

Abstract
CMYA1 (cardiomyopathy-associated protein 1, also termed Xin) localizes to the intercalated disks (ICDs) of the myocardium and functions to maintain ICD structural integrity and support signal transduction among cardiomyocytes. Our previous study showed that CMYA1 overexpression impairs the function of gap junction intercellular communication processes. Successful model generation was verified based on PCR, western blot analysis, immunohistochemistry, and immunofluorescence analysis. Myocardial CMYA1 expression was confirmed at both the mRNA and the protein levels in the CMYA1-OE transgenic mice. Masson’s trichrome staining and electron microscopy revealed myocardial fibrosis and uneven bead width or the interruption of ICDs in the hearts of the CMYA1-OE transgenic mice. Furthermore, the Cx43 protein level was reduced in the CMYA1-OE mice, and co-immunoprecipitation assays of heart tissue protein extracts revealed a physical interaction between CMYA1 and Cx43. Electrocardiogram analysis enabled the detection of an obvious ventricular bigeminy for the CMYA1-OE mice. In summary, analysis of our mouse model indicates that elevated CMYA1 levels may induce myocardial fibrosis, impair ICDs, and downregulate the expression of Cx43. The observed ventricular bigeminy in the CMYA1-OE mice may be mediated by the reduced Cx43 protein level.
Keywords: mice, cardiac structure, intercalated disk, CMYA1, connexin 43
Introduction
CMYA1 (cardiomyopathy-associated gene family-1), also known as XIRP1 (Xin actin-binding repeat containing protein-1), which is located on chromosome 3p22.2, was originally discovered as a downregulated gene during cardiac development in chicken embryos [1]. CMYA1 has been shown to be highly expressed in intercalated disks (ICDs) in mouse and pig hearts [2–5]. CMYA1 functions in the cyclization process of cardiac development and myocardial contractility [6]. CMYA1 knockout during mouse embryonic development demonstrated that CMYA1 also plays a role in mammalian myocardial wall development and morphogenesis [2].
Many subsequent studies explored the function of the CMYA1 gene and demonstrated that the CMYA1 protein, which localizes to the ICDs of the heart, functions in the regulation of postnatal cardiac development and growth [2,6–12]. The results from our previous study showed an increased expression level of CMYA1 in the heart tissues from patients with left ventricular noncompaction cardiomyopathy (LVNC), and this was accompanied by a decreased expression level of the connexin 43 (Cx43) protein [13]. Furthermore, CMYA1 overexpression in a cell model indicated that CMYA1 overexpression impairs the function of gap junction intercellular communication (GJIC) processes by inhibiting Cx43 expression [13], which offers a potential explanation for the abnormal heart development and arrhythmia that occur in LVNC.
In the present study, based on our previous observations from LVNC patients and a cardiomyocyte model of CMYA1 overexpression, we aimed to gain further insight into the effects of CMYA1 overexpression on cardiac structure and function by establishing a mouse model of CMYA1 overexpression.
Materials and Methods
Creation of CMYA1 overexpression transgenic mice
This project was approved by the Institutional Ethical Review Board of Fuwai Hospital (Beijing, China). Procedures for the creation of a transgenic mouse model were approved by the Institutional Animal Care and Use Committee of the Chinese Institute of Laboratory Animal Science. To create the transgenic mouse model of CMYA1 overexpression, the cDNA (615-bp fragment) of the human CMYA1 gene (GenBank No. V2 NM_001198621.3) was cloned into a commercialized expression plasmid under the α-MHC promoter (Hanheng, Shanghai, China). The specific primers for the human CMYA1 cDNA are as follows: CMYA1-V2-forward, 5′-GAAGTGGTCCCTGGTGATGTC-3′; CMYA1-V2-reverse, 5′-CCCTTCTTCTTTCTGTCGTTC-3′.
RT-PCR and quantitative real-time PCR analyses
The transgenic mouse lines were created by microinjecting the recombinant plasmid into the male pronuclei of fertilized mouse oocytes, which were then implanted into pseudo-pregnant females. The transgenic founder mice were mated with wild-type C57BL mice to produce F1 generation of transgenic mice. For genotype identification, transgenic mice of several generations were examined by PCR using specific primers for the human CMYA1 cDNA and genomic DNA from tail biopsies of CMYA1-OE transgenic and wild-type littermate control mice as a template [14,15].
Western blot analysis
The heart tissues were harvested and washed twice with PBS and then lysed with cool Radio-Immunoprecipitation Assay (RIPA) and centrifuged at 12,000 g at 4°C for 10 min. The supernatants were collected, and the protein content was assessed using a BCA assay kit (Biyuntian Biology, Beijing, China), and western blot analysis was performed as previously described [16–20]. Briefly, protein samples were separated by SDS-PAGE (7% non-gradient) and transferred to nitrocellulose (NC) membranes. The NC membranes were blocked with 5% skimmed milk in tris buffered saline tween-20 (TBST) for 1 h and then incubated with primary antibodies overnight at 4°C, followed by incubation with the corresponding HRP-conjugated secondary antibodies (1:5000; CST Biotechnology, Beverly, USA) for 2 h at room temperature. The primary antibodies used were as follows: rabbit polyclonal anti-CMYA1 (1:1000; Santa Cruz, Santa Cruz, USA), mouse monoclonal anti-GAPDH (1:1000; Proteintech, Rosemont, USA), mouse monoclonal anti-Cx43 (1:1000; Abcam, Cambridge, UK), and rabbit monoclonal anti-flag (1:1000; CST Biotechnology) antibodies.
