

Abstract
The sulfated glycolipid PG545 shows promising antitumor activity in various cancers. This study was conducted to explore the effects and the mechanism of PG545 action in endometrial cancer (EC). PG545 exhibited strong synergy as assessed by the Chou-Talalay-Method in vitro when combined with cisplatin, or paclitaxel in both type I (Hec1B) and type II (ARK2) EC cell lines. While PG545 showed antitumor activity as monotherapy, a combination of PG545 with paclitaxel and cisplatin was highly effective in reducing the tumor burden and significantly prolonged survival of both Hec1B and ARK2 xenograft bearing mice. Mechanistically, PG545 elicits ER stress as an early response with resultant induction of autophagy. Our data demonstrated an increase in pERK, Bip/Grp78, IRE1α, Calnexin and CHOP/GADD153 within 6–24 hrs of PG545 treatment in EC cells. In parallel, PG545 also blocked FGF2 and HB-EGF mediated signaling in EC cells. Moreover, melatonin-mediated ER stress inhibition reduced PG545-mediated autophagy and PG545 in combination with cisplatin further heightened this stress response. Collectively these data indicate that PG545 exhibits strong synergistic effects with chemotherapeutics in vitro and showed promising antitumor activity in vivo. Our preclinical data indicates that in future studies PG545 can be a useful adjunct to chemotherapy in endometrial cancer.
Keywords: Autophagy, Endometrial Cancer, ER stress, PG545, Synergy
1. Introduction
Endometrial Cancer (EC) is the most common malignancy of the female reproductive tract in developed countries with an estimated 10,920 deaths in the United States in 2017 [1]. There is an increase in the rate of EC in recent years. Two major risk factors, obesity and advanced age account for the increase in the rate of EC which is emerging as a major public health concern [2,3].
The majority of women with EC are diagnosed at an early stage and can be cured with resection, chemotherapy or radiation [4,5]. Still, there is a subset of patients with unfavorable histology, advanced disease and severe comorbidities for which long-term prognosis remains poor [6]. For these patients, chemotherapy represents an important treatment modality next to the surgery. The standard chemotherapy regimen includes platinum and taxol-based agents achieving at best moderate overall response of up to 50% percent in advanced-stage disease [7].
Endometrial cancer itself is a very heterogeneous disease [8], making patient-adjusted therapy extremely challenging. EC can be generally classified into two types showing distinct histological and molecular features [9]. Type I appears to be more differentiated with endometroid-like histology and slower disease progression. PTEN loss, K-Ras mutation and microsatellite instability are frequently seen in Type I [10,11]. Type II, however, is less differentiated and often resulting in a clear or papillary serous cell appearance. It commonly harbors alterations in p53, p16, HER2 and E-Cadherin and shows a more aggressive course of the disease. Additionally, upregulation of several heparin-binding growth factors (HB-GFs) such as FGF, VEGF, HGF and HB-EGF and their cognate receptors are significantly linked to enhanced tumor vascularization and poor survival outcome in EC implicating growth factors and their receptors as novel therapeutic targets [12–16]. However, response rates are at best moderate (6.8–28.9%), raising the question of whether targeted therapy of single pathways is the right course to pursue, specifically considering the tumor’s heterogeneity and evolving resistance mechanisms [17]. Combinations of different drugs or agents that simultaneously target multiple pathways may represent a reasonable and promising alternative.
Heparan sulfate (HS) proteoglycans (HSPG) are involved in several aspects of carcinogenesis [18]. HSPGs structure stabilizes the extra-cellular matrix (ECM) and provides binding sites to various molecules including the heparin-binding growth factors mentioned above [19]. Heparanase is a β-endoglucorinadase, which cleaves HSPGs and releases the molecules bound to them including VEGF, FGF and HB-EGF. Its expression is significantly increased in endometrial neoplasias [20]. Degradation of the ECM exacerbates invasion and dissemination of cancer cells in the surrounding tissue and the released growth factors induce further proliferation and angiogenesis of cancer [21]. In endometrial cancer, heparanase expression is associated with increased myometrial invasion, lymph-vascular space involvement and is correlated with less differentiated histological subtypes [22,23]. In addition to the increase in heparanase expression, HSPG levels on the cell surface are commonly upregulated in EC, indicating a more profound role of HSPGs in tumorigenesis [18,24]. Thus, the heparanase-HSPG system represents a promising therapeutic target for multi-targeted therapy in endometrial cancer.
PG545 is a fully sulfated glycolipid and is currently in Phase I clinical investigation [25]. It functions as an immune stimulator that activates NK cells via dendritic cells [26] and also inhibits heparanase and HS-mediated growth factor signaling [27]. PG545 has shown antiproliferative, antimetastatic and antiangiogenic properties in various carcinoma cell lines [28,29]. Our group further demonstrated that PG545 synergizes with Gemcitabine and Paclitaxel in pancreatic and ovarian cancer respectively, highlighting its potential use in combination therapy [30,31].
This study was conducted to evaluate the anti-tumor activity of PG545 in type I and type II EC cells alone and in combination with taxane and platin-based agents. By using both types I and II cells, we show that PG545 modulates GF signaling in vitro and can be used alone and in combination with chemotherapeutic agents to prolong survival in an in vivo model. This is the first report to show the potential use of PG545 in EC. Additionally, we also show that ER stress is an early response to trigger autophagy after PG545 treatment. Taken together, we show that PG545, in combination with chemotherapy can effectively elicit an ER stress-mediated increase in autophagy that can be therapeutically targeted. Thus combinations of PG545 with known chemotherapies may be promising options to be considered in future clinical trials of endometrial cancer.
2. Materials and methods
2.1. Chemical reagents
PG545 was generously provided by Zucero Therapeutics (Melbourne, Australia). Cisplatin was purchased from EMD Millipore (Calbiochem, Millipore, Billerica, MA) and paclitaxel (30 mg/5ml) from Hospira (Lake Forest, IL). All compounds were dissolved in phosphate buffer saline (PBS, Gibco, USA). Primary antibodies anti-cleaved PARP, anti-LC3BI/II, anti-phospho- and total-PERK, anti-Bip, anti-CHOP, anti-GAPDH, anti-IRE1α, anti-phospho- and total-ERK, anti-phospho- and total-AKT and secondary rabbit and mouse IgG were purchased from Cell Signaling Technology (Danvers, MA); anti-PCNA, anti-p62 and anti-Calnexin were purchased from Santa Cruz (Santa Cruz, CA).
2.2. Cell lines
Four cell lines representing different histological grades of EC were used for the in vitro studies [32]. Ishikawa (well-differentiated endometrioid, less aggressive), RL95-2 (poorly differentiated endometrioid) [both cell lines were kindly provided by Dr. Paul Goodfellow (Washington University, St. Louis, MO)] and Hec-1B (cisplatin-resistant) [purchased from American Type Culture Collection (ATCC)] were used as representatives for type EC. As type II cells we chose ARK2 [kindly gifted by Prof. Shi-Wen Jiang who was affiliated with Prof. (Dr.) Karl C. Podratz, (Mayo Clinic, Rochester, MN)] representing serous cell carcinoma. Hec1B, ARK2 and RL95-2 were cultured in DMEM/F12 media and Ishikawa cells were in DMEM media (Thermo Fisher, Waltham, MA), supplemented with 10% heat-inactivated Fetal Bovine Serum and 1% antibiotic–antimycotic (Thermo Fisher, Waltham, MA). All cells were incubated in a humidified atmosphere at 37 °C with 5% CO2.
2.3. Colony formation assay (CFA)
1×103 cells were plated in 6-well plates in triple replicates and incubated with increasing concentrations of PG545 (2.5, 5, 10, 20 and 40 μM) for 24 h. The media was removed after 24 h and replaced. Colonies were fixed in methanol (Thermo Fisher, Waltham, MA) and stained with 0.5% crystal violet (Millipore-Sigma USA) on day 7. Several colonies were assessed using Quantity One (Bio-Rad) software. Each experiment was repeated at least thrice.
2.4. Cell viability assay
MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (Millipore-Sigma USA) assay was used to assess the cell viability of PG545 treated cells. Five thousand cells per well were plated in a 96 well plate. After 24 h media was removed and the treatment regimen of various drug combinations was added for 48 h. After a 48-hour incubating period 20 μl of MTT was added. After four hours remaining media was removed and 200 μl Dimethyl Sulfoxide (DMSO) were added. Absorbance was measured at 490 nm wavelength in a microplate reader.
2.5. Synergy calculations
For all MTT experiments, five thousand cells per well of a 96 well plate were plated as replicates of five. The half-maximal Inhibitory Concentrations (IC50) were then determined for all drugs and cell lines used in synergy studies. Increasing concentrations of PG545, cisplatin, paclitaxel, chloroquine and quinacrine were added for 48 h and assessed via MTT assay. Out of these data sets, IC50 values were determined by Prism software (GraphPad Software, La Jolla, CA) as previously described in [33]. Constant ratio (multiples of IC50: IC50 ratios) were conducted between PG545, each of quine and Quinacrine. CI values were calculated by the CalcuSyn software (Biosoft, USA) using the Chou-Talalay method as previously described [34,35]. The Combination Index (CI) classifies the drug interaction into synergy, addition or antagonism. CI values < 0.9 indicate synergy, while a range of 0.3–0.7 can be considered as strong; 0.7–0.85 as moderate; 0.85–0.9 slight synergy. Values from 0.9 to 1.1 show additive effect, and values > 1.1 depict antagonism [34]. Non-constant ratio combinations, i.e. IC50 of PG545 with various Chloroquine concentrations were also determined via the Chou-Talalay-Method [34].
2.6. Immunoblot
Western Blots were performed as previously described [36]. Every western blot was performed at least three times and a representative blot image was shown in the figures. The densitometric quantification of the western blots was shown in the figures.
2.7. Migration (wound scratch) assay
80–90% confluent 1 × 105 EC cells were incubated with 0% serum medium and were scratched with a 200 μl pipette tip. Heparin-binding growth factors (HB-EGF at 50 ng/ml, FGF-2 at 10 ng/ml, FBS equal to 10% volume) (Gibco, USA) were added with or without 10 μM of PG545. The area of migration was then assessed via ImageJ software.
2.8. Invasion assay
Hec1B and ARK2 cells (1 × 105) were plated into Fluorimetric Invasion Assay (FIA) chambers (Corning, NY). The outer compartment contained FBS or 50 ng/mL HB-EGF as a chemoattractant. The plate was incubated for 48hrs letting cells invade the Matrigel layer and reach the surface membrane. Cells were fixed and stained with Coomassie Staining (BioRad, CA) and counted per well in replicates of three. EC cell invasion was assessed in the absence and presence of PG545.
2.9. Apoptosis staining via Annexin V/PI double staining
Apoptosis of EC cells treated with PG545 and/or cisplatin and paclitaxel was assessed as previously described [30]. EC cells were stained with an Apoptosis detection kit (Roche, USA) labeled with Annexin V-FITC and Propidium Iodide. cells were then quantified by FACSCalibur (Becton Dickinson, USA) measuring Annexin V-FITC and PI-positive cells.
