

Abstract
Being responsible for more than 90% of cellular functions, protein molecules are workhorses in all the life forms. In order to cater for such a high demand, proteins have evolved to adopt diverse structures that allow them to perform myriad of functions. Beginning with the genetically directed amino acid sequence, the classical understanding of protein function involves adoption of hierarchically complex yet ordered structures. However, advances made over the last two decades have revealed that inasmuch as 50% of eukaryotic proteome exists as partially or fully disordered structures. Significance of such intrinsically disordered proteins (IDPs) is further realized from their ability to exhibit multifunctionality, a feature attributable to their conformational plasticity. Among the coded amino acids, cysteines are considered to be “order-promoting” due to their ability to form inter- or intramolecular disulfide bonds, which confer robust thermal stability to the protein structure in oxidizing conditions. The co-existence of order-promoting cysteines with disorder-promoting sequences seems counter-intuitive yet many proteins have evolved to contain such sequences. In this chapter, we review some of the known cysteine-containing protein domains categorized based on the number of cysteines they possess. We show that many protein domains contain disordered sequences interspersed with cysteines. We show that a positive correlation exists between the degree of cysteines and disorder within the sequences that flank them. Furthermore, based on the computational platform, IUPred2A, we show that cysteine-rich sequences display significant disorder in the reduced but not the oxidized form, increasing the potential for such sequences to function in a redox-sensitive manner. Overall, this chapter provides insights into an exquisite evolutionary design wherein disordered sequences with interspersed cysteines enable potential modulatory protein functions under stress and environmental conditions, which thus far remained largely inconspicuous.
1. Introduction
Proteins are the workhorses of cellular functions. More than 90% of cellular activities involve proteins at some level, making them the functionally diverse class of biomolecules. Proteins also exhibit extraordinary diversity in their structures, which underpins their ability to carry out a plethora of cellular activities. Their ability to fold into hierarchically organized assemblies, from secondary to quaternary structures, is guided by the commands encoded in the primary sequence of amino acids derived from their corresponding genes. Dramatic examples for specialized functions achieved via specific protein structure include enzymes, which adopt well defined conformations to impart specificity and selectivity during catalysis. Numerous such examples exist, such as membrane bound receptors, channels, immunoglobulins, etc., wherein precisely folded conformations of the proteins lead to optimally specialized functions. Since the determination of the first protein structure (myoglobin) at an atomic level resolution by Kendrew and colleagues1 followed by that of hemoglobin by Perutz and colleagues in 1960,2 protein folding and structure have dominated the attention of structural biologists during the second half of the twentieth century. With the use of X-ray crystallography and nuclear magnetic resonance (NMR), numerous protein structures were solved at high resolutions that now dominate in protein data bank (PDB), with ~500,000 structures. A well-folded, albeit dynamic structure of proteins containing defined secondary and/or tertiary elements was considered to be the hallmarks for imparting functions.
The dynamics of the protein structure was not discounted though; for instance, the classical “lock and key” model for the enzyme-substrate interactions was modified to an “induced fit” one. However, unstructured, flexible regions of the proteins were largely ignored and considered unimportant both structurally and functionally. A paradigm shift in protein science occurred in the 1990s with the identification of parts of the proteins that were structurally ill-defined and those that deviated from the well-known structural motifs as functionally relevant and often times, even important.3–5 Such regions called the “intrinsically disordered regions” (IDRs) have emerged into prominence since then. Furthermore, many fully disordered proteins were discovered that underwent a “disorder to order” transition along with those that never gained structure. Protein that fell in the latter category are called “intrinsically disordered proteins” (IDPs) or “intrinsically unstructured proteins” (IUPs). There is now a compelling evidence suggesting that many functional protein regions, and even entire functional proteins, lack stable tertiary and/or secondary structure in solution, and instead exist as dynamic ensembles of interconverting structures. This indicates that the protein universe includes not only transmembrane, globular, and fibrous proteins, but also IDPs and IDRs. Understandably, the significance of these disordered proteins was heavily contested initially and were received with skepticism for it challenged the fundamental principles of protein structure and function of the time. With the passage of nearly three decades, it is now clear that the ordered proteins occupy only half of the proteome in various domains while it less than half in eukaryotic proteome.6 IDPs and IDRs play significant roles in both norm and pathology just as the ordered proteins do.7–9 Furthermore, IDPs are often associated with multiple functions and are represented in almost every major cellular process. Although these IDPs and IDRs are biologically active, they still fail to form specific 3D structures, and instead exist as extended dynamically mobile conformational or collapsed ensembles.5,7,10–15 These floppy proteins and regions have been known by a variety of names, including pliable, rheomorphic,16 flexible,17 mobile,18 partially folded,19 natively denatured,20 natively unfolded,11,21 natively disordered,13 intrinsically unstructured,7,12 intrinsically denatured,20 intrinsically unfolded,21 intrinsically disordered,5 vulnerable,22 chameleon,23 malleable,24 4D proteins,25 protein clouds,26 dancing proteins,27 and proteins waiting for partners28 among others29. A consensus regarding the naming of these proteins has been reached, and they are now called intrinsically disordered in most of the scientific publications.29 Unlike ordered proteins, whose 3-D structure is relatively stable and whose Ramachandran angles vary only slightly around their equilibrium positions with occasional cooperative conformational switches, IDPs/IDRs exist as structural ensembles, either at the secondary or at the tertiary level. These proteins prevail as dynamic ensembles whose atom positions and backbone Ramachandran angles vary greatly over time without reaching specific equilibrium values, and typically undergo nonco-operative conformational changes.
Remarkable progress has been made during the last decade in the prediction and identification of sequences that show disorder propensity and many numbers of precise computational prediction tools are available.30–32 Such tools also have revealed the precise contributions of individual amino acids for promoting disorder, and out of all the 20 naturally occurring amino acids, cysteines are considered to be the most order promoting (detailed further down). Moreover, oxidation of cysteines to disulfide bonds seem to make this amino acid counter-intuitive to disorder. Here, we analyze PDB to sieve through the proteins that contain both disorder promoting sequences as well as cysteines to varying degrees. We show that such sequences are not serendipitous occurrence but are evolved to accomplish protein functions that require both conformational flexibility as well as stability and order when needed. We conclude that presence of intrinsic disorder and cysteines is an elegant design for proteins to accomplish two widely differentiated attributes of flexibility and stability.
2. IDPs and IDRs
The data accumulated regarding the structural heterogeneity of IDPs suggests that representing these proteins as members of three well-defined structural classes; native molten globules, native premolten globules, and native coils5,13,33 is an oversimplification. IDPs may contain foldons, inducible foldons, semi-foldons, and nonfoldons; some disordered proteins might also have unfoldons, which are the regions that require an order-to-disorder transition to make the protein active.34 Therefore, the currently available data suggests that intrinsic disorder can have multiple faces and can affect different levels of a protein’s structural organization or entire proteins, and that different protein regions can be disordered to different degrees. As a result, it has been proposed that functional proteins are representative of a continuous spectrum of differently disordered conformations that extends from fully ordered to completely structure-less, and every stage in-between.34 It has also been shown that there is no distinct boundary between ordered proteins and IDPs, and the structure-disorder space of a protein should instead be viewed as a continuum.34 This immediately implied dramatic extension of protein functionality. In fact, IDPs/IDRs, with their capability to undergo disorder-to-order transitions, enrichment in various posttranslational modifications (PTMs), and high probability of being affected by alternative splicing, serve as a foundation of the proteoform concept, where proteoforms constitute a set of structurally and functionally distinct protein molecules encoded by a single gene.35,36 They are also a cornerstone of the “protein structure-function continuum” model, where any protein represents a dynamic conformational ensemble, and where multiple proteoforms have different structural features with various functions.35,37,38
A more detailed analysis to gain additional information on the compositional difference between a nonredundant set of ordered proteins with several datasets of disordered proteins (where proteins were grouped based on different techniques used to identify disorder, such as X-ray crystallography, NMR, and CD) revealed that IDPS/IDRs share at least some common sequence features over many proteins.3,4 In fact, the IDPS/IDRs were shown to be significantly depleted in bulky hydrophobic (Ile, Leu, and Val) and aromatic amino acid residues (Trp, Tyr, and Phe), which would normally form the hydrophobic core of a folded globular protein, and also possess a low amount of Cys and Asn residues. It has been proposed that these depleted residues, Trp, Tyr, Phe, Ile, Leu, Val, Cys, and Asn be called order-promoting amino acids. On the other hand, IDPs were shown to be substantially enriched in Ala, as well as in polar, disorder-promoting amino acids: Arg, Gly, Gln, Ser, Glu, and Lys, and also in the hydrophobic, but structure-breaking Pro.5,39–42 Based on their frequencies of being found in ordered or disordered proteins amino acid residues are arranged in the following sequence (from the most order-promoting residues to the most disorder-promoting ones): Cys, Trp, Tyr, Ile, Phe, Val, Leu, His, Thr, Asn, Ala, Gly, Asp, Met, Lys, Arg, Ser, Gln, Pro, and Glu.5,39–42 Note that these biases in the amino acid compositions of IDPs/IDRs are also consistent with the low overall hydrophobicity and high net charge characteristic of natively unfolded proteins.
Ordered and disordered proteins and regions are different from each other at multiple levels. In addition to amino acid composition, these differences include hydropathy, net charge, flexibility index, helix propensities, strand propensities, aromaticity (which is a combined Trp+Tyr+Phe content), and some other combined compositions of various groups of amino acids. Taking these observations into account, 265 property-based attribute scales39 and more than 6000 composition-based attributes (e.g., all possible combinations that have one to four amino acids in the group) have been compared.43 This comparison revealed that 10 of these attributes, including the 14Å contact number, flexibility, hydropathy, coordination number, β-sheet propensity, content of major disorder-promoting residues (Arg+Ser+Pro+Glu), volume, content of major order-promoting residues (Cys+Trp+Tyr+Phe), bulkiness, and net charge provide a fairly good discrimination between order and disorder.5 Later, 517 amino acid scales (including a variety of hydrophobicity scales, different measures of side chain bulkiness, polarity, volume, compositional attributes, the frequency of each single amino acid, etc.) were analyzed to construct a new amino acid attribute that discriminates between order and disorder44 This new scale out-performed the original 517 amino acid scales for discrimination of order and disorder, and provided a new ranking for the tendencies of amino acid residue to promote order or disorder: Trp, Phe, Tyr, Ile Met, Leu, Val, Asn, Cys, Thr, Ala, Gly, Arg, Asp, His, Gln, Lys, Ser, Glu, Pro.44
It was pointed out that the depletion of IDPs/IDRs in Cys is defined by a significant contribution of this amino acid residue to the protein conformational stability. In fact, cysteine residues can form disulfide bonds or be involved in the coordination of different prosthetic groups (e.g., Zn2+ ions in zinc-fingers), or be subjected to numerous PTMs. Furthermore, redox-sensing cysteine residues were shown to regulate functional behavior of target proteins in response to changes in the levels of the reactive oxygen species (ROS).45 In fact, this is in line with the well-known context-dependence of the structural behavior of IDPs/IDRs, where a disorder-to-order or order-to-disorder transition can be induced in a target protein as a result of its binding to specific macromolecular partners, as a consequence of the undergoing of posttranslational modifications, or as a response to changes in environmental factors such as pH and temperature.46 The redox-regulated conditionally disordered proteins play a number of important roles in response to various forms of oxidative stress or to naturally occurring changes in redox potential.45–47 The functions of these redox-regulated conditionally disordered proteins can be regulated by oxidative modifications of a specific cysteine thiol or through disulfide bond formation by redox-sensitive cysteine residues. In response to changes in the cellular redox status, formation of such disulfide bridges can promote both disorder-to-order and order-to-disorder transitions thereby affecting protein functionality and indicating importance of such thiol-switch proteins.45,47–49
3. Cysteines in structure–function relation of proteins
From an evolutionary perspective, cysteines are believed to be incorporated late as coded amino acids based on their prevalence, which is statistically biased toward higher organisms such as eukaryotes.50 Cysteine is unique among all the coded amino acids marked by its high reactivity and redox chemistry and constitutes the only amino acid that can form covalent bonds in the form of disulfides in oxidative environments that can be reversibly dissociated under reductive conditions. In addition, disulfide bond formation also has a significant effect on the fold and structure of the protein, and so despite being able to undergo myriad PTMs, it is this reversible disulfide bond formation that comes to the fore when looking at structure–function modification by cysteine. While these residues have captivated the attention for their redox-based regulation of protein functions, whether and how disulfide bond formation and dissociation modify the structural dynamics of the protein such as disorder to order and vice versa, have been underexplored. Cysteine residues utilize an elegant mechanism of shuffling between the free thiol, in orther words, reduced form, and the disulfide bonded oxidized form, in a reversible manner dictated by the cellular environment and/or environmental stress.51–53 This redox process has been exploited within proteins to fulfill myriad physiological functions, such as providing protection from oxidative stress, regulation of cell signaling pathways, and coordinating metals51,53,54 among many others. In such redox sensitive proteins, cysteines are found to be enriched and positionally conserved within functionally relevant regions,53,55,56 having significant implications for the structural aspects of the protein as elaborated below. The oxidation of cysteine residues to form disulfide bonds that covalently link two regions of the protein intramolecularly or two independent proteins or chains to form hetero-chain-linked proteins as seen in insulin or immunoglobulins. Along with forming a physical cross-link, this covalent bridge also imparts a degree of structural order to the molecule, due to which cysteines are considered to be order promoting amino acids. On the other hand, the reduced form of cysteines, where such disulfide bonds are abrogated, allows conformational flexibility and disorder. Consequently, widespread tertiary structural changes accompany the redox flux of cysteine in proteins.57,58 Interestingly, cysteine residues do not show any preference for a secondary structure, equally appearing in α-helices, β-strands along with coils, although are notably rare in 310 helices,59,60 indicating a role in maintaining the overall fold or topology of the molecule when bridged. In contrast, reduced forms of cysteine have free thiol groups, which owing to their highly reactive and polar nature can mediate myriad biochemical processes, as for instance exemplified by the catalytic triad of cysteine proteases. The gain in functional utility of the thiol may come at the expense of structural order of the protein typifying the delicate balance between the two in certain class of cysteine containing proteins.
