

Abstract
Cyclooxygenase-2 (COX-2) enzyme inhibitors have not eliminated the necessity for developed drugs not only in the nonsteroidal anti-inflammatory drug (NSAIDs) area, but also in other therapeutic applications including prevention of cancer and Alzheimer's disease. A series of novel substituted cyclic imides have been reported as selective COX-2 inhibitors. To understand the structural features responsible for their activity, a 3D validated pharmacophore and quantitative structure−activity relationship (QSAR) model have been developed. The values of enrichment factor (EF), goodness of hit score (GH), area under the ROC curve (AUC), sensitivity, and specificity refer to the good ability of the pharmacophore model to identify active compounds. Multiple linear regression (MLR) produced statistically significant QSAR model with (R2training = 0.763, R2test = 0.96) and predictability (Q2training = 0.66, Q2test = 0.84). Then, using the pharmacophore and QSAR models, eight authenticated botanicals in two herbal medicines and the ZINC compounds database, were virtually screened for ligands to COX-2. The retrieved hits which also obey lipinski's rule of five (RO5) were docked in the COX-2 3D structure to investigate their binding mode and affinity. Finally, based on the docking results, nine molecules were prioritized as promising hits that could be used as leads to discover novel COX-2 inhibitors. COX-2 inhibition of most of these hits has not been reported previously. Ten-nanosecond molecular dynamics simulation (10-ns MD) was performed on the initial structure COX-2 complex with ZINC000113253375 and ZINC000043170560 resulted from the docking. Our utilization of the 3D pharmacophore model, QSAR, molecular docking, and molecular dynamics simulation trials can be a potent strategy to successfully predict activity, efficiently design drugs, and screen large numbers of new compounds as active drug candidates.
Keywords: Cyclooxygenase-2, Docking, QSAR, Pharmacophore, Molecular dynamics simulation, Virtual screening
Cyclooxygenase-2; Docking; QSAR; Pharmacophore, Molecular dynamics simulation, Virtual screening.
1. Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are a commonly prescribed for their established anti-inflammatory, antipyretic, and analgesic properties [1]. NSAIDs inhibit cyclooxygenase enzyme (COX), which mediates the bioconversion of arachidonic acid to inflammatory prostaglandins (PGs). There are two COX isoenzymes (COX-1 and COX-2). COX-1 is responsible for the maintenance of physiological homeostasis, while COX-2 is induced in response to pro-inflammatory conditions.
In spite of their benefits, conventional NSAIDs have significant gastrointestinal toxicity and irritation since they inhibit both COX-1 and COX-2 isoforms. These adverse side effects encouraged the improvement of selective COX-2 inhibitors as promising gastro protective agents [2]. Later on, the long term use of certain COX-2 inhibitor causes ulcer exacerbation in high-risk patients, thrombosis, and kidney toxicity. Thus COX-2 inhibitors are still of interest to researchers and need more work and improvement in the NSAIDs area [3, 4, 5]. In addition, more investigations in the role of COX-2 inhibitors in cancer chemotherapy [6, 7] and neurological diseases such as Parkinson [8] and Alzheimer's diseases are still needed [9, 10]. Many valuable studies on COX-2 inhibitors have still published until now, as, these inhibitors are still subject to the interest of researchers worldwide [11, 12, 13, 14, 15, 16, 17].
Back to NSAIDs area, they are generally used for the treatment of pain and edema representing the choice of treatment for various inflammatory diseases such as Rheumatoid arthritis and osteoarthritis [18].
Since these drugs are related to negative side effects, new anti-inflammatory drugs are needed and complementary and alternative medicines are being sought [19].
These days about 70% of the world's population relies on herbal drugs for its primary pharmaceutical care [20]. Natural sources are the most promising pool for drug candidates as revealed by statistics [21].
Examples of medicinal plants used locally to treat arthritis and related disorders are Voltarit® and Rheumax®. Voltarit® contains 5 herbs including Apium graveolens (Celery), Crataegus laevigata (Hawthorn berries), Curcuma longa (Turmeric), Harpagophytum procumbens (Devil's claw), and Vaccinium myrtillus (Bilberry). Rheumax® contains 4 herbs including Curcuma longa (Turmeric), Boswellia serrata, Tinospora cordifolia, and Vitex negundo. These eight herbs contain 43 major active components, including steroids, terpenes, alkaloids, and, flavonoids [22]. It will be a good idea to screen these components to search for unknown COX-2 inhibitors.
In the race to design and develop new therapeutic molecules with the maximum number of pharmacophoric features, computational techniques have emerged as new tools to minimize the time and resources required for their chemical syntheses, in vitro, and in vivo testing. Computer-aided or in silico drug design (CADD) mainly uses computing power to facilitate and accelerate the process of drug design by selecting the most likely lead candidate for biological testing. The most common approaches of CADD include ligand-based drug design (pharmacophore and quantitative-structure activity relationship (QSAR))and target-based drug design (pharmacophore and molecular docking) [23, 24].
In this study, the Pharmacophore hypothesis and QSAR model were performed for a series of novel cyclic imide derivatives as COX-2 inhibitors. Then, the inhibitory activity towards COX-2 of 43 compounds (in-house library) and zinc “all now” (all purchasable in 2 weeks) database [25] was predicted relying on potent strategy which is the use of pharmacophore fit score (first filter), pIC50 predicted by QSAR model (second filter), docking score and also by compound-enzyme interactions (third filter). The compounds must pass all these filters to be considered as hits recommended for further bio studies. An additional 10-ns MD simulation was performed on the initial structure of the COX-2 complex with the two best compounds resulting from the docking. The root mean square deviations (RMSD) and the radius of gyration (Rg) of the enzyme were calculated to examine the stability of the system (enzyme, water, ions, etc.) [26].
The performance of our pharmacophore and QSAR models was validated by many strict parameters in order to use both of them effectively in virtual screening [27, 28]. We especially emphasize procedures used to define QSAR model applicability domain that should be used when the model is employed for the prediction of external compounds [29, 30]. In other words, we use machine learning to identify already-known chemical compounds as potential novel COX-2 inhibitors that have not yet been recognized as such.
2. Material and methods
2.1. Pharmacophore Model
2.1.1. Data set and pharmacophore modeling
A 3D ligand-based pharmacophore model was created using LigandScout v4.4.1 from five potent cyclic imide compounds collected from the literature. The values of the inhibitory activity of these COX-2 inhibitors ranged from 100 to–360 nM. The structures and experimental biological activities (IC50) of these compounds are listed in Table 1. The three dimensional (3D) structures of all compounds were constructed using Chemdraw. For pharmacophore modeling, LigandScout used espresso algorithm which has many stages including clustering and conformation generation [31].
Table 1.
Structures and their IC50 values of active compounds in pharmacophore model.
| Compound | Structure | IC50 (μm) |
|---|---|---|
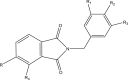 |
||
| 30 | R = NO2 | 0.1 |
| R1, R3 = H | ||
| R2 = SO2NH2 | ||
| R4 = H | ||
| 46 | R = NO2 | 0.18 |
| R1, R2, R3 = OCH3 | ||
| R4 = H | ||
| 47 | R = NO2 | 0.24 |
| R1, R3 = H | ||
| R2 = OCH3 | ||
| R4 = H | ||
| 49 | R1, R2, R3 = OCH3 | 0.28 |
| R, R4 = Cl | ||
| 50 | R1, R3 = H | 0.36 |
| R2 = OCH3 | ||
| R, R4 = Cl |
LigandScout's clustering method divides the ligands into test-set and training-set. The aim of this clustering is to choose compounds that are similar in terms of 3D pharmacophore characteristics and therefore bear a higher chance of delivering a large overlap of chemical features. The 3D clustering algorithm performs fast alignments and clusters based on a similarity value between 0 and 1. Since this algorithm basically performs combinatorial alignments of all conformations of all compounds, a low number of conformations is recommended. The cluster distance can be varied until the desired cluster size is reached. Conformations of the training-set molecules were generated. After ranking the molecules according to their number of conformations (flexibility), pharmacophore features were projected onto these molecules and all their conformations. All conformations of the two top-ranked (i.e. the least flexible) molecules are then aligned using Inte:Ligand's molecular alignment algorithm [32].
