

SUMMARY
Prdm12 is a key transcription factor in nociceptor neurogenesis. Mutations of Prdm12 cause congenital insensitivity to pain (CIP) from failure of nociceptor development. However, precisely how deletion of Prdm12 during development or adulthood affects nociception is unknown. Here, we employ tissue- and temporal-specific knockout mouse models to test the function of Prdm12 during development and in adulthood. We find that constitutive loss of Prdm12 causes deficiencies in proliferation during sensory neurogenesis. We also demonstrate that conditional knockout from dorsal root ganglia (DRGs) during embryogenesis causes defects in nociception. In contrast, we find that, in adult DRGs, Prdm12 is dispensable for most pain-sensation and injury-induced hypersensitivity. Using transcriptomic analysis, we find mostly unique changes in adult Prdm12 knockout DRGs compared with embryonic knockout and that PRDM12 is likely a transcriptional activator in the adult. Overall, we find that the function of PRDM12 changes over developmental time.
In brief
Landy et al. report that Prdm12 is necessary for nociceptor development. Embryonic sensory neuron-specific conditional knockout causes defects in nociception and pruriception, but adult conditional knockout has a limited effect on nociception. Transcriptional changes after adult knockout are distinctly different from those seen after embryonic loss of the gene.
Graphical Abstract
INTRODUCTION
Nociception is a critical warning system for the detection of tissue damage by noxious stimuli, but it can often go awry, resulting in either greatly increased or decreased pain sensation. Patients with congenital insensitivity to pain (CIP) are a prime example of the latter. These people cannot feel mechanical, thermal, or inflammatory pain or even discomfort associated with internal injuries. Cases of CIP have typically been associated with mutations of nerve growth factor (NGF) or its receptor Ntrk1 (TRKA), which cause failure of nociceptor development (Capsoni et al., 2011; Carvalho et al., 2011; Einarsdottir et al., 2004) or the voltage-gated sodium channels, Scn9a (Nav1.7) or Scn11a (Nav1.9) (Cox et al., 2006; Leipold et al., 2013). Recently, additional mutations have been identified that cause CIP (Imhof et al., 2020; Nahorski et al., 2015), including those of the transcription factor PRDM12 (PRDI-BF1-RIZ homologous domain-containing family) (Chen et al., 2015). As in other forms of CIP, patients with Prdm12-associated CIP are unable to feel pain from noxious chemical, thermal, or mechanical stimuli but retain normal touch, proprioception, and tickle sensations (Chen et al., 2015; Saini et al., 2017; Zhang et al., 2016). Therefore, similar to how drugs have been developed that target NGF and Nav1.7, Prdm12 or its downstream effectors may serve as potential novel analgesic targets (Hoffman et al., 2011).
The PRDM family of transcription factors are known to have essential roles in cell-fate transitions (Hohenauer and Moore, 2012), and many lines of evidence suggest that PRDM12 has an essential role in nociceptor development and maintenance. It is highly conserved from mouse to human, with 98% protein identity, suggesting a highly conserved function, opening the gateway for study in mouse models. Indeed, consistent with the idea of having a highly conserved function, loss of PRDM12 function in humans or its homologs in Drosophila, frog embryos, and mice leads to abnormalities in sensory neuron development (Bartesaghi et al., 2019; Chen et al., 2015; Desiderio et al., 2019; Moore et al., 2004; Nagy et al., 2015). Additionally, Prdm12 is specifically expressed in myelinated Aδ- and unmyelinated C-fiber nociceptors into adulthood (Chen et al., 2015; Kinameri et al., 2008; Matsukawa et al., 2015; Nagy et al., 2015; Sharma et al., 2020; Thélie et al., 2015; Usoskin et al., 2015). The highly conserved sequence, function, and expression pattern raise the possibility that PRDM12 is serving an important role in nociceptor biogenesis.
Structurally, PRDM12 consists of a PR domain, three zinc finger domains, and a polyalanine repeat. The PR domain is characteristic of all members of the PRDM family of proteins and bears weak homology to SET domains, which have histone methyltransferase (HMT) activity (Kinameri et al., 2008). However, PRDM12 itself is reported to have weak endogenous HMT activity and is thought to exert repressive activity predominantly through interaction with EHMT2 (euchromatic histone-lysine N-methyltransferase, also called G9a), which catalyzes repressive chromatin marks (Yang and Shinkai, 2013). This interaction was shown to be dependent on the second zinc finger domain (ZnF2), contained in exon V (Yang and Shinkai, 2013).
Thus far, most of the work surrounding Prdm12 has focused on its role in nociceptor neurogenesis. Early reports indicated that PRDM12 promoted expression of sensory neuronal markers (Kinameri et al., 2008; Matsukawa et al., 2015; Thélie et al., 2015; Yang and Shinkai, 2013). Consistent with this, Prdm12 was found to be necessary for the initiation and maintenance of tropomyosin receptor kinase A (TRKA) expression, a marker for early nociceptor development (Desiderio et al., 2019). In addition, in the absence of Prdm12, the entire nociceptor lineage failed to develop. However, the mechanism by which Prdm12 knockout (KO) leads to a deficiency in nociceptors is unclear. Work by Bartesaghi et al. (2019) found evidence for decreased proliferation in Prdm12 KO mice with no change in cell death, whereas work from Desiderio et al. (2019) found the reverse, that there was no change in proliferation, but there was an increase in cell death. Differences in the way proliferation and cell death were quantitated in these studies could account for these discrepancies.
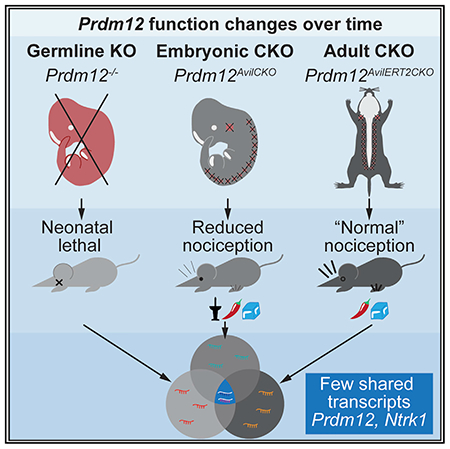
Therefore, in our study, we sought to further clarify the mechanism by which Prdm12 controls nociceptor development. Furthermore, we wanted to examine the behavioral defects in mice that lack Prdm12 during embryogenesis and determine whether it is an important component of pain sensation in mature sensory neurons. To do that, we generated three mouse models with which to study the effect of Prdm12 KO at different times: (1) Prdm12−/−, a constitutive KO to assess the early embryonic changes resulting from Prdm12 deletion; (2) Prdm12AvilCKO, a dorsal root ganglion (DRG)-specific conditional KO to assess pain sensation in mice lacking Prdm12 from around embryonic day 12.5 (E12.5) onward; and (3) Prdm12AvilERT2CKO, a tamoxifen-inducible DRG-specific conditional KO to investigate the role of Prdm12 in adult nociceptors. With these models, we confirm that Prdm12 expression is necessary for the development of nociceptors and show that its absence results in defects in proliferation during neurogenesis. Furthermore, we demonstrate that embryonic sensory neuron-specific KO of the gene results in mice with defects in mechanical, cold, chemical, and high-heat nociception as well as itch. Finally, we show that KO of Prdm12 in mature DRGs has very little effect on nociception, even under conditions of neuropathic injury or inflammation. However, we provide transcriptomic evidence for an alternate function of Prdm12 in these neurons compared with embryonic development and suggesting that it is an activating transcription factor in the adult, rather than a repressor.
RESULTS
Prdm12 exon V constitutive KO mice show selective loss of the developing nociceptor population
To study the role of Prdm12 during embryogenesis, we first generated homozygous null mice in which exon V was deleted from conception. This was accomplished by crossing mice expressing an exon V floxed Prdm12 allele (Figures 1A and 1B) (Chen et al., 2020) to germline CAG-Cre mice (Sakai and Miyazaki, 1997), generating heterozygous Prdm12+/− mice, which were then crossed to each other, producing Prdm12−/− homozygotes. Using in situ hybridization with a probe specific for exon V, we confirmed that expression of this sequence was eliminated from mutant DRGs at E11.5 (Figure 1C). Prdm12−/− embryos appeared grossly normal during development and were observed to move and breathe normally immediately after cesarean section at E18.5, but newborn pups die within hours of birth. On closer examination, lumbar DRGs from Prdm12−/− embryos were found to be smaller than those of control littermates (Figure 1D). The relative size of mutant DRGs to control DRGs shrank from ~68% at E11.5 to ~43% at E18.5 (Figure 1D).
Figure 1. DRGs from Prdm12−/− embryos are smaller and lack nociceptors.
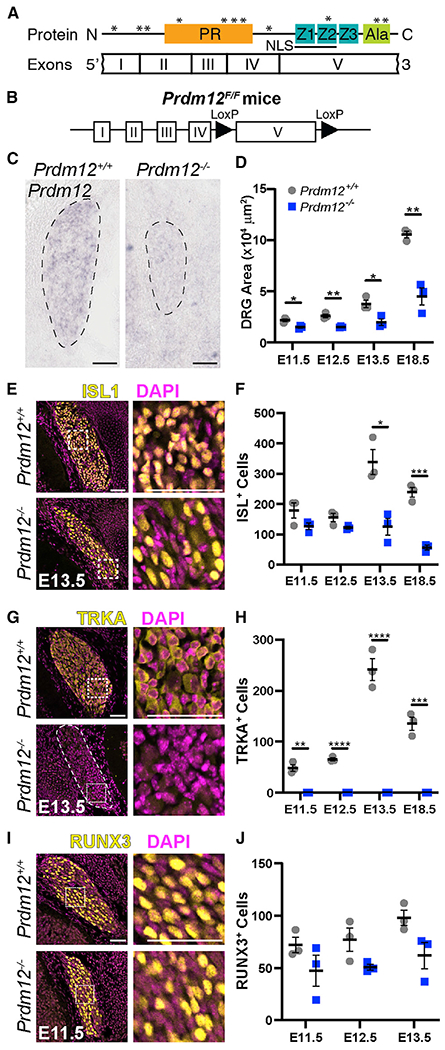
(A) PRDM12 protein domain structure with corresponding exons. *Human disease-causing mutations.
(B) Schematic of the Prdm12F/F allele.
(C) In situ hybridization with an exon-V-specific probe verified deletion of this transcript in Prdm12−/− embryos. Scale bar, 50 μm.
(D) Quantification of DRG area from immunofluorescence images reveals Prdm12−/− DRGs are smaller at all time points (E11.5, p = 0.017; E12.5, p = 0.003; E13.5, p = 0.023; E18.5, p = 0.003).
(E) Representative images of ISL1 immunohistochemistry, with inset shown on right. Scale bars, 50 μm.
(F) Quantification reveals a similar number of ISL1+ cells at E11.5 and E12.5 in control and KO tissue, but a significant reduction in counts at E13.5 (p = 0.012) and E18.5 (p = 0.0005) in KO embryos.
(G) Representative images of TRKA immunohistochemistry, with inset shown on right. Scale bars, 50 μm.
(H) Quantification reveals a complete absence of TRKA+ precursors in Prdm12−/− embryos at all time points (E11.5, p = 0.001; E12.5 and E13.5, p < 0.0001; E18.5, p = 0.0004).
(I) Representative images of RUNX3 immunohistochemistry, with inset shown on right. Scale bars, 50 μm.
(J) Quantification reveals no significant difference between control and KO DRGs at any time point. For all graphs, a data point represents the average cells or area/section across three DRGs taken from the lumbar region of a single embryo in n = 3 embryos. Control embryos were Prdm12+/+ or Prdm12+/−.
Results are presented as means ± SEM; statistical analysis performed with two-tailed Student’s t tests.
To identify what cellular changes occurred to result in smaller Prdm12−/− DRGs, we analyzed how the number and types of neurons were affected. We performed immunohistochemistry with the pan-sensory neuron transcription factor ISLET1 (ISL1), which defines differentiated sensory neurons, and TRKA, a specific marker for nociceptors (Figures 1E–1H) (Moqrich et al., 2004; Smeyne et al., 1994). We found that the number of neurons (ISL+) was unchanged at early embryonic time points (E11.5 and E12.5) but was significantly decreased at later embryonic time points (E13.5 and E18.5) (Figures 1E and 1F). In contrast, TRKA was completely absent at all time points in Prdm12−/− mice, indicating the entire nociceptor lineage was lost. Notably, the number of ISL1+ and TRKA+ cells in control DRGs increases at E13.5 but remains constant in KO embryos (Figures 1F and 1H). Given that myelinated neurons are born before the unmyelinated neurons during DRG neurogenesis, we surmise that the sudden increase of ISL1+ and TRKA+ neurons in control tissue at E13.5 is due to the differentiation of the main pool of nociceptive neurons (Kitao et al., 2002; Lawson and Biscoe, 1979; Ma et al., 1999).
