

Abstract
Magnesium ions play an essential role in many vital processes. To correctly describe their interactions in molecular dynamics simulations, an accurate parametrization is crucial. Despite the importance and considerable scientific effort, current force fields based on the commonly used 12–6 Lennard-Jones interaction potential fail to reproduce a variety of experimental solution properties. In particular, no parametrization exists so far that simultaneously reproduces the solvation free energy and the distance to the water oxygens in the first hydration shell. Moreover, current Mg2+ force fields significantly underestimate the rate of water exchange leading to unrealistically slow exchange kinetics. In order to make progress in the development of improved models, we systematically optimize the Mg2+ parameters in combination with the TIP3P water model in a much larger parameter space than previously done. The results show that a long-ranged interaction potential and modified Lorentz–Berthelot combination rules allow us to accurately reproduce multiple experimental properties including the solvation free energy, the distances to the oxygens of the first hydration shell, the hydration number, the activity coefficient derivative in MgCl2 solutions, the self-diffusion coefficient, and the binding affinity to the phosphate oxygen of RNA. Matching this broad range of thermodynamic properties, we present two sets of optimal parameters: MicroMg yields water exchange on the microsecond timescale in agreement with experiments. NanoMg yields water exchange on the nanosecond timescale facilitating the direct observation of ion-binding events. As shown for the example of the add A-riboswitch, the optimized parameters correctly reproduce the structure of specifically bound ions and permit the de novo prediction of Mg2+-binding sites in biomolecular simulations.
1. Introduction
Magnesium ions play a crucial role in a large variety of physiological processes such as ATP hydrolysis, cellular signaling, or the catalytic activity of enzymes and ribozymes.1−4 In particular, in nucleic acid systems, Mg2+ ions are essential to stabilize the tertiary structure, to drive folding or to enable catalytic reactions.3,5−11 Due to the biochemical importance of Mg2+, the modeling of these ions has received significant scientific attention.12−21 However, providing a quantitative description of their interactions and resolving their role in the folding and function of biomolecules is challenging. On the one hand, ab initio quantum mechanical approaches could provide unbiased insights but are limited to small system sizes. On the other hand, classical all-atom simulations allow us to treat much larger and biologically relevant systems but require accurate empirical force fields. Currently, in the most widely used force fields, Mg2+ ions are modeled as point charges and the electrostatic, dispersion, and excluded volume interactions are taken into account by a pairwise interaction potential. Hereby, the most common form of the nonelectrostatic interactions is the 12–6 Lennard-Jones (LJ) potential.22
In order to provide accurate Mg2+ models, the two parameters of the LJ potential are typically adjusted to reproduce experimental solution properties such as the solvation free energy ΔGsolv,17−20 the distance to the water oxygens in the first hydration shell R1,13,14,16,18,19 and the coordination number n1.13,16−20 In addition to thermodynamic and structural data, including kinetic properties in the parametrization is crucial to capture dynamical processes such as water exchange, ion-binding, or ion-pairing.16,20,23 However, including kinetic data in the optimization is quite demanding since water exchange is on the microsecond timescale24,25 and involves the concerted motion of two exchanging water molecules.26 Therefore, simple and computationally efficient methods based on transition state theory are insufficient to provide an accurate rate estimate.26
Moreover, force fields optimized based on ion–water properties alone frequently fail to reproduce thermodynamic and structural properties of electrolytes at nonvanishing salt concentrations.27,28 It is therefore essential to balance ion–water and ion–ion interactions by including experimental data for activity coefficient derivatives or osmotic pressures in the optimization.28−32
Finally, in order to improve the applicability of the parameters for biomolecular simulations, it has proven useful to consider the interactions between Mg2+ and specific ion-binding sites on RNA and proteins.33,34
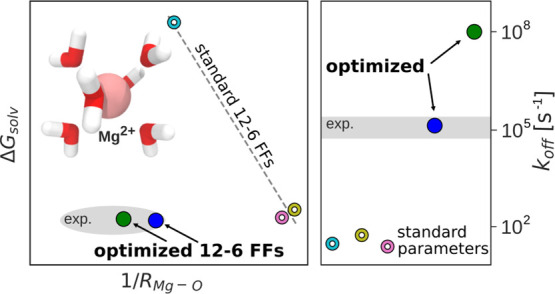
Reproducing this broad range of structural, thermodynamic, and kinetic properties by optimizing the force field parameters is tremendously challenging. Despite considerable scientific effort, none of the Mg2+ force fields from the literature based on the common 12–6 LJ interaction potentials is able to reproduce all properties with sufficient accuracy.23 For example, apparently, no parameter combination of the LJ parameters exists that simultaneously reproduces ΔGsolv and R1.17,18,20,35 In addition, current force fields underestimate the experimental rate of water exchange by several orders of magnitude leading to unrealistically slow exchange dynamics in biomolecular simulations.20,23,26 The too slow exchange kinetics has severe consequences since it governs any type of reaction involving the replacement of strongly bound hydration water. Therefore, important biochemical processes such as the transition from outer-to-inner sphere binding, chemical reactions in metalloenzymes or ribozymes, or the transport of ions across cell membranes24,25,36−39 become so rare that they cannot be simulated with sufficient statistics or, in many cases, not at all. In order to address this problem, Allnér et al. developed a set of Mg2+ parameters that reproduces the experimental exchange rate.16 However, similar to the ΔGsolv–R1 parametrization problem, the authors failed to simultaneously reproduce the solvation free energy and the water exchange rate.
The aim of our current work is to provide optimized Mg2+ parameters in combination with the TIP3P water model that accurately reproduce all the abovementioned thermodynamic and kinetic properties. However, due to the complexity of the optimization problem, the question arises whether simple 12–6 potentials are sufficient for an accurate description or whether additional terms in the interaction potential are required. In particular, classical nonpolarizable force fields do not include many-body quantum effects explicitly. While Mg2+ ions themselves have a low polarizability, they polarize their environment strongly. Therefore, charge-induced dipole interactions and charge transfer can become significant, rendering the interaction between the cation and water more attractive and long-ranged compared to ions with low charge density. It therefore seems appealing to include additional parameters in the interaction potential to account for such polarization effects explicitly. For example, 12–6–4 potentials include an additional r–4 term that mimics the charge-induced dipole interactions.19 This parametrization strategy has proven successful in simultaneously reproducing ΔGsolv, R1, and n1 for different metal ions.19 Another possibility is to modify the description of the electrostatic term.40,41 For example, Jungwirth and co-workers, introduced an additional charge scaling term to reproduce structural properties of aqueous MgCl2 from neutron-scattering experiments.15 Nevertheless, it is clear that any model with more parameters is expected to be in general more accurate if optimized properly with respect to all its parameters.
Here, we follow an alternative approach to improve the agreement with experimental properties without introducing more complex force fields. This approach is motivated by the fact that nonpolarizable force fields of the 12–6 type take polarizability into account implicitly. Therefore, instead of enlarging the parameter space of the interaction model, we explore the already existing force field parameter space in all depth. This includes an extensive optimization in an extended range of possible ion–water LJ parameters and a systematic optimization of the ion–ion and ion–RNA combination rules. Our optimization strategy is justified a posteriori by illustrating that the agreement with experimental results is similar or better compared to more complex force fields with additional terms in the interaction potential.
Our optimization procedure is done in three consecutive steps. In the first step, we optimize the ion–water interactions by selecting parameter combinations that reproduce ΔGsolv, R1, and n1. In the second step, we optimize the water-exchange kinetics by choosing the combination that reproduces the experimental water-exchange rate (microMg). In addition, we choose a second parameter set that yields accelerated water-exchange dynamics (nanoMg). In the last step, we optimize the ion–ion and ion–biomolecule interactions by introducing scaling factors in the combination rules.30 This allows us to reproduce the activity derivative of MgCl2 solutions over a broad concentration range. Furthermore, we balance the Mg2+–RNA interactions by tuning the pairwise interaction to reproduce the binding affinity toward the nonbridging oxygen atoms of the phosphate group on RNA. Finally, we test the performance of our optimized parameters for the add A-riboswitch. MicroMg leads to stable RNA structures and correctly reproduces the structure of two specifically bound ions. This parameter set is particularly suited to simulate Mg2+ in aqueous solutions and its interactions with biomolecules such as nucleic acids, proteins, and lipids. On the other hand, nanoMg yields accelerated water-exchange dynamics and is therefore particularly suited to investigate specific ion-binding including the de novo prediction of inner-sphere ion-binding sites on RNA.