In oxidized, disulfide bonded proteins cysteines are commonly observed to be present within conserved sequences or motifs and one that acquire higher degree of structural order and stability. However, cysteine-containing sequences in proteome show a statistical bias with human and viral proteomes showing the highest degree of enrichment in cysteine-containing sequences.47 This indicates that cysteines are acquired by the proteins to accomplish specialized and specific, complex roles. But even within the 30% sequence density, cysteines show remarkable variations in their distributions and sequence conservation that will be discussed in detail later in the chapter. As described earlier, IDPs and IDRs have come to bear significance in protein functions both in norm and pathology. One of the defining characteristics of a disordered protein is its conformational plasticity, which permits the protein to dynamically adopt multiple structures and perform a multitude of biological functions. Given that the presence of cysteines often leads to the formation of covalent bonds via disulfides, it seems counter-intuitive for cysteines to be abundantly present within IDPs and IDRs. In fact, it is the precise reason why cysteines are considered as order-promoting residues in many of the computational prediction programs. However, a closer look at the proteome reveals the presence of numerous cysteine-rich segments that are flanked by disordered regions even among evolutionarily unrelated but structurally and functionally similar proteins. As described in the rest of this chapter, such sequences reveal mainly redox-based regulation as a key function for IDRs and IDPs containing cysteines with a positive correlation with the density of cysteines present (Fig. 1).
Fig. 1.
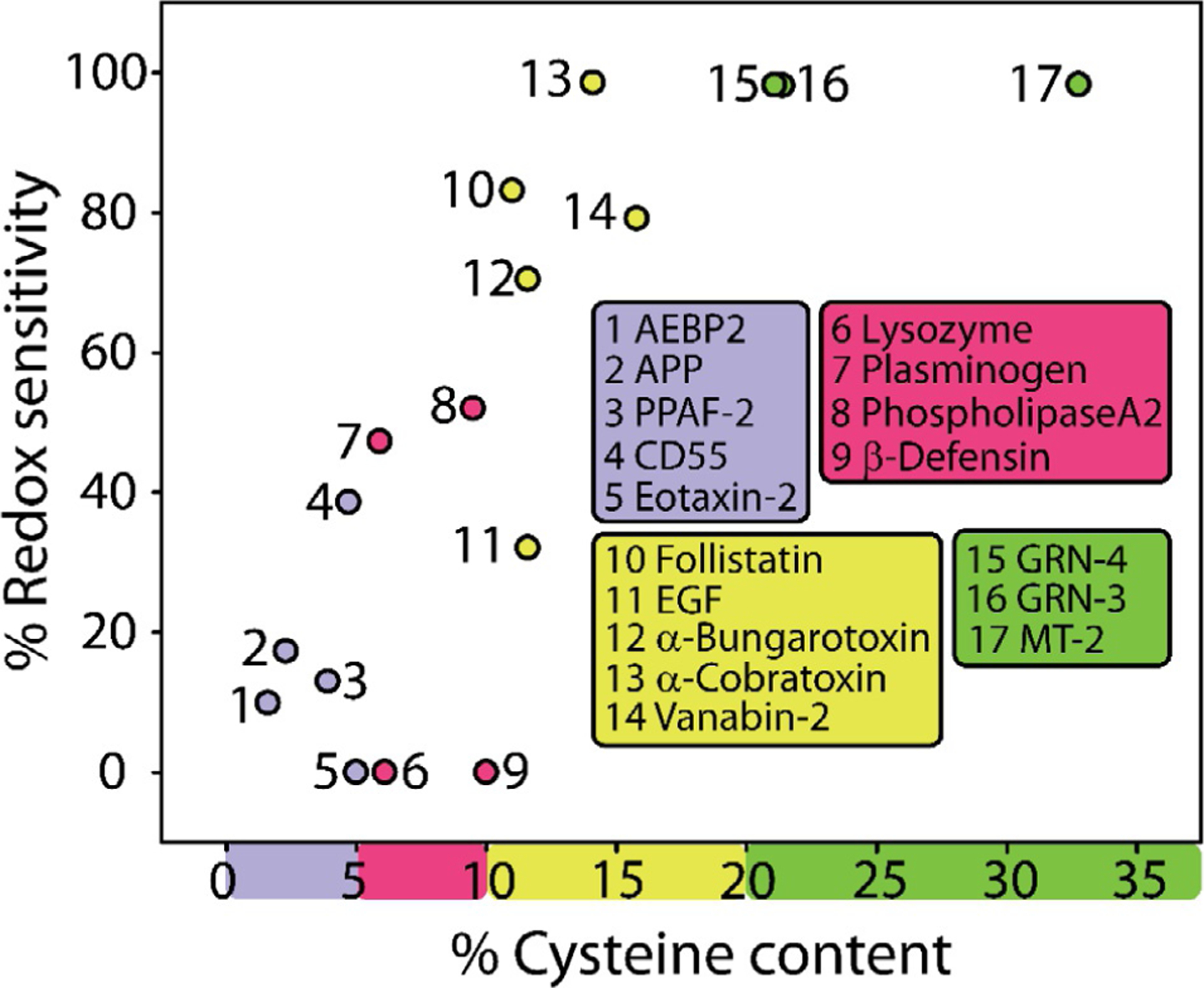
Positive correlation between the percentage of cysteines in the protein and their corresponding redox sensitivity based on IUPred2A predictions.
4. Correlations between cysteine containing protein families, disorder propensity and redox-functions
One of the key aspects involved in the regulation of cellular functions is the sensing and response to oxidative stress. Cells encounter a constant influx of oxidative stress that may occur due to an insult from endogenous or exogenous agents, or during immune response via reactive oxygen species (ROS). Cells have adapted to deal with oxidative stress in many ways, including genetic regulation utilizing regulatory proteins, but the one involving cysteine-based redox dynamics under posttranslational control is observed frequently.51,53,61 Cysteines form covalent bonds either with other cysteines to form disulfides in the oxidative state or interact with divalent metal cations in the reduced state. These reactions often lead to disorder-to-order structural transition and are responsible for cysteines being considered as the order-promoting amino acids in many computational algorithms.30,62,63 From the outset, the ability of thiol functionality in cysteines to undergo redox chemistry seems to be the primary evolutionary reason for the incorporation as the latest encoded amino acid, which is corroborated by their increased presence in Homo sapiens.50 However, the chemical instability of thiols under physiological environments and their inability to remain free is manifested in either their coordination with metal ions or posttranslational modifications.64 When free thiols in cysteines undergo modifications, often an order to disorder transition is favored that enables the potential exposure of unfolded, disordered regions to the solvent.45 Therefore, it is believed that the chemistry of cysteines could contribute to redox-induced folding and unfolding of protein to achieve specialized functions in the cell under normal or stress conditions. However, not many well characterized examples exist to understand this phenomenon fully.
As redox chemistry seems to be the center of cysteine functions, here forth we will discuss the density of cysteines within the protein sequence, as well as the sequences that flank them, in order to systematically analyze their significance. To compile the existing knowledge on the structure–function relation of cysteine domains and explore the redox-induced conformational dynamics within them, we sorted 17 proteins from the UniProt database using their cysteine content and presence of cysteine-containing domains as parameters to choose representative domains in broadly classified categories, the details of which are listed in Section 10. Here, we review some select representative proteins that are clubbed as a function of cysteine density in the protein sequence. The protein groups display both common, as well as unique cysteine-containing sequences that inform us about how this versatile amino acid is utilized in the presence of both disorder- and order-promoting sequences that flank them.
5. Sequences containing <5% cysteines
Highest number of cysteine-containing proteins fall in this category. Some of the commonly observed cysteine-containing domains within a majority of these proteins are discussed here. Among these, the amyloid precursor protein (APP) is a transmembrane protein that is well known for its role in Alzheimer’s disease (AD) pathology and for many hypothesized functional roles. Other proteins in this category include chemokines, serine proteases, nerve growth factors, etc., which utilize cysteine domains in an array of functional roles. APP is a ubiquitously expressed protein within most of the eukaryotic cells. Several isoforms of the protein have been identified that are generated from alternative splicing; APP695 (695 amino acids long), APP751, and APP770, being the most predominant ones. APP is homologous to NOTCH receptors, and its putative roles include promoting cellular proliferation, growth and differentiation, while in the brain, it has been linked to promoting neural outgrowth and synaptogenesis.65–67 Despite being extensively studied, the full spectrum of the functional roles of this protein remains unclear. APP, like many in this category, has a multidomain structure and undergoes extensive posttranslational processing by proteases to release soluble, extracellular and cytoplasmic proteins and peptides, the functions of which are not yet completely understood. Amyloid-β (Aβ) is one such extracellularly produced peptides derived from APP that is involved in AD pathogenesis. The unprocessed extracellular region of the protein is also involved in cell-to-cell signaling and mediates cell adhesion while the cytoplasmic fragments have hypothesized roles in multiple intracellular pathways.67,68 APP consists of three cysteine-containing domains, growth factor-like domain, copper binding domain and Kunitz-type inhibitor domain that are present in the N-terminal, extracellular side of the protein. We will discuss the copper-binding and Kunitz-type domains here, while the growth-factor-like domains are discussed further below.
(i). APP: Copper binding domain (CuBD):
(PDB accession code: 1OWT) CuBD constitutes residues 124–189 of APP. This region is involved in the binding of Cu(II) to APP, which has neuroprotective functions against oxidative stress.69 The domain consists of an α-helical segment (residues 147–159) sandwiched between three β-strands (β1:133–139, β2:162–167 and β3:181–188) that form a β-sheet (Fig. 2A: 1, left panel). Since this domain lies in the oxidizing extracellular milieu, six cysteines form three disulfide bonds. Cys133–Cys187 connects the strands β1 and β3, lying at extreme ends of the domain. Cys158–Cys186 bridges the α-helix to strand β3 while in the central disulfide bond, Cys144–Cys174 links two loops at either ends of the structure. The co-ordination of Cu(II) is performed by four ligands; His147, His151, Tyr168, and Met-170. The tetrahedrally coordinated Cu(II) is reduced to Cu(I). The coordination sphere and overall higher structure assumed by CuBD shows significant similarity to copper chaperones, despite thiols not being used as ligands.70 Cysteines play a structural role within this domain as well, remaining functionally inert. But as evident from Fig. 2A, right panel, this region shows the maximum disorder under reducing conditions and in turn suggests that conformational flexibility is achieved under reduced form for binding metal ions.
Fig. 2.
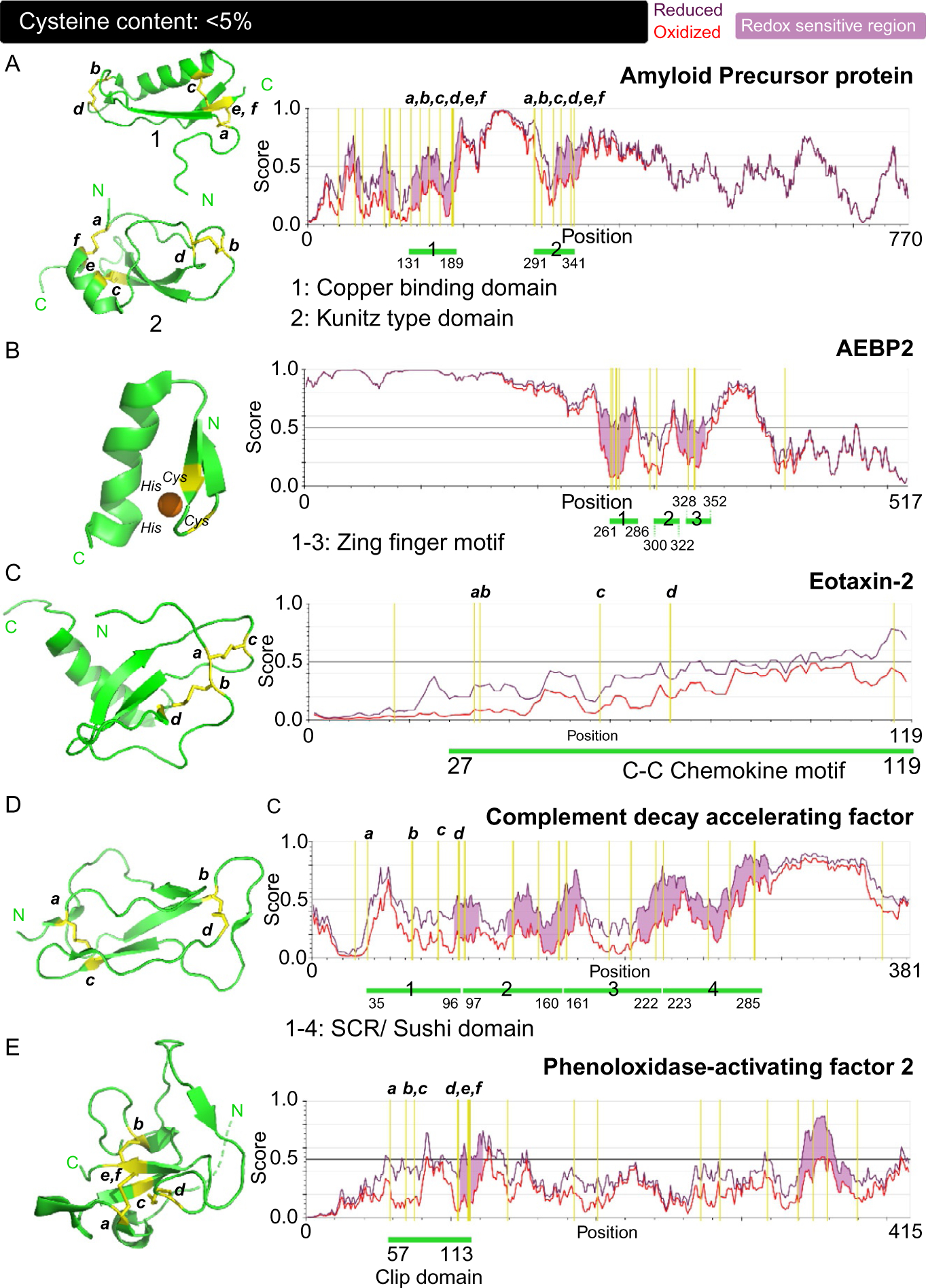
Correlation between the structure, disorder propensities and redox sensitivity in proteins with cysteine content: <5%. (A–E) PDB-derived structures of the cysteine-containing domains of the respective proteins are displayed in the left panels and the prediction of structural disorder within the proteins performed using IUPred2A computational platform are the right panels. The positions of cysteines in the domains are annotated and their corresponding location in the primary sequence are marked on the right panels. Shown is the disorder score for oxidized forms of the protein (red) with disulfide bonds and reduced forms (dark purple) where cysteine residues in the sequence are swapped with serine. A higher score (0–1) indicates disorder. All five proteins; amyloid precursor protein (A), AEBP2 (B), eotaxin-2 (C), complement decay accelerating factor (D) and phenoloxidase-activating factor-2 (E), have a cysteine content of less than 5%. The positions of cysteine residues within the sequences are indicated with a yellow vertical line while the position of the individual cysteine-rich domains is pointed using the green line. Redox sensitivity (colored light purple) of proteins is marked by regions where the oxidized and reduced forms display significant difference in structural disorder.