Immunofluorescence and immunohistochemistry assay
The mouse myocardial samples were fixed in 10% neutral buffered formalin. After that, a series of alcohol and xylene gradients were used for dehydration. Then, the samples were washed with PBS for 10 min and incubated with 5% normal goat serum (ZSGB-BIO, Beijing, China) in PBS for 1 h at room temperature, followed by washing with PBS for 10 min. After that, the samples were incubated with the primary antibody overnight at 4°C, followed by washing with PBS for 10 min. The tissues were incubated with fluorescein-conjugated secondary antibody for 1 h and washed with PBS for 10 min, followed by observation with a Leica Sp8 confocal laser scanning microscope (Leica, Wetzlar, Germany). The cell nuclei were counterstained with 0.1% 4′,6-diamidino-2-phenylindole. The antibodies and dilutions used were as follows: rabbit polyclonal anti-CMYA1 (1:100; Santa Cruz), mouse anti-Sarcomeric Alpha Actinin (1:100; Abcam), mouse monoclonal anti-CX43 (1:100; Abcam), Alexa 488-conjugated goat anti-mouse IgG (1:500; Yeasen Biology, Shanghai, China), Alexa 488-conjugated goat anti-rabbit IgG (1:500; Yeasen Biology), Alexa 594-conjugated goat anti-mouse IgG (1:500; Yeasen Biology), and Alexa 594-conjugated goat anti-rabbit IgG (1:500; Yeasen Biology).
The immunohistochemistry assay procedure was similar to that described above, but included blocking with 0.5% H2O2 (Yisheng Biology). Then the samples were washed with PBS for 10 min, and after that, the samples were incubated with the primary rabbit polyclonal anti-CMYA1 antibody (1:100; Santa Cruz) overnight at 4°C, followed by washing with PBS for 10 min. Then the tissues were incubated with HRP polymer for 30 min at room temperature, followed by washing with PBS for 10 min twice. And then 1 ml DAB plus substrate (Yisheng Biology) was added for 15 min. The immunohistochemistry images were acquired using a Leica DM750 microscope at a magnification of 400×.
Masson’s trichrome staining
Left ventricle sections were fixed in formalin for about 24 h and embedded in paraffin. After being deparaffinized and rehydrated, the sections were incubated in 0.1 mM potassium dichromate for at least 16 h. Tissue sections were then subject to Masson’s trichrome staining (Biyuntian Biology) and examined under a light microscope.
Transmission electron microscopy analysis
Once anesthetized, mouse hearts were excised immediately and cut into small pieces, then fixed in 5% glutaraldehyde solution, and processed according to standard protocols for transmission electron microscopy (TEM) sample preparation. Images were taken using a Hitachi H-7500 Transmission Electron Microscope (Tokyo, Japan).
Co-immunoprecipitation assays
Co-immunoprecipitation assays were performed using a Pierce Classic Magnetic IP/Co-IP Kit (88804; Thermo Scientific, Waltham, USA). Prior to lysis, heart tissues were pre-cooled to 4°C, followed by lysis and incubation of extracts at 4°C for at least 16 h with 5 μg of antibodies pre-bound to protein A/G-sepharose beads. After incubation, the beads were washed three times with cold washing buffer, followed by elution with 50 mM Tris-HCl (pH 6.8), 2% SDS, 0.1% bromophenol blue, 10% glycerol, and 10 mM dithiothreitol. The eluted sample was then subject to SDS-PAGE and western blot analysis. The immunoprecipitation (IP) antibody used was rabbit polyclonal anti-CMYA1 antibody (5 μg; Santa Cruz); the immunoblotting (IB) antibody was mouse monoclonal anti-Cx43 antibody (1:1000; Abcam).
Echocardiography assessment of cardiac function
After being anesthetized via inhalation of isoflurane, animals were placed on a warming platform in a supine position. The chests were cleaned using hair removal cream. Images were obtained using a Visual Sonics Vevo 770 high-resolution imaging system (Visual Sonics, Toronto, Canada). M-mode echocardiography of the left ventricle was recorded at the tip of the mitral valve apparatus using a 30-MHz transducer (707B), as previously described [21].