2. Methods
2.1. Molecular Dynamics Simulations
In the following, Mg2+ ions are modeled as point charges and the electrostatic, dispersion, and excluded volume interactions are taken into account by a pairwise interaction potential. Hereby, the most common form of the LJ potential is used with a repulsive r–12 and an attractive r–6 term. Overall, the interaction potential has the following form
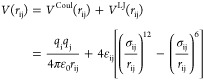 |
1 |
where qi and qj are the charges of atoms i and j, respectively, rij is the distance between them, and ϵ0 is the dielectric constant of vacuum. The parameters σij and εij describe the LJ diameter and interaction strength, respectively. We refrain from adjusting parameters of the Coulomb term while the two parameters of the LJ potential are free to be optimized. The Lorentz–Berthelot combination rules are used to describe the interactions between atoms i and j
| 2 |
We used the TIP3P water model42 with LJ parameters of σo = 0.315061 nm and εo = 0.6364 kJ/mol. This choice was motivated by the fact that this water model is frequently used in biomolecular simulations since the AMBER force fields for nucleic acids and proteins have been optimized in combination with TIP3P water.
In order to compare our newly optimized parameters (Table 1) to force fields from the literature, we performed simulations using the 12–6-based parameters by Allnér–Villa,16 Mamatkulov–Schwierz,20 and Li–Merz18 (HFE set) and the 12–6–4-based parameters by Li–Merz.19 For our optimization procedure and for simulations with the Mg2+ parameters by Mamatkulov–Schwierz,20 Cl– parameters were taken from Mamatkulov–Schwierz.20 In all other cases, the Cl– parameters were taken from Joung–Cheatham.43 The parameters of all force fields used are listed in Table S6. Simulations with force fields of the 12–6 type were performed with GROMACS44 (versions 5.1.4, 2018, 2020). Simulations with force fields of the 12–6–4 type were performed with AMBER45 (version 2020) since GROMACS does not support 12–6–4 interaction potentials. An overview over the various simulation setups can be found in Section S1.1 (Table S1).
Table 1. Optimized Force Field Parameters for Mg2+ for Simulations with the TIP3P Water Modela.
| microMg | nanoMg | ||
|---|---|---|---|
| σii | [nm] | 0.1019 | 0.1025 |
| εii | [kJ/mol] | 235.80 | 389.80 |
| σio | [nm] | 0.2085 | 0.2088 |
| εio | [kJ/mol] | 12.250 | 15.750 |
| λσCl | 1.8000 | 1.8000 | |
| λεCl | 0.1000 | 0.1000 | |
| σMgCl | [nm] | 0.4878 | 0.4884 |
| εMgCl | [kJ/mol] | 0.8181 | 1.0518 |
| λσRNA | 1.1375 | 1.1435 | |
| λεRNA | 0.3200 | 0.2500 | |
| σMgOP | [nm] | 0.2262 | 0.2277 |
| εMgOP | [kJ/mol] | 4.6061 | 4.6266 |
σii, εii, σio, and εio are the ion–ion and ion–water LJ parameters. λσX and λε are the scaling factors for the Lorentz–Berthelot combination rules (eq 3) for the interaction with Cl– or the RNA atoms, shown exemplary for the interaction between Mg2+ and OP. Note that the scaling factors are only valid in combination with the Cl– parameters from ref (20) and the parmBSC0χOL3 RNA parameters.50−52
Following the work by Fyta and Netz,30 we introduce adjustable scaling parameters λσX and λε in the Lorentz–Berthelot combination rules to describe the Mg2+–Cl– and the Mg2+–RNA interactions. With this, the Lorentz–Berthelot combination rules have the following form
| 3 |
where X denotes Cl– or the atoms of the RNA. Note that these additional scaling factors leave the ion–water interaction parameters unchanged. Therefore, the solvation free energy, the structural properties of the first hydration shell, and the rate of water exchange remain unchanged.
To optimize the ion–RNA interactions, dimethylphosphate (DMP) was used similar to previous work.16,33,46,47 The DMP molecule contains two nonbridging phosphate oxygen atoms that are considered to be the most important inner-sphere Mg2+-binding sites. The force field parameters for the DMP molecule are based on a parametrization with GAFF48 (see Section S1.2).
For the add A-riboswitch (PDB ID: 1Y26(49)), the parmBSC0χOL3 force field50−52 was used. Adenine was parameterized using GAFF (see Section S1.2).
The analysis was performed with the built-in GROMACS44 code and using the MDAnalysis package53,54 for python.
2.2. Optimization Procedure
In the first step, we optimize the ion–water interaction by performing a grid search in σio–εio space. Initially, all σio–εio parameter combinations are selected that match the experimental solvation free energy ΔGsolv, the Mg2+–oxygen distance in the first hydrations shell R1, and the coordination number n1.
In the second step, we optimize the water-exchange dynamics by calculating the rate of water exchange for the abovementioned parameter combinations. Two parameter sets were chosen: the microMg parameter set yields water exchange on the microsecond timescale and reproduces the experimental rate exactly. The nanoMg parameter set yields water exchange on the nanosecond timescale, thereby providing exchange dynamics that are 2 orders of magnitude faster while still reproducing thermodynamic and structural properties.
In the last step, we optimize the ion–ion and ion–RNA interactions by calculating activity coefficient derivatives and ion-binding affinities by performing a grid search in λσX and λε parameter space (eq 3). In particular, we used Kirkwood–Buff theory55 to calculate the activity coefficient derivatives acc and to select the scaling factors λσCl and λε that reproduce the experimental activity derivative over a broad range of MgCl2 concentrations. Subsequently, alchemical transformation calculations were used to calculate the binding affinity of Mg2+ toward one of the nonbridging phosphate oxygens of DMP. Finally, the scaling factors λσ,εRNA that reproduce the experimental binding affinity ΔGb and binding distance Rb toward the phosphate oxygen were selected.
2.3. Free-Energy Perturbation and Single-Ion Properties
The solvation free energy of neutral MgCl2 ion pairs ΔGsolv was calculated following the same procedure described in our previous work.20 Since the proton solvation free energy used for absolute solvation free energies can vary according to different sources, we use the more robust solvation free energy of neutral MgCl2 ion pairs.35 The parameter range used in our current study is σio = 0.16–0.24 nm and εio = 1.8–28 kJ/mol. Finite size, pressure, and surface effect corrections were applied and simulations with three different box sizes yielded the same result in agreement with previous findings.35,56 Further details can be found in refs (20, 35) and Section S1.3.
In addition to ΔGsolv, R1, and n1, the self-diffusion coefficient D0 was calculated (see Section S1.4).
2.4. Umbrella Sampling
One-dimensional free-energy profiles were calculated using umbrella sampling.57,58 The distance between Mg2+ and the leaving water molecule rMgOw1 or between Mg2+ and one of the two nonbridging phosphate oxygens of DMP rMgOP was used as the umbrella coordinate. Note that parameters defining partial charge, atom type, and angle potential of the two nonbridging phosphate oxygens of the DMP were adjusted to be identical to the AMBER RNA force field parameters (parmBSC0χOL3,50−52 see Section S1.2). The Mg2+ parameters and scaling factors are therefore directly transferable.
The two-dimensional free-energy profile was calculated as a function of the distance between Mg2+ and the two exchanging water molecules, rMgOw1 and rMgOw2. During the sampling, additional restraints have been applied (see ref (26) and Section S1.5).
2.5. Rate Constant of Water Exchange
The rate constant k of water exchange in the first hydration shell of Mg2+ is defined by24
| 4 |
where 6 is the coordination number of the first hydration shell and [Mg(H2O)62+] is the concentration of hexa-coordinated Mg2+ ions.
The most popular theory to calculate reaction rates is transition state theory (TST).59,60 In simple systems for which the reaction coordinate is exactly known, TST gives an accurate estimate of the rate. However, in complex many-body systems as the ones presented here, TST can fail due to the violation of the non-recrossing hypothesis which forms the cornerstone of the theory. Therefore, in the following, we use TST only to compare to results from the literature or to provide an upper estimate for the rate constant (see Sections S1.6 and S2.3).
In order to provide an accurate rate estimate, we use 1 μs long trajectories of a 1 M MgCl2 solution and calculate the rate from the number of transitions over time. Hereby, we follow each water molecule individually through the trajectory. The rate constant k is then given by
| 5 |
where NH2O is the number of water molecules in the simulation box and N is the total number of transitions for all water molecules (counting the exchange from first to second hydration shell and the reverse transition as individual events). tB = NMg × pB × tsim is the cumulative time the water molecule spends in the first hydration shell of any Mg2+ ion. NMg is the number of Mg2+ ions in the simulation box, pB = 6/(NH2O – 6) is the probability of water to be in the first hydration shell, and tsim is the total simulation time. The number of transitions is calculated from an indicator function which defines the bound and unbound state using two cutoff parameters. Different values for the cutoff parameters were tested and the calculated rates were found to be insensitive to the exact definition (for further details see Section S1.7). Errors are calculated from block averaging61 by dividing the trajectory into two blocks.
