

Abstract
Scope
Human milk oligosaccharides (hMOs) can attenuate inflammation by modulating intestinal epithelial cells, but the mechanisms of action are not well‐understood. Here, the effects of hMOs on tumor necrosis factor‐α (TNF‐α) induced inflammatory events in gut epithelial cells are studied.
Methods and results
The modulatory effects of 2’‐fucosyllactose, 3‐fucosyllactose (3‐FL), 6’‐sialyllactose, lacto‐N‐tetraose, lacto‐N‐neotetraose (LNnT), lactodifucotetraose (LDFT), and lacto‐N‐triaose (LNT2) on immature (FHs 74 Int) and adult (T84) intestinal epithelial cells with or without TNF‐α are determined. Interleukin‐8 (IL‐8) secretion in FHs 74 Int and T84 are quantified to determine hMO induced attenuation of inflammatory events by ELISA. 3‐FL, LNnT, and LDFT significantly attenuate TNF‐α induced inflammation in FHs 74 Int, while LNT2 induces IL‐8 secretion in T84. In addition, microscale thermophoresis assays and ELISA are used to study the possible mechanisms of interaction between effective hMOs and tumor necrosis factor receptor 1 (TNFR1). 3‐FL, LNnT, and LDFT exert TNFR1 ectodomain shedding while LNnT also shows binding affinity to TNFR1 with a Kd of 900 ± 660 nM.
Conclusion
The findings indicate that specific hMO types attenuate TNF‐α induced inflammation in fetal gut epithelial cells through TNFR1 in a hMO structure‐dependent fashion suggest possibilities to apply hMOs in management of TNF‐α dependent diseases.
Keywords: human milk oligosaccharides, inflammation, intestinal epithelial cells, tumor necrosis factor receptors, tumor necrosis factor‐α
Human milk oligosaccharides (hMOs) can attenuate inflammation by modulating intestinal epithelial cells, but the mechanisms are not well‐understood. The anti‐inflammatory effects of different hMOs are tested on tumor necrosis factor‐α induced inflammation in immature and mature intestine epithelial cells. This study shows that the anti‐inflammatory effects of hMOs are structure‐dependent and through interference with tumor necrosis factor receptor 1 signaling.

1. Introduction
Breastfeeding is the gold standard for infant nutrition as it offers complete nutrition and essential bioactive components for the development of the newborn.[ 1 ] Exclusive breastfeeding is therefore recommended for the first six months of life by the World Health Organization.[ 2 ] For a variety of reasons, over 70% of the infants cannot be exclusively breastfed and have to be fed with cow‐milk based infant formula,[ 3 ] which mimics the nutritional composition of breast milk.[ 4 , 5 ] However, these infant formulas do not contain the same bioactive molecules as human milk,[ 6 ] as a consequence, formula‐fed babies have a higher risk for infections and inflammatory diseases than babies solely fed with infant formula.[ 7 ] One of the most important bioactive components of mother milk are human milk oligosaccharides (hMOs), which are unique to humans and are not found in the same variety and composition in other mammals.[ 8 ] Recently, some hMOs can be produced in sufficiently high amounts via genetically engineered microorganisms and since recently applied in infant formulas.[ 9 , 10 ]
It has been shown that hMOs provide multiple health‐promoting effects, which include support of growth of beneficial bacteria,[ 11 ] anti‐pathogenic effects,[ 12 ] immune modulating effects,[ 13 ] enhancement of intestinal barrier function,[ 14 , 15 ] as well as, attenuation of systemic and intestinal inflammation.[ 16 ] It is however still unclear which and how individual hMOs contribute to processes such as prevention and attenuation of intestinal inflammatory events.[ 17 ] It has been reported that hMO can directly interact with intestinal cells and modulate immunity.[ 18 ] The majority of hMOs can reach the intestine without being digested and some undergo hydrolyzation at low pH during transit through the gastrointestinal tract.[ 19 ] This may lead to the formation of lacto‐N‐Triaose (LNT2), which is the acid hydrolysate of the tetra and higher hMOs such as lacto‐N‐tetraose (LNT) and lacto‐N‐neotetraose (LNnT).[ 19 , 20 ] How these acid hydrolysates impact the inflammatory responses through intestine epithelial cells is also not known.
hMOs are considered to guide immune development during early postnatal intestinal gut immune barrier development.[ 14 , 21 ] This early postnatal developing gut is very susceptible for inflammatory events.[ 22 ] Disturbances in intestinal immune development in neonates may lead to intestinal inflammatory diseases such as necrotizing enterocolitis (NEC) and inflammatory bowel diseases (IBD).[ 14 , 23 ] Tumor necrosis factor‐α (TNF‐α) plays an important role in these inflammatory diseases.[ 24 ] To induce inflammation, TNF‐α needs to bind to the TNF‐receptors on the cell surface. There are two different receptors for TNF‐α, TNF receptor 1 (TNFR1) and TNF receptor 2 (TNFR2).[ 24 ] These receptors have two distinct roles, TNFR1 mainly induces inflammatory signaling pathways, while TNFR2 mediates immune modulatory functions and promotes tissue homeostasis and regeneration.[ 24 ] Therefore, the blockage of TNFR1 signaling, either by binding with TNFR1 as antagonists or by increasing the shedding of soluble TNFR1, may attenuate the response of the cells to TNF‐α.[ 25 ] Whether hMOs interfere with these TNFR signaling pathways is subject of investigation in the current study.