2.10. Xenografts
All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee of Mayo Clinic (IACUC). Xenograft model of HEC-1b cells (Type I) and ARK2 (Type II) Endometrial Cancer were established injecting 2.5 × 106 Hec1B Cells and due to their more aggressive behavior 1.5 × 106 ARK2 subcutaneously into the right flank of 6–8 week old female athymic nude mice (Harlan Laboratories, Indianapolis, IN). Cells were suspended in a total volume of 0.2 ml PBS. Once tumors were measuring at least 3 × 3 mm in all groups, mice were randomized into four groups of ten animals each. Treatment was initiated in Hec1B and ARK2 cells on day 7 after injection. Tumor size was measured every four days with calipers for 28 days after which the xenografts were monitored for meeting endpoint criteria for an ongoing survival study. Endpoint criteria were defined as follows: 1. Ulceration of tumor site 2. The tumor is larger than 1200 mm3 3. Tumor diameter greater than 20 mm 4. Weight loss greater than 10%. For assessment of Tumor size the modified ellipsoid volume was calculated by the following formula: volume = π/6 × length × width2. All xenografts models of Hec1B and ARK2 were assigned to following treatment groups: Control: receiving 200 ml every third day as placebo control; Chemotherapy: Cisplatin plus paclitaxel via intraperitoneal injection at 4 mg/kg and 16 mg/kg, respectively on days 7, 12, and 16 as previously described in [31]; PG545 alone: 20 mg/kg every three days as previously described in [30,31]. Combination: cisplatin plus paclitaxel equally dosed as chemotherapy group) plus PG545 (20 mg/kg, same dosing as PG545 alone).
2.11. Immunohistochemistry
The Pathology Research Core Laboratory (Mayo Clinic, Rochester, MN) performed immunohistochemistry staining on all xenograft tumors as per standard protocol. Paraffin-embedded tissue sections were stained for Ki67, and CD31 and subsequently analyzed. CD31, a marker for vascular endothelium was used to assess intratumoral microvessel density (iMVD). Representative areas of three per tumor were taken with a Zeiss LSM 510 microscope. Microvessel structures were then counted in 20X magnification and compared among different treatment groups. Ki67 staining was assessed using freely available ImageJ Add-on ImmunoRatio as previously described in [37].
2.12. Detection of autophagosomes by Cyto-ID staining
Hec1B and ARK2 cells were plated in the 4-well chambered slides and treated with 0 μM, 10 μM or 20 μM of PG545. After 24 h, cells were washed 2× with PBS and a Cyto-ID autophagy detection kit (Enzo Life Sciences) was used to detect autophagic vesicles. Images were taken with a Zeiss LSM 510 microscope and fluorescence was analyzed via ImageJ software.
2.13. Detection of ER activity by ER tracker blue-white DPX staining
ER activity was visualized by fluorescence microscopy stained with ER-tracker blue/white DPX as stated earlier [38,39]. Briefly, Hec1B and ARK2 cells (5 × 103) were seeded in 8 chambered polystyrene culture slides [BD Falcon, USA] per well in DMEM/F12 medium supplemented with FBS (10%). After 24 hrs of seeding, cells were incubated with 1× HBSS [Gibco, USA] supplemented with Mg++ (0.10 gm/L) and Ca++ (0.14 gm/L) and subsequently exposed to PG545 (20 μM) along with vehicle control in a time-dependent manner. Cells were washed with HBSS and processed for ER staining. Pre-warmed ER-Tracker Blue-White DPX (Thermo Fisher, Waltham, MA) staining (500 nM) was added to the cells and incubated for 30 mins at 37 °C at 5% CO2 incubator. The loading solution was removed and cells were then washed in HBSS. The samples were visualized and images were recorded using an EVOS fluorescent microscope.
2.14. Statistical analysis
Results are depicted as mean ± standard deviation (S.D.). Unless mentioned otherwise data was gathered from three independent experiments. Graph Pad Prism software (San Diego, CA) was used for statistical analyses. Data sets were analyzed via paired t-test. Survival data were analyzed by using Log-Rank tests based on Kaplan Meyer curves. The significance level was set at 0.05 (p < 0.05) unless determined otherwise.
3. Results
3.1. PG545 inhibits growth in a dose-dependent manner in type I and type II EC
The IC50 values of PG545 in Hec1B and ARK2 cell lines as evaluated by MTT assay (24hr) were ~63.5 μM for Hec1B and 70.8 μM for ARK2 cells, respectively (Fig. 1A and B). Increasing concentrations of PG545 effectively inhibited colony-forming ability with maximal inhibition of PG545 at 20 μM in Hec1B and 40 μM in ARK2 cells (Fig. 1C and D).
Fig. 1.
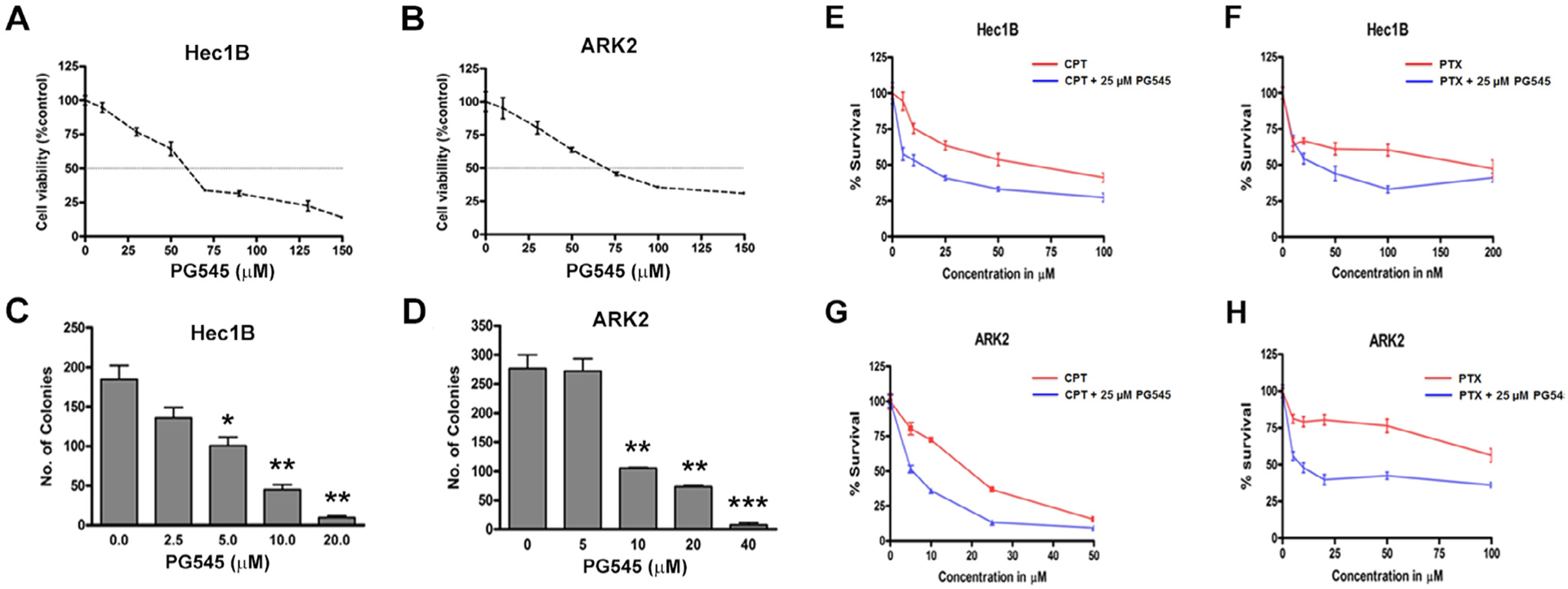
Cell viability assays show PG545 inhibits proliferation (A, B) and colony-forming abilities (C, D) of Hec1B and ARK2 cells in a dose-dependent manner respectively. E-H, ARK2 and Hec1B cells were incubated with increasing concentrations of cisplatin or paclitaxel alone (red) or in combination with 25 μM PG545. IC40 values for all four different combinations were significantly reduced by the addition of PG545 NS: Not significant; *p < 0.05, **p < 0.01, ***p < 0.001.
3.2. PG545 sensitizes and shows strong synergy with known chemotherapeutic drugs in EC cell lines
We have previously shown that PG545 synergizes with gemcitabine in pancreatic cancer and with cisplatin and paclitaxel in ovarian cancer [30,31]. Having determined that PG545 treatment inhibits the growth of EC cells, we then sought to determine if PG545 will sensitize and synergize with cisplatin and paclitaxel in EC. The study shows that PG545 (25 μM) sensitizes EC cells strongly to both cisplatin and paclitaxel (Fig. 1E–H) with the ARK2 cell line showing comparatively stronger synergy.
To this end, we evaluated Combination Indices (CI) between PG545 and the chemotherapeutic agents using the Chou-Talalay method as previously described [35]. Constant ratios of drug concentrations, in this case of the respective IC50 values for each compound, are plotted alone and in combination. The lower the Combination Index (CI) values at a given biological effect (Fraction affected or Inhibition in this case) indicates very strong synergy between the two tested compounds. Thus CI values between 0.1 and 0.3 indicate extremely strong synergy, 0.3–0.7 strong synergy, 0.7–0.85 moderate synergy, 0.85–0.9 slight synergy and CI values between 0.9 and 1.0 are considered nearly additive). Synergy was seen across all combinations. Very strong synergy was seen in Hec1B cells at low concentrations (Fig. 2A and B). In ARK2 cells, the CI values ranged from 0.3 to 0.5 and did not alter significantly with increasing Fraction affected (Fig. 2C and D) as seen in Hec1B cells. Since the curves represented in Fig. 2A–D are mathematical calculations, it should be noted that the experimental combination values determined by the CalcuSyn Software range from 0.171 to 0.984 in Hec1B and 0.220–0.64 in ARK2 cells. All experimental values were below 1 indicating that the combination is almost synergistic.
Fig. 2.

Synergism between PG545 with Cisplatin and Paclitaxel in equipotent combinations (IC50: IC50) was assessed using the Chou-Talalay method. Hec1B (A, B) and ARK2 (C, D) cells were treated alone and in combination with each drug for a time course of 48 h. Combination indices were calculated by the Calcusyn software and accordingly shown with GraphPad. CI between 0.3 and 0.7 shows strong synergism, 0.7–0.85 moderate synergism, 0.85–0.9 slight synergism, 0.9–1.10 nearly additive effect and greater than 1.10 antagonism. The area of additive effect is depicted by dotted lines at 0.9 and 1.1. E–J: Incubation with PG545 and or Cisplatin and or paclitaxel alone and in combination induces apoptosis Cells were stained with Annexin V- Propodium Iodine (PI) after 24 hr treatment with the given drug concentration and analyzed by flow cytometry. Graphs show the percentage of cells stained positive. P-values: ns = not significant, * < 0.05, ** < 0.01.
3.3. PG545 enhances apoptotic cell death of chemotherapy in vitro
Having established synergistic interaction between PG545 and the chemotherapeutic agents, we further investigated apoptotic cell death by Annexin V/PI assay after treating cells with PG545 (20 μM) alone or in combination with cisplatin (CPT, 20 μM) and/or paclitaxel (PTX, 50 nM) (Fig. 2E and H). While single treatments alone showed a mild increase in early and late apoptotic markers (9.02–10.56%), a combination of PG545 with cisplatin showed 35% of HEC1B and 22% of ARK2 cells undergoing cell death (Fig. 2F and G). The ARK2 cells were more sensitive to paclitaxel (Fig. 2J) and the combination of PTX with PG545 was more effective in inducing apoptosis compared to PTX treatment alone in both cell lines (p < 0.01) (Fig. 2I and J). Specifically, the addition of PG545 to PTX in Hec1B cells dramatically increased apoptosis from 11.79 to 39.32% (Fig. 2I).
3.4. PG545 alone and in combination with cisplatin and paclitaxel inhibits EC tumor growth in vivo and prolongs survival
To investigate the effects of PG545 in combination with chemotherapy in vivo, we used two xenograft models in female athymic nude mice: one with Hec1B cells and one with ARK2 cells (Fig. 3A). We injected 2.5 × 106 Hec1B and 1.25 × 106 ARK2 cells subcutaneously [37] in the right flank of female athymic nude mice. The mice were randomized into 4 groups before commencing the treatment, one week after tumor cell injection as described in the methods section. Tumor volume (in mm3) is shown until the first endpoint criteria were met in the survival study. PG545 (20 mg/kg, given twice a week till the end of the study) treatment as a single agent alone significantly reduced the tumor burden more compared to control and to the chemotherapy regimen alone (given on days 7. 14 and 21) (p = 0.001) (Fig. 3B and C). While the chemotherapy regimen alone also inhibited tumorigenesis (p = < 0.05), the combination of PG545 and chemotherapy was very effective in inhibiting tumor formation compared to either treatment alone (p = < 0001), (p < 0.05 in Hec1B and p < 0.01 in ARK2).