2.6. Kirkwood–Buff Theory
To optimize the scaling factors for ion–ion interactions, 150 ns long simulations were performed at finite salt concentration. The parameter range investigated was λσCl = 1–2.6 and λε = 0.01–1. The activity coefficient derivatives acc were calculated using Kirkwood–Buff theory.55 The optimization was done for a concentration of 0.25 M MgCl2. Additional simulations at MgCl2 concentrations of 0.25, 0.5, 1, and 2 M were performed for the final parameter sets. Errors were calculated from dividing the trajectories into three blocks and block averaging. Further details on the calculation of acc can be found in Section S1.8.
2.7. Alchemical Transformation
To optimize the scaling factors for ion–RNA interactions, the binding affinity was calculated from alchemical transformations (see Section S1.9 for simulation details). In particular, the binding affinity ΔGb0 and the binding distance Rb toward one of the nonbridging phosphate oxygens of DMP were calculated for the parameter range λσ = 0.97–1.23 and λεRNA = 0.08–1.04. Subsequently, the scaling factors that reproduced the experimental value for ΔGb62 and Rb(63) were selected. Note that in the experimental work by Sigel and Sigel,62 two sets of values are given for the binding affinity. The first value is the stability constant of the DMP (log K = 0.45 or equivalently ΔGb0 = −1.036 kBT). The second value (log K = 1.05 or ΔGb = −2.418 kBT) is the stability constant of a modular RNA model. The value takes into account the fourfold access of the phosphate oxygen-binding site on the backbone compared to the nucleobase-binding sites and is appropriate only within the context of the modular RNA model. For our simulations, hence, the first value (log K = 0.45) is appropriate and was used in the optimization.
To further validate the results from alchemical transformations, ΔGb0 and Rb were calculated independently from free-energy profiles obtained for the final parameter sets (Section S1.9). Both methods yielded identical results within error (Section S2.4). Similarly, ΔGb and Rb for the Allnér–Villa and Panteva–York m12–6–4 parameters33 were calculated from free-energy profiles. The free-energy profiles were obtained from refs (16, 33) with permission.
Errors were calculated from block averaging by dividing the trajectory of the alchemical transformation into three blocks.
2.8. Performance of MicroMg and NanoMg for the add A-Riboswitch
To test the performance of our optimized parameter sets in a biologically relevant RNA system, the add A-riboswitch was simulated for 100 ns. The simulations included five Mg2+ ions positioned as observed in the X-ray structure (PDB ID: 1Y26,49 resolution: 2.10 Å). A total of 30 additional Mg2+ ions were placed randomly into the simulation box to neutralize the charge of the riboswitch. The rmsd was calculated discarding the first 2 ns for equilibration.
In a second setup, used to predict inner-sphere binding sites with nanoMg, 10 replicas of 200 ns were simulated. Here, all Mg2+ ions were placed randomly in the simulation box.
Three-dimensional Mg2+ densities were obtained with GROmaρs.64 The density was visualized with PyMOL.65
3. Results and Discussion
In the following, we present the results from our optimization procedure. The optimization is performed in three sequential steps and is aimed to capture ion–water, ion–ion, and ion–RNA interactions. In particular, the optimization is designed to simultaneously reproduce the solvation free energy, the distance to oxygens in the first hydration shell, the hydration number, the activity coefficient derivative in MgCl2 solutions, the self-diffusion coefficient, and the binding affinity and distance to the phosphate oxygens of RNA (Tables 2 and 3).
Table 2. Results for Single-Ion, Ion–Ion, and Ion–RNA Properties for the Optimized Parameters in Direct Comparison with Experimental Resultsa.
| ΔGsolv [kJ/mol] | R1 [nm] | n1 | D0 [10–5 cm2/s] | ΔGb0 [kBT] | Rb [nm] | acc | |
|---|---|---|---|---|---|---|---|
| microMg | –2532.9 ± 1 | 0.207 ± 0.004 | 6 | 0.754 ± 0.006 | –0.633 ± 0.6 | 0.207 ± 0.004 | 0.93 ± 0.01 |
| nanoMg | –2532.0 ± 1 | 0.209 ± 0.004 | 6 | 0.750 ± 0.004 | –0.375 ± 0.1 | 0.207 ± 0.004 | 0.97 ± 0.01 |
| exp. | –253279 | 0.209 ± 0.00466 | 666 | 0.70679 | –1.03662 | 0.206–0.20863 | 0.9380 |
Solvation free energy of neutral MgCl2 ion pairs ΔGsolv, Mg2+–oxygen distance in the first hydration shell R1, coordination number of the first hydration shell n1, self-diffusion coefficient D0, binding affinity toward the phosphate oxygen of DMP ΔGb0, Mg2+–phosphate oxygen distance in the inner-sphere conformation Rb, and acc is the activity derivative of a MgCl2 solution at 0.25 M concentration. ΔGb is derived from the log stability constant of the DMP (log K = 0.45) given in ref (62).
Table 3. Properties of Water Exchange from Simulations and Experimentsa.
| N | k [s–1] | |
|---|---|---|
| microMg | 376 ± 56 | (8.04 ± 1.20) × 105 |
| nanoMg | 52,086 ± 120 | (1.11 ± 0.003) × 108 |
| Mamatkulov–Schwierz | 2 ± 2 | 24.0 ± 8.8 from ref (26) |
| Allnér–Villa | 2 ± 2 | <2.4 × 105 |
| Li–Merz (12–6) | 2 ± 2 | <3.5 × 104 |
| Li–Merz (12–6–4) | 6720 ± 160 | (1.44 ± 0.03) × 107 |
| exp. | 248,25 31424 | 5.3 × 105 from ref (24), 6.7 × 105 from ref (25) |
Number of transitions N in 1 μs for different force fields in 1 M MgCl2 solutions. The experimental value24,25 is obtained from eq 5. The rate constant k is calculated from the number of transitions for microMg, nanoMg, and Li–Merz (12–6–4). The value for Mamatkulov–Schwierz is taken from ref (26). For Allnér–Villa and Li–Merz (12–6), an upper estimate is given based on TST since the number of transitions is insufficient to calculate the rate from eq 5. The errors for N and k are obtained from block averaging.
Matching this broad range of structural and thermodynamic properties, we present two sets of optimal parameters: MicroMg yields water exchange on the microsecond timescale and matches the experimental exchange rate. NanoMg yields water exchange on the nanosecond timescale. Subsequently, we validate the performance of our optimized parameter sets for the example of the add A-riboswitch.
3.1. Optimization of Solvation Free Energy, Mg2+–Water Distance, and Coordination Number
In the first step, we optimize the ion–water interactions by adjusting the LJ parameters σio and εio to reproduce the experimental solvation free energy ΔGsolv, the distance to the oxygens in the first hydration shell R1, and the coordination number n1 (Figure S1). Since ΔGsolv includes the energy and entropy of ion hydration, it is considered the most important thermodynamic property in the development of accurate force field parameters.35 Moreover, Mg2+ is coordinated by six water molecules arranged in octahedral symmetry.66 In order to correctly capture the structure of the first hydration shell, we include R1 and n1 in our optimization.
Reproducing ΔGsolv and R1 with force fields of the 12–6 type is challenging. As illustrated in Figure 1A, in previous work, no parameter combination could be found that matches both properties simultaneously.16−18,20,35 In order to improve the agreement, we have considered a much larger σio and εio range in the optimization (Figure S1). As shown in Figures 1A and S1 and Table 2, this allows us to accurately reproduce ΔGsolv, R1, and n1. Hereby, the agreement with experimental results is comparable to 12–6–4 interaction potentials which have one additional adjustable parameter. Based on the results for ΔGsolv and R1, we conclude that additional terms in the interaction potential that mimic polarization effects are not strictly necessary. Nevertheless, charge-induced dipole interactions and charge transfer are particularly important for Mg2+ ions in aqueous solutions. Both effects render the interaction potential more attractive and long-ranged compared to metal cations with lower charge density. This becomes evident from Figure 1B: the interaction potential of our optimized parameter sets microMg and nanoMg and the 12–6–4 potential by Li–Merz are similar in shape and more attractive and long-ranged compared to previous 12–6-based force fields. Capturing the long-ranged interactions is therefore crucial to correctly describe ion–water interactions in general and ΔGsolv and R1 in particular.
Figure 1.