ECG measurements
Surface electrodes were attached to the skin beside the chest. ECGs (sp2006) were recorded in a 10-s rhythm strip that was obtained at a paper speed of 50 mm/s and a vertical ECG calibration of 20 mm/mV. Rhythm analysis was conducted by visual inspection. To avoid measuring the complexes immediately preceding or following a noted cardiac arrhythmia, each measure was recorded for three consecutive complexes, and the averaged heart rate was calculated based upon the average of all R to R interval measurements during the 10-s ECG rhythm strip.
Statistical analysis
All data were analyzed with SPSS 22.0 (SPSS Inc, Chicago, USA). Student’s t-tests were used to assess differences between groups. In all analyses, P<0.05 were considered to be of statistically significant difference.
Results
Verification of CMYA1 overexpression in CMYA1-OE transgenic mice
Pronuclei of fertilized wild-type C57 mouse oocytes were microinjected with a recombinant plasmid encoding the human CMYA1 cDNA fused with a FLAG tag to create the CMYA1 overexpression (CMYA1-OE) mice. Genotype identification was performed in 6- and 9 month-old mice of F2 generation by PCR using human-specific primers of CMYA1 cDNA. RT-PCR results showed that the expression level of the human CMYA1 gene was high in the CMYA1-OE transgenic mice, but the expression of CMYA1 gene was not detectable in the wild-type littermate control mice (Fig. 1A). We also observed an obviously higher expression level of CMYA1 protein in CMYA1-OE transgenic mice compared to that in wild-type mice based on western blot analyses using an anti-FLAG antibody (Fig. 1B). Moreover, both immunocytochemistry (Fig. 1C) and immunofluorescence (Fig. 1D) analyses further confirmed CMYA1 protein expression in CMYA1-OE transgenic mice and showed that the transgenic CMYA1 protein is localized at ICDs of the myocardium.
Figure 1.
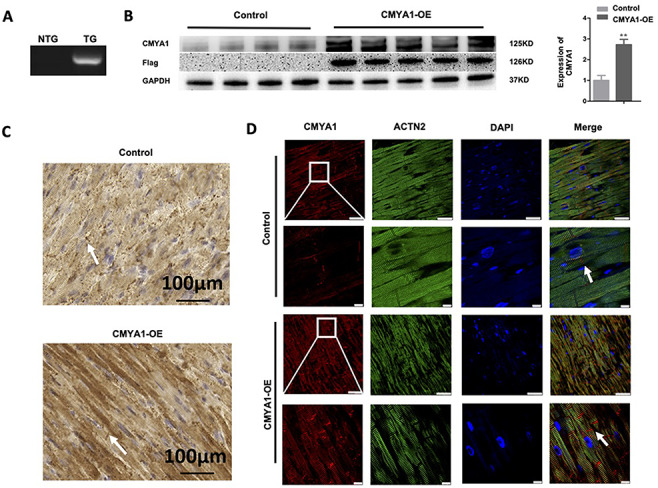
Verification of CMYA1 overexpression in CMYA1-positive transgenic mice (A) PCR-based genotyping of the wild-type littermate control (wild-type C57 background) and CMYA1-OE mice using gDNA as the template. RT-PCR–based analysis of the expression level for the human CMYA1 gene: CMYA1 expression was high in CMYA1-OE transgenic mice but was not detectable in the wild-type C57 mice. NTG: non-transgenic; TG: transgenic. (B) Western blot analyses using an anti-FLAG antibody showed the obviously increased expression level of the CMYA1 protein in the CMYA1-OE transgenic mice (n=5) compared to the wild-type mice (n=4). **P<0.01. GAPDH was used as the loading control. (C,D) Immunocytochemistry (scale bar: 100 μm; C) and immunofluorescence (scale bar: 25 μm in the upper panel and 10 μm in the lower panel; D) assays both showed the increased CMYA1 expression level in the CMYA1-OE transgenic mice and the localization of CMYA1 expression at the ICDs of the myocardium (marked by white arrows). The white boxed areas in the upper panels are enlarged in the lower panels. ACTN2: α-actinin 2.
Pathological changes in CMYA1 overexpression transgenic mice
Using Masson’s trichrome staining, we observed obvious fibrosis occurring in the heart tissues from CMYA1-OE transgenic mice; no fibrosis was observed in the wild-type mice (Fig. 2A). Notably, the extent of fibrosis was increased with time, as the fibrosis of 9-month-old CMYA1-OE transgenic mice was more severe than that of 6-month-old CMYA1-OE transgenic mice (Fig. 2A). But there was no significant heart morphology difference between the control and 9-month-old CMYA1-OE mice, and the hematoxylin-eosin (HE) staining also showed no difference between the two groups (Supplementary Fig. S1A). We also conducted a TEM–based analysis of the ultrastructure of the heart tissues. The heart tissues of the 9-month-old CMYA1-OE transgenic mice exhibited both uneven bead widths and interruption of ICDs; no such disruption was found in the age-matched wild-type mice (Fig. 2B).
Figure 2.