Fig. 3.
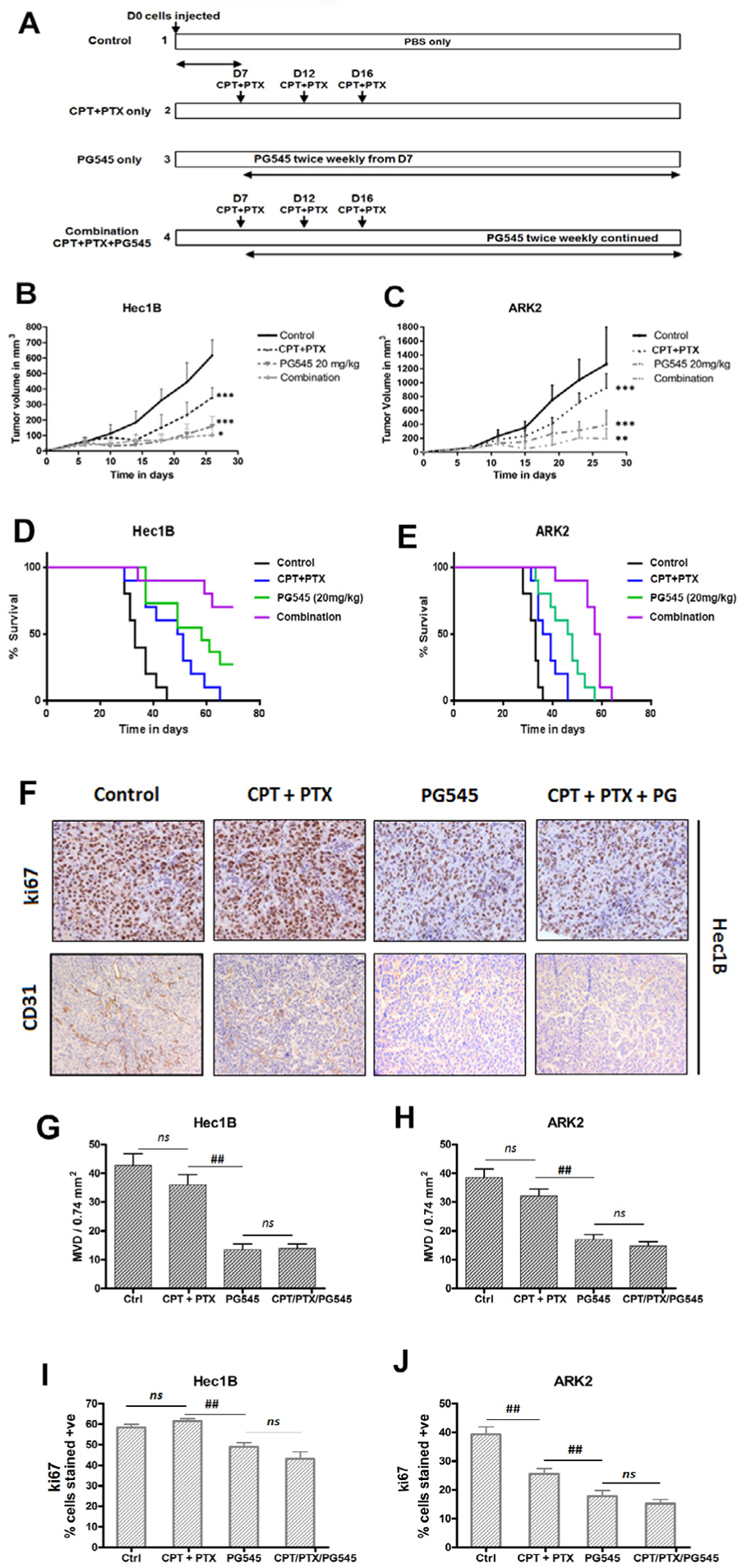
PG545 reduced tumor progression alone and in combination with chemotherapy in Hec1B and ARK2 cell line. Treatment is shown schematically (A). Each xenograft model, Hec1B and ARK2, was studied in four different groups. 2.5 × 106 Hec1B cells and 1.25 × 106 ARK2 were injected in 10 mice/group. PG545 20 mg/kg was injected intraperitoneally every 72 h beginning on D7. Cisplatin/paclitaxel was administered via i.p. injection at 20 mg/kg and 16 mg/kg, respectively on days 7, 12, and 16 as per Materials and Methods. Tumor volume was recorded every four days after treatment start and depicted until the first sacrifice of a xenograft. All treatment groups differ significantly from each other across both Hec1B (B) and ARK2 (C) xenografts. P-values: ns = not significant, * < 0.05, ** < 0.01, *** < 0.001. PG545 significantly prolongs survival alone and in combination with chemotherapy in type I and II endometrial cancer. Kaplan-Meier Curve depicts Hec1B xenograft (D) and ARK2 xenograft (E). Each xenograft model was studied in four different groups. 2.5 × 10^6 Hec1B cells and 1.25 × 10^6 ARK2 were injected in 10 mice/group. PG545 20 mg/kg was injected intraperitoneally every 72 h beginning on D7. Cisplatin/paclitaxel was administered via i.p. injection at 20 mg/kg and 16 mg/kg, respectively on days 7, 12, and 16 as per Materials and Methods. P-values: ns = not significant, * < 0.05, ** < 0.01, *** < 0.001. Immunohistochemical analysis of ki67 and CD31 staining in endometrial tumor tissues (F). Quantification of microvessels was performed in 20X magnification with 3 fields per analyte (G, H). Quantification of ki67 was performed using 5 fields per analyte using ImageJ software and ImmunoRatio Add-on (I, J). P-values: ns = not significant, # < 0.05, ## < 0.01.
We also evaluated the effect of PG545 and chemo (CPT + PTX) alone and the combination of PG545 and chemo (CPT + PTX) treated mice bearing Hec1B and ARK2 xenografts on the overall survival of these mice. All treatment groups in both cell xenograft models showed significantly better survival compared to the controls. In the Hec1B median survival was 33 days in the control group, this was followed by a median survival of 50 days in the chemotherapy group and 58 days in the PG545 group (Fig. 3D). PG545 treatment alone did not significantly prolong survival compared to the chemotherapy (p = 0.09) alone group. However, the combination of both regimens significantly increased survival compared to either treatment alone (p < 0.001). The survival study was terminated after 72 days. Two of the three mice in the Hec1B combination group died due to tumor ulceration and not due to tumor burden.
The median survival in the chemotherapy group was only 37.5 days and was comparatively less than PG545 alone with a median survival of 47 days (p < 0.05) or the combination of both regimens with a median survival of 58 days (p < 0.001). Still, after 63 days all ARK2 xenograft tumors met the sacrificing criteria(Fig. 3E).
We confirmed the enhancing effects of PG545 in mouse-derived xenografts reducing tumor volume and prolonging survival in combination with chemotherapy. Enhancing effects could not only be seen by analyzing the survival data but also upon monitoring tumor growth in the first 30 days (Fig. 3B and C).
3.5. PG545 downregulates the angiogenic CD31 and proliferative Ki67 markers upon treatment
Paraffin-embedded xenografts were stained with CD31 antibody, an established marker for angiogenesis in order to determine the intratumoral Micro Vessel Density (iMVD). The iMVD can be measured by counting CD31 positively stained angio-epithelial tissue [41]. The iMVD was significantly reduced in all PG545 treated groups compared to control or the chemotherapy groups (p < 0.001) (Fig. 3F and 3G–H). Chemotherapy alone had no significant impact on vascular formation in either Hec1B (p = 0.32) or ARK2 (p = 0.18) xenografts compared to the controls. Furthermore, the iMVD of the combination-treated groups were also not significantly reduced compared to the PG545 groups alone, indicating that the observed effect of CD31 downregulation is caused by PG545 treatment. The results are consistent with previous studies in other tumor types [29–31].
Similarly, expression of the proliferation marker Ki67 was downregulated by PG545 treatment in both xenograft models (Fig. 3F and I–J). Ki67 expression was significantly downregulated upon PG545 treatment compared to control in Hec1B (p < 0.01) and ARK2 (p < 0.01). Surprisingly, chemotherapy treatment did not alter Ki67 expression in Hec1B tumors (p = 0.15) compared to its control whereas it did in the ARK2 xenograft (p < 0.01). Similar to CD31 expression, the combination of PG545 and chemotherapy did not significantly reduce Ki67 expression compared to PG545 alone in either xenograft model, most likely due to the time elapsed between the last administration of chemotherapy and therefore the difference in treatment between the two groups.
3.6. PG545 induces autophagy in EC
In order to investigate the mechanistic basis of the synergy that we observed between PG545 and CPT and PTX, we assessed the impact of PG545 on autophagy. Western blot analysis of lysates from PG545-treated Hec1B and ARK2 cells showed an increase in the lipidated form of LC3BII in both the cell lines (Fig. 4A and B) respectively. The densitometric quantification provided in Fig. 4C reveals the upregulation of LC3BII over LC3BI. Consistent with the increase in LC3BII levels, immunofluorescence (IFC) analysis of autophagy using cyto-ID [42] staining also showed that PG545-induced autophagy in a dose-dependent manner (Fig. 4D). Fig. 4E shows the Corrected Total Cell Fluorescence (CTCF) of the fluorescence of the autophagosomes quantified using ImageJ software. CTCF values are significantly increased (p < 0.001) in ARK2 cells treated for 24 h in a dose-dependent manner (p = 0.07). Hec1B cells also showed a significant increase (p < 0.01, p < 0.001) in CTCF following PG545 treatment although it was less than that seen in the ARK2 cells (Fig. 4D and E).
Fig. 4.
