

Abstract
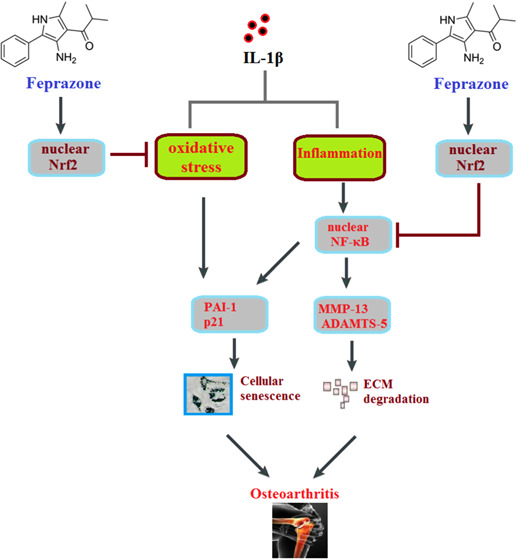
The proinflammatory cytokine interleukin-1 β (IL-1β)-mediated cellular senescence in chondrocytes is involved in the development and pathological progression of osteoarthritis (OA). Feprazone, a nonsteroidal anti-inflammatory drug (NSAID) and a cyclooxygenase (COX) inhibitor, is widely used in clinics. This study aims to investigate whether Feprazone has a protective effect against IL-1β-induced cellular senescence in human chondrocytes. In this study, C-28/I2 chondrocytes were stimulated with IL-1β (10 ng/mL) in the presence or absence of Feprazone (10 and 20 μM). Cellular senescence was assessed using senescence-associated β-galactosidase (SA-β-Gal) staining. The cell cycle was examined using flow cytometry. Gene and protein expressions were determined with real-time polymerase chain reaction (PCR) and western blot analysis. We found that treatment with Feprazone ameliorated IL-1β-induced increase in cellular senescence. Feprazone increased telomerase activity and prevented cell cycle arrest in the G0/G1 phase. We also found that Feprazone reduced the expressions of plasminogen activator inhibitor-1 (PAI-1) and p21, two important regulators of cellular senescence. Additionally, treatment with Feprazone reduced the expressions of matrix metalloprotein (MMP-13) and a disintegrin-like and metalloproteinase with thrombospondin type-1 motif-5 (ADAMTS-5). Interestingly, Feprazone prevented the activation of nuclear factor kappa-B (NF-κB) by preventing nuclear translocation of NF-κB p65 and the luciferase activity of the NF-κB promoter. The results also show that Feprazone increased nuclear levels of nuclear factor erythroid 2-related factor-2 (Nrf2) and reduced the production of reactive oxygen species (ROS). Importantly, silencing of Nrf2 abolished the protective effects of Feprazone against IL-1β-induced NF-κB activation and cellular senescence. These findings shed light on the potential use of Feprazone in the treatment of OA based on a novel mechanism.
Introduction:
Osteoarthritis (OA) is a joint disease commonly observed in the elderly population and sportsmen. It is a degenerative pathology of articular cartilage and is accompanied by the formation of intra-articular osteophytes and synovial inflammation around the joints.1 Clinically, OA is mainly characterized by joint pain, swelling, and stiffness of limbs, which restrict mobility and impact the normal lives of patients. With the upcoming aging society, the morbidity of OA has been increasing annually.2−4 Currently, the pathological mechanism of OA remains unknown. However, it is consistently considered that OA is not induced by a single factor but by multiple factors, including aging, gender, joint damage, inflammation, and heredity.5 Recently, the cell senescence in chondrocytes has been reported to be involved in the development and processing of OA.6,7 Cell senescence is an irreversible cellular process, which is mainly characterized by the decreased ability to proliferate.8 The cell senescence induced by external elements is defined as stress-induced senescence. These external factors include ultraviolet radiation, oxidative stress, activation of oncogenes, and chronic inflammation.9 The terminal structure of chromosomes is sensitive to external changes, such as the upregulation of matrix metalloproteinase (MMP)-13 and the excessive production of reactive oxygen species (ROS). Therefore, when the cells encounter injury during the process of replication, the structure of DNA is easily changed, further contributing to the shortening of telomeres and inducing cell senescence.10,11 The process of oxidative stress is mainly induced by the excessive accumulation of ROS and can be regulated by the Keap1/Nrf2 signaling pathway.12,13 In addition to oxidative stress, cell senescence of chondrocytes is also accompanied by the activation of inflammatory signaling pathways, such as the NF-κB signaling pathway.14 Excessive production of proinflammatory cytokines, such as IL-1β, has been associated with the progression of OA. IL-1β has been involved in a variety of cellular activities. For example, IL-1β influences the expressions of essential structural proteins, including collagen type II and aggrecan, thereby interfering with the activity of chondrocytes.15 Huang16 recently reported that the activated NF-κB signaling pathway and cellular senescence in rat chondrocytes can be induced by IL-1β, indicating that p-coumaric acid might be a promising agent for the treatment of OA. Therefore, alleviating inflammatory activation and IL-1β-induced cellular senescence might provide a novel therapeutic approach for the clinical treatment of OA.