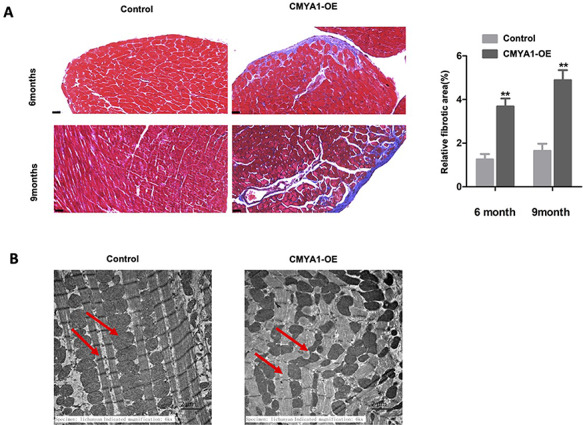
Pathological changes in CMYA1 overexpression transgenic mice (A) Masson’s trichrome staining revealed obvious fibrosis in heart tissues from the CMYA1-OE transgenic mice; no fibrosis was evident for the wild-type control mice. The extent of fibrosis increased with time between the 6- and 9-month age time points. **P<0.01, n=5 for each group. (B) The 9-month-old CMYA1-OE mice heart tissues showed uneven bead width or interruption. The red arrows represent the intercalated disks and the mitochondria, respectively.
Cx43 expression in CMYA1-overexpressing transgenic mice
Western blot analyses showed that the Cx43 protein level was decreased in the heart tissues of 9-month-old CMYA1-OE transgenic mice compared to age-matched littermate controls (Fig. 3A). Furthermore, both the decreased Cx43 expression and the expected localization of Cx43 at the ICDs of the myocardium were observed in the immunofluorescence analysis (Fig. 3B). We also conducted co-immunoprecipitation assays using protein extracts from heart tissues. The results revealed that CMYA1 and Cx43 can physically interact with each other (Fig. 3C).
Figure 3.
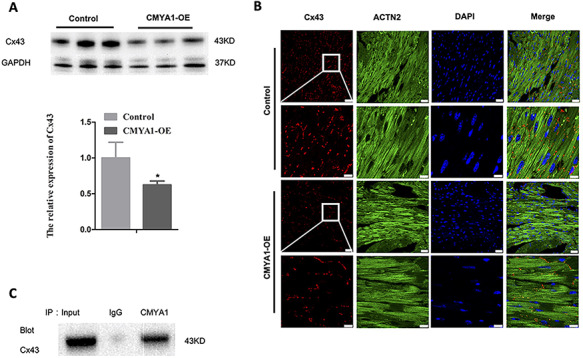
Cx43 expression in CMYA1 overexpression transgenic mice (A) Western blot analyses using anti-Cx43 antibody revealed a decrease in the level of the Cx43 protein in the heart tissues from 9-month-old CMYA1-OE transgenic mice compared with that in age-matched littermate control mice. *P<0.05, n=3. (B) Immunofluorescence analysis showed that the Cx43 protein is localized at ICDs of the myocardium. (C) Co-immunoprecipitation assays revealed a physical interaction between CMYA1 and Cx43 in the extracts from heart tissues.
Changes of cardiac function and electrophysiology in CMYA1-overexpressing transgenic mice
Echocardiography examination of both 6- and 9-month-old mice revealed no significant changes in the left ventricular ejection fraction of CMYA1-OE and control littermate mice (Fig. 4A). Other indexes of echocardiography are listed in the Supplementary Table S1. However, electrocardiogram measurements of 6-month-old mice revealed that CMYA1-OE transgenic mice but not littermate controls exhibited a ventricular bigeminy (Fig. 4B).
Figure 4.
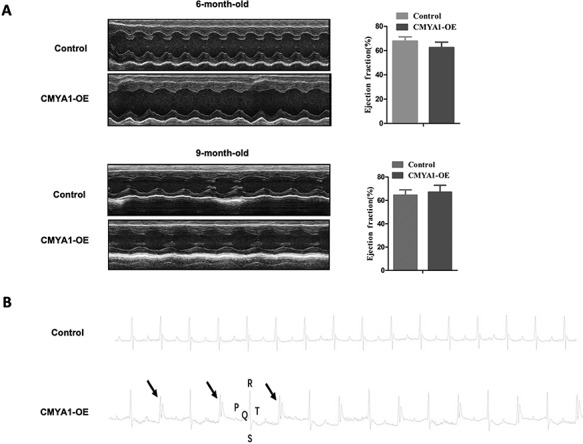
Changes of cardiac function and electrophysiology in CMYA1-OE transgenic mice (A) Echocardiography examination of 6-month-old and 9-month-old mice indicated no significant differences in the left ventricular ejection fraction (EF) between CMYA1-OE transgenic mice and age-matched littermate controls (n=3). (B) Electrocardiogram measurements of 6-month-old mice revealed a ventricular bigeminy in the CMYA1-OE transgenic mice (n=3).
