly, NAGs showed a strongly correlated fold change behaviour in vitro and in vivo during the epiblast transition from E4.5 to E5.5 (Fig 5C, E and F). Underscoring the special role of NAGs, this effect was not observed for a matched set of top differentially expressed genes in ESC differentiation in vitro. Furthermore, NAG orthologues showed significantly co‐regulated expression dynamics during macaque pre‐ to post‐implantation epiblast differentiation in vivo (Fig 5D–F), which further underscores their relevance. We thus propose that NAGs constitute an integral component of the naïve transcriptional network with potential functional relevance in vivo across mammalian species.
Deregulation of signalling cascades is a hallmark of differentiation delay
We next analysed the extent of deregulation of five key signalling pathways known to be active in pluripotent cells: LIF/Stat3, mTORC1, Wnt/β‐catenin, Fgf/ERK and Notch (Smith et al, 1988; Lowell et al, 2006; Watanabe et al, 2006; Kunath et al, 2007; Ying et al, 2008; Molotkov et al, 2017). Changes in the activity of a signalling pathway are not necessarily reflected in expression changes of pathway member transcripts. Thus, in order to quantify pathway activities, we employed “expression footprints”. We identified pathway‐specific marker gene sets, each containing 50 genes, reporting pathway activity changes. These marker sets were determined using N24‐derived KO transcription profiles of ESC lines deficient for key signalling pathway components. Thereby, we defined specific mTORC1 (affected by Tsc2 KO), Wnt/β‐catenin signalling (Tcf7l1 KO), Fgf/ERK (Fgfr1 and Ptpn11 KOs) and Notch (Rbpj KO) pathway targets (Appendix Fig S7A, Dataset EV4). Overlaps with available Tcf7l1 ChIP profiles (Martello et al, 2012) supported the reliability of this approach. For LIF signalling, we compared RC9 ESCs grown in 2i in the presence and absence of LIF for 24 h. This resulted in a list of LIF‐sensitive genes including known targets such as Socs3, Gbx2, Junb, Tfcp2l1, Klf4 and Klf5 (Martello et al, 2012; Ohtsuka et al, 2015) (Dataset EV4). In summary, the expression footprints present non‐overlapping sets of marker genes whose expression state is indicative of the activity of the respective signalling pathway.
Using the expression footprints, we then asked whether we could detect a preferential deregulation of one or more of these signalling footprints in specific KOs either in 2i (for LIF) or at the N24 time point (for all other pathway profiles) (Fig 6A, Dataset EV4). Surprisingly, we detected mis‐regulated LIF target genes in several KO ESC lines cultured in 2i in the absence of LIF (Fig 6A). Activation of such a “LIF‐like” profile in 2i was closely correlated with the extent of differentiation delay observed at N24 (Appendix Fig S7B). Notably, the presence of LIF together with 2i before induction of differentiation slows down naïve state exit (Appendix Fig S7C), suggesting a causative role of increased LIF likeness for the observed differentiation delays (Dunn et al, 2014; Mulas et al, 2017). Tsc2, Ptpn11, Raf1, Mapk1 and Trim71 KO ESCs showed the greatest similarity to the LIF profile in 2i; in contrast, Tcf7l1 KO ESCs, despite showing a pronounced differentiation defect, lack the “LIF footprint” (Appendix Fig S7B). Tsc2 depletion resulted in the strongest activation of a LIF footprint of all KOs, suggesting that a component of the LIF response is mediated through Akt (Appendix Fig S7B) (Watanabe et al, 2006; Niwa et al, 2009). Addition of a JAK inhibitor to several KOs showing a LIF‐like profile resulted in no or only minor amelioration of the differentiation defect. This suggests that these KOs do not directly activate Jak/Stat signalling, but that a LIF‐like expression profile reflects a consolidated naïve network that is resilient to dismantling.
Figure 6. Signalling pathways and gene expression modules regulated by multiple KO genes.

- Relationship between the KO expression profiles at N24 (or in 2i for the LIF pathway) to the expression footprints of the five signalling pathways, as indicated (see Appendix Fig S7A for details on derivation of pathway footprint genes). Relative pathway activity is set to −1 (red, indicating relative inactivity of pathway in pathway‐defining KO) for the Fgf/ERK (defined by Fgfr1 & Ptpn11 KOs), Wnt (Tcf7l1 KO) and Notch (Rbpj KO) pathways and + 1 (green, indicating relative activity in pathway‐defining KO or upon LIF supplementation) for mTORC1 (Tsc2 KO) and LIF pathways. Pathway activity is indicated by a colour gradient (green for activity, red for inactivity). All tiles with an absolute pathway activity smaller |0.3| are greyed out. The size of the tiles corresponds to the Spearman correlation of pathway footprints between each KO and the corresponding representative KOs. The colour gradient on the sidebar at the left indicates average log2FC of naïve marker gene expression for the corresponding KO at N24 (strength of differentiation phenotype).
- Multiple regression coefficients of pathway activity profiles across all 73 KOs were used to predict the differentiation delay phenotype (defined by average naïve marker log2FC at N24). Error bars indicate the standard error of the coefficients.
- The absolute correlation value (Pearson’s) between average expression change of cluster genes and expression change of naïve marker genes is plotted against the enrichment of NAGs in all clusters. Constitutive clusters are indicated by triangles, induced clusters are shown as circles. Adjusted P‐values (Fisher’s exact test) are indicated as colour gradient for upNAGs (e.g. formative genes; shades of red) and downNAGs (e.g. naïve TFs; shades of blue).
- GO enrichment analysis for constitutive KO clusters. Dot‐size scales with log2 enrichment. Multiple testing corrected P‐values (Fisher’s exact test) are colour coded in shades of red.
- As for (D) except GO enrichment analysis is for the N24‐induced clusters.
Tsc2 deficiency leads to constitutive activity of the mTORC1 pathway, which has previously been associated with an exit from naïve pluripotency phenotype (Villegas et al, 2019). KOs including Pten, Csnk1a1, Zfp281, Eed and Suz12 exhibited deregulated Tsc2 responsive genes. Surprisingly, deletion of Etv5, a core downstream TF in the Fgf/ERK pathway, also increased the set of Tsc2‐responsive genes. Genes downstream of the Notch pathway were most strongly affected in a group of mutants deficient for mRNA homeostasis (Smg5 and Smg6) and transcriptional regulators, including Zfp423, Jarid2, Mbd3 and L3mbtl3. The latter suggests an interaction between the Notch pathway (Rbpj) with the Polycomb and NuRD complexes to modulate network rewiring during the exit from naïve pluripotency. Co‐operativity between Rbpj and the Polycomb‐associated protein L3mbtl3 has been reported in Drosophila and C. elegans (Xu et al, 2017). As expected, Raf1, Fgfr2, Mapk1 and Etv5 KO ESCs showed an Fgf‐signalling footprint at N24. Mbd3, Mta3, Nsd1 and Ctbp2 KOs showed Fgf/ERK target deregulation similar to reference KOs, indicating an involvement of chromatin regulators in ERK target gene control (Fig 6A). The KOs for the beta‐catenin destruction complex member Apc and the previously discussed Jmjd1c showed the expected similarities with the Tcf7l1 profile. Interestingly, several KOs with strong phenotypes showed footprints similar to Tcf7l1 KOs at N24. This is in‐line with evidence that β‐catenin constitutes or regulates a major differentiation switch during the exit from naïve pluripotency and is influenced by multiple exit KO genes (Wray et al, 2011; Hoffman et al, 2013; Neagu et al, 2020). Rbpj and Tcf7l1 KOs did not show a correlation with each other or with Tsc2 KO, consistent with independent but cooperative mechanisms (Kalkan et al, 2019).
We then asked to what extent the alteration of any of the five signalling pathways was predictive for the strength of the differentiation defect. We found that whereas aberrant Fgf/ERK or mTORC1 activity were unreliable predictors, deregulation of Notch/Rbpj or Wnt/Tcf7l1 at N24, or deregulation of LIF target genes in 2i correlated with differentiation delay (Fig 6B). Together these data indicate that many KOs with a delay phenotype have altered activity of at least one of the five key signalling pathways. Strikingly, however, there was no single signalling pathway perturbed downstream of all KOs. We observed that KOs showing multiple pathway footprints are rare, but most KOs showing a delayed naïve marker gene downregulation at N24 deregulate at least one specific pathway. Trp53, Ncor1 and Usp9x constitute exceptions, which show appreciable differentiation defects without clear deregulating of any of the tested signalling cascades. We conclude that signalling pathways are harnessed in an independent fashion and that most KO genes contribute directly or indirectly to at least one pathway activity that promotes the exit from naïve pluripotency.
Identification of gene‐networks downstream of multiple KO genes
To explore common regulatory targets of multiple KO factors, we classified deregulated genes into two groups: (i) deregulated by a KO in both 2i and at N24, termed “constitutive KO response”; (ii) not, or only weakly, deregulated by a KO in 2i, but significantly deregulated at N24, termed “N24‐induced KO response”. Genes in both groups are affected by knockouts, but genes in the second group deviate from wild‐type expression only under conditions of differentiation. We identified all genes that showed either type of behaviour in at least one KO (Appendix Fig S7D; Dataset EV5). Genes deregulated exclusively in PRC2 core‐components (Eed or Suz12 KOs) or Csnk1a1 KOs were excluded from further analysis, because the deregulation of thousands of genes in these KOs had a disproportionate impact on the resulting gene lists. In total, the constitutive and the N24‐induced KO response groups contained 1,886 and 716 genes, respectively. Genes belonging to the N24‐induced KO response group were strongly enriched for factors that are dynamically regulated during normal differentiation, whereas genes of the constitutive response group were not (Appendix Fig S7E). To characterise the constitutive KO response genes, we grouped them based on their log2 fold changes between KO and WT at N24. This resulted in twelve clusters, ranging in size from 5 to 496 genes (Fig EV4A and Dataset EV5). Analysis of the N24‐induced response genes yielded 12 clusters ranging from 17 to 107 genes (Fig EV4B and Dataset EV5).