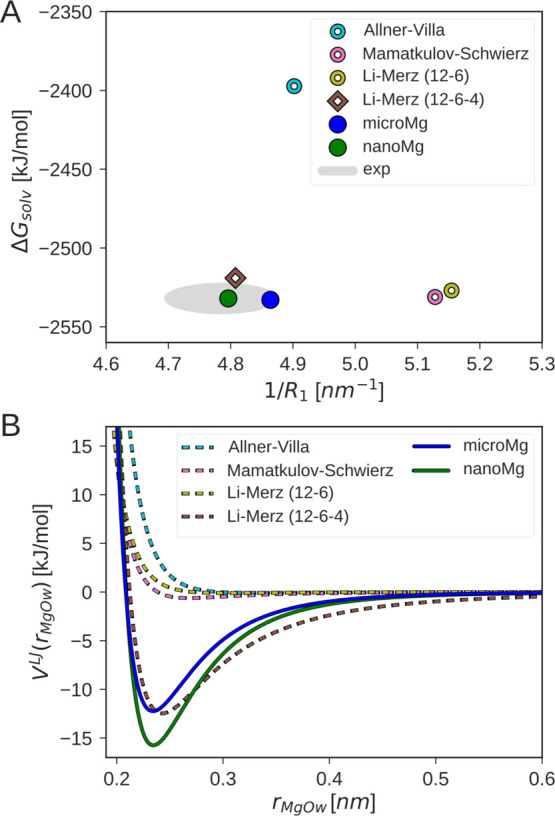
Comparison of the optimized parameter sets microMg and nanoMg with force fields from the literature16,18−20 and experimental data. (A) Solvation free energy ΔGsolv for neutral MgCl2 pairs in correlation with the inverse of the Mg2+–oxygen distance of the first hydration shell 1/R1. The gray area indicates the experimental results from refs (66, 70). (B) Lennard-Jones interaction potential VLJ as a function of the Mg2+–oxygen distance rMgOw for different Mg2+ force fields and TIP3P water.
3.2. Optimization of the Water-Exchange Rate
In aqueous solutions, water molecules from the first tightly bound hydration shell around Mg2+ exchange with the second hydration shell on the microsecond timescale.24,25 In the second step of our parametrization, we optimize the water-exchange dynamics by calculating the rate constant k of water exchange in the range of parameter combinations that reproduce ΔGsolv, R1, and n1 obtained in the previous optimization step. Hereby, special emphasis is placed on an accurate calculation of the rate constant. The reason is that the popular and frequently used transition state theory (TST) can fail due to the violation of the noncrossing hypothesis. In fact, recent work shows that for water exchange, TST based on the Mg2+–water distance overestimates the true rate by more than 2 orders of magnitude.26 In order to provide a more accurate rate estimate, we use 1 μs long simulations of 1 M MgCl2 solutions to calculate the rate from the number of transitions (eq 5). Based on this, two parameter combinations were selected (Table 3): the parameter set microMg yields water exchange on the microsecond timescale in agreement with experimental results.24,25 The parameter set nanoMg yields water exchange on the nanosecond timescale. Hereby, we chose the parameter combination that leads to the fastest exchange while still matching all other experimental properties.
Figure 2A compares the number of exchanges for different force fields with the 248–314 transitions expected from experiments.24,25 The results reveal that water exchange is several orders of magnitude too slow for the force fields by Allnér–Villa,16 Mamatkulov–Schwierz,20 and Li–Merz (12–6).18 Surprisingly, the rate calculated from the Allnér–Villa parameters deviates from the experimental results despite being optimized on the exchange kinetics. The reasons are twofold: the Allnér–Villa parameters were optimized with the mTIP water model,67 and transferring the parameters to TIP3P leads to deviations (Section S2.2). In addition, the rate used in the optimization was calculated from TST and might therefore deviate notably from the true rate as discussed above. The number of exchanges for the Li–Merz 12–6–4 parameters is significantly higher and thus the rate overestimates experiments by 1 order of magnitude (Table 3). As expected from the current optimization procedure, the number of transitions for microMg closely matches the experimental value while nanoMg yields the highest number of exchanges of all force fields considered here.
Figure 2.
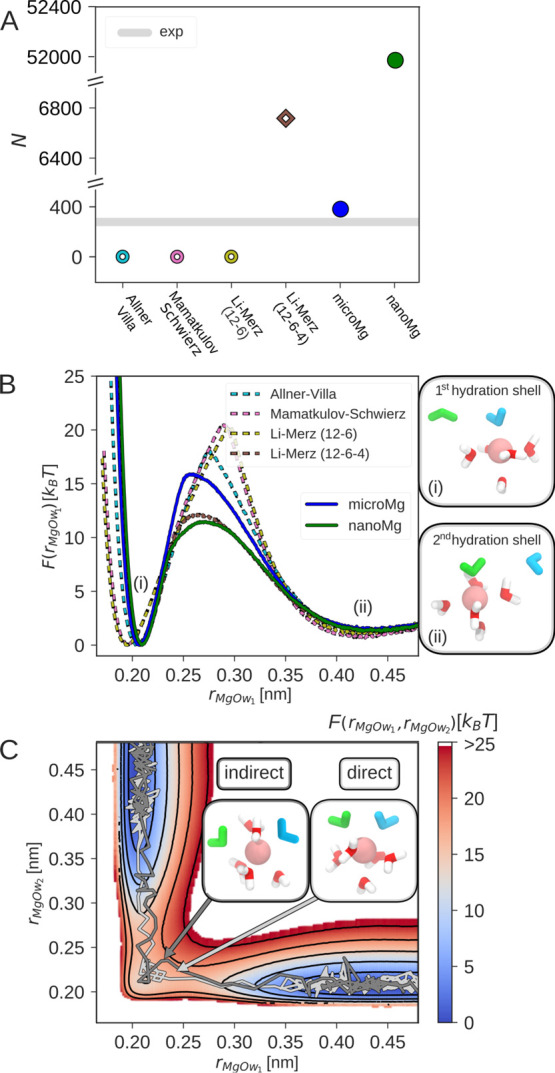
Water exchange in the first hydration shell of Mg2+. (A) Number of exchange events in 1 μs for different force fields in 1 M MgCl2 solutions. The gray horizontal line indicates the experimental values24,25 (Table 3). (B) One-dimensional free-energy profiles as a function of the distance between Mg2+ and the leaving water molecule rMgOw1 for different force fields. The snapshots show representative conformations in the two stable states: (i) before exchange: leaving water (Ow1 shown in blue) is in the first hydration shell and incoming water (Ow2 shown in green) is in the second hydration shell. (ii) After exchange: Ow1 is in the second and Ow2 is in the first hydration shell. (C) Two-dimensional free-energy profile as a function of the distances between Mg2+ and the two exchanging waters, rMgOw2 and rMgOw1, obtained with the microMg parameters. The insets show representative snapshots of the transition state along the direct (dark gray) and the indirect (light gray) exchange pathway. The energy contour spacing corresponds to 5 kBT.
Qualitative insights into the cause of the vastly different timescales of water exchange for the different force fields can be gained from the free-energy profiles along the Mg2+–water distance rMgOw1 (Figure 2B). The free-energy profiles have two minima corresponding to the first and second hydration shell [shown in snapshots (i,ii) of Figure 2B]. The two minima are separated by a high free-energy barrier which corresponds to the cost of removing one water from the tightly bound first hydration shell. The height of the barriers differs by more than 10 kBT for the different force fields. Force fields with slow exchange kinetics [Li–Merz (12–6), Mamatkulov–Schwierz, and Allnér–Villa] have high energetic barriers of 20.4, 20.7, and 18.0 kBT. Force fields with comparatively faster exchange kinetics [microMg, Li–Merz (12–6–4), and nanoMg] have lower energetic barriers of 15.9, 12.2, and 11.5 kBT, respectively.
Water-exchange dynamics is, however, more complex than the one-dimensional free-energy profiles might suggest. It involves the concerted motion of two exchanging water molecules in which the molecular void provoked by the leaving water is immediately filled by an entering water molecule.26 In order to provide a more realistic picture, Figure 2C shows the two-dimensional free-energy profile as a function of the distance of the two exchanging waters rMgOw1 and rMgOw2 obtained with the microMg parameters (for nanoMg see Figure S3). Two exchange pathways are shown corresponding to a direct and indirect exchange mechanism in agreement with our previous results.26
In summary, the optimized microMg parameters yield much closer agreement with experimental water exchange rates compared to force fields from the literature (Table 3). In addition, the self-diffusion coefficient D0 matches the experimental value without further optimization (Table 2).