Because TRKA+ nociceptors were completely absent from the DRG at all embryonic time points, we wanted to test whether there was a compensatory increase in alternate cell fates. To investigate the effect of Prdm12-KO on non-nociceptive sensory lineages, we stained DRGs for RUNX3 (runt-related transcription factor), which marks early proprioceptive neurons (Figure 1I) (Inoue et al., 2002; Kramer et al., 2006; Levanon et al., 2002). Although numbers of RUNX3+ cells trended lower in KO tissue at all time points, no significant differences between control and KO cells were noted. In fact, average RUNX3 levels were relatively constant from E11.5 to E13.5 (Figure 1J) (Lallemend and Ernfors, 2012). Therefore, proprioceptor development is unaffected, indicating that the fate of nociceptors do not switch to proprioceptors in Prdm12−/− mice. Overall, our data suggest that the absence of Prdm12 during neurogenesis results in selective loss of the nociceptor population, whereas proprioceptive DRG neurons develop normally, consistent with reports in Prdm12 exon II KO mice (Bartesaghi et al., 2019; Desiderio et al., 2019).
Nociceptors fail to proliferate in Prdm12−/− embryos
We next sought to examine the developmental mechanism by which nociceptors are lost in Prdm12−/− embryos. Two previous studies using a Prdm12 exon II KO mouse suggest two possibilities: (1) nociceptors die by apoptosis (Desiderio et al., 2019), or (2) precursors fail to proliferate (Bartesaghi et al., 2019). To address these possibilities in our mouse model, we examined the changes in apoptosis using the marker cleaved caspase-3 and in proliferation using a thymidine analog (Figure 2). We found that the total number of cells undergoing apoptosis was unchanged or even reduced at E12.5 in Prdm12−/− embryos compared with controls (Figure 2B). When normalized to DRG size, however, no significant differences were noted between control and Prdm12−/− mice, suggesting that the overall rate of apoptosis was similar between groups (Figure 2C). It is particularly notable that no difference was seen at E13.5 because an increase in apoptosis at that time point would specifically point to death of the newly ISL1+, TRKA+ cells normally present in controls (Figure 1). Thus, it does not appear that nociceptors or their precursors are dying in increased numbers.
Figure 2. Defects in proliferation but not cell death in Prdm12−/− mice.
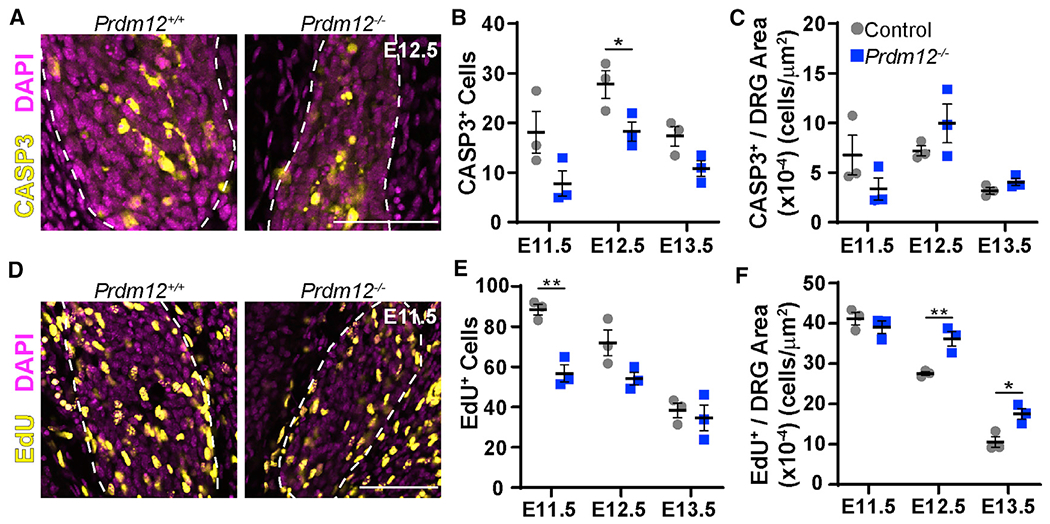
(A) Representative images of cleaved caspase-3 immunohistochemistry in E12.5 embryos. Scale bar, 50 μm.
(B) Quantification reveals a small but significant reduction of CASP3+ cells in Prdm12−/− embryos at E12.5 (p = 0.043).
(C) When normalized to DRG area, there is no significant difference in the number of CASP3+ cells at any time point.
(D) Representative images of DRGs labeled with EdU just before collection at E11.5. Scale bar, 50 μm.
(E) Quantification reveals a significant reduction in EdU-labeled cells at E11.5 (p = 0.003) in Prdm12−/− DRGs.
(F) When corrected for DRG size, counts indicate increased relative EdU labeling in Prdm12−/− DRGs at E12.5 (p = 0.009) and E13.5 (p = 0.021). For all graphs, a data point represents the average cells/section across three DRGs taken from the lumbar region of a single embryo for n = 3 embryos. Control embryos were Prdm12+/+ or Prdm12+/−.
Results are presented as means ± SEM; statistical analysis performed with two-tailed Student’s t tests.
We next looked at the effect of Prdm12 KO on proliferation of sensory neuron precursor cells. To do that, we intraperitoneally (i.p.) injected the thymidine analog 5-ethynyl-2′-deoxyuridine (EdU) into pregnant dams half an hour before collecting embryos to label proliferating cells at each time point. Using immunohistochemistry to visualize EdU-labeled cells, we found that the total number of EdU+ cells is significantly reduced in Prdm12−/− DRGs at E11.5 (Figures 2D and 2E). Thus, we infer that the progenitors that would make TRKA+ nociceptors are not present at E11.5, resulting in an overall decrease in the total number of proliferating cells (Figure 2E). Furthermore, total EdU levels are similar in control and Prdm12−/− DRGs at E12.5 and E13.5, indicating that non-nociceptive lineages are proliferating normally. When normalized to DRG size, there appears to be a significant increase in relative proliferation at E12.5 and E13.5 in Prdm12−/− tissue, but that is because the DRGs are smaller due to the absence of nociceptors (Figure 2F). Our findings are consistent with what Bartesaghi et al. (2019) describe in a constitutive Prdm12 exon II KO model showing a reduction in proliferation using phospho-histone H3 (pH3) staining. In summary, we have shown that Prdm12 likely has a role in the proliferation of progenitors that become nociceptors whereas proliferation of non-nociceptive populations is unchanged.
Prdm12AvilCKO mice have reduced nociception and pruriception
The above evidence, as well as the human phenotype of Prdm12-associated CIP suggests that loss of Prdm12 results in insensitivity to pain because of a failure of development of nociceptive neurons (Chen et al., 2015). To test that hypothesis, we examined whether deletion of Prdm12 leads to a painless phenotype. Because Prdm12−/− mice die neonatally, we used a conditional-KO approach to specifically target sensory neurons using AdvillinCre/+ knockin mice (Hasegawa et al., 2007; Zhou et al., 2010). Although Advillin protein is reported to be enriched in non-peptidergic isolectin B4 (IB4+) nociceptors (Hunter et al., 2018), our findings suggest the CRE recombinase in the AdvillinCre/+ knockin mice is broadly expressed in almost all DRG neurons, including those expressing Prdm12 mRNA (see Figure S1). Furthermore, although Advillin is reported to be expressed in other tissues, including endocrine cells, Merkel cells, and sympathetic ganglia (Hunter et al., 2018), Prdm12 is not expressed in those tissues and thus, deletion in those tissues should not affect a nociceptive phenotype (Chen et al., 2015; Kinameri et al., 2008; Matsukawa et al., 2015; Nagy et al., 2015). Therefore, we crossed AdvillinCre/+ mice heterozygous for Prdm12−/+ to the Prdm12F/F mice, and to a CRE-dependent fluorescent reporter (R26LSL-tdTomato, Ai14) (Madisen et al., 2010). The resulting Prdm12AvilCKO mice survive, in contrast to the neonatal lethality seen with germline KO. Prdm12AvilCKO mice were then tested to see whether loss of Prdm12 specifically in DRGs causes deficits in pain sensation.
To investigate the phenotypic consequence of sensory neuron-specific Prdm12 KO, we tested the sensation of Prdm12AvliCKO mice to a variety of stimuli. Notably, mechanical nociception was reduced in mutant mice in both pinprick and tail clip assays (Figures 3A and 3B). Nociceptive cold sensitivity to a 0–5°C cold plate was similarly reduced, reflecting that cold-thermal-sensing nociceptors are affected as well (Figure 3C). Sensitivity to intraplantar capsaicin injection was also reduced (Figure 3D). Curiously, although human Prdm12-associated CIP also causes complete heat insensitivity, latencies to respond to a focused beam of light were unchanged on the Hargreaves assay (Figure 3E). On hot plate, Prdm12AvilCKO mice have no differences in reflexive responses at 50°C, 52°C, or 55°C but do show a significant delay in response at 56°C, although lick times at both 50°C and 56°C are unchanged (Figure 3F). Thus, sensitivity to nociceptive heat stimuli appears to be largely intact, except for initial detection of very high temperatures. Pruriception was tested by intradermal injection of either chloroquine or histamine into the nape of the neck. Prdm12AvliCKO mice showed a significantly reduced scratch response to both stimuli, indicating that loss of Prdm12 during development affects itch sensation as well as nociception (Figures 3G and 3H). Finally, as with cases of human CIP, non-nociceptive sensation remained intact (Chen et al., 2015). Responses of Prdm12AvilCKO mice to low-threshold mechanical stimuli applied with von Frey hairs were similar to that of control mice, as were responses to dynamic stimuli applied with a brush stroke (Figures 3I and 3J). Control and mutant mice also had a similar latency in their responses to a piece of tape applied to the plantar surface of the hindpaw, again reflecting normal tactile sensation (Figure 3K).
Figure 3. Prdm12AvilCKO mice have reduced sensation to nociceptive stimuli and chemical pruritogens.
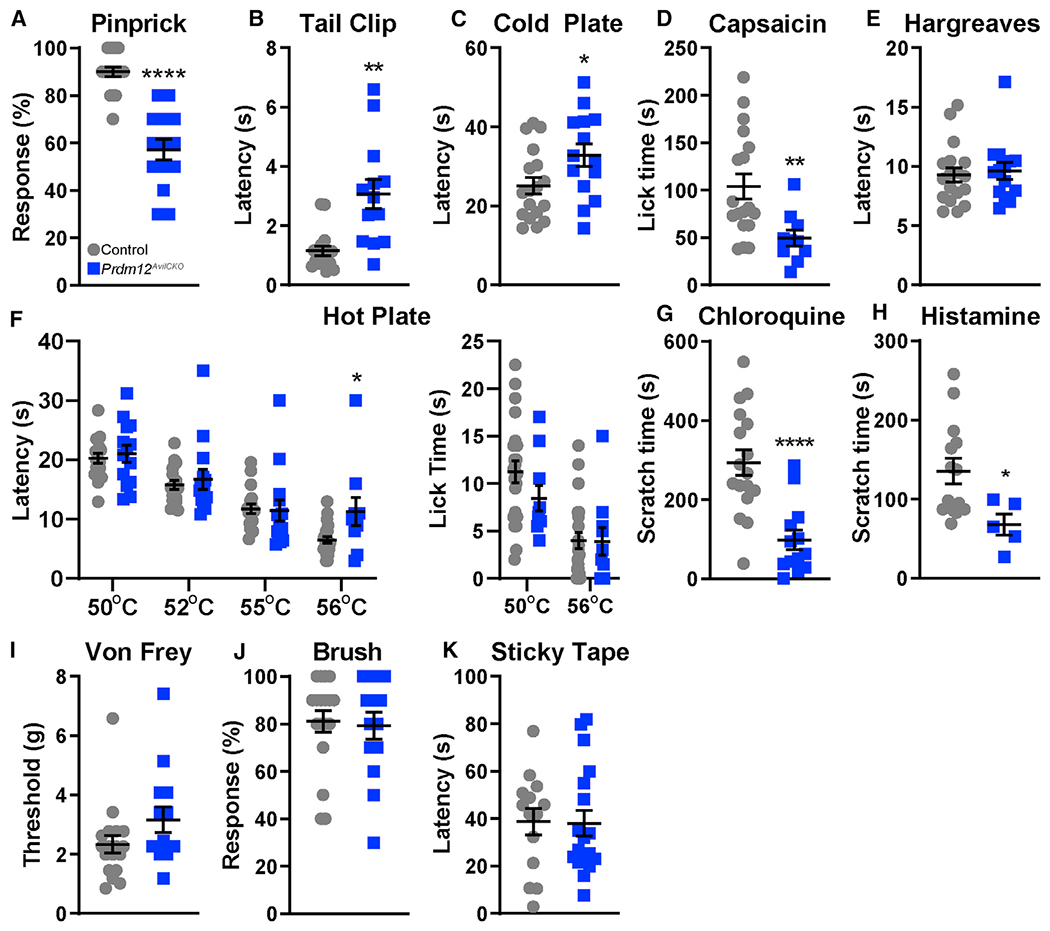
(A) Prdm12AvilCKO mice show reduced sensitivity to a sharp pin, p < 0.0001.