In the present study, we investigated the effects of six different hMOs (2’‐fucosyllactose: 2’‐FL, 3‐fucosyllactos: 3‐FL, 6’‐sialyllactose: 6’‐SL, lacto‐N‐tetraose: LNT, lacto‐N‐neotetraose: LNnT, lactodifucotetraose: LDFT) and one hMOs acid hydrolysate, that is, LNT2 on TNF‐α induced inflammatory events in gut epithelial cells. To this end, two types of gut epithelial cells were tested. An immature human primary fetal intestinal epithelial cell FHs 74 Int was applied as well as the adult colonic epithelial cell line T84 cells[ 18 ] to determine possible differences in the efficacy of hMOs in modulating inflammatory events in immature or adult cells. As the Interleukin‐8 (IL‐8) induction by intestinal epithelial cells strongly reflects the degree of inflammatory response after stimuli,[ 26 ] IL‐8 secretion in FHs 74 Int and T84 were measured to determine possible hMO induced attenuation of inflammatory events. In order to further explore the possible mechanisms of action, the interactions between effective hMOs and TNFR1 were also investigated, which include the possible binding between hMOs and TNFR1, and whether hMOs cause TNFR1 ectodomain shedding.
2. Experimental Section
2.1. Components
In the present study, 2’‐FL (provided by FrieslandCampina Domo, Amersfoort, the Netherlands), 3‐FL, 6’‐SL, LNT2, LNT, LNnT, and LDFT (provided by Glycosyn LLC, Woburn, MA, USA) were tested. An overview of the structure and components are shown in Table 1 .
Table 1.
Overview of the structure of selected hMOs
| Name (abbreviated) | Structure | Schematic diagram |
|---|---|---|
| 2’‐FL | Fucα1‐2Galβ1‐4Glc |

|
| 3‐FL |
Galβ1‐4Glc Fucα1‐3/ |
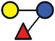
|
| 6’‐SL | NeuNAcα2‐6Galβ1‐4Glc |

|
| LNT2 | GlcNAcβ1‐3Galβ1‐4Glc |

|
| LNT | Galβ1‐3GlcNacβ1‐3Galβ1‐4Glc |

|
| LNnT | Galβ1‐4GlcNacβ1‐3Galβ1‐4Glc |

|
| LDFT |
Fucα1‐2Galβ1‐4Glc Fucα1‐3/ |
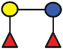
|
 Glucose;
Glucose;  Galactose;
Galactose;  Fucose;
Fucose;  Sialic Acid;
Sialic Acid;  N‐acetyglucosamine.
N‐acetyglucosamine.
2.2. Cell Culture and Reagents
Nontransformed human small intestinal epithelial FHs 74 Int cells (ATCC, Manassas, VA, USA) were maintained in Hybri‐Care medium (ATCC, Manassas, VA, USA) supplemented with 10% heat‐inactivated fetal bovine serum (Sigma–Aldrich, Zwijndrecht, The Netherlands), 50 µg mL−1 penicillin‐streptomycin solution (Sigma‐Aldrich, Zwijndrecht, The Netherlands), and 30 ng mL−1 EGF (ATCC, Manassas, VA, USA). Human colon carcinoma T84 cells were cultured in Dulbecco's Modified Eagle Medium:F‐12 (DMEM:F12) medium (Gibco, Life Technologies, Bleiswijk, The Netherlands) supplemented with 10% heat‐inactivated fetal bovine serum (Sigma‐Aldrich, Zwijndrecht, The Netherlands) and 50 mg mL−1 gentamicin (Lonza, Verviers, Belgium). Cells were cultured at 37 °C in 5% CO2 as recommended by the manufacturer. Recombinant human TNF‐α was obtained from PeproTech (Rocky Hill, NJ, USA).
2.3. Cell Viability and WST‐1 Assay
FHs 74 Int and T84 cells were resuspended in fresh culture medium at 1 × 105 cells mL−1, after which 200 µL of cell suspension was seeded per well in 96‐well plates (Corning, NY, USA). Cells were then cultured until reaching 70–80% confluence. Prior to treatment, cells were washed twice with 1× phosphate‐buffered saline (PBS; Lonza, Verviers, Belgium), after which, culture medium was replaced by 100 µL of fresh medium containing one of the ingredients. FHs 74 Int and T84 cells treated with 5 mg mL−1 of 2’‐FL, 3‐FL, 6’‐SL, LNT2, LNT, LNnT, and LDFT for 24 h. Cell viability was determined by WST‐1 assay following the manufacturer's instructions. Briefly, after 24 h treatment, 10 µL WST‐1 reagent (Sigma‐Aldrich, Zwijndrecht, The Netherlands) was added to the culture medium in 1:10 final dilution directly into the culture medium and incubated for 1 h at 37 °C and 5% CO2. Absorbance (450 nm) was measured using a Benchmark Plus Microplate Reader using Microplate Manager version 5.2.1 for data acquisition. The data for each sample was plotted as the percentage change compared to the negative control. The assays were performed with three technical replicates and each experiment was repeated five times.