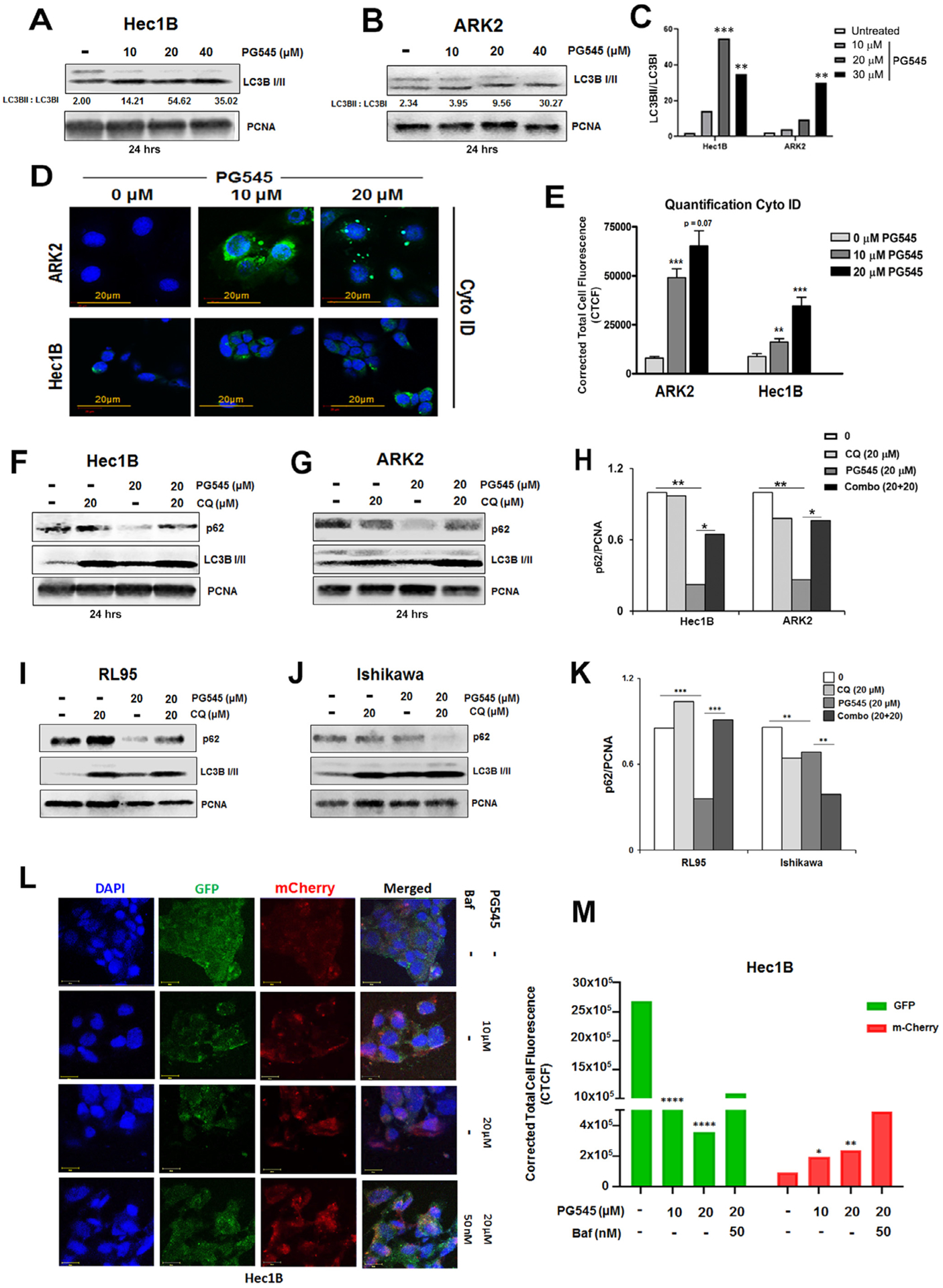
PG545 induces autophagy Hec1B (A) and ARK2 (B) cells. Immunoblot analyses are shown for Hec1B and ARK2 cell lysates treated with 10, 20 and 40 μM PG545 for 24 h. The ratio of LC3BII over LC3BI was measured in 4C. Cyto ID staining was used to demonstrate the autophagosome formation in ARK2 and Hec1B cells (4D). Quantification of Corrected Total Cell Fluorescence (CTCF) was conducted by using ImageJ software (E). Autophagic flux was also investigated in Hec1B (F) and ARK2 (G) cells. Cells were either treated with 20 μM PG545 and/or with chloroquine. Both compounds were added for 24 h. Cell lysates were analyzed by Western blot using antibodies to LC3BI/II, p62 and PCNA. The level of p62 over PCNA was measured in H. P values: ns = not significant, * < 0.05, ** < 0.01, *** < 0.001. Autophagic flux was investigated in RL95-2 (I) and Ishikawa (J) cells similarly to Fig. 4 F, G. The level of p62 over PCNA was measured in K. L, Transient expression of Cherry-GFP-LC3B (48 h of incubation), autophagic flux was determined in Hec1B cells incubated with 10 μM and 20 μM PG545 for 24 h and co-treated with Bafilomycin 1A for 12 h. Confocal microscopy analysis depicts alterations in the autolysosome formation. Quantification of Corrected Total Cell Fluorescence (CTCF) for both GFP and m-Cherry was conducted by using ImageJ software (M).
As the increase in LC3BII levels alone is not a sufficient indication that the cells are undergoing autophagy since the increase in LC3BII levels could be due to autophagy induction or inhibition of the final steps of autophagosome fusing with the lysosome, we determined the levels of p62 (SQSTM1), another important autophagy marker in the presence and absence of chloroquine (CQ), a well-known inhibitor of autophagy to determine if the cells were undergoing autophagic flux. PG545 treatment of Hec1B and ARK2 cells resulted in the downregulation of p62 and an increase in the levels of LC3BII, and CQ treatment rescued the expression of both these proteins, clearly indicating that the cells were undergoing autophagic flux (Fig. 4F and G). The densitometric quantification provided in Fig. 4H also demonstrates significant loss of p62 in PG545 treated Hec1B and ARK2 cells (without CQ), and rescue of p62 in the presence of CQ respectively. In parallel, endometrial cell line RL95 also showed a similar phenomenon, however, CQ could not rescue p62 in the Ishikawa cell line (Fig. 4I–K).
Although the increase in LC3BII and a decrease in p62 levels are indicative of autophagic flux, autophagic flux is reliably monitored with the use of fluorescent-tagged Cherry-GFP-LC3B construct [43,44] following autophagy induction. Since the pH-sensitive GFP signal is quenched in the lysosomes, the autophagosomes light up as both red and green (seen as orange to yellow in merged images), while autolysosomes light up as mostly red. Following transient transfection for 48 h of Hec1B cells with Cherry-GFP-LC3B, cells were treated with 10 and 20 μM PG545 resulted in an increase in the mCherry positive signals in the treated cells (Fig. 4L, rows 2–3, Fig. 4M respectively) compared to untreated control cells (Fig. 4L, row 1, Fig. 4M) clearly indicated the activation of autophagic flux. Importantly, co-treatment with Bafilomycin A that prevents the fusion of autophagosomes to lysosomes showed more GFP staining in the merged panel (Fig. 4L, row 4, Fig. 4M) indicating inhibition of lysosomal activity.
3.7. PG545 elicits ER stress as an early response
Our data suggest that PG545 induces autophagic flux in EC cells. In contrast to our studies, Shteingauz et al. have shown that PG545, by inhibiting heparanase-induced autophagy impedes cell growth in vitro and in vivo in cervical and glioblastoma cell lines [45]. To understand this contradictory result between the two studies, we set out to determine if autophagy could be induced as a defense mechanism to overcome ER stress-induced unfolded protein response (UPR) as previously reported in other cancers [46–48]. To understand whether inhibition of GF-mediated signaling by PG545 mechanistically could alter and/or activate ER stress, a known trigger of autophagy [49,50], we sought to determine if PG545-induced ER stress as an activator of adaptive response could play a role in PG545 induced autophagy. To study the activity of ER stress, Hec1B and ARK2 cells were stained with ER-Tracker Blue-White DPX dye following 20 μM PG545 treatment in a time-dependent manner (15, 30 and 60 mins). This photo-stable dye is specific for the ER and provides blue staining in live cells. PG545 treated cells exhibited significantly increased blue staining compared to the control untreated cells indicating an accelerated activity of ER leading to ER stress in vitro (Fig. 5A). The increase in fluorescent signal was again quantified as CTCF. A significant rise of CTCF was observed in both Hec1B and ARK2 cells (Fig. 5B).
Fig. 5.
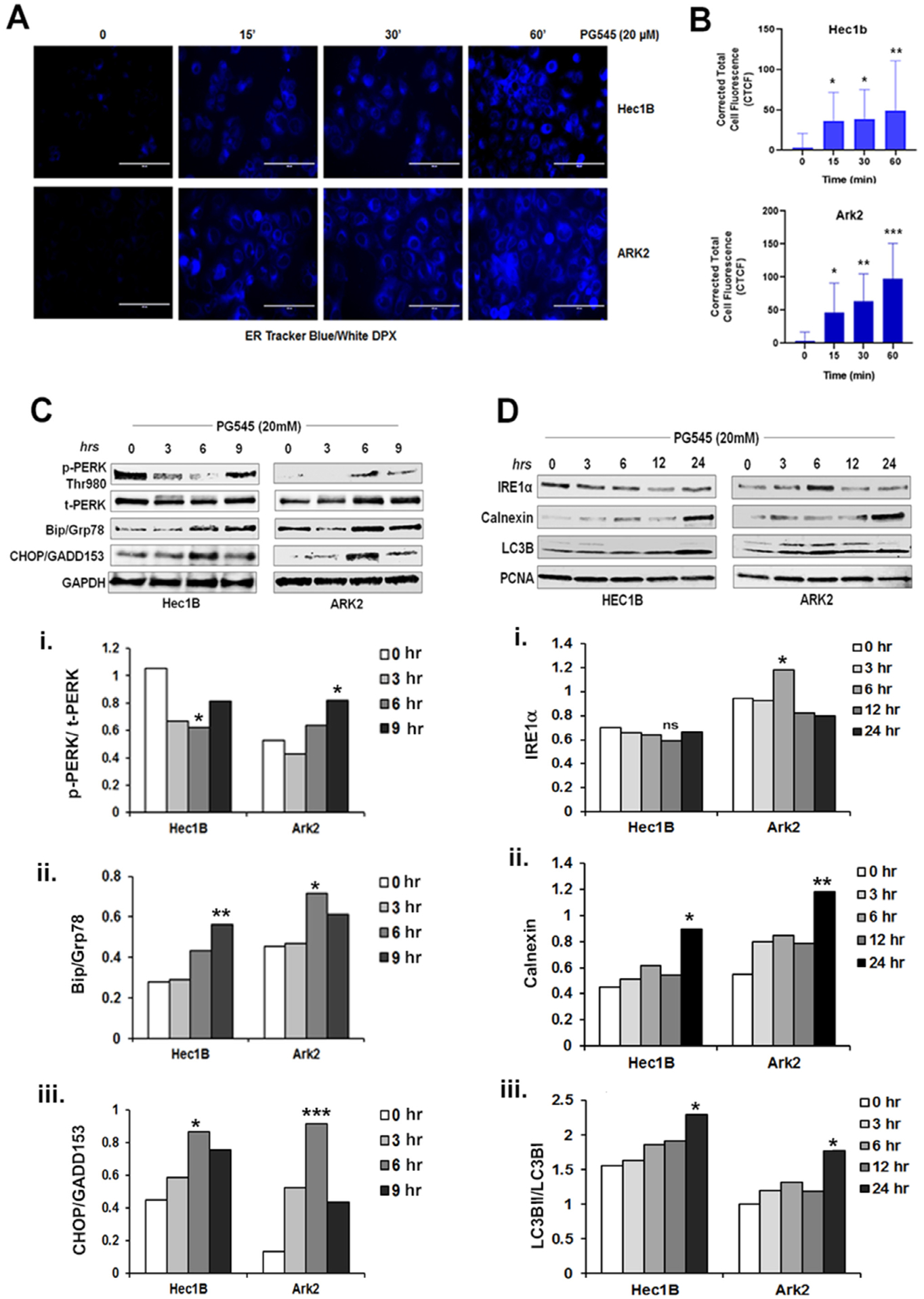
PG545 (20 μM, 0–60 mins) escalates ER activity as measured in blue-white DPX staining in Hec1B and ARK2 cells (A). Cells were incubated in Hanks balances salt solution and incubated with PG545 as mentioned in the materials and methods section. The corrected total cell fluorescence was shown in B, P values: * < 0.05, ** < 0.01. C-D. PG545 treated (20 μM) cell lysate was analyzed by Western blot using ER stress marker antibodies to p-PERK (Thr980), t-PERK, Bip/Grp78, CHOP/GADD153, GAPDH, IRE1α, Calnexin, LC3B and PCNA.
Given that PG545 treatment leads to increased ER abundance in both cell lines, we analyzed a number of ER stress-related molecular markers to assess if any were modulated by PG545. Data shown in Fig. 5C and D revealed that PG545 could modulate the three canonical ER stress pathways within 6–24 h. Protein kinase R-like endoplasmic reticulum kinase (PERK) mediated phosphorylation of the eukaryotic initiation factor 2 (eIF2) α subunit is a well-established mechanism to downregulate protein synthesis under a variety of stress conditions, which subsequently upregulates cellular stress-induced transcription factor C/EBP homologous protein (CHOP) or growth arrest and DNA damage 153 (GADD153) [51]. The activation of PERK by phosphorylation at Thr980 and upregulation and accumulation of CHOP/GADD153 level in the PG545 treated EC cells conferred the attenuation of the translational initiation and the commencement of apoptosis upon ER stress respectively. However, the PERK and IRE activation was comparably much pronounced in ARK2 cells Fig. 5C (i–iii) and D (i–iii). The synthesis of the major ER chaperone, GRP78/BiP, was markedly induced after PG545 treatment which suggests the accumulation of unfolded polypeptides in the ER. Another the Ca++ dependent chaperone, Calnexin, which is also substantially documented for involvement in apoptosis [52], was also upregulated at 24 h of PG545 treatment in both cell lines further confirmed prolonged ER stress.