Figure EV4. Signalling pathways and gene expression modules regulated by multiple KO genes.

- Heatmap showing average log2FCs (KON24 versus RC9N24) of constitutive KO response gene clusters (c1‐c12). For reference, average naïve marker log2FCs versus WT at N24 are shown for each KO in a separate row below.
- As for (A) for N24‐induced KO response gene clusters (i1‐i12).
- Fisher test‐based enrichments of ChIP targets, up/downNAGs and pathway‐defining gene sets in constitutive clusters are indicated by circles. Size indicates log2 fold over‐representation; the colour gradient encodes adjusted P‐values.
- Similar to Fig EV4C for N24‐induced clusters.
Gene expression patterns in 9 out of 12 N24‐induced clusters showed a strong correlation with the phenotypic strength across all KOs, indicating that they contain genes relevant for the naïve to formative transition (Fig 6C). All phenotype‐correlated N24‐induced clusters showed enrichment of up (N24‐induced clusters i4, i9, i12)‐ or downNAGs (N24‐induced clusters i1, i2, i5, i11).
Induced cluster 1 was enriched for genes involved in cell fate commitment and stem cell population maintenance and contained genes that are associated with signalling and transcriptional activation (Fig 6E, Dataset EV5, Appendix Figs S8–S20). Naïve marker genes were detected in N24‐induced cluster 1 (i1), with the notable exception of Klf2. Klf2 was part of cluster i5, within which more than half the genes were bound by Tcf7l1 (Martello et al, 2012) (Fig EV4D). This provides further support for the notion that Wnt/Tcf7l1 controls Klf2 expression, independently of the rest of the naïve network.
Cluster i4 was enriched for functional terms overlapping with cluster i1, indicating that these two clusters contain pivotal cell fate switch genes with opposing expression patterns during differentiation. Consistently, formative factors were detected in cluster i4, which was further enriched for several GO terms relating to neuroectodermal differentiation, indicating that i4 contains genes of the formative programme and the ectodermal “default” programme of ESC differentiation (Ying et al, 2003) (Fig 6E, Dataset EV5, Appendix Figs S8–S20). Average expression of genes in both clusters (downregulation in cluster i1, upregulation in cluster i4) showed strong correlations with the extent of differentiation defects across all KOs (Fig 6C).
Interestingly, cluster i11 with the second highest NAG enrichment and phenotype correlation is enriched for terms pertaining to cell adhesion, indicating the importance for cell–cell interaction for proper regulation of the exit from naïve pluripotency. Further enrichments for cell adhesion in clusters i5 and i7 underscore the importance of controlling cell adhesion properties for maintaining proper differentiation behaviour. Cluster i8, although neither strongly correlated with the exit defect nor enriched for NAGs, contained pivotal cell fate switch genes such as Tcf7l1, Fgf4 and Tfe3 (Betschinger et al, 2013).
A correlation with the differentiation defect was less pronounced in constitutive clusters and only detectable in constitutive clusters c6, c7 and c8, with an overall weaker effect than observed for N24‐induced clusters. Consistently, only five out of twelve constitutive clusters showed an enrichment for NAGs, including phenotype associated c5, c6 (downNAGs) and c8 (upNAGs). Constitutive cluster 6 genes were enriched for Bmp signalling and Fgf/ERK signalling‐related terms and constitutive cluster 7 for Fgf/ERK signalling and extracellular matrix related terms (Fig 6D and Dataset EV5). Overall, in contrast to the N24‐induced KO response, constitutive clusters showed a less cluster‐specific enrichment for GO terms (Fig 6D and E). In summary, constitutive and N24‐induced clusters define crucial genetic modules that are co‐regulated by multiple exit factors.
Wiring between regulatory pathways and gene expression clusters
To identify regulatory pathways potentially controlling the gene expression clusters from above, we measured signalling pathway activity using the pathway‐specific genes defined before across the 2h‐resolved 32h long WT differentiation time course (Fig 7A). mTORC1 and LIF signalling were downregulated in parallel, with slightly slower kinetics for LIF signalling. Notably, the Tcf7l1‐axis of Wnt signalling showed an early activation whereas Fgf/ERK signalling appeared to initiate strong activation only just before the 20h time point. This is in‐line with a reported sequential requirement of first Wnt and then Fgf signalling to transit cells to a formative state (Neagu et al, 2020). We then utilised pathway activities and cluster‐specific expression levels across all KOs to build a model predicting the impact of pathway activity on cluster‐gene expression (Figs 7B and EV5AB). We only considered those pathways‐to‐cluster connections in our model where activity changes of the regulatory pathways preceded the expression changes of their putative target clusters over the 32 h WT differentiation time course (Fig EV5C and D). In our model, the Notch and mTORC1 pathways showed high connectivity to N24‐induced clusters (7 out of 12 clusters, each), whereas Wnt/β‐catenin signalling was specifically correlated with clusters containing high levels of downNAGs (cluster i1, i5 and i11) and the only constitutive cluster containing an appreciable number of (down)NAGs (const c6), indicating a major role for Tcf7l1 in downregulation of the naïve GRN.
Figure 7. Wiring between regulatory pathways and gene expression clusters.

- Pathway activities (average expression of the 50 specific pathway‐defining genes) across a WT differentiation time course for the mTORC1, LIF, Fgf/Erk, Notch and Wnt pathways. Pathway activity is defined as the average log2FC of pathway‐defining genes relative to the mean expression throughout the time course.
- Visualisation of connections between pathways (central ellipses) and gene clusters (rectangles), based on multiple regression models. The colour gradient of the clusters indicates adjusted P‐values of enrichment for up‐ or downNAGs. Connecting arrows link pathways to clusters that fit time course validated regression models (see Fig EV5B–D). Connecting arrows are coloured by source pathway. Unconnected clusters are listed separately (constitutive, left; N24‐induced, right).
Figure EV5. Wiring between regulatory pathways and gene expression clusters.
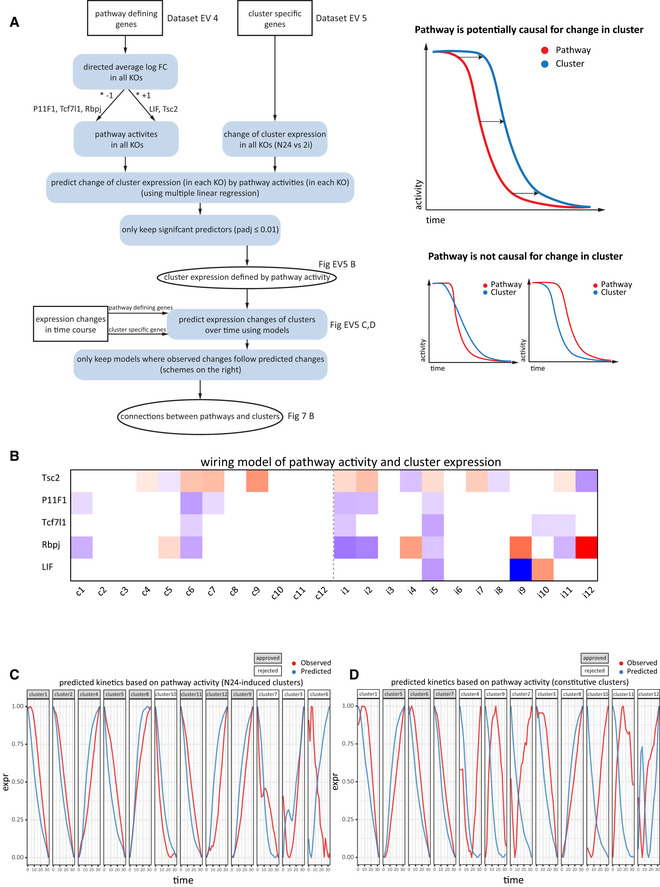
- Schematic overview of the workflow to model cluster‐pathway connections. White rectangles indicate input data from previous analysis steps, white ellipses indicate results from this analysis and blue rounded rectangles represent different processing steps and calculations to obtain indicated results. Schematic pathway to cluster relations over the time course is shown on the right. If the change in pathway activity is causal for a change in cluster expression, behaviour as shown in the top scheme is expected. The two schemes at the bottom exclude causality as the cluster changes precede changes in the pathway.
- Heatmap showing multiple regression models of pathway activities that predict cluster expression. Only significant (adjusted P ≤ 0.01) interactions are shown. Colour intensity represents the strength of the interaction. Red tiles indicate that higher activity of a certain pathway is linked to higher expression of the cluster, while blue tiles indicate the inverse correlation.
- Observed cluster expression change (red) and cluster expression change predicted by regression models (blue) throughout the 32 h WT differentiation time course for all induced clusters. Predicted cluster expression is calculated using pathway activities throughout the WT time course and significant (adj. P ≤ 0.01) interactions from the multiple regression models from (B). Graphs show log2FC of cluster expression relative to the mean cluster expression across the time course. Cluster–pathway connections that were not validated through this test (predicted expression did not follow the same trend AND/OR did not precede observed expression) were rejected, as indicated.