2.1.2. Pharmacophore model validation
The validation of a pharmacophore model is considered an important step before its use in virtual screening. The Predictive ability, specificity, and sensitivity of a pharmacophore model are earnest metrics for the reliability of performance. We assessed the predictive ability of the model on a decoy set to determine the accuracy of recognition of active and inactive compounds.
Sensitivity states how good the model correctly classifies compounds and specificity shows how well the model is able to exclude inactive compounds [28]. The Sensitivity (TPR) and specificity (TNR) can be measured using Eqs. (1) and (2), respectively.
| (1) |
| (2) |
Other metrics to validate the pharmacophore model are the receiver operating characteristic (ROC) curve which indicates how well a model can differentiate between active and inactive compounds [31] and the area under the ROC-curve (AUC) [33]. The ROC curve provides the true positive rate plotted against the false positive one of the hits. If the curve was sharp and then flattened, this means that the model ranked the active compounds higher than the inactive ones. The AUC value lies between 0 (bad classifier) when the model ranks all the inactive compounds first and 1 (excellent classifier) on the contrary situation. Hence, to evaluate the performance of the model, a set of 703 inactive compounds was obtained from DUD-E as a decoy set for 5 active and selective COX-2 inhibitors. The decoy and active compounds have similar physicochemical properties but different two dimensional topological ones [34, 35, 36]. Using the idbgen routine included in LigandScout, the compounds were converted into a LigandScout format [52]. In addition to these metrics, the statistical parameters of goodness of hit score (GH) [37], enrichment factor (EF), and accuracy (ACC) were determined to investigate the performance of the model. The formulas are written in Eqs. (3), (4), and (5) below:
| (3) |
| (4) |
| (5) |
Ht is the total number of hits and Ha is the number of active hits. The range of the GH score is (0–1) with a threshold value equal to 0.6 [38]. Ideally, when the model picks all the active compounds with no inactive ones, it will have a steep slope for the ROC curve, a high value of AUC, a high EF value, and the highest value of sensitivity and specificity which is 1 [39].
2.1.3. Pharmacophore-based virtual screening (PBVS)
Virtual screening, based on the best pharmacophore model as a query, was performed using Ligandscout v4.4.1 [40]. A part of the ZINC database “All Now” and a small dataset consisting of 43 active components in some herbal preparations were used for the screening study. The hit compounds were retrieved based on the Pharmacophore Fit Score values and then evaluated based on their drug-likeness properties using Lipinski's RO5 [41].
2.2. QSAR model
2.2.1. Data set
For QSAR studies a series of forty cyclic imide derivatives with their COX-2 reported IC50 values were compiled from recently published studies [42, 43, 44]. Various structures of dataset were selected. No large gaps were allowed between activity values. For QSAR modeling, the negative logarithm pIC50 of the biological activities IC50 was used. The pIC50 values ranged between 4.151 and 7.000 with an average of 5.401 [45]. The data set was divided randomly using MATLAB into a training-set (32 molecules) to build the model and the eight remaining molecules were used to test the performance of the model (test-set) [46]. The structures of the studied molecules and their corresponding experimental biological activities are listed in Table 2.
Table 2.
Structures of compounds used in the QSAR study, and pIC50 values.
| No | pIC50 | pIC50 predicted | |
|---|---|---|---|
| 3–4 | 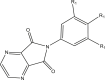 |
||
| 3t | R1 = MeO | 5.283 | 5.53 |
| 4r | R1 = Cl | 4.978 | 5.174 |
| 5–10 |  |
||
| 5r | R1 = MeO, R2 = H | 5.193 | 5.370 |
| 6r | R1 = Cl, R2 = H | 5.040 | 5.051 |
| 7r | R1 = MeO, R2 = 5-NO2 | 7.000 | 6.630 |
| 9r | R1 = MeO, R2 = 5-Me | 6.397 | 5.433 |
| 11t | R1 = MeO, R2 = 5-tert-But | 5.000 | 4.567 |
| 12r | R1 = Cl, R2 = 5-tert-But | 4.684 | 4.571 |
| 13r | R1 = MeO, R2 = 5,6- Dichloro | 4.906 | 5.003 |
| 14r | R1 = Cl, R2 = 5,6-Dichloro | 4.521 | 4.704 |
| 15r | R1 = MeO, R2 = 4,5,6,7-Tetrachloro | 4.347 | 5.064 |
| 18–19 |  |
||
| 18r | R1 = MeO | 6.346 | 6.747 |
| 19r | R1 = Cl | 6.000 | 5.990 |
| 20–21 | 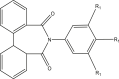 |
||
| 20r | R1 = MeO | 6.346 | 6.251 |
| 21t | R1 = Cl | 6.000 | 5.796 |
| 22–31 | 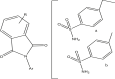 |
||
| 23r | R = H,Ar = b | 4.602 | 4.525 |
| 24t | R = 5-Me, Ar = a | 4.301 | 4.025 |
| 27r | R = 5-tert-But, Ar = b | 4.301 | 5.074 |
| 28t | R = 5,6-Dichloro, Ar = a | 6.397 | 6.299 |
| 30r | R = 5-nitro, Ar = a | 7.000 | 7.221 |
| 31r | R = 5-nitro, Ar = b | 6.522 | 6.239 |
| 32–33 | 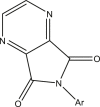 |
||
| 32r | Ar = a | 6.096 | 6.310 |
| 33r | Ar = b | 6.000 | 5.434 |
| 34–35 | 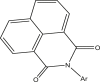 |
||
| 34r | Ar = a | 5.522 | 5.617 |
| 35r | Ar = b | 5.397 | 4.810 |
| 37r | 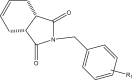 |
4.151 | |
| R1 = 3,4,5-trimethoxy | 4.336 | ||
| 38–52 | 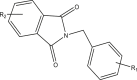 |
||
| 38r | R1 = 3,4,5-trimethoxy | 4.471 | |
| R2 = H | 5.597 | ||
| 41t | R1 = 3,4,5-trimethoxy | 4.619 | |
| R2 = CH3 | 4.289 | ||
| 42r | R1 = 4-methoxy | 4.442 | |
| R2 = CH3 | 4.720 | ||
| 44t | R1 = 3,4,5-trimethoxy | 4.492 | |
| R2 = tert-butyl | 4.295 | ||
| 45r | R1 = 4-methoxy | 4.536 | |
| R2 = tert-butyl | 4.388 | ||
| 46r | R1 = 3,4,5-trimethoxy | 6.744 | |
| R2 = NO2 | 6.500 | ||
| 47t | R1 = 4-methoxy | 6.619 | |
| R2 = NO2 | 6.448 | ||
| 48r | R1 = 4-fluoro | 4.507 | |
| R2 = NO2 | 5.752 | ||
| 50r | R1 = 4-methoxy | 6.443 | |
| R2 = 5,6-Dichloro | 5.408 | ||
| 52r | R1 = 4-methoxy | 5.387 | |
| R2 = 3,4,5,6-tetrachloro | 4.968 | ||
| 53r | 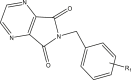 |
5.070 | |
| R1 = 3,4,5-trimethoxy | 4.860 | ||
| 55–56 | 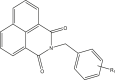 |
||
| 55r | R1 = 3,4,5-trimethoxy | 5.136 | 4.974 |
| 56r | R1 = 4-methoxy | 4.767 | 5.317 |
| 57r |  |
6.522 | 6.148 |
∗r: training, ∗t: test.
2.2.2. Optimization of compounds
The structures of the molecules were assembled using Hyperchem software (version 8.0; Hyperchem, Alberta, Canada) (13). The geometry of the compounds was optimized using Hyperchem. The molecular mechanics force field (MM+) then semi empirical method AM1 were applied [45].