(ii). APP: Kunitz-type protease inhibitor (KPI) domain:
(PDB accession code: 1ZJD) APP isoforms, APP751, and APP770 contain a protease inhibitor-like domain near the central region of the protein. This domain is formed by the residues 291–341. As observed in the other APP domains, six cysteines are involved in disulfide bond formation; Cys291-Cys341 form the first disulfide bond, linking the first and last residue making up the domain. Cys300–Cys324 and Cys316–Cys337 form the internal disulfide bridges (Fig. 2A: 2). KPI domain consists of five β-strands with an α-helix at the C-terminal end of the domain. This domain is involved in protease inhibiting function of APP.71,72 All three domains of APP involve oxidized forms of cysteine which play a structural role within the protein. The disorder propensities of the redox forms displayed in Fig. 2A: 2, left panel, indicate the crucial role the disulfide bonds play in imparting structural order to these domains while maintaining a functionally neutral role.
(iii). Zinc finger motifs:
(PDB accession code: 1ZNF). Proteins containing zinc finger motifs are known to bind DNA and regulate gene expression. Zinc finger motifs present in transcriptional regulators are crucial for DNA recognition. Zinc fingers were originally discovered in the transcription factor TFIIIA, which was found to contain nine zinc fingers in a row. Now there is an overwhelming number of zinc finger proteins in PDB and they come in different variations such as knuckles, treble clefs, ribbons, and other fanciful folds. AEBP2 is a transcriptional regulator that belongs to a diverse family of zinc fingers that contain a canonical C2H2 motif, which represents one of the first zinc fingers to be identified, and one that binds zinc in a tetra-coordinated complex.73 These are independently folded domains within the protein. Most of other zinc finger motifs identified also utilize a characteristic combination of cysteine and histidine residues to coordinate zinc. This zinc coordination by the cysteines and histidine allows the emergence of the structural element, which in turn enables interaction with DNA double helix. The motifs usually involve α-helices that adopt a structure that is reminiscent of fingers and hence, its name. Individual zinc finger motifs contain two secondary structural elements; a β-hairpin and an α-helix. The zinc ion is coordinated in between the two secondary structural elements by two cysteines from the hairpin and two histidines from the helix (Fig. 2B, left panel). In the apo form, the cysteines in this motif are present in the free thiol form to allow metal coordination.74–76 The zinc finger domain, although contains secondary structural elements, IUPread2A prediction shows that the sequence displays propensity for a degree of disorder.
(iv). Chemokine CC motifs:
(PDB accession code: 1EIG). The term “chemokine” (CK) is a portmanteau derived from their function as “chemotactic cytokines.” They are important components of the immune response, forming the secondary proinflammatory mediators. These molecules elicit the activation of leukocytes by binding to the chemokine receptors on their surface. CKs are grouped into two distinct categories based on the cysteine-motifs they display; CC or β-CKs, having a tandem cysteine repeat and CxC or α-CKs with the cysteines separated by an amino acid. Another rare CK subfamily contains CxxxC motif or γ-CK, where two cysteines are separated by three amino acids. Eotaxin (ET1–3) is a subgroup of CC CK family, which strongly activates eosinophils and basophils. Eotaxin-2 (ET2) is a 13 kDa protein that binds specifically to CC chemokine receptor-3 (CCR3) and forms a potent immune activating response against allergens, parasites and viruses. The immune system utilizes a startling array of these signaling molecules, all of which display a very similar topological, or tertiary structure, although do show variations in quaternary assemblages.77–79 The structure of ET2 has been determined using solution NMR and consists of the CC CK domain architecture.80
In ET-2, this domain extends from residues 27–119 region of this protein and consists of a disordered N-terminal region, a feature that is also shared by other CKs (Fig. 2C, left panel). A three stranded β-sheet is present in the central region of the domain and an α-helix that extends for 13 residues is present at the C-terminal end of the domain. This domain consists of four cysteine residues that form two disulfide bonds; Cys33–Cys58 and Cys34–Cys74. After the tandem cysteine motif (C33 and C34), is a loop made up of 10 residues that is followed by a 310 helix. This loop is termed as the N-loop and is a crucial mediator of CK functioning. This loop along with an adjacent strand β3 from the β-sheet, forms an extended cleft that contacts the CCR3 and is responsible for mediating the binding between the CK and its receptor. The characteristic cysteine motifs (CC, CxC, CxxxC) are crucial in maintaining the integrity of this N-loop fold by tethering the opposing ends of this region. The individual cysteine motifs also allow the formation of a distinct overall architecture for each leading to the variety in CK families. The stability of the core of this domain is largely in part to the disulfide bonds and the hydrophobic residues in the central β-sheet. It is noteworthy that although cysteines facilitate formation of a stable domain, the N-loop region also exhibits significant flexibility and both the disulfide bonds happen to connect disordered regions of the domain.80–82 However, as observed from IUPred2A predictions, the chemokine CC motif does not seem to be particularly sensitive to redox as observed in Fig. 2C, right panel. In addition, although disorder propensity is high for the reduced protein, the difference in disorder is not significant between those observed for oxidized and reduced forms. This observation points toward an inherent flexibility of this domain which is only mildly attenuated by disulfide bridges. The example also highlights the subtle interplay between conformational plasticity and rigidity in affording functionality to cysteine containing proteins.
(v). Short consensus repeat (SCR) domain/complement control protein (CCP) domain/sushi domains:
(PDB accession code: 1OJV). Complement decay accelerating factor (CD55): CD55 along with CD46, CD35, C4BP and factor H, belong to the regulators of complement activation (RCA) protein family. CD55 is a ~40 kDa protein that forms a part of the complement system within the innate immune response. RCA family is responsible for regulating the activation of the three complement pathways; classical, alternative and lectin pathways.83,84 They act at specific steps within the complement cascade and prevent their inappropriate or over activation. Specifically, CD55 interacts with C3 convertase and brings about its inactivation. Because of this crucial function, RCA proteins including CD55 are expressed ubiquitously as transmembrane, glycosylated proteins on cellular surfaces. RCA proteins consists of a characteristic cysteine-rich domain termed as short consensus repeat (SCR) domain, also termed as complement control protein (CCP) domain or sushi domain. CCPs’ domain structure is unique to the RCA proteins; they are also present in a wide array of molecules that are involved in molecular recognition and/or protein-protein interactions such as coagulation factors, cell adhesion molecules, cytokines, with the number of domains and their structural arrangement being the determinants of each class. Despite their differences, an invariant feature of this domain is the presence of two disulfide bonds formed between CysI–CysIII and CysII–CysIV and a tryptophan residue within the hydrophobic core of the domain. CD55 consists of four tandem repeats of the CCP domains.84–86 Overall, the CD55 constitutes a rigid molecule with CCP repeats giving it a string on a bead like appearance. Within the four CCPs of CD55, repeat 3 has been ascribed functional importance in immune response.83–85,87,88 CD55 consists of domains that are ~60 residues long with short intervening linkers between adjacent repeats. Each repeat displays a common structural architecture; five β-strand form a structure dominating β-sheet, where strands β1-β4 and strands β3-β5 are tethered by the aforementioned disulfide bonds which clasp a disordered, flexible N-terminus and the C-terminus lying at diametric ends of the domains. The abrogation of disulfide bonds within these domains renders the domain to be disordered as marked by differences in disorder propensities in Fig. 2D, right panel. A central hydrophobic region within the β-sheet is the common feature of these domains. Each fold is also punctuated by surface exposed loops that show variability. CCP domains also consist of glycosylation sites, usually one per domain, a modification that impart structural stability to this transmembrane protein and potential protection from proteases.
(vi). Clip domains:
(PDB accession code: 2B9L): Serine protease homologue (SPH) that is a member of the polyphenoloxidase activator family that is involved in cell signal dependent cascade pathways such as immune response, and embryonal development in insects. Phenoloxidase-activating factor 2 (PPAF-2) is a member of the SPH family. Their hallmark function is to activate prophenoloxidase system (proPO) as a part of innate immune response which is analogous to the complement system. This family thus forms the first line of defense within insect and other arthropod immune systems. SPHs are defined by the presence of the clip domain and the serine protease-like domain which lacks the catalytic protease activity. Similar to the trypsin protein family, SPHs are expressed in the inactive form as zymogens, requiring processing for activation. The clip domains are widely distributed in serine proteases, but their precise roles remain unknown.89–92 In PPAF-2, this domain extends from residues 57–113 and is located in the N-terminal region (Fig. 2E, left panel). The clip domain derives its name because of the resemblance it bears to a folded paper clip with disulfide bonds holding the fold in place. In terms of its overall structure, their fold is similar to that of antimicrobial defensins, which will be discussed later, although important distinctions exist. The clip domain in PPAF-2 consists of six cysteines that are involved in three disulfide bridges; Cys58–Cys112, Cys69–Cys105 and Cys75–Cys113 (Fig. 2E, left panel). The N-terminus of the domain is composed of a disordered, bulging loop that forms an outer rim. Central core of the domain is composed of a β-sheet with four β-strands. The two central β-strands β2-β3 have a parallel orientation, which is a distinction compared to the β-defensin fold. The disulfide bond function in fixing the disordered loops on the outer rims to the central β-sheet structure. The cleft formed from this arrangement of loops linked to the central residues is a common feature, observed in other clip domains as well.93,94 IUPred2A analysis of this domain indicates that relatively high disorder exists near the three cysteines and is predicted to have some roles under both reducing and oxidizing conditions (Fig. 2E, right panel). This seems to correlate with the degree of disorder present within the regions flanking the cysteine residues in the domain.
6. Sequences containing 5%–10% of cysteines
(vii). Defensin folds:
(PDB accession code: 1IJU). Antimicrobial peptides constitute a diverse class of small, amphipathic molecules that are part of the innate immune response of multicellular organisms. There is great diversity in the nature of these peptides; they have varying lengths, different amino acid constitutions and largely conserved disulfide bonds. Many antimicrobial peptides including neutrophil peptides and defensins contain a unique fold termed as the defensin fold. These peptides display antimicrobial activity via membrane interaction and subsequent disruption. They are cationic in nature and are secreted by leukocytes. Defensins mediate membrane disruption by electrostatic interactions with the charged phospholipid head groups. Human βD-1 is a 7.4 kDa peptide, active against gram-positive and negative bacteria, as well as adenovirus, although the mode of action is vastly different with bacteria and viruses due to obvious biochemical differences.95–98
The defensin fold represent a unique architecture that is common for most antimicrobial peptide families, where each assume a characteristic structure. Defensins are subgrouped based on the connectivity pattern among its six disulfide bonds; α-, β- and θ-defensins. α-defensins have a disulfide bonding pattern of CysI–CysVI, CysII–CysIV and CysIII–CysV, while β-defensins have CysI–CysV, CysII–CysIV and CysIII–CysVI. θ-Defensins have a disulfide pattern similar to α-defensins, but are unique due to their circularity, with the N-terminal and C-terminal connected to form a closed, ring-like structure. The defensin fold in βD-1 consists of short helix at the N-terminal region, covering residues 34–40, which is thought to mediate interactions with the target cell wall. This helical stretch is enriched in hydrophobic residues and is notably absent in α- and θ-defensins. This is followed by an antiparallel β-sheet core that is connected by three disulfide bonds; Cys37–Cys66, Cys44–Cys59 and Cys49–Cys67. This arrangement shares similarities with the clip domain described previously. The β-strands β2-β3 adopt a hairpin fold with β3, lying between β1 and β2. A hallmark feature of the defensin family is the presence of a β-bulge formed by the conserved GxC motif, after CysIV, and although the functional importance of this bulge is not yet known, it has been postulated to allow proper folding and disulfide bond formation. The C-terminal region displays a biased presence of cationic residues, possibly indicating another region crucial for interactions with the negative charges on the lipids of the cell wall. Although the central and the C-terminal regions of the fold show significant disorder in the reduced state (Fig. 3A, right panel), these regions are not predicted to be redox sensitive. The defensin fold represents yet another example of the structure- and possibly stability-inducing role for cysteines, although as can be seen in Fig. 3A, right panel, there is an internal disorder propensity to this protein without those tethering bridges. The defensin fold, although unique to this family of peptides, shows similarity to variety of motifs in toxins potentially hinting at a shared evolutionary relationship between the two functional protein families.97–100
Fig. 3.
