

Abstract
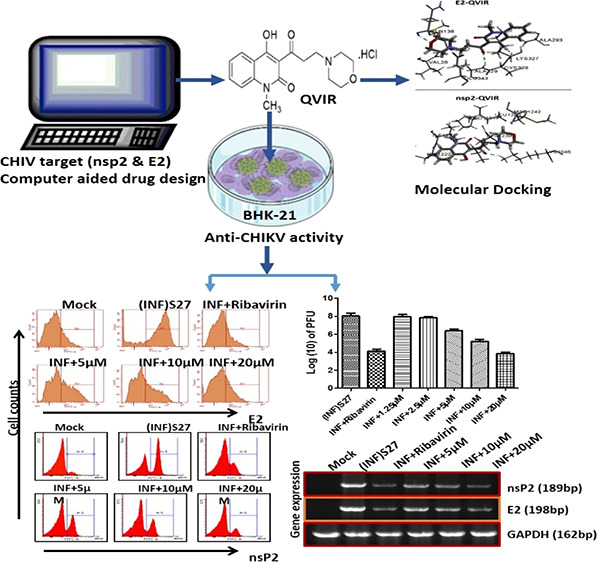
The re-emergence of Chikungunya virus (CHIKV) infection in humans with no approved antiviral therapies or vaccines is one of the major problems with global significance. In the present investigation, we screened 80 in-house quinoline derivatives for their anti-CHIKV activity by computational techniques and found 4-hydroxy-1-methyl-3-(3-morpholinopropanoyl)quinoline-2(1H)-one (QVIR) to have potential binding affinities with CHIKV nsP2 and E2 glycoproteins. QVIR was evaluated in vitro for its anti-CHIKV potential. QVIR showed strong inhibition of CHIKV infection with an EC50 (50% effective concentration) value of 2.2 ± 0.49 μM without significant cytotoxicity (CC50 > 200 μM) and was chosen for further elucidation of its antiviral mechanism. The infectious viral particle formation was abolished by approximately 72% at a QVIR concentration of 20 μM during infection in the BHK-21 cell line, and the CHIKV RNA synthesis was diminished by 84% for nsP2 as well as 74% for E2, whereas the levels of viral proteins were decreased by 69.9% for nsP2 and 53.9% for E2. Flow cytometry analysis confirmed a huge decline in the expression of viral nsP2 and E2 proteins by 71.84 and 67.7%, respectively. Time of addition experiments indicated that QVIR inhibited viral infection at early and late stages of viral replication cycle, and the optimal inhibition was observed at 16 h post infection. The present study advocates for the first time that QVIR acts as a substantial and potent inhibitor against CHIKV and might be as an auspicious novel drug candidate for the development of therapeutic agents against CHIKV infections.
1. Introduction
Chikungunya fever in humans caused by Chikungunya virus (CHIKV) is a vector-borne disease, spread by Aedes aegypti and Aedes albopictus mosquitoes. The major symptoms are high fever, nausea, rashes, polyarthralgia, myalgia, and headache.1−3 One of the significant clinical symptoms is polyarthralgia that may persist for months or years in a few patients.4 It is an arboviral disease, endemic in tropical and subtropical regions, where 2.5 billion people are at risk.5 As a promptly spreading recurring infectious disease, Chikungunya fever (CHIKF) has drawn a vast consideration. Vaccination is an effective way to control the outbreak of this illness including CHIKF, but there are no approved vaccine and specific treatment available for CHIKV at present, and the current therapeutic approach mainly depends on the analgesics, antipyretics, and anti-inflammatory agents to mitigate the patient’s symptoms.6,7 Therefore, there is an urgent need to discover new antiviral drugs against this illness. CHIKV is spherical in shape and the genome is single-stranded RNA that is approximately 12 kb long, which has a positive sense that contains two open reading frames (ORFs). The first ORF encodes for nonstructural proteins (nsP1 to nsP4) and the second ORF encodes for three major structural proteins (capsid and envelope glycoproteins E1 and E2) of CHIKV.8,9 The nonstructural protein nsP1 performs methyl- and guanyl-transferase activities, while nsP2 acts as a helicase/protease exercise, nsP3 plays an important role in the replication of RNA, and nsP4 works as RNA-dependent RNA polymerase.10−12 The CHIKV nonstructural protein nsP2 is essential due to its role in the separation of all four nonstructural proteins from their precursor protein.13 The C-terminal domain of nsP2 exhibits proteolytic action by catalyzing a reaction of deprotonation of a thiol group (−SH) on the cysteine residue at the active site that helps in precursor protein cleavage, which is crucial for CHIKV genome replication. These differential ways of catalysis by the viral nsP2 protein designate it as a pharmaceutically critical and challenging site to discover an appropriate inhibitor against it.14 The virus particle presents two surface proteins E2 and E1 that assist in cell entry; E2 helps in cell attachment and E1 acts as a fusion protein. Two N glycosylation sites are present in the E2 envelope protein. The E2 protein made up of domains A, B, and C is situated at the center of protein, in the distal end, and onto the viral membrane, respectively.15,16
The quinoline derivatives have shown broad-spectrum antiviral activity; for example, 9-aminoquinolines, like chloroquine and its hydroxy derivative (hydroxychloroquine), have potential antiviral activities against coronaviruses,17 human immunodeficiency virus (HIV),18,19 and respiratory syncytial virus.20,21 Quinoline derivatives exhibit proven activity against flavivirus replication,22−24 for example, against West Nile virus,25,26 T-cell lymphotropic virus (HTLV),27 Hepatitis C virus,28 Zika virus,29 Japanese Encephalitis virus,30 and dengue virus.31 The 8-hydroxyquinoline (8-HQ) derivative 5,7-dichloro-8-HQ is a potent inhibitor of DENV231 and West Nile virus.26 The 4-aminoquinoline derivative amodiaquine has activity against DENV2.32 Quinolone-N-acylhydrazone hybrids show activity against arboviruses Zika (ZIKV) and Chikungunya (CHIKV).33 RG7109 (a quinoline derivative) was found to be a potent NS5B polymerase inhibitor and has activity against the Hepatitis C virus.28 Chloroquine, an antimalarial drug, has been reported to inhibit CHIKV replication in a dose-dependent manner in Vero A cells with an IC50 value of 7.0 μM, having an IC90 of 15.0 μM and an SI of 37.14 μM.34 Moreover, quinine, also an antimalarial drug, inhibits CHIKV in vitro (IC50 = 0.1 μg/mL).34 It was suggested that quinine in high concentration inhibits replication by causing mutation in the CHIKV nonstructural protein nsP1.35,36 Chloroquine interferes with the endosome-mediated CHIKV internalization, raises the endosomal pH by interfering with the protonation of the endocytic vesicles, and therefore prevents the E1 fusion step needed for the release of CHIKV RNA into the cell cytoplasm, thus acting as a CHIKV entry inhibitor.37
Considering the importance of the antiviral activity of quinoline derivatives, we screened out our in-house library of 80 quinoline derivatives for their anti-CHIKV potential. These novel unreported 80 compounds include various chalcone, pyrazoline, pyrazolidine, thiadiazole, amide, and enaminone derivatives of the quinoline/quinolone scaffold. We identified 4-hydroxy-1-methyl-3-(3-morpholinopropanoyl)quinoline-2(1H)-one (QVIR) to have potential anti-CHIKV activity. In the present study, we demonstrated that QVIR acts as a potent antiviral agent against CHIKV in BHK-21 cells infected by the CHIKV strain S27 and studied the mechanism through which QVIR eradicates the CHIKV viral load. Our finding also demonstrated that QVIR strongly inhibits the synthesis of nsP2 and E2 proteins of CHIKV. This is the first investigation of QVIR against CHIKV in vitro.