2.2.3. Descriptors generation and features selection
For each compound, 1875 descriptors were calculated using PaDEL software was used to calculate 1875 descriptors [47]. The calculated descriptors which encoding different properties like physiochemical, electronic, and topological properties were analyzed for the existence of constant or near constant variables (standard deviation of 0.1 as a threshold) using MATLAB. The detected ones were then removed. Collinear descriptors (i.e. correlation coefficient between descriptors is greater than 0.9) were detected and the one displaying the most noteworthy correlation with the activity was held and others were expelled from the data. Finally, 191 descriptors remained. SPSS (version 13.0; SPSS Inc., Chicago, IL, USA) (15) statistical software was utilized to select the features that had the minimum number of descriptors (simplicity) and kept good performance [48]. The QSAR equation consisted of the descriptors (X) and the measured pIC50 (Y). To correlate between X and Y, Multiple Linear Regression (MLR) method was used. Different models were generated with different R squares and numbers of descriptors for each model. These values are crucial for the selection of acceptable models. No less than five compounds should be involved in the equation for each descriptor. The performance of MLR model was evaluated by calculating root-mean-square error (RMSE) and from R2 values. RMSE was calculated by Eq. (6):
| (6) |
yi is the wanted output observation, y0 is the predicted value, and ns is the number of the total compounds in the data set. MATLAB was used to do all the calculation for the training and test sets like calculating R2, RMSE, and Q2.
2.2.4. Validation
To confirm the robustness of the model, the dataset was divided into a training-set consists of 32 compounds and test-set (8 compounds) and statistical parameters (Eqs. (7), (8), (9), (10), (11), and (12)), in addition to R2 in accordance with Tropsha et al, Roy and Roy were calculated.
| Q2 > 0.5, | (7) |
| R2 > 0.6, | (8) |
| (9) |
| (10) |
| 0.85 ≤ k ≤ 1.15 and 0.85 ≤ k' ≤ 1.15 | (11) |
| (12) |
The value should be larger than 0.5 [49] to express that the model has good external prediction.
2.2.5. Applicability of domain
The applicability of domain (AD) is widely comprehended in QSAR field to estimate the unreliability and vulnerability in the prediction of a specific molecule based on how similar it is to the compounds used to build the model [50]. In this study, we used the Williams plot to evaluate the AD of our QSAR model. The Williams plot provides leverage values ( ) plotted against the standardized residuals. The leverage hi for each compound, was calculated by Eq. (13) to predict its activity by QSAR model [51]:
| (13) |
Where xi is the descriptor row vector of the query molecule and x is the k ×n matrix where k is the descriptor value for n molecule from the training-set. The diagonal elements in this matrix represent the leverage values (h) for the molecules in the dataset. The warning leverage, h∗, was fixed at 3p/n, where p is the number of descriptors plus one and n is the number of training samples. A molecule with leverage higher than h∗ may refer to unreliable predictions. The developed QSAR model could be used to predict the activity of certain compound only if it was inside the applicability domain as Williams plot showed.
MLR was used to determine the standardized residuals of inhibitory activity. The calculated leverage and standardized residuals values were used for definition the AD. The AD was identified as a square area between ±3 standard deviation and h∗. Leverage values for all of training-set and test-set compounds were calculated. The compound was considered inside of the applicability domain only if had standardized residual <3 standard deviation units and leverage value not exceeding h∗.
2.2.6. Prediction
QSAR is an important part in computational drug design [52]. The QSAR equation was applied to the compounds that we got it by pharmacophore virtual screening on ZINC “all now” database and the 43 herbal components in order to predict their inhibitory activity on the COX-2 enzyme. The expected inhibitory activity must be considered reliable just for inhibitors that fall within the AD pool on which the model was developed. Leverage values and the predicted pIC50 of the retrieved compounds are listed in Table 5.
Table 5.
Pharmacophore fit score and pIC50 predicted by QSAR each tested compound.
| Compounds | Pharmacophore fit score | leverage | AD | pIC50 predicted by QSAR |
|---|---|---|---|---|
| ZINC000029396226 | 96.92 | 0.257 | 1 | 5.850 |
| ZINC000000009029 | 92.25 | 0.258 | 1 | 5.894 |
| ZINC000019851284 | 82 | 0.178 | 1 | 4.009 |
| ZINC000114185151 | 92.93 | 0.162 | 1 | 5.303 |
| ZINC000113253375 | 79.65 | 0.403 | 1 | 7.496 |
| ZINC000043170560 | 86.40 | 0.235 | 1 | 6.789 |
| Astragalin | 59.75 | 0.171 | 1 | 5.233 |
| catechin | 55.76 | 0.072 | 1 | 4.538 |
| apigenin | 42.8 | 0.234 | 1 | 4.350 |
| Curcumin | 53.86 | 0.472 | 1 | 6.324 |
| cyanidin | 52.43 | 0.273 | 1 | 5.585 |
| epicatechin | 62.73 | 0.174 | 1 | 4.112 |
| harpagide | 51.56 | 0.699 | 1 | 1.971 |
| harpagoside | 52.31 | 0.151 | 1 | 0.366 |
| hyperoside | 55.36 | 0.113 | 1 | 4.843 |
| isoquercitrin | 49.93 | 0.273 | 1 | 5.071 |
| luteolin | 52.51 | 0.124 | 1 | 4.467 |
| quercitrin | 63.35 | 0.145 | 1 | 4.725 |
| agnuside | 59.22 | 0.198 | 1 | 4.520 |
| Ferulic acid | 54.29 | 0.195 | 1 | 3.508 |
| eugenol | 44.35 | 0.249 | 1 | 3.091 |
| procumbide | 55.29 | 0.387 | 1 | 2.010 |
| quecetin | 53.16 | 0.082 | 1 | 4.317 |
| celecoxib | 52.52 | 0.144 | 1 | 6.148 |
| rofecoxib | 42.52 | 0.106 | 1 | 5.084 |
| diclofinac | NA | NA | NA | 5.029 |
Ad:1 means the compound is inside the applicability domain of QSAR model.
NA: not available.
Later on, only the compounds that fall inside the AD and pass the QSAR filter will succeed to reach the docking study.
2.3. Molecular docking
All the compounds that achieved both good pharmacophore fit score in pharmacophore-virtual screening and good pIC50 predicted by QSAR model) besides celecoxib, rofecoxib, and diclofenac were docked to COX-2 (PDB code: 5KIR, 2.697 Å) [53]. The Schrodinger suite of software (Maestro, version 121) was used to do all the docking procedure with its calculations and scores.
Molecular docking was performed using the extra-precision (XP) mode of Glide (Grid-based Ligand Docking with Energetics). Glide algorithm searches for ligand orientations, positions, and conformations in the enzyme-binding pocket. The final evaluation of binding energy was done with Glide score (G Score). Glide performance was validated by a re-docking approach. Before docking the nine molecules, the bound ligand found in the X-ray crystal structure was docked back into the binding pocket of COX-2 enzyme. This was done to confirm that Glide could imitate the position and orientation of the inhibitors as observed in the crystal structure.
2.3.1. Ligand preparation
The two-dimensional (2D) structures of the studied inhibitors were sketched using the freeware ACD/ChemSketch version 12.01 (ACD/Labs Release, Canada) [54]. Then, they were converted into the standard structure-data file (SDF) format using the freely available open source toolbox, Open Babel [55].
Energy minimization was conducted using optimized potential for liquid simulations (OPLS3) force field [56, 57] using Lig Prep Module provided by Maestro version 12.1 (Schrödinger, LCC, New York, 2019) [58]. One three-dimensional (3D) conformer that has the lowest energy and correct chirality was generated for each ligand to be used during the docking procedure.
2.3.2. Protein preparation
The 3D structure of COX-2 (PDB code: 5KIR, 2.697 Å) was retrieved from the Protein Data Bank (PDB) and processed using Protein Preparation Wizard from Maestro version 12.1 (Schrödinger, LCC, New York, 2019). Protein preparation was executed for chain A only, while the other chain and extra molecules like water molecules, and arachidonic acid were removed. Steps of protein preparation involve adding explicit hydrogen atoms, assigning bond orders and formal charges, creating disulfide bonds, capping termini, and finding overlaps. The corrected structure was energetically optimized and minimized to relieve any strain and to fine-tune the placement of various groups [59]. Protein-energy minimization was conducted using OPLS3 force field.
2.3.3. Receptor grid generation
Glide Receptor Grid Generation platform as a part of Maestro was employed to create a rigid grid around the interacting residues of the active site from the prepared protein. The default grid box size of 20 Å was used and the size of the inner grid box was changed to (12 × 12 × 12 Å3). The centroid of grid box was located on the ligand. Grids for molecular docking with Glide16 were calculated with a hydrogen bond constraint to Arg 513 of COX-2 [60].