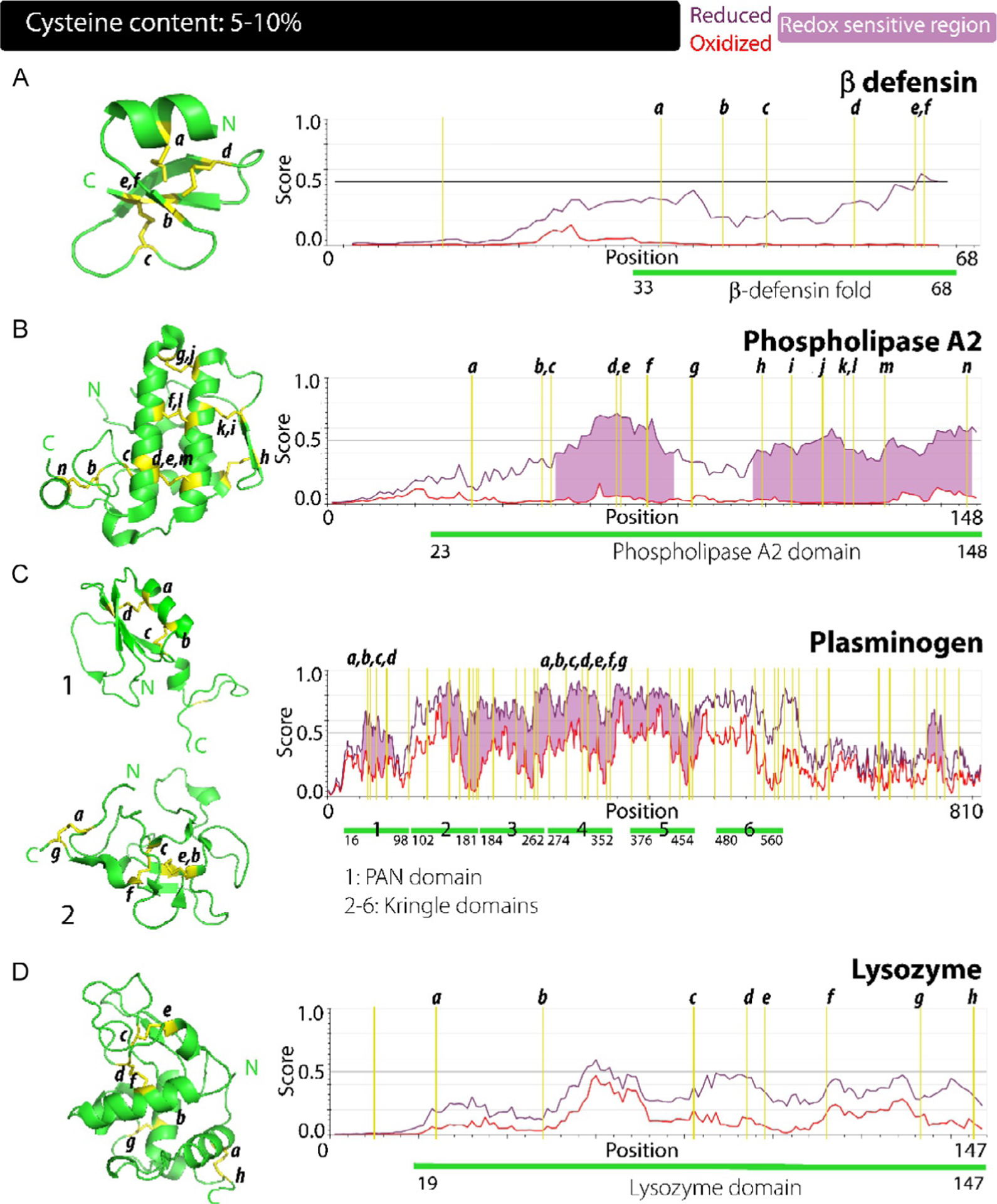
Correlation between the structure, disorder propensities and redox sensitivity in proteins with cysteine content: 5%–10%. (A–E) β defensin (A), phospholipase A2 (B), plasminogen (C) and lysozyme (D) have a cysteine content in the range of 5%–10%. β defensin (A) shows a lack of predicted redox sensitivity across its sequence due to relatively mild differences in disorder propensities of its redox forms while overall, more cysteine residues within these proteins impart greater redox sensitivity.
(viii). Phospholipase A2 domains:
(PDB accession code: 3ELO). Phospholipases represent one of the earliest enzymes to be identified and biochemically characterized from snake venom. Analogous enzymes have since been identified in mammals that carry out the same function of hydrolyzing the sn-2 acyl bond in glycerophospholipids. Initially isolated from pancreas, a vast number of phospholipases have since been added to this family that has been divided into a total of 16 groups and subgroups based on their cellular localization, their mode of action, and the disulfide bond connectivities.101,102 Among these, the secreted group of the enzyme (termed sPLA2) predominate. Different members of these groups also display heterogeneity in the residues utilized for their hydrolyzing action; some using the classical catalytic triad of Ser-His-Asp, while secreted forms utilize Ser-Asp. Similar to other proteases, they are produced as inactive, zymogen form, which then undergoes processing to become an active enzyme. Human pancreatic PLA2 G1B is a 16 kDa enzyme that is activated by trypsin processing and contains the characteristic secreted PLA2 domain structure.101–103 sPLA2s are calcium-dependent for their hydrolyzing activity and thus have a Ca2+ binding loop that is composed of the conserved sequence, xCGxGG within their members. The domain of this enzyme extends from residue 23–148 and is composed of four major α-helices forming the central core of the domain; one of the helices extends for 18 residues (residue 90–108) being the longest stretch of secondary structural element, while two minor helices are located at the c-terminal end along with a 310 helix (Fig. 3B, left panel). Two small antiparallel β-strands form a hairpin at the outer-rim of the domain. A total of 14 cysteines coordinate to form seven disulfide bridges that straddle and constrain the dynamics of the overall structure; Cys33– Cys99, Cys49–Cys146; that connects N- and C-termini, Cys51– Cys67, Cys66–Cys127, Cys73–Cys120, Cys83–Cys113, and Cys106–Cys118 (Fig. 3B, left panel). The disulfide-bond induced architecture of this domain has been shown to significantly enhance the Ca2+ binding affinity, thus indirectly enhancing the hydrolyzing activities as well, while also playing a role in mediating membrane interactions.104 The active site of sPLA2 members is composed of a His-Asp diad, which for PLA2 G1B constitutes residues His71 and Asp121. The presence of a functionally important loop was also determined, labeled as 69-loop, which is composed of residues Lys62, Asn67, Tyr69, Thr70, and His71. This highly flexible and disordered loop covers the active site and has been shown to be important initiator of interactions with cell wall.102,104,105 The PLA2 domain maintains its tertiary fold using its disulfide bonds, as noted in other examples so far, but assumes functionality in a large part to the disorder and plasticity encoded within the 69-loop. As observed in Fig. 3C, PLA2 also shows significant disorder propensity in absence of the stabilizing disulfide bonds under reducing conditions. It is interesting to observe that six out of the seven disulfide bonds connect well-formed secondary structure elements yet, most of these regions show disordered structure under reducing conditions.
(ix). PAN/apple domains:
(PDB accession code: 4DUU). Plasminogen is a quintessential example of PAN/apple domain containing protein. Plasminogen and plasmin represent a family of peptidases that mediate fibrinolysis and thus play a role in the regulation of blood coagulation.106 In addition, it is also known to be involved in other functions such as extracellular matrix formation, tissue repair and modeling, mediation of immune response, regulating inflammatory reactions, cell adhesion, migration and proliferation.106,107 This pleiotropic involvement in diverse cellular pathways has rendered this family of proteins to be a potential target for multiple therapeutic strategies, most prominently in cancer and blood clotting disorders.106,107 Plasminogen serves as the precursor, or zymogen form of plasmin, being usually present in the inactive, closed form. Interaction with fibrin brings about significant conformational changes in the protein that allow conversion to an active or open form of the enzyme. The open form is processed by enzymes like tissue plasminogen activator, urokinase plasminogen activator, kallikrein, and factor XII to release plasmin.108 Human plasminogen is a 90 kDa protein with seven domains; one PAN/apple domain, five kringle domains (detailed further down) in tandem and one serine protease domain at the C-terminal.106–110
This hairpin-loop assembly, termed PAN domain, extends from residues 16–98 in plasminogen. It resembles growth factor domains but is distinguished by the position of its four conserved cysteines and two disulfide bonds; CysI–CysIV and CysII–CysIII (Fig. 3C: 1, left panel). Overall, the domain adopts a globular structure that consists of a β-strand from residues 19–23 at N-terminal region of the domain, followed by an α-helix extending from residues 26–37, which precedes a β-sheet forming the central core. The β-sheet consists of four strands in antiparallel orientation that are followed by a short helix at the C-terminal of the domain. The disulfide bonds; Cys49–Cys73 and Cys53–Cys61, form the fulcrum of the central fold. A disordered, flexible loop constitutes the linker between the PAN domain and the first kringle module. This protein assumes a closed conformation when three basic residues, Lys50, Arg68 and Arg70 within the PAN module making contacts with a lysine-binding site in fourth and fifth kringle domains. For plasmin production, sequential cleavage occurs within this domain, first at Arg68–Met69, and secondly at Lys77–Lys78 which releases the closed conformation assumed.108–111 Fig 3C, right panel, highlights potential redox sensitive regions with differences in disorder propensity for the oxidized and reduced forms of this domain.
(x). Kringle domains:
(PDB accession code: 1B2I). Kringle domains are ~80 amino acid, cysteine-rich modules that have been found in clotting factors, apolipoproteins, and growth factors. Kringle domain derives its name from the Scandinavian pastry due to their overall structural resemblance. The structure of plasminogen kringle-4 domain comprises of residues from 376 to 454. It shows largely a disordered, not so well-defined structure which is dominated by loops held together by three disulfide bonds (Fig. 3C: 2, left panel). Out of the three, two disulfide bonds seem to anchor the domain; Cys377–Cys454 joining the two ends of the domain and Cys398–Cys437 lying in the central core. The domain spanning disulfide bond Cys377–Cys454 also shows a much less defined electron density indicating significant dynamics associated or in other words disorder within the region. Apart from the covalent disulfide bonds, the structure is further stabilized by the presence of a small segment of antiparallel β-sheet and connecting β-turns concentrated in the domain core. Other than this secondary structure, no other is present in the domain which is dominated by unstructured loops held in place by the three disulfide bonds. These disordered loops predominate and are hydrophilic in nature. The lysine binding site that stabilizes the closed conformation of plasminogen is located within this domain and is in the form of a cleft composed of the segments; His407–Lys411, Pro429–Lys433, Pro435–Phe438, and Arg445– Cys449.108,112–114 The highly disordered domain is also reflected in IUPred2A computations which also shows high degree of disorder and the propensity to function under redox flux (Fig. 3C, right panel). Plasminogen is a classic example for the key roles of cysteines in the structure and function of a protein. Cysteine disulfide bonds constrain and induce a conformational fold that is functionally latent. The overall higher order structure of the protein looks akin to a spiral when viewed from top. In the presence of fibrin, the required closed state is disrupted, and the active state arises out of large-scale structural changes and processing of plasminogen. Fig 3D shows the disorder propensities of the cysteine rich PAN and kringle domains, where disulfide bonds reign-in the intrinsic disorder and allow defined folds to form.108,111
(xi). Lysozyme folds:
(PDB accession code: 1AKI). As the name indicates, the lysozyme is an enzyme family that functions as an important degradative as well as antimicrobial protein molecule by lysing the polysaccharide cell wall but also have roles in immune activation and as such, they are part of the innate immune response of plants and higher organisms.115 Lysozyme was first identified by Alexander Fleming in the bodily fluids of humans such as tears, saliva, milk and blood.116 It was the first enzyme to have its structure elucidated and since then has served as the prototypical model for structural investigations of proteins with over 800 structures deposited in the PDB databank. For the past few decades, the chicken egg lysozyme has also been thoroughly studied in lieu of delineating the protein folding process. Structural analogy has been established between the lysozyme family and α-lactalbumin with both containing similar topology held together by disulfide bonds.117,118 The lysozyme fold has a significant globular character that is rich in secondary structural elements. Two distinct domains are identifiable; An α-helix rich α-domain which is spread across the distal ends of the N and C-terminals containing a total of four α-helices, and a domain containing of a β-hairpin and a long, disordered loop that dominates this part of the structure, which is somewhat incongruously termed as β-domain. The two-domain assembly contains four disulfide bonds; Cys6–Cys127, Cys30–Cys115 both present within the α-domain while Cys64–Cys80 in the β-domain connects the disordered loops and Cys76–Cys94 links the large disordered loop in the β-domain with the α-domain (Fig. 3D: 1, left panel). The active site of the enzyme is located in a cleft between the two domains of the protein and consists of two acidic residues crucial in the mechanism; Asp52 and Glu35, with the latter lying in a helix in the α-domain. Role of disulfide bonds in maintaining structural integrity was investigated in this protein and was found to be crucial in mitigating undue conformational fluctuations within and between domains.119 The study also revealed that although most of individual secondary structural elements were preserved, the cooperativity in inducing a tertiary fold was lost.59,118–122 In addition to assisting proper folding of the protein just like in many other proteins, cysteines in lysozyme seems to be crucial for the structural compactness and stability of the β-domain and for the inter-domain integrity. The regions flanking cysteines in the β-domain show significant level of disorder with disulfide bonding stabilizing the loops (Fig. 3D). Analysis using IUPred2A although showed significant level of disorder, did not predict redox sensitivity in the region (Fig. 3D; right panel).