3.3. Optimization of Activity Derivative and Binding Affinity
In the last step of our parametrization, we sequentially optimize the ion–ion and the ion–RNA interactions. In order to correctly capture ion-pairing, a proper balance between ion–water and ion–ion interactions is required. This balance is achieved by optimizing the parameters based on experimental activity coefficients.28 Following the seminal work by Fyta and Netz,30 we introduce scaling parameters in the Lorentz–Berthelot combination rules (eq 3). The advantage of this approach is that it allows us to reproduce the experimental activity coefficient derivative acc over a broad concentration range for MgCl2 solutions (Figure 3A) without changing ΔGsolv, R1, n1, D0, or k. In fact, with the standard combination rules (eq 2), acc was found to be too small for all parameter combinations investigated, in agreement with previous findings.17,20,30 Since no solid foundation for the standard combination rules exists, it is not surprising that they fail to capture cation–anion interactions. Targeting this interaction in the optimization therefore appears to be the natural choice. The resulting scaling factors (Table 1) reflect that without modifications, the Mg2+–Cl– interactions are too attractive. This problem can easily be corrected by increasing the effective diameter of the outer-sphere ion pair via λσCl and by reducing the cation–anion LJ energy via λε.
Figure 3.
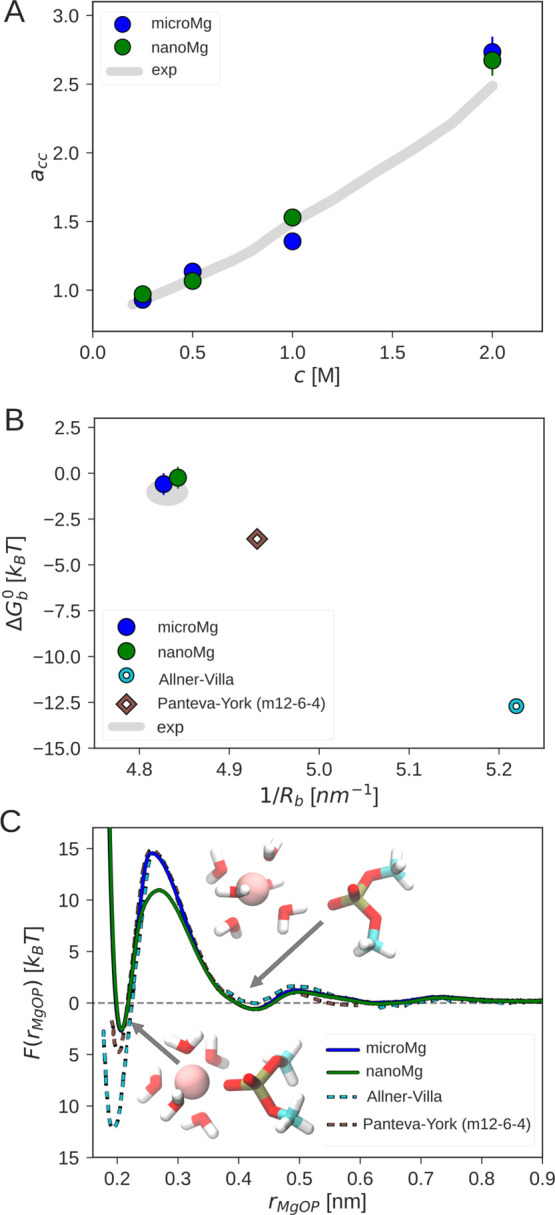
Optimization of ion–ion and the ion–RNA interactions. (A) Activity derivative acc as a function of the MgCl2 salt concentration with the optimized scaling factors for microMg and nanoMg (Table 1) and from experiments.80 (B) Binding affinity ΔGb0 in correlation with inverse of the Mg2+–phosphate oxygen distance 1/Rb. The experimental values (gray) are taken from refs (62, 63). (C) Free-energy profiles as a function of the distance between Mg2+ and the phosphate oxygen of DMP, rMgOP, for different force fields. The Allnér–Villa and Panteva–York free-energy profiles were obtained from refs (16, 33) with permission. The insets show Mg2+ in the inner- and outer-sphere conformation.
So far, the optimization was performed based on bulk ion properties. However, this does not guarantee that the interactions of Mg2+ and specific ion-binding sites on biomolecules are described correctly. For example, the 12–6–4 Li–Merz parameters significantly overestimate the Mg2+–RNA interactions.33 In order to solve this problem, modified m12–6–4 parameters were developed by Panteva and co-workers to reproduce experimental site-specific binding affinities.33
Here, we introduce ion–RNA scaling parameters in the Lorentz–Berthelot combination rules (eq 3) that are optimized based on experimental binding affinities62 and structural properties of the inner-sphere conformation.63 Similar to previous work,16,33,46,47 DMP was used. The DMP molecule contains two nonbridging phosphate oxygen atoms that are considered to be the most important inner-sphere Mg2+-binding sites on larger RNA molecules.3,62,68,69 Similar to acc, the unmodified combination rules result in Mg2+–RNA interactions that are too attractive, possibly reflecting the small excess polarizability of Mg2+. Again, this can be corrected by increasing the effective diameter via λσRNA and by reducing the cation–RNA LJ energy via λε (Table 1, Section S2.4).
Figure 3B summarizes the binding affinity ΔGb0 and binding distance Rb for different force fields from the literature. As expected, the Allnér–Villa parameters show the largest deviations from the experimental results. The Panteva–York m12–6–4 parameters provide significant improvement. Finally, the optimized ion–RNA scaling factors for microMg and nanoMg provide the closest agreement with experimental results.
Initial insights into the process of ion-binding to RNA is obtained from the free-energy profiles as a function of the distance between Mg2+ and the phosphate oxygen of DMP for different force fields (Figure 3C). The free-energy profiles have two stable states corresponding to the inner-sphere and the outer-sphere conformation (inset of Figure 3C). In the inner-sphere conformation, Mg2+ forms a direct contact with the phosphate oxygen of the DMP. In the outer-sphere conformation, the contact is mediated by a water molecule from the first hydration shell. There are clear deviations between the free-energy profiles from different force fields. The different depths of the first minimum reflect the different binding affinities as discussed above. Consequently, force fields that overestimate the binding affinity have a higher free-energy barrier and therefore slower dissociation kinetics. Similarly, the minimum in the free-energy profile is shifted to the left for force fields that underestimate Rb.
The free-energy barrier for nanoMg is 3.5 kBT lower compared to microMg while ΔGb0 and Rb are identical. Removing a water molecule from the first hydration shell for nanoMg requires less work as reflected in the higher water-exchange rate (Table 3). Mg2+ association and dissociation is therefore 2–4 orders of magnitude faster compared to other force fields.
In summary, matching ΔGb0, Rb, and the rate of water exchange is crucial to describe the kinetics of ion binding and the structure of specifically bound Mg2+ ions. In this respect, microMg is particularly suited to reproduce the distribution and exchange of Mg2+ as closely as possible. On the other hand, the ion-binding kinetics for nanoMg is significantly enhanced. This opens up the possibility to use nanoMg to predict inner-sphere ion-binding sites from straightforward simulations without the necessity to use enhanced sampling techniques.70
3.4. Performance of MicroMg and NanoMg for the add A-Riboswitch
In order to evaluate the performance of the optimized parameter sets, the add A-riboswitch was simulated (Figure 4A). This riboswitch is particularly suited to validate the parameters since experimental49,71−73 and simulation results16,74 exist.
Figure 4.

Representative snapshot of the add A-riboswitch from simulations with the nanoMg parameter set and three-dimensional Mg2+ probability density. The probability density is low in the blue regions (diffusive ions) and high in the red regions (specifically bound ions). Selected inner-sphere (pink) and outer-sphere (black) Mg2+ ions are shown including the water molecules in their first hydration shell. Snapshots (i–iii) show the most probable ion-binding sites predicted from simulations with nanoMg. Snapshots (i,ii) coincide with the two inner-sphere binding sites reported in the X-ray structure.49 The experimental electron density is shown as gray mesh.
The experimental X-ray structure49 includes five Mg2+ ions (PDB ID: 1Y26,49 resolution: 2.10 Å). Two ions are bound in inner-sphere conformation to phosphate oxygens, two ions are involved in outer-sphere interactions, while the remaining ion is a crystallization artifact due to crystal packing.