2. Results
2.1. Structure-Based Virtual Screening (SBVS)
The workflow for SBVS is presented in Figure 1. Eighty in-house unreported quinoline/quinolone derivatives were used for SBVS against Chikungunya targets. These 80 novel compounds include various chalcone, pyrazoline, pyrazolidine, thiadiazole, amide, and enaminone derivatives of the quinoline/quinolone scaffold. The ligands were prepared by LigPrep 2.6 with Epik 2.4, modules of Schrödinger, which includes the generation of protonation, and tautomeric states of ligands and then energy were minimized using the OPLS_2005 force field. As a result of this, a docking library of 156 compounds was obtained. The prepared docking library (156 compounds) was subjected to XP docking with three different CHIKV targets: the mature envelope E2 glycoprotein (PDB ID: 3N42; resolution, 3.0 Å), the nonstructural protein nsP2 (PDB ID: 3TRK; resolution, 2.397 Å), and the nonstructural protein nsP3 (PDB ID: 4TU0; resolution, 2.3 Å). Ligands were docked within the cavity of the CHIKV envelope glycoprotein complex, between E1 and E2 (allosteric site). The catalytic dyad amino acid residues (Cys1013 and His1083) of the tunnel-shaped active site of nsp2 (PDB: 3TRK) were used and centered for grid generation and hence for docking calculations. The top 10 hits for each target were retained based on their Glide XP score. Out of 10 hits, three structurally varied hits were chosen for each target based on their binding pattern in the active site and binding free energy. The docking scores (XP Gscore) and binding free energy of the top three selected compounds are listed in Table 1. Out of these three hits, against each of the three target proteins, one common hit (QVIR) is selected for in vitro studies. QVIR showed potential binding affinity with two CHIKV targets, the mature envelope E2 glycoprotein and the nonstructural protein nsP2, while it showed weak binding potential with the nonstructural protein nsP3. QVIR showed high binding affinity within the cavity of the CHIKV envelope glycoprotein complex, between E1 and E2 (allosteric site), and was predicted computationally as a viral entry inhibitor. The 3D representation of docked conformation of the selected compound QVIR and standard drugs ribavirin and chloroquine, with the envelope E2 glycoprotein and the nonstructural protein nsP2, obtained after XP docking is depicted in Figure 2A,B, respectively.
Figure 1.

Workflow of the structure-based virtual screening (SBVS).
Table 1. XP Gscore and Binding Free Energy Estimates of the Top 3 Hits along with Ribavirin and Chloroquine, Obtained after Extra Precision (XP) Docking.
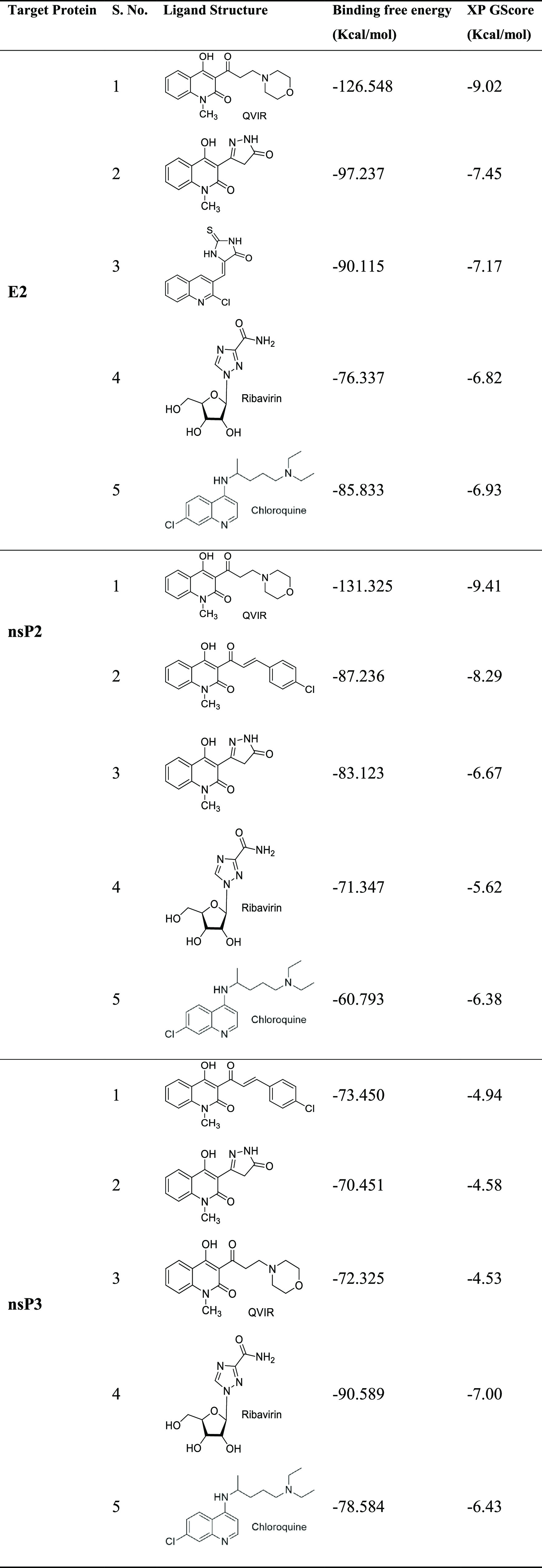
Figure 2.

Three-dimensional representation of docked conformation of (A) envelope E2 glycoprotein and (B) nonstructural protein nsP2 with ligands QVIR, ribavirin, and chloroquine obtained after Glide XP docking. Green dashed lines represent conventional hydrogen bonds with the interacting amino acid residues.
2.2. Synthesis and Chemistry of Compounds
Compound 3 (QVIR) was synthesized as per the scheme provided in Figure 3 with high purity and yield. The pyrone (methyl-substituted pyranoquinolinedione, 1) was achieved by the cycloaddition reaction of N-methyl aniline with diethyl malonate in diphenyl ether as a solvent (yield: 66%).38 Compound 1 on subsequent hydrolysis in a basic medium afforded 3-acetyl-4-hydroxy-1-methyl quinolin-2(1H)-one (2) (yield: 93%).38 The title compound 3, QVIR (β-amino carbonyl compound), was prepared by amino alkylation of compound 2 by formaldehyde and morpholine in the presence of HCl in an ethanol medium (yield: 80%).39 The purity of the compounds was confirmed by UPLC–MS/MS and was found to be more than 95%. QVIR was obtained in the form of a pale yellow fluffy solid, which has a melting point of 297–300 °C. In the IR spectrum of compound 1, the characteristic absorption bands at 3447, 1742, and 1665 cm–1 were attributed to the hydroxyl (−OH) group, ketone (—C=O) function of the lactone ring, and ketone (—C=O) function of amide, respectively. The 1H-NMR spectrum of compound 1 exhibited a characteristic signal at δ 3.56 and 5.68, as a singlet was due to −N–CH3 and a pyrano proton integrating for three and one protons, respectively. The signals of aromatic proton of the compound were detected at δ 7.3–7.73, while the hydroxyl proton appeared as a broad singlet at δ 13.10. The spectral data of 2 exhibit IR bands at 1739 cm–1 due to the presence of an acetyl carbonyl group and 1652 cm–1 due to the carbonyl group (—C=O) of amide. The 1H-NMR spectrum of compound 2 displays a characteristic signal at δ 2.80, demonstrating the formation of an acetyl (−COCH3) group. A peak at δ 3.76 displays the presence of −N–CH3 and, at δ 7.34–8.62, showed the presence of aromatic protons, while the hydroxyl proton came into view as a broad singlet at δ 14.18. Moreover, the mass spectrum of this prototype compound 2 showed the molecular ion peak, i.e., m/z (M+) at 217.07, which confirmed its successful synthesis. The IR spectrum of compound 3 (QVIR) showed stretching bands at 1653, 1738, and 3472 cm–1 due to characteristic amide carbonyl, acetyl carbonyl, and hydroxyl groups, respectively. In the 1H-NMR spectrum, the singlet appearing at δ 15.10 showed the presence of a D2O-exchangeable hydroxyl proton. Two triplets of eight protons of morpholine appeared at δ 2.65–2.70 and 3.72–3.77, while a singlet at δ 3.58 showed N–CH3 protons. Two triplets at δ 3.40–3.45 and 3.62–3.67 were assigned to four protons of −CO–CH2 and −N–CH2, respectively. All four aromatic protons of the quinolone ring appeared to be resonating at δ 7.40–7.75 as a multiplet. Also, the 13C-NMR spectrum of QVIR is in agreement with the structure. The UPLC–MS/MS of QVIR with m/z peaks at 316.19 (M+) and 317.20(M+ + H) further verified its successful synthesis (Figure 4).
Figure 3.

Route of synthesis of the target compound 4-hydroxy-1-methyl-3-(3-morpholinopropanoyl)quinoline-2(1H)-one hydrochloride (QVIR).
Figure 4.

(a) Chromatogram and retention time and (b) mass spectrum of the compound QVIR in UPLC–MS/MS analysis.
2.3. Effect of QVIR on BHK-21 Cells
To determine the adverse effect of the compound QVIR on BHK-21 cells, after 24 h of treatment with QVIR (5, 10, and 20 μM) and ribavirin (5 μM), the treated and untreated cells were analyzed in a microscope for morphological changes. QVIR exhibited its inertness against BHK-21 cells at the tested dose levels (5, 10, and 20 μM) (Figure 5a). This study was performed in triplicate and similar observation was observed in each replicate. The reference drug ribavirin was used as a control, as it was reported to have anti-CHIKV potential.40 Further, MTT assay was performed to determine the cytotoxicity of QVIR (10 to 200 μM) on BHK-21 cells. QVIR showed no significant evidence of cytotoxicity even at a higher dose (200 μM); hence, the CC50 value was considered to be >200 μΜ (Figure 5b). These results confirmed that the drug did not exhibit a cytotoxic effect on BHK-21 cells at a concentration of 200 μM for 24 h duration; therefore, based on the above finding, we determined a dose-dependent inhibition of QVIR on CHIKV infection.