Feprazone is a kind of nonsteroidal anti-inflammatory drug (NSAID) with promising antipyretic, analgesic, and anti-inflammatory effects.17 The dramatic therapeutic property of Feprazone against rheumatic arthritis and rheumatoid arthritis has been clinically proven.18,19 Currently, no further fundamental investigations are being performed on Feprazone to explore its potential anti-inflammatory property on novel adaptation diseases. In the present study, the anti-oxidative stress and antisenescence effects of Feprazone on the IL-1β-treated chondrocytes will be investigated to explore the potential therapeutic property of Feprazone against OA.
Results
Feprazone Ameliorated Cellular Senescence in IL-1β-Challenged C-28/I2 Chondrocytes
To evaluate the effect of Feprazone on the cell senescence of chondrocytes, the cells were stimulated with IL-1β (10 ng/mL) in the presence or absence of Feprazone (10 and 20 μM) for 14 days. As shown in Figure 1, significantly elevated positive SA-β-Gal staining was observed in the chondrocytes stimulated with IL-1β but it was dramatically suppressed by treatment with Feprazone in a dose-dependent manner. For further confirmation, the telomerase activity in the treated chondrocytes was evaluated. As shown in Figure 2, the telomerase activities in the control, IL-1β, IL-1β + 10 μM Feprazone, and IL-1β + 20 μM Feprazone were 21.8, 12.5, 16.7, and 19.2 IU/L, respectively. These data indicate that the cell senescence induced by IL-1β was alleviated by Feprazone.
Figure 1.
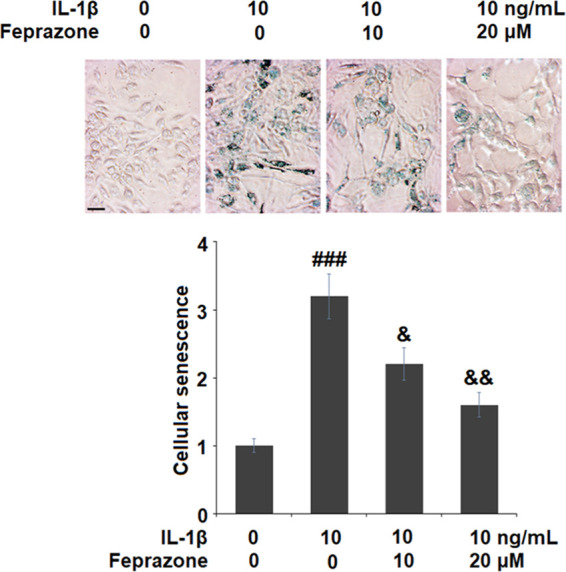
Feprazone ameliorated cellular senescence in IL-1β-challenged C-28/I2 chondrocytes. Cells were stimulated with IL-1β (10 ng/mL) in the presence or absence of Feprazone (10 and 20 μM) for 14 days. Cellular senescence was assessed using senescence-associated β-galactosidase (SA-β-Gal) staining. Scale bar, 100 μm (n = 5; ###, P < 0.001 vs the vehicle group; &, P < 0.05 vs the IL-1β group; &&, P < 0.01 vs the IL-1β group).
Figure 2.

Feprazone increased telomerase activity in IL-1β-challenged C-28/I2 chondrocytes. Cells were stimulated with IL-1β (10 ng/mL) in the presence or absence of Feprazone (10 and 20 μM) for 14 days. Telomerase activity (n = 5 or 6; ###, P < 0.001 vs the vehicle group; &, P < 0.05 vs the IL-1β group; &&, P < 0.01 vs the IL-1β group) was assayed.
Feprazone Prevented Cell Cycle Arrest in the G0/G1 Phase in IL-1β-Challenged C-28/I2 Chondrocytes
As shown in Figure 3, the cell fraction at the G0/G1 phase elevated and the cell fraction at the S phase decreased by stimulation with IL-1β but greatly reversed by the administration of Feprazone, indicating a promising acceleratory effect of Feprazone on the replication of chondrocytes.