3.4.1. Stability and Specific Ion-Binding Sites with MicroMg and NanoMg
Initially, we investigate that microMg and nanoMg do not affect the stability of the tertiary structure. In 100 ns simulations, both parameter sets yield stable structures that are close to the experimental structure with an rmsd of 0.28 (microMg) and 0.24 nm (nanoMg). Both values are slightly smaller compared to the rmsd of 0.30 nm obtained with the Allnér–Villa parameters.16
With microMg, both inner-shell ions of the X-ray structure remain bound over the duration of the simulation. The distance of Mg2+ to the phosphate oxygen of the first binding site (Figure 4B) is 0.208 ± 0.005 nm in close agreement with the 0.210 nm in the X-ray structure. The average distance of Mg2+ to the oxygen phosphate of the second binding site (Figure 4C) is 0.209 ± 0.005 nm. This value is smaller compared to the 0.244 nm in the X-ray structure. Note, however, that the experimental value is located in the exclusion range and might be over-rated as judged by the assignment criterion of 0.206–0.208 nm.63 With nanoMg, the distances are similar with 0.209 ± 0.012 nm for both binding sites. Here, however, the ions exchange with bulk due to the faster exchange kinetics discussed above. After 100 ns, 9 Mg2+ ions are observed in inner-sphere conformation with nonbridging phosphate oxygens. No inner-sphere contacts other than with phosphate oxygens are observed in agreement with experimental results, reproducing that nucleobase nitrogens and carbonyls are weak inner-sphere Mg2+-binding sites.63,75
In addition, about 20 outer-sphere contacts are formed with microMg and nanoMg, similar to the results with the Allnér–Villa parameters.16 The outer-sphere interactions are mediated by a water molecule and involve besides phosphate oxygens also the oxygen and nitrogen atoms of the nucleobases and the ribose oxygens (Section S2.5).
3.4.2. Identification and de novo Prediction of Inner-Sphere Binding Sites with NanoMg
With nanoMg, the ion-binding kinetics is 2–4 orders of magnitude faster compared to other force fields. Consequently, the formerly rare events of Mg2+ association and dissociation can now be observed directly in straightforward simulations. To evaluate this behavior, in a second step, all ions were placed randomly within the simulation box. Figure 4 shows the distribution of Mg2+ ions around the add A-riboswitch after a cumulative duration of 2 μs. The riboswitch is surrounded by highly mobile, diffusive ions leading to a low probability density (blue regions). In addition, about 16 Mg2+ ions bind in inner-sphere conformation leading to regions of high probability density (red regions). The three ion-binding sites with the highest probability density are shown in Figure 4B–D. Interestingly, the two most probable binding sites correspond to the two inner-sphere binding sites from the X-ray structure. Hereby, the experimental electron density is on top of the probability density calculated from the simulations (Figure 4B,C). This illustrates that the nanoMg parameters are particularly useful to predict inner-sphere binding sites. One such prediction, corresponding to the third most likely binding site, is shown in Figure 4D (see Section S2.6 for additional predictions). In addition, the simulations provide an unique atomistic description of the binding site including the exact coordination chemistry of Mg2+, RNA, and hydration water.
4. Conclusions
The importance of Mg2+ in biological systems has driven the development of force field parameters for molecular dynamics simulations. However, Mg2+ force fields in combination with TIP3P water have fundamental drawbacks in reproducing a broad range of structural, thermodynamic, and kinetic properties. Our current work shows that the effects of polarizability that are presumably the cause of the deviations can be included by tuning the parameters of the 12–6 Lennard-Jones potential in an enlarged parameter range and by modifying the standard combination rules. Our results show that this allows us to accurately reproduce the experimental solvation free energy, the distances to the oxygens of the first hydration shell, the hydration number, the activity derivative in MgCl2 solutions, the self-diffusion coefficient, and the binding affinity and distance to the phosphate oxygens of RNA.
In particular, by increasing the LJ interaction strength between Mg2+ and water, the interaction potential becomes more attractive and long-ranged, thereby mimicking the charge-induced dipole and charge-transfer effects that Mg2+ ions cause in their environment. This in turn allows us to correctly describe ion–water interactions as quantified by the solvation free energy and the structure of the first hydration shell.
The activity coefficient derivative and the binding affinity to RNA reveal that the Mg2+–Cl– and Mg2+–RNA interactions described by the standard combination rules are too attractive. The reason is that the combination rules do not reflect the law of matching water affinities according to which ions with high charge density have more tightly bound first hydration shells compared to ions with low charge density.76 This problem can easily be solved by introducing scaling factors that reduce the LJ energy of Mg2+–Cl– pairs or the Mg2+–RNA interactions while simultaneously increasing their effective diameters. The advantage of this approach is that it leaves the ion–water interactions unchanged and can therefore be transferred to other anions or other binding sites on biomolecules. On RNA, for instance, the nucleobase nitrogens and carbonyl atoms could be considered as additional Mg2+-binding sites.62 However, special care must be taken since the Mg2+ ions are unlikely to bind in the inner-sphere binding mode63,77,78 and the binding affinity of the inner- and outer-sphere conformation can differ significantly.69
In progressing toward improved force fields, special emphasis is placed on an accurate calculation of the water-exchange rate, circumventing the shortcomings of transition state theory.26 Matching the abovementioned broad range of thermodynamic properties, we present two sets of optimal parameters: microMg which yields water exchange on the microsecond timescale in agreement with experiments and nanoMg which yields water exchange on the nanosecond timescale. As shown for the example of the add A-riboswitch, microMg yields stable RNA structures and reproduces the structure of specifically bound ions. NanoMg yields accelerated water exchange and ion-binding dynamics and is therefore particularly suited for the de novo prediction of Mg2+-binding sites on biomolecules.
In summary, the Mg2+ parameters presented here provide an efficient and highly accurate model for the simulation of Mg2+ ions in aqueous solutions and their distribution and exchange around biomolecules such as nucleic acids, proteins, or lipids.
Acknowledgments
We acknowledge financial support from the DFG (Emmy Noether program, grant no. 315221747). LOEWE CSC and GOETHE HLR are acknowledged for supercomputing access. N.S. thanks Roland R. Netz and Dominik Horinek for inspiring and fruitful discussions. K.K.G. thanks Angélica Sandoval-Pérez and Jürgen Köfinger for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jctc.0c01281.
Additional information regarding the methods presented in the main manuscript are presented. Additional results including a study of box size dependencies on the calculation of solvation free energies, a brief discussion of the performance of Allnér–Villa in combination with mTIP3P water, a study of the dependency of the number of transitions on calculation parameters, a comparison of the two-dimensional free-energy profiles of microMg and nanoMg, a comparison of different methods to obtain binding affinities and distances, a brief discussion of outer-sphere ion-binding sites, and additional de novo-predicted inner-sphere ion-binding sites are presented (PDF)
The authors declare no competing financial interest.
Notes
The parameter files are freely available at https://github.com/bio-phys/Magnesium-FFs.