Discussion
The ICD between neighboring cardiomyocytes is fundamental to both mechanical and electrical coupling, as well as transduction of signals among cardiomyocytes [22]. Abnormalities in the ICD structure or components have been implicated in some types of cardiomyopathies, arrhythmias, and heart failure in patients and in animal models of heart disease [23–33].
CMYA1 is highly and specifically expressed in striated muscles, where it localizes to the ICDs of cardiomyocytes [2–5,12]. The CMYA1 protein functions to maintain both the structural and functional integrity of ICDs [10,31,34,35]. Accordingly, the abnormal expression of CMYA1 may affect the normal development, structure, and/or function of the heart. Treatment with CMYA1 antisense oligonucleotides during chicken embryonic development was shown to cause abnormal cardiac morphogenesis, including myocardium thickening and multiple invaginations into the heart cavity [7]. Work with a CMYA1-knockout mouse model showed that mice lacking normal murine CMYA1 function exhibited a disordered myocardium, abnormally sized heart, and lack of a ventricular septum [9]. The findings from these previous studies together suggest that CMYA1 is essential for proper cardiac morphogenesis and development and that CMYA1 dysfunction may functionally contribute to the development of certain cardiomyopathies.
Studies have revealed that there is a significant upregulation of CMYA1 expression in the early stage of acute myocardial infarction, and similar elevations have been observed in ischemia–reperfusion, pressure overload–induced cardiac hypertrophy, and inflammatory dilated cardiomyopathy [13]. Conversely, a downregulation of CMYA1 expression was detected in failing hearts from patients with heart failure, idiopathic dilated cardiomyopathy, and ischemic cardiomyopathy [22]. LVNC is one of the most prevalent genetic cardiomyopathies, and LVNC is associated with the abnormal embryonic development of the heart, which is characterized by increased myocardial trabeculations and recesses.
The most common clinical presentations of LVNC are congestive heart failure, cardiac arrhythmias, and thromboembolism [36]. The pathogenic mechanism of LVNC is not well understood. Previously, we observed that the hearts of LVNC patients have higher expression levels of the CMYA1 protein compared to those of control subjects [13]. Of note, the upregulation of CMYA1 in the hearts of LVNC patients is not congruent with previous findings from some of the aforementioned cardiomyopathies (wherein CMYA1 downregulation is presumed to drive the pathology), suggesting the possibility of distinct CMYA1 pathogenic mechanisms.
We here successfully established a transgenic mouse model of CMYA1 overexpression, which can facilitate further investigation of the impacts of CMYA1 overexpression on cardiac phenotypes at the animal level. After confirming the successful expression and ICD localization of the human CMYA1 protein in our murine model, we detected myocardial fibrosis and uneven bead width or interruption of ICDs in the heart tissues from CMYA1-OE transgenic mice. Of note, these pathological phenotypes in CMYA1-OE hearts are similar to the presentations in the hearts of LVNC patients. Myocardial fibrosis is the major cause of heart failure, arrhythmia, and even sudden cardiac death [13,37,38]. Our results suggest that elevated CMYA1 levels may drive myocardial fibrosis that promotes heart failure or arrhythmia in LVNC, although the direct causal relationship between elevated CMYA1 levels and myocardial fibrosis requires to be established in further studies. Disruption of the ICD structure is known to be one of the major causal factors for hypertrophied human myocardium and dilated cardiomyopathy [39,40]. Thus, the abnormal ICD shapes we observed in our CMYA1-OE murine model support our understanding that the disruption of the ICD structures resulting from elevated CMYA1 levels may contribute to LVNC pathogenesis.
Deficiencies in ICD components have been reported to lead to many types of cardiomyopathy, arrhythmias, and heart failure in human patients and in various genetically engineered animal models [23–31]. ICDs are known to contain adherens junctions, desmosomes, and gap junctions, which collectively maintain the integrity of the association between cardiomyocytes and also enable the myocardium to function in synchrony [34]. Abnormalities in the structure and function of gap junctions also commonly lead to arrhythmias [41–44]. Cx43, a major gap junction protein in the ventricular myocytes [45], is known to maintain conduction velocity in ventricles. A study of Cx43 gene-deficient mice reported that the ventricle conduction velocity was reduced by 38% when the ventricular Cx43 level was reduced by 50% [46]. The results from our previous study showed an increase in the CMYA1 level in the heart tissue from LVNC patients, which was accompanied by a decrease in the Cx43 level. That study also included experiments with a cell model, showing that CMYA1 overexpression impairs the function of GJIC processes and revealing that this results from the inhibition of Cx43 expression [13]. Based on our previous results, we hypothesized that decreased Cx43 levels may at least partially explain the abnormal cardiac morphology and arrhythmia that occur in LVNC patients.
In the present study, we indeed observed that CMYA1 overexpression reduced Cx43 levels and led to a ventricular bigeminy (a type of arrhythmia), which further supports the aforementioned hypothesis. In summary, we demonstrated that CMYA1 overexpression in mice can induce myocardial fibrosis, impair ICDs, and downregulate the expression of Cx43. The ventricular bigeminy we observed in the CMYA1-OE mice may be mediated by reduced Cx43 expression. Thus, our results deepen our understanding of the influence of CMYA1 on the pathogenesis of LVNC and raise new questions about how the protein–protein interactions in ICDs lead to the occurrence of arrhythmogenesis.
