

Abstract

Pure antiestrogens, or selective estrogen receptor degraders (SERDs), have proven to be effective in treating breast cancer that has progressed on tamoxifen and/or aromatase inhibitors. However, the only FDA-approved pure antiestrogen, fulvestrant, is limited in efficacy by its low bioavailability. The search for orally bioavailable SERDs has continued for nearly as long as the clinical history of the injection-only fulvestrant. Oral SERDs that have been developed and tested in patients ranged from nonsteroidal ER binders containing an acrylic acid or amino side chain to bifunctional proteolysis-targeting chimera (PROTAC) pure ER degraders. Structural evolution in the development of oral SERD molecules has been closely associated with quantifiable ER-degrading potency, as seen in the structural comparison analysis of acrylic acid and basic amino side-chain-bearing SERDs. Failure to improve on fulvestrant in the clinical trials by numerous acidic SERDs and early basic SERDs is blamed on tolerability and/or insufficient efficacy, which will likely be overcome by the new-generation basic SERD molecules and PROTAC ER degraders with improved oral bioavailability, low toxicity, and superior efficacy of receptor degradation.
Introduction
The estrogen receptor (ER) is an estrogen-inducible transcription factor that regulates the expression of target genes involved in metabolism, development, and reproduction. In the absence of estrogen, the receptor is associated with heat shock proteins that stabilize and protect the receptor and maintain the DNA binding region in an inactive state. Upon binding an estrogen, the receptor undergoes a conformational change that enables the dissociation from heat shock proteins and facilitates the formation of a receptor dimer.1 The homodimer creates new surfaces that recognize and bind to the estrogen-response elements (EREs) of the DNA to activate cell-specific transcriptional responses in coordination with coregulatory proteins in a given cell.
The estrogen receptor is expressed in approximately 75% of all breast cancers which are dependent on estrogen stimulation for tumor growth. Tamoxifen became the first targeted therapy for breast cancer as an antagonist of the ER to block estrogen-stimulated proliferation of breast tumor cells. However, it soon became clear that tamoxifen has tissue-selective agonist properties,2a and clinical evidence of an estrogen-like stimulation of tumors by tamoxifen has been observed in breast cancer patients in the beginning of tamoxifen treatment.2b,2c This partial agonist activity limits the expression of antagonism and calls into question whether the therapeutic efficacy of tamoxifen could also be limited by its mixed action toward the estrogen receptor in different tissues.2a,2d−2f Indeed, many of the side effects of tamoxifen were believed to result from its partial agonist activity observed in the clinic as well as in laboratory animal models. Complete endocrine ablation by novel molecules that are only antagonistic ER binders devoid of agonistic activity would overcome tamoxifen resistance in breast cancer therapy.
Pure Antiestrogens
In efforts to synthesize pure antiestrogens that have high affinity toward the estrogen receptor but with little or no agonist activity, a program of medicinal chemistry at ICI (Imperial Chemical Industries, now AstraZeneca) utilized the molecular scaffold of 7α- substituted estradiol reported by the French researchers as an effective ER-binding absorbent, which led to the discovery of 7α-estradiol analogues with long-chain alkyl substituents that have the desired profile of activity.3a Chemical structures of the four compounds reported from their study are illustrated in Figure 1. These compounds were shown to be devoid of estrogenic activity and achieved a complete antagonism of estrogen action. The most potent analogue, ICI 164,384, blocked the uterotrophic action of both estradiol and tamoxifen in female rats.3a−3c They also found that in MCF-7 and ZR-75-1 breast cancer cells ICI 164,384 was a more potent inhibitor of cell growth, consistent with the greater binding affinity of ICI 164,384 for the rat uterus estrogen receptor than that of tamoxifen.
Figure 1.
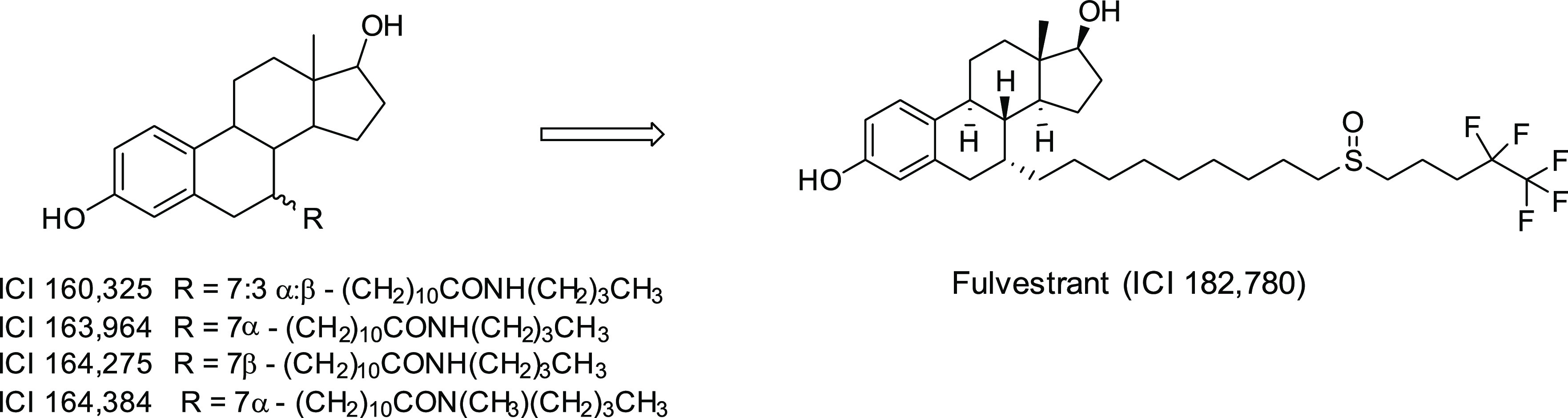
Structures of pure antiestrogens.
While the properties of ICI 164,384 satisfy key criteria which define pure antiestrogens, the ICI laboratory went on to identify a more potent pure antiestrogen, 7-α-[9-(4,4,5,5,5-pentafluoropentylsulfinyl)nonyl]estra-1,3,5-(10)-triene-3,17-β-diol, or ICI 182,780 (fulvestrant, Figure 1).4 Compared to ICI 164,384, this new antiestrogen was found to have ∼5-fold higher ER binding affinity and antiproliferative activity, 10-fold greater antiuterotrophic potency, and significantly greater efficacy in blocking xenograft tumor growth in mice. In this first report of fulvestrant, the poor oral bioavailability was already noted, and a parenteral depot formulation in arachis oil with an extended duration of action was used to demonstrate antitumor efficacy in xenograft models.
The unique action of estrogen receptor downregulation by the pure antiestrogens was discovered shortly after the primary candidate, ICI 182,780, was developed for clinical trials. In the investigation of mechanism of action of the antiestrogen ICI 164,384, Korach and co-workers5a used a mouse model system to reveal the effects on uterine function, as measured by DNA and protein syntheses, the temporal pattern of ICI binding to the ER, and the DNA-binding capacity of the native uterine ICI–ER complexes. Measurement of uterine nuclear ER and cytosolic levels by exchange binding assay indicated a reduction in total ER levels within 0.5 h after ICI treatment, which remained below 20% for 24 h.5a In another mechanistic study to investigate whether ICI 164,384 prevented DNA binding, Parker and co-workers5b found that ICI 164,384 treatment caused a decrease in cellular content of estrogen receptor protein by markedly reducing its half-life from about 5 h in the presence of estradiol to <1 h by ICI 164,384. They proposed that this might be caused by impaired receptor dimerization.5b The study concluded that (1) the effect of ICI 164,384 is not on estrogen receptor mRNA but on the receptor protein itself; (2) the “pure” antiestrogen ICI 164,384 reduces the cellular content of the estrogen receptor by increasing its turnover; (3) ICI 164,384 binds to the same region of the receptor and sterically interferes with ER dimerization; and (4) a side-chain length of 16–18 atoms was optimal for both the inhibitory effects of antiestrogens on dimerization and DNA binding.
The degradation effect of pure antiestrogens on ER protein was soon confirmed in clinical trials. In the first trial to test the tolerance, pharmacokinetics, and short-term biological effects of seven daily doses of a short-acting formulation of ICI 182,780 in postmenopausal women prior to surgery,6 treatment with ICI 182,780 was associated with significant reductions in the tumor expression of ER (median ER index, 0.72 before versus 0.02 after treatment; P < 0.001), progesterone receptor (median progesterone receptor index, 0.50 before versus 0.01 after treatment; P < 0.05), and Ki67 (median Ki67 labeling index, 3.2 before versus 1.1 after treatment; P < 0.05). Treatment with ICI 182,780 also resulted in a significant reduction in pS2 expression (P < 0.05), but this appeared unrelated to tumor ER status.
Subsequent clinical trials confirmed fulvestrant efficacy in treating patients with recurring disease upon tamoxifen treatment, leading to FDA approval of the drug in 2002 as a second line endocrine therapy for metastatic or advanced breast cancer.7a−7e In this setting, pure antagonism and receptor degradation were proven to be as effective as aromatase inhibitors (AIs) which shut off peripheral production of estrogen in postmenopausal patients. Moreover, when breast cancer progresses on AI treatment, response to fulvestrant was also clinically demonstrated,8a−8c further establishing the clinical utilities of fulvestrant after tamoxifen and AI treatment failures.