2.4. Cell Stimulation
FHs 74 Int and T84 cells were resuspended and seeded at 1 × 105 cells mL−1 in a flat bottom 96 wells plate at 200 µL per well. Cells were then cultured until reaching 70–80% confluence. Prior to treatment, cells were washed twice with 1× phosphate‐buffered saline (PBS; Lonza, Verviers, Belgium), after which culture medium was replaced by 200 µL of fresh medium containing one of the ingredients, the pH were tested with pH indicator paper, no pH change were observed. For IL‐8 induction, FHs 74 Int, and T84 cells were treated with 5 mg mL−1 of 2’‐FL, 3‐FL, 6’‐SL, LNT2, LNT, LNnT, and LDFT in the absence or presence of 10 ng mL−1 TNF‐α for 24 h. After 24 h of incubation, the secretion of the proinflammatory cytokine IL‐8 was measured in the supernatant by ELISA (R&D SYSTEM, Minneapolis, MN, USA) according to the manufacturer's protocol. For soluble TNFR1 measurement, FHs 74 Int cells were treated with 5 mg mL−1 of 3‐FL, LNnT, and LDFT in the absence or presence of 10 ng mL−1 TNF‐α for 24 h. After 24 h of incubation, the soluble TNFR1 was measured in the supernatant by ELISA (R&D SYSTEM, Minneapolis, MN, USA) according to the manufacturer's protocol.
2.5. Western Blot
FHs 74 Int and T84 cells were resuspended and seeded at 1 × 105 cells mL−1 in a 6 wells plate at 2 mL per well for 48 h. After that, the culture medium was replaced by 2 mL of fresh medium in the absence or presence of 10 ng mL−1 TNF‐α for 24 h. After the treatments, total protein extracts were obtained from cells. Cells were harvested in ice‐cold PBS and lysed with RIPA lysis and extraction buffer (Thermo Scientific, MA, USA) supplemented with protease inhibitor cocktails (Sigma‐Aldrich, Zwijndrecht, The Netherlands). Lysates were sonicated (5 s twice) and centrifuged (12 000 × g, 20 min, 4 °C). Subsequently the supernatants were collected, and protein yield was quantified using Pierce BCA Protein Assay Kit (Thermo Scientific, MA, USA). Normalized 30 µg of protein samples were prepared with Laemmli sample buffer containing β‐mercaptoethanol and electrophoresed on an SDS‐polyacrylamide gel. Proteins were transferred to a PVDF membrane (Sigma‐Aldrich, Zwijndrecht, The Netherlands). After blocking for 1 h with 1:1 mixture of Licor blocking buffer (LI‐COR Biosciences) and 1× PBS for the blocking solution, the membrane was probed with the primary antibody TNFR1 (1:500, Abcam, Cambridge, UK) and TNFR2 (1:1000, Abcam, Cambridge, UK) overnight at 4 °C. After that, the membrane was washed four times in PBS‐T and followed by incubation with the secondary antibody for 1 h at room temperature. Immunoreactivity was visualized by the Odyssey Imaging System (LI‐COR Biosciences). Signal intensity was analyzed by using Image J (National Institutes of Health, Bethesda, MD).
2.6. Tumor Necrosis Factor Receptor 1 Binding Assay
The binding affinity between hMOs and TNFR1 was quantified with the microscale thermophoresis (MST) assay. MST experiments were performed on a NanoTemper Monolith NT.115 with blue/red filter (NanoTemper Technologies GmbH, Munich, Germany) according to the manufacturer's protocol. After optimization, the final concentration of His‐tagged TNFR1 (Abcam, Cambridge, UK) was kept constant at 50 nM, 10 µL of the 5 mm 3‐FL, 5 mm LDFT, and 250 µM LNnT were diluted 1:1 in 10 µL PBS‐T buffer (PBS 1×; 0.05% Tween 20) to make a 16‐sample dilution series. The 50 nM His‐tagged TNFR1 was incubated with dye for 30 min, after that, 16 different concentrations of samples were incubated with 50 nM His‐tagged TNFR1 with dye for 2 h. Pre‐incubated samples and protein mixtures were loaded into standard capillaries (NanoTemper Technologies GmbH, Munich, Germany), measurements were performed at 25 °C using 40% MST power with 80% excitation power. All experiments were repeated at least three times. Data analyses were performed using the NanoTemper analysis software.
2.7. Statistical Analysis
The results were analyzed using GraphPad Prism. Normal distribution of the data was confirmed using the Kolmogorov‐Smirnov test. Parametric data are expressed as mean ± standard deviation (SD). Statistical comparisons of parametric distributed data were performed using one‐way ANOVA with Dunnett multiple comparison tests or two‐way ANOVA for group analysis. p < 0.05 was considered as statistically significant (#, *p < 0.05, ##, **p < 0.01, ###, ***p < 0.001, ####, ****p < 0.0001).
3. Results
3.1. Human Milk Oligosaccharides and a Human Milk Oligosaccharide's Acid Hydrolysis Product did not Influence Cell Viability
To exclude toxic effects of the tested hMOs on cell viability of FHs 74 Int and the human colon carcinoma cell line T84, cells were treated with 5 mg mL−1 of 2’‐FL, 3‐FL, 6’‐SL, LNT2, LNT, LNnT, and LDFT for 24 h after which the cell viability was quantified. As shown in Figure 1 , 5 mg mL−1 of 2’‐FL, 3‐FL, 6’‐SL, LNT2, LNT, LNnT, and LDFT did not have a statistically significant negative effect on the cell viability of both FHs 74 Int and T84 cells.