Figure 3.
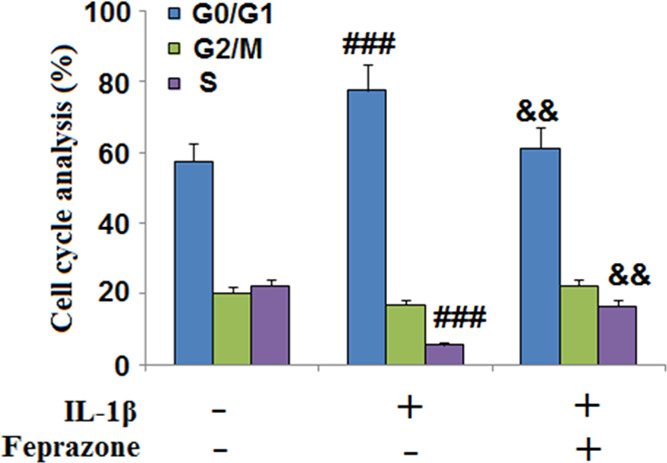
Feprazone prevented cell cycle arrest in the G0/G1 phase in IL-1β-challenged C-28/I2 chondrocytes. Cells were stimulated with IL-1β (10 ng/mL) in the presence or absence of Feprazone (20 μM) for 14 days. The cell fraction in the G0/G1 phase, G2/M phase, and S phase was calculated (n = 5; ###, P < 0.001 vs vehicle group; &, P < 0.05 vs IL-1β group; &&, P < 0.01 vs IL-1β group).
Results shown in Figure 4 indicate that the expressions of PAI-1 and p21 were significantly elevated by stimulation with IL-1β but greatly suppressed by treatment with Feprazone, indicating an inhibitory effect of Feprazone against the expression of senescence-related proteins.
Figure 4.

Feprazone reduced the expressions of p53 and p21 in IL-1β-challenged C-28/I2 chondrocytes. Cells were stimulated with IL-1β (10 ng/mL) in the presence or absence of Feprazone (20 μM). (A) mRNAs of PAI-1 and p21 and (B) proteins of PAI-1 and p21 (n = 5; ###, P < 0.001 vs the vehicle group; &, P < 0.05 vs the IL-1β group; &&, P < 0.01 vs the IL-1β group).
Feprazone Reduced the Expressions of MMP-13 and ADAMTS-5 in IL-1β-Challenged C-28/I2 Chondrocytes
MMP-13 and ADAMTS-5 play an important role in mediating IL-1β-induced degradation of the extracellular matrix (ECM). As shown in Figure 5A, MMP-13 and ADAMTS-5 were significantly upregulated by incubation with IL-1β but dramatically downregulated by the administration of Feprazone. As shown in Figure 5B, the concentrations of MMP-13 in the control, IL-1β, IL-1β + 10 μM Feprazone, and IL-1β + 20 μM Feprazone were 153.5, 525.6, 366.8, and 278.9 pg/mL, respectively. Approximately 103.6, 305.7, 214.2, and 156.5 pg/mL concentrations were detected in the chondrocytes treated with blank medium, IL-1β, IL-1β + 10 μM Feprazone, and IL-1β + 20 μM Feprazone, respectively.
Figure 5.
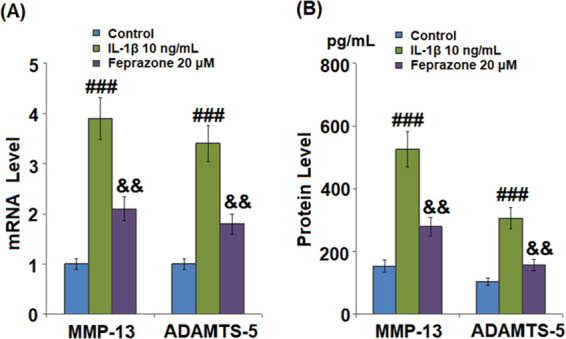
Feprazone reduced the expressions of MMP-13 and ADAMTS-5 in IL-1β-challenged C-28/I2 chondrocytes. Cells were stimulated with IL-1β (10 ng/mL) in the presence or absence of Feprazone (20 μM) for 24 h. (A) mRNAs of MMP-13 and ADAMTS-5 and (B) proteins of MMP-13 and ADAMTS-5 (n = 5; ###, P < 0.001 vs the vehicle group; &, P < 0.05 vs the IL-1β group; &&, P < 0.01 vs the IL-1β group).