Oral SERDS: Acidic and Basic SERDS
While the mode of action by a pure antiestrogen devoid of agonist activity in any tissue was proven clinically effective in patients progressing on tamoxifen or aromatase inhibitors, much was still left to be fulfilled. At the approved dose of a 250 mg monthly injection, fulvestrant was similar but not superior to tamoxifen or AI (anastrozole), possibly due to insufficient drug exposure and its inherent pharmacokinetic limitations arising from the unique molecular feature of a steroid with a long hydrophobic aliphatic derivative. The search continued for novel antiestrogens that have better antagonist/agonist profiles and are not cross-resistant to tamoxifen. The development of second- and third-generation ER antagonists, now collectively termed selective estrogen receptor modulators (SERMs), greatly expanded the structural diversity of ER binding molecules and deepened our understanding of ER-mediated carcinogenesis and therapeutic intervention strategies. These new-generation SERMs, such as the benzothiophene-based raloxifene, indole-based bazedoxifene, and tetrahydronaphthalene-based lasofoxifene, share several important functions including acting as an antagonist in the breast tissue, the lack of uterotrophic property, and protecting the bone. Unfortunately, all exhibited cross-resistance to tamoxifen and failed to show superiority over tamoxifen in the clinic.9a−9c
One SERM, GW5638 (Figure 2), discovered through tissue-selective screening of synthetic triphenylethylenes10a showed distinct pharmacology of full agonist activity in bone but antagonist activity in the rat uterus. GW5638 induces a unique structural change in the ER distinct from that induced by tamoxifen and is not cross resistant to tamoxifen.10b Upon binding to the ER LBD, GW5638 relocates the carboxy-terminal helix (H12) to the known coactivator-docking site and repositions residues in H12, increasing the exposed hydrophobic surface of ER LBD. The resulting destabilization of ER may explain GW5638’s ability to induce degradation of the ER, although less effectively than ICI 182,780.10c In a study of GW7604, the more active hydroxylated metabolite of GW5638, it was found that the extent to which GW7604-bound ER was ubiquitinated was not significantly different from the basal level, whereas the ICI-bound ER was heavily ubiquitinated.10d The mechanism by which GW7604 mediates degradation is different from that of ICI 182,780 and suggests that other factors besides ubiquitination and transcriptional activation can influence the rate at which ER degradation occurs. The antiestrogenic and ER degradation activities of GW5638 and the lack of cross-resistance to tamoxifen in a tamoxifen-like molecule draw analogy to the reversal of function from estrogen to fulvestrant, an estrogen derivative in that the 7α-alkyl substitution of estrogen changed the molecule to a pure antiestrogen. Indeed, a study by Fan et al. showed that the acrylic acid moiety in GW5638 was key to impart the ER downregulation activity.10e
Figure 2.
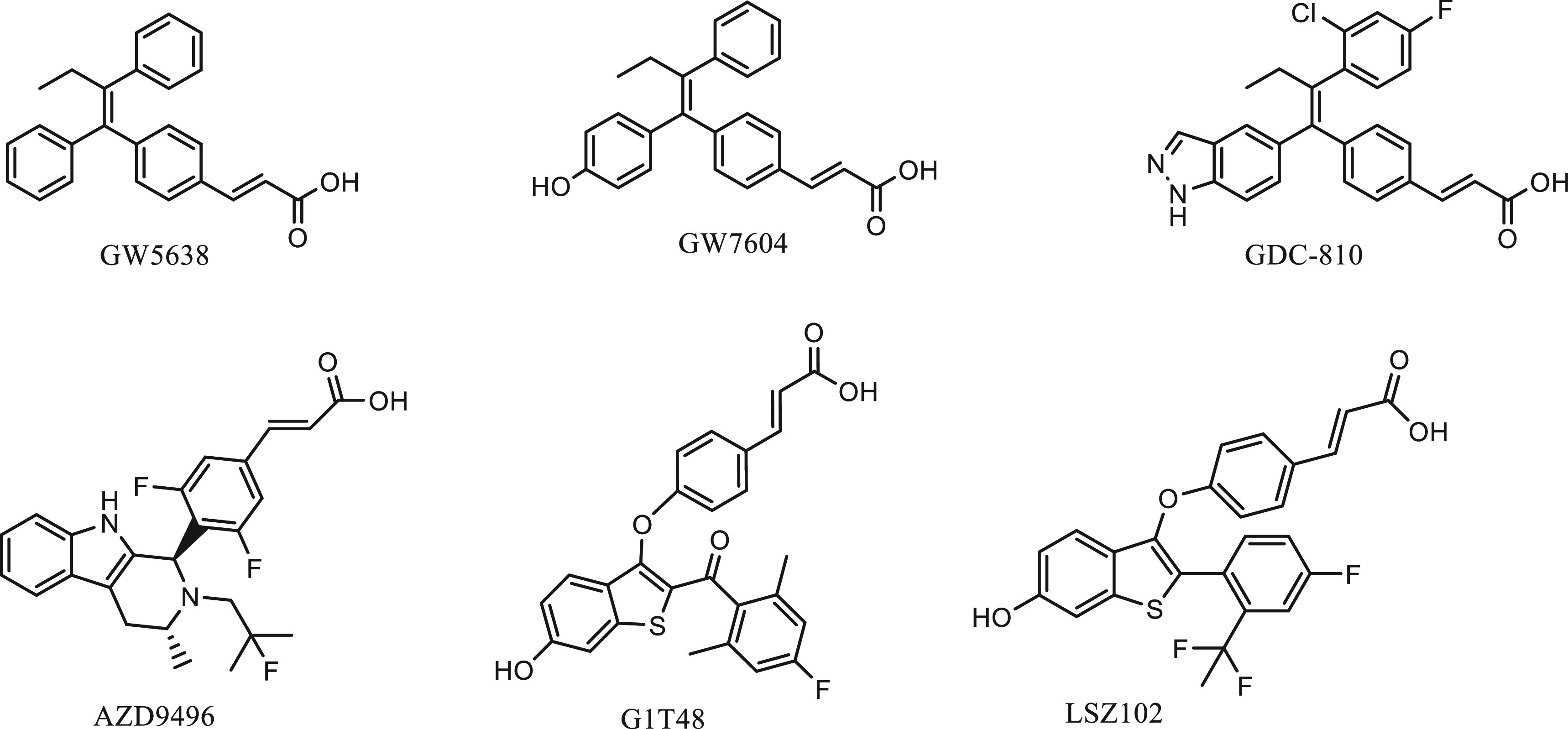
Structures of orally available SERDs with an acrylic acid functional group.
These findings informed further development of structurally similar, preclinically improved GW5638-like molecules (Figure 2) that were now collectively referred to as oral SERDs,11 a reflection of the well-established therapeutic modality of fulvestrant as an effective ER degrader and the urgent need to improve its lack of oral bioavailability and low drug exposure. These GW5638-like molecules were shown to have greater antiestrogenic and ER degradation potency than GW5638 and are non-cross-resistant to tamoxifen and AI in various breast tumor models. GDC-0810 and AZD9496 were the first two to enter clinical trials in 2013 (NCT01823835, NCT02248090), well over a decade after the inconclusive clinical trial of GW5638.11
The GDC-0810 molecule retained the core structure of GW5638 with modifications on the triphenylethylene moiety to achieve similar activities but greater drug exposure compared to GW7604, the more active metabolite of GW5638.12a However, when GDC-0810 was compared with fulvestrant in a phase 2 trial (NCT02569801), it failed to show comparable or superior efficacy, and the study was terminated. AZD9496 employed a novel binding motif of substituted aryl indole to achieve greater binding affinity to both wild-type and mutant ER than fulvestrant. In a diverse panel of clinically relevant breast tumor models, AZD9496 was shown to inhibit ER+ breast cancer cell proliferation and block tumor growth in endocrine-resistant, ESR1 mutant breast cancer models more efficaciously than fulvestrant.12b−12d Despite this preclinically observed advantage over fulvestrant, in a randomized window of opportunity study comparing AZD9496 with fulvestrant in patients with ER+, HER2– primary breast cancer, the oral SERD was inferior to fulvestrant in both anticancer efficacy and reduction of ER and PR expression.12e Other similar oral SERD candidates, such as LSZ102 and G1T48, were soon found unable to move beyond phase 1 studies in the clinical trials. An important clinical observation also emerged that the acidic SERDS all presented a gastrointestinal tolerability issue in early phase studies.
At the time when several clinical trials of GDC-0810 and AZD9496 showed early signs of difficulty in meeting primary end points, a basic SERD, RAD1901 (elacestrant), was showing promising results in its phase 1 studies.13a,13b Attention and hope for a clinically viable oral SERD quickly shifted to compounds with a basic side chain replacing the acrylic acid in these recently tested molecules.14a,14b Partial ER-degrading activities have been observed in some of the new-generation SERMs like bazedoxifene and RAD1901 (Figure 3) but not raloxifene or lasofoxifene.11,15a−15c Bazedoxifene was shown to downregulate WT, Y537S, and D538G somatic mutant ERs in MCF-7 cells by inducing a conformational change in ER that is distinct from fulvestrant or GW5638.15a,16 Recent reviews have covered current progress in SERDs, both steroidal derivatives and nonsteroidal molecules as SERDs for the treatment of breast cancer.17 The SERD properties of bazedoxifene are thought to arise from its disruption of helix 12 which appears displaced out of the AF-2 cleft into a less stable orientation.16Figure 4 shows the repositioning of H12 upon bazedoxifene (antagonist) binding to the ligand binding domain of ERα compared to the estradiol-bound (agonist) ER. In an antagonist-bound conformation, H12 is reoriented to occupy the LXXLL motif-mediated coactivator binding site within the ligand binding domain and reduces or blocks the ability to recruit coactivators and their normal functioning.