Figure 1.

hMOs and the hMOs acid hydrolysate LNT2 did not alter cell viability of FHs 74 Int and T84. A) FHs 74 Int and B) T84 cells were treated with 5 mg mL−1 of 2’‐FL, 3‐FL, 6’‐SL, LNT2, LNT, LNnT, and LDFT for 24 h, cells treated with culture medium served as negative control. Results are presented as percentage change against negative control. Data are presented as mean ± SD (n = 5), significant differences compared to the negative control were determined by using one‐way analysis of variance with Dunnett multiple comparisons test and indicated by *(p < 0.05), **(p < 0.01), ***(p < 0.001), or by ****(p < 0.0001).
3.2. Human Milk Oligosaccharide's Acid Hydrolysis Lacto‐N‐Triaose Induces IL‐8 Production in T84 Cells
First, the impact of the tested hMOs and the hMO's acid hydrolysis product LNT2 on IL‐8 production was tested as IL‐8 is one of the primary gut‐epithelial cell derived chemokine responsible for inducing inflammation.[ 27 ] To this end, FHs 74 Int and T84 were treated with 5 mg mL−1 of 2’‐FL, 3‐FL, 6’‐SL, LNT2, LNT, LNnT, and LDFT for 24 h.
As shown in Figure 2A, hMOs and LNT2 were not able to modulate IL‐8 secretion in the fetal cell line FHs 74 Int. Also, in T84 the tested hMOs did not alter IL‐8 production but the hMO's acid hydrolysis product LNT2 significantly induced IL‐8 secretion in T84 cells (Figure 2B). LNT2 significantly increased IL‐8 secretion to 1.4‐fold compared to the untreated control (p < 0.0001).
Figure 2.

hMO's acid hydrolysate LNT2 induce IL‐8 production in T84 cells under homeostatic conditions. A) FHs 74 Int and B) T84 cells were incubated with 5 mg mL−1 of 2’‐FL, 3‐FL, 6’‐SL, LNT2, LNT, LNnT, and LDFT for 24 h, cells treated with culture medium served as negative control. Results are presented as fold change against negative control. Data are presented as mean ± SD (n = 6), significant differences compared to the negative control were determined by using one‐way analysis of variance with Dunnett multiple comparisons test and indicated by *(p < 0.05), **(p < 0.01), ***(p < 0.001), or by ****(p < 0.0001).
3.3. Human Milk Oligosaccharides Attenuates Tumor Necrosis Factor‐α‐Induced IL‐8 Secretion in FHs 74 Int in a Chemical Structure‐Dependent Way
In order to investigate the ability of hMOs and LNT2 to attenuate inflammatory responses in fetal and adult intestine epithelial cells, we investigated the effects of hMOs and LNT2 on IL‐8 secretion after exposure of the epithelial cells to the proinflammatory cytokine TNF‐α. To this end, FHs 74 Int and T84 cells were treated with 10 ng mL−1 TNF‐α for 24 h with 5 mg mL−1 of either 2’‐FL, 3‐FL, 6’‐SL, LNT2, LNT, LNnT, LDFT. After that, the concentration of the proinflammatory cytokine IL‐8 in the supernatant was measured. As shown in Figure 3 , TNF‐α significantly increased IL‐8 secretion in both FHs 74 Int and T84 (p < 0.0001). Interestingly, the hMO's inhibiting effects on TNF‐α induced IL‐8‐secretion was restricted to the fetal FHs 74 Int cells. 3‐FL, LNnT, and LDFT reduced TNF‐α induced IL‐8‐secretion with 70% (p < 0.05), 38% (p < 0.0001), and 64% (p<0.01), respectively (Figure 3A). The HMOs 2’‐FL, 6’‐SL, LNT and the hydrolysis product LNT2 did not reduce the TNF‐α induced IL‐8 secretion. Results were different with the adult gut epithelial cell line. With the adult cell line T84, only the hMO's acid hydrolysis product LNT2 impacted TNF‐α induced IL‐8‐secretion but this was an enhancement instead of an attenuation (p < 0.001).
Figure 3.

3‐FL, LNnT, and LDFT attenuate TNFα‐induced IL‐8 secretion in FHs 74 Int cells. A) FHs 74 Int and B) T84 cells were stimulated with 5 mg mL−1 of 2’‐FL, 3‐FL, 6’‐SL, LNT2, LNT, LNnT, and LDFT in presence of TNFα (10 ng mL−1) for 24 h. Cells treated with culture medium served as negative control. Results are presented as fold change against positive control. Data are presented as mean ± SD (n = 6), statistical significance was analyzed using one‐way analysis of variance with Dunnett multiple comparisons test (*vs medium; # vs TNFα; #, *p < 0.05; ##, **p < 0.01; ###, ***p < 0.001; ####, ****p < 0.0001).
The above results suggest that hMOs were able to suppress TNF‐α induced IL‐8‐secretion in the fetal gut epithelial cell FHs 74 Int in a structural‐dependent way, and it also confirmed that hMOs and LNT2 have different regulatory patterns on fetal cells and adult cells.