3.8. PG545-induced ER stress triggers autophagy in EC cell lines
ER stress more recently has been implicated as a potent initiator of autophagy, a self-degradative procedure that also has an early adaptive function [46]. In order to determine whether PG545 induced ER stress is driving autophagy, EC cells were pretreated for 30 mins with 1 μM of melatonin, which is involved in ER homeostasis [26], with and without subsequent cotreatment of 20 μM PG545 for 2 h. The live cells were visualized under EVOS fluorescent microscope Fig. 6A. A representative image of this study showed enhanced ER activity after PG545 treatment both in Hec1B (Fig. 6A panel 2) and ARK2 (Fig. 6C panel 2), which is substantially decreased in melatonin pretreated cells. The significant differences in integrated fluorescence density among the experimental groups are shown in Fig. 6B and D for Hec1B and ARK 2 respectively. Correspondingly, western blot data also showed PG545 mediated upregulation of BiP and CHOP consequently reduced in melatonin treated cells Fig. 6E (i–ii). These data further confirmed that PG545 mediated ER stress is a potential autophagy inducer in EC cells 6E (iii).
Fig. 6.
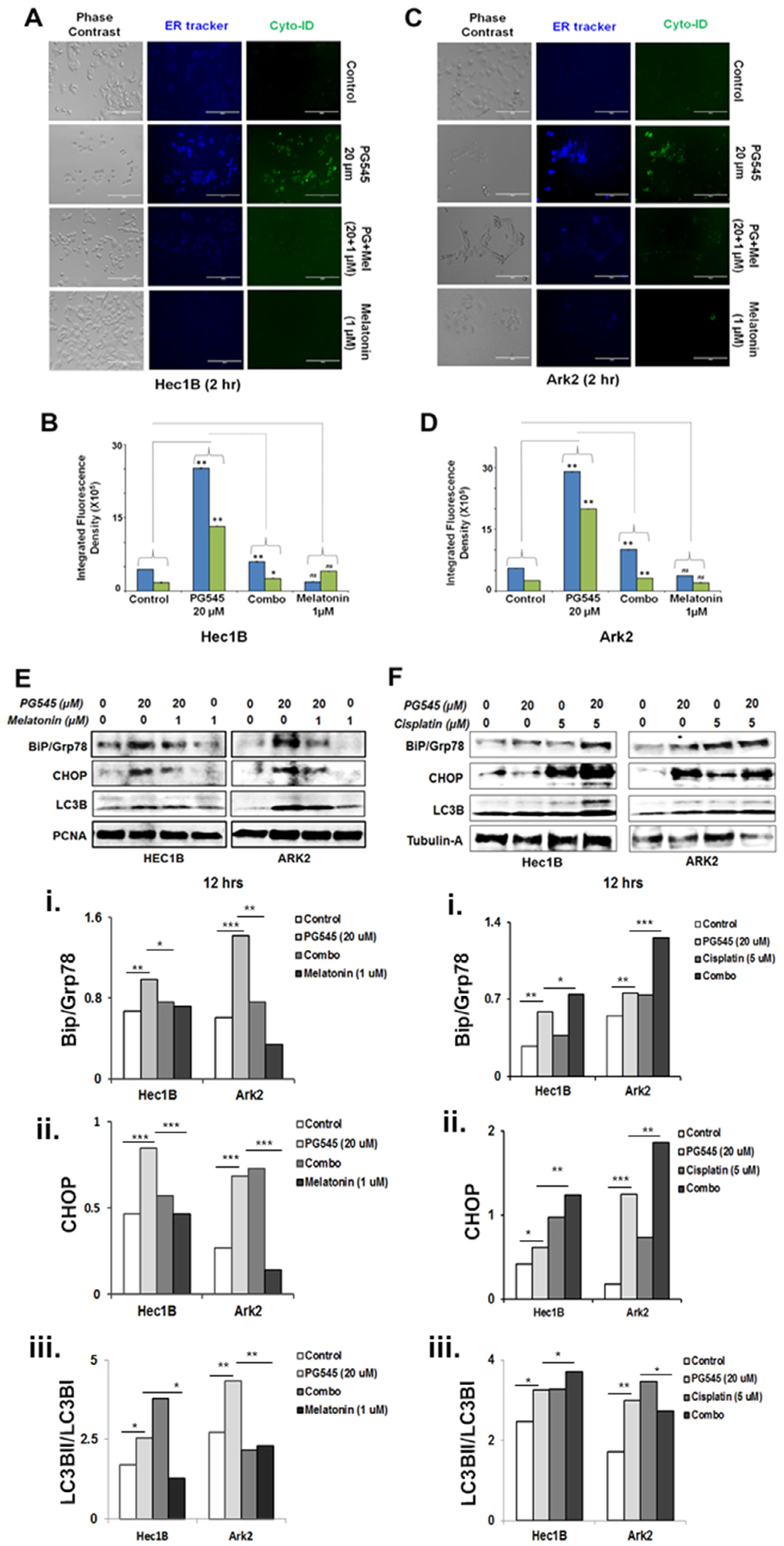
PG545 mediated ER stress triggers autophagy. PG545 (20 μM, 2 hrs) escalates ER activity (blue-white DPX staining) and autophagy (cyto-ID staining) which were subsequently inhibited by melatonin (1 μM, 30 mins) pre-incubation as measured in Hec1B and ARK2 cells (A and C). The integrated fluorescence density was analyzed using ImageJ software (B and D). P values: ns = not significant, * < 0.05, ** < 0.01. Western blot analysis in melatonin pretreated (1 μM) Hec1B and ARK2 cells demonstrated inhibition of Bip/Grp78, CHOP/GADD153 and LC3BII in figure E. PG545 treated (20 μM) EC cells in combination with cisplatin (5 μM) enhances ER stress as evident in western blot analysis using Bip/Grp78, CHOP/GADD153 and LC3BII (F).
3.9. PG545 in combination with cisplatin escalates ER stress in EC cell lines
We have so far established that PG545 synergizes with chemotherapeutic drugs, triggers ER stress and consequently promotes autophagy in EC cell lines. Therefore, we hypothesized that this compound may escalate ER stress and autophagy in combination with known chemotherapeutics. PG545 in combination with cisplatin treatment for 12 h demonstrated heightened ER stress and autophagy in EC cell lines. Both BiP and CHOP were upregulated along with LC3BII (Fig. 6F i–iii).
3.10. PG545 blocks growth factor-dependent signaling of EC cell lines
Similar to our previous reports in ovarian cancer, PG545 attenuated heparan sulfate-binding growth factor signaling both in Hec1B treated with FGF2 and ARK2 treated HB-EGF under serum-starved conditions (Fig. 7A and B). PG545 treatment resulted in decreased phosphorylation of ERK at Thr202/Tyr204 or of Akt at Ser473 after the addition of growth factors. Activation of ERK and Akt was observed after ten minutes of HB-EGF treatment in ARK2 cell lines, whereas full activation of these two proteins was noted after 1 h of FGF2 stimulation of Hec1B cells. Upon PG545 treatment activation of these pathways was significantly attenuated in both cell lines at all examined time points [Fig. 7A (i–ii) and 7B (i–ii)].
Fig. 7.
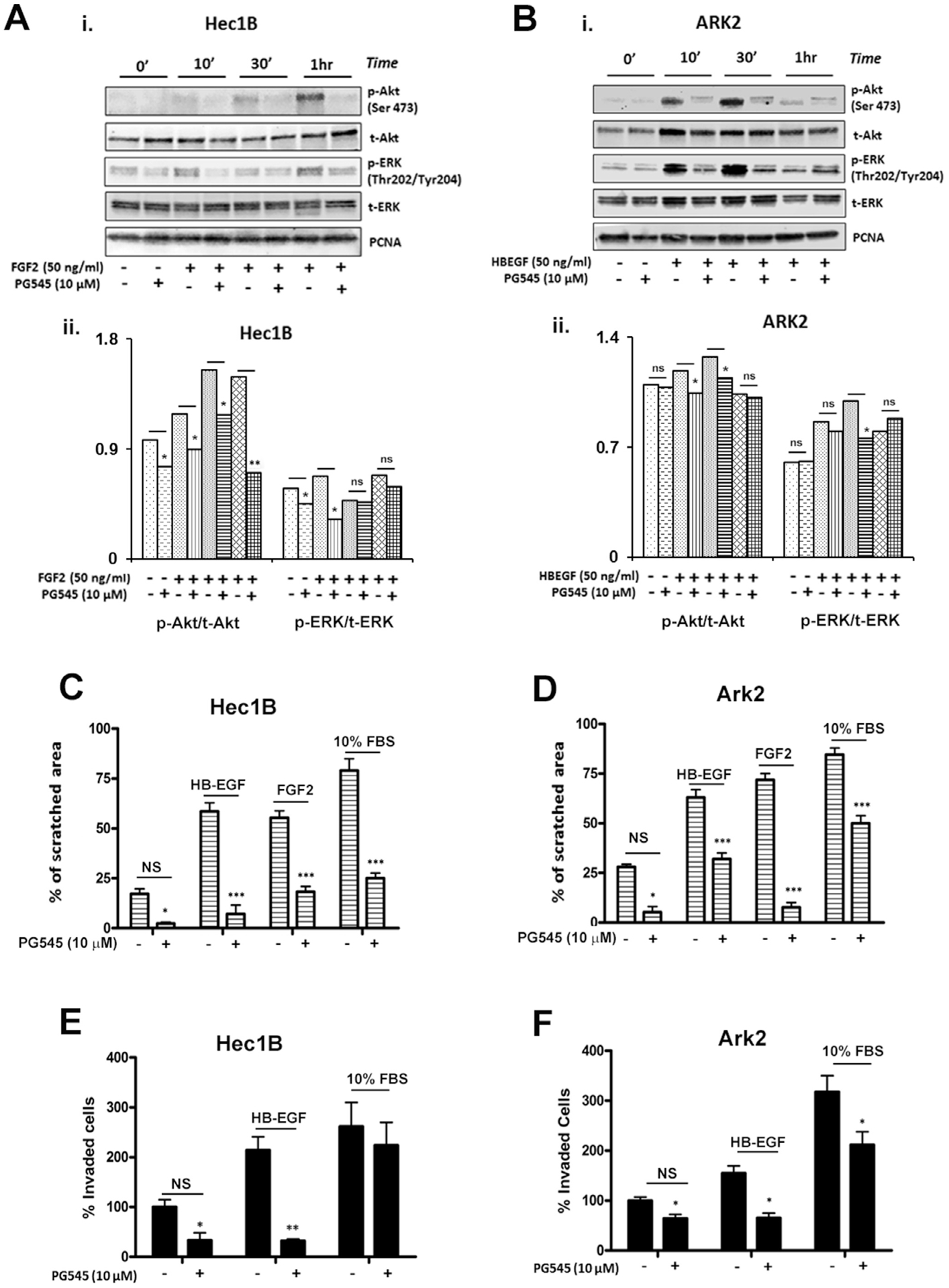
A and B: PG545 inhibits growth factor-mediated signaling. The cells were grown in a growth factor free medium for 12 h. 50 ng/ml of FGF2 (A i.) or HB-EGF (B i.) were then added in the presence and absence of PG545 to Hec1B and ARK2 cells respectively. Cells were harvested at specified time points. Western blot analysis showed reduced phosphorylation of ERK and AKT (A ii–B ii). PCNA, total ERK and total Akt are shown as loading controls. PG545 reduced migration, evaluated by wound scratch assay (C-D), and invasion (E-F) significantly reduced growth factor-induced cell motion in Hec1B and ARK2.