Figure 3.
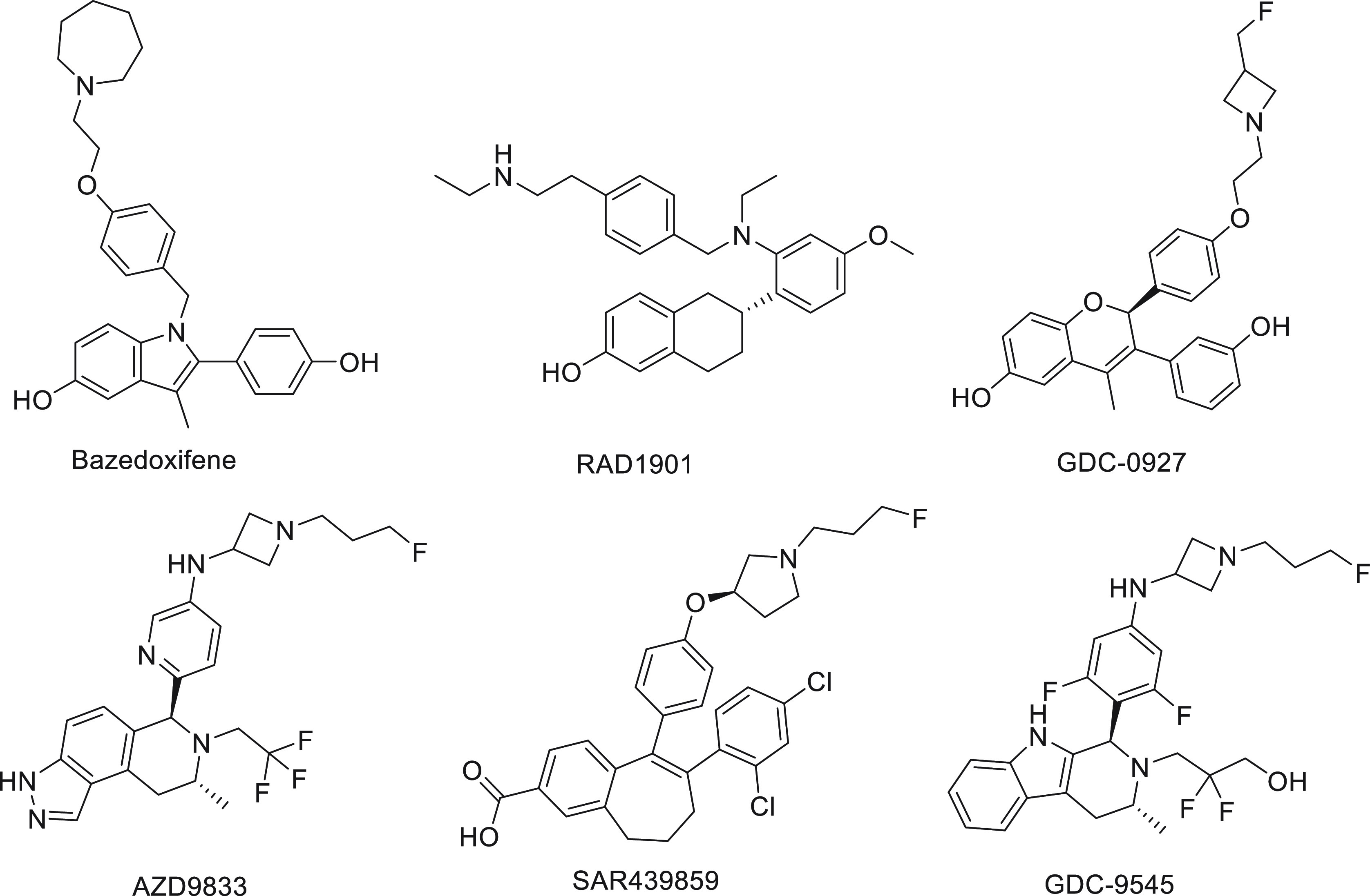
Structures of oral SERDs with a basic side chain.
Figure 4.
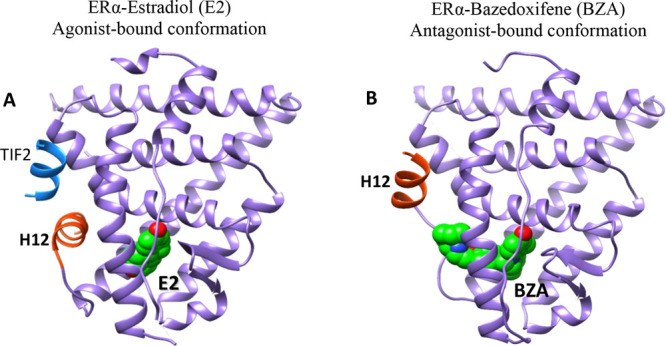
Comparison of the crystal structures of ERα in active (agonist-bound) and inactive (antagonist-bound) conformations. (A) Active form when bound to Estradiol (E2) and a short peptide from TIF2 transcriptional coactivator bearing canonical LXXLL motif (PDB code: 1GWR) and (B) inactive form when bound to bazedoxifene (BZA) (PDB code: 4XI3). In the antagonist-bound conformation, H12 is repositioned to occupy the coactivator binding groove.
Optimization of the binding motif and the helix-12-destabilizing side-chain structure led to the discovery of a diverse group of orally bioavailable SERDs bearing an amino side chain. These novel molecules showed greater ER degradation and antiestrogen activities than the first-generation nonsteroidal SERDs like GDC-0810 and the SERM/SERD compounds like RAD1901 and bazedoxifene. For example, Genentech’s GDC-9545 (Figure 3) was developed to address the poor clinical performance of the acrylic acid SERD GDC-0810 (unmet efficacy end point and adverse effects) and the company’s first-generation basic SERD GDC-0927 (bioavailability). It is highly potent in competing with estradiol for binding and in driving an antagonist conformation within the ER ligand binding domain, induces ER turnover, and suppresses ER transcriptional activity, resulting in robust antiproliferative activity (Table 1). GDC-9545 was shown to have greater in vivo efficacy compared to GDC-0927 and fulvestrant.18 GDC-9545 is currently being evaluated in multiple phase 1 and phase 2 clinical trials (NCT03332797, NCT04436744, and NCT04576455).
Table 1. Selected SERD Properties.
| agonist/antagonist
profile |
|||||||
|---|---|---|---|---|---|---|---|
| molecular type | DC50 MCF-7 | Dmax MCF-7 | IC50 (antiproliferation) MCF-7 | breast | uterus (% of control w. wt) | bone | references |
| Steroidal | |||||||
| ICI 164,383 | >95% | 39 nM | antagonist | 40–50% | antagonist | (3c, 5a, 22a) | |
| ICI 182,780 | 0.4 nM | >95% | 0.6 nM | antagonist | 42% | antagonist | (5a, 5b, 12a, 12c) |
| Acrylic Acids | |||||||
| GW5638/GW7604 | 390 nM/1.7 nM | 82%/86% | 985 nM/5 nM | antagonist | weak antagonist | agonist | (12a) |
| GDC-0810 | 0.7 nM | 87% | 2.5 nM | antagonist | weak antagonist | agonist | (12a) |
| AZD9496 | 0.14 nM | >95% | 0.04 nM | antagonist | weak agonist | unknown | (12b, 12c) |
| Basic Side Chain | |||||||
| RAD1901 | 1.5 nM | ∼70% | 8.9 nM | antagonist/agonist | weak agonist | unknown | (15b, 22b) |
| bazedoxifene | 10 nM | ∼70% | 0.24 nM | antagonist | weak agonist | agonist | (15c, 16) |
| GDC-0927 | 0.3 nM | 97% | 0.2 nM | antagonist | ∼60% | unknown | (22c, 22d) |
| GDC-9545 | 0.04 nM | 84% | 0.26 nM | antagonist | ∼50% | unknown | (23a) |
| AZD9833 | 0.16 nM | 99% | 5.0 nM | antagonist | not reported | unknown | (19b) |
| PROTACs | |||||||
| ARV-471 | 0.9 nM | >95% | not reported | antagonist | ∼45% | unknown | (23b) |
| ERD-308 | 0.17 nM | >99% | 0.77 nM | antagonist | not reported | unknown | (23c) |
Compared to acrylic-acid-containing oral SERDs that do not degrade ER equally in different ER+ cell lines, the basic SERDs were optimized to deliver maximal ERα degradation across multiple ER+ cell lines, a feature possessed by fulvestrant.19a,19b Improving on the preceding oral SERD, AZD9496, AstraZeneca’s new compound, is a potent ER degrader in not only MCF-7 cells but also CAMA-1, T47D, and BT474 cells that express ER. In several patient-derived and cell line xenograft models, including models with clinically relevant ESR1 mutations, AZD9833 was shown to block tumor growth more efficaciously than fulvestrant. Furthermore, in an ESR1 wild-type and an ESR1 D538G PDX model, AZD9833 demonstrated benefits in combination with palbociclib. AZD9833 has progressed into a multistage monotherapy and palbociclib combination for the first time in patient clinical trials, SERENA-1 (NCT03616587).