3.4. FHs 74 Int and T84 have Different Expression Patterns of Tumor Necrosis Factor‐α Receptors
As 3‐FL, LNnT, and LDFT attenuate TNF‐α induced IL‐8 secretion on fetal cells, we hypothesized that hMOs might have anti‐inflammatory effects on fetal intestine epithelial cells through interfering in TNF‐α induced proinflammatory pathway. TNF‐α has two distinct cell surface receptors, TNF receptor 1 (TNFR1) and TNF receptor 2 (TNFR2). To determine whether the receptors are differently expressed in FHs 74 Int and T84, we compared the expression level of TNFR1 and TNFR2 after western blot. To this end, FHs 74 Int and T84 were cultured in the presence TNF‐α for 24 h. Cells cultured in normal medium served as controls. As shown in Figure 4A, FHs 74 Int and T84 showed different protein expression patterns of TNFR1 and TNFR2 but was not influenced by TNF‐α. The protein expression of TNFR1 in fetal cells was significantly higher than in the adult cell line T84 (p < 0.0001, Figure 4B). However, the expression of TNFR2 in fetal FHs 74 Int cells was significantly lower than in T84 (p < 0.001, Figure 4C). Neither TNFR1 nor TNFR2 was significantly altered by TNF‐α (Figure 4B,C).
Figure 4.

FHs 74 Int and T84 showed different expression pattern of TNFR1 and TNFR2. FHs 74 Int and T84 were incubated with or without 10 ng mL TNFα for 24 h. A) The TNFR1 and TNFR2 expression in western blot. Western blot results were analyzed by using Image J gradation analysis of B) TNFR1 and C) TNFR2. Results are represented as mean ± SD (n = 5). Significant differences compared between medium and TNFα, FHs 74 Int, and T84 were determined by using two‐way ANOVA and indicated by * (p < 0.05), ** (p < 0.01), *** (p < 0.001), or by **** (p < 0.0001).
3.5. Lacto‐N‐Neotetraose Attenuate Tumor Necrosis Factor‐α‐Induced IL‐8 Secretion by Interacting with the Tumor Necrosis Factor Receptor 1 in Fetal Gut Epithelial Cells
As 3‐FL, LNnT, and LDFT only inhibited TNF‐α induced IL‐8 secretion in the fetal cell line FHs 74 Int, and as FHs 74 Int has high expression of TNFR1 and low expression of TNFR2 compared to T84, we hypothesized that hMOs might have its anti‐inflammatory effects through interaction with TNFR1. To study the interaction with hMOs and TNFR1, the binding affinity of 3‐FL, LNnT, LDFT with TNFR1 was determined with MST. A wide concentration range of 3‐FL (0.00061 µM to 5 mm), LNnT (0.00763 µM to 250 µM), and LDFT (0.00061 µM to 5 mm) were incubated with His‐tagged TNFR1 in a constant range (50 nM) at room temperature for 2 h. Subsequently the binding affinity was measured by MST. As shown in Figure 5 , a ligand‐dependent binding effect was detected but only LNnT interacts with TNFR1. There was no detectable binding of 3‐FL and LDFT to TNFR1 (Figure 5A,C). LNnT was shown to bind TNFR1 with a K d of 900 ± 660 nM (Figure 5B).
Figure 5.

LNnT has binding affinity to TNFR1. Dose‐response curve for the binding interaction between A) 3‐FL, B) LNnT, C) LDFT and TNFR1. Values on the X‐axis represent the ligand (3‐FL, LNnT, and LDFT) concentration, Y‐axis represent the normalized fluorescence. The binding affinity is observed for the LNnT‐TNFR1 interaction, which was a 900 ± 660 nM K d. All binding curves were determined in at least triplicate by MST and are represented as the mean ± SD.
The above results confirm binding of LNnT to TNFR1 but also suggests that 3‐FL and LDFT inhibit TNF‐α induced IL‐8 secretion via another mechanisms.
3.6. 3‐FL, Lacto‐N‐Neotetraose, and LDFT Cause Ectodomain Shedding of Tumor Necrosis Factor Receptor 1 and Thereby Inhibit Tumor Necrosis Factor‐α Induced Inflammation
Another possible explanation for attenuation of TNF‐α induced IL‐8 secretion by 3‐FL, LNnT, and LDFT is ectodomain shedding of TNFR1. TNF‐α needs to bind to the receptors on the cell surface to induce downstream pro‐inflammatory effects in epithelial cells.[ 28 ] Ectodomain shedding is a process in which the ectodomain of TNFR1 is detached from the cell‐source.[ 29 ] This can be done by several mechanism and might be influenced by bioactive molecules,[ 30 ] such as hMOs. Shedding will reduce the number of receptors on the cell surface and might serve as soluble decoy protein that competes with cell‐surface bound TNFR1 thereby decreasing the response of the cells to TNF‐α.[ 29 ] The quantification of the soluble receptor is a measure for the degree of shedding of the ectodomain of TNFR1.[ 30 ] To test whether 3‐FL, LNnT, and LDFT induce ectodomain shedding of TNFR1 on FHs 74 Int, we incubated FHs 74 Int in the absence or presence of 10 ng mL−1 TNF‐α for 24 h with either 5 mg mL−1 of 3‐FL, LNnT, and LDFT. After that, the concentration of TNFR1 in the supernatant was measured. As shown in Figure 6A, 3‐FL, LNnT, and LDFT did not modulate the soluble TNFR1 under homeostatic conditions. As expected, TNF‐α‐exposure induced a significant decrease of soluble TNFR1 (p < 0.001), while 3‐FL (p < 0.05), LNnT (p < 0.0001), and LDFT (p < 0.01) significantly prevented this TNF‐α induced TNFR1 concentration increase in the medium (Figure 6B). TNF‐α inhibited TNFR1 ectodomain shedding to 70% compared to the medium control (p < 0.001), and treatment with 3‐FL, LNnT, and LDFT restored TNFR1 to 87% (p < 0.05), 108% (p < 0.0001), and 92% (p < 0.01) respectively. This suggests that 3‐FL, LNnT, and LDFT could attenuate TNF‐α induced inflammation by TNFR1 ectodomain shedding in fetal cell line FHs 74 Int.