2.3.4. Extra precision (XP) molecular docking
To predict the binding pose of ligands COX-2, extra precision (XP) Glide docking procedure was utilized to dock the ligands into the generated receptor grid. XP refines the predicted docking modes using an anchor-and-grow algorithm to more thoroughly sample ligand degrees of freedom [61, 62]. Glide workflow from Maestro version 12.1 (Schrödinger, LCC, New York, 2019) was used during this step. Ligands were docked flexibly into the rigid docking box using Glide's internal conformation generator and the “Alternate Protocol 2” which is the process of applying constraints in a flexible ligand docking experiment. For each ligand, one docking hit with the lowest Glide docking score was generated and used to analyze docking results [63].
2.4. Molecular dynamics simulation of COX-2
The MD simulations were performed using the GROMACS 2019.1 [64]. The topology parameters of COX-2 were created. The interaction parameters were computed using the charmm36 force field. The system was immersed in a cubic water box (9.906 ∗ 9.906 ∗ 9.90618 nm3) of extended simple point charge (SPC) water molecules. The solvated system was neutralized by adding one chloride ions in the simulation, and the entire system was composed of 8790 atoms of COX-2, 1 Cl- counterions, and 28997 solvent atoms. The energy was minimized using the steepest descent method of 1000 steps. Then, simulations were performed in a constant number of molecules, constant pressure, and constant temperature (NPT) ensemble with coupled temperature and pressure at 310 K and 1 bar, respectively. In the next step, the solute (protein and counterion) was fixed and the position-restrained dynamics simulation of the system, in which the atom positions of COX-2 were restrained at 300 K. The water and the counterion permitted to relax about the protein. The relaxation time of water was 20 ps. Finally, the full system was subjected to 10-ns MD at 300 K temperature and 1 bar pressure. The MD simulation and results analysis were performed on the Ubuntu 18.04.3 LTS Linux on an Intel® Xeon(R) CPU E5-2650 v4 @ 2.20GHz × 24 and 64 GB of RAM.
2.5. Molecular dynamics simulation on COX-2 complex with 375 and 560
The complex of ZINC000113253375 (375) and ZINC000043170560 (560) with COX-2 was chosen for MD simulation. The topology parameters of 375 and 560 were built by the CHARMM General Force Field (CGenFF) program [65], and were embedded to the topology parameters of COX-2. An additional 10-ns MD simulation was performed on the initial structure COX-2 complex with 375 and 560 resulted from the docking. All the simulation processes were performed by the GROMACS 2019.1 and the same as previous section and at the end of simulation results of the complex of 375 and 560 with COX-2 were analyzed.
3. Results and discussions
3.1. Pharmacophore
3.1.1. Pharmacophore modeling
The aim of this study is finding new COX-2 inhibitors by ligand-based pharmacophore modeling. Based on five potent and diverse cyclic imides collected from the literature (mentioned in Table 1 with their biological activities IC50) [42,43], a pharmacophore model was created. The pharmacophore model of the most active compound is displayed in Figure 1. The eleven features of the generated pharmacophore were two aromatic (R) showed as blue sphere, one hydrophobic (H) colored by yellow sphere, one H-bond donors (D) showed as green sphere, six H-bond acceptors (A) showed as red sphere, one positive charge (P) colored by blue lines. The Pharmacophore Fit Score values are used to measure the overlapping between the features of pharmacophore and chemical functionalities of the compound and listed in Table 3. The values of Pharmacophore Fit Score of the compounds in training-set are high (103.87–106.94) and indicate good mapping with the model. Accordingly, the model could be a hopeful query in virtual screening in order to find promising active hits so screening decoy and active sets was done to evaluate the pharmacophore model.
Figure 1.

Mapping the most active training compound on the model in 2D (a) and 3D (b).
Table 3.
The Pharmacophore-Fit Score values of the training and test compounds.
| Compounds | COX-2 IC50 μM | Pharmacophore- Fit score | Matched Feature Pairs |
|---|---|---|---|
| 30 | 0.1 | 106.943 | 11 |
| 46 | 0.18 | 100.663 | 10 |
| 47 | 0.24 | 103.873 | 10 |
| 49t | 0.28 | 71.428 | 7 |
| 50t | 0.36 | 73.634 | 7 |
| Celecoxib | 0.3 | 52.520 | 6 |
t: test compound.
3.1.2. Pharmacophore model validation
Two datasets including five COX-2 inhibitors and 703 inactive compounds were used to validate the model. The values of enrichment factor (EF) and goodness of hit score (GH) refer to the good ability of the model to identify active compounds. The receiver operating characteristic (ROC) curve of the pharmacophore model is shown in Figure 2. The retrieved hits by the model are 3 and the area under the ROC curve (AUC) value is 0.8%. The pAUC (partial area under the curve) values are 1.00, 1.00, and 1.00 at 1%, 5% and 10% of the screened database respectively. Also, the model is sensitive because it retrieved 3 active compounds from 5 ones (60% of the total active compounds) and very specific because it selected no decoys. This high selectivity and sensitivity indicates that our pharmacophore is a very good filter for recognizing COX-2 inhibitors [27]. All the parameters of validation process are shown in Table 4 and state good quality of the pharmacophore model so it can be used successfully in virtual screening.
Figure 2.

ROC plot of the pharmacophore model.
Table 4.
Statistical factors of the best pharmacophore model.
| Features | AAAAAAHDRRP |
|---|---|
| Ha | 3 |
| Ht | 3 |
| A | 5 |
| D | 703 |
| TPR | 60% |
| FPR | 0% |
| AUC | 0.8 |
| ACC | 0.997 |
| Sensitivity | 0.6 |
| specificity | 1 |
| GH | 0.6 |
| EF | 141.6 |
Features: A: H-bond acceptors, H: hydrophobic groups, D: H-bond donors, R: aromatic ring, P: positive charge.
Ha: active hits, Ht: total hits, A: total number of actives in the dataset, D: total number of decoys in the dataset, TP: true positive, FP: false positive, EF: enrichment factor, GH: goodness of hit, AUC: area under ROC curve, ACC: accuracy.
3.1.3. Pharmacophore-based virtual screening (PBVS)
The most active training compound (30), which has high Pharmacophore Fit Score (106.94) mapped on the model in Figure 1. Virtual screening was done to identify new inhibitors of COX-2 by the validated pharmacophore model. ZINC “all now” database containing of 5002 molecules were converted into a LigandScout format using (idbgen) function. Six molecules of ZINC database were selected by screening using the model. The values of Pharmacophore Fit Score of the hits ranged from 79.65 to 96.92. In addition, small herbal components data contained 43 compounds was exposed to all mentioned steps. Table 5 shows the pharmacophore fit scores of all the retrieved hits. 17 active components have had pharmacophore fit scores as good as the pharmacophore fit score of celecoxib and/or rofecoxib. These 23 compounds obey lipinski's RO5 and will be prepared to pass our QSAR model and its applicability domain filter in the next step. The COX inhibitors in the studied herbs might be responsible, at least in part, for the anti-inflammatory activity of the studied medicinal plants.
3.2. QSAR study
3.2.1. QSAR model
The best QSAR model was built using 7 descriptors such as extended topochemical atom, information content, auto correlation, RDF, BCUT, barysz matrix, atom type electrotopological state and molecular linear free energy relation. For the selection of the most important descriptors, stepwise MLR method was used.
The MLR analysis with a stepwise selection was carried out to relate the pIC50 to a 7 set of descriptors. The SPSS software (version 13.0; SPSS Inc., Chicago, IL, USA) (15) was used for the MLR analysis). It is defined by Eq. (14):
| pIC50 = -6.2128 (±2.496) +0.1658 (±0.038) ETA-beta +2.2784 (±0.491) IC1+0.9179 (±0.239) GATS8m –0.0017 (±0.000) VR1_Dzs +0.1979 (±0.056) nHdsCH -0.0402 (±0.011) RDF70s -0.3436 (±0.159) BCUTp-1l | (14) |
The built model produced good results for the training-set and the test-set. The 7 descriptors with their contribution to the model and their statistical parameters are shown in Table 6.
Table 6.