Compared to AZD9496, the basic amino side chain bearing AZD9833 is a better ERα degrader. Figure 5 shows an overlay of the crystal structure of ERα-AZD9496 and a docked structure of ERα-AZD9833 complexes. The model of the ERα in complex with AZD9833 was built using the coordinates of ERα from the crystal structure of the ERα-AZD9496 complex (PDB: 5ACC). After adding the missing loops and side chains during the protein preparation setup, docking studies were performed using the Glide software20a and employing the OPLS3e force field20b with a flexible ligand sampling and a standard precision mode. The core ligand structures of both compounds bind in a very similar manner. The long basic amino side chain of AZD9833 is pushed further against the N-terminal of H12 reaching up to Leu539, including Val534 and Pro535. Distances between the side chain δC of Leu539 and the side chains’ fluorine of AZD9833 and carboxyl of AZD9496 are 3.5 and 6.7 Å, respectively. This closer interaction with AZD9833 could propagate to H12 and displace it out of the AF-2 cleft into a less stable orientation.
Figure 5.
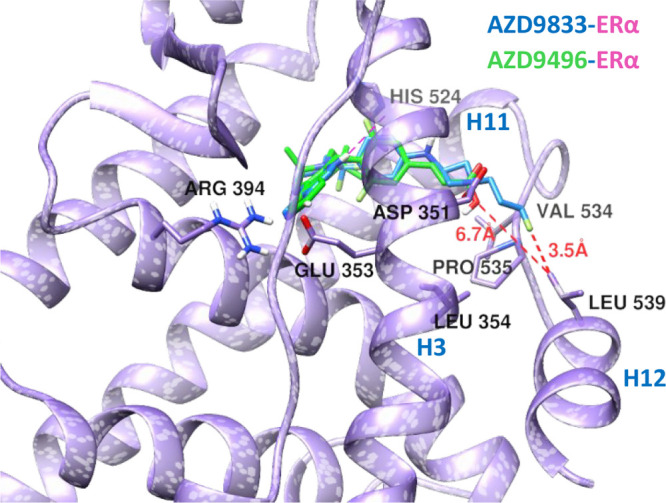
Overlay of the X-ray crystal structure of ERα in complex with AZD9496 (green compound and purple ribbon) (5ACC) and docked model of ERα in complex with AZD9833 (blue compound and purple ribbon). Amino acids that make hydrogen bonds with the protein and key hydrophobic residues on H12 and H3 that are in the hydrophobic interface are shown in the stick model. Distances between the side chain δC of Leu539 and the side chains of AZD compounds are 3.5 and 6.7 Å, for the fluorine atom of AZD9833 and the carboxyl of AZD9496, respectively.
To understand the structural basis for the increased degradation of ERα by AZD9833 when compared to the same by AZD9496, explicit solvent all-atom molecular dynamics (MD) simulations were carried out with the Desmond program20c for 100 ns using the crystal structure of ERα-AZD9496 and docked structure of ERα-AZD9833 solvated with the SPC water model and neutralized by adding counterions (i.e., Na+/Cl–) in an orthorhombic box under periodic boundary conditions. The default Desmond protocol was used for minimization and relaxation using the OPLS3e force field. MD simulations were run for 100 ns in the NPT ensemble with a 300 K Nose–Hoover thermostat and 1 atm pressure, by saving trajectories at a 50 ps interval. The backbone root-mean-square deviations (RMSDs) of the proteins in both systems are below 2.7 Å during the entire 100 ns simulation (Figure 6A). However, in the AZD9833 complex the RMSD is about 0.5 Å larger than that in AZD9496 during the last 20 ns, which indicates that the protein is slightly more flexible when bound to AZD9833 than to AZD9496. Root-mean-square fluctuations (RMSFs) of aligned residues show that the fluctuations mainly arise from the loop regions in the protein (Figure 6B).
Figure 6.

(A) Backbone root-mean-square deviations of the protein in the 100 ns MD simulations for ERα-AZD9833 (maroon) and ERα-AZD9496 (blue). (B) Root-mean-square fluctuations of ERα LBD residues in the 100 ns MD simulations for ERα-AZD9833 (maroon) and ERα-AZD9496 (blue).
Comparison of the initial and final structures showed (figure not included) that the basic amino side chain of AZD9833 continues to push against the N-terminal H12, especially against Leu539 (Leu539:δC-AZD9833:F (fluoropropyl) distance 3.5–3.7 Å vs Leu539:δC-AZD9496:O (carboxyl) distance 6.7–8.6 Å). In both systems, H12 and the N-terminals of H11 and H3 were found to move but less in AZD9496-bound ERα compared to the AZD9833-bound ERα. The concerted movement of these helices can disturb the hydrophobic surface. Lys362 from H3 plays a key role in the antagonistic activity of ERα.21a In an antagonist-bound ER, relocation of H12 to the coactivator binding site prevents the recruitment of the transcription complex by blocking the critical residue, K362, required for coactivator recruitment.21b−21d Movement of helix 12 during the MD simulations has shown to provide access to Lys362 for any possible ubiquitination.
This comparison study shows that the level of exposure of the hydrophobic surface seems to depend upon the length and type of the side chain and its interaction with the N-terminus of H12. This effect is prominent when side chains are extended as in the case of AZD9833 that could disrupt the beneficial positioning of H12 by steric hindrance. The distal functional group in the core ligand, like the bulky 2-fluoro-2-methyl group in AZD9496 and 2,2,2,-trifluoroethyl in AZD9833, seems to play a role in the movement of N-terminal H11 that can ease the recognition of ubiquitin by the ubiquitin binding domain on H8 of ERα that is parallel to H11. Any change in the positioning of H11 can also affect the dimerization of ERα because H11 is part of the ERα dimer interface. Thus, the disruption of H12, inhibition of dimerization, and easy access of the UBD and Lys362 for ubiquitination followed by proteasomal degradation seem to be responsible for the antagonistic activity of SERDs. Based on the comparison studies and the literature,17 SERDs with a basic amino side chain carrying a hydrocarbon chain, preferably with a terminal fluoropropyl, like in AZD9833, GDC0927, SAR439859, GDC9545, and GNE149, seem to be more suitable for increasing the surface hydrophobicity, thereby to engage H12 more efficiently. The comparative computational docking and molecular dynamics studies of AZD9833 and AZD9496 showed a closer interaction with H12 for the basic amino side chain bearing AZD9833 (Leu539:δC-AZD9833:F (fluoropropyl) distance = 3.5–3.7 Å) than for the acrylic acid side chain bearing AZD9496 (Leu539:δC-AZD9496:O (carboxyl) distance = 6.7–8.6 Å). ERα surface hydrophobicity could further be increased by increasing the hydrophobic chain length of the amino side chain by 1 or 2 carbon lengths without hindering H12. This could be achieved either by changing the fluoropropyl to fluorobutyl or fluoropentyl or by changing the four-membered azetidine ring to the five-membered pyrrolidine or six-membered piperidine ring. These chemical modifications likely engage H12 more efficiently to further increase the surface hydrophobicity and in turn the ERα degradation potency.
The SERDs with a basic side chain now appear to be better degraders of the ER than the acrylic acid analogues. They also exhibit a more desirable agonist/antagonist profile than the acidic SERDs (Table 1). How these new SERDs act in bone is yet unclear, and thus these compounds remain to be classified pure antiestrogen as defined by the steroidal antiestrogens. While high potency partial antagonism can be achieved with nondegrading SERMs like 4-OHT and lasofoxifene,24a−24d a pure antiestrogen that lacks agonistic activity across ER expression tissues without simultaneously degrading the ER has yet to exist, which poses an important, consequential question: can pure antagonism be achieved by sustainable ER degradation only? In other words, can a pure ER degrader function as a pure antiestrogen?
Pure Degraders
The question seems to have found an answer in the emerging targeted protein degradation technology called proteolysis-targeting chimeras (PROTACs). ER PROTACs are heterobifunctional molecules comprising an ER-binding warhead linked to an E3 ligase binding motif that facilitates the ubiquitination and subsequent degradation of ER via the proteasome. The ER-binding warhead can be a SERM moiety which does not induce hydrophobic surface exposure and results in receptor degradation (Figure 7). Rather, the PROTAC molecule engages an E3 ligase to ubiquitinate ER and degrades it in a catalytic manner. Thus, pure antagonism of ER is realized by elimination of the receptor, rather than conformational changes of ER to block recruitment of cofactors required for ER transcriptional activation. In this case, complete blockade of all stimulatory actions of estrogens is achieved by degradation of the receptor. PROTAC ER degraders have been shown to rapidly and completely eliminate intracellular levels of the receptor, thereby completely abrogating ER signaling.23b,23c They can degrade wild-type as well as mutant ER, provided the ER binding ligand retains sufficient affinity for both.25a The rapid progress in ER PROTAC development culminated in a first-in-class, orally bioavailable ER degrading agent, ARV-471, that entered clinical trials in 2019 (NCT04072952).