7. Sequences containing 10%–20% cysteines
(xii). Three-finger toxin folds:
(PDB accession codes: α-bungarotoxin 1ABT, α-cobratoxin 1CTX). This toxin fold is commonly observed in toxins from many species including metazoans, nematodes and several vertebrates. Structurally, the three-finger toxin fold belong to the snake venom superfamily; α-bungarotoxin (αB) and α-cobratoxin are two prominent examples of these proteins, whose solved structures are represented in Fig. 4A and B. αB is a 7.9 kDa protein found in the venom of the Taiwanese banded krait. It functions as a potent antagonist of the skeletal muscle-type nicotinic acetylcholine receptor. Snake toxins are classified into different classes based on their mechanism of action and targets; phospholipases, serine proteases, metalloproteases and three-finger peptides. Although they share a common fold, this family contains a variety of pathologically relevant molecules with diverse activities such as inhibition of adrenoreceptors, ion channels, platelet receptors. The efficacy of these toxins is partly down to their resistance to degradation. Apart from their toxic effects, these molecules have also shown promise in therapeutic applications and palliative care. αB belongs to the three-finger peptide category, which is defined by the presence of a cysteine-rich, three-finger fold. In terms of its functionality, αB shows remarkable binding affinity to acetylcholine receptor and thus has been used in studies to identify and purify them from putative sources.123–125
Fig. 4.
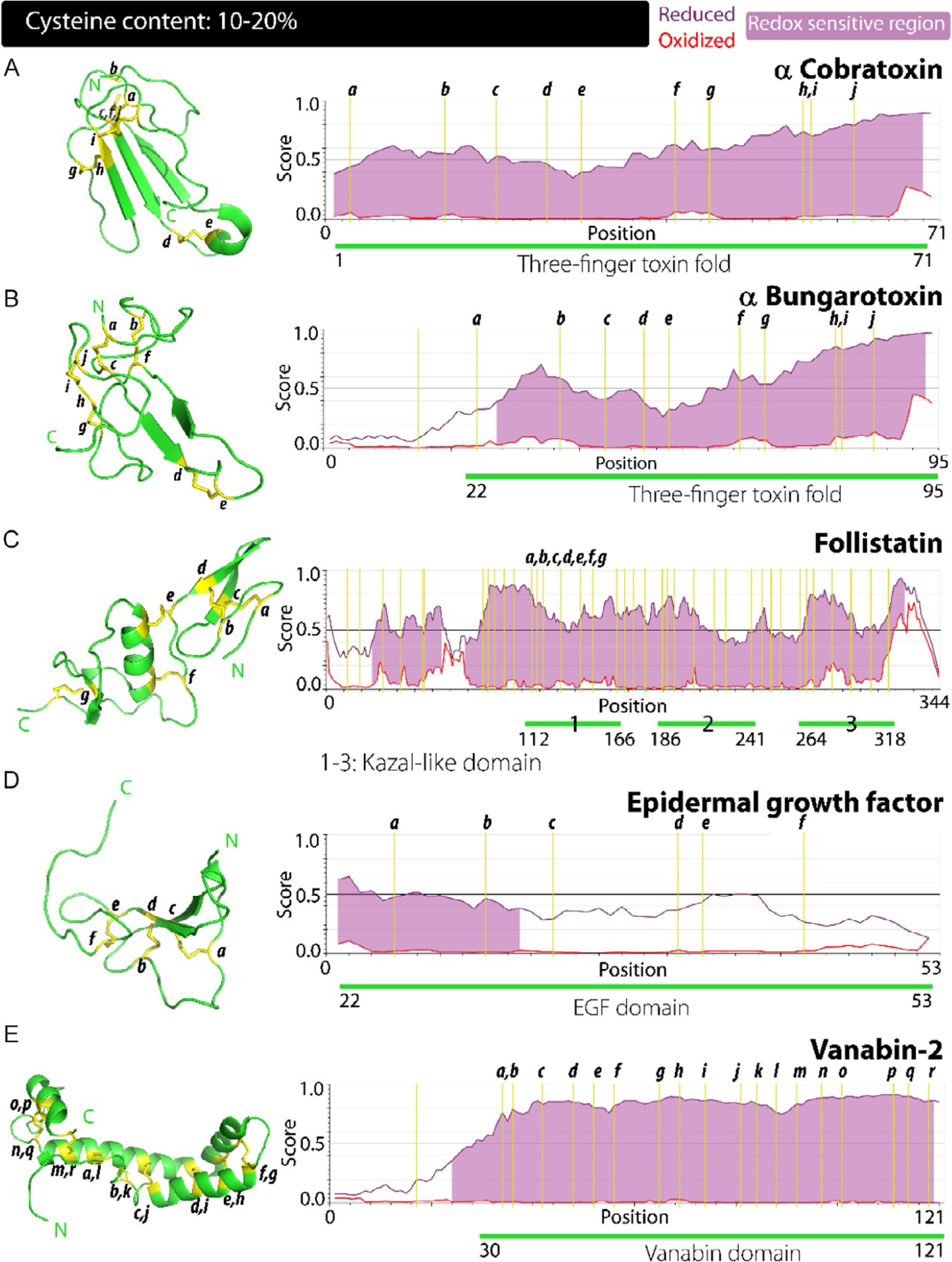
Correlation between the structure, disorder propensities and redox sensitivity in proteins with cysteine content: 10%–20%. (A–E) Cysteine residues account for 10%–20% of the sequence in α cobratoxin (A), α bungarotoxin (B), follistatin (C), epidermal growth factor (D) and vanabin-2 (E). Significant differences are observed between the disorder scores of the oxidized and reduced forms of these proteins highlighting a clear trend of increased redox sensitivity with increased cysteine content along with an increase in the loops and coils within the structure as evidenced in the left panels.
The distinguishable feature of the three-finger toxin fold is the spacing of eight conserved cysteines and their connectivities which adopt the following pattern of CI–CIII; CII–CIV, CV–CVI, CVII–CVIII. The structure of α-bungarotoxin (αB) comprises of three antiparallel β-strands that make up finger-1, a central core that constitute finger-2 and the C-terminal loop assembly which is finger-3. The scaffold for this entire assembly is held together by the five disulfide bridges involving 10 cysteine residues in αB; Cys24–Cys44, Cys37–Cys65, Cys50–Cys54, Cys69–Cys80 and Cys81–Cys86 (Fig. 4B, left panel). The central finger-2 containing two disulfide bridges supporting which are surrounded by sequences with significant disorder (Fig. 4A, right panel). This overall assembly is almost flat with a central concave cleft, with all three fingers lying in approximately the same plane. There is remarkable positional conservation of the cysteines conserved in members of the three-finger toxin family preserving its overall topology as observed in the three-finger domain of α-cobratoxin (Fig. 4A, left panel). The variations in functions among the members of this superfamily of toxins seems arise from the length and constitution of the disordered loops, as portrayed in Figs. 2 and 3 of the study by Kessler et al.126 Thus, the diverse interaction specificities of individual members arise out of differences in conformational flexibility present within the disordered loops. Such a structural plasticity within these domains is akin to three fingers of a hand moving autonomously to one another, justifying the aptness of the domain name.123,124 The degree of disorder within the loops is also predicted by IUPred2A algorithm, which shows significant disorder in the three-finger toxin fold of α-cobratoxin and αB, covering almost 80% of the sequence under reduced state (Fig. 4A and B, right panels). This also suggests a potential for this family of proteins to be functional under redox conditions providing a possible evolutionary emergence of a domain containing conserved cysteines within a sequence with high degree of disorder. The three-finger toxin fold is present in functionally divergent molecules indicating the versatility achieved by this fold in its interactions.
(xiii). Kazal-like domains:
(PDB accession code: 2KCX). Kazal domains are widely distributed in animals, apicomplexans and oomycetes. Human Follistatin-related protein 3 (FSTL-3) is a representative of the family of proteins containing Kazal-like domains. FSTL-3 is an 8 kDa protein involved in regulating the reproductive cycle in humans and other higher organisms and shares structural and functional similarity with follistatin. Recent studies have revealed follistatin to be a multifunctional protein that plays important roles in postnatal processes and embryonic development.127,128 Its functions within the reproductive pathways are mediated by its interaction with and subsequent inhibition of activin, a protein that activates the production and secretion of growth hormone, prolactin and adrenocorticotropic hormone. It can also suppress the activity of follicle stimulating hormone, thus transitioning an organism to a nonreproductive phase.128 Follistatin also has putative roles in stress response of cells such as in nutrient deprivation and oxidative stress. Studies have shown increased follistatin expression in response to oxidative stress. Although the protein is hypothesized to play a neuroprotective role within such environment, the exact molecular mechanism for its action has still not been elucidated. Structurally, follistatin and FSTL-3 consist of Kazal-like domains.128–131
The Kazal-like domain is widely found to be present in proteins with protease inhibitor activity, specifically against serine proteases. In follistatin, there are three repeats of this domain with each extending for about ~50 amino acids. Structurally this domain consists eight cysteines forming four intra-domain disulfide bonds (Fig. 4C, left panel). In Kazal-like domain-1 which covers residues from 112 to 166, this represents the bonds Cys118–Cys150, Cys122–Cys143 and Cys132–Cys164. The domain also contains four antiparallel β-strands interspersed with two α-helices. The N-terminal β-strands form a hairpin structure. Based on homologous domains observed in other protease inhibiting proteins, Kazal-like domains in follistatin are hypothesized to form an extended binding cleft for proteases comprising the CysI and β-strand located near the N-terminal of the domain.127,132,133 The observed protective role of follistatin in oxidative stress can potentially be attributed to the presence of multiple cysteine rich domains within, as observed in Fig. 4C, right panel. These domains can potentially modulate their redox state based on cellular cues, acting as an oxidative stress buffer.
(xiv). Epidermal growth factor (EGF) domains:
(PDB accession code: 1EGF). The family of EGF domains consists of a ~6 kDa protein sequence that are present either modularly or in tandem and are involved in a wide array of signaling mechanisms. These domains are present mainly in the extracellular milieu and function as key regulators of growth and development. Since its discovery, the characteristic EGF like domains has been found in a diverse range of growth factors such as transforming growth factor-α (TGF-α), epiregulin, amphiregulin, among others. EGF domains are also present within transmembrane proteins such as APP and Notch.66,134,135 Once formed, the EGF protein then acts as a paracrine molecule where these ligands show remarkable binding affinity to specific receptors termed as EGF receptors. These factors activate an intracellular signaling cascades of the MAPK and Raf signaling pathways.136–138
The EGF domain is the most widely recognized structure involved in protein–protein interactions mediating many signal-transduction pathways. Structurally, the defining feature of this domain family is that they are stabilized by three intramolecular disulfide bonds with conserved cysteines making the specific CI–CIII, CII–CIV and CV–CVI connectivities.138 The domain consists of a central antiparallel β-hairpin structure formed by two β-strands, which happens to be the only defined secondary structure (Fig. 4D, left panel). The rest of the domain is devoid of any secondary structural features and is rich in loops and turns. The tertiary arrangement of this domain is crucial to its interactions with the receptor and its maintenance is predominantly down to the three disulfide bonds holding the assembly. This domain show resemblance to the short repeat domain of CD55 discussed previously, with both these domains relying on their disulfide bridges to maintain their functionally relevant fold.137–141 As the IUPred2A predictions indicate, the sequences surrounding the cysteines show significant disorder in the reduced state and that the N-terminal region could be active in redox states (Fig. 4D, right panel).
(xv). Vanabin domain:
(PDB accession number: 1VFI). Vanadium binding proteins (VBP) represent a family of metalloproteins that, as the name suggests, can coordinate vanadium ions. These class of proteins are predominantly observed within marine life where the element is much more abundant than on land. These protein domains utilize the oxidation chemistry of vanadium to bring about redox-based processes at the cellular level.142 The invertebrate organism ascidians especially display staggering concentrations of vanadium inside their cells, reaching sometimes up to 350 mM.143,144 The enriched metal is then utilized by multiple enzymes as cofactors; VBPs being the most prominent. From ascidian cells, four different VBPs; Vanabins 1–4, have been characterized so far although analogous proteins have been identified. All four vanabins display a conserved motif of Cx2–5C within their sequence.144–146 Other metal cations are able to bind to vanabins, such as copper, iron and zinc which might be inhibitory to protein functions, however the overwhelming concentrations of vanadium in the intracellular milieu ensures it occupies the relevant sites.145,146 Vanabins, along with some other cysteine-rich proteins do not display a canonical fold or higher order structure that is ubiquitously found and as such, there is no recognizable vanadium binding protein domain, being present only in the representative proteins. Still, they do display interesting structural traits despite being disulfide-rich proteins.147
Van-2 is a 13 kDa member of the vanabin family and was one of the first VBPs identified and characterized. It consists of 19 cysteine residues that form nine disulfide bonds (Fig. 4E). The structure is composed of four α-helices that are present throughout the protein; helix α1 composed of; 43–55, α2; 62–73, α3; 75–95 and α4; 109–117 giving an overall appearance reminiscent of a bow shape with helix α1 lying on one side, and α2–4 lying on the other, connected by a turn. The nine-disulfide bond bridge the two sides of this bow forming covalent links between them. NMR studies identified the presence of a lysine rich, highly basic surface of the protein that was involved in vanadium binding. Interestingly, reduced cysteines in the form of thiols appear to be inhibitory to vanadium coordination, while disulfide bonded formation restores the vanadium binding capabilities of the protein. This hints at the disulfide engendered tertiary fold to be crucial for these interactions, perhaps by stabilizing it and allowing a positively charged face to form where lysine and arginine residues cluster together and coordinate the metal.147 Another intriguing aspect of this protein is the formation of disulfide bonds and their stable sustenance within the reducing, cytoplasmic environment.147 Van-2 represents a unique case study in cysteine-rich proteins, their redox state and the consequences on the structure-function of proteins. Here a key question that remains unanswered is whether vanadium dependent redox processes have any effect on the redox state of the thiol. Can the reduced, thiol-free form of the protein become oxidized due to interaction with vanadium leading to disulfide bond formation in the cytoplasm? The answer to the question can reveal the puzzling disulfide-rich structure of this cytoplasmic family. As Fig. 4E reveals, these proteins tend to be disordered when disulfide are reduced but attain significant structural order when oxidized, where almost the entire stretch of the protein is predicted to be redox-sensitive (Fig. 4E; right panel).