3.11. PG545 inhibits growth factor-stimulated migration and invasion in EC
Type I and Type II EC cell lines, Hec1A and ARK2, also demonstrated growth factor triggered migration and invasion. The effect of PG545 on migration was examined by a scratch wound cell migration assay. As shown in Fig. 7C and D, maximum inhibition of wound closure was observed in HB-EGF-stimulated Hec1B (~60%) and FGF2-stimulated ARK2 (~65%) after 10 μM PG545 treatment.
The effects of PG545 on cell invasion were assessed by an in vitro matrigel cell invasion assay. ARK2 cells were found to be highly invasive in comparison to Hec1B. A significant inhibition (~200% in Hec1B and ~ 75% in ARK2) of cell invasion was also observed after PG545 (10 μM) treatment (Fig. 7E and F).
4. Discussion
Considerable evidence associates increased heparanase activity with almost all aspects of tumor development such as tumor initiation, progression, metastasis, chemoresistance and relapse [53,54]. Furthermore, heparanasehigh cancer patients had a shorter life span in comparison to heparanaselow patients [54,55]. Together, these reports suggest that heparanase is generally involved in cancer progression and hence may be an effective target for anticancer drug development.
PG545 is a fully sulfated glycolipid immunomodulatory agent and inhibitor of heparanase and angiogenesis [27], which is undergoing early clinical investigation for the treatment of cancer. This is the first preclinical study to describe the effects of PG545 in endometrial cancer. Heparan mimetics have received increasing attention in recent years due to their therapeutic potential. PG545 displayed significant anticancer effects in pre-clinical models of a variety of cancers [29,31,56]. Given its multi-targeting approach of sequestering heparin-binding growth factors (HBGFs), blocking heparanase and blocking growth factor-mediated signaling, PG545 is a promising candidate for targeting a heterogeneous disease such as endometrial cancer. In our study, PG545 displayed strong anti-tumorigenic potential in both Types I and Type II EC and effectively blocked FGF2 and HB-EGF mediated signaling in lower concentrations and also significantly interfered with FGF2- and HB-EGF-induced migration and invasion of EC cells in vitro. PG545 also significantly reduced tumor growth in vivo in both cancer types. Important clinical markers, namely Ki67 and the iMVD (assessed with CD31 staining) were markedly reduced by PG545 treatment potentially inhibiting FGF2, VEGF or HB-EGF -mediated angiogenesis [11,57].
Another important aspect of the therapeutic potential of PG545 seems to be its effect to enhance the chemotherapeutic regimen. Similar to reports in other cancers [31,58] we found that PG545 synergizes with paclitaxel in vitro and with cisplatin both in vitro and in vivo. Notably, PG545 synergized with paclitaxel and cisplatin in Hec1B cells, commonly referred to as cisplatin-resistant type I cancer cells, and also in ARK2 cells, representative of the more aggressive and usually more chemoresistant type II cancer [59]. Besides its synergizing effects on cell viability and proliferation, we also showed that the addition of PG5455 significantly increases the apoptotic effects of cisplatin and paclitaxel in vitro.
Mechanistically, we have uncovered that autophagy is initiated in EC cells in response to PG545-induced ER stress. The cross-talk between drug-induced ER stress and the induction of autophagy to maintain ER homeostasis is well documented [48]. Exploring the mechanism behind PG545-induced autophagy seen in EC cells, we discovered that PG545 induces autophagy as a defense mechanism to clear unfolded proteins. This observation is consistent with the previously reported finding that inhibition of heparanase enzymatic activity can stimulate ER stress in cancer cells [60]. In this study, we have established that PG545 elicits ER stress as an early response with resultant induction of autophagy. Autophagy is now known as a two-edged sword. Apparently, autophagy is a cellular recycling process mainly leads to cell survival in nutrient-deficient condition [61]. However, in cancer cells, many variations of autophagy-dependent cell death are evident. A recent review clearly demonstrates the different modes of autophagy and apoptosis [62] including a coordinated action of both apoptosis and autophagy in parallel. In this context, we showed that PG545, as a potential heparinase inhibitor, triggers ER stress and subsequent autophagy as an adaptive response which terminally leads to apoptotic cell death. To prove it partially i.e. PG545 induced autophagy is ER stress-dependent, we introduced Melatonin, a pan ER stress inhibitor, which consequently reduced autophagy as evident in less LC3BII in Fig. 6E. Likewise, our observation, melatonin, an antioxidant and gross ER stress inhibitor, secreted from the pineal gland, has a suppressive function on autophagy [63,64]. A functional inhibitor and a close homolog of heparanase-1, heparanase-2 (Hpa2), can also promote tumor suppression via inhibition of heparanase enzymatic activity and stimulation of ER stress.
Our data also highlight that a likely consequence of excess ER stress is cell death since the combination of PG545 with cisplatin in EC cells induces significantly more ER stress in vitro compared to treatment of PG545 and/or cisplatin alone.
Substantial increase in autophagy as evident with concurrent upregulation of LC3BII and downregulation of p62 signifying that this intracellular mechanism, similar to ER stress, is also evoked after PG545 treatment in EC cells. While PG545 has been shown to inhibit autophagy in HeLa cells to inhibit growth [45], our data add to the more accepted notion that drug-induced autophagy is context- and more importantly cell type-dependent. Autophagy is supposed to aid cell survival during starvation, and usually, the basal level of autophagy is aberrantly high compared to normal cells [65]. However, unlike a brief and transient surge in ER stress and concomitant autophagy which exhibits cytoprotective role with potential survival benefits, a prolonged and profound ER stress-induced autophagy, beyond the basal threshold, can initiate apoptotic cell death. Therefore, inhibition of autophagy with CQ treatment also can overwhelm the cells with unprocessed protein burden which can trigger apoptosis (data not shown). In addition, we also observed PG545 triggers excessive ER stress in combination with known chemotherapeutics in EC.
In a previous study, we showed that PG545 interferes with Wnt/βcatenin pathway and attenuates tumor growth in pancreatic cancer [30]. This observation further supported that impaired mitochondrial ATP production, hypoxia or deubiquitinase inhibitor can downregulate Wnt signaling via the reduction of β-catenin through elevated ER stress [66–68]. In parallel, inhibition of FGF2 signaling can also drive the EC cells to the escalation of ER stress [69] which substantiates the dichotomy of the crosstalks between PG545-mediated inhibition of growth factor signaling and activation of ER stress with subsequent autophagy.
Nevertheless, there are a few limitations to this study. PG545 is effective in the EC xenograft model of type I and II EC, but the subcutaneous model we used in both cell lines is a monocellular model, only using one distinct cell line per xenograft. Therefore, it does not account for intratumoral heterogeneity, limiting its predictive value. PG545’s effects on autophagy in vivo also remain unknown, since reliable in vivo autophagy marker is yet to be established. Furthermore, PG545, which is currently in phase I clinical trials, has a variety of anti-cancer effects and recently also emerged as an immunomodulatory drug, showing potential in activating natural killer cells [26]. Our xenograft models were nude mice, not capable of mobilizing a T cell immune response. Also, autophagy itself is known to have multiple immunomodulatory effects in the microenvironment further complicating the picture [70]. It seems reasonable to explore the effects of PG545 patient-derived xenograft models in the future.
In summary, PG545 exhibited synergistic effects when combined with the chemotherapeutic agents, paclitaxel and cisplatin in vitro and in vivo through inhibition of growth factor-stimulated signaling and extended ER stress-mediated autophagy (Fig. 8). This is the first study to show PG545’s efficacy alone or in combination with chemotherapy in type I and type II endometrial cancer. These findings support the future exploration of PG545 alone or in combination with chemotherapy in phase I/II clinical trials.
Fig. 8.
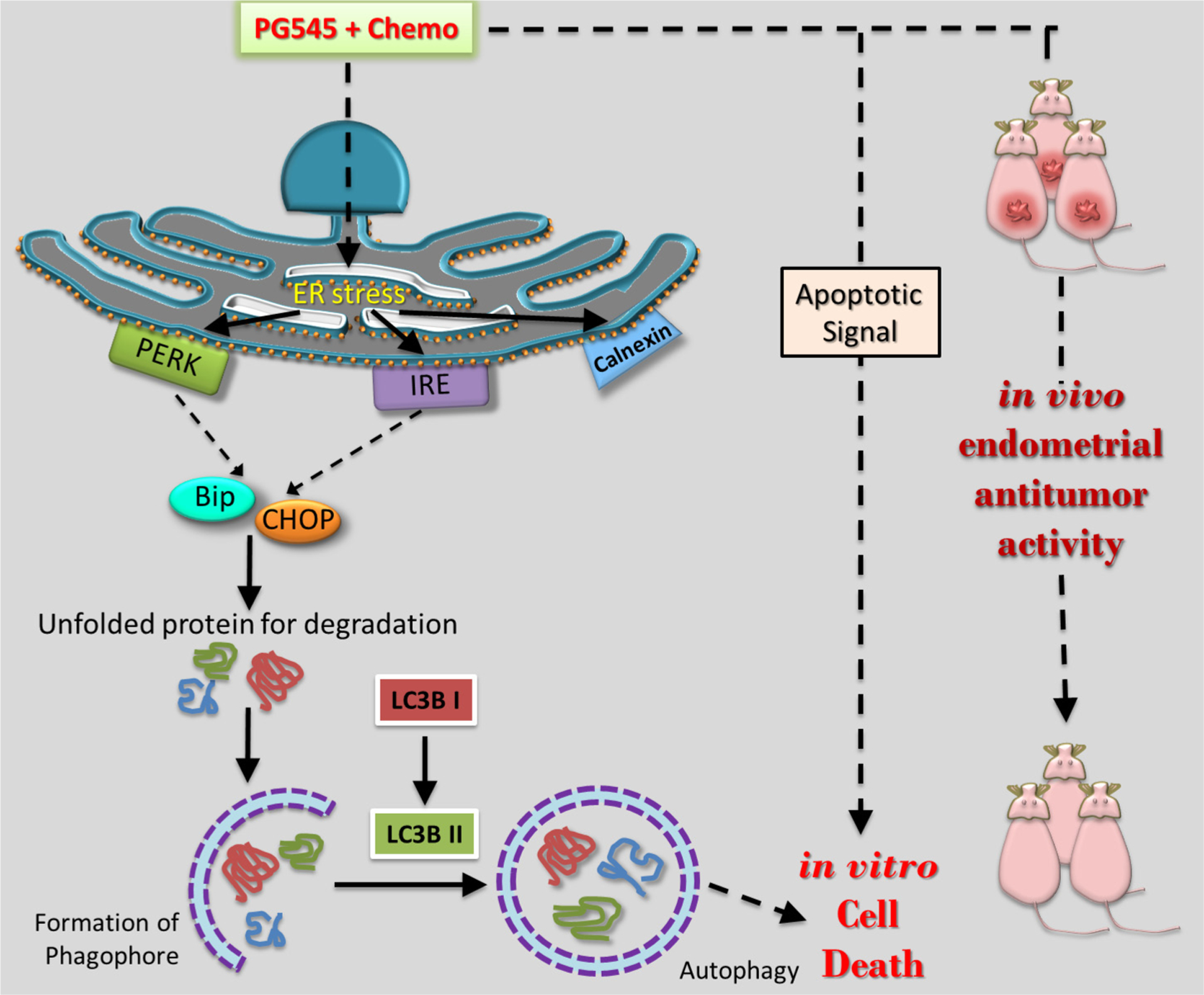
Schematic model of human endometrial cancer cell death mechanism activated by PG545 in combination with known chemotherapeutics.