Supplementary Material
Contributor Information
Chunyan Li, State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Disease, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, China; Department of Clinical Laboratory, Beijing Jishuitan Hospital, Beijing 100032, China.
Hongliang Zhang, Department of Cardiology, The First Affiliated Hospital of Jiamusi University, Jiamusi 154002, China.
Yuanyuan Xie, State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Disease, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, China.
Shenghua Liu, State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Disease, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, China.
Ranxu Zhao, State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Disease, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, China.
Jian Huang, State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Disease, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, China.
Jie Huang, State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Disease, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, China.
Yingjie Wei, State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Disease, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, China.
Supplementary Data
Supplementary data is available at Acta Biochimica et Biophysica Sinica online.
Funding
This work was supported by a grant from the Chinese Academy of Medical Sciences (CAMS) Initiative for Innovation Fund for Medical Science (No. CAMS-I2M, 2016-I2M-1-015, to Y.W.).
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1. Wang DZ, Hu X, Lin JL, Kitten GT, Solursh M, Lin JJ. Differential displaying of mRNAs from the atrioventricular region of developing chicken hearts at stages 15 and 21. Front Biosci 1996, 1: a1–a15. doi: 10.2741/A100 [DOI] [PubMed] [Google Scholar]
- 2. Sinn HW, Balsamo J, Lilien J, Lin JJ. Localization of the novel Xin protein to the adherens junction complex in cardiac and skeletal muscle during development. Dev Dyn 2002, 225: 1–13. doi: 10.1002/dvdy.10131 [DOI] [PubMed] [Google Scholar]
- 3. Jain SK, Schuessler RB, Saffitz JE. Mechanisms of delayed electrical uncoupling induced by ischemic preconditioning. Circ Res 2003, 92: 1138–1144. doi: 10.1161/01.RES.0000074883.66422.C5 [DOI] [PubMed] [Google Scholar]
- 4. Schulz R, Gres P, Skyschally A, Duschin A, Belosjorow S, Konietzka I, Heusch G. Ischemic preconditioning preserves connexin 43 phosphorylation during sustained ischemia in pig hearts in vivo. FASEB J 2003, 17: 1355–1357. doi: 10.1096/fj.02-0975fje [DOI] [PubMed] [Google Scholar]
- 5. Solan JL, Marquez-Rosado L, Sorgen PL, Thornton PJ, Gafken PR, Lampe PD. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J Cell Biol 2007, 179: 1301–1309. doi: 10.1083/jcb.200707060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jung-Ching Lin J, Gustafson-Wagner EA, Sinn HW, Choi S, Jaacks SM, Wang DZ, Evans S, et al. Structure, expression, and function of a novel intercalated disc protein, Xin. J Med Sci 2005, 25: 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang DZ, Reiter RS, Lin JL, Wang Q, Williams HS, Krob SL, Schultheiss TM, et al. Requirement of a novel gene, Xin, in cardiac morphogenesis. Development 1999, 126: 1281–1294. [DOI] [PubMed] [Google Scholar]
- 8. Choi S, Gustafson-Wagner EA, Wang Q, Harlan SM, Sinn HW, Lin JL, Lin JJ. The intercalated disk protein, mXinalpha, is capable of interacting with beta-catenin and bundling actin filaments [corrected]. J Biol Chem 2007, 282: 36024–36036. doi: 10.1074/jbc.M707639200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Otten J, van der Ven PF, Vakeel P, Eulitz S, Kirfel G, Brandau O, Boesl M, et al. Complete loss of murine Xin results in a mild cardiac phenotype with altered distribution of intercalated discs. Cardiovasc Res 2010, 85: 739–750. doi: 10.1093/cvr/cvp345 [DOI] [PubMed] [Google Scholar]