Feprazone Prevented the Activation of NF-κB in IL-1β-Challenged C-28/I2 Chondrocytes
NF-κB is a central regulator of the inflammatory signaling pathway. We further investigated the activity of NF-κB in the treated chondrocytes. As shown in Figure 6A, the nuclear level of NF-κB p65 was significantly elevated by stimulation with IL-1β but greatly suppressed by the treatment with Feprazone. The luciferase activity of NF-κB in the chondrocytes was also dramatically elevated by stimulation with IL-1β but greatly suppressed by treatment with Feprazone. These data indicate that the activation of NF-κB induced by IL-1β was significantly ameliorated by Feprazone.
Figure 6.

Feprazone prevented the activation of NF-κB in IL-1β-challenged C-28/I2 chondrocytes. Cells were stimulated with IL-1β (10 ng/mL) in the presence or absence of Feprazone (20 μM) for 6 h. (A). Nuclear levels of NF-κB p65 and (B) luciferase activity of NF-κB (n = 5; ###, P < 0.001 vs vehicle group; &, P < 0.05 vs IL-1β group; &&, P < 0.01 vs IL-1β group).
Feprazone Promoted the Activation of Nrf2 in IL-1β-Challenged C-28/I2 Chondrocytes
To further investigate the effect of Feprazone on oxidative stress, the nuclear level of Nrf2 and the production of ROS were evaluated. As shown in Figure 7A, the suppressed nuclear level of Nrf2 in the chondrocytes induced by IL-1β was significantly elevated by treatment with Feprazone. In addition, the production of intracellular ROS (Figure 7B) was dramatically elevated by stimulation with IL-1β but greatly suppressed by treatment with Feprazone. These data indicate that the oxidative stress induced by IL-1β was significantly alleviated by Feprazone.
Figure 7.

Feprazone promoted the activation of Nrf2 in IL-1β-challenged C-28/I2 chondrocytes. Cells were stimulated with IL-1β (10 ng/mL) in the presence or absence of Feprazone (20 μM) for 6 h. (A) Nuclear levels of Nrf2 and (B) intracellular ROS was measured using DCFH-DA staining. Scale bar, 100 μm (n = 4 or 5; ##, P < 0.01 vs the vehicle group; ###, P < 0.001 vs the vehicle group; &&, P < 0.01 vs the IL-1β group).
Silencing of Nrf2 Abolished Feprazone-Induced NF-κB Activation and Cellular Senescence
To explore the possible mechanism underlying the anti-inflammatory and antisenescence effects of Feprazone, the chondrocytes were transfected with Nrf2 siRNA, followed by stimulation with IL-1β (10 ng/mL) in the presence or absence of Feprazone (20 μM). As shown in Figure 8A, the expression of Nrf2 was significantly suppressed by the introduction of Nrf2 siRNA, indicating a successful establishment of Nrf2 knockdown chondrocytes. As shown in Figure 8B, the elevated luciferase activity of NF-κB in the chondrocytes induced by IL-1β was significantly suppressed by the introduction of Feprazone but greatly promoted by the transfection with Nrf2 siRNA. The increased percentage of positive SA-β-Gal staining (Figure 8C) induced by stimulation with IL-1β was dramatically inhibited by the introduction of Feprazone but greatly elevated by the transfection with Nrf2 siRNA.
Figure 8.

Silencing of Nrf2 abolished Feprazone-induced NF-κB activation and cellular senescence. Cells were transfected with Nrf2 siRNA, followed by stimulation with IL-1β (10 ng/mL) in the presence or absence of Feprazone (20 μM). (A) Western blot analysis revealed successful knockdown of Nrf2, (B) luciferase activity of NF-κB, and (C) cellular senescence (n = 5; ####, P < 0.0001 vs vehicle group; &&, P < 0.01 vs IL-1β group; $$, P < 0.01 vs IL-1β + Feprazone group).