8. Sequences containing >20% cysteines
(xvi). Granulin domains (Knottin-like domains):
(representative PDB accession number: 2YJE). Granulins (GRNs) are short, ~6 kDa peptides that display an abundance of cysteine within their sequences (~20%). In humans, seven GRN (1–7) modules are present as tandem repeats within the precursor protein called progranulin (PGRN). Each GRN domain consists of 12 cysteines at conserved locations. PGRN is a 68 kDa extracellular molecule that is involved in neuronal growth, differentiation and survival. Furthermore, PGRN is also found to play roles in immunomodulation, inflammation and angiogenesis. The proteolytic cleave of PGRN releases the individual GRN domains, which in the extracellular environment is performed by proteases such as neutrophil elastase, proteinase-3, MMPs, while its intracellular processing within the lysosome is carried out by cathepsin-L. However, roles for individual GRNs have not been established, although these cysteine-rich proteins have been observed to play a role in immune response, as growth factors, and inflammation, in some instances having a contrary action to the precursor. Within the context of a redox role, both the redox forms of GRN-3 have been shown to be functional despite a stark structural difference them.148–152
Structurally, the GRN domain broadly belongs to the knottin-II class of domains which are known to form eight disulfide bridges in a crossover pattern. GRNs represent a unique cysteine rich fold that is defined by the presence of its 12 cysteine at conserved locations. The exact redox state of the cysteines in GRNs has not been established although the disulfide bonded form is hypothesized to be the native state due to the extracellular localization that has been well documented so far. However, recent studies have identified an intracellular presence for these modules suggesting a potential redox sensing role for the cysteine residues. Members of the GRN family have a consensus sequence of x2–3Cx5–6Cx5CCx8CCx6CCxDxxHCCPx4Cx5–6Cx. Among the GRNs, NMR structure of GRN-4, a 57 amino acids long member of the family has been solved. GRN-4 shows a well-defined and ordered N-terminal region, while C-terminal region is disordered (Fig. 5). It consists of two prominent β-strands within the central region of the protein forming a β-hairpin structure. Rest of the protein shows a complete lack of secondary structure and is dominated by coils. The disulfide bonding pattern for GRN-4 was identified to be; CI–CIII, CII–CV, CIV–CVII, CVI–CIX, CVIII–CXI, CX–CXII. The similarity in the sequences of GRNs suggests adoption of similar structural features by the other members where a central β-hairpin is held together by a network of disulfide rungs. Recent studies into the role of disulfide bonds in the structure of GRN-3 revealed that these covalent bridges provide a semblance of order to a protein that otherwise behaves as in intrinsically disordered protein in the fully reduced form, while also enhancing its thermal stability.149–151,153,154 More importantly, it was shown that GRN-3 is functional in both redox forms,148,149 which support the IUPred2A analysis that predicts a redox function for the entire protein (Fig. 5A, right panel). More importantly, the protein is also predicted to be fully disordered under reduced condition which is confirmed by experiments with GRN-3 and further corroborated in its computational modeling results, as displayed by the I-TASSER modeling algorithm (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) in Fig. 5A, left panel. Other GRNs in the family are also predicted to be fully disordered in line with that displayed in the disorder plots of GRN-3 and 4 (Fig. 5A and B, right panels).
Fig. 5.
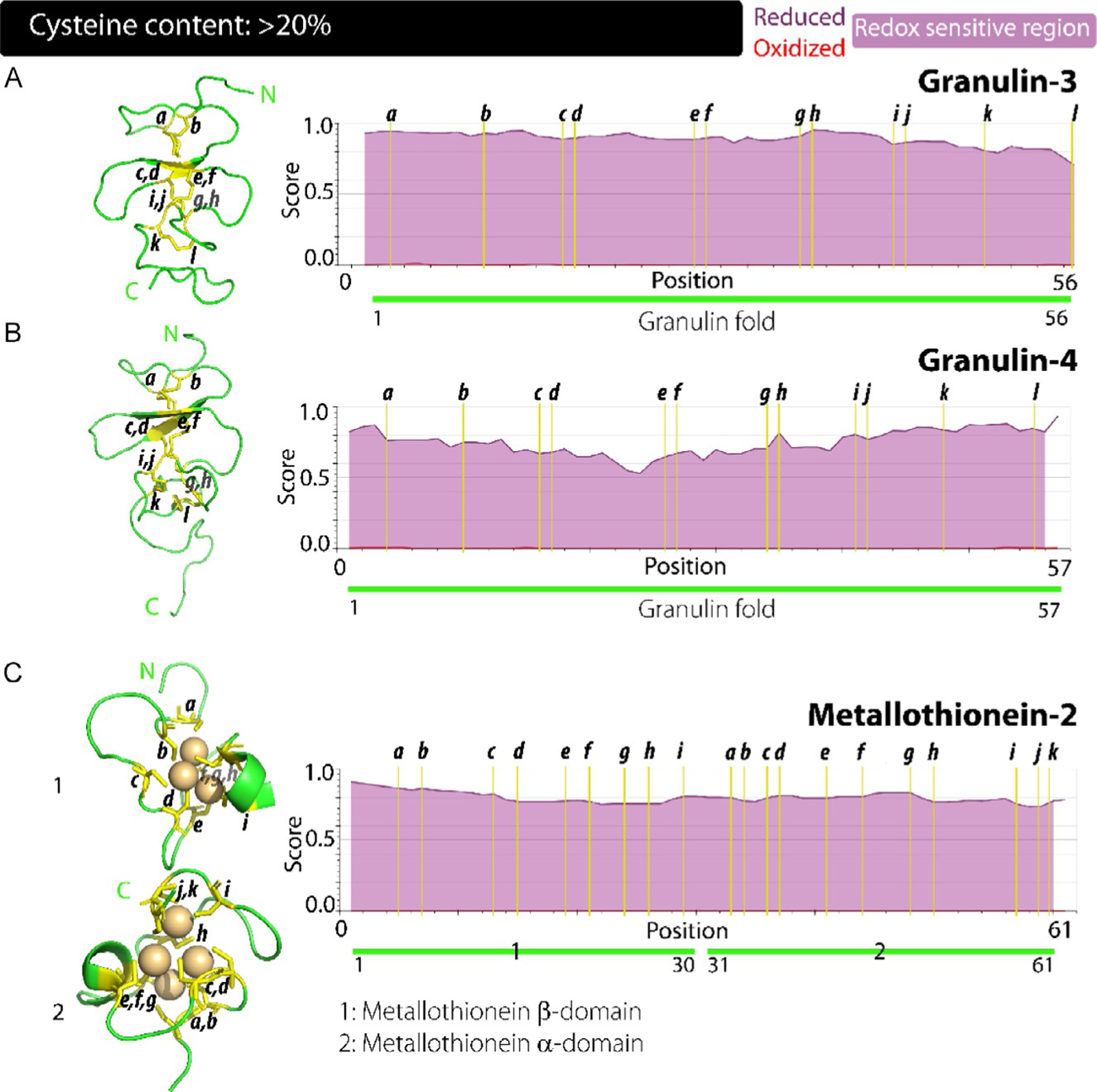
Correlation between the structure, disorder propensities and redox sensitivity in proteins with cysteine content: > 20%. (A–C) GRN-3 (A), GRN-4 and MT-2 (C) have sequences significantly enriched in cysteine residues accounting for more than 20% of the total. These proteins also show stark contrast for disorder propensity among their redox states, with the entire length of the sequence being redox sensitive along with a structure dominated by loops and coils.
(xvii). Metallothionein domain:
(PDB accession code: β-domain: 2MHU, α-domain: 1MHU). Structural disorder within cysteine-rich proteins is exemplified by the MT family. The MTs are small, ~6 kDa proteins, whose sequence is dominated by cysteine residues (about 30%). This family is widespread among plant and animal kingdoms, as well as in microorganisms.155,156 In humans, the MT family consists of four members; MT1–4, that are ubiquitously expressed within the body. These proteins function as metal scavengers within the cellular environment and are known to chelate ions of Zn, Cd, Hg, Cu, Ag, Au, Bi, As, Co, Fe, Pb, Pt, Tc and even U.157–159 This marks them as a unique protein family that functions as a reservoir of specific metal ions. Based on the ability to chelate metals, they play important role as regulators of redox responses in the cell.160,161 Expression of MTs is enhanced in the cells under the conditions of oxidative and metallic stress, especially Cd overload.160,162 MTs thus maintain metalostasis by coordinating or releasing respective metal ions from its bound clusters depending on cellular needs. They are also known to mediate metal-transfer with other proteins such as serum albumin, ferritin, α-synuclein Aβ, certain transcription factors and metalloenzymes.163,164 Such functional roles assign further importance to this protein family as orchestrators of multiple cellular pathways.155,157,159,161–165
The MT family members represent a very unique cysteine-rich domain structure. All four MTs contain 20 cysteine resides, all of which are present in reduced form. These proteins thus utilize the chemical functionality bestowed by the free thiol group. MT-2 is 61 amino acid long and is comprised of two distinct domains within its sequence; β-domain that extends from residues 1–30 and an α-domain that covers the residues 31–61 (Fig. 5C, right panel). This cysteine-rich protein family lacks any secondary structure and thus provides tremendous challenges for structural characterizations. The metal chelated forms have attenuated flexibility due to the constrained tertiary fold assumed and are more amenable to study. The 20 cysteines with MT-2 are distributed unevenly across its two domains; β-domain has nine cysteines, while α-domain contains 11. These cysteines occur in conserved motifs of CxxC, CC, and CxC. The free thiols of cysteine residues serve as excellent ligands and allow coordination clusters to form within the individual domains; β-domain capable of coordinating three and α-domain chelating four divalent metal cations.159,166,167 The MT family attests further to the versatility of cysteine residues in modulating the structure-function dynamic of proteins. Cysteines in reduced form are structurally inert but functionally crucial for the protein in binding metal ions and this renders the protein completely disordered as observed in (Fig. 5C, right panel).
9. Conclusions and future perspectives
The aforementioned sections detailing the descriptions of protein domains containing cysteine-rich sequences and their representative protein structures bring forth the underlying significance of cysteines in the structure and function. In addition, the domains discussed above show significant levels of disorder within the sequence that correlates positively toward higher percentage of cysteine content in sequences. In other words, increased presence of cysteines in sequences also correlates with increased disorder propensity within the sequences that flank the cysteines. At first, this seems to be at odds with the generality of amino acid preferences within folded and unfolded proteins wherein cysteines are considered to be order-promoting residues. This could mean two possible attributes that add uniqueness to these cysteine-containing sequences: decrease structural dynamics of the protein under oxidized conditions while increasing conformational plasticity under reducing conditions. This also envisages the fact that cysteines can act as redox-controlled modulation of protein functions in intra or extracellular environments or while the cell is under redox-induced stress as described above.48,51,53,57,61 Moreover, versatility of such cysteine-rich, disorder-promoting sequences is readily appreciable when one factors in the ability of cysteines to chelate metal ions in their reduced, free-thiol forms. This feature is observed in many metal sequestering proteins such as MTs and zinc fingers. Increasing number of examples are emerging in which redox-based thiol switches lead to conformational transitions either from order-to-disorder or vice versa.
Despite the existing paucity in experimental support for the potential redox sensitive functions among cysteine-rich proteins, limited but important clues together with the IUPred2A predictions conclusively reveal that proteins with high cysteine content show a strong correlation between structural plasticity and redox sensitivity. These redox sensitive regions thus seem to function as sensors for cellular cues and bring about structural transitions that modulate the functional state of the protein. It has been reported that approximately 5% of the proteome corresponds to redox-sensitive, cysteine-rich protein sequences in yeast, the fraction of such proteins is as high as 30% in human genome.47 This striking observation offers clues to the evolution of disorder promoting sequences with cysteines interspersed between them—as the species complexity increases such in the case of humans, so does the complexity of cellular functions. Therefore, proteins capable of multiple functions and adaptable to different environments and stress conditions have to be generated to cater the demand. For example, extracellular proteins transported into the cells, receptors that sense complex extracellular milieu and initiate signaling cascades, and cytosolic proteins that are transported in and out of various subcellular organelles, have to endure significant environmental changes including those in pH, ionic strength and oxidizing conditions. Therefore, adaptation for such proteins is a key attribute that needs to be engrained in their biogenesis. In the last decade, we have understood that IDPs and IDRs are able to achieve many of such attributes due to conformational plasticity. However, disordered sequences interspersed with cysteines provide an additional yet crucial layer of capability for the protein to endure significant redox changes, structural and stability modulations as well as the ability sense metal ions.
The significance of cysteines in disordered sequences is also comprehended by the mutations that occur in the redox-sensitive regions of the proteins that directly lead to pathology. It has been recognized that mutations to cysteine residues are the second most (arginine being the first) to occur in the human genome.73 Cysteine mutations that occur on the extracellular proteins predominantly affect the disulfide bonding patterns leading to incorrect folds168,169 while those of the intracellular proteins lead to the mitigated metal binding capability and/or stability of the protein leading to misfolding and formation of toxic aggregates.170,171 These evidences envisage the significance of cysteines and disulfide bonds especially when present within a disordered sequence in both norm and pathology. Unfortunately, dearth of both experimental and theoretical data on IDPs and IDRs with interspersed cysteines have limited our understanding of such a remarkable design among proteins which enable them to behave as modulatory proteins under extreme environmental and stress conditions. Nevertheless, with the existing evidence described in this chapter, it would be appropriate to conclude that the potential for the proteins to perform the delicate orchestration between conformational plasticity, stability and modulatory functions stems from an exquisite design of using disorder-promoting amino acids with classical order-promoting cysteines interspersed between them. Further studies to understand this phenomenon in detail in the coming years will enable the ways to comprehend protein functions, molecular design and recognition.