(B and C) Prdm12AvilCKO mice show a delayed response to a clip attached to the tail (B) (p = 0.002), and when placed on a cold plate (C) (0–5°C, p = 0.033).
(D) Mutant mice spend less time licking after intraplantar injection of capsaicin, p = 0.0017.
(E) Prdm12AvilCKO mice showed no differences in response to heat stimuli in the Hargreaves test.
(F) Mutant mice showed similar latencies to response on hot plate at 50°C, 52°C, and 55°C but had a delayed response at 56°C, p = 0.0112. Lick times at 50°C and 56°C were unchanged.
(G) Prdm12AvilCKO mice show reduced scratch time after intradermal chloroquine injection in the nape of the neck, p < 0.0001.
(H) Scratch time is similarly reduced in Prdm12AvilCKO mice after intradermal histamine injection in the nape of the neck, p = 0.029.
(I–K) Light touch is normal in Prdm12AvilCKO mice, which show similar withdrawal thresholds to von Frey hairs (I), responses to dynamic light touch (J), and latency to removal of a piece of tape applied to the plantar surface of the hindpaw (K).
For (A)–(K), n = 18 control and 14 mutant with 50/50 M:F, except for (D) and lick time in (F) (n = 16 control, 16 mutant, 50/50 M:F) and (H) (n = 14 control, 50/50 M:F, 5 mutant, 4F:1M). Data analyzed by two-tailed Student’s t test; results are presented as means ± SEM.
Developmental loss of Prdm12 reduces the number of nociceptors in the DRG
Having revealed a behavioral phenotype resulting from loss of Prdm12, we next assessed what molecular changes occurred in Prdm12AvilCKO DRGs. To confirm that ourgenetic manipulation was successful, we performed RNAscope using a probe specifically targeting exon V of Prdm12. As expected, the probe detected mRNA transcripts as multiple, distinct puncta in control tissue but not in mutant DRGs, indicating successful KO of that region (Figure 4A).
Figure 4. Nociceptor populations are reduced in Prdm12AvilCKO mice.
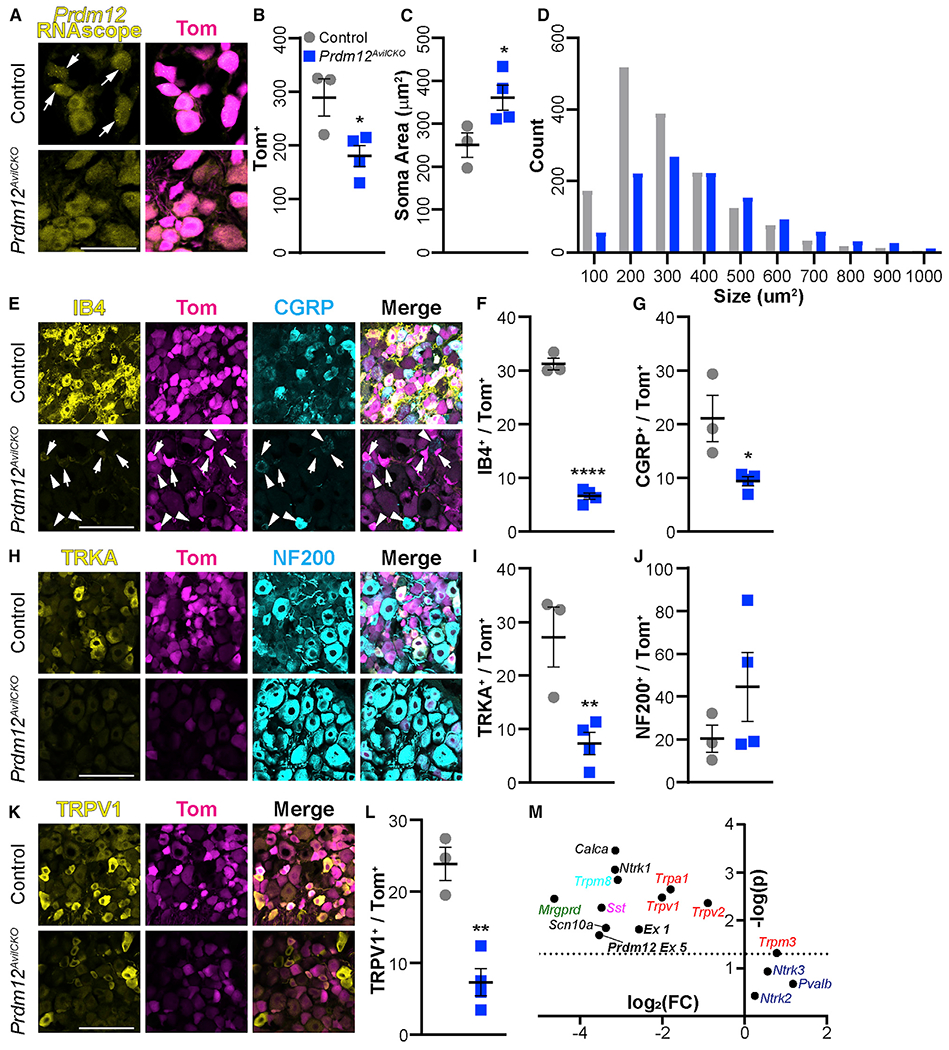
(A) RNAscope using exon-V-specific probes confirmed knockout of Prdm12 from mutant DRGs. Scale bar, 50 μm.
(B) Prdm12AvilCKO mice have a reduction of Tom+ neurons, p = 0.0326.
(C and D) The remaining neurons in Prdm12AvilCKO are larger, on average (C) (p = 0.0466), because of selective loss of cells ≤ 300 μm2 (D).
(E) Representative image showing number of IB4+ (arrows) and CGRP+ (arrowheads) nociceptors are reduced in Prdm12AvilCKO DRGs. Scale bar, 100 μm.
(F and G) Quantification of IB4+ (F) (p < 0.0001) and CGRP+ (G) (p = 0.027) nociceptors as a percentage of the total Tom+ population of sensory neurons.
(H) Representative image of TRKA+ nociceptors and NF200+ myelinated neurons in Prdm12AvilCKO and control mice. Scale bar, 100 μm.
(I) Quantification showing significant reduction of TRKA+ nociceptors, p = 0.0135.
(J) Quantification of NF200+ neurons showing a wide range in Prdm12AvilCKO mice, but no significant change from control littermates, p = 0.278.
(K and L) Representative image (K) and quantification (L) of the reduction in number of TRPV1+ nociceptors, p = 0.0025. Scale bar, 100 μm.
(M) qRT-PCR results plotted showing —log(p) versus log2 (fold change) reveal significant reductions in many nociceptive gene transcripts, including Prdm12, but a significant increase in Trpm3, and no change in touch and proprioceptive genes. The dotted line represents p = 0.05; points above it differ significantly between control and mutant mice. Each point represents the analysis of significance and fold change from average values of n = 3 biological replicates.
All analysis was completed using DRGs from lumbar levels 2 through 5. Each data point in (B), (C), (F), (G), (I), (J), and (L) represents the average count across three DRGs from control (n = 3) or Prdm12AvilCKO (n = 4) mice taken after behavior analysis around 10 weeks of age. All quantification analyzed by two-tailed Student’s t test; results are presented as means ± SEM.
Upon further analysis, DRGs from Prdm12AvilCKO mice were found to contain fewer total neurons (Figure 4B), with selective loss of smaller (≤300 μm2) soma, resulting in increased average soma size (Figures 4C and 4D). Indeed, histological assessment revealed that Prdm12AvilCKO DRGs were missing most of their small-diameter nociceptive neurons. The population of non-peptidergic IB4+ C fibers was reduced by ~80% in mutant DRG sections (Figures 4E and 4F), and the number of peptidergic CGRP+ neurons was similarly reduced by ~50% (Figure 4G). Unlike the findings in the constitutive KO (Prdm12−/−), Prdm12AvilCKO mice still have TRKA+ neurons, although that population was reduced by ~75% (Figures 4H and 4I). With such a drastic loss of unmyelinated C fibers, we expected most cells remaining in the DRG to be myelinated. However, we found a wide variation in the percentage of NF200+ cells (18%–85%), with some mutant DRGs showing no change in the relative number of NF200+ cells (Figure 4J). Finally, TRPV1 was reduced by ~70% (Figures 4K and 4L).
The reduction in TRPV1, although consistent with the reduced sensitivity to intraplantar capsaicin, led us to question whether heat sensitivity in Prdm12AvilCKO mice was largely retained because of compensation by other heat-sensitive ion channels. In particular, previous studies have shown that TRPA1 and TRPM3 have an overlapping role with TRPV1 in heat sensation (Vandewauw et al., 2018). Using qRT-PCR, we found Trpv1 and Trpa1 transcript levels to be decreased by 75% and 71%, respectively, whereas Trpm3 transcript was increased by 72% (Figure 4M). We also found that the transcript of Trpv2, an extreme heat sensor, was reduced by 46% (Figure 4M) (Caterina, 1999). Therefore, we conclude that elevated levels of Trpm3 compensate for lost Trpv1 and Trpa1, allowing for normal heat sensation at 50–55°C, but that the reflexive response at 56°C is stunted, possibly because of reduced Trpv2 expression (see Figure 3F). Levels of other transcripts were also consistent with our behavioral and histological findings, including reductions in Prdm12 (Prdm12 exons 1 and 5) and other nociceptive markers (Ntrk1, Calca, Trpm8, Mrgprd, Sst, and Scn10a), whereas touch and proprioceptive markers (Ntrk2, Ntrk3, and Pvalb) were unchanged (Figure 4M).
Adult KO of Prdm12 does not affect nociception in naive or injured animals
Although our results so far add to the field of knowledge regarding the role of Prdm12 during nociceptor development, very little is known yet about the function of this transcription factor in adulthood. Prdm12 continues to be expressed in nociceptors of mature DRGs (Figure 4A) and through late adulthood (Chen et al., 2015; Kinameri et al., 2008; Sharma et al., 2020; Usoskin et al., 2015). We set out to investigate the adult role of Prdm12 by crossing Prdm12F/F mice with the AvilCreERT2BAC transgenic strain heterozygous for Prdm12−/+ (Lau et al., 2011) to generate Prdm12AvilERT2CKO mice. The CreERT2 allows for temporal control of recombination because it only becomes nuclear localized with exposure to tamoxifen.
To test whether loss of Prdm12 from mature nociceptors affects pain sensation, Prdm12AvilERT2CKO mice and control littermates were injected with tamoxifen at 8 weeks of age to delete Prdm12 from sensory neurons (Figure 5A). After 4 weeks, several nociceptive assays were performed. Prdm12AvilERT2CKO mice had no differences in the cold plate assay, the reflexive component of hot plate, von Frey, cold plantar assay (CPA), Hargreaves, or after intraplantar injection of either capsaicin or formalin (Figures 5B–5K). Curiously, mutant mice did spend significantly less time licking during prolonged exposure to a 50°C hot plate (Figure 5C).