- Similar to Fig EV5C for constitutive clusters.
Together, this shows that N24‐induced clusters which contain genes dynamically regulated during the exit from naïve pluripotency are largely under the control of five key signalling pathways gating differentiation in a cooperative manner by co‐regulation of the majority of differentiation‐specific gene modules.
Discussion
Ordered progression through pluripotency requires the shutdown of the naïve TF network and concomitant large‐scale rewiring to establish the formative GRN (Buecker et al, 2014; Kalkan & Smith, 2014; Kalkan et al, 2017). Following Harald Weintraub’s pioneering work with muscle differentiation and Thomas Graf’s work on haematopoiesis, the ability of certain transcription factors to change cell identity without a developmental context gained central prominence with somatic cell reprogramming to pluripotency (Lassar et al, 1986; Xie et al, 2004; Takahashi & Yamanaka, 2006). However, most of these studies involved forced expression of selected transcription factors to achieve synthetic cell state transitions in vitro. Here, we utilised the remarkable properties of ESCs to recapitulate the pivotal embryonic transition from naïve to formative pluripotency. We show that this developmental cell state transition is not based on the action of few master transcription factors. Although crucial for defining lineage trajectories, transcription factors do not initiate cell fate decisions. Instead, the robust stability of naïve pluripotent stem cells is sustained and dismantled by multiple crosstalking components and by at least four diverse signalling inputs.
Our dissection of an authentic cell state transition indicates that the exit from naïve pluripotency is largely guided along a trajectory constrained by signalling pathways and funnelled through a handful of genetic modules. These adaptations of cellular networks establish the opportunity for TFs to initiate and consolidate cell identity. In the specific case of the exit from naïve pluripotency, expression of Oct4 and Sox2, which are key TFs for both the naïve and formative states, supports the maintenance of both the naïve and formative regulatory networks to ensure lineage fidelity during differentiation. Transcription factor circuits are crucial to maintain cell identity and to ensure proper lineage identity, but our data suggest that lineage‐specific TFs do not stand at the top of cell fate decision hierarchies.
Several signalling cascades have been implicated in ESC self‐renewal and naïve pluripotency exit, including pathways controlled by LIF, Akt/mTORC1, Wnt, Fgf/ERK and Notch. We derived transcription footprints of these pathways and scrutinised the extent to which they were deregulated by the different KOs to find that virtually all KOs showing a strong differentiation defect also showed deregulation of at least one of these five pathways. Notably, however, we observed segregation in terms of pathway deregulation. Thus, KOs that induce, e.g. a Tcf7l1‐like profile were less likely to show a signature of another pathway. We also confirm a previously reported sequential activity of Wnt and Fgf/ERK signalling during the naïve to formative transition (Neagu et al, 2020). Our data suggest that the exit from naïve pluripotency involves co‐ordinated activity changes of independent but functionally partially redundant pathways under the control of separable genetic networks. The observation that single gene depletion is not sufficient to prevent exit from the naïve state is consistent with this interpretation. Our analyses exposed discrete pathway and GRN features that mediate timely and robust mammalian cell state transitions. The finding that multiple sub‐networks with distinct expression patterns can be identified indicates the existence of several independent but cooperative molecular programmes that control differentiation.
By examining co‐regulation with the core naïve network across all KOs at N24, we identified the NAGs. NAGs obey strikingly similar expression dynamics in vitro and in vivo. Moreover, in the macaque pre‐ to post‐implantation transition, orthologous NAGs show similar behaviour as during mESC exit from naïve pluripotency. Therefore, this cohort of genes constitutes a layer of the pluripotency network that is tightly linked to, and likely acts in conjunction with, the core naïve TF network across mammalian species (Dunn et al, 2014). We suggest that collective modulation of NAG expression will propel naïve cells into formative differentiation and, conversely, that collective NAG deregulation will delay proper differentiation in exit factor KOs. The control of NAG expression appears to be interwoven with the signalling pathways involved in controlling the naïve to formative transition. The N24‐induced clusters enriched for NAGs showed high levels of deregulation in KOs exhibiting a strong differentiation delay and an overall high connectivity to all tested pathways. The functional significance of NAGs likely extends beyond self‐renewal and naïve identity. Indeed, most NAGs are not transcription factors. The growth factor Pdgfa is a case in point. Pdgfa is the most strongly associated NAG, but has no known activity on ESCs, which do not express the cognate receptor, Pdfgra. Within the ICM, Pdgfa expression is restricted to the naïve epiblast, whereas Pdgfra is present exclusively in the primitive endoderm. This reciprocal expression pattern is consistent with the known paracrine action of Pdgfa to promote primitive endoderm segregation (Artus et al, 2013). Thus, conserved linkage of NAGs to the naïve TF network may mediate paracrine communication to other lineages within the blastocyst as well as consolidate naïve epiblast fate.
Whereas most naïve TFs were tightly co‐regulated across the 73 KOs, Klf2 expression was uncoupled from other members of the core network. Interestingly, while Klf2 is highly expressed in mouse ESCs and naïve epiblast, it is very lowly expressed in primate blastocysts, and also in the porcine pluripotent compartment (Ramos‐Ibeas et al, 2019). Klf2 may therefore be a rodent‐specific addition to the naïve TF network. A further significant species difference is the very low expression of TCF7L1 in primate naïve cells, which underlies the differential responsiveness of mouse and human naïve cells to GSK3 or Wnt pathway inhibition (Bredenkamp et al, 2019; Rostovskaya et al, 2019). Klf2 is a genomic target of Tcf7l1 (Martello et al, 2012). Together this suggests a rodent‐specific linkage between Klf2 and Tcf7l1 as regulatory module to sustain and extinguish the naïve to formative transition, respectively.
For most KOs, the degree of failure in naïve TF downregulation and the neutralisation of global differentiation‐related transcription changes are comparable. However, the relatively weak Trim71 phenotype despite specific upregulation of some naïve TF genes suggests that upregulation solely of core naïve factors must reach a certain threshold to stably maintain naïve pluripotency, consistent with negative feedback constraints within the network (Chambers & Tomlinson, 2009; Dunn et al, 2014; Niwa, 2018).
The remarkable advantages of ESCs as an experimental venue include the utilisation of well‐defined culture conditions to recapitulate key features of in utero developmental progression (Nichols & Smith, 2012; Boroviak & Nichols, 2014; Kalkan et al, 2017; Smith, 2017). Our high‐resolution transcriptome analysis confirms that differentiation over 24 h in vitro mimics the peri‐implantation transition from naïve to formative epiblast in utero. These data provide rich resources for further studies. In particular, many strong differentiation delay KO transcriptome profiles retained transcriptome resemblance to the pre‐implantation epiblast. This relationship held true in 2i and at N24 and indicates that the KO of these genes perturb the cell state transition machinery operative in vivo. In general, we surmise that stalling the activity of the exit machinery is critical to in vitro capture and self‐renewal of naïve stem cells, consistent with empirical requirements for signal inhibition in both mouse and human (Ying et al, 2008; Bredenkamp et al, 2019). Interestingly, deficiency for some differentiation regulators increases similarity of 2i profiles to the in vivo pre‐implantation epiblast. Therefore, we propose that some of the mechanisms driving exit from naïve pluripotency contribute to the lack of complete identity between in vivo E4.5 epiblast and ES cells in 2i. This is in‐line with ESCs representing an engineered interruption in developmental progression and suggests that state‐of‐the‐art self‐renewal culture conditions may not yet capture the naïve state perfectly (Smith, 2017).
Our analyses provide a comprehensive inventory of genetic factors and regulatory networks governing the major cell fate transition from pre‐ to post‐implantation epiblast. We present evidence that mammalian in vivo development employs similar regulatory networks operating with similar mechanisms (Boroviak & Nichols, 2017; Rossant & Tam, 2017). These programmed cell state transitions in development do not rely upon instructions delivered by one or two master transcription factors. Rather, a cloud of activity involving multiple co‐ordinated and partially redundant inputs serves to destabilise an existing, multiply stabilised GRN. Thereby, a consolidated cell state can be rapidly dismantled by a combination of signals, triggering transition to the next developmental waystation.