Figure 7.
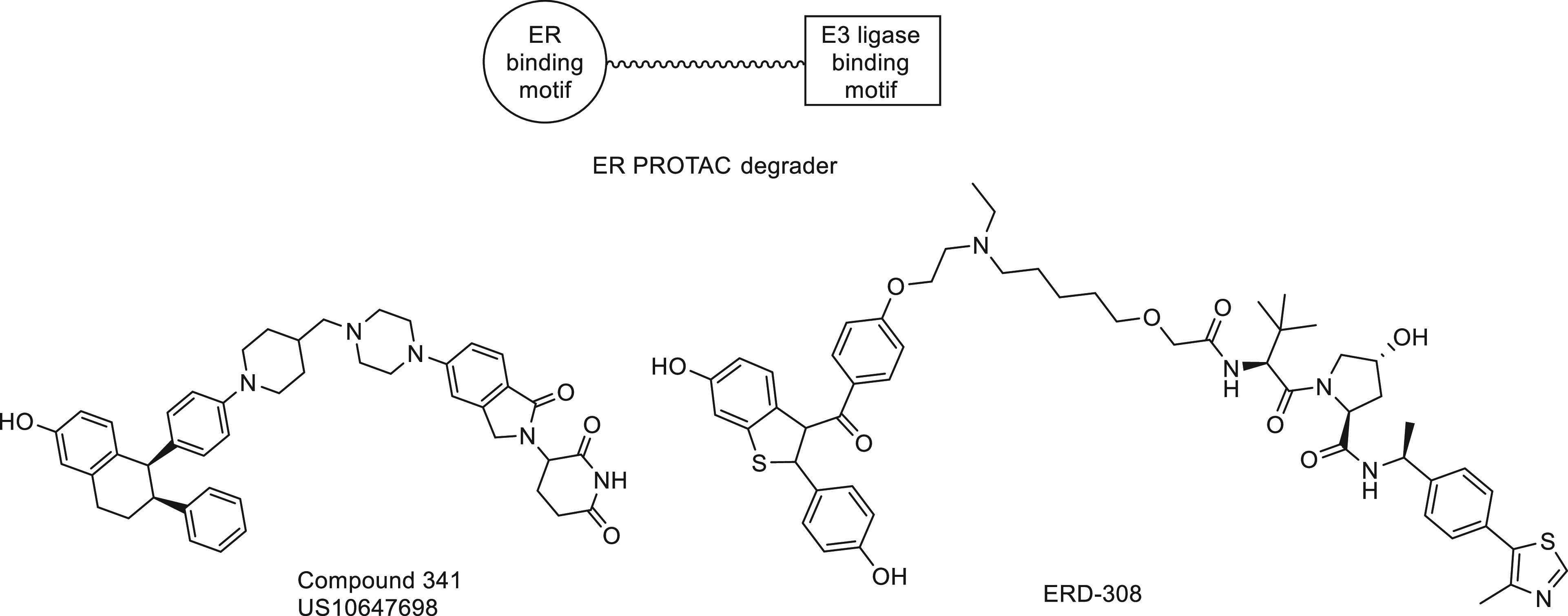
PROTAC ER degraders.
The unique mechanism underlying the PROTAC approach offers several pharmacological advantages that could be translated to clinical benefits in ER-targeted therapy. PROTAC-induced rapid and complete degradation of ER protein eliminates any ligand-dependent (AF2) or ligand-independent (AF1) agonism. PROTAC action is event-driven as opposed to occupancy-driven in the inhibitory setting; thus, only a transient binding event is required for degradation, and the PROTAC molecules can cycle through multiple rounds of activity, removing substoichiometric quantities of proteins. These promising attributes of a PROTAC ER degrader appear to be borne out in the first clinical trial results where ER degradation and clinical benefits are observed in heavily pretreated patients.25b
Perspective
In perspective, the effort to overcome tamoxifen resistance began with the search for a pure antiestrogen that was quickly identified in the estradiol-modified molecule known as ICI 182,780. The steroidal molecule was found to have no agonist activities in any tissue and increased rapid turnover of the receptor, yet also lacked significant oral bioavailability. The ensuing quest for oral SERDs looked for two desired properties: (1) oral bioavailability and (2) high potency in ER antagonism and degradation. A large number of novel molecules have since been discovered and tested in breast cancer models that meet the oral SERD criteria, only to fail in clinical trials, either due to tolerability or insufficient efficacy. Improving on ER degrading and toxicity profile, some of the latest oral SERDs have gone further in human trials where safety and efficacy are still under evaluation. In a parallel development, PROTAC molecules have emerged as potent antiestrogens by effectively degrading the ER. As the PROTAC molecules utilize the known and clinically tested ER-binding motifs like raloxifene and lasofoxifene, clinical data have so far indicated no toxicity liability; thus, it is likely that the decades-long quest for a pure antiestrogen that has oral bioavailability and ER degrading pharmacology may have found the solution in the form of a PROTAC ER degrader.
Acknowledgments
This work was supported by the NIH RCMI program at Xavier University of Louisiana through Grant U54MD007595.
Biographies
Dr. Madhusoodanan Mottamal received his Ph.D. in Chemistry (Computational) from IIT Bombay (India, 1995). He did his postdoctoral research first at the Bar-Ilan University (Israel, 1996–1998) and then at the IBMS, Academia Sinica (Taiwan 1998–2001). Subsequently, he did research work at the City College of New York, Boston College, and Hormel Institute. Currently, he is a core scientist at the RCMI Cancer Research Center at Xavier University of Louisiana. His scientific interests are mainly focused on computer-aided drug discovery and molecular modeling to design novel potential drug candidates for the treatment of various diseases, such as cancer and other diseases.
Dr. Borui Kang obtained his Ph.D. in Medicinal Chemistry from Xi’an Jiaotong University, China, in 2016. He joined Xavier University of Louisiana in 2017 and has been working as a medicinal chemist. His research interests include design and synthesis of small-molecule anticancer drugs comprising kinase inhibitors, receptor antagonists, and PROTAC compounds and structure–activity relationship (SAR) analysis.
Dr. Xianyou Peng received his Ph.D. in Organic Chemistry from the Institute of Chemistry, Chinese Academy of Science (China), in 2012. He worked as a Senior Team Leader (2012–2015) and as an Associate Director (2015–2019) for Pharmaron, China. He is currently a research scientist at the RCMI Cancer Research Center at Xavier University of Louisiana. His main research interest is on the design and synthesis of small-molecule drugs for various cancer treatments.
Dr. Guangdi Wang is a Professor of Chemistry at Xavier University of Louisiana. He received his Ph.D. in Chemistry from the University of New Orleans in 1995. His current research focuses on three main areas: (1) therapeutic development for the treatment of breast cancer, (2) drug metabolism and disposition, and (3) proteomic studies on mechanisms of drug action and for the discovery of novel therapeutic targets in cancer.
The authors declare no competing financial interest.
This paper was published ASAP on March 30, 2021, with an incorrect compound listed in the “Oral SERDS: Acidic and Basic SERDS” section. The corrected version was posted on April 13, 2021.