Figure 5.

BHK-21 cells were treated at different doses of the compound QVIR (5, 10, and 20 μM) for 24 h. (a) The treated and untreated cells were analyzed in a microscope for morphological changes under a 20× objective. (b) BHK-21 cells were cultured in 96-well plates and exposed with increasing concentration (from 10 to 200 μM) of QVIR at 37 °C with 5% CO2 for 24 h. (c) Inhibition of CHIKV infection by the compound QVIR. BHK-21 cells were infected with the CHIKV prototype strain S27 (MOI, 0.01). Different doses of the drugs (1.25, 2.5, 5, 10, and 20 μM) were added to the experimental samples. Ribavirin (10 μM) was used as a positive control. The supernatants as well as cells were collected from all the experimental samples at 24 h post treatments and plaque assay was performed to determine the number of infectious particles of CHIKV. The bar diagram represents the virus titer in the log10 scale for all the experimental samples from three independent experiments. (d) BHK-21 cells were infected by CHIKV on an MOI of 0.01 following treatment with 20 μM QVIR at different time intervals (0, +2, +4, +8, +12, +16, +20, and +24 hpi). Ribavirin (10 μM) was used as a positive control. The results were assessed by plaque assay for the determination of the number of active virus particles. The bar graph represents the percentage of virus particle release, and the open bar and solid bar display the QVIR- and ribavirin-treated groups, respectively. (e) The line graph represents the percentage of plaque-forming unit and the EC50 of QVIR. The statistical analysis of the experimental data was presented as mean ± SEM from three independent experiments.
2.4. QVIR Inhibited the CHIKV Infection in BHK-21 Cells
To determine the effect of QVIR on the reduction level of mature infectious viral particle production after treatment, the BHK-21 cells were infected with the CHIKV prototype strain S27 at a multiplicity of infection of 0.01. Plaque assay was carried out on CHIKV-infected BHK-21 cells after treatment with QVIR at concentrations of 1.25, 2.5, 5, 10, and 20 μM. Cells were observed under a microscope to investigate the cytopathic effect (CPE). At 24 h post infection (hpi), the supernatant was collected and used to infect BHK-21 cells, and the plaque numbers were calculated after 4 days. The plaque numbers were changed to log10 of PFU/mL and presented in the form of a bar diagram (Figure 5c). The plaque numbers were reduced by 99.15% after treatment with 10 μM ribavirin as a positive control (Figure 5c,e), whereas the plaque numbers were reduced by 82.62, 97.86, and 99.61% with 5, 10, and 20 μM QVIR post treatment, respectively (Figure 5c). The EC50 value of the compound QVIR was found to be 2.2 ± 0.49 μM (Figure 5e).
2.5. Inhibition of Infectious Viral Particle Release
Time of drug addition assay was performed to investigate the inhibition of infectious viral particle replication. Figure 5d shows that QVIR inhibits approximately 83% of infectious viral particle release even after addition of drug (20 μM) at 8 hpi. Unlike QVIR, ribavirin showed almost 70% inhibition in viral particle release at 8 hpi. At 12 and 16 hpi, QVIR suppressed the release of infectious viral particles by around 65 and 49%, respectively, whereas ribavirin inhibited the release of virus particles up to 48 and 36% at 12 and 16 hpi, respectively (Figure 5d). The inhibitory effects of QVIR on the release of infectious virus particles were greater than those of the ribavirin control. The results advocated that QVIR might interfere in multiple stages of the CHIKV life cycle.
2.6. QVIR Inhibited CHIKV RNA and Protein Levels
It was seen during our investigation that QVIR was able to cause reduction in CHIK particle production in BHK-21 cells. We were keen to find out the level of CHIKV RNA and protein, which is important in viral life cycle and might have been impaired by this compound. To ascertain this, BHK-21 cells after infection and drug treatment were collected at 24 h post treatment. Reverse transcriptase PCR was performed to check the level of CHIKV RNA, and protein levels were determined by the western blotting technique. The structural and nonstructural proteins E2 and nsP2 were preferred as mentioned earlier. The RNA levels of nsP2 were decreased by 52.1, 67.5, and 84.1% after the treatment with 5, 10, and 20 μM QVIR, respectively. Similarly, the RNA levels of E2 were diminished by 39, 55.6, and 74%, respectively, as shown in Figure 6. Ribavirin-treated virus-infected BHK-21 cells exhibited 75.8 and 82.7% reduction in the E2 and nsP2 CHIKV RNA levels, respectively. The RNA level was estimated by normalizing the GAPDH quantity. The relative band strength in Figure 6a is plotted in Figure 6b as a bar graph for comparison. The protein levels of nsP2 were reduced to 36.4, 63.4, and 69.9% after treatment with QVIR at concentrations of 5, 10, and 20 μM, respectively. Similarly, QVIR at doses of 5, 10, and 20 μM suppresses the E2 protein levels of CHIKV by 22.8, 45.8, and 53.9%, respectively (Figure 6c,d). Thus, our data demonstrated that the level of structural and nonstructural RNA and protein (E2 and nsP2) was reduced significantly by the compound QVIR.
Figure 6.

Effect of the compound QVIR in CHIKV RNA and protein levels. (a) CHIKV (S27) infection with an MOI of 0.01 and treatment with ribavirin (positive control) and the compound QVIR (5, 10, and 20 μM). Whole-cell RNA was isolated from the CHIKV-infected samples at 24 h post treatment and CHIKV nsP2 and E2 genes were amplified using respective primers by RT-PCR. (b) Bar diagrams showing the relative band intensities in the viral RNA expression pattern in infected and drug-treated samples as obtained through PRISM software. The open bar and solid bar represent E2 and nsP2, respectively. Data represented as mean ± SEM from three independent experiments. p ≤ 0.01 was considered to be statistically significant. (c) As mentioned above, infected and treated BHK-21 cell lysates were separated in 10% SDS PAGE, and the viral protein expression pattern was assessed by western blot using antibodies against the CHIKV nonstructural protein nsP2 and envelope E2 glycoproteins. In both, GAPDH was used as a loading control. (d) Bar diagrams showing the relative band intensities in the viral protein expression pattern in infected and drug-treated samples as obtained through PRISM software; the open bar and solid bar represent E2 and nsP2, respectively. Data represented as mean ± SEM from three independent experiments. p ≤ 0.01 was considered statistically significant (**p ≤ 0.01; ***p ≤ 0.001).
2.7. Flow Cytometry Observation of the Viral Protein Suppression Level
To demonstrate the effect of the compound QVIR on the level of CHIKV protein expression, flow cytometry investigation was performed for the E2 and nsP2 proteins. The treated and untreated virus-infected cells were prepared for the analysis by flow cytometry. The numbers of cells exhibiting the expression of nsP2 protein were reduced by 43.25, 54.04, and 71.48% at concentrations of 5, 10, and 20 μM, respectively. Similarly, the percentages of infected cells showing E2 expression were reduced by 44.60, 51.22, and 67.7% after treatment with 5, 10, and 20 μM QVIR, respectively (Figure 7a–d). Remarkably, the viral protein (nsP2 and E2) levels were decreased after ribavirin treatment, which was comparable with the compound of interest.
Figure 7.

Flow cytometric analysis depicting the inhibition of CHIKV envelope E2 glycoprotein expression after treatment with the compound QVIR. (a) Histogram analysis showing percent positive cells for envelope E2 glycoproteins: mock + infection (INF) S27 and infection (INF) + compound QVIR (5, 10, and 20 μM). (c) Graphical representation showing percent positive cells for mock + infection (INF) S27 and infection (INF) + compound QVIR (5, 10, and 20 μM). Similarly, the expression profile of nonstructural protein nsP2 was presented (b) as a histogram and (d) by graphical representations. Student’s t-test was performed to calculate the p values. p < 0.05 was considered to be statistically significant between groups (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001).