Figure 6.

hMOs cause ectodomain shedding of TNFR1. FHs 74 Int cells were incubated with 5 mg mL−1 of 3‐FL, LNnT, LDFT in the A) absence or B) presence of 10 ng mL−1 TNFα for 24 h. Cells treated with culture medium served as negative control. Data are presented as mean ± SD (n = 5), statistical significance was measured using one‐way analysis of variance with Dunnett multiple comparisons test (*vs medium; # vs TNFα; #, *p < 0.05; ##, **p < 0.01; ###, ***p < 0.001; ####, ****p < 0.0001).
4. Discussion
Previous studies have shown that hMOs in human milk can attenuate intestinal inflammation,[ 16 ] however, how individual hMO's currently applied in infant formula impact inflammatory responses in intestinal epithelial cells and which mechanisms are involved is still largely unknown. Here we studied, to the best of our knowledge for the first time, the attenuating effects of six different hMOs and one hMOs acid hydrolysate on TNF‐α induced inflammatory responses in fetal and adult intestinal epithelial cells. TNF‐α is a key‐cytokine in NEC and IBD.[ 24 ] We show that the modulatory effects of individual hMOs are strongly structure‐dependent and that attenuating effects on inflammatory responses are mainly observed in immature fetal epithelial cells that express more TNFR1. Especially 3‐FL, LNnT, and LDFT significantly attenuated TNF‐α induced IL‐8 secretion in fetal cells FHs 74 Int. The anti‐inflammatory effects of effective hMOs were strongly related to TNFR1 through different mechanisms, 3‐FL, LNnT, and LDFT exerted TNFR1 ectodomain shedding while LNnT showed binding affinity to TNFR1.
Kuntz et al. reported that acidic and neutral hMOs isolated from human milk may impact the viability of intestinal epithelial cells under homeostatic conditions[ 31 ] and thereby decrease cell‐responses such as release of cytokines. To exclude such an impact on viability in our study we tested the impact of the hMOs on viability of FHs 74 Int and T84 cells and confirmed that the tested hMOs had no negatively impact on cell‐survival in the concentration range applied. Also, we confirmed that under homeostatic condition in the absence of an inflammatory challenge, the six tested hMOs did not alter IL‐8 secretion. Surprisingly we found that only the hMOs acid hydrolysate LNT2 increased IL‐8 secretion in T84 cells under homeostatic condition but also under TNF‐α stimulation. This might be explained by the strong activation effects of LNT2 on Toll‐like receptors (TLRs) signaling.[ 13 ] Cheng et al. showed that LNT2 significantly activated TLRs signaling and induced cytokine production in THP‐1 macrophages.[ 13 ] T84 constitutively express TLRs,[ 32 , 33 ] which could be responsible for the increased production of the proinflammatory cytokine IL‐8 when exposed to LNT2.
3‐FL, LNnT, and LDFT attenuated TNF‐α induced IL‐8 secretion in immature epithelial cells, while 2’‐FL, 6’‐SL, and LNT did not change the inflammatory response. These data underpin our previous notion that seemingly minor differences, such as substituent‐linkage position to the lactose moiety or the linkage within monosaccharide residues in the substituent, can have a significant impact on their biological actions.[ 13 ] As effects were related to attenuation of the effects of TNF‐α, we focused on possible interactions of the hMOs with the two receptors for TNF‐α. The immature intestinal epithelial cell FHs 74 Int expressed more TNFR1 than T84, while the more immune regulatory receptor TNFR2[ 24 ] was lower in FHs 74 Int than in T84 (Figure 4). As FHs 74 Int is a primary immature cell type known to keep its immature status in the culture, T84 is originally isolated from an adult and known to have a mature phenotype which might explain the maintained difference in expression. Also, this might explain the more pronounced impact of hMOs on the immature fetal cells than on the adult cells and the higher susceptibility of fetal cells for inflammation.[ 22 ] Beyond the current experimental setting, organoid 3d models using immature cells from babies and adults might contribute to strengthening of this hypothesis. However, for differentiating the progenitor cells specific growth hormones are applied that interfere with keeping cells in an immature state. This was the reason to work with the two cell types in the current study.