References
- Klein-Hitpass L.; Tsai S. Y.; Greene G. L.; Clark J. H.; Tsai M. J.; O’Malley B. W. Specific binding of estrogen receptor to the estrogen response element. Mol. Cell. Biol. 1989, 9 (1), 43–49. 10.1128/MCB.9.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Furr B. J.; Jordan V. C. The pharmacology and clinical uses of tamoxifen. Pharmacol. Ther. 1984, 25 (2), 127–205. 10.1016/0163-7258(84)90043-3. [DOI] [PubMed] [Google Scholar]; b Tormey D. C.; Simon R. M.; Lippman M. E.; Bull J. M.; Myers C. E. Evaluation of tamoxifen dose in advanced breast cancer: a progress report. Cancer Treat. Rep. 1976, 60 (10), 1451–1459. [PubMed] [Google Scholar]; c McIntosh I. H.; Thynne G. S. Tumour stimulation by anti-oestrogens. Br. J. Surg. 1977, 64 (12), 900–901. 10.1002/bjs.1800641218. [DOI] [PubMed] [Google Scholar]; d Gottardis M. M.; Jordan V. C. Development of tamoxifen-stimulated growth of MCF-7 tumors in athymic mice after long-term antiestrogen administration. Cancer Res. 1988, 48 (18), 5183–5187. [PubMed] [Google Scholar]; e Gottardis M. M.; Robinson S. P.; Satyaswaroop P. G.; Jordan V. C. Contrasting actions of tamoxifen on endometrial and breast tumor growth in the athymic mouse. Cancer Res. 1988, 48 (4), 812–815. [PubMed] [Google Scholar]; f Howell A.; Dodwell D. J.; Anderson H. New endocrine approaches to breast cancer. Baillieres Clin. Endocrinol. Metab. 1990, 4 (1), 67–84. 10.1016/S0950-351X(05)80316-7. [DOI] [PubMed] [Google Scholar]
- a Wakeling A. E.; Bowler J. Steroidal pure antioestrogens. J. Endocrinol. 1987, 112 (3), R7–10. 10.1677/joe.0.112R007. [DOI] [PubMed] [Google Scholar]; b Wakeling A. E.; Bowler J. Biology and mode of action of pure antioestrogens. J. Steroid Biochem. 1988, 30 (1–6), 141–147. 10.1016/0022-4731(88)90086-6. [DOI] [PubMed] [Google Scholar]; c Wakeling A. E.; Bowler J. Novel antioestrogens without partial agonist activity. J. Steroid Biochem. 1988, 31 (4B), 645–653. 10.1016/0022-4731(88)90014-3. [DOI] [PubMed] [Google Scholar]
- Wakeling A. E.; Bowler J. Development of novel oestrogen-receptor antagonists. Biochem. Soc. Trans. 1991, 19 (4), 899–901. 10.1042/bst0190899. [DOI] [PubMed] [Google Scholar]
- a Gibson M. K.; Nemmers L. A.; Beckman W. C. Jr.; Davis V. L.; Curtis S. W.; Korach K. S. The mechanism of ICI 164,384 antiestrogenicity involves rapid loss of estrogen receptor in uterine tissue. Endocrinology 1991, 129 (4), 2000–2010. 10.1210/endo-129-4-2000. [DOI] [PubMed] [Google Scholar]; b Dauvois S.; Danielian P. S.; White R.; Parker M. G. Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc. Natl. Acad. Sci. U. S. A. 1992, 89 (9), 4037–4041. 10.1073/pnas.89.9.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFriend D. J.; Howell A.; Nicholson R. I.; Anderson E.; Dowsett M.; Mansel R. E.; Blamey R. W.; Bundred N. J.; Robertson J. F.; Saunders C.; Baum M.; Walton P.; Sutcliffe F.; Wakeling A. E. Investigation of a new pure antiestrogen (ICI 182780) in women with primary breast cancer. Cancer Res. 1994, 54 (2), 408–414. [PubMed] [Google Scholar]
- a Robertson J. F.; Nicholson R. I.; Bundred N. J.; Anderson E.; Rayter Z.; Dowsett M.; Fox J. N.; Gee J. M.; Webster A.; Wakeling A. E.; Morris C.; Dixon M. Comparison of the short-term biological effects of 7alpha-[9-(4,4,5,5,5-pentafluoropentylsulfinyl)-nonyl]estra-1,3,5, (10)-triene-3,17beta-diol (Faslodex) versus tamoxifen in postmenopausal women with primary breast cancer. Cancer Res. 2001, 61 (18), 6739–6746. [PubMed] [Google Scholar]; b Vergote I. Faslodex; Investigators, Fulvestrant versus anastrozole as second-line treatment of advanced breast cancer in postmenopausal women. Eur. J. Cancer 2002, 38 (Suppl 6), S57–S58. [DOI] [PubMed] [Google Scholar]; c Howell A.; DeFriend D. J.; Robertson J. F.; Blamey R. W.; Anderson L.; Anderson E.; Sutcliffe F. A.; Walton P. Pharmacokinetics, pharmacological and anti-tumour effects of the specific anti-oestrogen ICI 182780 in women with advanced breast cancer. Br. J. Cancer 1996, 74 (2), 300–308. 10.1038/bjc.1996.357. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Howell A.; Robertson J. F.; Quaresma Albano J.; Aschermannova A.; Mauriac L.; Kleeberg U. R.; Vergote I.; Erikstein B.; Webster A.; Morris C. Fulvestrant, formerly ICI 182,780, is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J. Clin. Oncol. 2002, 20 (16), 3396–3403. 10.1200/JCO.2002.10.057. [DOI] [PubMed] [Google Scholar]; e Osborne C. K.; Pippen J.; Jones S. E.; Parker L. M.; Ellis M.; Come S.; Gertler S. Z.; May J. T.; Burton G.; Dimery I.; Webster A.; Morris C.; Elledge R.; Buzdar A. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: results of a North American trial. J. Clin. Oncol. 2002, 20 (16), 3386–3395. 10.1200/JCO.2002.10.058. [DOI] [PubMed] [Google Scholar]
- a Ingle J. N.; Suman V. J.; Rowland K. M.; Mirchandani D.; Bernath A. M.; Camoriano J. K.; Fishkin P. A.; Nikcevich D. A.; Perez E. A. North Central Cancer Treatment Group Trial, N., Fulvestrant in women with advanced breast cancer after progression on prior aromatase inhibitor therapy: North Central Cancer Treatment Group Trial N0032. J. Clin. Oncol. 2006, 24 (7), 1052–1056. 10.1200/JCO.2005.04.1053. [DOI] [PubMed] [Google Scholar]; b Perey L.; Paridaens R.; Hawle H.; Zaman K.; Nole F.; Wildiers H.; Fiche M.; Dietrich D.; Clement P.; Koberle D.; Goldhirsch A.; Thurlimann B. Clinical benefit of fulvestrant in postmenopausal women with advanced breast cancer and primary or acquired resistance to aromatase inhibitors: final results of phase II Swiss Group for Clinical Cancer Research Trial (SAKK 21/00). Ann. Oncol. 2007, 18 (1), 64–69. 10.1093/annonc/mdl341. [DOI] [PubMed] [Google Scholar]; c Chia S.; Gradishar W.; Mauriac L.; Bines J.; Amant F.; Federico M.; Fein L.; Romieu G.; Buzdar A.; Robertson J. F.; Brufsky A.; Possinger K.; Rennie P.; Sapunar F.; Lowe E.; Piccart M. Double-blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor-positive, advanced breast cancer: results from EFECT. J. Clin. Oncol. 2008, 26 (10), 1664–1670. 10.1200/JCO.2007.13.5822. [DOI] [PubMed] [Google Scholar]
- a Serrano D.; Lazzeroni M.; Gandini S.; Macis D.; Johansson H.; Gjerde J.; Lien E.; Feroce I.; Pruneri G.; Sandri M.; Bassi F.; Brenelli F.; Luini A.; Cazzaniga M.; Varricchio C.; Guerrieri-Gonzaga A.; DeCensi A.; Bonanni B. A randomized phase II presurgical trial of weekly low-dose tamoxifen versus raloxifene versus placebo in premenopausal women with estrogen receptor-positive breast cancer. Breast Cancer Res. 2013, 15 (3), R47. 10.1186/bcr3439. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Johnston S. R.Endocrine manipulation in advanced breast cancer: recent advances with SERM therapies Clin. Cancer Res. 2001, 7, 4376s–4387s; discussion 4411s–4412s. [PubMed] [Google Scholar]; c Deshmane V.; Krishnamurthy S.; Melemed A. S.; Peterson P.; Buzdar A. U. Phase III double-blind trial of arzoxifene compared with tamoxifen for locally advanced or metastatic breast cancer. J. Clin. Oncol. 2007, 25 (31), 4967–4973. 10.1200/JCO.2006.09.5992. [DOI] [PubMed] [Google Scholar]