Name, type, and meaning of descriptors with their coefficient, error, and contribution to the model.
| Name | Type of descriptor | Meaning | Coefficient | Error | contribution |
|---|---|---|---|---|---|
| ETA-beta | Extended topochemical atom | A measure of electronic features of the molecule | 0.1658 | 0.038 | + |
| IC1 | Information content | Information content index (neighborhood symmetry of 1-order) | 2.2784 | 0.491 | + |
| GATS8m | Autocorrelation | Geary autocorrelation - lag 8/weighted by mass | 0.9179 | 0.239 | + |
| VR1_Dzs | Barysz matrix | Coefficient sum of the last eigenvector from Barysz matrix/weighted by I-state | 0.0017 | 0.000 | - |
| nHdsCH | Atom type electrotopological state | Count of atom-type H E-State: =CH- | 0.1979 | 0.056 | + |
| RDF70s | RDF | Radial distribution function - 070/weighted by relative I-state | 0.0402 | 0.011 | - |
| BCUTp-1l | BCUT | nhigh lowest polarizability weighted BCUTS | 0.3436 | 0.159 | - |
| Intercept | 6.2128 | 2.496 | - |
3.2.2. Validation results
Results show that this model has a cross-validated correlation coefficient (Q2) value higher than 0.5 (0.8407). Q2 is important parameter but not enough to judge the power of the model. Indeed, the true predictive power of a QSAR model can be established only through model validation procedure which consists of prediction of activities of compounds in test set (i.e. not included in model building). A strong validation should be performed to insure good reliable, prediction, and conclusion of the QSAR equation. This implies quantitative evaluation and a set of statistical criteria. Such as R2 which is a measure of the degree of the fit between the predicted outcomes and the experimental ones. Other statistical parameters were performed to insure good quality of the model [29, 30].
R2 (calculated for the test set) value larger than 0.6. R20 is the correlation coefficient for regressions between predicted against experimental values through the origin. R′20 is the correlation coefficient for regressions between experimental against predicted values through the origin. an additional parameter was specified as a worthy parameter of the external prediction. The value of here is 0.8778 (larger than 0.5) and that means the model has good external prediction [49].
The values of Slopes (k and k') of regression lines through the origin are very close to 1 and fall in the acceptable range between 0.85 and 1.15. It could be seen from the results that all criteria were satisfied thus giving power and trust for the developed model. Statistical parameters of the external test set for the MLR model are given in Table 7. The predicted pIC50 values versus their experimental values were plotted in Figure 3 for the training-set and test-set.
Table 7.
Statistical parameters of the test set.
| Parameter | MLR |
|---|---|
| Q2 | 0.8407 |
| R2 | 0.9605 |
| R02 | 0.9531 |
| R0|2 | 0.9415 |
| 0.0077 | |
| 0.0197 | |
| R2m | 0.8778 |
| R/2m | 0.8281 |
| K | 0.9694 |
| K/ | 1.0301 |
Figure 3.

Plot of predicted pIC50 versus their experimental values.
3.2.3. Applicability of domain
The warning leverage, h∗, was fixed at 3p/n (h∗ = 0.75) in this study, where p is the number of descriptors plus one and n is the number of training samples.
The applicability domain (AD) was defined as a square region between leverage threshold h∗ of 0.75 and ±3 standard deviation. Leverage values for all compounds in training-set and test-set were calculated (Figure 4). As mentioned before, the compound was considered inside of the AD only if it had leverage value ≤0.75 and standardized residual <3 standard deviation units. It can be concluded from the plot that all compounds of training-set and the test-set are situated inside the assigned domain. One of the inhibitors with response over the threshold of ±3 standard deviation unit of standardized residual.
Figure 4.

William plot for standardized residual versus leverage.
Concerning the previously conditions, the suggested model can be used with a high level of confidence right now.
3.2.4. Prediction
Twenty-three molecules from ZINC database and our in-house library were selected by virtual screening using the pharmacophore model. Then only nine compounds from those twenty-three passed our QSAR model and its applicability domain filter. These nine compounds were ZINC000029396226, ZINC000000009029, ZINC000114185151, ZINC000113253375, ZINC000043170560, Astragalin, Curcumin, cyanidin and isoquercitrin. Curcumin is the main bioactive compound isolated from rhizomes of curcuma longa. Isoquercitrin and astragalin, naturally occurring flavonoids, have been identified in a variety of medicinal plants such as the eight herbs in Voltarit® and Rheumax®. Cyanidin is the most commonly anthocyanin that identified in Vitex negundo (Rheumax®), Billberry and Hawthorn berries (Voltarit®) [22]. The pIC50 of all tested compounds listed before in Table.5.
3.3. Docking results and discussion
Finally, to confirm the discovery of new lead compounds, we finished with the docking study of the compounds retrieved from ZINC and herbal data to choose the best hits that have the best glide docking score. For validation the reliability of docking, the heavy-atom root mean squared deviation (RMSD) value was determined between the crystal ligand and re-docked ligand using Schrodinger. The value of RMSD equal to 0.5 Å (no more than 2 Å) and that reveal good agreement between the experimental and predicted binding pose [66]. The hits that showed good pharmacophore score with good predicted pIC50 from QSAR model (5 compounds from ZINC and 4 compounds from herbal data that are listed in Table 8) were exposed to docking with the 3D structure of COX-2 (PDB code: 5KIR, 2.697 Å) by GLIDE.
Table 8.
Types of interactions of the hits, celecoxib and rofecoxib with the binding site of COX-2.
| Compound | Glide docking score (kcal/mol) | Interaction type with Arg513 | Distance Aº | Hydrophobic interactions | Hydrogen bonding with residues |
|---|---|---|---|---|---|
| ZINC000029396226 | -7.956 | H-bond and positive charge | 2.347 | VAL523, TYR 385, TYR 348, ILE 517, ALA 516, PHE 518, | ARG 513 PHE 518 HIE90 |
| ZINC000000009029 | -8.715 | H-bond and positive charge | 2.237 | VAL523, TYR 385, TYR 348, ILE 517, TYR355, PHE 518, | ARG 513 |
| ZINC000114185151 | -7.279 | H-bond and positive charge | 2.287 | VAL523, TYR 385, TYR 348, ILE 517, ALA 516, PHE 518, VAL349, ALA 527 | ARG 513 PHE 518 |
| ZINC000113253375 | -9.293 | positive charge | 2.761 | VAL523, TYR 385, TYR 348, ILE 517, ALA 516, PHE 518, VAL349, ALA 527, LEU 352 | PHE 518 |
| ZINC000043170560 | -9.764 | positive charge | 2.282 | VAL 116, TYR385, TYR348, TYR355, LEU 352, PHE 381, PHE518 | - |
| Astragalin | -9.185 | H-bond and positive charge | 2.172 | TYR385, TYR348, TYR355, VAL 116, VAL 349, PHE 518, ALA 516, LEU352, LEU384, VAL 523 | ARG513 PHE 518 GLN 192 |
| Curcumin | -9.096 | H-bond and positive charge | 2.417 | VAL523, TYR355, ALA516, VAL 116, VAL 349, ILE 517, LEU 352, MET 113 | ARG513 TYR355 |
| Cyanidin | -6.360 | positive charge | 2.179 | VAL 116, VAL 523, PHE 518, ILE 517, ALA 516, TYR 355, VAL 349, LEU359, LEU352 | PHE 518 |
| Isoquercitrin | -7.470 | positive charge | 2.063 | VAL 523, TYR385, TYR348, TYR355, LEU384, LEU 359, PHE 518, VAL349, ALA 516, VAL 116 | ARG513 SER530 |
| Celecoxib | -10.452 | H-bond and positive charge | 2.474 | VAL349, PHE 518, TYR385, TYR 355, LEU352, VAL 116, ALA 516 | ARG 513 PHE 518 GLU192 |
| Rofecoxib | -9.734 | H-bond and positive charge | 2.192 | VAL 523, TYR385, TYR348, TYR355, LEU384, LEU 359, PHE 518, ILE 517, ALA 516 | ARG513 |
| Diclofinac | -7.839 | H-bond | TYR385, TYR348, LEU 531, VAL 523, TYR355 | SER 530 |
The structure of the catalytic domain, which is the largest region in COX enzyme, is similar between COX-1 and COX-2 (RMS deviation of 0.4 A°). The Arg120, Tyr355, and Glu524 residues form the entrance of the active site while we find Tyr385 and Tyr348 in the apex of the active site. The main differences between both active sites are at the positions 513, 434 and 523. In other words, Ile434 and Ile523 in COX-1 are replaced by valine residues in COX-2. The hydrophobic His513 in COX-1 was replaced by the positively charged arginine in COX-2. These differences are the key points of selective inhibitors [67, 68]. The residue 513 plays a vital role for ligands to distinguish the structural difference between the active sites of COX isoforms [69]. The reason mentioned above elucidates one way in which many hit compounds of the present study selectively inhibits COX-2.