3. Discussion and Conclusions
Development of appropriate vaccine and effective antiviral drugs is urgently needed for the management of widespread incidences of CHIKV infection. Due to the absence of potent antiviral molecules for CHIKV infection, a structure-based virtual screening (SBVS) technique was applied to screen an in-house library of quinoline/quinolone derivatives to find out antiviral compounds against CHIKV. The workflow for SBVS is presented in Figure 1. Eighty unreported novel quinoline/quinolone derivatives were used for SBVS against three different CHIKV targets: the mature envelope E2 glycoprotein, the nonstructural protein nsP2, and the nonstructural protein nsP3. Ligands were docked within the cavity of the CHIKV envelope glycoprotein complex, between E1 and E2 (allosteric site). The catalytic dyad amino acid residues (Cys1013 and His1083) of the tunnel-shaped active site of nsp2 were used for docking calculations. The top 10 hits for each target were retained and three structurally diverse hits were chosen for each target based on their binding pattern in the active site and binding free energy. The docking scores (XP Gscore) and binding free energy of the top three compounds docked to E2, nsP2, and nsP3 are listed in Table 1. Out of these three hits, QVIR showed potential binding affinity with two CHIKV targets, the mature envelope E2 glycoprotein and the nonstructural protein nsP2, while it showed weak binding potential with the nonstructural protein nsP3. QVIR showed high binding affinity within the cavity of the CHIKV envelope glycoprotein complex, between E1 and E2 (allosteric site), and was predicted computationally as a viral entry inhibitor. The 3D representation of docked conformation of the selected compound QVIR and standard drugs ribavirin and chloroquine, with the envelope E2 glycoprotein and the nonstructural protein nsP2, obtained after XP docking is depicted in Figure 2A,B, respectively. QVIR was selected and synthesized for in vitro studies to assess its anti-CHIKV efficacy.
Targeting the CHIKV nsP2 protease actions within the C-domain would have an inhibitory activity on the viral replication. Singh et al. identified four potential protease inhibitors of nsP2 protease by screening a library of compounds by the application of a developed homology model for nsP2.41 Ideally, the replication cycle would be stopped by blocking the protein function when the drug binds to this active site (C-domain of nsP2 protease). A similar type of work was reported by Bassetto et al. through in silico identification of CHIKV nsP2 inhibitors by means of virtual screening of a large compound library by employing the developed homology model for the CHIKV nsP2.42
Infection caused by alphaviruses could be suppressed in vitro by blocking the intracellular furin-mediated cleavage of viral structural envelope proteins: E2E3 or p62 precursor. This blocking has been determined by showing the inhibitory effect of an irreversible furin-inhibiting peptide, decanoyl-RVKR-chloromethyl ketone, on in vitro CHIKV infection.43 This peptide significantly decreased the processing of CHIKV structural proteins E3E2 in infected myoblast cell cultures in vitro and led to the development of immature viral particles and impaired viral spreading among the cells. Thus, the chemical structure of the furin-inhibiting peptide may be a starting point for developing novel generations of active peptidomimetics utilizing the ligand-based drug design approach, targeting the intracellular furin cleavage step.
The glycoproteins E2 of CHIKV help in the binding of CHIKV to the host cell.44 The binding motifs in both the domains A and B of E2 glycoprotein promote interaction.45,46 A structural study of QVIR in the active site of E2 glycoprotein clearly showed the presence of three polar interactions (hydrogen bonds) with key amino acid residues Lys327, Cys328, and Gln138, while ribavirin interacts and forms two hydrogen bonds with Lys327 and Thr288. Similarly, chloroquine forms two hydrogen bonds with Lys327 and Pro26. Ser25, Pro26, Met27, Tyr137, Gln138, Ala286, Thr288, Gly326, Ala329, and Glu343 had been recognized to be the interacting residues present in the binding site of E2 for the potential activity of QVIR against the E2 glycoprotein of CHIKV.
The CHIKV nonstructural protein nsP2 play a multifunctional role with a C-terminal proteolytic domain (cysteine protease). It is an important drug target for antiviral development.41,42 The cysteine protease mainly consists of 324 residues, and deprotonation of the thiol group of cysteine triggers catalysis in the active site utilizing basic amino acids such as histidine.47 Due to its multifunctional role, the cysteine protease is broadly accepted as an important target for the development of antiviral drugs for CHIKV. Earlier, the cysteine protease was reported to be papain-like, with a catalytic pair composed of His and Cys residues.48 The structural analysis of the QVIR–nsP2 complex showed the presence of two hydrogen bonds with key amino acid residues Lys1045 and His1222, while ribavirin interacts to form three hydrogen bonds with Lys1045, Ala1046, and Glu1204. Meanwhile, chloroquine interacts to form only one hydrogen bond with Glu1204. QVIR has been found to be fixed in the catalytic dyad of nsP2 protease (His1083 and Cys1013). In addition, it is surrounded by a number of aromatic residues of Tyr1047, Tyr1079, Tyr1078, Trp1014, Trp104, His1083, and His1222, which can contribute to π–π stacking and increase the stability in the active site of nsP2.
The quinoline derivative chloroquine was first reported >35 years ago as an antiviral agent.49,50 Concerning the alphaviruses, chloroquine was established to be effective in vitro.51−53 In CHIKF, chloroquine and chloroquine phosphate were used as a cure and as anti-inflammatory agents against chronic CHIKV arthritis.54 Another antimalarial drug, quinine, a derivative of quinoline, also exhibited antiviral effects in vitro at an IC50 concentration (IC50 = 0.1 μg/mL) lesser than that of chloroquine (1.1 μg/mL).35 This prompted us to screen our in-house quinolone derivatives and evaluate them in vitro for their potential antiviral properties against CHIKV. It was found that QVIR exhibited an antiviral effect against CHIKV infection significantly in BHK-21 cells with insignificant cytotoxicity. The plaque was inhibited up to 99.61% at a QVIR concentration of 20 μM, and the EC50 value of the compound was found to be 2.2 ± 0.49 μM. Suramin, an antiparasitic drug, was also reported to have antiviral properties against Sindbis virus as well as Semliki Forest virus, and it is a strong inhibitor of CHIKV RNA synthesis.55 CHIKV structural and nonstructural RNA levels (81.02% for nsP2 and 75.08% for E2) were decreased after the treatment with QVIR. On the other hand, the structural (E2) and nonstructural (nsP2) protein levels of CHIKV were reduced by 69.1 and 53.8%, respectively. The antiviral properties of QVIR were confirmed by flow cytometry, and the CHIKV structural (E2) and nonstructural proteins were found to be suppressed at a significant level. Finally, to identify the potential antiviral mechanism of QVIR, time of addition assay was performed, and the drug was added at different time intervals post infection. The result showed that QVIR inhibited viral replication up to 99% at 0 hpi and this effect was maintained even up to 8 hpi. The release of infectious virus particles was inhibited to an extent of around 50% after the addition of drug within 20 hpi. This observation advocated that QVIR suppressed CHIKV infection at early and late stages of viral replication. It was concluded that QVIR is a potential antiviral molecule that has significant anti-CHIKV properties with no adverse cytotoxic effect when tested in vitro in BHK-21 cells. Significant inhibition in CHIKV RNA levels and a noticeable suppression of viral protein levels also suggested that QVIR may have multiple targets. This advocated the significance of QVIR as an antiviral drug candidate for further investigation against CHIKV infection.
4. Experimental Section
4.1. Materials
Chemicals and reagents were purchased from Sigma-Aldrich (India), Merck, and S. D. Fine Chemicals. Thin-layer chromatography (TLC) was used for the assessments of the confirmation of the reaction and purity of the compounds. Melting points (M.P.) of the synthesized compounds were obtained by using open capillary tubes on a melting point apparatus. The infrared (IR) spectrum was recorded using a Shimadzu FT-IR spectrometer in the 4000–400 cm–1 range by using KBr (potassium bromide) pellets. The Bruker 300 MHz NMR instrument was used for the recording of the 1H-NMR spectrum of compounds in the solvent DMSO-d6. UPLC–MS (Q-TOF-ESI) (Waters Corp., USA) with an ESI method was performed for molecular mass detection (m/z) and purity of the synthesized compounds.