Materials and Methods
Cloning
Guide RNAs (gRNAs) were designed using the computational pipeline described below, or by http://crisprscan.org. To clone the gRNAs for ESC KO of the 73 selected genes, we used RecET recombineering to insert the guide RNA protospacer encoding sequence between the U6 promoter and the universal sgRNA scaffold in pBR322‐U6‐cm‐ccdB‐sgRNA‐amp, as described previously (Baker et al, 2016). In brief, the plasmid was linearised at the point of insertion by BstZ17I digestion, purified using the Invitrogen Charge Switch PCR Purification Kit and dissolved in water; 80‐mer oligonucleotides containing the protospacer sequence, flanked by 30 bp of homology to the target plasmid were dissolved in water. E. coli GB05 transformed with the pSC101‐Prha‐ETgA‐tet plasmid (Baker et al, 2016) were cultured to OD600 ~0.5 in deep‐well 96‐well plates and then induced for 1 h for expression of the ETgA operon with L‐rhamnose. Electroporation with 200 ng (0.1 pmol) of the linearised vector and 50 pmol of oligonucleotide, using BTX 96‐well electroporation plates (MOS96, 2 mm gap,) and the BTX ECM630 electroporator with HT‐200 adapter, was described previously (Sarov et al, 2012). After 1 h of recovery time, 100 µl (1/10th) of the culture was transferred to a new plate with 900 µl of LB media plus 100 µg/ml ampicillin. After overnight selection, the saturated cultures were plated for single colonies on L‐agar + 100 micrograms/ml ampicillin. A single colony for each construct was picked and the clones were grown under selection in 96‐well plates, plasmid DNA was isolated, and Sanger sequenced from both directions using the M13F and M13R primers (flanking the U6‐protospacer‐sgRNA scaffold cassette, Dataset EV6). Sequence analysis confirmed the correct engineering of the plasmids. Alternatively, annealed oligonucleotides (Dataset EV6) were cloned into a BsaI site of a gRNA expression vector (Addgene Plasmid number 41824) (Mali et al, 2013) and correct insertion was determined by Sanger sequencing with the SP6 primer (Dataset EV6). For generating rescue cell lines, indicated coding sequences were cloned into a pCAG‐3xFLAG‐empty‐pgk‐hph vector (Betschinger et al, 2013) after PCR amplification. Correct insertion was detected by restriction digest and Sanger sequencing with the 3xFlag_seq primer (Dataset EV6).
Cell culture
Diploid (biparental) ES cells were routinely cultured in gelatin‐coated 25 cm2 flasks in DMEM supplemented with 15% FCS (batch tested, Biowest), 1× Penicillin‐Streptomycin (Sigma), 0.1 mM NEEA, 1 mM sodium pyruvate, 1 mM l‐glutamine, 0.05 mM 2‐mercaptoethanol (Gibco), 10 ng/ml LIF (batch tested, in‐house), 1.5 µM CHIRON and 0.5 µM PD0325901 (termed ESDMEM‐2i) on gelatin‐coated plates. The basal medium during all differentiation assays and for haploid ES cell culture (N2B27) consisted of a 1:1 mixture of DMEM/F12 and Neurobasal medium supplemented with 1× B27 (Gibco), 0.5× N2 (homemade), 0.1 mM NEAA, 1 mM l‐glutamine, 1× Penicillin‐Streptomycin and 0.05 mM 2‐mercaptoethanol. Haploid ES cells were routinely cultured in N2B27 supplemented with 3 µM CHIR99201, 1 µM PD0325901 and 10ng/ml LIF and regularly sorted based on FSC/SSC parameters by FACS (Leeb et al, 2015). Haploid Rex1::GFPd2‐IRES‐BSD (hRex1‐GFPd2) (Leeb et al, 2012) and haploid Rex1::mKO2‐IRES‐BSD/pOct4‐GFP‐IRES‐Puro ESCs (generated in this study from a double reporter mouse) were derived from activated oocytes in 2i/LIF medium and used for screens. Biparental diploid ES cells expressing a destabilised GFP transgene (GFPd2) under control of the endogenous Rex1 promoter and carrying an EF1alpha‐driven Cas9 cassette targeted to the Rosa26 locus (RC9 cells, (Li et al, 2018)) served as the parental cell line for the KO lines generated in this study. A list of KO cell lines can be found in Dataset EV6.
Transposon‐based saturation screen for key players of the exit from pluripotency
The gene‐trap vectors 5’‐PTK‐3’, TNN, TNP (Horie et al, 2011) together with haploid Rex1::GFPd2 ES cells (hRex1‐GFPd2, Leeb et al, 2014) and haploid Rex1::mKO2‐IRES‐BSD/pOct4‐GFP‐IRES‐Puro ES cells (hOR, this study) were used in 35 independent experiments to drive the screen to saturation. Mutant pools were generated by electroporation of 107 haploid ESCs using a GenePulser Xcell (270 V, 500 μF, ∞ Ω, Bio‐Rad) with 0.5 μg gene‐trap plasmid and 10 μg hyperactive transposase (hyPBase) (Yusa et al, 2011). 24 h after electroporation, selection was started for 4 days using 1µg/ml puromycin. Thereafter, cells were plated at a density of 104 cells/cm2 in N2B27 medium in the absence of LIF or inhibitors to allow differentiation. After 7–10 days in differentiation conditions, GFP‐positive cells were sorted and replated at a density of 104 cells/cm2. After culture in N2B27 medium for a further 7–10 days, GFP‐positive ESCs were sorted, expanded for 48 h in 2i/LIF medium and DNA was isolated using the PureGene kit (Qiagen) according to the manufacturer’s instructions. Sequencing libraries were prepared using an optimised Splinkerette PCR protocol (Leeb et al, 2014) with a modified and improved set of adapters and primers (see Dataset EV6). Adapters were annealed at 50 µM each in T4 DNA ligase buffer by incubating at 97.5°C for 150 s followed by a temperature decrease of 0.1°C/5 s for 775 cycles. Ready‐to‐use adapters were stored at −20°C. Genomic DNA was isolated using the PureGene kit (Qiagen) according to the manufacturer’s recommendations and quantified on the Qubit 2 fluorometer (Life Technologies) using the Qubit dsDNA Broad Range Assay (Life Technologies, Q32853). 2 µg of genomic DNA was diluted to a total volume of 120 µl with Low TE buffer (10 mM Tris–HCl, pH 8.0, 0.1 mM EDTA) and sheared to 250 bp on a Covaris S‐series Sample Preparation System with the following settings: Duty Cycle—20%, Intensity—5, Cycles per Burst—200, Time—60 s, Temperature—4–7°C. After shearing, DNA was isolated with the QIAquick PCR purification kit (Qiagen) and quality assessed using the Agilent Bioanalyzer with the DNA high sensitivity kit (Agilent). End repair was performed using the NEBNext End Repair Module (E6050S) by mixing 100 µl sheared DNA, 11.6 µl buffer and 6 µl enzyme followed by incubation at 20°C for 60 min. After cleanup with the PureLink PCR Purification Kit (K3100‐01), 42 µl of the end repaired DNA was A‐tailed with the NEBNext dA‐Tailing Module (E6053S) with 5 µl buffer and 3 µl enzyme at 37°C for 60 min. Following PureLink purification of the A‐tailed DNA, the annealed adapters were ligated using the NEBNext Quick Ligation (E6056S) kit at 20°C for 30 min. Products were cleaned up using Ampure XP beads at a 1:1 ratio. Libraries were amplified using Kapa HIFI (Roche) with 1 µM PB5_pr1 and 1 µM SplAP1 during 18 cycles of 20 s 98°C, 20 s 63°C and 40 s 72°C with an initial denaturation at 95°C for 2 min and a final extension at 72°C for 5min. Samples were cleaned up using Ampure XP beads at a ratio of 0.8. A second round of PCR was conducted with 24 µl of PCR1 using the Kapa HIFI polymerase with 0.6 µM SplAP2 and 0.6 µM index primer for 13 cycles at an annealing temperature of 60°C with all other settings as in PCR1. Final libraries were quantified using the KAPA SYBR Fast qPCR Mix with primers SYB7 and SYB5 (Dataset EV6) at 0.5 µM each, and a final quality control was conducted with the Agilent Bioanalyzer DNA High Sensitivity Assay. Integration sites were mapped as in (Leeb et al, 2014) and described below.
Generation of KO ESCs
Direct comparison between KOs necessitates a similar culture and passaging history. We therefore established an experimental pipeline to parallelise the generation of multiple KO ESC lines per week. 2 × 105 RC9 cells were transfected in a 6‐well format with a set of two gRNA‐containing vectors (Dataset EV6; (1 µg each), together with 0.5 µg of pCAG‐dsRed by Lipofectamine2000 (3 µl per reaction). 12–16 h after transfection, medium was replaced. To enrich for transfectants, cultures were sorted for GFP/dsRed double‐positive cells on a BD FACS Aria III 48h after transfection and plated at clonal density in 6‐cm dishes (100–800 cells per dish) in ESDMEM‐2i. Approximately 1 week after sorting, 48 colonies per gene KO were picked into 96‐well plates, trypsinised and split into one expansion‐plate and one plate for PCR genotyping after boiling lysis and proteinase K treatment (see PCR genotyping). Identified KO clones were expanded to 12‐well or 6‐well plates after 3 days of growth, and the remaining plate was frozen by adding 100 µl 2× freezing medium (80% FCS/20% DMSO) to 100 µl cell suspension as a backup. Clones were frozen from 12‐ or 6‐well plates in freezing medium containing 50% FCS and 10% DMSO. The 73 cell lines generated by this strategy and analysed by RNAseq are listed in Dataset EV6.
PCR genotyping
Crude DNA lysates were generated by pelleting half of a picked colony in a PCR plate at 500 g at 4°C, followed by two washes with PBS. 25 µl of water were added and cell pellets were boiled for 5min at 95°C. After cooling, Proteinase K was added at a final concentration of 3 µg/µl and incubated at 65°C for 1 h followed by inactivation at 95°C for 10min. Successful KO generation was confirmed by PCR, employing a three‐primer strategy. Specific reverse primers for the deletion event and a possible wild‐type allele were used in combination with a common forward primer (Dataset EV6) in separate PCRs. Cell lines with indels caused by a NHEJ event around the exonic gRNA1 target site were detected by Sanger sequencing of the “wild‐type”‐like band. Verification of the KO events was performed by the same PCR strategy on DNA, isolated using the PureGene DNA isolation kit (Qiagen) according to the manufacturer recommendations. For genotyping PCR, we used OneTaq (NEB) or JumpStart RedTaq (Sigma) PCR Master Mix according to the manufacturer’s recommendations for cycling and a standard annealing temperature of 55°C and 35–40 cycles.
qPCR genotyping
Cells grown in ESDMEM‐2i were harvested in RLT buffer and isolated with the RNeasy mini kit (Qiagen) according to the manufacturer’s recommendations. cDNA was transcribed using the SensiFAST cDNA Synthesis Kit (Bioline). Expression of exonic regions upstream of the deletion, the deleted exon and an intronic region downstream of the mutated exon was determined for the tested KO by qPCR using the Sensifast SYBR No Rox‐Kit (Bioline). Primers are listed in Dataset EV6.