Figure 5. Knockout of Prdm12 in adulthood does not substantially reduce pain sensitivity in naive or injured mice.
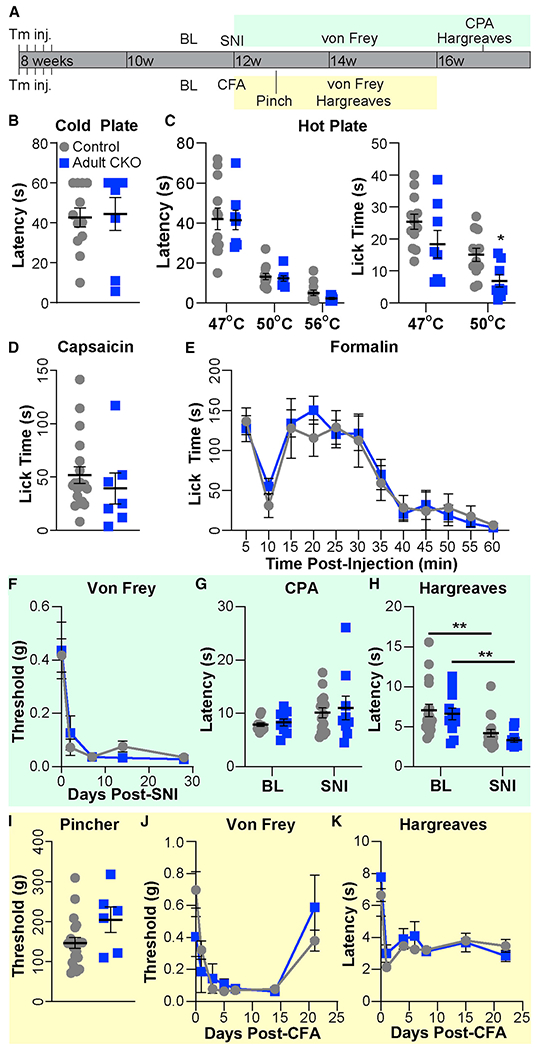
(A) Schematic showing experimental timeline.
(B) No difference was observed in reaction time on a cold plate between Prdm12AvilERT2CKO (adult conditional gene knockout [CKO], n = 8) and control (n = 12) mice (both 50/50 M:F).
(C) Although no difference was observed at 47°C, adult CKO mice spent significantly less time licking on a 50°C hot plate, p = 0.0126. No differences were found in the reflexive response on hot plate at any temperature.
(D and E) No difference was observed between Prdm12AvilERT2CKO (n = 7, 3:4 M:F) and control (n = 19, 11:8 M:F) in the time spent licking after capsaicin (D) or formalin (E) injection into the hindpaw. (F–H) Behavioral results before and after SNI.
(F) Time course of withdrawal thresholds for Prdm12AvilERT2CKO (n = 12, 5:7 M:F) and control (n = 18, 8:10 M:F) mice showing both groups developed mechanical allodynia after SNI.
(G) Responses of control (n = 15, 7:8 M:F) and Prdm12AvilERT2CKO (n = 9, 5:4 M:F) mice to cold plantar assay (CPA) did not differ significantly at baseline or 4 weeks after SNI.
(H) Both groups experienced heat hyperalgesia 4 weeks after SNI but did not differ from each other (control versus adult CKO) at either time point. Same n as (F), control p = 0.0044, Prdm12AvilERT2CKO p = 0.0078.
(I–K) Behavioral results after CFA injection.
(I) No difference was observed in the withdrawal threshold to paw pinch between Prdm12AvilERT2CKO (n = 6, 3:3 M:F) and control (n = 21, 12:9 M:F) mice, which was tested 3 days after CFA injection. Note that withdrawal thresholds are several-fold higher because of the larger area over which pressure is applied with the rodent pincher compared with von Frey filaments.
(J) Time course of withdrawal thresholds showing both groups developed tactile allodynia after CFA and recovered over the same time period. Same n as (I). (K) Both groups developed heat hyperalgesia after CFA injection. Same n as (I).
Statistical analysis by 2-way ANOVA with post hoc Tukey tests when a significant difference was detected for (E)–(H), (J), and (K); two-tailed Student’s t tests for other datasets. Results are presented as means ± SEM.
Although we found limited changes in nociception in Prdm12AvilERT2CKO mice, PRDM12 has been implicated in chromatin remodeling complexes with EHMT2 (Yang and Shinkai, 2013). EHMT2 has been found to mediate mechanical allodynia and heat hyperalgesia upon nerve injury through repression of potassium (K+) channels (Laumet et al., 2015; Liang et al., 2016). Therefore, we hypothesized that PRDM12 may also have a role in sensitization after injury through its interactions with EHMT2. We assessed the effect on allodynia and hyperalgesia in Prdm12AvilERT2CKO mice in the setting of both nerve and inflammatory injury.
Spared nerve injury (SNI) was performed on Prdm12AvilERT2CKO mice and littermate controls 4 weeks after tamoxifen injection, and nociceptive behavior was reassessed. Von Frey testing was performed at several time points to establish a time course of responses. Surprisingly, no reduction in mechanical allodynia was found because both control and Prdm12AvilERT2CKO mice became hypersensitive after SNI, with no difference in paw withdrawal thresholds between the groups at any time point (Figure 5F). Neither controls nor mutants developed cold allodynia after SNI, as measured with the cold plantar assay. This is likely because the posture adopted by injured mice lifts the hypersensitized region away from the glass, preventing direct application of the cold stimulus. Both groups of mice experienced similar degrees of heat hyperalgesia, as measured by Hargreaves, but again, no difference emerged between controls and mutants (Figure 5H). These findings indicate that Prdm12 does not affect hypersensitivity of mature nociceptors after nerve injury.
We next wanted to determine whether loss of Prdm12 would confer protection after inflammatory injury. To test that, we injected complete Freund’s adjuvant (CFA) into the hindpaw of control and mutant mice and measured the responses to mechanical and thermal stimuli over the next 3 weeks (Figures 5I–5K). Again, no differences in responses were noted between control and mutant animals. Both groups developed similar levels of tactile allodynia, which faded approximately 3 weeks after injection (Figures 5I and 5J). As an additional measure, we also tested sensitivity to pinch in the inflamed paw 3 days after CFA injection and found no difference between cohorts (Figure 5I). Similarly, no differences in thermal hyperalgesia were noted between the two groups (Figure 5K). Overall, our results show that loss of Prdm12 function during adulthood does not significantly alter pain sensation or affect the development of allodynia and hyperalgesia after injury.
Molecular and transcriptional changes following Prdm12-KO in adult
To look for molecular changes that may illuminate the observed lack of phenotype, we assessed transcriptional and histological changes in the DRG. Lumbar DRGs were taken from control and Prdm12AvilERT2CKO mice 4 weeks after SNI, so that we could investigate changes after both tamoxifen injection and neuropathic injury. We first verified successful KO of Prdm12 using the exon-V-specific RNAscope probe and found it be absent from Prdm12AvilERT2CKO DRG neurons (Figure 6A). We again confirmed that finding with qRT-PCR, which also revealed reductions in Trpm8 and Ntrk1 (63% and 47%, respectively) (Figure 6B) similar to the embryonic conditional KO, Prdm12AvilCKO. However, transcripts of many genes that were downregulated in Prdm12AvilCKO mice were unchanged in Prdm12AvilERT2CKO mice, such as Trpa1, Trpv1, Ssf, Scn10a, and Mrgprd (Figure 6B). Similarly, immunohistological assessment of nociceptor populations within the DRG found no significant differences in numbers of IB4, CGRP, TRKA, NF200, or TRPV1 between groups (Figures 6D–6I). Additionally, no significant changes in those markers were noted between DRGs ipsilateral and contralateral to SNI, suggesting that nerve injury also does not alter the numbers of those nociceptor populations at the protein level (Figures 6D–6I), although transcripts of some nociceptive markers appear to change after injury (Renthal et al., 2020).
Figure 6. DRG nociceptor populations are unchanged after Prdm12 knockout and/or SNI.
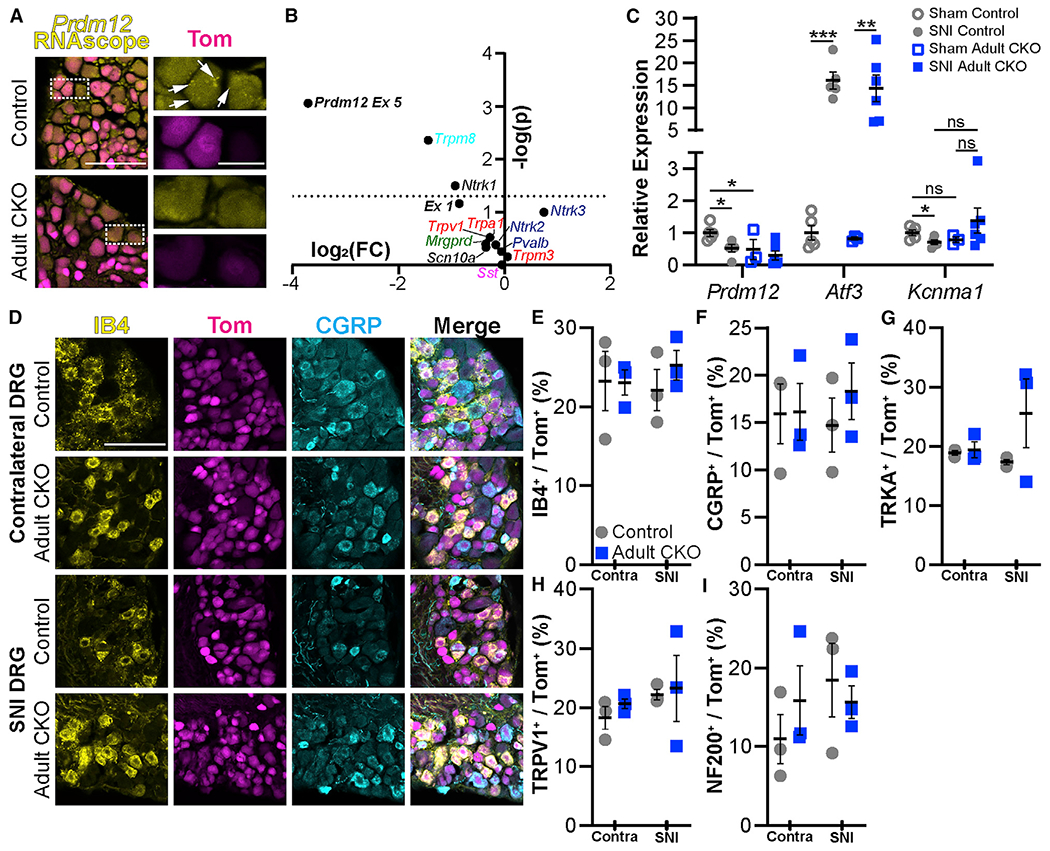
(A) Exon-V-specific RNAscope verified loss of mRNA transcript in Prdm12AvilERT2CKO (adult CKO) mice. Scale bar, 100 μm. Inset arrows indicate mRNA puncta detected by the probe; inset scale bar, 25 μm.
(B) qRT-PCR in Prdm12AvilERT2 mice after tamoxifen injection revealed reductions in expression of exon 5 of Prdm12, as well as Trpm8 and Ntrk1; other genes were unaffected. The dotted line represents p = 0.05; points above it differ significantly between control and mutant mice. Each point represents the analysis of significance and fold change from average values of n = 6 biological replicates.
(C) Relative expression of select genes after SNI or sham surgery specifically from L4–5 DRG. Prdm12 is reduced after SNI in control animals (p = 0.0356) and in sham mutants (p = 0.0482). Additionally, Atf3 expression is increased in both control (p = 0.0001) and mutant (p = 0.0010) mice, but Kcnma1 is reduced only in control animals (p = 0.0239).
(D) Representative images of lumbar DRGs from control and Prdm12AvilERT2CKO DRGs contralateral to and ipsilateral to SNI with immunohistochemistry for IB4 and CGRP. Scale bar, 100 μm.
(E–I) Quantification of these images revealed no changes in the number of IB4+ (E), CGRP+ (F), TRKA+ (G), TRPV1+ (H), or NF200+ (I) neurons after SNI in either control or Prdm12AvilERT2CKO mice or between those two groups. Each data point in (E)–(I) represents the average counts/section across three DRGs taken from the L2–L5 region of n = 3 mice. DRGs were collected after behavior assessment at 18 weeks.