- a Willson T. M.; Henke B. R.; Momtahen T. M.; Charifson P. S.; Batchelor K. W.; Lubahn D. B.; Moore L. B.; Oliver B. B.; Sauls H. R.; Triantafillou J. A.; Wolfe S. G.; Baer P. G. 3-[4-(1,2-Diphenylbut-1-enyl)phenyl]acrylic acid: a non-steroidal estrogen with functional selectivity for bone over uterus in rats. J. Med. Chem. 1994, 37 (11), 1550–1552. 10.1021/jm00037a002. [DOI] [PubMed] [Google Scholar]; b Connor C. E.; Norris J. D.; Broadwater G.; Willson T. M.; Gottardis M. M.; Dewhirst M. W.; McDonnell D. P. Circumventing tamoxifen resistance in breast cancers using antiestrogens that induce unique conformational changes in the estrogen receptor. Cancer Res. 2001, 61 (7), 2917–2922. [PubMed] [Google Scholar]; c Wu Y. L.; Yang X.; Ren Z.; McDonnell D. P.; Norris J. D.; Willson T. M.; Greene G. L. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol. Cell 2005, 18 (4), 413–424. 10.1016/j.molcel.2005.04.014. [DOI] [PubMed] [Google Scholar]; d Wijayaratne A. L.; McDonnell D. P. The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J. Biol. Chem. 2001, 276 (38), 35684–35692. 10.1074/jbc.M101097200. [DOI] [PubMed] [Google Scholar]; e Fan M.; Rickert E. L.; Chen L.; Aftab S. A.; Nephew K. P.; Weatherman R. V. Characterization of molecular and structural determinants of selective estrogen receptor downregulators. Breast Cancer Res. Treat. 2007, 103 (1), 37–44. 10.1007/s10549-006-9353-2. [DOI] [PubMed] [Google Scholar]
- McDonnell D. P.; Wardell S. E.; Norris J. D. Oral Selective Estrogen Receptor Downregulators (SERDs), a Breakthrough Endocrine Therapy for Breast Cancer. J. Med. Chem. 2015, 58 (12), 4883–4887. 10.1021/acs.jmedchem.5b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lai A.; Kahraman M.; Govek S.; Nagasawa J.; Bonnefous C.; Julien J.; Douglas K.; Sensintaffar J.; Lu N.; Lee K. J.; Aparicio A.; Kaufman J.; Qian J.; Shao G.; Prudente R.; Moon M. J.; Joseph J. D.; Darimont B.; Brigham D.; Grillot K.; Heyman R.; Rix P. J.; Hager J. H.; Smith N. D. Identification of GDC-0810 (ARN-810), an Orally Bioavailable Selective Estrogen Receptor Degrader (SERD) that Demonstrates Robust Activity in Tamoxifen-Resistant Breast Cancer Xenografts. J. Med. Chem. 2015, 58 (12), 4888–4904. 10.1021/acs.jmedchem.5b00054. [DOI] [PubMed] [Google Scholar]; b De Savi C.; Bradbury R. H.; Rabow A. A.; Norman R. A.; de Almeida C.; Andrews D. M.; Ballard P.; Buttar D.; Callis R. J.; Currie G. S.; Curwen J. O.; Davies C. D.; Donald C. S.; Feron L. J.; Gingell H.; Glossop S. C.; Hayter B. R.; Hussain S.; Karoutchi G.; Lamont S. G.; MacFaul P.; Moss T. A.; Pearson S. E.; Tonge M.; Walker G. E.; Weir H. M.; Wilson Z. Optimization of a Novel Binding Motif to (E)-3-(3,5-Difluoro-4-((1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-2,3,4,9-tetra hydro-1H-pyrido[3,4-b]indol-1-yl)phenyl)acrylic Acid (AZD9496), a Potent and Orally Bioavailable Selective Estrogen Receptor Downregulator and Antagonist. J. Med. Chem. 2015, 58 (20), 8128–8140. 10.1021/acs.jmedchem.5b00984. [DOI] [PubMed] [Google Scholar]; c Weir H. M.; Bradbury R. H.; Lawson M.; Rabow A. A.; Buttar D.; Callis R. J.; Curwen J. O.; de Almeida C.; Ballard P.; Hulse M.; Donald C. S.; Feron L. J.; Karoutchi G.; MacFaul P.; Moss T.; Norman R. A.; Pearson S. E.; Tonge M.; Davies G.; Walker G. E.; Wilson Z.; Rowlinson R.; Powell S.; Sadler C.; Richmond G.; Ladd B.; Pazolli E.; Mazzola A. M.; D’Cruz C.; De Savi C. AZD9496: An Oral Estrogen Receptor Inhibitor That Blocks the Growth of ER-Positive and ESR1-Mutant Breast Tumors in Preclinical Models. Cancer Res. 2016, 76 (11), 3307–3318. 10.1158/0008-5472.CAN-15-2357. [DOI] [PubMed] [Google Scholar]; d Toy W.; Weir H.; Razavi P.; Lawson M.; Goeppert A. U.; Mazzola A. M.; Smith A.; Wilson J.; Morrow C.; Wong W. L.; De Stanchina E.; Carlson K. E.; Martin T. S.; Uddin S.; Li Z.; Fanning S.; Katzenellenbogen J. A.; Greene G.; Baselga J.; Chandarlapaty S. Activating ESR1Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discovery 2017, 7 (3), 277–287. 10.1158/2159-8290.CD-15-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Robertson J. F. R.; Evans A.; Henschen S.; Kirwan C. C.; Jahan A.; Kenny L. M.; Dixon J. M.; Schmid P.; Kothari A.; Mohamed O.; Fasching P. A.; Cheung K. L.; Wuerstlein R.; Carroll D.; Klinowska T.; Lindemann J. P. O.; MacDonald A.; Mather R.; Maudsley R.; Moschetta M.; Nikolaou M.; Roudier M. P.; Sarvotham T.; Schiavon G.; Zhou D.; Zhou L.; Harbeck N. A Randomized, Open-label, Presurgical, Window-of-Opportunity Study Comparing the Pharmacodynamic Effects of the Novel Oral SERD AZD9496 with Fulvestrant in Patients with Newly Diagnosed ER(+) HER2(−) Primary Breast Cancer. Clin. Cancer Res. 2020, 26 (16), 4242–4249. [DOI] [PubMed] [Google Scholar]
- a de Vries E.; Venema C.; Glaudemans A.; Jager A.; Menke-van der Houven van Oordt C.; Neven P.; Jiang H.; Wang D.; O’Neill A.; Patki A.; Aftimos P. Abstract P1-10-04: Elacestrant, a novel oral selective estrogen receptor degrader (SERD), decreases tumoral 18F-FES uptake in a phase 1 study of ER+, HER2 -, advanced breast cancer patients. Cancer Res. 2018, 78, P1-10-04. [Google Scholar]; b Bardia A.; Aftimos P.; Bihani T.; Anderson-Villaluz A. T.; Jung J.; Conlan M. G.; Kaklamani V. G. EMERALD: Phase III trial of elacestrant (RAD1901) vs endocrine therapy for previously treated ER+ advanced breast cancer. Future Oncol. 2019, 15 (28), 3209–3218. 10.2217/fon-2019-0370. [DOI] [PubMed] [Google Scholar]
- a Nagasawa J.; Govek S.; Kahraman M.; Lai A.; Bonnefous C.; Douglas K.; Sensintaffar J.; Lu N.; Lee K.; Aparicio A.; Kaufman J.; Qian J.; Shao G.; Prudente R.; Joseph J. D.; Darimont B.; Brigham D.; Maheu K.; Heyman R.; Rix P. J.; Hager J. H.; Smith N. D. Identification of an Orally Bioavailable Chromene-Based Selective Estrogen Receptor Degrader (SERD) That Demonstrates Robust Activity in a Model of Tamoxifen-Resistant Breast Cancer. J. Med. Chem. 2018, 61 (17), 7917–7928. 10.1021/acs.jmedchem.8b00921. [DOI] [PubMed] [Google Scholar]; b Kahraman M.; Govek S. P.; Nagasawa J. Y.; Lai A.; Bonnefous C.; Douglas K.; Sensintaffar J.; Liu N.; Lee K.; Aparicio A.; Kaufman J.; Qian J.; Shao G.; Prudente R.; Joseph J. D.; Darimont B.; Brigham D.; Heyman R.; Rix P. J.; Hager J. H.; Smith N. D. Maximizing ER-alpha Degradation Maximizes Activity in a Tamoxifen-Resistant Breast Cancer Model: Identification of GDC-0927. ACS Med. Chem. Lett. 2019, 10 (1), 50–55. 10.1021/acsmedchemlett.8b00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wardell S. E.; Nelson E. R.; Chao C. A.; McDonnell D. P. Bazedoxifene exhibits antiestrogenic activity in animal models of tamoxifen-resistant breast cancer: implications for treatment of advanced disease. Clin. Cancer Res. 2013, 19 (9), 2420–2431. 10.1158/1078-0432.CCR-12-3771. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wardell S. E.; Nelson E. R.; Chao C. A.; Alley H. M.; McDonnell D. P. Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader. Endocr.-Relat. Cancer 2015, 22 (5), 713–724. 10.1530/ERC-15-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wardell S. E.; Ellis M. J.; Alley H. M.; Eisele K.; VanArsdale T.; Dann S. G.; Arndt K. T.; Primeau T.; Griffin E.; Shao J.; Crowder R.; Lai J. P.; Norris J. D.; McDonnell D. P.; Li S. Efficacy of SERD/SERM Hybrid-CDK4/6 Inhibitor Combinations in Models of Endocrine Therapy-Resistant Breast Cancer. Clin. Cancer Res. 2015, 21 (22), 5121–5130. 10.1158/1078-0432.CCR-15-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning S. W.; Jeselsohn R.; Dharmarajan V.; Mayne C. G.; Karimi M.; Buchwalter G.; Houtman R.; Toy W.; Fowler C. E.; Han R.; Laine M.; Carlson K. E.; Martin T. A.; Nowak J.; Nwachukwu J. C.; Hosfield D. J.; Chandarlapaty S.; Tajkhorshid E.; Nettles K. W.; Griffin P. R.; Shen Y.; Katzenellenbogen J. A.; Brown M.; Greene G. L. The SERM/SERD bazedoxifene disrupts ESR1 helix 12 to overcome acquired hormone resistance in breast cancer cells. eLife 2018, 7, e37161. 10.7554/eLife.37161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shagufta; Ahmad I.; Mathew S.; Rahman S. Recent progress in selective estrogen receptor downregulators (SERDs) for the treatment of breast cancer. RSC Med. Chem. 2020, 11 (4), 438–454. 10.1039/C9MD00570F. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lu Y.; Liu W. Selective estrogen receptor degraders (SERDs): A promising strategy for estrogen receptor positive endocrine-resistant breast cancer. J. Med. Chem. 2020, 63 (24), 15094–15114. 10.1021/acs.jmedchem.0c00913. [DOI] [PubMed] [Google Scholar]; c Wang L.; Sharma A. The quest for orally available selective estrogen receptor degraders (SERDs). ChemMedChem 2020, 15 (22), 2072–2097. 10.1002/cmdc.202000473. [DOI] [PubMed] [Google Scholar]