Immunoblotting
Whole cell fractions were isolated using RIPA buffer, and protein concentrations were determined using a Bradford Assay (Bio‐Rad). 20 µg whole cell lysate were separated on 8‐12% SDS–PAGE gels (depending on the molecular weight of the target proteins) and subsequently blotted on 0.2 µm nitrocellulose membranes (Amersham). Membranes were blocked in TBS or PBS containing 5% milk and 0.1% Tween‐20 or 3% BSA and 0.1% Tween‐20 for 1h. Primary antibodies were incubated overnight at 4°C, at a dilution of 1/1,000 with the following exceptions: Anti‐Gapdh 1/25,000, Anti‐Tubulin 1/5,000, Anti‐Vinculin 1/5,000, Anti‐Smg5 1/200 and Smg7 1/2,000. Secondary goat anti‐mouse IgG HRP (1:10,000) or goat anti‐rabbit IgG HRP (1:15,000) were incubated at RT for 1h in blocking solution. Antibody binding was detected using the ECL select detection kit (Amersham). A list of antibodies can be found in Dataset EV6.
Parallel differentiation
ES cells were differentiated in seven batches, always including WT controls, control KO ESCs (no phenotype expected) and a range of ESCs with expected weak to strong phenotypes. Where applicable two independent KO clones were used, other KO ESCs were cultured in replicates before initiation of differentiation. 24 h before starting the differentiation assay, the medium was changed from ESDMEM‐2i to N2B27‐based 2i/LIF medium. Cells were then trypsinised, counted and plated in 2i (without LIF) at a density of 104 cells/cm2 in two replicate wells of a 6‐well plate for differentiation, one 6‐well as undifferentiated control and 2 wells of a 12‐well plate (undifferentiated control and differentiated sample) to monitor differentiation by FACS. 12h after plating, cells were washed twice with PBS and medium was changed to unsupplemented N2B27. For the undifferentiated controls, medium was replaced with fresh 2i. 24 h later, at N24, cultures were harvested in RLT buffer and stored at −80°C before isolation of RNA using the RNeasy Mini Kit (Qiagen). mRNA was isolated from 1 ug total RNA by poly‐dT enrichment using the NEBNext Polya mRNA Magnetic Isolation Module according to the manufacturer’s instructions. Samples were eluted in 15 µl 2x first strand cDNA synthesis buffer (NEBnext, NEB). After chemical fragmentation by incubating for 15 min at 94°C, the sample was directly subjected to the workflow for strand specific RNA‐Seq library preparation (Ultra Directional RNA Library Prep, NEB). For ligation, custom adapters were used (Dataset EV6). After ligation, adapters were depleted by an Ampure‐XP bead purification (Beckman Coulter) adding beads in a ratio of 1:1, followed by an index PCR (15 cycles) using Illumina compatible index primer. After double Ampure‐XP beads purifications (with beads added in a ratio 1:1), libraries were quantified on a Fragment Analyzer run with a NGS Assay Kit (Agilent) and loaded on a HiSeq 3000 flowcell with 50 cycles single end sequencing by pooling the samples based on molarity aiming for 30 mio. reads per sample.
2h resolved differentiation time course
Medium was changed from ESDMEM‐2i to N2B27‐2i/LIF 24h before initiating the assay. Cells were trypsinised, counted and plated at a density of 104 cells/cm2 in 6‐well plates in 2i (without LIF). 12h later, medium was changed to unsupplemented N2B27 (or 2i for the undifferentiated controls), and cells were harvested in buffer RLT every 2 h for the next 32h (until N32). RNA was isolated using the RNeasy Mini Kit (Qiagen). All libraries were generated from 500 ng RNA using the TruSeq Stranded mRNA Library Prep Kit (Illumina) and analysed on a HiSeq4000 machine to generate paired end reads at 75 bp read length.
LIF signature
1x104 cells/cm2 were plated in N2B27‐2i and N2B27‐2iLIF and grown for 24 h after which RNA was isolated using the RNeasy Mini Kit (Qiagen). Libraries were made from 500ng high quality RNA using the Quantseq 3′ mRNA‐Seq Library Prep Kit FWD for Illumina (Lexogen) according to the manufacturer’s instructions. Libraries were quantified using qPCR according to the Quantseq Library Prep Kit and with the included primers, pooled and quality checked using an Agilent Bioanalyzer DNA High Sensitivity Assay (Agilent). Multiplexed samples were sequenced on a HiSeq4000 machine to generate single end reads of 50bp read length.
RNAi experiments
For RNAi FlexiTube, siRNAs against Klf2 were used with AllStars Negative Control siRNAs (Qiagen). 20 ng siRNAs/4 × 104 cells were transfected in 2i using DharmaFect (Dharmacon). 12 h later, the medium was changed to N2B27 after two PBS washes. At N24 and N32, Rex1‐GFPd2 expression was determined by flow cytometry (see below).
Flow cytometry
Cells were dissociated in 0.25% trypsin/EDTA, and trypsin was neutralised with DMEM supplemented with 15% FCS. After passing through a 40 µm mesh, Rex1‐reporter activity was measured using a BD Fortessa machine. High‐throughput measurements were performed in 96‐well plates using the HTS unit of the BD Fortessa. Data were analysed using FlowJo software.
Data analysis
Statistical analysis
Established standard tests (e.g. for differential expression testing, gene set enrichment) were used throughout. Correction for multiple testing was performed whenever necessary. Details of individual statistical tests are provided in the following sections.
Transposon mutagenesis screen in haploid murine embryonic stem cells
Integration mapping was performed as previously described (Leeb et al, 2014). Most gene‐trap insertions were found in intronic sequences, consistent with the relative length distributions of genomic features. However, after normalising to available TTAA sites per region, TTAA sites in 5’UTRs and promoters (defined as < 500 bp upstream of the TSS) were overrepresented, whereas TTAA sites in introns were relatively depleted. 7,760 independent integrations passing cut‐off criteria were mapped to 3,469 genes. Of those, 232 Genes (2,474 independent integration sites) were hit in five or more independent screens, indicating that those are integrations causative for the detected differentiation defect. To complement the candidate list and correct for gene‐specific biases, we further developed a strategy to assign statistical strength to candidate genes by assessing the number of integrations per gene in relation to available TTAA transposon integration sites. We utilised the number of different TTAA sites within each gene to calculate the probability of having integrations in k or more different locations by chance. All TTAA sites in mm10 were assessed using bowtie, and the analysis was restricted to genes classified as protein‐coding, long non‐coding RNAs and micro‐RNAs. We computed the probability that a gene is hit k times given that it contains n TTAA transposon integration sites assuming a binomial distribution (while k is the observed number of integrations).
p0 was then calculated by
We applied this test to genes with at least 26 TTAA sites, as genes with fewer TTAA sites would be significant (P‐value < 0.05) with a single integration. Candidate gene lists between both analyses showed overlapping results and top hits were largely identical. This resulted in a list of 421 significant genes (adjusted P‐value < 0.01 and hit in at least two independent screens).
Guide RNA design criteria
To automate the design of sgRNAs as far as possible, we used the R package CRISPRseek (Zhu et al, 2014) to score the efficiency and likely off‐target binding of candidate sgRNA sequences. For a selected gene, the algorithm proceeds as follows:
Retrieve the sequence of the first protein‐coding exon.
Sequence has to be at least 50nt long.
Find sgRNAs within the sequence and score them.
If no appropriate sgRNA could be found, move to next exon.
Still in the first third of the coding sequence?
Find a second sgRNA in an intron at least 5kb, but no further than 30kb downstream.
Score the sgRNAs.
A minimal efficiency of 0.2 and an off‐target score < 100 was required.
PCR primer design strategy for simplified knockout validation
The surrounding regions of the sgRNAs (min distance 20 bp) were used as input for PRIMER3 primer predictions. Primers were designed to have a length of 18–27 bases with an optimum of 20 and to have melting temperatures between 57 and 63°C with an optimum of 60°C. All primers were checked for off‐target binding sites by using Blastn. Blastn was called with the parameters:
‐task megablast ‐use_index false ‐word_size 7 ‐perc_identity 65 –evalue 30000 ‐max_target_seqs 50 ‐num_threads 4 –outfmt 7.
RNA quantification, RNAseq
Quality control was performed using fastQC (version 0.11.5). Transcripts were mapped to the mm10 mouse reference genome and counts determined using STAR (version 2.5.3). FPKM values were calculated using DESeq2 (version 1.24.0).
RNA quantification, QuantSeq
Transcripts from RC9 cells in 2i and 2i/LIF medium were measured using QuantSeq (Lexogen) and quantified as described. Reads were trimmed with bbduk (version 35.92). Quality control was performed using fastQC (version 0.11.5). Transcripts were mapped to the mm10 mouse reference genome using STAR (version 2.5.3). After indexing with samtools (version 1.3), reads in genes were counted using HTSeq‐count (version 0.6.0). Counts were subjected to differential expression analysis.