Graphs show means ± SEM; statistical analysis two-tailed Student’s t test (B) or 2-way ANOVA with post hoc Tukey tests when a significant difference was detected (C–I).
We next wanted to test how PRDM12 affects K+ channel expression after SNI in control and mutant mice given its reported interaction with EHMT2, which represses K+ channel expression after injury (Cao et al., 2010; Chen et al., 2009; Laumet et al., 2015; Liang et al., 2016). First, we verified that Atf3 was increased after injury (Laedermann et al., 2014; Tsujino et al., 2000) by about 16-fold in both control and Prdm12AvilERT2CKO DRGs, indicating Atf3 expression is independent of Prdm12 (Figure 6C). Surprisingly, levels of Prdm12 itself were reduced by 58% after nerve injury in control animals, close to detectable levels in the KO mice (Figure 6C), in contrast to EHMT2, whose protein expression is increased after injury (Laumet et al., 2015; Liang et al., 2016). Interestingly, although levels of the potassium ion channel Kcnma1 were reduced upon SNI in control animals as expected, its expression was unchanged in mutant DRGs and trending toward an increase in expression after SNI (Figure 6C). This is reminiscent of the prevention of reduction in K+ channel expression after inhibition or loss of EHMT2 function (Laumet et al., 2015).
To investigate broader transcriptional changes that occur after the loss of Prdm12, we performed bulk mRNA sequencing (mRNA-seq) of DRGs harvested from Prdm12AvilERT2CKO and control mice 2 weeks after tamoxifen injection. SNI was not performed on these animals. As expected, exon V is specifically knocked out in Prdm12AvilERT2CKO mice (Figure 7A). Surprisingly, however, we found that most of the differentially expressed genes (DEGs) were decreased in KO mice, suggesting PRDM12 acts as a transcriptional activator (Figure 7B). This is in contrast to prior evidence suggesting that histone methyltransferase activity associated with Prdm12 is repressive (Kinameri et al., 2008; Thélie et al., 2015).
Figure 7. Transcriptional changes in Prdm12AvilERT2CKO mice.
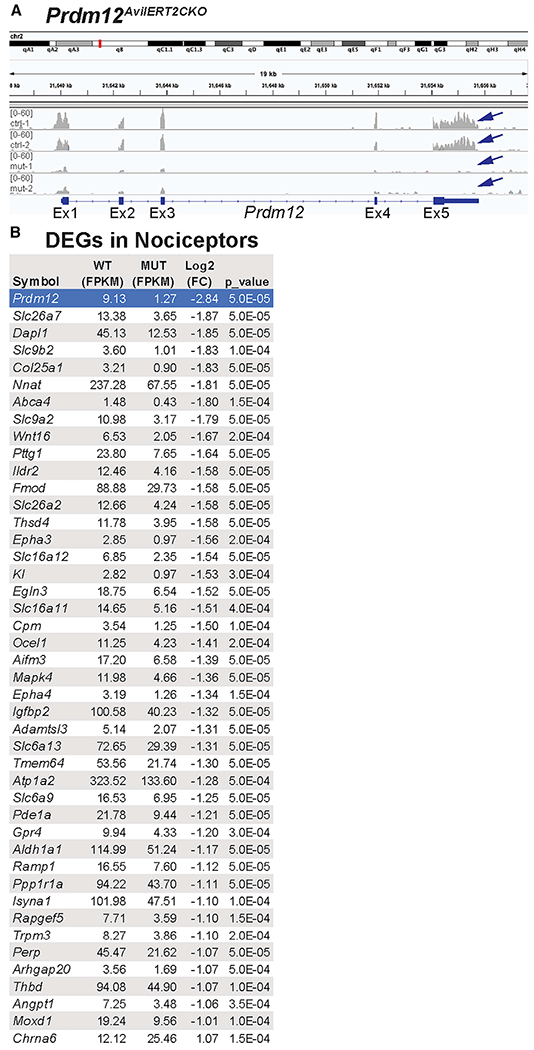
(A) Sequencing reads from two control (ctrl-1 and −2) and two mutant (mut-1 and −2) samples show that exon V is specifically knocked out (arrows).
(B) 43 genes expressed in nociceptors are decreased in the mutant (negative log2 [fold change]), and one gene is increased (Chrna6). FPKM values are averaged across both samples.
Because mRNA was harvested from all cells in the DRG, we cross-referenced the initial list of 150 DEGs to a published dataset from single-cell RNA sequencing (scRNA-seq) of DRG neurons (Sharma et al., 2020) (Figure 7B; Table S1; see Table S2 for complete dataset); 44 of those genes were expressed in nociceptors–43 were decreased after the loss of Prdm12, and only 1 gene, Chrna6, was increased (Figure 7B). Most of the nociceptive genes identified in our dataset are not classically used to define nociceptor cell types. Overall, despite the lack of overt differences in pain sensation in Prdm12AvilERT2CKO mice, it appears that loss of function of Prdm12 does have a role in transcriptional control of adult nociceptors, but the exact nature of that role requires further investigation.
DISCUSSION
In this study, we explore the role of Prdm12, both during embryonic development and in the adult. We find that, although it is necessary for nociceptor neurogenesis, its function in mature nociceptors is unclear. We find that constitutive deletion of Prdm12 exon V precludes development of the entire nociceptor lineage, normally marked by TRKA expression, resulting in smaller DRGs with fewer differentiated neurons. Moreover, our findings suggest that this is due, in part, to reduced precursor proliferation. Furthermore, we show that in an embryonic conditional KO model, mice have reduced sensitivity to certain modalities of pain and itch, with correspondingly reduced nociceptor populations in the DRGs, but curiously, heat sensation is mostly spared. Lastly, we provide evidence that the function of Prdm12 differs in adult DRGs compared with neurogenesis because KO did not significantly alter pain phenotype or nociceptor populations, even in the context of injury and inflammation.
Prdm12 is required for nociceptor development
We find that constitutive deletion of Prdm12 exon V alone impedes nociceptor development. As with prior reports using an exon II KO, we find that expression of TRKA is completely absent from DRGs of Prdm12−/− embryos at all embryonic time points, supporting a key role in specification of this lineage (Bartesaghi et al., 2019; Desiderio et al., 2019). Our findings indicate that nociceptors fail to develop in Prdm12−/− mice, likely because of defects in proliferation and/or specification of progenitors that are likely to become nociceptors and not because of an increase in cell death or respecification to proprioceptors.
Conditional KO of Prdm12 from sensory neurons recapitulates aspects of human CIP
Here, we show that Prdm12AvilCKO mice bred to selectively remove Prdm12 from DRGs during neurogenesis have deficiencies in several sensory modalities. As with patients with CIP, they have reduced nociception (mechanical, cold, and capsaicin injection), while sparing light touch (Indo, 2014). These nociceptive behavioral differences are accompanied by losses of IB4+, CGRP+, TRKA+, and TRPV1+ nociceptors from the DRGs. Furthermore, although mice in our study also showed reductions of pruriception, people with Prdm12-associated CIP report normal itch sensation, and those with poly-alanine expansions in Prdm12 have seemingly excessive itch leading to midface toddler excoriation syndrome (MiTES) (Moss et al., 2018; Nahorski et al., 2015; Srinivas et al., 2017; Zhang et al., 2016).
The nociceptive phenotype in Prdm12AvilCKO mice bear a striking resemblance to the phenotype observed after ablation of Nav1.8+ postmitotic sensory neurons (Abrahamsen et al., 2008). These mice also show reduced IB4+ and CGRP+ nociceptor populations, as well as reduced TRKA and TRPV1 expression, and defects in mechanical and cold nociception as, well as capsaicin-injection, but not heat when tested up to 55°C. These similarities suggest that deletion of Prdm12 and ablation of Nav1.8+ neurons affect comparable nociceptor populations. We found that levels of Trpm3 were increased, which may compensate for the loss of Trpv1 and Trpa1, helping to explain why thermosensation is preserved at 55°C and below (Vandewauw et al., 2018). Curiously, we did find a significant reduction in reflexive heat sensitivity at 56°C, possibly because of a corresponding reduction in expression of the extreme heat sensor Trpv2, which was not assessed after Nav1.8 ablation.
Residual pain in Prdm12AvilCKO and Prdm12AvilERT2CKO mice may indicate autonomous PR domain function
Comparing and contrasting the phenotype between the Prdm12 exon II (Bartesaghi et al., 2019; Desiderioet al., 2019) and exon V (shown here) KO mouse models can give us some clues as to the function of different domains within the PRDM12 protein. In the exon II KO, exons II–V, which code for all functional domains for PRDM12 including the PR domain, are deleted (Bartesaghi et al., 2019; Desiderio et al., 2019). In the exon V KO, the coding sequence for the three zinc finger domains, polyalanine repeat, and a nuclear localization sequence, is deleted. We found in our adult Prdm12AvilERT2CKO mice that transcripts for exons I–IV, which include the PR domain, are still expressed albeit at reduced levels (~35%–55%) (Figures 6B and 7A). Therefore, although the putative interaction with EHMT2 that occurs through ZnF2 is disrupted in both the exon II and exon V KO mouse models (Yang and Shinkai, 2013), the PR domain is potentially still expressed in the Prdm12 exon V mouse model.
At the phenotypic level, subtle differences are seen in the Prdm12 exon II and exon V sensory neuron-specific conditional KO mice. In the KO of Prdm12 exon II, also using the AvilCre/+ strain, mice were observed to develop eye opacities, as well as tail and facial scratches and wounds, similar to clouding of the eye and self-mutilating injuries seen in patients with CIP (Chen et al., 2015; Desiderio et al., 2019), but no such phenotype was noted in the Prdm12AvilCKO mice presented here. It is possible that the PR domain is translated in the Prdm12AvilCKO mice, carrying out an as-yet undescribed function independent of EHMT2, leading to a less-severe phenotype than that seen with the sensory-specific conditional KO of Prdm12 exon II (Desiderio et al., 2019). Strain differences could also be another variable because our model was on a mixed background, whereas the exon II KO was in C57BL/6J mice. Furthermore, the retention of exons I-IV in the adult Prdm12AvilERT2CKO mice might contribute to the mild nociceptive phenotype. Further studies directly comparing the nociceptive behaviors in the exon II and exon V conditional KO mice will lend further insight into the potential role of the PR domain.
Prdm12 may function as both an activator and a repressor with distinct functions in adult nociceptors
As a whole, the PRDM family of transcription factors can have multifaceted roles, including being an activator of cellular lineages and repressor of alternative fates, even within the same cell (Hohenauer and Moore, 2012). Early evidence suggested PRDM12 functions primarily as a repressor because of its interactions with EHMT2 and its role in repressing neighboring progenitor domains of the V1 progenitor population in the developing spinal cord (Fog et al., 2012; Kinameri et al., 2008; Thélie et al., 2015). However, the RNA-seq data we report here points to PRDM12 serving as a transcriptional activator in the adult context, given that almost every transcript identified was downregulated in Prdm12AvilERT2CKO mice. Although it is possible that PRDM12 could be repressing a repressor, we are unable to identify a repressor that is upregulated after Prdm12 KO. Furthermore, overexpression of PRDM12 with NEUROG1 in Xenopus laevis explants induces expression of several other genes essential for sensory neurogenesis, such as TRKA, although germline KO in mice resulted in both increases and decreases of downstream targets during development (Desiderio et al., 2019; Nagy et al., 2015). Thus, evidence suggests that PRDM12 may act as either a repressor or activator and that this activity may change over developmental time, with PRDM12 being an activator in the adult mouse.
It is also notable that, with the exception of Prdm12 and Ntrk1, there is almost no overlap in DEGs identified in our work and that by Desiderio et al. (2019). Developmentally, Prdm12 regulates an array of genes involved in the generation of spinal cord interneurons, as well as transcriptional regulators of sensory neuron differentiation (Desiderio et al., 2019). Most DEGs identified in our dataset are not obviously related to nociceptor neurogenesis, and they are not generally used to define nociceptor cell types; however, some are nociceptor-specific (Sharma et al., 2020). Given that PRDM12 is proposed to induce Ntrk1 expression through interactions with NEUROG1, which is only present during DRG embryogenesis, the lack of overlap between datasets is not surprising but, again, points to alternate roles of Prdm12 during development and in adult DRGs.