4.2. Structure-Based Virtual Screening (SBVS)
Computations were accomplished on an Intel Core i7, 4.90 GHz processor with 16 GB memory and 2 GB graphics running with a Windows 10 operating system. The Maestro 9.4 graphical user interface (GUI) of Schrödinger was utilized as the computational platform. Glide in Maestro 9.4 was utilized for extra precision (XP) docking of ligands. The X-ray crystal structures of CHIKV envelope E2 glycoprotein (PDB ID: 3N42; resolution, 3 Å) and nonstructural proteins nsP2 (PDB ID: 3TRK; resolution, 2.397 Å) and nsP3 (PDB ID: 4TU0; resolution, 2.3 Å) were retrieved from Protein Data Bank (PDB) and applied for all of the computational studies.45 The preparation of protein structure like deletion of water molecules, assignment of bond orders, inclusion of hydrogen atoms, and treatment of formal charges were done by Protein Preparation Wizard in Maestro 9.4. The hydrogen bonding network was optimized through the exhaustive sampling option. The energy of the protein structure was minimized to an RMSD of 0.3 Å by utilizing the Impref module with the OPLS_2005 force field. Glide scoring grids (docking grid box of 20 × 20 × 20 Å) were generated by defining the active binding site residues in the protein structure. Before docking simulation with Glide XP, an in-house library of 80 quinoline compounds was screened based on Lipinski’s “Rule of 5” to figure out compounds with potential drug candidate likeness.56 Structurally similar compounds (Tanimoto coefficient < 0.1) were clustered in a single molecule. LigPrep 2.6 with Epik 2.4 had been utilized to expand protonation, and tautomeric states of ligands at pH 7.0 ± 2.0 and then energy were minimized by utilizing the OPLS_2005 force field. Therefore, a docking library of 156 compounds was constructed. The docking simulation of the prepared ligand library was performed by Glide XP docking.57 The binding free energy of the docked ligands was evaluated by MM-GBSA (VSGB 2.0).58 The top hits were chosen depending on their contacts with the critical amino acid residues in the binding region of the target protein and binding free energy assessment of the docked complex.
4.3. Synthesis of 4-Hydroxy-6-methyl-2H-pyrano[3,2-c]quinoline-2,5(6H)-dione (Compound 1)
Diethylmalonate (32.1 g, 0.2 mol) was added in a round-bottom flask having N-methyl aniline (10.7 g, 0.1 mol) and diphenyl ether (30 g), equipped with a distillation unit with a vigreux column. About 22 mL of ethanol was distilled during the reflux time of 10 h. The temperature of the reaction mixture was then reduced to 100 °C, 50 mL of dioxane was added, and the mixture was kept overnight. The precipitate was collected by filtration, and diphenyl ether was removed from the precipitate by washing with dioxane and diethyl ether. The product was recrystallized with dimethyl formamide as a light brown powder. Yield: 16 g (66%); M.P.: 256–258 °C; IR (KBr, cm–1): 1665 cm–1 (—C=O amide), 1742 cm–1 (—C=O lactone), 3050 cm–1 (−CH str.), 3447 cm–1 (−OH); 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 3.56 (s, 3H, N-CH3), 5.68 (s, 1H, pyrano H), 7.38–7.73 (m, 4H, Ar-H), 13.10 (s, 1H, OH); MS (ESI): m/z 243.09 (M+).
4.4. Synthesis of 3-Acetyl-4-hydroxy-1-methylquinolin-2(1H)-one (Compound 2)
Lactone (compound 1, 9.73 g, 0.04 mol) in ethylene glycol (90 mL) was treated with NaOH solution (80%, 10 mL), and the mixture was gently boiled. After 2 h, 350 mL of ice water was added to the reaction mixture and then slowly acidified with conc. HCl (25 mL). The precipitated solid product was filtered and washed thoroughly with water and dried. The synthesized compound was recrystallized with toluene as a pale yellow powder. Yield: 8.05 g (93%); M.P.: 155–157 °C; IR (KBr, cm–1): 1652 cm–1 (—C=O amide), 1739 cm–1 (—C=O acetyl), 2955 cm–1 (−CH str.), 3472 cm–1 (−OH); 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 2.80 (s, 3H, CH3 of COCH3), 3.76 (s, 3H, N-CH3), 7.34–8.62 (m, 4H, Ar-H), 14.18 (s, 1H, OH); MS (ESI): m/z 217.11 (M+).
4.5. Synthesis of 4-Hydroxy-1-methyl-3-(3-morpholinopropanoyl)quinoline-2(1H)-one Hydrochloride (Compound 3, QVIR)
Compound 2 (3-acetyl-4-hydroxy-1-methylquinolin-2(1H)-one; 6.5 g, 30 mmol), morpholine (3.5 g, 40 mmol), and paraformaldehyde (1.2 g, 13.2 mmol) were taken in a 100 mL round-bottom flask, attached to a reflux condenser. In this mixture, conc. HCl (1 mL) in 50 mL absolute ethanol was added and then refluxed for 6 h. To the reaction mixture, acetone (50 mL) was added while still warm, allowed to cool to room temperature, and then chilled in a refrigerator overnight. The crystalline solid product was filtered and washed with acetone. The product was recrystallized with a mixture of ethanol and acetone as a pale yellow fluffy solid. Yield: 8.45 g (80%); M.P.: 297–300 °C; IR (KBr, cm–1): 1653 (C=O amide), 1738 (C=O acetyl), 3042 (Ar C-H), 3472 cm–1 (−OH); 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 2.65–2.70 (t, 4H, NCH2, morpholine, J = 7.5 Hz), 3.40–3.45 (t, 2H, COCH2, J = 7.5 Hz), 3.58 (s, 3H, NCH3), 3.62–3.67 (t, 2H, NCH2, J = 7.5 Hz), 3.72–3.77 (t, 4H, OCH2, morpholine, J = 7.5 Hz), 7.40–7.75 (m, 4H, Ar-H), 15.10 (s, 1H, OH); 13C-NMR (125 MHz, DMSO-d6) δ (ppm): 206.52, 177.99, 160.36, 141.49, 135.85, 125.24, 121.06, 116.84, 113.02, 105.49, 69.51, 57.57, 43.52, 35.48, 32.95; MS (ESI): m/z 316.19 (M+), 317.20 (M+ + H).
4.6. UPLC–MS/MS Analysis of QVIR
UPLC analysis of QVIR was performed on a Waters ACQUITY UPLC system (Waters Corp., MA, USA) equipped with a binary solvent delivery system, an autosampler, a column manager, and a tunable synapt MS detector (Synapt; Waters, U.K.). UPLC chromatographic separation was performed on a Waters ACQUITY UPLC BEH C18 column (100 × 2.1 mm, 1.7 μm particle size) with a Waters ACQUITY Van Guard precolumn (5 × 2.1 mm, 1.7 μm particle size). The mobile phase consisted of acetonitrile:2 mmol of ammonium acetate buffer (90:10, v/v) at a column temperature of 40 °C, an autosampler temperature at 4 °C, and a flow rate of 0.2 mL/min. The injection volume was 10 μL in full loop mode. The total chromatographic run time was 3.0 min. Synapt mass spectrometric detection was carried out on a UPLC–MS/MS (Q-TOF-ESI) (Waters Corp., USA) with an electrospray ionization (ESI) technique. The ESI source was used in positive ionization mode. A micro channel plate (MCP) was used as a detector plate. The optimal MS parameters were as follows: capillary voltage, 3.60 kV; cone voltage, 21 V; extraction cone voltage, 7.40 V; source temperature, 120 °C; desolvation temperature, 350 °C; cone gas flow, 50 L/h; desolvation gas flow, 500 L/h; collision energy, 16.5. Nitrogen was used as the desolvation and cone gas. Argon was used as the collision gas at a flow of 1.5 mL/min. All data collected in centroid mode were acquired and processed using Mass Lynx (ver. 4.1) software.
4.7. Cell Lines and Viruses
BHK-21 (baby hamster kidney) cells were maintained in an EMEM medium (Sigma-Aldrich) supplemented with 10% heat-inactivated FBS and 1% penicillin–streptomycin solution, incubated in a humidified incubator at 37 °C with 5% CO2, and subcultured every 3 days to maintain subconfluency. The CHIKV S27 prototype strain was used in this study. The propagation of the isolate was carried out in BHK-21 cells following the standard virus adsorption technique.
4.8. Antiviral Drug Assays and RT-PCR
BHK-21 cells were planted in six-well cell culture plates and allowed to adhere for 24 h. The confluent monolayers of the BHK-21 cells were infected with CHIKV at a multiplicity of infection (MOI) of 0.01. The virus became allowed to adsorb onto cells for 1 h, and then a viral inoculum was removed from the wells. To remove unbound viruses, the wells were washed twice with phosphate-buffered saline. Next, the infected cells were treated with DMSO, ribavirin (10 μM), and different doses of QVIR (5, 10, and 20 μM) prepared in the medium. The culture plates were incubated in a CO2 incubator at 37 °C supplemented with 5% CO2. The cells were then collected at 24 h post infection (hpi), and the RNA was isolated by using the TRIzol (Invitrogen Corp., Carlsbad, CA) method of RNA extraction. Using an equal amount of RNA (1 μg), complementary DNA was synthesized from the extracted RNA through reverse transcription in the presence of random hexamers. This cDNA was used to amplify CHIKV nonstructural and structural genes (nsP2 and E2)59 (Qiagen RT-PCR kit). Primers for nsP2, E2, and GAPDH were designed with the help of online tools (http://bioinfo.ut.ee/primer3-0.4.0/). The designed primers and their respective amplified fragments are mentioned in Table 2. Primers for GAPDH were used to test for the adequacy of the specimen, extraction, and reverse transcriptase (RT) procedure. The nsP2 and E2 primers, forward and reverse, were used to amplify their respective gene fragments (Table 2). PCR was performed using Qiagen RT-PCR kit in a 25 μL reaction volume with cycling condition mentioned in Table 2.