The extra precision glide docking scores for the hits ranged from -7.279 to -9.764. The glide docking score for sodium diclofenac, rofecoxib and celecoxib and as a reference potent COX-2 inhibitors were -7.839, -9.735 and -10.452 respectively. Analysis of the binding poses disclosed that the compounds oriented in the COX-2 binding cavity in a similar way; i.e. binding with residues from both the membrane-binding domain (MBD) and the catalytic domain as summarized in Table 8. Moreover, binding modes of all studied compounds were close to that of the selective COX-2 inhibitors rofecoxib and celecoxib, especially the interaction with Arg513 which is essential for selective COX-2 inhibition [70]. However, celecoxib binds to Arg513 through positive charge interaction between the sulfonamide and guanidinium. The hits interact with Arg513 via positive charge and/or hydrogen bond. Access of celecoxib and hits to the adjunct pocket was through hydrophobic interaction with Tyr91. Other types of interactions like polar, hydrogen bonding, negative/positive charges, etc. with other residues in the binding site were also involved in the binding pose and will be explained for only the significant COX-2 inhibitors [71]. In the side pocket of the COX-2 channel, the methyl sulfone moiety of rofecoxib binds to it and the phenyl ring reaches the side chain of Tyr385. Rofecoxib contacts COX-2 channel's residues 42 times. All the interactions are hydrophobic ones and only one hydrophilic placed between O atoms of the methyl sulfone moiety of the inhibitor and the side-chain N atoms of His90 and Arg513 found in the base of the side pocket [72].
ZINC000043170560, ZINC000113253375 and Astragalin (as a plant component) had the lowest docking score amongst the hits as shown in Table 8 suggesting good binding poses and stable ligand-enzyme complexes. Figure 5 shows that ZINC000113253375 binds to the residues His90, Phe518 and Arg513 sited at the base of the side pocket (polar interaction with His90, hydrogen bond and hydrophobic interaction with Ph518, positive charge with Arg513). Figure 6 shows that Astragalin also binds to residues His90, Phe518 and Arg513 (polar interaction with His90, hydrogen bond and hydrophobic interaction with Ph518, positive charge and hydrogen bond with Arg513). They also bind to the hydrophobic side pocket contained of residues Leu352, Ser353, Ile517, and Phe518 (polar interaction with Ser353 and hydrophobic ones with the rest residues). We also see the hydrophobic interaction with residues Tyr355 lie at the mouth of the COX active site, and the catalytic Tyr385 located at the apex of the hydrophobic channel.
Figure 5.

Schematic representation of the interactions between compound ZINC000113253375 and COX-2 active site.
Figure 6.

Schematic representation of the interactions between Astragalin and COX-2active site.
Astragalin binds to the residue Gln192 that contributes to the outer shell of the side pocket with polar interaction and hydrogen bond and also binds to Tyr385 with hydrogen bond [71, 72].
3.4. Molecular dynamics simulation
The stability of the system (enzyme, water, ions, etc.) was examined by the calculation of root mean square deviations (RMSD) and the radius of gyration (Rg) of the enzyme with respect to its initial structure.
Figure 7 shows the time history of RMSD for COX-2, COX-2-375 and COX-2-560. Analysis of this figure indicates that the RMSD of these systems reaches equilibration and oscillates around in average value after 3.5 ns. The RMSD values indicated that conformation of COX-2 has been equilibrated after 3.5 ns in a water environment. Finally Rg values of COX-2, COX-2-375 and COX-2-560, plotted in Figure 8.
Figure 7.

RMSD values of protein backbone for COX-2 and COX-2 complex with compound 375 and 560 during 10 ns MD simulation.
Figure 8.

Time evolution of the radius of gyration (Rg) for COX-2 and COX-2 complex with compound 375 and 560 during 10-ns of MD simulation.
The Rg results clearly indicate that the conformation of the COX-2 in the presence of compound 375 and 560 has not changed and these complexes are stable.
4. Conclusions
Computational studies were carried out for the search of novel COX-2 inhibitors as new lead compounds from ZINC database and in-house natural product library. Firstly, both of these libraries were screened by validated 3D pharmacophore model. Then, QSAR model was constructed with a verified cross-validated and test-validated high degree of accuracy. The applicability domain of this model was determined. Finally, all hits that passed our pharmacophore and QSAR filters were docked to COX-2 3D structure using Schrodinger as a final filtering. Only those hits that obtain high enough scores in docking and desired ligands enzyme interactions were selected. The MD simulation study showed that the complexes of the COX-2 with the best two hits were stable.
The final results suggested nine novel lead compounds as COX-2 inhibitors that passed all filtering steps. Five compounds from the ZINC database are ZINC000029396226, ZINC000000009029, ZINC000043170560, ZINC000114185151, and ZINC000113253375. Four compounds from the in-house library are Astragalin, Curcumin, cyanidin, and isoquercitrin. Previous studies demonstrated the inhibitory activity of COX-2 by ZINC000029396226, ZINC000000009029, Curcumin, cyanidin, and isoquercitrin [73, 74, 75, 76, 77, 78, 79].
The inhibitory activity of COX-2 of the compounds ZINC000043170560, ZINC000113253375, ZINC000114185151 and Astragalin (the direct inhibition of COX-2 not through mediators) have not been reported before. We find our contribution significant for future in vitro studies of these compounds or their derivatives.
Declarations
Author contribution statement
Nathalie Moussa: Performed the experiments; Analyzed and interpreted the data; Wrote the paper.
Ahmad Hassan; Sajjad Gharaghani: Conceived and designed the experiments; Analyzed and interpreted the data.
Funding statement
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Data availability statement
Data will be made available on request.
Declaration of interests statement
The authors declare no conflict of interest.
Additional information
No additional information is available for this paper.
Acknowledgements
We thank University of Vienna for providing an opportunity to learn many approaches and techniques used in computational drug design during EUROPIN Summer School on Drug Design 2019.
Contributor Information
Nathalie Moussa, Email: nathalie86.moussa@damascusuniversity.edu.sy.
Ahmad Hassan, Email: aha226@gmail.com.