Indeed, the role of Prdm12 in adult DRGs remains elusive. We found that exon V deletion in 8-week-old Prdm12AvilERT2CKO had very little effect on the nociceptive phenotype under baseline, inflammatory, or neuropathic conditions. This was surprising for two reasons. First, as already described, PRDM12 is thought to interact with EHMT2. The latter methyltransferase is normally upregulated after nerve injury, and developmental KO or inhibition of EHMT2 reduces tactile allodynia and thermal hypersensitivity after neuropathic injury (Laumet et al., 2015; Liang et al., 2016). We hypothesized that PRDM12 would have a role in sensitization after nerve injury through its interactions with EHMT2, but that was refuted by the lack of a difference in sensitivity in Prdm12AvilERT2CKO mice after SNI. In addition, Prdm12 mRNA transcript was surprisingly downregulated after injury, whereas the EHMT2 protein has been shown to be upregulated after injury, indicating that the potential interactions between the two might be complicated. Still, we do see that the reduction in K+ channel expression that normally occurs after nerve injury is prevented in Prdm12AvilERT2CKO mice, as it is after EHMT2 KO, suggesting that the particular K+ channel we tested, Kcnma1, is not sufficient to block neuropathic allodynia or hyperalgesia. It will be interesting to see, in future work, how Prdm12 and Ehmt2 potentially synergistically affect transcriptional changes after injury. Second, overexpression of Chrna6, which encodes the α6 subunit of the nicotinic acetylcholine receptor (nAChR), is protective against tactile allodynia in both neuropathic and inflammatory injury models (Wieskopf et al., 2015). Chrna6 was also the only gene found to be upregulated in Prdm12AvilERT2CKO mice, about 2-fold over control levels. Given the absence of a phenotype in our mice, it is possible that a further increase in expression level is required to achieve protection against allodynia.
It is clear that Prdm12 has an integral role in the development of sensory neurons, and further study is needed to clarify the precise mechanisms requiring PRDM12 that specify the nociceptive lineage. Beyond development, however, Prdm12 remains a specific marker of nociceptive neurons. Although our study demonstrates that direct inhibition of its activity may not provide significant analgesic relief, we do find transcriptional changes resulting from its loss and contend that further study is needed to define the precise nature of those changes and whether they present a different angle for development of novel analgesics.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Helen Lai (Helen.Lai@UTSouthwestern.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
The accession number for the RNA-seq data reported in this paper is GEO: GSE167468.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
The following mouse strains were used: Prdm12F/F (Chen et al., 2020), CAG-Cre (Sakai and Miyazaki, 1997), AdvillinCre/+ (JAX#032536) (Hasegawa et al., 2007), R26LSL-tdTomato/+ (Ai14, JAX#007908) (Madisen et al., 2010), AvilCreERT2BAC (JAX#032027) (Lau et al., 2011), R26LSL-EYFP/+ (Ai3, JAX#007903). All mice were outbred and thus are mixed strains (at least C57BL/6J, C57BL/ 6N, and ICR). Both male and female mice were used for all studies. No sex differences were noted for any quantified data, therefore sexes of the same genotype were pooled for analysis. Mice crossed to AdvillinCre/+ always included a fluorescent reporter and were screened for “dysregulated” expression. Normal fluorescence is visible in the trigeminal and dorsal root ganglia of pups within the first three days after birth, while dysregulation results in patchy fluorescence of the whole body. Mice expressing AvilCreERT2BAC were injected with tamoxifen (Sigma) at eight weeks of age. Injections were given over a period of five days (1/day, 120mg/kg delivered intraperitoneally (i.p.) as a 40mg/ml solution dissolved in sunflower oil with 10% ethanol) (Lau et al., 2011; Sikandar et al., 2018). All animal experiments were approved by the Institutional Animal Care and Use Committee at UT Southwestern.
METHOD DETAILS
Behavior assessments
For all behavioral tests, animals were habituated for 30 min one day before testing, and again immediately prior to testing. A single experimenter conducted all tests and was blinded to genotype. The subsequent statistical analyses included all data points; no methods were used to predetermine sample sizes. Littermates were used as controls.
von Frey mechanical sensitivity
von Frey withdrawal thresholds were determined using the simplified up-down method (Bonin et al., 2014). After acclimation in plastic chambers with wire mesh flooring, mice were tested with graded filaments from 0.008 to 2.0 g applied for ~3 s to the plantar hindpaw with at least 5 min between each application. Responses on both the left and right paw were recorded. Toe spreading, flinching, or licking was recorded as a positive response.
Tail clip and pinprick
Mechanical nociception was assessed using the tail clip and pinprick assays. For tail clip, electrical tape was wrapped around the jaws of a 1 cm binder clip, which was then attached to the tail, about 1 cm from the rostral end. The latency to response (biting or clawing at the clip, or otherwise trying to remove it) was recorded for a single trial, with a cutoff of 10 s. Pinprick was performed using 0.2 mm insect pins (FST 26002-20) to deliver a sharp mechanical stimulus. Mice were again acclimated in plastic chambers, then challenged 10 times on each paw, with 10 min between each trial. Positive responses (paw flinching, licking, or vocalization) was recorded and reported as a percentage of total trials.
Rodent pincher
The inflamed paw in the CFA inflammation model was tested with the Rodent Pincher Analgesia Meter (Bioseb). Mice were restrained by wrapping the mouse inside a paper towel with the inflamed paw exposed. The pincher was used to apply ramping pressure until a response (paw withdrawal or flicking, or vocalization) was observed. Three recordings were made per mouse, spaced at least 10 min apart.
Heat sensitivity (hot plate and Hargreaves)
For hot plate, mice were placed directly on the plate (IITC) set to the designated temperature. The latency to response (hindpaw licking or flicking, or jumping) was recorded and averaged over three trials. Cutoff times were used to prevent injury as follows: 1 min for 50°C, 45 s for 52°C, and 30 s for 55°C and 56°C. To study affective pain, mice were placed on the hot plate at 47°C, 50°C, or 56°C and recorded for 3 minutes, 1 minute, or 30 s, respectively. The videos were then analyzed to determine total time spent licking the hindpaws. For Hargreaves, mice were acclimated on a heated (30°C) glass surface (IITC), then exposed to a beam of radiant heat following the standard Hargreaves method. Beam intensity was adjusted to result in latency of ~10 s in wild-type animals. Paw withdrawal latency was recorded for 3 exposures per paw, with at least a 5 min interval between exposures. A cutoff time of 30 s was used to prevent tissue damage.
Cold sensitivity assays
Cold nociception was measured using either the cold plate or cold plantar assay. For cold plate, a cooling block was chilled at −20°C, then allowed to warm until the surface temperature reached 0°C as measured by an infrared thermometer. Mice were placed on the plate, and the latency to response (hindpaw licking or flicking, or jumping) was recorded and averaged over three trials. A cutoff time of 60 s was used to prevent injury. The cold plantar assay was performed using dry ice loaded into a syringe to stimulate the hindpaw (Brenner et al., 2012). Mice were placed on a thin, single pane of glass, and the tip of the dry ice pellet was pressed against the glass under the hindpaw. Withdrawal latency was recorded for 3 exposures per paw, with at least 5 min interval between exposures. A cutoff time of 30 s was used to prevent tissue damage.
Itch assays
Itch sensation was measured by pruritogen injection into the nape of the neck (Kuraishi et al., 1995; Shimada and LaMotte, 2008). The injection area was shaved one day prior to testing. On the day of testing, mice were habituated in cylindrical containers for 30 minutes, then injected with 20 μL of histamine (100 μg/μL) orchloroquine (200 μg/μL) dissolved in PBS. The mice were video recorded for 30 min following pruritogen injection, and the videos were subsequently scored to determine total time spent scratching the injected area.
Capsaicin test
The capsaicin test was performed by intraplantar injection to one hindpaw of 10 μL containing 0.3 μg/μL capsaicin (Sigma M2028) in 0.9% saline/10% ethanol/10% Tween-20 following acclimation. Mice were then video recorded for 10 minutes, and the videos were subsequently scored to determine time spent licking the injected paw.
Formalin test
The formalin test was performed by intraplantar injection to one hindpaw of 20 μL containing 5% formalin in 0.9% saline. Mice were then video recorded for 1 hour, and the videos were subsequently scored to determine time spent licking the injected paw. Times were binned into 5 minute intervals to identify the acute and chronic phases following injection.
Touch assays
Non-nociceptive touch sensation was measured using the dynamic brush and sticky tape assays. For dynamic brush assay, were again acclimated in von Frey chambers. The tip of a cotton tipped applicator (Henry Schein) was teased apart to “fluff” it up and ensure no filaments were sticking straight up. The swab was then lightly brushed across the plantar surface of the hindpaw (about 1 s from heel to toe) 10 times per paw, with 10 min between each trial. Positive responses (paw flicking or withdrawal) are reported as a percentage of total trials. For sticky tape, a 5 mm × 5 mm piece of lab tape (Fisher) was adhered to the plantar surface of the hindpaw, and the mouse was allowed to freely explore its enclosure. Latency to removal of the tape was recorded and averaged across two trials per paw.
Injury and inflammation
SNI surgery was performed as previously described (Decosterd and Woolf, 2000). Briefly, under 3% isoflurane anesthesia, the left sciatic nerve was exposed where it branches into the sural, tibial, and common peroneal nerves. The tibial and common peroneal nerves were tightly ligated using 5.0 silk suture, and a ~3 mm section of nerve was removed just distal to the knot. Mice were allowed to recover for at least 48 hours prior to testing. For CFA-induced inflammation, 20 μL of Complete Freund’s Adjuvant (Sigma F5881) was injected into the plantar surface of the left hindpaw. Mice were initially tested 6 hours post-injection to allow for development of the inflammatory response and then periodically over 3 weeks post-injection.
Tissue processing
Pregnant dams were injected with 0.5 mg/mL EdU (5-ethynyl-2′-deoxyuridine, Carbosynth) at a dose of 10 μg EdU/g mouse 30 minutes prior to CO2 euthanasia for collection of embryos (Wang et al., 2011). Embryos fixed in 4% paraformaldehyde (PFA) in PBS for 2 hours at 4°C, washed overnight in PBS, and cryoprotected in 30% sucrose. Adult mice were anesthetized with Avertin (2,2,2-Tribromoethanol) (0.025 – 0.030 mL of 0.04 M Avertin in 2-methyl-2-butanol and distilled water/g mouse) and transcardially perfused, first with 0.012% w/v Heparin/PBS and then 4% PFA/PBS. A dorsal or ventral laminectomy exposed the spinal cord and DRGs for a post-fix in 4% PFA (2 hours at 4°C). Tissue was then washed in PBS and cryoprotected in 30% sucrose before the laminectomy was performed on the reverse side, allowing DRGs to be removed and embedded in OCT (Tissue-Tek Optimal Cutting Temperature Compound). All tissue was sectioned using a Leica CM1950 cryostat.
Immunohistochemistry and confocal imaging
Cryosections (20–30 μm were blocked with PBS/1% normal goat or donkey serum (Jackson labs)/0.3% Triton X-100 (Sigma) for up to 1 hr at room temperature (RT), then incubated overnight with primary antibody at 4°C. Sections were washed 3 times in PBS, then the appropriate secondary antibody (Alexa 488, 567, and/or 647, Invitrogen) was incubated for an hour at RT, and sections were again washed 3 times in PBS. For development of EdU signal, sections were then re-permeabilized in 0.5% Triton X-100 for 30 min at RT, then incubated in EdU detection solution (100 mM Tris pH 7.5, 4 mM CuSO4, 100 mM sodium ascorbate, 5 μM sulfo-Cy3 azide (Lumiprobe)) for 30 min at RT, and rinsed 3 times in PBS. Slides were mounted with Aqua-Poly/Mount (Polysciences Inc.), and coverslipped (Fisher). The following primary antibodies and dilutions were used: mouse anti-Islet1/2 (1:20,000; DSHB 39.4D5), goat anti-TRKA (1:20; R&D Systems AF1056), rabbit anti-RUNX3 (1:50,000; gift from Thomas Jessell), rabbit anti-CASP3 (1:50; BD PharMingen 557035), IB4-488 (1:500, Invitrogen I21411), rabbit anti-CGRP (1:1000; Immunostar 24112), rabbit anti-NF200 (1:500; Sigma N4142), rabbit anti-TRPV1 (1:500; Alomone ACC-030).