Discussion
The pathogenesis of OA is closely related to the degradation of the chondrocyte extracellular matrix and articular cartilage tissues and the sustainable inflammation induced by senescent chondrocytes.20 Cell senescence can be triggered by multiple complicated factors, such as the shortening of telomerase, genomic damage, and oxidative stress.21 SA-β-Gal is currently considered as the most reliable biomarker for identifying cell senescence. Li reported that SA-β-Gal is highly expressed in the cartilage tissues isolated from OA patients, while no expression of SA-β-Gal was observed in the cartilage tissues of healthy people. Severer OA clinical symptoms are observed as the expression of SA-β-Gal increases.22 The length of the telomere is another biomarker of cell senescence and is regulated by the activity of telomerase. With the increase of the activity of telomerase, the cellular self-repairment ability of chondrocytes is elevated to slow down the progression of cell senescence.23 The rate of cell senescence is mediated by inflammation. The NF-κB signaling pathway can be activated by DNA injury, further inducing the excessive production of proinflammatory factors, such as IL-6, IL-1β, and TNF-α. As a consequence, the process of cell senescence is accelerated by these proinflammatory factors through inhibiting the cell cycle and the inflammatory cascade signaling in the inflammatory cells.24 In the present study, an in vitro cell senescence model in the human chondrocytes was successfully established by stimulation with IL-1β and verified by the upregulation of SA-β-Gal, activated telomerase activity, G0/G1 phase arrest, elevated expressions of senescence-related proteins (PAI-1 and p21), and the activated NF-κB signaling pathway. By the introduction of Feprazone, we found that the cell senescence in the chondrocytes was significantly alleviated. We further investigated the expressions of MMP-13 and ADAMTS-5, which are the inducers of the degradation of the extracellular matrix (ECM) and reported to be involved in the development and progression of OA.25,26 We found that the elevated expressions of MMP-13 and ADAMTS-5 in the chondrocytes induced by IL-1β were significantly reversed by Feprazone, indicating a promising inhibitory effect of Feprazone against the degradation of ECM. However, in vivo experiments will be performed to verify the anti-OA property of Feprazone in our future work.
Under the physiological state of hypoxia, chondrocytes can produce ROS to maintain cellular functions. However, excessive production of ROS is induced under the OA pathological state, further inhibiting the synthesis of proteoglycan and type II collagens via activating the serine-derived activated protein kinase. As a result, the ECM is destroyed and the degeneration of cartilage tissues is accelerated. The expression of matrix metalloproteinase is also elevated by ROS, further inducing the excretion of proinflammatory factors and contributing to the development of OA.27 Oxidative stress can be triggered by the excessive production of ROS, which is negatively expressed by the Nrf2 signaling pathway. Activated Nrf2 is released from the complex of Keap1–Nrf2 and transferred into the nucleus to upregulate the expression of HO-1, further suppressing the production of ROS.12,28 In the present study, we found that the activated oxidative stress induced by IL-1β was significantly suppressed by Feprazone, accompanied by the upregulation of Nrf2. When Nrf2 was knocked down in the chondrocytes, the antisenescence and anti-inflammatory effects of Feprazone were abolished, indicating that Feprazone might exert antisenescence and anti-inflammatory effects through upregulating Nrf2 to suppress oxidative stress.
Taken together, our data indicate that Feprazone mitigates IL-1β-induced cellular senescence in chondrocytes through upregulating Nrf2.
Materials and Methods
Cell Culture and Treatment
The human chondrocytes, C-28/I2 cells, were cultured in DMEM/F12 medium containing 10% fetal bovine serum. A total of 4 × 106 cells were cultured in T-75 flasks and incubated in an incubator at 37 °C and 5% CO2. For the treatment experiment, cells were stimulated with IL-1β (10 ng/mL)29 in the presence or absence of Feprazone (10, 20 μM). Feprazone was commercially purchased from GLFBIO (CAT#GC40565), and the powder was dissolved in DMSO and then stored in a −20 °C freezer before use.
SA-β-Gal Staining
A total of 1.2 × 106 cells were seeded on a six-well cell culture plate. Then, the treated C-28/I2 cells were fixed with a senescence β-galactosidase staining kit (Cat#9860, Cell Signaling Technology) for approximately 15 min at room temperature, followed by being stained using the β-gal dye for 12 h. After washing three times, a phase-contrast microscope was used to take images of the stained chondrocytes to determine the percentage of positive cells.
Telomerase Activity Evaluation
A total of 5 × 104 cells were seeded on a 96-well cell culture plate. A telomerase ELISA kit (Cat#11854666910, Roche) was used to detect the telomerase activity in the treated chondrocytes as previously reported.30 Briefly, the treated chondrocytes were lysed with a RIPA lysis buffer (Beyotime, Shanghai, China), followed by mixing the protein supernatant and the reaction buffer to detect the telomerase activity according to the instructions of the manufacturer. Subsequently, the absorbance values at 450 nm were read using a microreader (Thermo, Massachusetts). Based on the standard curve, the telomerase activity (IU/L) of the treated chondrocytes was calculated.