References
- 1.Abouzid K., Frohberg P., Lehmann J., Decker M. 6-Aryl-4-oxohexanoic acids: synthesis, effects on eicosanoid biosynthesis, and anti-inflammatory in vivo-activities. Med. Chem. 2007;3:433–438. doi: 10.2174/157340607781745393. [DOI] [PubMed] [Google Scholar]
- 2.Ulbrich H., Fiebich B., Dannhardt G. Cyclooxygenase-1/2 (COX-1/COX-2) and 5-lipoxygenase (5-LOX) inhibitors of the 6,7-diaryl-2,3-1H-dihydropyrrolizine type. ChemInform. 2003;34(20):953–959. doi: 10.1016/s0223-5234(02)01418-6. [DOI] [PubMed] [Google Scholar]
- 3.Dannhardt G., Laufer S. Structural approaches to explain the selectivity of COX-2 inhibitors: is there a common pharmacophore? Curr. Med. Chem. 2000;7(11):1101–1112. doi: 10.2174/0929867003374237. [DOI] [PubMed] [Google Scholar]
- 4.Kim H.-J., Chae C.H., Yi K.Y., Park K.-L., Yoo S. Computational studies of COX-2 inhibitors: 3D-QSAR and docking. Bioorg. Med. Chem. 2004;12(7):1629–1641. doi: 10.1016/j.bmc.2004.01.027. [DOI] [PubMed] [Google Scholar]
- 5.Almansa C., de Arriba A.F., Cavalcanti F.L., Gómez L.A., Miralles A., Merlos M., Forn J. Synthesis and SAR of a new series of COX-2-selective inhibitors: pyrazolo [1,5-a] pyrimidines. J. Med. Chem. 2001;44(3):350–361. doi: 10.1021/jm0009383. [DOI] [PubMed] [Google Scholar]
- 6.Tsujii M., Kawano S., Tsuji S., Sawaoka H., Hori M., DuBois R.N. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93(5):705–716. doi: 10.1016/s0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- 7.Tsujii M., Kawano S., DuBois R.N. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. PNAS USA. 1997;94(7):3336–3340. doi: 10.1073/pnas.94.7.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asanuma M., Miyazaki I. Nonsteriodal anti-inflammatory drugs in experimental Parkinsonian models and Parkinson’s disease. Curr. Pharmaceut. Des. 2008;14:1428–1434. doi: 10.2174/138161208784480153. [DOI] [PubMed] [Google Scholar]
- 9.Breitner J.C., Zandi P.P. Do nonsteroidal anti inflammatory drugs reduce the risk of Alzheimer's disease? N. Engl. J. Med. 2001;345:1567–1568. doi: 10.1056/NEJM200111223452110. [DOI] [PubMed] [Google Scholar]
- 10.McGeer P.L., Rogers J., McGeer E.G. Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J. Alzheimers Dis. 2006;9:271–276. doi: 10.3233/jad-2006-9s330. [DOI] [PubMed] [Google Scholar]
- 11.Abu-Hashem Al-Hussain, Zaki Synthesis of novel benzodifuranyl; 1,3,5-triazines; 1,3,5-oxadiazepines; and thiazolopyrimidines derived from visnaginone and khellinone as anti-inflammatory and analgesic agents. Molecules. 2020;25(1):220. doi: 10.3390/molecules25010220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y., Zheng Y., Shi W., Guo Y., Xu T., Li Z.…Li J. Design, synthesis and investigation of the potential anti-inflammatory activity of 7-O-amide hesperetin derivatives. Molecules. 2019;24(20):3663. doi: 10.3390/molecules24203663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maghraby M.T.-E., Abou-Ghadir O.M.F., Abdel-Moty S.G., Ali A.Y., Salem O.I.A. Novel class of benzimidazole-thiazole hybrids: the privileged scaffolds of potent anti-inflammatory activity with dual inhibition of cyclooxygenase and 15-lipoxygenase enzymes. Bioorg. Med. Chem. 2020;28(7):115403. doi: 10.1016/j.bmc.2020.115403. [DOI] [PubMed] [Google Scholar]
- 14.Hanafy Metwally N., Said Mohamed M. New imidazolone derivatives comprising a benzoate or sulfonamide moiety as anti-inflammatory and antibacterial inhibitors: design, synthesis, selective COX-2, DHFR and molecular-modeling study. Biol. Chem. 2019;99(5):103438. doi: 10.1016/j.bioorg.2019.103438. [DOI] [PubMed] [Google Scholar]
- 15.Khan A., Diwan A., Thabet H.K., Imran M. Synthesis of novel N-substitutedphenyl-6-oxo-3-phenylpyridazine derivatives as cyclooxygenase-2 inhibitors. Drug Dev. Res. 2020 doi: 10.1002/ddr.21655. [DOI] [PubMed] [Google Scholar]
- 16.Arefi H., Naderi N., Shemirani A.B.I., Kiani Falavarjani M., Azami Movahed M., Zarghi A. Design, synthesis, and biological evaluation of new 1,4-diarylazetidin-2-one derivatives (β-lactams) as selective cyclooxygenase-2 inhibitors. Archiv Der Pharmazie. 2020;353(3):1900293. doi: 10.1002/ardp.201900293. [DOI] [PubMed] [Google Scholar]
- 17.Venugopala Al-Attraqchi, Tratrat Nayak, Morsy Aldhubiab.…Odhav Novel series of methyl 3-(substituted benzoyl)-7-substituted-2-phenylindolizine-1-carboxylates as promising anti-inflammatory agents: molecular modeling studies. Biomolecules. 2019;9(11):661. doi: 10.3390/biom9110661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao H., Yu R., Choi Y., Ma Z.Z., Zhang H., Xiang W., Lee D.Y.W., Berman B.M., Moudgil K.D., Fong H.H.S. Discovery of cyclooxygenase inhibitors from medicinal plants used to treat inflammation. Pharmacol. Res. 2010;61:519–524. doi: 10.1016/j.phrs.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barnes P.M., Powell-Griner E., McFann K., Nahin R.L. Complementary and alternative medicine use among adults: United States, 2002. Adv. Data. 2004;343:1–19. [PubMed] [Google Scholar]
- 20.Tringali C. 2rd ed. Taylor & Francis; London, England: 2001. Bioactive Compounds from Natural Sources; p. 98. [Google Scholar]
- 21.Newman D.J., Cragg G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 22.Lesley B., Marc C. 3ed ed. Churchill Livingstone; Australia: 2010. Herbs and Natural Supplements an Evidence-Based Guide. [Google Scholar]
- 23.Kapetanovic I.M. Computer-aided drug discovery and development (CADDD): in silico-chemico-biological approach. Chem. Biol. 2008;171(2):165–176. doi: 10.1016/j.cbi.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marshall G.R. Computer-aided drug design. Annu. Rev. Pharmacol. Toxicol. 1987;27(1):193–213. doi: 10.1146/annurev.pa.27.040187.001205. [DOI] [PubMed] [Google Scholar]
- 25.Irwin S., Mysinger B., Coleman ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012;52(7):1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin X., Li X., Lin X. A review on applications of computational methods in drug screening and design. Molecules. 2020;25(6):1375. doi: 10.3390/molecules25061375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fawcett T. An introduction to ROC analysis. Pattern Recogn. Lett. 2006;27(8):861–874. [Google Scholar]
- 28.Elmira N., Sajjad Gh. Toward a hierarchical virtual screening and toxicity risk analysis for identifying novel CA XII inhibitors. Biosystems. 2017;162:35–43. doi: 10.1016/j.biosystems.2017.09.005. [DOI] [PubMed] [Google Scholar]
- 29.Tropsha A., Gramatica P., Gombar V.K. The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb. Sci. 2003;22:69–77. [Google Scholar]
- 30.Golbraikh A., Tropsha A. Beware of q2. J. Mol. Graph. Model. 2002;20:269–276. doi: 10.1016/s1093-3263(01)00123-1. [DOI] [PubMed] [Google Scholar]
- 31.Vuorinen A., Schuster D. Methods for generating and applying pharmacophore models as virtual screening filters and for bioactivity profiling. Methods. 2015;71:113–134. doi: 10.1016/j.ymeth.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 32.Gerhard W., LigandScout Thierry L. 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005;45(1):160–169. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- 33.Ambure P., Kar S., Roy K. Pharmacophore mapping-based virtual screening followed by molecular docking studies in search of potential acetylcholinesterase inhibitors as anti-Alzheimer’s agents. Biosystems. 2014;116:10–20. doi: 10.1016/j.biosystems.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 34.Mysinger M.M., Carchia M., Irwin J.J., Shoichet B.K. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 2012;55(14):6582–6594. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang N., Shoichet B.K., Irwin J.J. Benchmarking sets for molecular docking. J. Med. Chem. 2006;49(23):6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karaboga A.S., Planesas J.M., Petronin F., Teixidó J., Souchet M., Pérez-Nueno V.I. Highly specific and sensitive pharmacophore model for identifying CXCR4 antagonists. Comparison with docking and shape-matching virtual screening performance. J. Chem. Inf. Model. 2013;53(5):1043–1056. doi: 10.1021/ci400037y. [DOI] [PubMed] [Google Scholar]
- 37.Seal A., Yogeeswari P., Sriram D., Consortium O., Wild D.J. Enhanced ranking of PknB Inhibitors using data fusion methods. J. Cheminf. 2013;5(1):2. doi: 10.1186/1758-2946-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chitranshi N., Chitranshi N., Gupta S., Tripathi P.K., Seth P.K. New molecular scaffolds for the design of Alzheimer’s acetylcholinesterase inhibitors identified using ligand-and receptor-based virtual screening. Med. Chem. Res. 2013;22(5):2328–2345. [Google Scholar]
- 39.Lu S.H., Lu Shin Hua, Wu Josephine W., Hsuan Liang L., Jian Hua Z., Kung Tien L. The discovery of potential acetylcholinesterase inhibitors: acombination of pharmacophore modeling, virtual screening, and molecular docking studies. J. Biomed. Sci. 2011;18(1):1. doi: 10.1186/1423-0127-18-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Z. Pharmacophore-based virtual screening versus docking-based virtual screening: a benchmark comparison against eight targets. Acta Pharmacol. Sin. 2009;30(12):1694–1708. doi: 10.1038/aps.2009.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lipinski C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods. 2000;44(1):235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 42.Al-Suwaidan I.A., Alanazi A.M., El-Azab A.S., Al-Obaid A.M., ElTahir K.E.H., Maarouf A.R., Abdel-Aziz A.A.-M. Molecular design, synthesis and biological evaluation of cyclic imides bearing benzene sulfonamide fragment as potential COX-2 inhibitors. Part 2. Bioorg. Med. Chem. Lett. 2013;23(9):2601–2605. doi: 10.1016/j.bmcl.2013.02.107. [DOI] [PubMed] [Google Scholar]
- 43.Alanazi A.M., El-Azab A.S., Al-Suwaidan I.A., ElTahir K.E.H., Asiri Y.A., Abdel-Aziz N.I., Abdel-Aziz A.A.-M. Structure-based design of phthalimide derivatives as potential cyclooxygenase-2 (COX-2) inhibitors: anti-inflammatory and analgesic activities. Eur. J. Med. Chem. 2015;92:115–123. doi: 10.1016/j.ejmech.2014.12.039. [DOI] [PubMed] [Google Scholar]
- 44.Abdel-Aziz A.A.-M., ElTahir K.E.H., Asiri Y.A. Synthesis, anti-inflammatory activity and COX-1/COX-2 inhibition of novel substituted cyclic imides. Part 1: molecular docking study. Eur. J. Med. Chem. 2011;46(5):1648–1655. doi: 10.1016/j.ejmech.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 45.Fatemi M.H., Gharaghani S. A novel QSAR model for prediction of apoptosis-inducing activity of 4-aryl-4-H-chromenes based on support vector machine. Bioorg. Med. Chem. 2007;15(24):7746–7754. doi: 10.1016/j.bmc.2007.08.057. [DOI] [PubMed] [Google Scholar]
- 46.MATLAB. MATLAB, Version 7. The MathWorks, Inc., Natick, MA.