Acknowledgments
We acknowledge Prof. Shi-Wen Jiang and Prof. Karl C. Podratz, (Mayo Clinic, Rochester, MN) for ARK2 cell line and Dr. Paul Goodfellow (Washington University, St. Louis, MO) for kindly providing us the Ishikawa and RL95 cell lines.
Grant support
This work was supported by the Department of Laboratory Medicine and Pathology, (VS), Mayo Clinic, Rochester.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2017, Cancer J. Clin 67 (1) (2017) 7–30. [DOI] [PubMed] [Google Scholar]
- [2].Henley SJ, Miller JW, Dowling NF, Benard VB, Richardson LC, Uterine cancer incidence and mortality – United States, 1999–2016, MMWR Morb. Mortal Wkly. Rep 67 (48) (2018) 1333–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Raglan O, Kalliala I, Markozannes G, Cividini S, Gunter MJ, Nautiyal J, Gabra H, Paraskevaidis E, Martin-Hirsch P, Tsilidis KK, Kyrgiou M, Risk factors for endometrial cancer: an umbrella review of the literature, Int. J. Cancer 145 (7) (2019) 1719–1730. [DOI] [PubMed] [Google Scholar]
- [4].Creasman WT, Odicino F, Maisonneuve P, Quinn MA, Beller U, Benedet JL, Heintz AP, Ngan HY, Pecorelli S, Carcinoma of the corpus uteri. FIGO 26th annual report on the results of treatment in gynecological cancer, Int. J. Gynaecol. Obstet 95 (Suppl. 1) (2006) S105–S143. [DOI] [PubMed] [Google Scholar]
- [5].Amant F, Moerman P, Neven P, Timmerman D, Van Limbergen E, Vergote I, Endometrial cancer, Lancet 366 (9484) (2005) 491–505. [DOI] [PubMed] [Google Scholar]
- [6].Dellinger TH, Monk BJ, Systemic therapy for recurrent endometrial cancer: a review of North American trials, Expert Rev. Anticancer Ther 9 (7) (2009) 905–916. [DOI] [PubMed] [Google Scholar]
- [7].Fleming GF, Brunetto VL, Cella D, Look KY, Reid GC, Munkarah AR, Kline R, Burger RA, Goodman A, Burks RT, Phase III trial of doxorubicin plus cisplatin with or without paclitaxel plus filgrastim in advanced endometrial carcinoma: a Gynecologic Oncology Group Study, J. Clin. Oncol. J. Am. Soc. Clin. Oncol 22 (11) (2004) 2159–2166. [DOI] [PubMed] [Google Scholar]
- [8].McAlpine J, Leon-Castillo A, Bosse T, The rise of a novel classification system for endometrial carcinoma; integration of molecular subclasses, J. Pathol 244 (5) (2018) 538–549. [DOI] [PubMed] [Google Scholar]
- [9].Bokhman JV, Two pathogenetic types of endometrial carcinoma, Gynecol. Oncol 15 (1) (1983) 10–17. [DOI] [PubMed] [Google Scholar]
- [10].Hecht JL, Mutter GL, Molecular and pathologic aspects of endometrial carcinogenesis, J. Clin. Oncol. J. Am. Soc. Clin. Oncol 24 (29) (2006) 4783–4791. [DOI] [PubMed] [Google Scholar]
- [11].Bansal N, Yendluri V, Wenham RM, The molecular biology of endometrial cancers and the implications for pathogenesis, classification, and targeted therapies, Cancer Control J. Moffitt Cancer Center 16 (1) (2009) 8–13. [DOI] [PubMed] [Google Scholar]
- [12].Kamat AA, Merritt WM, Coffey D, Lin YG, Patel PR, Broaddus R, Nugent E, Han LY, Landen CN Jr., Spannuth WA, Lu C, Coleman RL, Gershenson DM, Sood AK, Clinical and biological significance of vascular endothelial growth factor in endometrial cancer, Clin. Cancer Res 13 (24) (2007) 7487–7495. [DOI] [PubMed] [Google Scholar]
- [13].Dobrzycka B, Mackowiak-Matejczyk B, Kinalski M, Terlikowski SJ, Pretreatment serum levels of bFGF and VEGF and its clinical significance in endometrial carcinoma, Gynecol. Oncol 128 (3) (2013) 454–460. [DOI] [PubMed] [Google Scholar]
- [14].Felix AS, Edwards RP, Stone RA, Chivukula M, Parwani AV, Bowser R, Linkov F, Weissfeld JL, Associations between hepatocyte growth factor, c-Met, and basic fibroblast growth factor and survival in endometrial cancer patients, Br. J. Cancer 106 (12) (2012) 2004–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Scambia G, Benedetti Panici P, Ferrandina G, Battaglia F, Distefano M, D’Andrea G, De Vincenzo R, Maneschi F, Ranelletti FO, Mancuso S, Significance of epidermal growth factor receptor expression in primary human endometrial cancer, Int. J. Cancer J. Int. Cancer 56 (1) (1994) 26–30. [DOI] [PubMed] [Google Scholar]
- [16].Nogami Y, Banno K, Kisu I, Yanokura M, Umene K, Masuda K, Kobayashi Y, Yamagami W, Nomura H, Tominaga E, Susumu N, Aoki D, Current status of molecular-targeted drugs for endometrial cancer (Review), Mol. Clin. Oncol 1 (5) (2013) 799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Papa A, Zaccarelli E, Caruso D, Vici P, Benedetti Panici P, Tomao F, Targeting angiogenesis in endometrial cancer – new agents for tailored treatments, Expert Opin. Investig. Drugs 25 (1) (2016) 31–49. [DOI] [PubMed] [Google Scholar]
- [18].Sasisekharan R, Shriver Z, Venkataraman G, Narayanasami U, Roles of heparin-sulphate glycosaminoglycans in cancer, Nat. Rev. Cancer 2 (7) (2002) 521–528. [DOI] [PubMed] [Google Scholar]
- [19].Sanderson RD, Heparan sulfate proteoglycans in invasion and metastasis, Sem. Cell Dev. Biol 12 (2) (2001) 89–98. [DOI] [PubMed] [Google Scholar]
- [20].Canaani J, Ilan N, Back S, Gutman G, Vlodavsky I, Grisaru D, Heparanase expression increases throughout the endometrial hyperplasia-cancer sequence, Int. J. Gynaecol. Obstetr. Organ Int. Federation Gynaecol. Obstetr 101 (2) (2008) 166–171. [DOI] [PubMed] [Google Scholar]
- [21].Vlodavsky I, Friedmann Y, Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis, J. Clin. Invest 108 (3) (2001) 341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hasengaowa, Kodama J, Kusumoto T, Seki N, Matsuo T, Ojima Y, Nakamura K, Hongo A, Hiramatsu Y, Heparanase expression in both normal endometrium and endometrial cancer, Int. J. Gynecol. Cancer 16 (3) (2006) 1401–1406. [DOI] [PubMed] [Google Scholar]
- [23].Inamine M, Nagai Y, Hirakawa M, Mekaru K, Yagi C, Masamoto H, Aoki Y, Heparanase expression in endometrial cancer: analysis of immunohistochemistry, J. Obstetr. Gynaecol. J. Inst. Obstetr. Gynaecol 28 (6) (2008) 634–637. [DOI] [PubMed] [Google Scholar]
- [24].Oh JH, Lee HS, Park SH, Ryu HS, Min CK, Syndecan-1 overexpression promotes tumor growth and angiogenesis in an endometrial cancer xenograft model, Int. J. Gynecol. Cancer 20 (5) (2010) 751–756. [DOI] [PubMed] [Google Scholar]
- [25].Ferro V, Liu L, Johnstone KD, Wimmer N, Karoli T, Handley P, Rowley J, Dredge K, Li CP, Hammond E, Davis K, Sarimaa L, Harenberg J, Bytheway I, Discovery of PG545: a highly potent and simultaneous inhibitor of angiogenesis, tumor growth, and metastasis, J. Med. Chem 55 (8) (2012) 3804–3813. [DOI] [PubMed] [Google Scholar]
- [26].Brennan TV, Lin L, Brandstadter JD, Rendell VR, Dredge K, Huang X, Yang Y, Heparan sulfate mimetic PG545-mediated antilymphoma effects require TLR9-dependent NK cell activation, J. Clin. Invest 126 (1) (2016) 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dredge K, Hammond E, Davis K, Li CP, Liu L, Johnstone K, Handley P, Wimmer N, Gonda TJ, Gautam A, Ferro V, Bytheway I, The PG500 series: novel heparan sulfate mimetics as potent angiogenesis and heparanase inhibitors for cancer therapy, Investig. New Drugs 28 (3) (2010) 276–283. [DOI] [PubMed] [Google Scholar]
- [28].Dredge K, Hammond E, Handley P, Gonda TJ, Smith MT, Vincent C, Brandt R, Ferro V, Bytheway I, PG545, a dual heparanase and angiogenesis inhibitor, induces potent anti-tumour and anti-metastatic efficacy in preclinical models, Br. J. Cancer 104 (4) (2011) 635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ostapoff KT, Awasthi N, Cenik BK, Hinz S, Dredge K, Schwarz RE, Brekken RA, PG545, an angiogenesis and heparanase inhibitor, reduces primary tumor growth and metastasis in experimental pancreatic cancer, Mol. Cancer Ther 12 (7) (2013) 1190–1201. [DOI] [PubMed] [Google Scholar]
- [30].Jung DB, Yun M, Kim EO, Kim J, Kim B, Jung JH, Wang E, Mukhopadhyay D, Hammond E, Dredge K, Shridhar V, Kim SH, The heparan sulfate mimetic PG545 interferes with Wnt/beta-catenin signaling and significantly suppresses pancreatic tumorigenesis alone and in combination with gemcitabine, Oncotarget 6 (7) (2015) 4992–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Winterhoff B, Freyer L, Hammond E, Giri S, Mondal S, Roy D, Teoman A, Mullany SA, Hoffmann R, von Bismarck A, Chien J, Block MS, Millward M, Bampton D, Dredge K, Shridhar V, PG545 enhances anti-cancer activity of chemotherapy in ovarian models and increases surrogate biomarkers such as VEGF in preclinical and clinical plasma samples, Eur. J. Cancer 51 (7) (2015) 879–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mullany SA, Moslemi-Kebria M, Rattan R, Khurana A, Clayton A, Ota T, Mariani A, Podratz KC, Chien J, Shridhar V, Expression and functional significance of HtrA1 loss in endometrial cancer, Clin. Cancer Res 17 (3) (2011) 427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chien J, Aletti G, Baldi A, Catalano V, Muretto P, Keeney GL, Kalli KR, Staub J, Ehrmann M, Cliby WA, Lee YK, Bible KC, Hartmann LC, Kaufmann SH, Shridhar V, Serine protease HtrA1 modulates chemotherapy-induced cytotoxicity, J. Clin. Investig 116 (7) (2006) 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chou TC, Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies, Pharmacol. Rev 58 (3) (2006) 621–681. [DOI] [PubMed] [Google Scholar]
- [35].Chou TC, Drug combination studies and their synergy quantification using the Chou-Talalay method, Cancer Res. 70 (2) (2010) 440–446. [DOI] [PubMed] [Google Scholar]
- [36].Khurana A, Liu P, Mellone P, Lorenzon L, Vincenzi B, Datta K, Yang B, Linhardt RJ, Lingle W, Chien J, Baldi A, Shridhar V, HSulf-1 modulates FGF2- and hypoxia-mediated migration and invasion of breast cancer cells, Cancer Res. 71 (6) (2011) 2152–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kalogera E, Roy D, Khurana A, Mondal S, Weaver AL, He X, Dowdy SC, Shridhar V, Quinacrine in endometrial cancer: repurposing an old antimalarial drug, Gynecol. Oncol 146 (1) (2017) 187–195. [DOI] [PubMed] [Google Scholar]