Differential expression analysis
Differential expression analysis was carried out using limma (version 3.40.6), after transforming transcript counts using the included voom function. We then fitted a linear model to sample measurements from each combination of knockouts (including RC9 control) and conditions (2i and N24). RNAseq batches were included in the model as a confounding factor and batch‐corrected using removeBatchEffect for visualisation. Contrasts were fitted to determine the expression differences between (i) each knockout sample in 2i and the RC9 control, (ii) each knockout sample in N24 and the RC9 control, and (iii)the difference between the log fold changes calculated in (i) and (ii) (interaction effect). We also fitted a contrast to determine changes during normal differentiation, i.e. the differential expression between RC9‐N24 versus RC9‐2i. All fitted contrasts were tested for differential expression using moderated t‐statistics through the limma function treat (H0 |log2FC| < log2(1.5)). P‐values were corrected for multiple testing using the Benjamini–Hochberg method (FDR). For subsequent analyses, the more stringent P‐values and FDR values as calculated by treat were used (unless noted otherwise). Differential expression analysis of 2i/LIF versus 2i RC9 measurements was carried out separately, but in an identical manner.
GO enrichment analysis
Mouse gene GO annotations were extracted from the R package org.Mm.eg.db (version 3.4.0). GO terms that included between 5 and 500 genes were selected for further analysis. For each gene list of interest, significance of functional enrichment of GO terms compared with the background list was determined using Fisher’s exact tests. The background was restricted to genes whose transcripts were detected at a median count above 5. To reduce redundancy of functional categories, we clustered GO terms that differed in 5 or fewer genes of interest (using the R base function hclust on the L1‐distance of the binary membership matrix). The smallest GO term by total annotations was selected independently from Fisher test results as the primary, i.e. most specific term to represent each cluster. We then carried out correction for multiple hypothesis testing using the Benjamini–Hochberg method on all primary GO terms to obtain adjusted P‐values.
Pathway enrichment was performed in the same way as described above for GO terms with Reactome pathway annotations taken from the reactome.db R package (version 1.68.0).
Naïve marker dependency of N24 KO expression patterns (NAGs)
To quantify the similarity of each gene’s log2 fold change across KOs at N24 with the log2 fold change of seven core pluripotency markers (Nanog, Esrrb, Tbx3, Tfcp2l1, Klf4, Prdm14 and Zfp42), multiple regression analysis was performed. That is, each gene’s DE pattern was modelled as a linear combination of marker DE patterns, quantifying the strength of association as their shared variance (multiple‐R 2). NAGs were defined as having a naïve marker R 2 of ≥ 0.65.
Cluster analysis (constitutive and N24‐induced)
Based on the differential expression analysis of all KOs (excluding Suz12, Eed & Csnk1a1 KOs) in both N24 and 2i, and their interactions with the RC9 control, we defined two main categories of KO‐responsive genes: (i) the constitutive knockout response: genes that were significantly changed (adj. P ≤ 0.05) in the same direction in both N24 and 2i, in at least one knockout condition. Genes from the N24‐induced KO response category were excluded. (ii) N24‐induced knockout response: genes that were significantly changed in both N24 and the knockout:RC9 interaction term, in at least one knockout condition. To determine whether genes in the N24‐induced knockout response subset were overall more correlated with naïve pluripotency markers, we checked the distribution of naïve marker multiple‐R2 values in the N24‐induced knockout response set compared with both the background distribution and genes classified as part of the constitutive KO response.
To determine whether distinct functional clusters of genes that may be activated in all, single or sub‐groups of knockouts, we clustered genes in either of the categories (constitutive and N24‐induced response) based on their KON24 versus RC9N24 log fold changes, using the R base package hclust with the “Ward.D2” method. We determined the total within‐cluster variance at different numbers of clusters. The number of clusters was chosen such that the model improved strongly by increasing the number of clusters up to that value, but only marginal gains result from increasing it further. Thereby, 12 gene clusters in the constitutive response and 12 clusters in the induced response were identified. GO enrichment analysis was carried out as described above for member genes of each cluster. Here, we required at least five genes of a cluster to be enriched in a GO term to be considered.
Differentiation pathway correlation analysis
We defined downstream responses of key differentiation pathways using the most specific responses to the knockouts of their upstream regulators: Tsc2 (mTORC1), Ptpn11 & Fgfr1 (Fgf/Erk signalling), Tcf7l1 (Wnt signalling) and Rbpj (Notch signalling). In addition, LIF‐specific changes were defined as the response of genes in RC9 cells in the 2i/LIF compared with the 2i condition. To ensure specificity, we required that the log fold change of each response gene for a given regulator (in N2B27 medium) exceeded that observed in the remaining conditions by a factor of two. In the case of Ptpn11 and Fgfr1, this criterion had to be met by the lower of the two log fold changes for each gene. Additionally, the genes were required to satisfy a FPKM (CPM in case of LIF) cut‐off of at least 1 in either the KO or the WT. From all remaining, significantly expressed genes (FDR ≤ 0.05) we then selected, by absolute log fold change in descending order, the top 50 most specific genes for each pathway (Appendix Fig S7A).
Pathway activity was then determined across all KOs at N24. For each pathway, the log2 fold changes of the 50 pathway‐specific response genes from the associated regulator knockout (KON24 versus RC9N24) were selected. We then correlated these log fold changes to the log fold changes of the same genes in all other knockouts at N24 (and in 2i for the LIF pathway footprint, which was derived by the contrast 2i versus 2i/LIF). A positive correlation indicates that the pathway is disrupted in a manner similar to knocking out its regulator, whereas a negative correlation indicates the opposite. In the case of LIF‐specific genes, a positive correlation indicates an effect similar to that of adding LIF. P‐values of correlation values were calculated based on Fisher’s Z transform. P‐values were then corrected for multiple testing using the Bonferroni method.
To determine how predictive the calculated correlations were of the differentiation phenotype (mean naïve marker log2FC versus RC9 at N24), we carried out multiple linear regression and extracted the coefficients of the model to establish the relative contribution of each pathway signature to naïve marker deregulation.
Quantification of differentiation delay in knockouts
Raw RNA‐seq data were aligned to the mm10 genome, and read counts were obtained using the STAR aligner (version2.5.2b). Gene expression for the following biotypes was quantified: protein_coding, misc_RNA, miRNA, scaRNA, scRNA, snoRNA, snRNA and sRNA. Read counts were normalised using DESeq2 (version 1.22.2) and subsequently the log2fc between all samples and the mean expression of the 2i samples was calculated. This resulted in expression profiles for each gene over 32 h of differentiation.
Gaussian process regression was used to smoothen the profiles of each gene (R package tgp version 2.4‐14). The expression profiles of 73 knockouts at N24 were mapped onto the differentiation axis from the time course to quantify differentiation delays per KO, i.e. we aimed to quantify how many hours the KO expression pattern is “behind” the expected differentiation in WT cells. This “mapping” to the time axis was done first computing the log2FCs between N24 versus 2i for each KO. The resulting log2FC profiles were compared with the log2FCs at each time point during the WT differentiation. We assumed that the time point at which the difference between the WT profile and the KO profile is minimised best reflects the molecular differentiation state of the respective KO.
We used the Euclidean distance for this purpose. To make distances better interpretable between the knockouts the distances were set to:
where the maximum and minimum distances refer to the respective maximum and minimum Euclidean distances of the respective KO across all time points (i.e. the worst and the best matching time point). Thus, the best matching time point will get a distance score of 0.
In order to further increase the time resolution, additional time points were imputed via linear interpolation. Here, we interpolated expression profiles every 15 min between the two neighbouring time points of the time point with the smallest Euclidean distance. Timing of knockouts was repeated on the interpolated time points, and the new minimal distance was used to quantify the differentiation delay.
Prediction of cluster expression by pathway activity
Pathway activities for each pathway in all KOs were calculated using following formulas:
The combinations of pathway activities and average cluster expression changes over all KOs were used to build a multiple regression model to predict the expression changes in each cluster by the five pathway activities. Connections between pathway activities and cluster expression changes not passing a significance cut‐off (adj. P‐value ≤ 0.01) of the multiple regression models were excluded from each model. The remaining models were tested in the time course data. Connections from clusters where prediction of expression changes based on pathway activities did follow or contradict the observed expression changes in the time course were excluded.
Embryo comparison
Sequencing data were obtained from the European Nucleotide Archive (Toribio et al, 2017) from single‐cell mouse embryo profiling studies (Deng et al, 2014; Mohammed et al, 2017). The macaque FKPM expression data set was provided by (Nakamura et al, 2016). Orthologues genes mapping in 1‐to‐1 fashion were used during comparative analysis. Alignments to gene loci were quantified with htseq‐count (Anders et al, 2014) based on annotation from Ensembl 87 (Toribio et al, 2017). Principal component and cluster analysis were based on log2 FPKM values computed with the Bioconductor package DESeq (Anders & Huber, 2010), custom scripts and FactoRmineR packages (Lê et al, 2008). Highly variable genes were calculated by fitting a non‐linear regression curve between average log2 FPKM and the square of coefficient of variation. Specific thresholds were applied along the x‐axis (log2FPKM) and y‐axis (coefficient of variation) to identify the most variable genes. Fractional identity between the bulk RNAseq (2i, N24) and the mouse, macaque embryo or human capacitation data set were computed using R package DeconRNASeq (Gong & Szustakowski, 2013).