- Metcalfe C.; Ingalla E.; Blake R.; Chang J.; Daemen A.; De Bruyn T.; Giltnane J.; Guan J.; Hafner M.; Hartman S.; Kategaya L.; Kleinheinz T.; Liang J.; Mody V.; Nannini M.; Oeh J.; Ubhayakar S.; Wertz I.; Young A.; Zbieg J.; Zhou W.; Sampath D.; Friedman L.; Wang X. Abstract P5-04-07: GDC-9545: A novel ER antagonist and clinical candidate that combines desirable mechanistic and pre-clinical DMPK attributes. Cancer Res. 2019, 79, P5-04-07. [Google Scholar]
- a Scott J. S.; Moss T.; Stokes S.; Nissink W. M.; Morrow C. J.; Lawson M.; Cureton N.; Gangl E.; Gutierrez P. M.; Mather R.; Lindemann J. P.; Sykes A.; Fisher D.; Polanski R.; Carroll D.; Twomey A. B.; Klinowska T. Abstract 5674: Discovery of AZD9833, an oral small molecule selective degrader of the estrogen receptor (SERD). Cancer Res. 2020, 80, 5674. [Google Scholar]; b Scott J. S.; Moss T. A.; Balazs A.; Barlaam B.; Breed J.; Carbajo R. J.; Chiarparin E.; Davey P. R. J.; Delpuech O.; Fawell S.; Fisher D. I.; Gagrica S.; Gangl E. T.; Grebe T.; Greenwood R. D.; Hande S.; Hatoum-Mokdad H.; Herlihy K.; Hughes S.; Hunt T. A.; Huynh H.; Janbon S. L. M.; Johnson T.; Kavanagh S.; Klinowska T.; Lawson M.; Lister A. S.; Marden S.; McGinnity D. F.; Morrow C. J.; Nissink J. W. M.; O’Donovan D. H.; Peng B.; Polanski R.; Stead D. S.; Stokes S.; Thakur K.; Throner S. R.; Tucker M. J.; Varnes J.; Wang H.; Wilson D. M.; Wu D.; Wu Y.; Yang B.; Yang W. Discovery of AZD9833, a Potent and Orally Bioavailable Selective Estrogen Receptor Degrader and Antagonist. J. Med. Chem. 2020, 63 (23), 14530–14559. 10.1021/acs.jmedchem.0c01163. [DOI] [PubMed] [Google Scholar]
- a Schrodinger Release 2020-2: Glide, Schrodinger, LLC: New York, NY, 2020. [Google Scholar]; b Harder E.; Damm W.; Maple J.; Wu C.; Reboul M.; Xiang J. Y.; Wang L.; Lupyan D.; Dahlgren M. K.; Knight J. L.; Kaus J. W.; Cerutti D. S.; Krilov G.; Jorgensen W. L.; Abel R.; Friesner R. A. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12 (1), 281–296. 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]; c Schrodinger Release 2020-2: Desmond molecular dynamics system; D.E. Shaw Research: New York, NY, 2020. Maestro-Desmond interoperability tools; Schrodinger: New York, NY, 2020. [Google Scholar]
- a Watanabe C.; Fukuzawa K.; Tanaka S.; Aida-Hyugaji S. Charge clamps of lysines and hydrogen bonds play key roles in the mechanism to fix helix 12 in the agonist and antagonist positions of estrogen receptor alpha: intramolecular interactions studied by the ab initio fragment molecular orbital method. J. Phys. Chem. B 2014, 118 (19), 4993–5008. 10.1021/jp411627y. [DOI] [PubMed] [Google Scholar]; b Shiau A. K.; Barstad D.; Loria P. M.; Cheng L.; Kushner P. J.; Agard D. A.; Greene G. L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95 (7), 927–937. 10.1016/S0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]; c Brzozowski A. M.; Pike A. C.; Dauter Z.; Hubbard R. E.; Bonn T.; Engstrom O.; Ohman L.; Greene G. L.; Gustafsson J. A.; Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389 (6652), 753–758. 10.1038/39645. [DOI] [PubMed] [Google Scholar]; d Pike A. C.; Brzozowski A. M.; Hubbard R. E.; Bonn T.; Thorsell A. G.; Engstrom O.; Ljunggren J.; Gustafsson J. A.; Carlquist M. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J. 1999, 18 (17), 4608–4618. 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Thompson E. W.; Katz D.; Shima T. B.; Wakeling A. E.; Lippman M. E.; Dickson R. B. ICI 164,384, a pure antagonist of estrogen-stimulated MCF-7 cell proliferation and invasiveness. Cancer Res. 1989, 49, 6929–6934. [PubMed] [Google Scholar]; b Garner F.; Shomali M.; Paquin D.; Lyttle C. R.; Hattersley G. RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anti-Cancer Drugs 2015, 26 (9), 948–956. 10.1097/CAD.0000000000000271. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Spoerke J.; Daemen A.; Chang C.-W.; Giltnane J.; Metcalfe C.; Dickler M.; Bardia A.; Perez Fidalgo J.; Mayer I.; Boni V.; Winer E.; Hamilton E.; Bellet M.; Urruticoechea A.; Gonzalez Martin A.; Cortes J.; Martin M.; Gates M.; Cheeti S.; Fredrickson J.; Wang X.; Friedman L.; Liu L.; Li R.; Chan I.; Mueller L.; Milan S.; Lauchle J.; Humke E.; Lackner M. Abstract P5–11–01: Phamacodynamic and circulating tumor DNA evaluation in a phase I study of GDC-0927, a selective estrogen receptor antagonist/ degrader (SERD). Cancer Res. 2019, 79, P5-11-01. [Google Scholar]; d Guan J.; Zhou W.; Hafner M.; Blake R. A.; Chalouni C.; Chen I. P.; De Bruyn T.; Giltnane J. M.; Hartman S. J.; Heidersbach A.; Houtman R.; Ingalla E.; Kategaya L.; Kleinheinz T.; Li J.; Martin S. E.; Modrusan Z.; Nannini M.; Oeh J.; Ubhayakar S.; Wang X.; Wertz I. E.; Young A.; Yu M.; Sampath D.; Hager J. H.; Friedman L. S.; Daemen A.; Metcalfe C. Therapeutic Ligands Antagonize Estrogen Receptor Function by Impairing Its Mobility. Cell 2019, 178 (4), 949. 10.1016/j.cell.2019.06.026. [DOI] [PubMed] [Google Scholar]
- a Daemen A.; Spoerke J. M.; Zhou W.; Guan J.; Ingalla E.; Young A.; Hafner M.; Aimi J.; Chang C.-W.; Giltnane J. M.; Gates M.; Mayer I. A.; Azaro A.; Winer E. P.; Loi S.; Jhaveri K.; Lauchle J.; Gendreau S.; Humke E. W.; Metcalfe C. Abstract P2–11–05: ER pathway activity signature as a biomarker for endocrine agent GDC-9545. Cancer Res. 2020, 80, P2-11-05. [Google Scholar]; b Flanagan J.; Qian Y.; Gough S.; Andreoli M.; Bookbinder M.; Cadelina G.; Bradley J.; Rousseau E.; Willard R.; Pizzano J.; Crews C.; Crew A.; Taylor I.; Houston J. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer. Cancer Res. 2019, 79, P5-04-18. [Google Scholar]; c Hu J.; Hu B.; Wang M.; Xu F.; Miao B.; Yang C. Y.; Wang M.; Liu Z.; Hayes D. F.; Chinnaswamy K.; Delproposto J.; Stuckey J.; Wang S. Discovery of ERD-308 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Estrogen Receptor (ER). J. Med. Chem. 2019, 62 (3), 1420–1442. 10.1021/acs.jmedchem.8b01572. [DOI] [PubMed] [Google Scholar]
- a Grese T. A.; Sluka J. P.; Bryant H. U.; Cullinan G. J.; Glasebrook A. L.; Jones C. D.; Matsumoto K.; Palkowitz A. D.; Sato M.; Termine J. D.; Winter M. A.; Yang N. N.; Dodge J. A. Molecular determinants of tissue selectivity in estrogen receptor modulators. Proc. Natl. Acad. Sci. U. S. A. 1997, 94 (25), 14105–14110. 10.1073/pnas.94.25.14105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gennari L. Lasofoxifene: a new type of selective estrogen receptor modulator for the treatment of osteoporosis. Drugs Today 2006, 42 (6), 355–367. 10.1358/dot.2006.42.6.973583. [DOI] [PubMed] [Google Scholar]; c Andreano K. J.; Baker J. G.; Park S.; Safi R.; Artham S.; Oesterreich S.; Jeselsohn R.; Brown M.; Sammons S.; Wardell S. E.; Chang C. Y.; Norris J. D.; McDonnell D. P. The Dysregulated Pharmacology of Clinically Relevant ESR1Mutants is Normalized by Ligand-activated WT Receptor. Mol. Cancer Ther. 2020, 19 (7), 1395–1405. 10.1158/1535-7163.MCT-19-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Peterson G. M.; Naunton M.; Tichelaar L. K.; Gennari L. Lasofoxifene: selective estrogen receptor modulator for the prevention and treatment of postmenopausal osteoporosis. Ann. Pharmacother. 2011, 45 (4), 499–509. 10.1345/aph.1P604. [DOI] [PubMed] [Google Scholar]
- a Flanagan J. J.; Neklesa T. K. Targeting Nuclear Receptors with PROTAC degraders. Mol. Cell. Endocrinol. 2019, 493, 110452. 10.1016/j.mce.2019.110452. [DOI] [PubMed] [Google Scholar]; b Clinical Program Update: ARV-471 & ARV-110. https://ir.arvinas.com/static-files/ae52b7dd-e872-483a-bd26-070bae7d56b8 (accessed December 14, 2020).