Cell Cycle Analysis
Flow cytometry was used to detect the cell cycle of the treated chondrocytes. Briefly, cold ethanol was used to fix the cells overnight, followed by being stained with 50 μg/mL PI solution (Beyotime, Shanghai, China) for half an hour; then, flow cytometry was used to analyze each cell cycle phase (G0/G1, G2/M, S) of the cell fraction.
ELISA Assay
A total of 5 × 104 cells were seeded on a 96-well cell culture plate. The concentrations of MMP-13 and ADAMTS-5 in the treated chondrocytes were evaluated using the ELISA assay. Briefly, cells lysed with the lysis buffer were incubated with 5% bovine serum albumin (BSA) to remove the nonspecific binding proteins. The antibodies against MMP-13 and ADAMTS-5 were immobilized onto the 96-well microtiter plates, followed by adding the samples for approximately 30 min. Subsequently, the plates were added with HRP-conjugated anti-mouse immunoglobulin, washed, and further incubated with a TMB substrate solution for 30 min to terminate the reaction. Finally, the absorbance at 450 nm was measured using a spectrophotometer (Thermo, Massachusetts).
Real-Time PCR Analysis
The TRI reagent (Sigma, Massachusetts), an RNA isolation reagent, was used to extract total RNA from the treated chondrocytes, followed by being transformed into cDNA using a first-strand cDNA synthesis kit (Pharmacia LKB, Uppsala, Sweden). In the present study, a TaqMan system (Thermo, Massachusetts) was used to perform the real-time PCR. An ABI PRISM 7300 sequence detection system (Thermo, Massachusetts) was utilized to conduct the PCR amplification and product detection. The relative gene expression was normalized to GAPDH, and the 2–ΔΔCt method was used to calculate the relative expression. The following primers were used: ADAMTS-5: forward, 5′-GCAGAACATCGACCAACTCTACTC-3′, reverse, 5′-CCAGCAATGCCCACCGAAC-3′; MMP-3: forward, 5′-CCTCTATGGACCTCCCACAGAATC-3′, reverse, 5′-GGTGCTGACTGCATCGAAGGACAAA-3′; PAI-1: forward, 5′-AGCTCCTTGTACAGATGCCG-3′, reverse, 5′-ACAACAGGA GGAGAAACCCA-3′; p21: forward, 5′-GCGCCATGTCAGAACCGGCTGG-3′, reverse: 5′-TTAGGGCTTCCTCTTGGAGA-3′; and GAPDH: forward, 5′-ACT GGCGTCTTCACCACCAT-3′, reverse, 5′-AAGGCCATGCCAGTGAGCTT-3′.
Western Blot Assay
The protein in the treated chondrocytes was isolated using a cell lysis buffer (Beyotime, Shanghai, China), which was quantified with a BCA kit (Beyotime, Shanghai, China). Approximately 50 μg of protein was loaded and separated with sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), followed by being transferred onto a PVDF membrane (Thermo, Massachusetts). Subsequently, the membrane was incubated with 5% BSA to remove the nonspecific binding proteins, followed by incubation with the primary antibodies against PAI-1 (1:1000, #11907, CST, USA), p21 (1:2000, #2947, CST, USA), NF-κB p65 (1:2000, #8242, CST, USA), Nrf2 (1:2000, #12721, CST, USA), or β-actin (1:10 000, CST, USA), followed by incubation with HRP-linked rabbit (1:2000, #7074, CST, USA) or mouse (1:2000, #7076, CST, USA) IgG secondary antibodies. After being washed using the TBST buffer, the membrane was incubated with ECL solution and exposed using a Tanon 5200 system (Tanon, Shanghai, China). Finally, the densitometric analysis was performed using Image J software (National Institutes of Health, USA).
Luciferase Activity Assay
A total of 1.2 × 106 cells were seeded on a 6-well cell culture plate. A luciferase assay system (Promega, Wisconsin) was used to determine the luciferase activity of NF-κB in the treated chondrocytes. The NF-κB promoter plasmid was transfected into C-28/I2 cells. After treatment, the cell lysates were centrifuged at 4 °C for 5 min at 13 200 rpm, followed by being collected and seeded onto an opaque 96-well plate. Subsequently, each sample was added with 100 μL of a luciferase assay substrate (Promega, Wisconsin), followed by measuring the luminescence over 5 s using a microplate luminometer with an automatic injector (PolarStar, Medina).