GENEES (Genetic Network Explorer for Embryonic Stem Cell Differentiation) Application
To both visualise and interrogate gene expression analysis of all KO ES cells, we have set up the online tool GENEES (Genetic Network Exploration of Embryonic Stem Cell Differentiation). It is accessible at [http://shiny.cecad.uni‐koeln.de:3838/stemcell_network/]. The app integrates multiple visualisations and summary statistics to make all information directly available for exploratory analysis. The app has 3 different tabs for inspection of single genes, pre‐computed clusters and custom gene sets. Single genes can be inspected in the “Genes tab”, while previously described constitutive clusters and induced clusters can be further inspected in the “Pre‐computed clusters” tab. In the “Select custom geneset” tab, a set of genes of interest subjected to analysis can be defined and analysed.
Author contributions
Conceptualisation of study: ML, AB, AS and AFS. Study design: ML and AB. Experiments: ML, AL, AB, RS, MG and FT‐T. Wet‐laboratory experiments: AL, MH, JR, PvdL, HFT, LS, EG, ML and MS. Bioinformatic analyses: RS, FT‐T, MG, GGS, MR and AL. Manuscript writing: ML, AB, AS, AFS, AL, RS, MG, MH, FT‐T and GG. Funding: ML, AB, AFS and AS.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Acknowledgements
We thank Johanna Stranner, Thomas Sauer and Andy Riddell for help with flow cytometry, Meng Li and Kosuke Yusa for sharing RC9 cells, Andreas Dahl for NGS‐sequencing support and Christa Bücker for critical comments on the manuscript. ML is funded by a WWTF‐VRG grant (VRG14‐006). This study was funded by an FWF/DFG DACH grant to AB and ML (FWF grant number: I 3786; DGF grant number 398882498). AB and MG were supported by the BMBF (Sybacol). RS received support by the Cologne Graduate School of Ageing Research. LS is part of the FWF doctoral programme SMICH and supported by an Austrian Academy of Sciences DOC Fellowship. Wellcome and the Medical Research Council provide core support for the Wellcome‐MRC Cambridge Stem Cell Institute. AS received funding from the Medical Research Council (MR/P00072X/1) and is an MRC Professor. This project was initiated within the EU FP7 integrated project SyBoSS (Grant agreement: 242129) co‐ordinated by AFS and AB including partners ML and AS.
The EMBO Journal (2021) 40: e105776.
Contributor Information
Andreas Beyer, Email: andreas.beyer@uni-koeln.de.
Martin Leeb, Email: martin.leeb@univie.ac.at.
Data availability
The data sets produced in this study are available on the Gene Expression Omnibus database under: GSE145653 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145653).
References
- Acampora D, Omodei D, Petrosino G, Garofalo A, Savarese M, Nigro V, Di Giovannantonio LG, Mercadante V, Simeone A (2016) Loss of the Otx2‐binding site in the nanog promoter affects the integrity of embryonic stem cell subtypes and specification of inner cell mass‐derived epiblast. Cell Rep 15: 2651–2664 [DOI] [PubMed] [Google Scholar]
- Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W (2014) HTSeq—a Python framework to work with high‐throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artus J, Kang M, Cohen‐Tannoudji M, Hadjantonakis A‐K (2013) PDGF signaling is required for primitive endoderm cell survival in the inner cell mass of the mouse blastocyst. Stem Cells 31: 1932–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker O, Gupta A, Obst M, Zhang Y, Anastassiadis K, Fu J, Stewart AF (2016) RAC‐tagging: recombineering And Cas9‐assisted targeting for protein tagging and conditional analyses. Sci Rep 6: 25529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betschinger J, Nichols J, Dietmann S, Corrin PD, Paddison PJ, Smith A (2013) Exit from pluripotency is gated by intracellular redistribution of the bHLH transcription factor Tfe3. Cell 153: 335–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroviak T, Loos R, Bertone P, Smith A, Nichols J (2014) The ability of inner‐cell‐mass cells to self‐renew as embryonic stem cells is acquired following epiblast specification. Nat Cell Biol 16: 516–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroviak T, Nichols J (2014) The birth of embryonic pluripotency. Philos Transact Roy Soc B: Biol Sci 369(1657): 20130541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroviak T, Nichols J (2017) Primate embryogenesis predicts the hallmarks of human naïve pluripotency. Development 144: 175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroviak T, Stirparo GG, Dietmann S, Hernando‐Herraez I, Mohammed H, Reik W, Smith A, Sasaki E, Nichols J, Bertone P (2018) Single cell transcriptome analysis of human, marmoset and mouse embryos reveals common and divergent features of preimplantation development. Development 145: dev167833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredenkamp N, Yang J, Clarke J, Stirparo GG, von Meyenn F, Dietmann S, Baker D, Drummond R, Ren Y, Li D et al (2019) Wnt inhibition Facilitates RNA‐mediated reprogramming of human somatic cells to naive pluripotency. Stem Cell Rep 13: 1083–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buecker C, Srinivasan R, Wu ZX, Calo E, Acampora D, Faial T, Simeone A, Tan MJ, Swigut T, Wysocka J (2014) Reorganization of enhancer patterns in transition from naive to primed pluripotency. Cell Stem Cell 14: 838–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers I, Tomlinson SR (2009) The transcriptional foundation of pluripotency. Development 136: 2311–2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega V, Wong E, Orlov Y, Zhang W, Jiang J et al (2008) Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133: 1106–1117 [DOI] [PubMed] [Google Scholar]
- Deng Q, Ramsköld D, Reinius B, Sandberg R (2014) Single‐cell RNA‐seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343: 193–196 [DOI] [PubMed] [Google Scholar]
- Dunn SJ, Martello G, Yordanov B, Emmott S, Smith AG (2014) Defining an essential transcription factor program for naive pluripotency. Science 344: 1156–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elling U, Taubenschmid J, Wirnsberger G, O'Malley R, Demers SP, Vanhaelen Q, Shukalyuk AI, Schmauss G, Schramek D, Schnuetgen F et al (2011) Forward and reverse genetics through derivation of haploid mouse embryonic stem cells. Cell Stem Cell 9: 563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong T, Szustakowski JD (2013) DeconRNASeq: a statistical framework for deconvolution of heterogeneous tissue samples based on mRNA‐Seq data. Bioinformatics 29: 1083–1085 [DOI] [PubMed] [Google Scholar]
- Guo G, Huang Y, Humphreys P, Wang X, Smith A (2011) A PiggyBac‐based recessive screening method to identify pluripotency regulators. PLoS One 6: e18189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J, Guo G, Wray J, Eyres I, Nichols J, Grotewold L, Morfopoulou S, Humphreys P, Mansfield W, Walker R et al (2009) Oct4 and LIF/Stat3 additively induce Kruppel factors to sustain embryonic stem cell self‐renewal. Cell Stem Cell 5: 597–609 [DOI] [PubMed] [Google Scholar]
- Hoffman JA, Wu CI, Merrill BJ (2013) Tcf7l1 prepares epiblast cells in the gastrulating mouse embryo for lineage specification. Development 140: 1665–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie K, Kokubu C, Yoshida J, Akagi K, Isotani A, Oshitani A, Yusa K, Ikeda R, Huang Y, Bradley A et al (2011) A homozygous mutant embryonic stem cell bank applicable for phenotype‐driven genetic screening. Nat Methods 8: 1071–1077 [DOI] [PubMed] [Google Scholar]
- Kalkan T, Bornelöv S, Mulas C, Diamanti E, Lohoff T, Ralser M, Middelkamp S, Lombard P, Nichols J, Smith A (2019) Complementary activity of ETV5, RBPJ, and TCF3 drives formative transition from naive pluripotency. Cell Stem Cell 24: 785–801.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkan T, Olova N, Roode M, Mulas C, Lee HJ, Nett I, Marks H, Walker R, Stunnenberg HG, Lilley KS et al (2017) Tracking the embryonic stem cell transition from ground state pluripotency. Development 144: 1221–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkan T, Smith A (2014) Mapping the route from naive pluripotency to lineage specification. Philos Transact Roy Soc B: Biol Sci 369(1657): 20130540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokubu C, Takeda J (2014) When half is better than the whole: advances in haploid embryonic stem cell technology. Cell Stem Cell 14: 265–267 [DOI] [PubMed] [Google Scholar]
- Kunath T, Saba‐El‐Leil MK, Almousailleakh M, Wray J, Meloche S, Smith A (2007) FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self‐renewal to lineage commitment. Development 134: 2895–2902 [DOI] [PubMed] [Google Scholar]
- Lassar AB, Paterson BM, Weintraub H (1986) Transfection of a DNA locus that mediates the conversion of 10T1 2 fibroblasts to myoblasts. Cell 47: 649–656 [DOI] [PubMed] [Google Scholar]
- Lê S, Josse J, Husson F (2008) FactoMineR: an R package for multivariate. Analysis 2008 25: 18 [Google Scholar]
- Leeb M, Dietmann S, Paramor M, Niwa H, Smith A (2014) Genetic exploration of the exit from self‐renewal using haploid embryonic stem cells. Cell Stem Cell 14: 385–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb M, Perry ACF, Wutz A (2015) Establishment and use of mouse haploid ES cells. Curr Protoc Mouse Biol 5: 155–185 [DOI] [PubMed] [Google Scholar]