10. Methods
Compilation of proteins with cysteine-rich domains.
The representative proteins containing cysteine-rich domains were assembled using the UniProt database (https://www.uniprot.org/). Briefly, using annotations we directed our search for disulfide-bond containing proteins within the Swiss-Prot database, which contains curated and annotated repository of UniProt database. Search was further refined by sorting for proteins displaying cysteine-domains and ones identified were finally manually grouped based on their cysteine content (% of cysteines) yielding a final count of 17 representative proteins that contained common and some unique cysteine-rich domains.
Prediction of structural disorder and redox sensitivity in respective proteins.
Analysis of disorder propensities of the selected proteins was performed using the IUPred2A computational platform (https://iupred2a.elte.hu/). Redox sensitivity of the proteins was calculated using the same platform with an additional Redox State parameter with the IUPred2 long disorder algorithm. The % redox sensitivity was calculated by determining number of redox sensitive regions within the entire protein, based on the data generated by the Redox State IUPred2 algorithm. This % redox sensitivity was correlated by with % cysteine content of respective proteins and plotted using Origin 8.5 graphing program.
Domain structures.
The structures of the respective cysteine-rich domain were obtained from the RCSB PDB database (https://www.rcsb.org/). The obtained structures were then rendered using the molecular visualization tool PyMOL 2.4 (https://pymol.org/2/).
References
- 1.Kendrew JC, Bodo G, Dintzis HM, Parrish RG, Wyckoff H, Phillips DC. A three-dimensional model of the myoglobin molecule obtained by X-ray analysis. Nature. 1958;181:662–666. [DOI] [PubMed] [Google Scholar]
- 2.Perutz MF, Rossmann MG, Cullis AF, Muirhead H, Will G, North ACT. Structure of hæmoglobin: a three-dimensional fourier synthesis at 5.5-Å, Resolution, obtained by X-ray analysis. Nature. 1960;185:416–422. [DOI] [PubMed] [Google Scholar]
- 3.Romero P, Obradovic Z, Kissinger CR, et al. Thousands of proteins likely to have long disordered regions. Pac Symp Biocomput. 1998;437–448. [PubMed] [Google Scholar]
- 4.Garner E, Cannon P, Romero P, Obradovic Z, Dunker AK. Predicting disordered regions from amino acid sequence: common themes despite differing structural characterization. Genome Inform Ser Workshop Genome Inform. 1998;9:201–213. [PubMed] [Google Scholar]
- 5.Dunker AK, Lawson JD, Brown CJ, et al. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. [DOI] [PubMed] [Google Scholar]
- 6.Uversky VN. Intrinsically disordered proteins and their “mysterious” (meta)physics. Front Phys. 2019;7: Article 10 (18 pages). [Google Scholar]
- 7.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–331. [DOI] [PubMed] [Google Scholar]
- 8.Garza AS, Ahmad N, Kumar R. Role of intrinsically disordered protein regions/ domains in transcriptional regulation. Life Sci. 2009;84:189–193. [DOI] [PubMed] [Google Scholar]
- 9.Tompa P The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005;579:3346–3354. [DOI] [PubMed] [Google Scholar]
- 10.Dunker AK, Garner E, Guilliot S, et al. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac Symp Biocomput. 1998;473–484. [PubMed] [Google Scholar]
- 11.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. [DOI] [PubMed] [Google Scholar]
- 12.Tompa P Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. [DOI] [PubMed] [Google Scholar]
- 13.Daughdrill GW, Pielak GJ, Uversky VN, Cortese MS, Dunker AK. Natively disordered proteins. In: Buchner J, Kiefhaber T, eds. Handbook of Protein Folding. Weinheim, Germany: Wiley-VCH, Verlag GmbH & Co. KGaA; 2005:271–353. [Google Scholar]
- 14.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804:1231–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uversky VN, Dunker AK. The case for intrinsically disordered proteins playing contributory roles in molecular recognition without a stable 3D structure. F1000 Biol Rep. 2013;5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holt C, Sawyer L. Caseins as rheomorphic proteins: interpretation of primary and secondary structures of the αs1-, β-, and κ-caseins. J Chem Soc Faraday Trans. 1993;89:2683–2692. [Google Scholar]
- 17.Pullen RA, Jenkins JA, Tickle IJ, Wood SP, Blundell TL. The relation of polypeptide hormone structure and flexibility to receptor binding: the relevance of X-ray studies on insulins, glucagon and human placental lactogen. Mol Cell Biochem. 1975;8:5–20. [DOI] [PubMed] [Google Scholar]
- 18.Cary PD, Moss T, Bradbury EM. High-resolution proton-magnetic-resonance studies of chromatin core particles. Eur J Biochem. 1978;89:475–482. [DOI] [PubMed] [Google Scholar]
- 19.Linderstrom-Lang K, Schellman JA. Protein structure and enzyme activity. In: Boyer PD, Lardy H, Myrback K, eds. The Enzymes. 2nd Ed. New York: Academic Press; 1959:443–510. [Google Scholar]
- 20.Schweers O, Schonbrunn-Hanebeck E, Marx A, Mandelkow E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem. 1994;269:24290–24297. [PubMed] [Google Scholar]
- 21.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT Jr. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. [DOI] [PubMed] [Google Scholar]
- 22.Chen J, Liang H, Fernandez A. Protein structure protection commits gene expression patterns. Genome Biol. 2008;9:R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uversky VN. A protein-chameleon: conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J Biomol Struct Dyn. 2003;21:211–234. [DOI] [PubMed] [Google Scholar]
- 24.Fuxreiter M, Tompa P, Simon I, Uversky VN, Hansen JC, Asturias FJ. Malleable machines take shape in eukaryotic transcriptional regulation. Nat Chem Biol. 2008;4:728–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsvetkov P, Asher G, Paz A, et al. Operational definition of intrinsically unstructured protein sequences based on susceptibility to the 20S proteasome. Proteins. 2008;70: 1357–1366. [DOI] [PubMed] [Google Scholar]
- 26.Dunker AK, Uversky VN. Drugs for ‘protein clouds’: targeting intrinsically disordered transcription factors. Curr Opin Pharmacol. 2010;10:782–788. [DOI] [PubMed] [Google Scholar]
- 27.Livesay DR. Protein dynamics: dancing on an ever-changing free energy stage. Curr Opin Pharmacol. 2010;10:706–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janin J, Sternberg MJ. Protein flexibility, not disorder, is intrinsic to molecular recognition. F1000 Biol Rep. 2013;5:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dunker AK, Babu M, Barbar E, et al. What’s in a name? Why these proteins are intrinsically disordered. Intrinsically Disord Proteins. 2013;1:e24157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB. Protein disorder prediction: implications for structural proteomics. Structure. 2003;11:1453–1459. [DOI] [PubMed] [Google Scholar]
- 31.Nielsen JT, Mulder FAA. Quality and bias of protein disorder predictors. Sci Rep. 2019;9:5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He B, Wang K, Liu Y, Xue B, Uversky VN, Dunker AK. Predicting intrinsic disorder in proteins: an overview. Cell Res. 2009;19:929–949. [DOI] [PubMed] [Google Scholar]
- 33.Uversky VN. Protein folding revisited. A polypeptide chain at the folding-misfoldingnonfolding cross-roads: which way to go? Cell Mol Life Sci. 2003;60:1852–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uversky VN. Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta. 2013;1834:932–951. [DOI] [PubMed] [Google Scholar]
- 35.Uversky VN. p53 proteoforms and intrinsic disorder: an Illustration of the protein structure-function continuum concept. Int J Mol Sci. 2016;17:1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith LM, Kelleher NL, Consortium for Top Down Proteomics. Proteoform: a single term describing protein complexity. Nat Methods. 2013;10:186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uversky VN. Protein intrinsic disorder and structure-function continuum. Prog Mol Biol Transl Sci. 2019;166:1–17. [DOI] [PubMed] [Google Scholar]
- 38.Fonin AV, Darling AL, Kuznetsova IM, Turoverov KK, Uversky VN. Multi-functionality of proteins involved in GPCR and G protein signaling: making sense of structure-function continuum with intrinsic disorder-based proteoforms. Cell Mol Life Sci. 2019;76:4461–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams RM, Obradovi Z, Mathura V, et al. The protein non-folding problem: amino acid determinants of intrinsic order and disorder. Pac Symp Biocomput. 2001;89–100. [DOI] [PubMed] [Google Scholar]
- 40.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. [DOI] [PubMed] [Google Scholar]
- 41.Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. Intrinsic disorder and functional proteomics. Biophys J. 2007;92:1439–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vacic V, Uversky VN, Dunker AK, Lonardi S. Composition profiler: a tool for discovery and visualization of amino acid composition differences. BMC Bioinformatics. 2007;8:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li X, Obradovic Z, Brown CJ, Garner EC, Dunker AK. Comparing predictors of disordered protein. Genome Inform Ser Workshop Genome Inform. 2000;11:172–184. [PubMed] [Google Scholar]
- 44.Campen A, Williams RM, Brown CJ, Meng J, Uversky VN, Dunker AK. TOP-IDP-scale: a new amino acid scale measuring propensity for intrinsic disorder. Protein Pept Lett. 2008;15:956–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reichmann D, Jakob U. The roles of conditional disorder in redox proteins. Curr Opin Struct Biol. 2013;23:436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jakob U, Kriwacki R, Uversky VN. Conditionally and transiently disordered proteins: awakening cryptic disorder to regulate protein function. Chem Rev. 2014;114:6779–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Erdos G, Meszaros B, Reichmann D, Dosztanyi Z. Large-scale analysis of redox-sensitive conditionally disordered protein regions reveals their widespread nature and key roles in high-level eukaryotic processes. Proteomics. 2019;19:e1800070. [DOI] [PubMed] [Google Scholar]
- 48.Groitl B, Jakob U. Thiol-based redox switches. Biochim Biophys Acta. 2014;1844:1335–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Messens J, Collet JF. Thiol-disulfide exchange in signaling: disulfide bonds as a switch. Antioxid Redox Signal. 2013;18:1594–1596. [DOI] [PubMed] [Google Scholar]
- 50.Miseta A, Csutora P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol Biol Evol. 2000;17:1232–1239. [DOI] [PubMed] [Google Scholar]
- 51.Fra A, Yoboue ED, Sitia R. Cysteines as redox molecular switches and targets of disease. Front Mol Neurosci. 2017;10:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klomsiri C, Karplus PA, Poole LB. Cysteine-based redox switches in enzymes. Antioxid Redox Signal. 2011;14:1065–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poole LB. The basics of thiols and cysteines in redox biology and chemistry. Free Radic Biol Med. 2015;80:148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poole LB, Nelson KJ. Discovering mechanisms of signaling-mediated cysteine oxidation. Curr Opin Chem Biol. 2008;12:18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Terziyska N, Grumbt B, Kozany C, Hell K. Structural and functional roles of the conserved cysteine residues of the redox-regulated import receptor Mia40 in the intermembrane space of mitochondria. J Biol Chem. 2009;284:1353–1363. [DOI] [PubMed] [Google Scholar]
- 56.Fomenko DE, Xing W, Adair BM, Thomas DJ, Gladyshev VN. High-throughput identification of catalytic redox-active cysteine residues. Science (New York, NY). 2007;315:387–389. [DOI] [PubMed] [Google Scholar]
- 57.Reichmann D, Xu Y, Cremers CM, et al. Order out of disorder: working cycle of an intrinsically unfolded chaperone. Cell. 2012;148:947–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fomenko DE, Marino SM, Gladyshev VN. Functional diversity of cysteine residues in proteins and unique features of catalytic redox-active cysteines in thiol oxidoreductases. Mol Cells. 2008;26:228–235. [PMC free article] [PubMed] [Google Scholar]
- 59.Costantini S, Colonna G, Facchiano AM. Amino acid propensities for secondary structures are influenced by the protein structural class. Biochem Biophys Res Commun. 2006;342:441–451. [DOI] [PubMed] [Google Scholar]
- 60.Otaki JM, Tsutsumi M, Gotoh T, Yamamoto H. Secondary structure characterization based on amino acid composition and availability in proteins. J Chem Inf Model. 2010;50:690–700. [DOI] [PubMed] [Google Scholar]
- 61.van der Reest J, Lilla S, Zheng L, Zanivan S, Gottlieb E. Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat Commun. 2018;9:1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Atkins JD, Boateng SY, Sorensen T, McGuffin LJ. Disorder prediction methods, their applicability to different protein targets and their usefulness for guiding experimental studies. Int J Mol Sci. 2015;16:19040–19054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta. 2010;1804: 996–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Giles NM, Watts AB, Giles GI, Fry FH, Littlechild JA, Jacob C. Metal and redox modulation of cysteine protein function. Chem Biol. 2003;10:677–693. [DOI] [PubMed] [Google Scholar]