DCFH-DA Staining Assay
The treated C-28/I2 cells were plated in a 24-well plate, followed by replacing the medium with DCFH-DA (Invitrogen, Paisley, U.K.) solution at a concentration of 10 μmol/L for 30 min. Subsequently, the chondrocytes were washed with phosphate-buffered saline (PBS) buffer, and the production of ROS was detected using a fluorescence microscope (Thermo, Massachusetts). Software Image J was used to quantify ROS. First, we characterized the regions of interest (ROI). We then assessed the integrated density value (IDV) of target cells. The number of cells (N) presented in the ROI was calculated. Average levels of ROS = IDV/N.
Statistical Analysis
Data are shown as mean ± standard deviation (S.D.). GraphPad Prism Software version 7.00 (San Diego, CA) was used for statistical analysis. The method of statistical analysis used was one-way analysis of variance (ANOVA). P < 0.05 was regarded as statistically significant.
Acknowledgments
This study was supported by the Fuzhou Second Hospital of Xiamen University.
The authors declare no competing financial interest.
References
- Samuels J.; Krasnokutsky S.; Abramson S. B. Osteoarthritis: a tale of three tissues. Bull. NYU Hosp. Jt. Dis. 2008, 66, 244–250. [PubMed] [Google Scholar]
- Loeser R. F. Age-related changes in the musculoskeletal system and the development of osteoarthritis. Clin. Geriatr. Med. 2010, 26, 371–386. 10.1016/j.cger.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmick C. G.; Felson D. T.; Lawrence R. C.; Gabriel S.; Hirsch R.; Kwoh C. K.; Liang M. H.; Kremers H. M.; Mayes M. D.; Merkel P. A.; Pillemer S. R.; Reveille J. D.; Stone J. H. and National Arthritis Data W. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis Rheumatol. 2008, 58, 15–25. 10.1002/art.23177. [DOI] [PubMed] [Google Scholar]
- Lawrence R. C.; Felson D. T.; Helmick C. G.; Arnold L. M.; Choi H.; Deyo R. A.; Gabriel S.; Hirsch R.; Hochberg M. C.; Hunder G. G.; Jordan J. M.; Katz J. N.; Kremers H. M.; Wolfe F. and National Arthritis Data W. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheumatol. 2008, 58, 26–35. 10.1002/art.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felson D. T. Risk factors for osteoarthritis: understanding joint vulnerability. Clin. Orthop. Relat. Res. 2004, 427, S16–S21. 10.1097/01.blo.0000144971.12731.a2. [DOI] [PubMed] [Google Scholar]
- Song W.; Zhang Y.; Wang J.; Ma T.; Hao L.; Wang K. Antagonism of cysteinyl leukotriene receptor 1 (cysLTR1) by montelukast suppresses cell senescence of chondrocytes. Cytokine 2018, 103, 83–89. 10.1016/j.cyto.2017.12.021. [DOI] [PubMed] [Google Scholar]
- McCulloch K.; Litherland G. J.; Rai T. S. Cellular senescence in osteoarthritis pathology. Aging Cell 2017, 16, 210–218. 10.1111/acel.12562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora E.; Bielak-Zmijewska A.; Mosieniak G. What is and what is not cell senescence. Postepy Biochem. 2018, 64, 110–118. 10.18388/pb.2018_120. [DOI] [PubMed] [Google Scholar]
- Loeser R. F. Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthritis Cartilage 2009, 17, 971–979. 10.1016/j.joca.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance B.; Sies H.; Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- Kauppila T. E. S.; Kauppila J. H. K.; Larsson N. G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. 10.1016/j.cmet.2016.09.017. [DOI] [PubMed] [Google Scholar]
- Bellezza I.; Giambanco I.; Minelli A.; Donato R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta, Mol. Cell Res. 2018, 1865, 721–733. 10.1016/j.bbamcr.2018.02.010. [DOI] [PubMed] [Google Scholar]
- Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela-Eirin M.; Varela-Vazquez A.; Guitian-Caamano A.; Paino C. L.; Mato V.; Largo R.; Aasen T.; Tabernero A.; Fonseca E.; Kandouz M.; Caeiro J. R.; Blanco A.; Mayan M. D. Targeting of chondrocyte plasticity via connexin43 modulation attenuates cellular senescence and fosters a pro-regenerative environment in osteoarthritis. Cell Death Dis. 2018, 9, 1166 10.1038/s41419-018-1225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojdasiewicz P.; Poniatowski ŁA.; Szukiewicz D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflammation 2014, 2014, 561459 10.1155/2014/561459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; You Y.; Xi Y.; Ni B.; Chu X.; Zhang R.; You H. p-Coumaric Acid Attenuates IL-1beta-Induced Inflammatory Responses and Cellular Senescence in Rat Chondrocytes. Inflammation 2020, 43, 619–628. 10.1007/s10753-019-01142-7. [DOI] [PubMed] [Google Scholar]
- Ozkaya-Bayazit E.; Akar U. Fixed drug eruption induced by trimethoprim-sulfamethoxazole: evidence for a link to HLA-A30 B13 Cw6 haplotype. J. Am. Acad. Dermatol. 2001, 45, 712–7. 10.1067/mjd.2001.117854. [DOI] [PubMed] [Google Scholar]
- Sturrock R.; Isaacs A.; Hart F. D. Feprazone compared with indomethacin in the management of rheumatoid arthritis. Practitioner 1975, 215, 94–97. [PubMed] [Google Scholar]
- Harkness A. J.; Burry H. C.; Grahame R. A trial of feprazone in ankylosing spondylitis. Rheumatology 1977, 16, 158–161. 10.1093/rheumatology/16.3.158. [DOI] [PubMed] [Google Scholar]
- Ashraf S.; Cha B. H.; Kim J. S.; Ahn J.; Han I.; Park H.; Lee S. H. Regulation of senescence associated signaling mechanisms in chondrocytes for cartilage tissue regeneration. Osteoarthritis Cartilage 2016, 24, 196–205. 10.1016/j.joca.2015.07.008. [DOI] [PubMed] [Google Scholar]
- Liu J.; Wang L.; Wang Z.; Liu J. P. Roles of Telomere Biology in Cell Senescence, Replicative and Chronological Ageing. Cells 2019, 8, 54. 10.3390/cells8010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Zhu H.; Yan X.; Gu H.; Gu Z.; Liu F. Endoplasmic reticulum stress participates in the progress of senescence and apoptosis of osteoarthritis chondrocytes. Biochem. Biophys. Res. Commun. 2017, 491, 368–373. 10.1016/j.bbrc.2017.07.094. [DOI] [PubMed] [Google Scholar]
- Wilson B.; Novakofski K. D.; Donocoff R. S.; Liang Y. X.; Fortier L. A. Telomerase Activity in Articular Chondrocytes Is Lost after Puberty. Cartilage 2014, 5, 215–220. 10.1177/1947603514537518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivieri F.; Albertini M. C.; Orciani M.; Ceka A.; Cricca M.; Procopio A. D.; Bonafe M. DNA damage response (DDR) and senescence: shuttled inflamma-miRNAs on the stage of inflamm-aging. Oncotarget 2015, 6, 35509–35521. 10.18632/oncotarget.5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma P.; Dalal K. ADAMTS-4 and ADAMTS-5: key enzymes in osteoarthritis. J. Cell Biochem. 2011, 112, 3507–3514. 10.1002/jcb.23298. [DOI] [PubMed] [Google Scholar]
- Zamolo G.; Grahovac M.; Zauhar G.; Vucinic D.; Kovac L.; Brajenic N.; Grahovac B. Matrix metalloproteinases MMP-1, MMP-2, and MMP-13 are overexpressed in primary nodular melanoma. J. Cutaneous Pathol. 2020, 47, 139–145. 10.1111/cup.13603. [DOI] [PubMed] [Google Scholar]
- Drevet S.; Gavazzi G.; Grange L.; Dupuy C.; Lardy B. Reactive oxygen species and NADPH oxidase 4 involvement in osteoarthritis. Exp. Gerontol. 2018, 111, 107–117. 10.1016/j.exger.2018.07.007. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Davies K. J. A.; Forman H. J. Oxidative stress response and Nrf2 signaling in aging. Free Radicals Biol. Med. 2015, 88, 314–336. 10.1016/j.freeradbiomed.2015.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F. F.; Hu P. F.; Xiong Y.; Bao J. P.; Qian J.; Wu L. D. Tricetin Protects Rat Chondrocytes against IL-1 β-Induced Inflammation and Apoptosis. Oxid. Med. Cell. Longevity 2019, 2019, 4695381 10.1155/2019/4695381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K. H.; Guo Y.; Li L.; Qu S.; Zhao W.; Lu Q. T.; Mo Q. Y.; Yu B. B.; Zhou L.; Lin G. X.; Sun Y. C.; Zhu X. D. Cancer stem cell-like characteristics and telomerase activity of the nasopharyngeal carcinoma radioresistant cell line CNE-2R. Cancer Med. 2018, 7, 4755–4764. 10.1002/cam4.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]