Table 2. PCR Profile.

4.9. Cellular Cytotoxicity Assay
To determine the changes in the cellular morphology of BHK-21 cells as a result of QVIR treatment, the cells were observed microscopically. Briefly, the cells were incubated in a CO2 incubator at 37 °C in the absence or presence of QVIR for 24 h at concentrations of 5, 10, and 20 μM and observed under a 20× objective using a phase-contrast microscope. This assay was performed three times independently in triplicate. To evaluate the adverse toxicity of QVIR on BHK-21 cells, the cells were cultured in 96-well plates (Corning) and, at 90% confluency, the cells were exposed with increasing concentration (from 10 to 200 μM) of QVIR at 37 °C with 5% CO2 for 24 h. The cells without any treatment were taken as a control. The MTT (3-{4,5-dimethylthiazol-2-yl}-2,5-diphenyltetrazolium bromide) method was used to determine the cellular toxicity of the drug. At 24 h post drug treatment, MTT was added at a concentration of 5 mg/mL (HiMedia) for 5 h at 37 °C. The formazan crystals formed were dissolved with 100 μL of DMSO for 15 min at 37 °C, and the absorbance was recorded at 570 nm using an ELISA plate reader. The percentage of cell viability was evaluated and CC50 was determined.60 The MTT analysis was carried out three times separately.
4.10. Plaque Assay
For quantification of the infectious viral titer, plaque assay was carried out to find out the inhibitory activity of QVIR against infection of CHIKV. The supernatant was collected from infected and treated BHK-21 cells at 24 h post treatments. Each sample was subjected to serial dilution (from 10–1 to 10–5) in a medium with 2% FBS. The diluted virus suspension was used to infect BHK-21 cells after the confluency reached approximately 90% in six-well tissue culture plates and incubated for 2 h with shaking at 37 °C with 5% CO2. The unbound viruses were removed by washing twice with 1× PBS and overlaid with 1% carboxymethyl cellulose (CMC) prepared in DMEM and incubated at 37 °C with 5% CO2 for 4 days. CMC was then removed and the cells were fixed with 4% paraformaldehyde and stained with 1% crystal violet solution for the visualization and enumeration of plaque. Viral titers were subsequently expressed as PFU per milliliter.61
4.11. Time of Addition Assay
To find out the possible mechanism of action for the QVIR drug on CHIKV replication, a time of addition experiment was performed. BHK-21 cells were infected with CHIKV at an MOI of 0.01 and 20 μM. QVIR was added at 0, 2, 4, 8, 12, 16, 20, and 24 h post infection. Ribavirin was used as a positive control (10 μM). The viral replication was measured by plaque assay. Titration at all times was performed at the end of the experiment.
4.12. Western Blot
As described earlier,61 BHK-21 cells infected with CHIKV were harvested after 24 h of drug treatment as per the procedure mentioned in Section 4.8. Harvested cells were lysed with RIPA buffer as well as with the help of an ultrasonicator. After quantification, protein-containing cell lysate samples were separated on 10% SDS-polyacrylamide gel by loading 50 μg of protein in each well and run for 3 h at 100 V. Separated proteins were subsequently transferred onto a polyvinylidene difluoride (PVDF) membrane. To determine the CHIKV structural and nonstructural proteins, the PVDF membrane was blocked with 5% skim milk and washed and then the membrane was incubated with E2 (The Native Antigen Company) and nsP2 (Antibody Research Corporation) primary antibodies at a dilution factor of 1:2500. The membrane was then washed three times with washing buffer, and GAPDH (GAPDH antibody was obtained from GeneTex) was used as a loading control. The PVDF membrane was then subsequently incubated for 2 h with secondary antibody (goat anti-mouse HRP-conjugated) at a dilution of 1:5000. The membrane was then washed and blots were developed with developing reagents. The band intensities of the CHIKV structural and nonstructural proteins were measured from three independent experiments.
4.13. Flow Cytometric Analysis (FACS)
The BHK-21 cells were divided into four groups: mock cells, CHIKV-infected cells, CHIKV-infected cells treated with different concentrations of QVIR (5, 10, and 20 μM), and CHIKV-infected cells treated with ribavirin. Treated and untreated cells at 24 h post treatment were removed from cell culture plates with the help of the trypsinization method. Cells were then washed with PBS and then fixed in 4% PFA for 15 min at room temperature. Before intracellular staining of CHIKV E2 and nsP2 antigens, the treated and untreated cells were permeabilized first with the help of permeabilization buffer (1× PBS + 0.5% BSA + 0.2% saponin + 0.01% NaN3) and then blocked with 1% bovine serum albumin in permeabilization buffer for 1 h at room temperature. The cells were then incubated with anti-E2 and nsP2 antibodies for 45 min at room temperature. The cells were then washed twice to remove free E2 and nsP2 antibodies with the help of permeabilization buffer and then finally incubated with Alexa Fluor (AF) 488 anti-mouse antibodies (Invitrogen, USA). The cells were washed and resuspended in FACS buffer and approximately 10,000 cells from each sample were acquired on a BD LSR II flow cytometer (Becton Dickinson). This assay was performed two times in triplicate. Statistical analysis was performed by using GraphPad Prism.
4.14. Statistical Analysis
The statistical analysis between groups was conducted by using one-way ANOVA in GraphPad Prism 5.0 software. Error bars represent the standard error of the mean (SEM). Results are from one of three representative experiments.
Acknowledgments
M.I. thanks the Department of Science and Technology-SERB, Government of India, for providing financial assistance (DST no.: PDF/2016/003753). M.I. thanks Japanese Society for the Promotion of Science (JSPS), Tokyo, Japan, for providing the fellowship during the experimental design of the study (JSPS/OF322, ID no. P19108). O.A., A.S.A.A., and M.A.A. also would like to thank Prince Sattam Bin Abdulaziz University for providing the necessary resources to conduct this research. We are indebted to Dr. Mohd Aslam (Advisor, Department of Biotechnology, Govt. of India) for the scientific advice in this study. The author expresses their sincere thanks to Dr. Ajai Kumar, Senior Technical Assistant, AIRF, JNU (New Delhi), for the UPLC–MS/MS analysis. We are thankful to BD FACS Academy, Jamia Hamdard, New Delhi (India), for providing flow cytometry facilities.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c00447.
1H-NMR and 13C-NMR spectra of the compound QVIR (4-hydroxy-1-methyl-3-(3-morpholinopropanoyl)quinoline-2(1H)-one hydrochloride) (PDF)