- Leeb M, Walker R, Mansfield B, Nichols J, Smith A, Wutz A (2012) Germline potential of parthenogenetic haploid mouse embryonic stem cells. Development 139: 3301–3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb M, Wutz A (2011) Derivation of haploid embryonic stem cells from mouse embryos. Nature 479: 131–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Yu JSL, Tilgner K, Ong SH, Koike‐Yusa H, Yusa K (2018) Genome‐wide CRISPR‐KO screen uncovers mTORC1‐mediated Gsk3 regulation in naive pluripotency maintenance and dissolution. Cell Rep 24: 489–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell S, Benchoua A, Heavey B, Smith AG (2006) Notch promotes neural lineage entry by pluripotent embryonic stem cells. PLoS Biol 4: e121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA‐guided human genome engineering via Cas9. Science 339: 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martello G, Smith A (2014) The nature of embryonic stem cells. Annu Rev Cell Dev Biol 30: 647–675 [DOI] [PubMed] [Google Scholar]
- Martello G, Sugimoto T, Diamanti E, Joshi A, Hannah R, Ohtsuka S, Gottgens B, Niwa H, Smith A (2012) Esrrb is a pivotal target of the Gsk3/Tcf3 axis regulating embryonic stem cell self‐renewal. Cell Stem Cell 11: 491–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer D, Stadler MB, Rittirsch M, Hess D, Lukonin I, Winzi M, Smith A, Buchholz F, Betschinger J (2020) Zfp281 orchestrates interconversion of pluripotent states by engaging Ehmt1 and Zic2. EMBO J 39: e102591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed H, Hernando‐Herraez I, Savino A, Scialdone A, Macaulay I, Mulas C, Chandra T, Voet T, Dean W, Nichols J et al (2017) Single‐Cell landscape of transcriptional heterogeneity and cell fate decisions during mouse early gastrulation. Cell Rep 20: 1215–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molotkov A, Mazot P, Brewer JR, Cinalli RM, Soriano P (2017) Distinct requirements for FGFR1 and FGFR2 in primitive endoderm development and exit from pluripotency. Dev Cell 41: 511–526.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira S, Seo C, Gordon V, Xing S, Wu R, Polena E, Fung V, Ng D, Wong CJ, Larson B et al (2018) Endogenous BioID elucidates TCF7L1 interactome modulation upon GSK‐3 inhibition in mouse ESCs. bioRxiv 10.1101/431023 [PREPRINT] [DOI]
- Mulas C, Kalkan T, Smith A (2017) NODAL secures pluripotency upon embryonic stem cell progression from the ground state. Stem Cell Rep 9: 77–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Okamoto I, Sasaki K, Yabuta Y, Iwatani C, Tsuchiya H, Seita Y, Nakamura S, Yamamoto T, Saitou M (2016) A developmental coordinate of pluripotency among mice, monkeys and humans. Nature 537: 57–62 [DOI] [PubMed] [Google Scholar]
- Neagu A, van Genderen E, Escudero I, Verwegen L, Kurek D, Lehmann J, Stel J, Dirks RAM, van Mierlo G, Maas A et al (2020) in vitro capture and characterization of embryonic rosette‐stage pluripotency between naive and primed states. Nat Cell Biol 22: 534–545 [DOI] [PubMed] [Google Scholar]
- Nichols J, Smith A (2012) Pluripotency in the embryo and in culture. Cold Spring Harb Perspect Biol 4: a008128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H (2018) The principles that govern transcription factor network functions in stem cells. Development 145: dev157420 [DOI] [PubMed] [Google Scholar]
- Niwa H, Ogawa K, Shimosato D, Adachi K (2009) A parallel circuit of LIF signalling pathways maintains pluripotency of mouse ES cells. Nature 460: 118–122 [DOI] [PubMed] [Google Scholar]
- Ohtsuka S, Nakai‐Futatsugi Y, Niwa H (2015) LIF signal in mouse embryonic stem cells. JAK‐STAT 4: e1086520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plusa B, Piliszek A, Frankenberg S, Artus J, Hadjantonakis A‐K (2008) Distinct sequential cell behaviours direct primitive endoderm formation in the mouse blastocyst. Development 135: 3081–3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu D, Ye S, Ruiz B, Zhou X, Liu D, Zhang Q, Ying Q‐L (2015) Klf2 and Tfcp2l1, two Wnt/β‐catenin targets, act synergistically to induce and maintain naive pluripotency. Stem Cell Rep 5: 314–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos‐Ibeas P, Sang F, Zhu Q, Tang WWC, Withey S, Klisch D, Wood L, Loose M, Surani MA, Alberio R (2019) Pluripotency and X chromosome dynamics revealed in pig pre‐gastrulating embryos by single cell analysis. Nat Commun 10: 500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossant J, Tam PPL (2017) New insights into early human development: lessons for stem cell derivation and differentiation. Cell Stem Cell 20: 18–28 [DOI] [PubMed] [Google Scholar]
- Rossant J, Tam PPL (2018) Exploring early human embryo development. Science 360: 1075–1076 [DOI] [PubMed] [Google Scholar]
- Rostovskaya M, Stirparo GG, Smith A (2019) Capacitation of human naïve pluripotent stem cells for multi‐lineage differentiation. Development 146: dev172916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarov M, Murray JI, Schanze K, Pozniakovski A, Niu W, Angermann K, Hasse S, Rupprecht M, Vinis E, Tinney M et al (2012) A genome‐scale resource for in vivo tag‐based protein function exploration in C. elegans . Cell 150: 855–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shy BR, Wu C‐I, Khramtsova GF, Zhang JY, Olopade OI, Goss KH, Merrill BJ (2013) Regulation of Tcf7l1 DNA binding and protein stability as principal mechanisms of Wnt/β‐catenin signaling. Cell Rep 4: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A (2017) Formative pluripotency: the executive phase in a developmental continuum. Development 144: 365–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AG, Heath JK, Donaldson DD, Wong GG, Moreau J, Stahl M, Rogers D (1988) Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature 336: 688–690 [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: 663–676 [DOI] [PubMed] [Google Scholar]
- Toribio AL, Alako B, Amid C, Cerdeno‐Tarraga A, Clarke L, Cleland I, Fairley S, Gibson R, Goodgame N, Ten Hoopen P et al (2017) European nucleotide archive in 2016. Nucleic Acids Res 45: D32–D36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursu A, Illich DJ, Takemoto Y, Porfetye AT, Zhang M, Brockmeyer A, Janning P, Watanabe N, Osada H, Vetter IR et al (2016) Epiblastin A induces reprogramming of epiblast stem cells into embryonic stem cells by inhibition of casein kinase 1. Cell Chem Biol 23: 494–507 [DOI] [PubMed] [Google Scholar]
- Villegas F, Lehalle D, Mayer D, Rittirsch M, Stadler MB, Zinner M, Olivieri D, Vabres P, Duplomb‐Jego L, De Bont E et al (2019) Lysosomal signaling licenses embryonic stem cell differentiation via inactivation of Tfe3. Cell Stem Cell 24: 257–270.e8 [DOI] [PubMed] [Google Scholar]
- Watanabe S, Umehara H, Murayama K, Okabe M, Kimura T, Nakano T (2006) Activation of Akt signaling is sufficient to maintain pluripotency in mouse and primate embryonic stem cells. Oncogene 25: 2697–2707 [DOI] [PubMed] [Google Scholar]
- Wray J, Kalkan T, Gomez‐Lopez S, Eckardt D, Cook A, Kemler R, Smith A (2011) Inhibition of glycogen synthase kinase‐3 alleviates Tcf3 repression of the pluripotency network and increases embryonic stem cell resistance to differentiation. Nat Cell Biol 13: 838–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Ye M, Feng R, Graf T (2004) Stepwise reprogramming of B Cells into macrophages. Cell 117: 663–676 [DOI] [PubMed] [Google Scholar]
- Xu T, Park S‐S, Giaimo BD, Hall D, Ferrante F, Ho DM, Hori K, Anhezini L, Ertl I, Bartkuhn M et al (2017) RBPJ/CBF1 interacts with L3MBTL3/MBT1 to promote repression of Notch signaling via histone demethylase KDM1A/LSD1. EMBO J 36: 3232–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Blelloch R (2014) Regulation of pluripotency by RNA binding proteins. Cell Stem Cell 15: 271–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo JC, Jiang J, Tan ZY, Yim GR, Ng JH, Goke J, Kraus P, Liang H, Gonzales KA, Chong HC et al (2014) Klf2 is an essential factor that sustains ground state pluripotency. Cell Stem Cell 14: 864–872 [DOI] [PubMed] [Google Scholar]
- Ying Q‐L, Stavridis M, Griffiths D, Li M, Smith A (2003) Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat Biotechnol 21: 183–186 [DOI] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle‐Morera L, Doble B, Woodgett J, Cohen P, Smith A (2008) The ground state of embryonic stem cell self‐renewal. Nature 453: 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusa K, Zhou L, Li MA, Bradley A, Craig NL (2011) A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci USA 108: 1531–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LJ, Holmes BR, Aronin N, Brodsky MH (2014) CRISPRseek: a bioconductor package to identify target‐specific guide RNAs for CRISPR‐Cas9 genome‐editing systems. PLoS One 9: e108424 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Data Availability Statement
The data sets produced in this study are available on the Gene Expression Omnibus database under: GSE145653 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145653).