In vivo, most of the proinflammatory effects of TNF‐α are exerted via TNFR1 (Figure 7A).[ 34 ] As shown in our current study 3‐FL, LNnT, and LDFT all attenuated TNF‐α induced inflammation by interfering with this TNF‐α binding to cell‐surface bound TNFR1. During TNF‐α stimulation, 3‐FL, LNnT, and LDFT could induce shedding of the TNFR1 ectodomain which subsequently serve as soluble decoy protein that competes with cell‐surface bound TNFR1 as well as by decreasing the number of cell‐surface bound TNFR1 available for ligand binding (Figure 7B). Besides induced ectodomain shedding of TNFR1, LNnT could also bind TNFR1 which inhibited the TNF‐α/TNFR1 signaling pathway (Figure 7C). The multifold ways of inhibition of LNnT coincides with the observation that LNnT had a stronger inhibiting effect than 3‐FL and LDFT on TNF‐α induced IL‐8 secretion.
Figure 7.

Schematic representation of the mechanisms of action of hMOs attenuate TNF‐α induced inflammation via TNFR1. A) TNF‐α signaling via its receptors TNFR1. B) hMOs induce shedding of cell‐surface TNFR1 increase soluble TNFR1 (sTNFR1) and inhibit the inflammatory response. C) hMOs binding to TNFR1, inhibit the binding of TNF‐α and inflammatory response.
In the present study, we provide evidence that some hMOs attenuate TNF‐α induced inflammation by directly influencing TNFR1 signaling, and subsequently inhibiting inflammatory responses. It therewith might also be instrumental in preventing or attenuating inflammatory events in TNF‐α dependent diseases such as rheumatoid arthritis and IBD.[ 35 ] Currently, these diseases are treated with TNF‐α‐inhibitors that block both TNFR1 and TNFR2, and not only TNFR1 signaling which is the main proinflammatory receptor.[ 35 ] This is undesired as TNFR2 is involved in tissue repair and immune modulation and should therefore not be suppressed.[ 25 ] Here we show that specific chemical hMO structures may specifically block TNFR1 signaling which might be beneficial for the treatment of the aforementioned diseases.[ 35 ]
5. Conclusions
In conclusion, we demonstrate that specific hMO types inhibit TNF‐α induced inflammatory responses in fetal gut epithelial cells in a structure‐dependent fashion. Especially 3‐FL, LNnT, and LDFT can effectively attenuate TNF‐α induced inflammation by interacting with the TNFR1 receptor which is highly expressed in the fetal cells compared to adult gut epithelial cells. Our findings not only contribute to better understanding of the structure‐function relationship of hMOs, but opens new venues to explore hMOs in management of TNF‐α dependent diseases as the hMOs have more specificity for the proinflammatory pathways than currently applied TNF‐α‐inhibitors. Understanding how and which hMOs have anti‐inflammatory effects could contribute to the future design of hMO containing products with predictable beneficial effects in specific target groups. A possible application of the current knowledge is application of 3‐FL, LNnT, and LDFT in infant formula for premature neonates that are more prone to NEC or other inflammatory disorders than term born babies.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
L.C., M.B.G.K., and P.d.V. conceived and designed the experiments. L.C., C.K., and W.W. performed the experiments. L.C. analyzed data. L.C. and P.d.V. wrote the original draft. All authors reviewed and edited the manuscript.
Acknowledgements
The project was financially supported by the China Scholarship Council (CSC) (grant number: 201505990304). This research was performed within the framework of the Sino‐Dutch Doctoral Program on Sustainable Dairy coordinated by the Carbohydrate Competence Center (CCC, www.cccresearch.nl).
Cheng L., Kong C., Wang W., Groeneveld A., Nauta A., Groves M. R., Kiewiet M. B. G., de P. Vos, The Human Milk Oligosaccharides 3‐FL, Lacto‐N‐Neotetraose, and LDFT Attenuate Tumor Necrosis Factor‐α Induced Inflammation in Fetal Intestinal Epithelial Cells In Vitro through Shedding or Interacting with Tumor Necrosis Factor Receptor 1. Mol. Nutr. Food Res. 2021, 65, 2000425. 10.1002/mnfr.202000425
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Le Doare K., Holder B., Bassett A., Pannaraj P. S., Front. Immunol. 2018, 9, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walker A., J. Pediatr. 2010, 156, S3. [DOI] [PubMed] [Google Scholar]
- 3. Heymann J., Raub A., Earle A., Bull. W. H. O. 2013, 91, 398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coulet M., Phothirath P., Allais L., Schilter B., Regul. Toxicol. Pharmacol. 2014, 68, 59. [DOI] [PubMed] [Google Scholar]