Author Contributions
# M.I. and O.A. contributed equally to this work.
Author Contributions
M.I., O.A., and S.P. conceived and designed the experiments. M.I., O.A., and S.P. performed the experiments and wrote the manuscript. M.I., O.A., S.P., W.H.K., M.H., and I.H. carried out data acquisition and the interpretation and analysis of results. O.A., A.S.A.A., and M.A.A. performed the synthesis and analysis of the compounds and contributed to reviewing of the manuscript. K.K. designed the experiments and contributed to reviewing of the manuscript. The final manuscript has been read and approved by all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Robinson M. C. An epidemic of virus disease in Southern Province, Tanganyika Territory, in 1952-53. I. Clinical features. Trans. R. Soc. Trop. Med. Hyg. 1955, 49, 28–32. 10.1016/0035-9203(55)90080-8. [DOI] [PubMed] [Google Scholar]
- Lam S. K.; Chua K. B.; Hooi P. S.; Rahimah M. A.; Kumari S.; Tharmaratnam M.; Chuah S. K.; Smith D. W.; Sampson I. A. Chikungunya infection--an emerging disease in Malaysia. Southeast Asian J. Trop. Med. Public Health 2001, 32, 447–451. [PubMed] [Google Scholar]
- Ledoux A.; Cao M.; Jansen O.; Mamede L.; Campos P. E.; Payet B.; Clerc P.; Grondin I.; Girard-Valenciennes E.; Hermann T.; Litaudon M.; Vanderheydt C.; Delang L.; Neyts J.; Leyssen P.; Frédérich M.; Smadja J. Antiplasmodial, anti-chikungunya virus and antioxidant activities of 64 endemic plants from the Mascarene Islands. Int. J. Antimicrob. Agents 2018, 52, 622–628. 10.1016/j.ijantimicag.2018.07.017. [DOI] [PubMed] [Google Scholar]
- Weaver S. C.; Lecuit M. Chikungunya Virus Infections. N. Engl. J. Med. 2015, 373, 94–95. 10.1056/NEJMc1505501. [DOI] [PubMed] [Google Scholar]
- Cecilia D. Current status of dengue and chikungunya in India. WHO South-East Asia J. Public Health 2014, 3, 22–27. 10.4103/2224-3151.206879. [DOI] [PubMed] [Google Scholar]
- Powers A. M. Vaccine and Therapeutic Options To Control Chikungunya Virus. Clin. Microbiol. Rev. 2018, 31, e00104–e00116. 10.1128/CMR.00104-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougeron V.; Sam I. C.; Caron M.; Nkoghe D.; Leroy E.; Roques P. Chikungunya, a paradigm of neglected tropical disease that emerged to be a new health global risk. J. Clin. Virol. 2015, 64, 144–152. 10.1016/j.jcv.2014.08.032. [DOI] [PubMed] [Google Scholar]
- Strauss J. H.; Strauss E. G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. 10.1128/MR.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss E. G.; Rice C. M.; Strauss J. H. Complete nucleotide sequence of the genomic RNA of Sindbis virus. Virology 1984, 133, 92–110. 10.1016/0042-6822(84)90428-8. [DOI] [PubMed] [Google Scholar]
- LaStarza M. W.; Lemm J. A.; Rice C. M. Genetic analysis of the nsP3 region of Sindbis virus: evidence for roles in minus-strand and subgenomic RNA synthesis. J. Virol. 1994, 68, 5781–5791. 10.1128/JVI.68.9.5781-5791.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp J. C.; Sokoloski K. J.; Gebhart N. N.; Hardy R. W. Alphavirus RNA synthesis and non-structural protein functions. J. Gen. Virol. 2015, 96, 2483–2500. 10.1099/jgv.0.000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; Goonawardane N.; Ward J.; Tuplin A.; Harris M. Multiple roles of the nonstructural protein 3 (nsP3) alphavirus unique domain (AUD) during Chikungunya virus genome replication and transcription. PLoS Pathog. 2019, 15, e1007239 10.1371/journal.ppat.1007239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelnabi R.; Jacobs S.; Delang L.; Neyts J. Antiviral drug discovery against arthritogenic alphaviruses: tools and molecular targets. Biochem. Pharmacol. 2020, 174, 113777. 10.1016/j.bcp.2019.113777. [DOI] [PubMed] [Google Scholar]
- Seyedi S. S.; Shukri M.; Hassandarvish P.; Oo A.; Shankar E. M.; Abubakar S.; Zandi K. Computational Approach Towards Exploring Potential Anti-Chikungunya Activity of Selected Flavonoids. Sci. Rep. 2016, 6, 24027. 10.1038/srep24027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini E. J.; Clarke M.; Gowen B. E.; Rutten T.; Fuller S. D. Cryo-electron microscopy reveals the functional organization of an enveloped virus, Semliki Forest virus. Mol. Cell 2000, 5, 255–266. 10.1016/S1097-2765(00)80421-9. [DOI] [PubMed] [Google Scholar]
- Lescar J.; Roussel A.; Wien M. W.; Navaza J.; Fuller S. D.; Wengler G.; Wengler G.; Rey F. A. The Fusion glycoprotein shell of Semliki Forest virus: An icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 2001, 105, 137–148. 10.1016/S0092-8674(01)00303-8. [DOI] [PubMed] [Google Scholar]
- Vincent M. J.; Bergeron E.; Benjannet S.; Erickson B. R.; Rollin P. E.; Ksiazek T. G.; Seidah N. G.; Nichol S. T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. 10.1186/1743-422X-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolain J. M.; Colson P.; Raoult D. Recycling of chloroquine and its hydroxyl analogue to face bacterial, fungal and viral infections in the 21st century. Int. J. Antimicrob. Agents 2007, 30, 297–308. 10.1016/j.ijantimicag.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinson J. A.; Montoya C. J.; Usuga X.; Ronquillo R.; Landay A. L.; Desai S. N. Chloroquine modulates HIV-1-induced plasmacytoid dendritic cell alpha interferon: Implication for T-cell activation. Antimicrob. Agents Chemother. 2010, 54, 871–881. 10.1128/AAC.01246-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Trani L.; Savarino A.; Campitelli L.; Norelli S.; Puzelli S.; D’Ostilio D.; Vignolo E.; Donatelli I.; Cassone A. Different pH requirements are associated with divergent inhibitory effects of chloroquine on human and avian influenza A viruses. Virol. J. 2007, 4, 39. 10.1186/1743-422X-4-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X.; Wang L.; Wang B.; Miao K.; Xiang K.; Feng S.; Gao L.; Shen H. C.; Yun H. Discovery of Piperazinyl quinoline Derivatives as Novel Respiratory Syncytial Virus Fusion Inhibitors. ACS Med. Chem. Lett. 2016, 7, 558–562. 10.1021/acsmedchemlett.5b00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph V. B.; Winkler G.; Stollar V. Acidotropic amines inhibit proteolytic processing of flavivirus prM protein. Virology 1990, 174, 450–458. 10.1016/0042-6822(90)90099-D. [DOI] [PubMed] [Google Scholar]
- Savarino A.; Boelaert J. R.; Cassone A.; Majori G.; Cauda R. Effects of chloroquine on viral infections: An old drug against today’s diseases?. Lancet Infect. Dis. 2003, 3, 722–727. 10.1016/S1473-3099(03)00806-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarino A.; Di Trani L.; Donatelli I.; Cauda R.; Cassone A. New insights into the antiviral effects of chloroquine. Lancet Infect. Dis. 2006, 6, 67–69. 10.1016/S1473-3099(06)70361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodell J. R.; Puig-Basagoiti F.; Forshey B. M.; Shi P.-Y.; Ferguson D. M. Identification of Compounds with Anti-West Nile Virus Activity. J. Med. Chem. 2006, 49, 2127–2137. 10.1021/jm051229y. [DOI] [PubMed] [Google Scholar]
- Ezgimen M.; Lai H.; Mueller N. H.; Lee K.; Cuny G.; Ostrov D. A.; Padmanabhan R. Characterization of the 8-hydroxyquinoline scaffold for inhibitors of West Nile virus serine protease. Antiviral Res. 2012, 94, 18–24. 10.1016/j.antiviral.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi F.; Guimarães Corrêa A. B.; Mascarenhas R. E.; Galvão B.; Séon-Méniel B.; Schmidt F.; Franck X.; Hocquemiller R.; Figadère B.; Fournet A. Quinoline compounds decrease in vitro spontaneous proliferation of peripheral blood mononuclear cells (PBMC) from human T-cell lymphotropic virus (HTLV) type-1-infected patients. Biomed. Pharmacother. 2008, 62, 430–435. 10.1016/j.biopha.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Talamas F. X.; Abbot S. C.; Anand S.; Brameld K. A.; Carter D. S.; Chen J.; Davis D.; de Vicente J.; Fung A. D.; Gong L.; Harris S. F.; Inbar P.; Labadie S. S.; Lee E. K.; Lemoine R.; Le Pogam S.; Leveque V.; Li J.; McIntosh J.; Nájera I.; Park J.; Railkar A.; Rajyaguru S.; Sangi M.; Schoenfeld R. C.; Staben L. R.; Tan Y.; Taygerly J. P.; Villaseñor A. G.; Weller P. E. Discovery of N-[4-[6-tert-butyl-5-methoxy-8-(6-methoxy-2-oxo-1H-pyridin-3-yl)-3 quinolyl] phenyl] methanesulfonamide (RG7109), a potent inhibitor of the hepatitis C virus NS5B polymerase. J. Med. Chem. 2014, 57, 1914–1931. 10.1021/jm401329s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa-Lima G.; Moraes A. M.; da Araújo A. S.; da Silva E. T.; de Freitas C. S.; Vieira Y. R.; Marttorelli A.; Neto J. C.; Bozza P. T.; de Souza M. V. N.; Souza T. M. L. 2,8-Bis(trifluoromethyl)quinoline analogs show improved anti-Zika virus activity, compared to mefloquine. Eur. J. Med. Chem. 2017, 127, 334–340. 10.1016/j.ejmech.2016.12.058. [DOI] [PubMed] [Google Scholar]