- 10. Wang Q, Lin JL, Reinking BE, Feng HZ, Chan FC, Lin CI, Jin JP, et al. Essential roles of an intercalated disc protein, mXinbeta, in postnatal heart growth and survival. Circ Res 2010, 106: 1468–1478. doi: 10.1161/CIRCRESAHA.109.212787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang Q, Lu TL, Adams E, Lin JL, Lin JJ. Intercalated disc protein, mXinalpha, suppresses p120-catenin-induced branching phenotype via its interactions with p120-catenin and cortactin. Arch Biochem Biophys 2013, 535: 91–100. doi: 10.1016/j.abb.2012.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feng HZ, Wang Q, Reiter RS, Lin JL, Lin JJ, Jin JP. Localization and function of Xinalpha in mouse skeletal muscle. Am J Physiol Cell Physiol 2013, 304: C1002–C1012. doi: 10.1152/ajpcell.00005.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xie Y, Liu S, Hu S, Wei Y. Cardiomyopathy-associated gene 1-sensitive PKC-dependent connexin 43 expression and phosphorylation in left ventricular noncompaction cardiomyopathy. Cell Physiol Biochem 2017, 44: 828–842. doi: 10.1159/000485348 [DOI] [PubMed] [Google Scholar]
- 14. Gordon JW, Ruddle FH. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science 1981, 214: 1244–1246. doi: 10.1126/science.6272397 [DOI] [PubMed] [Google Scholar]
- 15. Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques 2000, 29: 52–54. doi: 10.2144/00291bm09 [DOI] [PubMed] [Google Scholar]
- 16. Liu F, Shen XH, Su S, Cui H, Fang YH, Wang T, Zhang LF, et al. Fc gamma receptor I-coupled signaling in peripheral nociceptors mediates joint pain in a rat model of rheumatoid arthritis. Arthritis Rheumatol 2020, 72: 1668–1678. doi: 10.1002/art.41386 [DOI] [PubMed] [Google Scholar]
- 17. Liu F, Wang ZY, Qiu Y, Wei M, Li CY, Xie YK, Shen L, et al. Suppression of MyD88-dependent signaling alleviates neuropathic pain induced by peripheral nerve injury in the rat. J Neuroinflam 2017, 14: 70. doi: 10.1186/s12974-017-0822-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu F, Xu LB, Chen NZ, Zhou M, Li CY, Yang Q, Xie YK, et al. Neuronal Fc-epsilon receptor I contributes to antigen-evoked pruritus in a murine model of ocular allergy. Brain Behav Immun 2017, 61: 165–175. doi: 10.1016/j.bbi.2016.11.017 [DOI] [PubMed] [Google Scholar]
- 19. Liu Z, Liu F, Liu XW, Ma C, Zhao J. Surgical incision induces learning impairment in mice partially through inhibition of the brain-derived neurotrophic factor signaling pathway in the hippocampus and amygdala. Mol Pain 2018, 14: 1744806918805902. doi: 10.1177/1744806918805902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang ZY, Liu F, Wei M, Qiu Y, Ma C, Shen L, Huang YG. Chronic constriction injury-induced microRNA-146a-5p alleviates neuropathic pain through suppression of IRAK1/TRAF6 signaling pathway. J Neuroinflammation 2018, 15: 179. doi: 10.1186/s12974-018-1215-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu D, Ma Y, Zhang W, Bao D, Dong W, Lian H, Huang L, et al. Knockdown of cytochrome P450 2E1 inhibits oxidative stress and apoptosis in the cTnT(R141W) dilated cardiomyopathy transgenic mice. Hypertension 2012, 60: 81–89. doi: 10.1161/HYPERTENSIONAHA.112.191478 [DOI] [PubMed] [Google Scholar]
- 22. Wang Q, Lin JL, Erives AJ, Lin CI, Lin JJ. New insights into the roles of Xin repeat-containing proteins in cardiac development, function, and disease. Int Rev Cell Mol Biol 2014, 310: 89–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res 2010, 107: 700–714. doi: 10.1161/CIRCRESAHA.110.223412 [DOI] [PubMed] [Google Scholar]
- 24. Li J, Swope D, Raess N, Cheng L, Muller EJ, Radice GL. Cardiac tissue-restricted deletion of plakoglobin results in progressive cardiomyopathy and activation of {beta}-catenin signaling. Mol Cell Biol 2011, 31: 1134–1144. doi: 10.1128/MCB.01025-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lombardi R, Marian AJ. Molecular genetics and pathogenesis of arrhythmogenic right ventricular cardiomyopathy: a disease of cardiac stem cells. Pediatr Cardiol 2011, 32: 360–365. doi: 10.1007/s00246-011-9890-2 [DOI] [PubMed] [Google Scholar]
- 26. Noorman M, van der Heyden MA, van Veen TA, Cox MG, Hauer RN, de Bakker JM, van Rijen HV. Cardiac cell-cell junctions in health and disease: electrical versus mechanical coupling. J Mol Cell Cardiol 2009, 47: 23–31. doi: 10.1016/j.yjmcc.2009.03.016 [DOI] [PubMed] [Google Scholar]
- 27. Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res 2008, 80: 9–19. doi: 10.1093/cvr/cvn133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sheikh F, Ross RS, Chen J. Cell-cell connection to cardiac disease. Trends Cardiovasc Med 2009, 19: 182–190. doi: 10.1016/j.tcm.2009.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Swope D, Cheng L, Gao E, Li J, Radice GL. Loss of cadherin-binding proteins beta-catenin and plakoglobin in the heart leads to gap junction remodeling and arrhythmogenesis. Mol Cell Biol 2012, 32: 1056–1067. doi: 10.1128/MCB.06188-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van Tintelen JP, Hofstra RM, Wiesfeld AC, van den Berg MP, Hauer RN, Jongbloed JD. Molecular genetics of arrhythmogenic right ventricular cardiomyopathy: emerging horizon? Curr Opin Cardiol 2007, 22: 185–192. doi: 10.1097/HCO.0b013e3280d942c4 [DOI] [PubMed] [Google Scholar]