- 5. Piemontese P., Giannì M. L., Braegger C. P., Chirico G., Grüber C., Riedler J., Arslanoglu S., van Stuijvenberg M., Boehm G., Jelinek J., Roggero P., PLoS One 2011, 6, e28010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aly E., Ali Darwish A., Lopez‐Nicolas R., Frontela‐Saseta C., Ros‐Berruezo G., Bioactive Components of Human Milk: Similarities and Differences between Human Milk and Infant Formula, in Selected Topics in Breastfeeding, IntechOpen, London: 2018. 10.5772/intechopen.73074. [Google Scholar]
- 7. Ladomenou F., Moschandreas J., Kafatos A., Tselentis Y., Galanakis E., Arch. Dis. Child. 2010, 95, 1004. [DOI] [PubMed] [Google Scholar]
- 8. Bode L., Glycobiology 2012, 22, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vandenplas Y., Berger B., Carnielli V. P., Ksiazyk J., Lagström H., Luna M. S., Migacheva N., Mosselmans J. M., Picaud J. C., Possner M., Singhal A., Wabitsch M., Nutrients 2018, 10, 1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheng L., Akkerman R., Kong C., Walvoort M. T. C., de Vos P., Crit. Rev. Food Sci. Nutr. 2020, 1. 10.1080/10408398.2020.1754756. [DOI] [PubMed] [Google Scholar]
- 11. Asakuma S., Hatakeyama E., Urashima T., Yoshida E., Katayama T., Yamamoto K., Kumagai H., Ashida H., Hirose J., Kitaoka M., J. Biol. Chem. 2011, 286, 34583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morrow A. L., Ruiz‐Palacios G. M., Altaye M., Jiang X., Guerrero M. L., Meinzen‐Derr J. K., Farkas T., Chaturvedi P., Pickering L. K., Newburg D. S., J. Pediatr. 2004, 145, 297. [DOI] [PubMed] [Google Scholar]
- 13. Cheng L., Kiewiet M. B. G., Groeneveld A., Nauta A., de Vos P., J. Funct. Foods 2019, 59, 174. [Google Scholar]
- 14. Wu R. Y., Li B., Koike Y., Määttänen P., Miyake H., Cadete M., Johnson‐Henry K. C., Botts S. R., Lee C., Abrahamsson T. R., Landberg E., Pierro A., Sherman P. M., Mol. Nutr. Food Res. 2019, 63, 1. [DOI] [PubMed] [Google Scholar]
- 15. Cheng L., Kong C., Walvoort M. T. C., Faas M. M., de Vos P., Mol. Nutr. Food Res. 2020, 64, 1900976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. He Y., Liu S., Leone S., Newburg D. S., Mucosal Immunol. 2014, 7, 1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bode L., Contractor N., Barile D., Pohl N., Prudden A. R., Boons G. J., Jin Y. S., Jennewein S., Nutr. Rev. 2016, 74, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He Y. Y., Liu S. B., Kling D. E., Leone S., Lawlor N. T., Huang Y., Feinberg S. B., Hill D. R., Newburg D. S., Gut 2016, 65, 33. [DOI] [PubMed] [Google Scholar]
- 19. Gnoth M. J., Kunz C., Kinne‐Saffran E., Rudloff S., J. Nutr. 2000, 130, 3014. [DOI] [PubMed] [Google Scholar]
- 20. Bidart G. N., Rodríguez‐Díaz J., Yebra M. J., Appl. Environ. Microbiol. 2016, 82, 570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Figueroa‐Lozano S., de Vos P., Compr. Rev. Food Sci. Food Saf. 2019, 18, 121. [DOI] [PubMed] [Google Scholar]
- 22. Yu J. C., Khodadadi H., Malik A., Davidson B., Salles É. d. S. L., Bhatia J., Hale V. L., Baban B., Front. Immunol. 2018, 9, 1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schirbel A., Fiocchi C., Expert Rev. Gastroenterol. Hepatol. 2011, 5, 33. [DOI] [PubMed] [Google Scholar]
- 24. Bradley J. R., J. Pathol. 2008, 214, 149. [DOI] [PubMed] [Google Scholar]
- 25. Fischer R., Kontermann R. E., Maier O., Antibodies 2015, 4, 48. [Google Scholar]
- 26. Nanthakumar N., Meng D., Goldstein A. M., Zhu W., Lu L., Uauy R., Llanos A., Claud E. C., Walker W. A., PLoS One 2011, 6, e17776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cotton J. A., Platnich J. M., Muruve D. A., Jijon H. B., Buret A. G., Beck P. L., Int. J. Interferon, Cytokine Mediator Res. 2016, 8, 13. [Google Scholar]
- 28. Dostert C., Grusdat M., Letellier E., Brenner D., Physiol. Rev. 2019, 99, 115. [DOI] [PubMed] [Google Scholar]
- 29. Bartsch J. W., Wildeboer D., Koller G., Naus S., Rittger A., Moss M. L., Minai Y., Jockusch H., J. Neurosci. 2010, 30, 12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang W. S., Moon S. Y., Lee M. J., Park S. K., Cell. Physiol. Biochem. 2016, 38, 1963. [DOI] [PubMed] [Google Scholar]
- 31. Kuntz S., Rudloff S., Kunz C., Br. J. Nutr. 2008, 99, 462. [DOI] [PubMed] [Google Scholar]
- 32. Melmed G., Thomas L. S., Lee N., Tesfay S. Y., Lukasek K., Michelsen K. S., Zhou Y., Hu B., Arditi M., Abreu M. T., J. Immunol. 2003, 170, 1406. [DOI] [PubMed] [Google Scholar]
- 33. Cario E., Rosenberg I. M., Brandwein S. L., Beck P. L., Reinecker H.‐C., Podolsky D. K., J. Immunol. 2000, 164, 966. [DOI] [PubMed] [Google Scholar]
- 34. Horiuchi T., Mitoma H., Harashima S. I., Tsukamoto H., Shimoda T., Rheumatology 2010, 49, 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Steeland S., Libert C., Vandenbroucke R. E., Int. J. Mol. Sci. 2018, 19, 1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.