- 47.Todeschini, R milano chemometrics and QSPR group. 2000. http://www.disat.unimib.it/vhml Available from: URL:
- 48.SPSS for windows, statistical package for IBM PC, SPSS Inc. http://www.spss.com Available from: URL:
- 49.Roy P., Roy K. On some aspects of variable selection for partial least squares regression models. QSAR Comb. Sci. 2008;27:302–313. [Google Scholar]
- 50.Gharaghani S., Khayamian T., Ebrahimi M. Molecular dynamics simulation study and molecular docking descriptors in structure-based QSAR on acetylcholinesterase (AChE) inhibitors. SAR QSAR Environ. Res. 2013;24(9):773–794. doi: 10.1080/1062936X.2013.792877. [DOI] [PubMed] [Google Scholar]
- 51.Liu R., Sun H., So S.S. Development of quantitative structure-property relationship models for early ADME evaluation in drug discovery. 2. Blood-brain barrier penetration. J. Chem. Inf. Comput. Sci. 2001;41:1623–1632. doi: 10.1021/ci010290i. [DOI] [PubMed] [Google Scholar]
- 52.Tropsha A., Golbraikh A. Predictive QSAR modeling workflow model applicability domains and virtual screening. Curr. Pharmaceut. Des. 2007;13:3494–3504. doi: 10.2174/138161207782794257. [DOI] [PubMed] [Google Scholar]
- 53.Lyne P.D., Lamb M.L., Saeh J.C. Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J. Med. Chem. 2006;49(16):4805–4808. doi: 10.1021/jm060522a. [DOI] [PubMed] [Google Scholar]
- 54.CD/ChemSketch freeware version, ACD/labs Release 12.0, product Version12.01. 10 Feb 2009. http://www.acdlabs.com
- 55.Open Babel. http://openbabel.org
- 56.Harder E., Damm W., Maple J., Wu C., Reboul M., Xiang J.Y. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theor. Comput. 2016;12(1):281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- 57.Storer J.W., Giesen D.J., Cramer C.J., Truhlar D.G. Class IV charge models: a new semi empirical approach in quantum chemistry. J. Comput. Aided Mol. Des. 1995;9(1):87–110. doi: 10.1007/BF00117280. [DOI] [PubMed] [Google Scholar]
- 58.Maestro 9.9. Schrödinger, LLC.; New York: 2019. https://www.schrodinger.com/maestro [Google Scholar]
- 59.Madhavi S.G., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27(3):221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 60.Du J., Sun H., Xi L., Li J., Yang Y., Liu H. Molecular modeling study of checkpoint kinase 1 inhibitors by multiple docking strategies and prime/MMGBSA calculation. J. Comput. Chem. 2011;32(13):2800–2809. doi: 10.1002/jcc.21859. [DOI] [PubMed] [Google Scholar]
- 61.Halgren T.A., Murphy R.B., Friesner R.A., Beard H.S., Frye L.L., Pollard W.T., Banks J.L. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004;47(7):1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 62.Halgren T.A. Extra precision glide: docking and dcoring incorporating amodel of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 63.Friesner R.A., Banks J.L., Murphy R.B., Halgren T.A., Klicic J.J., Mainz D.T., Shenkin P.S. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 64.https://zenodo.org/record/2564764
- 65.Vanommeslaeghe K., MacKerell A.D. Automation of the CHARMM general force field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model. 2012;52(12):3144–3154. doi: 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Esposito E.X. Docking of sulfonamides to carbonic anhydrase II and IV. J. Mol. Graph. Model. 2000;18(3):283–289. doi: 10.1016/s1093-3263(00)00040-1. [DOI] [PubMed] [Google Scholar]
- 67.Luong C., Miller A., Barnett J., Chow J., Ramesha C., Browner M.F. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat. Struct. Mol. Biol. 1996;3(11):927–933. doi: 10.1038/nsb1196-927. [DOI] [PubMed] [Google Scholar]
- 68.Rimon G., Sidhu R.S., Lauver D.A., Lee J.Y., Sharma N.P., Yuan C. Coxibs interfere with the action of aspirin by binding tightly to one monomer of cyclooxygenase-1. Proc. Natl. Acad. Sci. Unit. States Am. 2010;107(1):28–33. doi: 10.1073/pnas.0909765106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ferreira L.G., Dos Santos R.N., Oliva G., Andricopulo A.D. Molecular docking and structure-based drug design strategies. [Review] Molecules. 2015;20(7):13384–13421. doi: 10.3390/molecules200713384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Picot D., Loll P.J., Garavito R.M. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature. 1994;367(6460):243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- 71.Smith W.L., Garavito R.M., DeWitt D.L. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and −2. J. Biol. Chem. 1996;271(52):33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 72.Orlando B.J., Malkowski M.G. Crystal structure of rofecoxib bound to human cyclooxygenase-2. Acta Crystallogr. F. 2016;72(10):772–776. doi: 10.1107/S2053230X16014230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.ZINC15. http://zinc.docking.org/substances/ZINC000029396226/ Available online:
- 74.ZINC15. http://zinc.docking.org/substances/ZINC000000009029/ Available online:
- 75.Sharma R.A., Gescher A.J., Steward W.P. Curcumin: the story so far. Eur. J. Canc. 2005;41:1955–1968. doi: 10.1016/j.ejca.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 76.González R., Ballester I., López-Posadas R., Suárez M.D., Zarzuelo A., Martínez-Augustin O., Medina F.S.D. Effects of flavonoids and other polyphenols on inflammation. Crit. Rev. Food Sci. Nutr. 2011;51(4):331–362. doi: 10.1080/10408390903584094. [DOI] [PubMed] [Google Scholar]
- 77.Soberón J.R., Sgariglia M.A., Sampietro D.A., Quiroga E.N., Vattuone M.A. Free radical scavenging activities and inhibition of inflammatory enzymes of phenolics isolated from Tripodanthus acutifolius. J. Ethnopharmacol. 2010;130(2):329–333. doi: 10.1016/j.jep.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 78.Noreen Y., Serrano G., Perera P., Bohlin L. Flavan-3-ols isolated from some medicinal plants inhibiting COX-1 and COX-2 catalysed prostaglandin biosynthesis. Planta Med. 1998;64(6):520–524. doi: 10.1055/s-2006-957506. [DOI] [PubMed] [Google Scholar]
- 79.Gibbons S. Bioactive compounds from natural sources isolation, characterization and biological properties. Phytother Res. 2002;16(6) 600–600. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.