Fluorescent images were taken on a Zeiss LSM880 confocal microscope with a 3 μm optical slice and the 20× objective. Images were pseudocolored with a magenta/yellow/cyan color scheme using Adobe Photoshop (Adobe) or Fiji. Cell counts were conducted manually using the built-in cell counter plugin on Fiji.
In situ hybridization
A probe for ISH was generated targeting a 482 bp sequence entirely within exon V of Prdm12 (see Table S3). ISH was performed as per standard protocols. Detailed protocol is available upon request. Briefly, DRG sections (30 μm) were dried at 50°C for 15 min then fixed in 4% PFA in DEPC-PBS for 20 min at RT. The sections were washed in DEPC-PBS for 5 min at RT then incubation in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% Na deoxycholate, 0.1% SDS, 1 mM EDTA, 50 mM Tris pH 8.0) for 60 min. The sections were then washed in DEPC-water followed by acetylation (500 μL of acetic anhydride in 200 mL of 0.1 M RNase-free triethanolamine-HCl at pH 8.0), washed in DEPC-PBS for 5 min., and prehybridized for 4 h at 64°C. Sections were incubated overnight at 64°C with 1–2 ng/μL of fresh Prdm12 probe. The following day, a series of low and high stringency washes in 2x and 0.2X SSC as well as treatment with RNaseA and RNase T1 were performed. The sections were blocked in 10% inactivated sheep serum for 1 h followed by overnight incubation with 1:1000 anti-digoxygenin (DIG) antibody (Roche). The sections were washed in PBT and incubated with NBT/BCIP (Roche) staining solution until the blue precipitate formed. The slides were then washed in PBS and coverslipped with Aqua-Poly/Mount (Polysciences Inc.) mounting media.
The RNAscope Fluorescent Multiplex Assay (Advanced Cell Diagnostics Inc., Hayward, CA) was performed according to the manufacturer’s instructions using a Prdm12 exon V-specific probe (ACDBio, 547081-C3). All incubation steps were performed in a HybEZ™ II oven set to 40°C. The slides were washed with distilled water three times and incubated with Protease III for 40 s. Slides were then washed with distilled water three times and incubated with the probe targeting Prdm12 for 2 hours. The slides were washed two times thoroughly using 1X wash buffer for 2 min, then incubated with Amp 1-Fl for 30 minutes. The same process (washing then treatment) was repeated for Amp 2-Fl, Amp 3-Fl and Amp 4-Fl for 15, 30 and 15 minutes, respectively. Slides were washed three times in PBS for 10 minutes and coverslipped with Aqua-Poly/Mount (Polysciences, Inc.) mounting media.
RNA-sequencing
RNA was extracted from lumbar DRGs 2-5 following the manufacturer’s protocol with a Direct-zol RNA Miniprep Plus kit (Zymo R2071). RNA libraries were prepared and sequenced by the UTSW Microarray core facility on an Illumina NextSeq SE-75 sequencer at 40 million reads/sample. RNA-seq reads were mapped to mouse genome (mm10) and junctions were identified using tophat(v2.1.2) (Kim et al., 2013). Differential expression analysis was performed using cufflinks (v.2.2.1) (Trapnell et al., 2013). Both alignment and differential expression analysis were performed using default parameters. From differential expression results, genes showing expression of > = 1 FPKM in either of the conditions cutoff was used in addition to FDR (5%) and fold change (2-fold up/down) cutoffs.
RT-qPCR
Mouse DRGs were dissected and transferred into Trizol reagent (Invitrogen 15596026), then homogenized in a dounce homogenizer with 20 strokes of pestle A followed by 20 strokes of pestle B. RNA was extracted using the Arcturus PicoPure RNA Isolation Kit (Applied Biosystems KIT0204), and cDNA was synthesized using the SuperScript VILO cDNA Synthesis Kit (Invitrogen 11754250). Real-time RT-qPCR was performed in triplicate using the 7500 Fast real-time PCR system (Applied Biosystems) and PowerUp SYBR Green Master Mix (Applied Biosystems A25776). Results were analyzed by calculating ΔCt with respect to Hprt1 to determine relative expression levels and assess for statistical significance, then calculating ΔΔCt to determine fold-change (FC) in mutants compared to control. All primer pairs used were validated using a 1:5 dilution series in triplicate, and used only if the slope of the standard curve was −3.3 ± 10% and R2 > 0.99. Primer pairs are listed in Table S3.
QUANTIFICATION AND STATISTICAL ANALYSIS
Cell counts were averaged across sections from 3 unique lumbar (L2-L5) DRGs per specimen, with 3 embryos or mice per time point or condition as indicated. Where applicable, counts were taken as a fraction of DRG area (measured in Fiji), or as a percentage of TOM+ cells. A two-tailed Student’s t test was used to analyze cell counts, most behavior results, and gene expression between control and mutant samples. If more than two variables were being compared, such as when analyzing data over time, or molecular changes in control and mutant with and without injury (Figures 6C and 6E–6I), a 2-way ANOVA was used, with post hoc Tukey tests as applicable. All data and graphs were processed in Microsoft Excel 2015 and GraphPad Prism 9. Mean ± SEM is reported throughout the manuscript.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | ||
|---|---|---|---|---|
| Antibodies | ||||
| Goat polyclonal anti-TRKA | R&D Systems | Cat#AF1056; RRID:AB_2283049 | ||
| Rabbit polyclonal anti-RUNX3 | Gift from Thomas Jessell | N/A | ||
| Rabbit polyclonal anti-CASP3 | PharMingen | Cat#557035; RRID:AB_396558 | ||
| Rabbit polyclonal anti-CGRP | Immunostar | Cat#24112; RRID:AB_572217 | ||
| Rabbit anti-TRPV1 | Alomone | Cat#ACC-030; RRID:AB_2313819 | ||
| Rabbit polyclonal anti-NF200 | Sigma | Cat#N4142; RRID:AB_477272 | ||
| Mouse monoclonal anti anti-Islet1/2 | DSHB | Cat#39.4D5; RRID:AB_2314683 | ||
| Chemicals, peptides, and recombinant proteins | ||||
| Chloroquine | Sigma | Cat#C6628 | ||
| Histamine | Sigma | Cat#H7250 | ||
| Capsaicin | Sigma | Cat#M2028 | ||
| Formaldehyde solution | Sigma | Cat#252549 | ||
| Complete Freund’s Adjuvant | Sigma | Cat#F5881 | ||
| 5-ethynyl-2′-deoxyuridine (EdU) | Carbosynth | Cat#NE08701 | ||
| Sulfo-Cy3 azide | Lumiprobe | Cat#C1330 | ||
| IB4-488 Conjugate | Invitrogen | Cat#I21411 | ||
| RNAscope Probe-Mm-Prdm12-O1-C3 | ACDBio | Cat#547081-C3 | ||
| TRIzol Reagent | Invitrogen | Cat#15596018 | ||
| PowerUp SYBR Green MasterMix | Applied Biosystems | Cat#A25776 | ||
| Critical commercial assays | ||||
| RNAScope Fluorescent Multiplex Reagent Kit |
ACDBio | Cat#320850 | ||
| Direct-zol RNA Miniprep Plus Kit | Zymo Research | Cat#R2071 | ||
| Arcturus PicoPure RNA Isolation Kit | Applied Biosystems | KIT0204 | ||
| SuperScript VILO cDNA Synthesis Kit | Invitrogen | Cat#11754250 | ||
| Deposited data | ||||
| RNA-seq Data | This Paper | GEO: GSE167468 | ||
| Experimental models: organisms/strains | ||||
| Prdm12F/F Mice | Chen et al., 2020 | N/A | ||
| CAG-Cre Mice | Sakai and Miyazaki, 1997 | N/A | ||
| AdvilinlCre/+ Mice | The Jackson Laboratory | JAX: 032536 | ||
| AdvillinCreERT2/+ Mice | The Jackson Laboratory | JAX: 032027 | ||
| R26LSL-tdTomato Mice | The Jackson Laboratory | JAX: 007908 | ||
| R26LSL-EYFP Mice | The Jackson Laboratory | JAX 007903 | ||
| Oligonucleotides | ||||
| Primers for genotyping and RT-qPCR, Table S3 | This Paper | N/A | ||
| Software and algorithms | ||||
| GraphPad Prism 9 | GraphPad | RRID:SCR_002798 | ||
| Other | ||||
| Von Frey Filaments | Bioseb | Bio-VF-M | ||
| 0.2 mm Insect Pins | FST | Cat#26002-20 | ||
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | ||
| Rodent pincher analgesia meter | Bioseb | Bio-RP-M | ||
| Hot plate analgesia meter | IITC | Cat#39 | ||
| Hargreaves Apparatus | IITC | Cat#390 | ||
Highlights.
Prdm12 is necessary for the development of nociceptive neurons
Embryonic Prdm12 conditional knockout causes sensory defects
Adult Prdm12 conditional knockout has a limited effect on nociception
Transcriptional changes differ between adult and embryonic knockout
ACKNOWLEDGMENTS
This work was supported by F31 NS111796 and the William F. and Grace H. Kirkpatrick award to M.A.L.; NIH R01 DK114036 to C.L.; the Rita Allen Foundation Award in Pain; the Welch Foundation; the President’s Research Council Award; the Kent Waldrep Foundation; and NIH R01 NS100741 to H.C.L. We thank Thomas Jessell for the RUNX3 antibody; Tou Yia Vue for help with EdU staining; Fan Wang for the AdvillinCre/+ mice; the UTSW Microarray Core Facility, UTSW Transgenic Core Facility, Neuroscience Microscopy Facility supported by the UTSW Neuroscience Department, and the UTSW Peter O’Donnell, Jr., Brain Institute; Stephanie Shiers, Rahul K. Kollipara, and the Johnson laboratory for technical assistance; Saida Hadjab and Vanja Nagy for helpful discussions; and Ted Price and Jane Johnson for critical reading of the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108913.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects.