- 31. Wang Q, Lin JL, Wu KH, Wang DZ, Reiter RS, Sinn HW, Lin CI, et al. Xin proteins and intercalated disc maturation, signaling and diseases. Front Biosci (Landmark Ed) 2012, 17: 2566–2593. doi: 10.2741/4072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang S, Ren J. Role of autophagy and regulatory mechanisms in alcoholic cardiomyopathy. Biochim Biophys Acta Mol Basis Dis 2018, 1864: 2003–2009. doi: 10.1016/j.bbadis.2018.03.016 [DOI] [PubMed] [Google Scholar]
- 33. Zhang Y, Ren J. Epigenetics and obesity cardiomyopathy: from pathophysiology to prevention and management. Pharmacol Ther 2016, 161: 52–66. doi: 10.1016/j.pharmthera.2016.03.005 [DOI] [PubMed] [Google Scholar]
- 34. Gustafson-Wagner EA, Sinn HW, Chen YL, Wang DZ, Reiter RS, Lin JL, Yang B, et al. Loss of mXinalpha, an intercalated disk protein, results in cardiac hypertrophy and cardiomyopathy with conduction defects. Am J Physiol Heart Circ Physiol 2007, 293: H2680–H2692. doi: 10.1152/ajpheart.00806.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Q, Lin JL, Chan SY, Lin JJ. The Xin repeat-containing protein, mXinbeta, initiates the maturation of the intercalated discs during postnatal heart development. Dev Biol 2013, 374: 264–280. doi: 10.1016/j.ydbio.2012.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pantazis AA, Elliott PM. Left ventricular noncompaction. Curr Opin Cardiol 2009, 24: 209–213. doi: 10.1097/HCO.0b013e32832a11e7 [DOI] [PubMed] [Google Scholar]
- 37. Liu T, Song D, Dong J, Zhu P, Liu J, Liu W, Ma X, et al. Current understanding of the pathophysiology of myocardial fibrosis and its quantitative assessment in heart failure. Front Physiol 2017, 8: 238. doi: 10.3389/fphys.2017.00238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li C, Liu F, Liu S, Pan H, Du H, Huang J, Xie Y, et al. Elevated myocardial SORBS2 and the underlying implications in left ventricular noncompaction cardiomyopathy. EBioMedicine 2020, 53: 102695. doi: 10.1016/j.ebiom.2020.102695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maron BJ, Ferrans VJ. Significance of multiple intercalated discs in hypertrophied human myocardium. Am J Pathol 1973, 73: 81–96. [PMC free article] [PubMed] [Google Scholar]
- 40. Perriard JC, Hirschy A, Ehler E. Dilated cardiomyopathy: a disease of the intercalated disc? Trends Cardiovasc Med 2003, 13: 30–38. doi: 10.1016/S1050-1738(02)00209-8 [DOI] [PubMed] [Google Scholar]
- 41. Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, Kleber AG, et al. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res 2000, 87: 656–662. doi: 10.1161/01.RES.87.8.656 [DOI] [PubMed] [Google Scholar]
- 42. Dupont E, Ko Y, Rothery S, Coppen SR, Baghai M, Haw M, Severs NJ. The gap-junctional protein connexin40 is elevated in patients susceptible to postoperative atrial fibrillation. Circulation 2001, 103: 842–849. doi: 10.1161/01.CIR.103.6.842 [DOI] [PubMed] [Google Scholar]
- 43. Hagendorff A, Schumacher B, Kirchhoff S, Luderitz B, Willecke K. Conduction disturbances and increased atrial vulnerability in Connexin40-deficient mice analyzed by transesophageal stimulation. Circulation 1999, 99: 1508–1515. doi: 10.1161/01.CIR.99.11.1508 [DOI] [PubMed] [Google Scholar]
- 44. Kanagaratnam P, Rothery S, Patel P, Severs NJ, Peters NS. Relative expression of immunolocalized connexins 40 and 43 correlates with human atrial conduction properties. J Am Coll Cardiol 2002, 39: 116–123. doi: 10.1016/S0735-1097(01)01710-7 [DOI] [PubMed] [Google Scholar]
- 45. Kanter HL, Saffitz JE, Beyer EC. Cardiac myocytes express multiple gap junction proteins. Circ Res 1992, 70: 438–444. doi: 10.1161/01.RES.70.2.438 [DOI] [PubMed] [Google Scholar]
- 46. Thomas SA, Schuessler RB, Berul CI, Beardslee MA, Beyer EC, Mendelsohn ME, Saffitz JE. Disparate effects of deficient expression of connexin43 on atrial and ventricular conduction: evidence for chamber-specific molecular determinants of conduction. Circulation 1998, 97: 686–691. doi: 10.1161/01.CIR.97.7.686 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.