Supplementary Material
References
- Williams N. H. Magnesium Ion Catalyzed ATP Hydrolysis. J. Am. Chem. Soc. 2000, 122, 12023–12024. 10.1021/ja0013374. [DOI] [Google Scholar]
- Cowan J. A. Structural and catalytic chemistry of magnesium-dependent enzymes. BioMetals 2002, 15, 225–235. 10.1023/a:1016022730880. [DOI] [PubMed] [Google Scholar]
- Pyle A. Metal ions in the structure and function of RNA. J. Biol. Inorg Chem. 2002, 7, 679–690. 10.1007/s00775-002-0387-6. [DOI] [PubMed] [Google Scholar]
- Sigel R. K. O.; Pyle A. M. Alternative Roles for Metal Ions in Enzyme Catalysis and the Implications for Ribozyme Chemistry. Chem. Rev. 2007, 107, 97–113. 10.1021/cr0502605. [DOI] [PubMed] [Google Scholar]
- Pan J.; Thirumalai D.; Woodson S. A. Magnesium-dependent folding of self-splicing RNA: Exploring the link between cooperativity, thermodynamics, and kinetics. Proc. Natl. Acad. Sci. 1999, 96, 6149–6154. 10.1073/pnas.96.11.6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirumalai D.; Lee N.; Woodson S. A.; Klimov D. Early events in RNA folding. Annu. Rev. Phys. Chem. 2001, 52, 751–762. 10.1146/annurev.physchem.52.1.751. [DOI] [PubMed] [Google Scholar]
- Freisinger E.; Sigel R. K. O. From nucleotides to ribozymes-A comparison of their metal ion binding properties. Coord. Chem. Rev. 2007, 251, 1834–1851. 10.1016/j.ccr.2007.03.008. [DOI] [Google Scholar]
- Laederach A.; Shcherbakova I.; Jonikas M. A.; Altman R. B.; Brenowitz M. Distinct contribution of electrostatics, initial conformational ensemble, and macromolecular stability in RNA folding. Proc. Natl. Acad. Sci. 2007, 104, 7045–7050. 10.1073/pnas.0608765104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferner J.; Villa A.; Duchardt E.; Widjajakusuma E.; Wöhnert J.; Stock G.; Schwalbe H. NMR and MD studies of the temperature-dependent dynamics of RNA YNMG-tetraloops. Nucleic Acids Res. 2008, 36, 1928–1940. 10.1093/nar/gkm1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederiksen J. K.; Li N.-S.; Das R.; Herschlag D.; Piccirilli J. A. Metal-ion rescue revisited: biochemical detection of site-bound metal ions important for RNA folding. RNA 2012, 18, 1123–1141. 10.1261/rna.028738.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi R.; Noetzel J.; Marx D. Exposing catalytic versatility of GTPases: taking reaction detours in mutants of hGBP1 enzyme without additional energetic cost. Phys. Chem. Chem. Phys. 2019, 21, 859–867. 10.1039/c8cp06343e. [DOI] [PubMed] [Google Scholar]
- Åqvist J. Ion-water interaction potentials derived from free energy perturbation simulations. J. Phys. Chem. 1990, 94, 8021–8024. 10.1021/j100384a009. [DOI] [Google Scholar]
- Babu C. S.; Lim C. Empirical Force Fields for Biologically Active Divalent Metal Cations in Water†. J. Phys. Chem. A 2006, 110, 691–699. 10.1021/jp054177x. [DOI] [PubMed] [Google Scholar]
- Mayaan E.; Moser A.; MacKerell A. D. Jr; York D. M. CHARMM Force Field Parameters for Simulation of Reactive Intermediates in Native and Thio-Substituted Ribozymes. J. Comput. Chem. 2007, 28, 495–507. 10.1002/jcc.20474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duboué-Dijon E.; Mason P. E.; Fischer H. E.; Jungwirth P. Hydration and Ion Pairing in Aqueous Mg2+ and Zn2+ Solutions: Force-Field Description Aided by Neutron Scattering Experiments and Ab Initio Molecular Dynamics Simulations. J. Phys. Chem. B 2018, 122, 3296–3306. 10.1021/acs.jpcb.7b09612. [DOI] [PubMed] [Google Scholar]
- Allnér O.; Nilsson L.; Villa A. Magnesium Ion-Water Coordination and Exchange in Biomolecular Simulations. J. Chem. Theory Comput. 2012, 8, 1493–1502. 10.1021/ct3000734. [DOI] [PubMed] [Google Scholar]
- Mamatkulov S.; Fyta M.; Netz R. R. Force fields for divalent cations based on single-ion and ion-pair properties. J. Chem. Phys. 2013, 138, 024505. 10.1063/1.4772808. [DOI] [PubMed] [Google Scholar]
- Li P.; Roberts B. P.; Chakravorty D. K.; Merz K. M. Rational Design of Particle Mesh Ewald Compatible Lennard-Jones Parameters for +2 Metal Cations in Explicit Solvent. J. Chem. Theory Comput. 2013, 9, 2733–2748. 10.1021/ct400146w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.; Merz K. M. Taking into Account the Ion-Induced Dipole Interaction in the Nonbonded Model of Ions. J. Chem. Theory Comput. 2014, 10, 289–297. 10.1021/ct400751u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamatkulov S.; Schwierz N. Force fields for monovalent and divalent metal cations in TIP3P water based on thermodynamic and kinetic properties. J. Chem. Phys. 2018, 148, 074504. 10.1063/1.5017694. [DOI] [PubMed] [Google Scholar]
- Nguyen H. T.; Hori N.; Thirumalai D. Theory and simulations for RNA folding in mixtures of monovalent and divalent cations. Proc. Natl. Acad. Sci. 2019, 116, 21022–21030. 10.1073/pnas.1911632116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J. E. On the Determination o f Molecular Fields. — II. From the Equation o f State o f a Gas. Proc. R. Soc. London, Ser. A 1924, 106, 463–477. 10.1098/rspa.1924.0082. [DOI] [Google Scholar]
- Panteva M. T.; Giambaşu G. M.; York D. M. Comparison of structural, thermodynamic, kinetic and mass transport properties of Mg2+ion models commonly used in biomolecular simulations. J. Comput. Chem. 2015, 36, 970–982. 10.1002/jcc.23881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely J.; Connick R. Rate of Water Exchange from Hydrated Magnesium Ion. J. Am. Chem. Soc. 1970, 92, 3476–3478. 10.1021/ja00714a048. [DOI] [Google Scholar]
- Bleuzen A.; Pittet P.-A.; Helm L.; Merbach A. E. Water exchange on magnesium(II) in aqueous solution: a variable temperature and pressure17O NMR study. Magn. Reson. Chem. 1997, 35, 765–773. 10.1002/(sici)1097-458x(199711)35:11<765::aid-omr169>3.0.co;2-f. [DOI] [Google Scholar]
- Schwierz N. Kinetic pathways of water exchange in the first hydration shell of magnesium. J. Chem. Phys. 2020, 152, 224106. 10.1063/1.5144258. [DOI] [PubMed] [Google Scholar]
- Hess B.; Holm C.; van der Vegt N. Modeling Multibody Effects in Ionic Solutions with a Concentration Dependent Dielectric Permittivity. Phys. Rev. Lett. 2006, 96, 147801. 10.1103/physrevlett.96.147801. [DOI] [PubMed] [Google Scholar]
- Weerasinghe S.; Smith P. E. A Kirkwood-Buff derived force field for sodium chloride in water. J. Chem. Phys. 2003, 119, 11342–11349. 10.1063/1.1622372. [DOI] [Google Scholar]
- Fyta M.; Kalcher I.; Dzubiella J.; Vrbka L.; Netz R. R. Ionic force field optimization based on single-ion and ion-pair solvation properties. J. Chem. Phys. 2010, 132, 024911. 10.1063/1.3292575. [DOI] [PubMed] [Google Scholar]
- Fyta M.; Netz R. R. Ionic force field optimization based on single-ion and ion-pair solvation properties: Going beyond standard mixing rules. J. Chem. Phys. 2012, 136, 124103. 10.1063/1.3693330. [DOI] [PubMed] [Google Scholar]
- Gee M. B.; Cox N. R.; Jiao Y.; Bentenitis N.; Weerasinghe S.; Smith P. E. A Kirkwood-Buff Derived Force Field for Aqueous Alkali Halides. J. Chem. Theory Comput. 2011, 7, 1369–1380. 10.1021/ct100517z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashefolgheta S.; Vila Verde A. Developing force fields when experimental data is sparse: AMBER/GAFF-compatible parameters for inorganic and alkyl oxoanions. Phys. Chem. Chem. Phys. 2017, 19, 20593–20607. 10.1039/c7cp02557b. [DOI] [PubMed] [Google Scholar]
- Panteva M. T.; Giambaşu G. M.; York D. M. Force Field for Mg2+, Mn2+, Zn2+, and Cd2+Ions That Have Balanced Interactions with Nucleic Acids. J. Phys. Chem. B 2015, 119, 15460–15470. 10.1021/acs.jpcb.5b10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B.; van der Vegt N. F. A. Cation specific binding with protein surface charges. Proc. Natl. Acad. Sci. 2009, 106, 13296–13300. 10.1073/pnas.0902904106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horinek D.; Mamatkulov S. I.; Netz R. R. Rational design of ion force fields based on thermodynamic solvation properties. J. Chem. Phys. 2009, 130, 124507. 10.1063/1.3081142. [DOI] [PubMed] [Google Scholar]
- Helm L.; Merbach A. E. Water exchange on metal ions: experiments and simulations. Coord. Chem. Rev. 1999, 187, 151–181. 10.1016/s0010-8545(99)90232-1. [DOI] [Google Scholar]
- Born B.; Weingärtner H.; Bründermann E.; Havenith M. Solvation Dynamics of Model Peptides Probed by Terahertz Spectroscopy. Observation of the Onset of Collective Network Motions. J. Am. Chem. Soc. 2009, 131, 3752–3755. 10.1021/ja808997y. [DOI] [PubMed] [Google Scholar]
- Ostmeyer J.; Chakrapani S.; Pan A. C.; Perozo E.; Roux B. Recovery from slow inactivation in K+ channels is controlled by water molecules. Nature 2013, 501, 121–124. 10.1038/nature12395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachura M.; Chakraborty S.; Gottberg A.; Ruckthong L.; Pecoraro V. L.; Hemmingsen L. Direct Observation of Nanosecond Water Exchange Dynamics at a Protein Metal Site. J. Am. Chem. Soc. 2017, 139, 79–82. 10.1021/jacs.6b11525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leontyev I.; Stuchebrukhov A. Accounting for electronic polarization in non-polarizable force fields. Phys. Chem. Chem. Phys. 2011, 13, 2613–2626. 10.1039/c0cp01971b. [DOI] [PubMed] [Google Scholar]
- Kohagen M.; Mason P. E.; Jungwirth P. Accurate description of calcium solvation in concentrated aqueous solutions. J. Phys. Chem. B 2014, 118, 7902–7909. 10.1021/jp5005693. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Joung I. S.; Cheatham T. E. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 2008, 112, 9020–9041. 10.1021/jp8001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Belfon K.; Ben-Shalom I. Y.; Brozell S. R.; Cerutti D. S.; Cheatham T. E. I.; Cruzeiro V. W. D.; Darden T.; Duke R. E.; Giambasu G.; Gilson M. K.; Gohlke H.; Goetz A. W.; Harris R.; Izadi P. A.; Izmailov S.; Kasavajhala K.; Kovalenko A.; Krasny R.; Kurtzman T.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Liu J.; Luchko T.; Luo R.; Man V.; Merz K. M.; Miao Y.; Mikhailovskii O.; Monard G.; Nguyen H.; Onufriev A.; Pan F.; Pantano S.; Qi R.; Roe D. R.; Roitberg A.; Sagui C.; Schott-Verdugo S.; Shen J.; Simmerling C. L.; Skrynnikov N. R.; Smith J.; Swails J.; Walker R. C.; Wang J.; Wilson L.; Wolf R. M.; Wu X.; Xiong Y.; Xue Y.; York D. M.; Kollman P. A.. Amber 2020, 2020. https://ambermd.org/AmberTools.php.