- Huang S. H.; Lien J. C.; Chen C. J.; Liu Y. C.; Wang C. Y.; Ping C. F.; Lin Y. F.; Huang A. C.; Lin C. W. Antiviral Activity of a Novel Compound CW-33 against Japanese Encephalitis Virus through Inhibiting Intracellular Calcium Overload. Int. J. Mol. Sci. 2016, 17, 1386. 10.3390/ijms17091386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai H.; Sridhar P. G.; Padmanabhan R. Characterization of 8-hydroxyquinoline derivatives containing aminobenzothiazole as inhibitors of dengue virus type 2 protease in vitro. Antiviral Res. 2013, 97, 74–80. 10.1016/j.antiviral.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonyasuppayakorn S.; Reichert E. D.; Manzano M.; Nagarajan K.; Padmanabhan R. Amodiaquine, an antimalarial drug, inhibits dengue virus type 2 replication and infectivity. Antiviral Res. 2014, 106, 125–134. 10.1016/j.antiviral.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra R. K. F.; Kümmerle A. E.; Guedes G. P.; Barros C. S.; Gomes R. S. P.; Cirne-Santos C. C.; Paixão I. C. N. P.; Neves A. P. Quinolone-N-acylhydrazone hybrids as potent Zika and Chikungunya virus inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 126881. 10.1016/j.bmcl.2019.126881. [DOI] [PubMed] [Google Scholar]
- Khan M.; Santhosh S. R.; Tiwari M.; Lakshmana Rao P. V.; Parida M. Assessment of in Vitro Prophylactic and Therapeutic Efficacy of Chloroquine Against Chikungunya Virus in Vero Cell. J. Med. Virol. 2010, 82, 817. 10.1002/jmv.21663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deLamballerie X.; Ninove L.; Charrel R. Antiviral treatment of chikungunya virus infection. Infect. Disord. Drug Targets 2009, 9, 101–104. 10.2174/187152609787847712. [DOI] [PubMed] [Google Scholar]
- Rashad A. A.; Mahalingam S.; Keller P. A. Chikungunya virus: emerging targets and new oppertunities for medicinal chemistry. J. Med. Chem. 2013, 57, 1147–1166. 10.1021/jm400460d. [DOI] [PubMed] [Google Scholar]
- Hucke F. I. L.; Bugert J. J. Current and Promising Antivirals Against Chikungunya Virus. Front. Public Health 2020 Dec 15, 8, 618624. 10.3389/fpubh.2020.618624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roschger P.; Fiala W.; Stadlbauer W. Nucleophilic Substitution and Ring Closure Reactions of 4-Chloro-3-nitro-2-quinolones [1]. J. Heterocycl. Chem. 1992, 29, 225–231. 10.1002/jhet.5570290141. [DOI] [Google Scholar]
- Thompson B. B. The Mannich Reaction. Mechanistic and Technological Considerations. J. Pharm. Sci. 1968, 57, 715–733. 10.1002/jps.2600570501. [DOI] [PubMed] [Google Scholar]
- Kaur P.; Thiruchelvan M.; Lee R. C. H.; Chen H.; Chen K. C.; Ng M. L.; Chu J. J. H. Inhibition of chikungunya virus replication by harringtonine, a novel antiviral that suppresses viral protein expression. Antimicrob. Agents Chemother. 2013, 57, 155–167. 10.1128/AAC.01467-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K. D.; Kirubakaran P.; Nagarajan S.; Sakkiah S.; Muthusamy K.; Velmurgan D.; Jeyakanthan J. Homology modeling, molecular dynamics, e-pharmacophore mapping and docking study of chikungunya virus nsP2 protease. J. Mol. Model. 2012, 18, 39–51. 10.1007/s00894-011-1018-3. [DOI] [PubMed] [Google Scholar]
- Bassetto M.; De Burghgraeve T.; Delang L.; Massarotti A.; Coluccia A.; et al. Computer-aided identification, design and synthesis of a novel series of compounds with selective antiviral activity against chikungunya virus. Antiviral Res. 2013, 98, 12–18. 10.1016/j.antiviral.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Ozden S.; Lucas-Hourani M.; Ceccaldi P. E.; Basak A.; Valentine M.; Benjannet S.; Hamelin J.; Jacob Y.; Mamchaoui K.; Mouly V.; Desprès P.; Gessain A.; Butler-Browne G.; Chrétien M.; Tangy F.; Vidalain P. O.; Seidah N. G. Inhibition of Chikungunya virus infection in cultured human muscle cells by furin inhibitors: impairment of the maturation of the E2 surface glycoprotein. J. Biol. Chem. 2008, 283, 21899–21908. 10.1074/jbc.M802444200. [DOI] [PubMed] [Google Scholar]
- Ashbrook A. W.; Burrack K. S.; Silva L. A.; Montgomery S. A.; Heise M. T.; Morrison T. E.; Dermody T. S. Residue 82 of the Chikungunya virus E2 attachment protein modulates viral dissemination and arthritis in mice. J. Virol. 2014, 88, 12180–12192. 10.1128/JVI.01672-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss J. E.; Vaney M. C.; Duquerroy S.; Vonrhein C.; Girard-Blanc C.; Crublet E.; Thompson A.; Bricogne G.; Rey F. A. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 2010, 468, 709–712. 10.1038/nature09555. [DOI] [PubMed] [Google Scholar]
- Li L.; Jose J.; Xiang Y.; Kuhn R. J.; Rossmann M. G. Structural changes of envelope proteins during alphavirus fusion. Nature 2010, 468, 705–708. 10.1038/nature09546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudkowska M.; Zagdańska B. Multifunctional role of plant cysteine proteinases. Acta Biochim. Pol. 2004, 51, 609–624. 10.18388/abp.2004_3547. [DOI] [PubMed] [Google Scholar]
- Rashad A. A.; Mahalingam S.; Keller P. A. Chikungunya virus: Emerging targets and new opportunities for medicinal chemistry. J. Med. Chem. 2014, 57, 1147–1166. 10.1021/jm400460d. [DOI] [PubMed] [Google Scholar]
- Inglot A. D. Comparison of the antiviral activity in vitro of some non-steroidal anti-inflammatory drugs. J. Gen. Virol. 1969, 4, 203–214. 10.1099/0022-1317-4-2-203. [DOI] [PubMed] [Google Scholar]
- Shimizu Y.; Yamamoto S.; Homma M.; Ishida N. Effect of chloroquine on the growth of animal viruses. Arch. Gesamte. Virus forsch. 1972, 36, 93–104. 10.1007/BF01250299. [DOI] [PubMed] [Google Scholar]
- Coombs K.; Mann E.; Edwards J.; Brown D. T. Effects of chloroquine and cytochalasin B on the infection of cells by sindbis virus and vesicular stomatitis virus. J. Virol. 1981, 37, 1060–1065. 10.1128/JVI.37.3.1060-1065.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A.; Marsh M.; White J. Inhibition of Semliki forest virus penetration by lysosomotropic weak bases. J. Gen. Virol. 1982, 58, 47–61. 10.1099/0022-1317-58-1-47. [DOI] [PubMed] [Google Scholar]
- Cassell S.; Edwards J.; Brown D. T. Effects of lysosomotropic weak bases on infection of BHK-21 cells by sindbis virus. J. Virol. 1984, 52, 857–864. 10.1128/JVI.52.3.857-864.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brighton S. W. Chloroquine phosphate treatment of chronic chikungunya arthritis. An open pilot study. S. Afr. Med. J. 1984, 66, 217–218. [PubMed] [Google Scholar]
- Ho Y. J.; Wang Y. M.; Lu J. W.; Wu T. Y.; Lin L. I.; et al. Suramin Inhibits Chikungunya Virus Entry and Transmission. PLoS One 2015, 10, e0133511 10.1371/journal.pone.0133511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 23, 23–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Li J.; Abel R.; Zhu K.; Cao Y.; Zhao S.; Friesner R. A. The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. 10.1002/prot.23106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das I.; Basantray I.; Mamidi P.; Nayak T. K.; Pratheek B. M.; Chattopadhyay S.; Chattopadhyay S. Heat shock protein 90 positively regulates Chikungunya virus replication by stabilizing viral non-structural protein nsP2 during infection. PLoS One 2014, 9, e100531 10.1371/journal.pone.0100531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelnabi R.; Staveness D.; Near K. E.; Wender P. A.; Delang L.; Neyts J.; Leyssen P. Comparative analysis of the anti-chikungunya virus activity of novel bryostatin analogs confirms the existence of a PKC-independent mechanism. Biochem. Pharmacol. 2016, 120, 15–21. 10.1016/j.bcp.2016.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S.; Kumar A.; Mamidi P.; Nayak T. K.; Das I.; Chhatai J.; Basantray I.; Bramha U.; Maiti P. K.; Singh S.; Suryawanshi A. R.; Chattopadhyay S. Development and characterization of monoclonal antibody against non-structural protein-2 of Chikungunya virus and its application. J. Virol. Methods 2014, 199, 86–94. 10.1016/j.jviromet.2014.01.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.