- [38].Sarkar Bhattacharya S, Mandal C, Albiez RS, Samanta SK, Mandal C, Mahanine drives pancreatic adenocarcinoma cells into endoplasmic reticular stress-mediated apoptosis through modulating sialylation process and Ca(2+)-signaling, Sci. Rep 8 (1) (2018) 3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sarkar Bhattacharya S, Thirusangu P, Jin L, Roy D, Jung D, Xiao Y, Staub J, Roy B, Molina JR, Shridhar V, PFKFB3 inhibition reprograms malignant pleural mesothelioma to nutrient stress-induced macropinocytosis and ER stress as independent binary adaptive responses, Cell Death Dis. 10 (10) (2019) 725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Al-Najar A, Al-Sanabani S, Korda JB, Hegele A, Bolenz C, Herbst H, Jonemann KP, Naumann CM, Microvessel density as a prognostic factor in penile squamous cell carcinoma, Urol. Oncol 30 (3) (2012) 325–329. [DOI] [PubMed] [Google Scholar]
- [42].Chan LL, Shen D, Wilkinson AR, Patton W, Lai N, Chan E, Kuksin D, Lin B, Qiu J, A novel image-based cytometry method for autophagy detection in living cells, Autophagy 8 (9) (2012) 1371–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Farkas T, Hoyer-Hansen M, Jaattela M, Identification of novel autophagy regulators by a luciferase-based assay for the kinetics of autophagic flux, Autophagy 5 (7) (2009) 1018–1025. [DOI] [PubMed] [Google Scholar]
- [44].Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin LS, Choi A, Chu CT, Chung J, Clarke PG, Clark RS, Clarke SG, Clave C, Cleveland JL, Codogno P, Colombo MI, Coto-Montes A, Cregg JM, Cuervo AM, Debnath J, Demarchi F, Dennis PB, Dennis PA, Deretic V, Devenish RJ, Di Sano F, Dice JF, Difiglia M, Dinesh-Kumar S, Distelhorst CW, Djavaheri-Mergny M, Dorsey FC, Droge W, Dron M, Dunn WA Jr., Duszenko M, Eissa NT, Elazar Z, Esclatine A, Eskelinen EL, Fesus L, Finley KD, Fuentes JM, Fueyo J, Fujisaki K, Galliot B, Gao FB, Gewirtz DA, Gibson SB, Gohla A, Goldberg AL, Gonzalez R, Gonzalez-Estevez C, Gorski S, Gottlieb RA, Haussinger D, He YW, Heidenreich K, Hill JA, Hoyer-Hansen M, Hu X, Huang WP, Iwasaki A, Jaattela M, Jackson WT, Jiang X, Jin S, Johansen T, Jung JU, Kadowaki M, Kang C, Kelekar A, Kessel DH, Kiel JA, Kim HP, Kimchi A, Kinsella TJ, Kiselyov K, Kitamoto K, Knecht E, Komatsu M, Kominami E, Kondo S, Kovacs AL, Kroemer G, Kuan CY, Kumar R, Kundu M, Landry J, Laporte M, Le W, Lei HY, Lenardo MJ, Levine B, Lieberman A, Lim KL, Lin FC, Liou W, Liu LF, Lopez-Berestein G, Lopez-Otin C, Lu B, Macleod KF, Malorni W, Martinet W, Matsuoka K, Mautner J, Meijer AJ, Melendez A, Michels P, Miotto G, Mistiaen WP, Mizushima N, Mograbi B, Monastyrska I, Moore MN, Moreira PI, Moriyasu Y, Motyl T, Munz C, Murphy LO, Naqvi NI, Neufeld TP, Nishino I, Nixon RA, Noda T, Nurnberg B, Ogawa M, Oleinick NL, Olsen LJ, Ozpolat B, Paglin S, Palmer GE, Papassideri I, Parkes M, Perlmutter DH, Perry G, Piacentini M, Pinkas-Kramarski R, Prescott M, Proikas-Cezanne T, Raben N, Rami A, Reggiori F, Rohrer B, Rubinsztein DC, Ryan KM, Sadoshima J, Sakagami H, Sakai Y, Sandri M, Sasakawa C, Sass M, Schneider C, Seglen PO, Seleverstov O, Settleman J, Shacka JJ, Shapiro IM, Sibirny A, Silva-Zacarin EC, Simon HU, Simone C, Simonsen A, Smith MA, Spanel-Borowski K, Srinivas V, Steeves M, Stenmark H, Stromhaug PE, Subauste CS, Sugimoto S, Sulzer D, Suzuki T, Swanson MS, Tabas I, Takeshita F, Talbot NJ, Talloczy Z, Tanaka K, Tanida I, Taylor GS, Taylor JP, Terman A, Tettamanti G, Thompson CB, Thumm M, Tolkovsky AM, Tooze SA, Truant R, Tumanovska LV, Uchiyama Y, Ueno T, Uzcategui NL, van der Klei I, Vaquero EC, Vellai T, Vogel MW, Wang HG, Webster P, Wiley JW, Xi Z, Xiao G, Yahalom J, Yang JM, Yap G, Yin XM, Yoshimori T, Yu L, Yue Z, Yuzaki M, Zabirnyk O, Zheng X, Zhu X, Deter RL, Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes, Autophagy 4 (2) (2008) 151–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shteingauz A, Boyango I, Naroditsky I, Hammond E, Gruber M, Doweck I, Ilan N, Vlodavsky I, Heparanase enhances tumor growth and chemoresistance by promoting autophagy, Cancer Res. 75 (18) (2015) 3946–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yan MM, Ni JD, Song D, Ding M, Huang J, Interplay between unfolded protein response and autophagy promotes tumor drug resistance, Oncol. Lett 10 (4) (2015) 1959–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Senft D, Ronai ZA, UPR, autophagy, and mitochondria crosstalk underlies the ER stress response, Trends Biochem. Sci 40 (3) (2015) 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Rashid HO, Yadav RK, Kim HR, Chae HJ, ER stress: autophagy induction, inhibition and selection, Autophagy 11 (11) (2015) 1956–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K, Autophagy is activated for cell survival after endoplasmic reticulum stress, Mol. Cell. Biol 26 (24) (2006) 9220–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yorimitsu T, Nair U, Yang Z, Klionsky DJ, Endoplasmic reticulum stress triggers autophagy, J. Biol. Chem 281 (40) (2006) 30299–30304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Oyadomari S, Mori M, Roles of CHOP/GADD153 in endoplasmic reticulum stress, Cell. Death Differ 11 (4) (2004) 381–389. [DOI] [PubMed] [Google Scholar]
- [52].Guerin R, Arseneault G, Dumont S, Rokeach LA, Calnexin is involved in apoptosis induced by endoplasmic reticulum stress in the fission yeast, Mol. Biol. Cell 19 (10) (2008) 4404–4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Vlodavsky I, Singh P, Boyango I, Gutter-Kapon L, Elkin M, Sanderson RD, Ilan N, Heparanase: From basic research to therapeutic applications in cancer and inflammation, Drug Resist. Updat 29 (2016) 54–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ilan N, Elkin M, Vlodavsky I, Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis, Int. J. Biochem. Cell Biol 38 (12) (2006) 2018–2039. [DOI] [PubMed] [Google Scholar]
- [55].Vlodavsky I, Beckhove P, Lerner I, Pisano C, Meirovitz A, Ilan N, Elkin M, Significance of heparanase in cancer and inflammation, Cancer Microenviron. 5 (2) (2012) 115–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cassinelli G, Favini E, Dal Bo L, Tortoreto M, De Maglie M, Dagrada G, Pilotti S, Zunino F, Zaffaroni N, Lanzi C, Antitumor efficacy of the heparan sulfate mimic roneparstat (SST0001) against sarcoma models involves multi-target inhibition of receptor tyrosine kinases, Oncotarget 7 (30) (2016) 47848–47863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Byron SA, Pollock PM, FGFR2 as a molecular target in endometrial cancer, Fut. Oncol 5 (1) (2009) 27–32. [DOI] [PubMed] [Google Scholar]
- [58].Fukuda T, Oda K, Wada-Hiraike O, Sone K, Inaba K, Ikeda Y, Miyasaka A, Kashiyama T, Tanikawa M, Arimoto T, Kuramoto H, Yano T, Kawana K, Osuga Y, Fujii T, The anti-malarial chloroquine suppresses proliferation and overcomes cisplatin resistance of endometrial cancer cells via autophagy inhibition, Gynecol. Oncol 137 (3) (2015) 538–545. [DOI] [PubMed] [Google Scholar]
- [59].Van Nyen T, Moiola CP, Colas E, Annibali D, Amant F, Modeling Endometrial cancer: past, present, and future, Int. J. Mol. Sci 19 (8) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Vlodavsky I, Gross-Cohen M, Weissmann M, Ilan N, Sanderson RD, Opposing functions of heparanase-1 and heparanase-2 in cancer progression, Trends Biochem. Sci 43 (1) (2018) 18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Codogno P, Meijer AJ, Autophagy and signaling: their role in cell survival and cell death, Cell Death Differ. 12 (Suppl. 2) (2005) 1509–1518. [DOI] [PubMed] [Google Scholar]
- [62].Denton D, Kumar S, Autophagy-dependent cell death, Cell Death Differ. 26 (4) (2019) 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kim CH, Kim KH, Yoo YM, Melatonin protects against apoptotic and autophagic cell death in C2C12 murine myoblast cells, J. Pineal Res 50 (3) (2011) 241–249. [DOI] [PubMed] [Google Scholar]
- [64].Chang CF, Huang HJ, Lee HC, Hung KC, Wu RT, Lin AM, Melatonin attenuates kainic acid-induced neurotoxicity in mouse hippocampus via inhibition of autophagy and alpha-synuclein aggregation, J. Pineal Res 52 (3) (2012) 312–321. [DOI] [PubMed] [Google Scholar]
- [65].Wang M, Law ME, Castellano RK, Law BK, The unfolded protein response as a target for anticancer therapeutics, Crit. Rev. Oncol. Hematol 127 (2018) 66–79. [DOI] [PubMed] [Google Scholar]
- [66].Xia Z, Wu S, Wei X, Liao Y, Yi P, Liu Y, Liu J, Liu J, Hypoxic ER stress suppresses beta-catenin expression and promotes cooperation between the transcription factors XBP1 and HIF1alpha for cell survival, J. Biol. Chem 294 (37) (2019) 13811–13821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Costa R, Peruzzo R, Bachmann M, Monta GD, Vicario M, Santinon G, Mattarei A, Moro E, Quintana-Cabrera R, Scorrano L, Zeviani M, Vallese F, Zoratti M, Paradisi C, Argenton F, Brini M, Cali T, Dupont S, Szabo I, Leanza L, Impaired mitochondrial ATP production downregulates Wnt signaling via ER stress induction, Cell Rep. 28 (8) (2019) 1949–1960 e6. [DOI] [PubMed] [Google Scholar]
- [68].Ding Y, Chen X, Wang B, Yu B, Ge J, Deubiquitinase inhibitor b-AP15 activates endoplasmic reticulum (ER) stress and inhibits Wnt/Notch1 signaling pathway leading to the reduction of cell survival in hepatocellular carcinoma cells, Eur. J. Pharmacol 825 (2018) 10–18. [DOI] [PubMed] [Google Scholar]
- [69].Li B, Pi Z, Liu L, Zhang B, Huang X, Hu P, Chevet E, Yi P, Liu J, FGF-2 prevents cancer cells from ER stress-mediated apoptosis via enhancing proteasome-mediated Nck degradation, Biochem. J 452 (1) (2013) 139–145. [DOI] [PubMed] [Google Scholar]
- [70].Puleston DJ, Simon AK, Autophagy in the immune system, Immunology 141 (1) (2014) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]