- Petrov A. S.; Pack G. R.; Lamm G. Calculations of Magnesium–Nucleic Acid Site Binding in Solution. J. Phys. Chem. B 2004, 108, 6072–6081. 10.1021/jp037517s. [DOI] [Google Scholar]
- Petrov A. S.; Funseth-Smotzer J.; Pack G. R. Computational study of dimethyl phosphate anion and its complexes with water, magnesium, and calcium. Int. J. Quantum Chem. 2005, 102, 645–655. 10.1002/qua.20442. [DOI] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Serganov A.; Yuan Y.-R.; Pikovskaya O.; Polonskaia A.; Malinina L.; Phan A. T.; Hobartner C.; Micura R.; Breaker R. R.; Patel D. J. Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem. Biol. 2004, 11, 1729–1741. 10.1016/j.chembiol.2004.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez A.; Marchán I.; Svozil D.; Sponer J.; Cheatham T. E.; Laughton C. A.; Orozco M. Refinement of the AMBER Force Field for Nucleic Acids: Improving the Description of α/γ Conformers. Biophys. J. 2007, 92, 3817–3829. 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banáš P.; Hollas D.; Zgarbová M.; Jurečka P.; Orozco M.; Cheatham T. E.; Šponer J.; Otyepka M. Performance of molecular mechanics force fields for RNA simulations: Stability of UUCG and GNRA hairpins. J. Chem. Theory Comput. 2010, 6, 3836–3849. 10.1021/ct100481h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgarbová M.; Otyepka M.; Šponer J.; Mládek A.; Banáš P.; Cheatham T. E.; Jurečka P. Refinement of the Cornell et al. Nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J. Chem. Theory Comput. 2011, 7, 2886–2902. 10.1021/ct200162x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud-Agrawal N.; Denning E. J.; Woolf T. B.; Beckstein O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowers R.; Linke M.; Barnoud J.; Reddy T.; Melo M.; Seyler S.; Domański J.; Dotson D.; Buchoux S.; Kenney I.; Beckstein O.. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. Proceedings of the 15th Python in Science Conference, 2016; pp 98–105.
- Kirkwood J. G.; Buff F. P. The Statistical Mechanical Theory of Solutions. I. J. Comp. Phys. 1951, 19, 774. 10.1063/1.1748352. [DOI] [Google Scholar]
- Hummer G.; Pratt L. R.; García A. E. Free Energy of Ionic Hydration. J. Phys. Chem. 1996, 100, 1206–1215. 10.1021/jp951011v. [DOI] [Google Scholar]
- Torrie G. M.; Valleau J. P. Monte Carlo free energy estimates using non-Boltzmann sampling: Application to the sub-critical Lennard-Jones fluid. Chem. Phys. Lett. 1974, 28, 578–581. 10.1016/0009-2614(74)80109-0. [DOI] [Google Scholar]
- Torrie G. M.; Valleau J. P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. 10.1016/0021-9991(77)90121-8. [DOI] [Google Scholar]
- Wynne-Jones W. F. K.; Eyring H. The Absolute Rate of Reactions in Condensed Phases. J. Chem. Phys. 1935, 3, 492–502. 10.1063/1.1749713. [DOI] [Google Scholar]
- Wigner E. The Transition State Method. Trans. Faraday Soc. 1937, 34, 29–41. 10.1039/tf9383400029. [DOI] [Google Scholar]
- Frenkel D.; Berend S.. Understanding Molecular Simulation: From Algorithms to Applications, 2nd ed.; Elsevier Science, 2001. [Google Scholar]
- Sigel R. K. O.; Sigel H. A stability concept for metal ion coordination to single-stranded nucleic acids and affinities of individual sites. Acc. Chem. Res. 2010, 43, 974–984. 10.1021/ar900197y. [DOI] [PubMed] [Google Scholar]
- Leonarski F.; D’Ascenzo L.; Auffinger P. Mg2+ions: do they bind to nucleobase nitrogens?. Nucleic Acids Res. 2017, 45, 987–1004. 10.1093/nar/gkw1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briones R.; Blau C.; Kutzner C.; de Groot B. L.; Aponte-Santamaría C. GROmaρs: A GROMACS-Based Toolset to Analyze Density Maps Derived from Molecular Dynamics Simulations. Biophys. J. 2019, 116, 4–11. 10.1016/j.bpj.2018.11.3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger L.The PyMOL Molecular Graphics System, version 1.8., 2015.
- Marcus Y. Ionic Radii in Aqueous solutions. Chem. Rev. 1988, 88, 1475–1498. 10.1021/cr00090a003. [DOI] [Google Scholar]
- Neria E.; Fischer S.; Karplus M. Simulation of activation free energies in molecular systems. J. Chem. Phys. 1996, 105, 1902–1921. 10.1063/1.472061. [DOI] [Google Scholar]
- Klein D. J.; Moore P. B.; Steitz T. A. The contribution of metal ions to the structural stability of the large ribosomal subunit. RNA 2004, 10, 1366–1379. 10.1261/rna.7390804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-León S.; Schwierz N. Hofmeister Series for Metal-Cation-RNA Interactions: The Interplay of Binding Affinity and Exchange Kinetics. Langmuir 2020, 36, 5979–5989. 10.1021/acs.langmuir.0c00851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha R. A.; Bussi G. Unraveling Mg2+-RNA binding with atomistic molecular dynamics. RNA 2017, 23, 628–638. 10.1261/rna.060079.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L.-Z.; Zhang D.; Chen S.-J. Theory and Modeling of RNA Structure and Interactions with Metal Ions and Small Molecules. Annu. Rev. Biophys. 2017, 46, 227–246. 10.1146/annurev-biophys-070816-033920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Ferré-D’Amaré A. R. Dramatic improvement of crystals of large RNAs by cation replacement and dehydration. Structure 2014, 22, 1363–1371. 10.1016/j.str.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noeske J.; Schwalbe H.; Wöhnert J. Metal-ion binding and metal-ion induced folding of the adenine-sensing riboswitch aptamer domain. Nucleic Acids Res. 2007, 35, 5262–5273. 10.1093/nar/gkm565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z.; Zhao Y.; Chen C.; Xiao Y. Role of Ligand Binding in Structural Organization ofAddA-riboswitch Aptamer: A Molecular Dynamics Simulation. J. Biomol. Struct. Dyn. 2011, 29, 403–416. 10.1080/07391102.2011.10507394. [DOI] [PubMed] [Google Scholar]
- Leonarski F.; D’Ascenzo L.; Auffinger P. Nucleobase carbonyl groups are poor Mg2+inner-sphere binders but excellent monovalent ion binders-a critical PDB survey. RNA 2019, 25, 173–192. 10.1261/rna.068437.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins K. Ions from the Hofmeister series and osmolytes: Effects on proteins in solution and in the crystallization process. Methods 2004, 34, 300–311. 10.1016/j.ymeth.2004.03.021. [DOI] [PubMed] [Google Scholar]
- Auffinger P.; Grover N.; Westhof E.. Metal Ions in Life Science; Springer, 2011; Vol. 9; pp 1–35. [PubMed] [Google Scholar]
- Zheng H.; Cooper D. R.; Porebski P. J.; Shabalin I. G.; Handing K. B.; Minor W. CheckMyMetal: A macromolecular metal-binding validation tool. Acta Crystallogr., Sect. D: Struct. Biol. 2017, 73, 223–233. 10.1107/s2059798317001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus Y.Ion Properties; Marcel Dekker, Inc.: New York, Basel, 1997. [Google Scholar]
- Robinson R. A.; Stokes R. H.. Electrolyte Solutions, 2nd ed.; Dover: New York, 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.