REFERENCES
- Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, Martinez-Barbera JP, Nassar MA, Dickenson AH, and Wood JN (2008). The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 321, 702–705. [DOI] [PubMed] [Google Scholar]
- Bartesaghi L, Wang Y, Fontanet P, Wanderoy S, Berger F, Wu H, Akkuratova N, Bouçanova F, Médard J-JJ, Petitpré C, et al. (2019). PRDM12 is required for initiation of the nociceptive neuron lineage during neurogenesis. Cell Rep 26, 3484–3492.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonin RP, Bories C, and De Koninck Y (2014). Molecular pain: a simplified up-down method (SUDO) for measuring mechanical nociception in rodents using von Frey filaments A simplified up-down method (SUDO) for measuring mechanical nociception in rodents using von Frey filaments. Mol. Pain 10, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner DS, Golden JP, and Gereau RW IV. (2012). A novel behavioral assay for measuring cold sensation in mice. PLoS One 7, e39765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao XH, Byun HS, Chen SR, Cai YQ, and Pan HL (2010). Reduction in voltage-gated K+ channel activity in primary sensory neurons in painful diabetic neuropathy: role of brain-derived neurotrophic factor. J. Neurochem 114, 1460–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capsoni S, Covaceuszach S, Marinelli S, Ceci M, Bernardo A, Minghetti L, Ugolini G, Pavone F, and Cattaneo A (2011). Taking pain out of NGF: a “painless” NGF mutant, linked to hereditary sensory autonomic neuropathy type V, with full neurotrophic activity. PLoS ONE 6, e17321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho OP, Thornton GK, Hertecant J, Houlden H, Nicholas AK, Cox JJ, Rielly M, Al-Gazali L, and Woods CG (2011). A novel NGF mutation clarifies the molecular mechanism and extends the phenotypic spectrum of the HSAN5 neuropathy. J. Med. Genet 48, 131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SR, Cai YQ, and Pan HL (2009). Plasticity and emerging role of BKCa channels in nociceptive control in neuropathic pain. J. Neurochem 110, 352–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina Michael, et al. (1999). A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 398, 436–441. 10.1038/18906. [DOI] [PubMed] [Google Scholar]
- Chen YC, Auer-Grumbach M, Matsukawa S, Zitzelsberger M, Themistocleous AC, Strom TM, Samara C, Moore AW, Cho LT, Young GT, et al. (2015). Transcriptional regulator PRDM12 is essential for human pain perception. Nat. Genet 47, 803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Wyler SC, Li L, Arnold AG, Wan R, Jia L, Landy MA, Lai HC, Xu P, and Liu C (2020). Comparative transcriptomic analyses of developing melanocortin neurons reveal new regulators for the anorexigenic neuron identity. J. Neurosci 40, 3165–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, et al. (2006). An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decosterd I, and Woolf CJ (2000). Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87, 149–158. [DOI] [PubMed] [Google Scholar]
- Desiderio S, Vermeiren S, Van Campenhout C, Kricha S, Malki E, Richts S, Fletcher EV, Vanwelden T, Schmidt BZ, Henningfeld KA, et al. (2019). Prdm12 directs nociceptive sensory neuron development by regulating the expression of the NGF receptor TrkA. Cell Rep 26, 3522–3536.e5. [DOI] [PubMed] [Google Scholar]
- Einarsdottir E, Carlsson A, Minde J, Toolanen G, Svensson O, Solders G, Holmgren G, Holmberg D, and Holmberg M (2004). A mutation in the nerve growth factor beta gene (NGFB) causes loss of pain perception. Hum. Mol. Genet 13, 799–805. [DOI] [PubMed] [Google Scholar]
- Fog CK, Galli GG, and Lund AH (2012). PRDM proteins: important players in differentiation and disease. BioEssays 34, 50–60. [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Abbott S, Han B-X, Qi Y, and Wang F (2007). Analyzing somatosensory axon projections with the sensory neuron-specific Advillin gene. J. Neurosci 27, 14404–14414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EM, Zhang Z, Anderson MB, Schechter R, and Miller KE (2011). Potential mechanisms for hypoalgesia induced by anti-nerve growth factor immunoglobulin are identified using autoimmune nerve growth factor deprivation. Neuroscience 193, 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenauer T, and Moore AW (2012). The Prdm family: expanding roles in stem cells and development. Development 139, 2267–2282. [DOI] [PubMed] [Google Scholar]
- Hunter DV, Smaila BD, Lopes DM, Takatoh J, Denk F, and Ramer MS (2018). Advillin is expressed in all adult neural crest-derived neurons. eNeuro 5, eneuro.0077-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imhof S, Kokotović T, and Nagy V (2020). PRDM12: new opportunity in pain research. Trends Mol. Med 26, 895–897. [DOI] [PubMed] [Google Scholar]
- Indo Y (2014). Nerve growth factor, pain, itch and inflammation : lessons from congenital insensitivity to pain with anhidrosis. Expert Rev. Neurother 10, 1707–1724. [DOI] [PubMed] [Google Scholar]
- Inoue K, Ozaki S, Shiga T, Ito K, Masuda T, Okado N, Iseda T, Kawaguchi S, Ogawa M, Bae SC, et al. (2002). Runx3 controls the axonal projection of proprioceptive dorsal root ganglion neurons. Nat. Neurosci 5, 946–954. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinameri E, Inoue T, Aruga J, Imayoshi I, Kageyama R, Shimogori T, and Moore AW (2008). Prdm proto-oncogene transcription factor family expression and interaction with the Notch-Hes pathway in mouse neurogenesis. PLoS ONE 3, e3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao Y, Robertson B, Kudo M, and Grant G (2002). Proliferation patterns of dorsal root ganglion neurons of cutaneous, muscle and visceral nerves in the rat. J. Neurocytol 31, 765–776. [DOI] [PubMed] [Google Scholar]
- Kramer I, Sigrist M, de Nooij JC, Taniuchi I, Jessell TM, and Arber S (2006). A role for Runx transcription factor signaling in dorsal root ganglion sensory neuron diversification. Neuron 49, 379–393. [DOI] [PubMed] [Google Scholar]
- Kuraishi Y, Nagasawa T, Hayashi K, and Satoh M (1995). Scratching behavior induced by pruritogenic but not algesiogenic agents in mice. Eur. J. Pharmacol 275, 229–233. [DOI] [PubMed] [Google Scholar]
- Laedermann CJ, Pertin M, Suter MR, and Decosterd I (2014). Voltagegated sodium channel expression in mouse DRG after SNI leads to re-evaluation of projections of injured fibers. Mol. Pain 10, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemend F, and Ernfors P (2012). Molecular interactions underlying the specification of sensory neurons. Trends Neurosci 35, 373–381. [DOI] [PubMed] [Google Scholar]
- Lau J, Minett MS, Zhao J, Dennehy U, Wang F, Wood JN, and Bogdanov YD (2011). Temporal control of gene deletion in sensory ganglia using a tamoxifen-inducible Advillin-Cre-ERT2 recombinase mouse. Mol. Pain 7, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumet G, Garriga J, Chen SR, Zhang Y, Li DP, Smith TM, Dong Y, Jelinek J, Cesaroni M, Issa JP, and Pan HL (2015). G9a is essential for epigenetic silencing of K+ channel genes in acute-to-chronic pain transition. Nat. Neurosci 18, 1746–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson SN, and Biscoe TJ (1979). Development of mouse dorsal root ganglia: an autoradiographic and quantitative study. J. Neurocytol 8, 265–274. [DOI] [PubMed] [Google Scholar]
- Leipold E, Liebmann L, Korenke GC, Heinrich T, Giesselmann S, Baets J, Ebbinghaus M, Goral RO, Stödberg T, Hennings JC, et al. (2013). A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat. Genet 45, 1399–1404. [DOI] [PubMed] [Google Scholar]
- Levanon D, Bettoun D, Harris-Cerruti C, Woolf E, Negreanu V, Eilam R, Bernstein Y, Goldenberg D, Xiao C, Fliegauf M, et al. (2002). The Runx3 transcription factor regulates development and survival of TrkC dorsal root ganglia neurons. EMBO J 21, 3454–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Gu X, Zhao JY, Wu S, Miao X, Xiao J, Mo K, Zhang J, Lutz BM, Bekker A, and Tao YX (2016). G9a participates in nerve injury-induced Kcna2 downregulation in primary sensory neurons. Sci. Rep 6, 37704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Fode C, Guillemot F, and Anderson DJ (1999). Neurogenin1 and neurogenin2 control two distinct waves of neurogenesis in developing dorsal root ganglia. Genes Dev 13, 1717–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukawa S, Miwata K, Asashima M, and Michiue T (2015). The requirement of histone modification by PRDM12 and Kdm4a for the development of pre-placodal ectoderm and neural crest in Xenopus. Dev. Biol 399, 164–176. [DOI] [PubMed] [Google Scholar]
- Moore AW, Roegiers F, Jan LY, and Jan Y (2004). Conversion of neurons and glia to external-cell fates in the external sensory organs of Drosophila hamlet mutants by a cousin-cousin cell-type respecification. Genes Dev 18, 623–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moqrich A, Earley TJ, Watson J, Andahazy M, Backus C, Martin-zanca D, Wright DE, Reichardt LF, and Patapoutian A (2004). Expressing TrkC from the TrkA locus causes a subset of dorsal root ganglia neurons to switch fate. Nat. Neurosci 7, 812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss CD, Srinivas SM, Sarveswaran N, Nahorski M, Gowda VK, Browne FM, Woods GD, and Moss C (2018). Midface toddler excoriation syndrome (MiTES) can becaused by autosomal recessive biallelic mutations in a gene for congenital insensitivity to pain, PRDM12. Br. J. Dermatol 179, 1135–1140. [DOI] [PubMed] [Google Scholar]
- Nagy V, Cole T, Van Campenhout C, Khoung TM, Leung C, Vermeiren S, Novatchkova M, Wenzel D, Cikes D, Polyansky AA, et al. (2015). The evolutionarily conserved transcription factor PRDM12 controls sensory neuron development and pain perception. Cell Cycle 14, 1799–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahorski MS, Chen YC, and Woods CG (2015). New Mendelian disorders of painlessness. Trends Neurosci 38, 712–724. [DOI] [PubMed] [Google Scholar]
- Renthal W, Tochitsky I, Yang L, Cheng YC, Li E, Kawaguchi R, Geschwind DH, and Woolf CJ (2020). Transcriptional reprogramming of distinct peripheral sensory neuron subtypes after axonal injury. Neuron 108, 128–144.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini AG, Padmanabh H, Sahu JK, Kurth I, Voigt M, and Singhi P (2017). Hereditary sensory polyneuropathy, pain insensitivity and global developmental delay due to novel mutation in PRDM12 gene. Indian J. Pediatr 84, 332–333. [DOI] [PubMed] [Google Scholar]
- Sakai K, and Miyazaki Ji. (1997). A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem. Biophys. Res. Commun 237, 318–324. [DOI] [PubMed] [Google Scholar]
- Sharma N, Flaherty K, Lezgiyeva K, Wagner DE, Klein AM, and Ginty DD (2020). The emergence of transcriptional identity in somatosensory neurons. Nature 577, 392–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada SG, and LaMotte RH (2008). Behavioral differentiation between itch and pain in mouse. Pain 139, 681–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikandar S, Minett MS, Millet Q, Santana-Varela S, Lau J, Wood JN, and Zhao J (2018). Brain-derived neurotrophic factor derived from sensory neurons plays a critical role in chronic pain. Brain 141, 1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeyne RJ, Klein R, Schnappt A, Long LK, Bryant S, Lewin A, Lira SA, and Barbacid M (1994). Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature 368, 17–20. [DOI] [PubMed] [Google Scholar]
- Srinivas SM, Gowda VK, Owen CM, Moss C, and Hiremagalore R (2017). Mid-face toddler excoriation syndrome (MiTES): a new paediatric diagnosis. Clin. Exp. Dermatol 42, 68–71. [DOI] [PubMed] [Google Scholar]
- Thélie A, Desiderio S, Hanotel J, Quigley I, Van Driessche B, Rodari A, Borromeo MD, Kricha S, Lahaye F, Croce J, et al. (2015). Prdm12 specifies V1 interneurons through cross-repressive interactions with Dbx1 and Nkx6 genes in Xenopus. Development 142, 3416–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujino H, Kondo E, Fukuoka T, Dai Y, Tokunaga A, Miki K, Yonenobu K, Ochi T, and Noguchi K (2000). Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: a novel neuronal marker of nerve injury. Mol. Cell Neurosci 182, 170–182. [DOI] [PubMed] [Google Scholar]
- Usoskin D, Furlan A, Islam S, Abdo H, Lönnerberg P, Lou D, Hjerling-Leffler J, Haeggström J, Kharchenko O, Kharchenko PV, et al. (2015). Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci 18, 145–153. [DOI] [PubMed] [Google Scholar]
- Vandewauw I, De Clercq K, Mulier M, Held K, Pinto S, Van Ranst N, Segal A, Voet T, Vennekens R, Zimmermann K, et al. (2018). A TRP channel trio mediates acute noxious heat sensing. Nature 555, 662–666. [DOI] [PubMed] [Google Scholar]
- Wang L, Bluske KK, Dickel LK, and Nakagawa Y (2011). Basal progenitor cells in the embryonic mouse thalamus - their molecular characterization and the role of neurogenins and Pax6. Neural Dev 6, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieskopf JS, Mathur J, Limapichat W, Post MR, Al-Qazzaz M, Sorge RE, Martin LJ, Zaykin DV, Smith SB, Freitas K, et al. (2015). The nicotinic α6 subunit gene determines variability in chronic pain sensitivity via cross-inhibition of P2X2/3 receptors. Sci. Transl. Med 7, 287ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C-M, and Shinkai Y (2013). Prdm12 is induced by retinoic acid and exhibits anti-proliferative properties through the cell cycle modulation of P19 embryonic carcinoma cells. Cell Struct. Funct 38, 197–206. [DOI] [PubMed] [Google Scholar]
- Zhang S, Malik Sharif S, Chen Y-C, Valente E-M, Ahmed M, Sheridan E, Bennett C, and Woods G (2016). Clinical features for diagnosis and management of patients with PRDM12 congenital insensitivity to pain. J. Med. Genet 53, 533–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wang L, Hasegawa H, Amin P, Han B-X, Kaneko S, He Y, and Wang F (2010). Deletion of PIK3C3/Vps34 in sensory neurons causes rapid neurodegeneration by disrupting the endosomal but not the autophagic pathway. Proc. Natl. Acad. Sci. USA 107, 9424–9429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq data reported in this paper is GEO: GSE167468.