- 65.Small DH, Nurcombe V, Reed G, et al. A heparin-binding domain in the amyloid protein precursor of Alzheimer’s disease is involved in the regulation of neurite outgrowth. J Neurosci. 1994;14:2117–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rossjohn J, Cappai R, Feil SC, et al. Crystal structure of the N-terminal, growth factor-like domain of Alzheimer amyloid precursor protein. Nat Struct Biol. 1999;6:327–331. [DOI] [PubMed] [Google Scholar]
- 67.Dawkins E, Small DH. Insights into the physiological function of the β-amyloid precursor protein: beyond Alzheimer’s disease. J Neurochem. 2014;129:756–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reinhard C, Hébert SS, De Strooper B. The amyloid-beta precursor protein: integrating structure with biological function. EMBO J. 2005;24:3996–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.White AR, Multhaup G, Galatis D, et al. Contrasting, species-dependent modulation of copper-mediated neurotoxicity by the Alzheimer’s disease amyloid precursor protein. J Neurosci. 2002;22:365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barnham KJ, McKinstry WJ, Multhaup G, et al. Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis. J Biol Chem. 2003;278:17401–17407. [DOI] [PubMed] [Google Scholar]
- 71.Navaneetham D, Jin L, Pandey P, et al. Structural and mutational analyses of the molecular interactions between the catalytic domain of factor XIa and the Kunitz protease inhibitor domain of protease nexin 2. J Biol Chem. 2005;280:36165–36175. [DOI] [PubMed] [Google Scholar]
- 72.Menéndez-González M, Pérez-Pinera P, Martínez-Rivera M, Calatayud MT, Blázquez Menes B. APP processing and the APP-KPI domain involvement in the amyloid cascade. Neurodegene Dis. 2005;2:277–283. [DOI] [PubMed] [Google Scholar]
- 73.Kluska K, Adamczyk J, Krężel A. Metal binding properties, stability and reactivity of zinc fingers. Coord Chem Rev. 2018;367:18–64. [Google Scholar]
- 74.Cassandri M, Smirnov A, Novelli F, et al. Zinc-finger proteins in health and disease. Cell Death Dis. 2017;3:17071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sun A, Li F, Liu Z, et al. Structural and biochemical insights into human zinc finger protein AEBP2 reveals interactions with RBBP4. Protein Cell. 2018;9:738–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Patel A, Hashimoto H, Zhang X, Cheng X. Characterization of how DNA modifications affect DNA binding by C2H2 zinc finger proteins. Methods Enzymol. 2016;573:387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Graves DT, Jiang Y. Chemokines, a family of chemotactic cytokines. Crit Rev Oral Biol Med. 1995;6:109–118. [DOI] [PubMed] [Google Scholar]
- 78.Zlotnik A, Yoshie O, Nomiyama H. The chemokine and chemokine receptor super-families and their molecular evolution. Genome Biol. 2006;7:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fernandez EJ, Lolis E. Structure, function, and inhibition of chemokines. Annu Rev Pharmacol Toxicol. 2002;42:469–499. [DOI] [PubMed] [Google Scholar]
- 80.Mayer KL, Stone MJ. NMR solution structure and receptor peptide binding of the CC chemokine eotaxin-2. Biochemistry. 2000;39:8382–8395. [DOI] [PubMed] [Google Scholar]
- 81.Crump MP, Spyracopoulos L, Lavigne P, Kim KS, Clark-lewis I, Sykes BD. Backbone dynamics of the human CC chemokine eotaxin: fast motions, slow motions, and implications for receptor binding. Protein Sci. 1999;8:2041–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ye J, Kohli LL, Stone MJ. Characterization of binding between the chemokine eotaxin and peptides derived from the chemokine receptor CCR3. J Biol Chem. 2000;275:27250–27257. [DOI] [PubMed] [Google Scholar]
- 83.Williams P, Chaudhry Y, Goodfellow IG, et al. Mapping CD55 function: the structure of two pathogen-binding domains at 1.7Å. J Biol Chem. 2003;278:10691–10696. [DOI] [PubMed] [Google Scholar]
- 84.Lublin DM, Atkinson JP. Decay-accelerating factor: biochemistry, molecular biology, and function. Annu Rev Immunol. 1989;7:35–58. [DOI] [PubMed] [Google Scholar]
- 85.Lukacik P, Roversi P, White J, et al. Complement regulation at the molecular level: the structure of decay-accelerating factor. Proc Natl Acad Sci U S A. 2004;101:1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ojha H, Ghosh P, Singh Panwar H, et al. Spatially conserved motifs in complement control protein domains determine functionality in regulators of complement activation-family proteins. Commun Biol. 2019;2:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ezekowitz RAB, Hoffmann JA. Innate immunity. Curr Opin Immunol. 1996;8:1–2. [DOI] [PubMed] [Google Scholar]
- 88.Norman DG, Barlow PN, Baron M, Day AJ, Sim RB, Campbell ID. Three-dimensional structure of a complement control protein module in solution. J Mol Biol. 1991;219:717–725. [DOI] [PubMed] [Google Scholar]
- 89.Ma TH, Benzie JA, He JG, Sun CB, Chan SF. PmPPAF is a pro-phenoloxidase activating factor involved in innate immunity response of the shrimp Penaeus monodon. Dev Comp Immunol. 2014;44:163–172. [DOI] [PubMed] [Google Scholar]
- 90.Kanost MR, Jiang H. Clip-domain serine proteases as immune factors in insect hemolymph. Curr Opin Insect Sci. 2015;11:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lu A, Zhang Q, Zhang J, et al. Insect prophenoloxidase: the view beyond immunity. Front Physiol. 2014;5:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kwon TH, Kim MS, Choi HW, Joo CH, Cho MY, Lee BL. A masquerade-like serine proteinase homologue is necessary for phenoloxidase activity in the coleopteran insect, Holotrichia diomphalia larvae. Eur J Biochem. 2000;267:6188–6196. [DOI] [PubMed] [Google Scholar]
- 93.Piao S, Jung JY, Park JW, Lee J, Lee BL, Ha NC. Preliminary X-ray crystallographic analysis of the catalytic domain of prophenoloxidase activating factor-I. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:771–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Piao S, Song YL, Kim JH, et al. Crystal structure of a clip-domain serine protease and functional roles of the clip domains. EMBO J. 2005;24:4404–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Smith JG, Nemerow GR. Mechanism of adenovirus neutralization by Human alpha-defensins. Cell Host Microbe. 2008;3:11–19. [DOI] [PubMed] [Google Scholar]
- 96.Pazgier M, Hoover DM, Yang D, Lu W, Lubkowski J. Human β-defensins. Cell Mol Life Sci. 2006;63:1294–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Torres AM, Kuchel PW. The β-defensin-fold family of polypeptides. Toxicon. 2004;44:581–588. [DOI] [PubMed] [Google Scholar]
- 98.Taylor K, Barran PE, Dorin JR. Structure-activity relationships in beta-defensin peptides. Biopolymers. 2008;90:1–7. [DOI] [PubMed] [Google Scholar]
- 99.Hoover DM, Chertov O, Lubkowski J. The structure of human β-defensin-1: new insights into structural properties of β-defensins. J Biol Chem. 2001;276: 39021–39026. [DOI] [PubMed] [Google Scholar]
- 100.Kouno T, Mizuguchi M, Aizawa T, et al. A novel β-defensin structure: big defensin changes its N-terminal structure to associate with the target membrane. Biochemistry. 2009;48:7629–7635. [DOI] [PubMed] [Google Scholar]
- 101.Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res. 2009;50(suppl):S237–S242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dennis EA, Cao J, Hsu Y-H, Magrioti V, Kokotos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev. 2011;111:6130–6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Murakami M, Kudo I. Phospholipase A2. J Biochem. 2002;131:285–292. [DOI] [PubMed] [Google Scholar]
- 104.Yu B-Z, Pan YH, Janssen MJW, Bahnson BJ, Jain MK. Kinetic and structural properties of disulfide engineered phospholipase A2: insight into the role of disulfide bonding patterns. Biochemistry. 2005;44:3369–3379. [DOI] [PubMed] [Google Scholar]
- 105.Xu W, Yi L, Feng Y, Chen L, Liu J. Structural insight into the activation mechanism of human pancreatic prophospholipase A2. J Biol Chem. 2009;284:16659–16666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Law RHP, Abu-Ssaydeh D, Whisstock JC. New insights into the structure and function of the plasminogen/plasmin system. Curr Opin Struct Biol. 2013;23:836–841. [DOI] [PubMed] [Google Scholar]
- 107.Didiasova M, Wujak L, Wygrecka M, Zakrzewicz D. From plasminogen to plasmin: role of plasminogen receptors in human cancer. Int J Mol Sci. 2014;15:21229–21252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Novokhatny VV, Kudinov SA, Privalov PL. Domains in human plasminogen. J Mol Biol. 1984;179:215–232. [DOI] [PubMed] [Google Scholar]
- 109.Marshall JM, Brown AJ, Ponting CP. Conformational studies of human plasminogen and plasminogen fragments: evidence for a novel third conformation of plasminogen. Biochemistry. 1994;33:3599–3606. [DOI] [PubMed] [Google Scholar]
- 110.Xue Y, Bodin C, Olsson K. Crystal structure of the native plasminogen reveals an activation-resistant compact conformation. J Thromb Haemost. 2012;10:1385–1396. [DOI] [PubMed] [Google Scholar]
- 111.Law RHP, Caradoc-Davies T, Cowieson N, et al. The X-ray crystal structure of full-length human plasminogen. Cell Rep. 2012;1:185–190. [DOI] [PubMed] [Google Scholar]
- 112.Mulichak AM, Tulinsky A, Ravichandran KG. Crystal and molecular structure of human plasminogen kringle 4 refined at 1.9-.ANG. resolution. Biochemistry. 1991; 30:10576–10588. [DOI] [PubMed] [Google Scholar]
- 113.Wu TP, Padmanabhan K, Tulinsky A, Mulichak AM. The refined structure of the. epsilon.-aminocaproic acid complex of human plasminogen kringle 4. Biochemistry. 1991;30:10589–10594. [DOI] [PubMed] [Google Scholar]
- 114.Castellino FJ, McCance SG. The kringle domains of human plasminogen. Ciba Found Symp. 1997;212:46–60. discussion 60–45. [DOI] [PubMed] [Google Scholar]
- 115.Ragland SA, Criss AK. From bacterial killing to immune modulation: recent insights into the functions of lysozyme. PLoS Pathog. 2017;13:e1006512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fleming A, Wright AE. On a remarkable bacteriolytic element found in tissues and secretions. Proc R Soc Lond Ser B. 1922;93:306–317. [Google Scholar]
- 117.Wohlkönig A, Huet J, Looze Y, Wintjens R. Structural relationships in the lysozyme superfamily: significant evidence for glycoside hydrolase signature motifs. PLoS One. 2010;5:e15388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McKenzie HA, White FH. Lysozyme and α-Lactalbumin: structure, function, and interrelationships. In: Anfinsen CB, Richards FM, Edsall JT, Eisenberg DS, eds. Advances in Protein Chemistry. Academic Press; 1991:173–315. [DOI] [PubMed] [Google Scholar]
- 119.Muttathukattil AN, Singh PC, Reddy G. Role of disulfide bonds and topological frustration in the kinetic partitioning of lysozyme folding pathways. J Phys Chem B. 2019;123:3232–3241. [DOI] [PubMed] [Google Scholar]
- 120.Held J, van Smaalen S. The active site of hen egg-white lysozyme: flexibility and chemical bonding. Acta Crystallogr D Biol Crystallogr. 2014;70:1136–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yokota A, Hirai K, Miyauchi H, et al. NMR characterization of three-disulfide variants of lysozyme, C64A/C80A, C76A/C94A, and C30A/C115AA marginally stable state in folded proteins. Biochemistry. 2004;43:6663–6669. [DOI] [PubMed] [Google Scholar]
- 122.Fischer B Folding of lysozyme. EXS. 1996;75:143–161. [DOI] [PubMed] [Google Scholar]
- 123.Juan HF, Hung CC, Wang KT, Chiou SH. Comparison of three classes of snake neurotoxins by homology modeling and computer simulation graphics. Biochem Biophys Res Commun. 1999;257:500–510. [DOI] [PubMed] [Google Scholar]
- 124.Tsetlin V Snake venom alpha-neurotoxins and other ‘three-finger’ proteins. Eur J Biochem. 1999;264:281–286. [DOI] [PubMed] [Google Scholar]
- 125.Ferraz CR, Arrahman A, Xie C, et al. Multifunctional toxins in snake venoms and therapeutic implications: from pain to hemorrhage and necrosis. Front Ecol Evol. 2019;7: Article 218 (19 pages).31667165 [Google Scholar]
- 126.Kessler P, Marchot P, Silva M, Servent D. The three-finger toxin fold: a multifunctional structural scaffold able to modulate cholinergic functions. J Neurochem. 2017; 142(suppl 2):7–18. [DOI] [PubMed] [Google Scholar]
- 127.Phillips DJ, de Kretser DM. Follistatin: a multifunctional regulatory protein. Front Neuroendocrinol. 1998;19:287–322. [DOI] [PubMed] [Google Scholar]
- 128.Patel K. Follistatin. Int J Biochem Cell Biol. 1998;30:1087–1093. [DOI] [PubMed] [Google Scholar]
- 129.Keutmann HT, Schneyer AL, Sidis Y. The role of follistatin domains in follistatin biological action. Mol Endocrinol. 2004;18:228–240. [DOI] [PubMed] [Google Scholar]
- 130.Hansen JS, Plomgaard P. Circulating follistatin in relation to energy metabolism. Mol Cell Endocrinol. 2016;433:87–93. [DOI] [PubMed] [Google Scholar]
- 131.Zhang L, Liu K, Han B, Xu Z, Gao X. The emerging role of follistatin under stresses and its implications in diseases. Gene. 2018;639:111–116. [DOI] [PubMed] [Google Scholar]
- 132.Schlott B, Wöhnert J, Icke C, et al. Interaction of kazal-type inhibitor domains with serine proteinases: biochemical and structural studies. J Mol Biol. 2002;318:533–546. [DOI] [PubMed] [Google Scholar]
- 133.Innis CA, Hyvönen M. Crystal structures of the heparan sulfate-binding domain of follistatin. Insights into ligand binding. J Biol Chem. 2003;278:39969–39977. [DOI] [PubMed] [Google Scholar]
- 134.Fleming RJ. Structural conservation of Notch receptors and ligands. Semin Cell Dev Biol. 1998;9:599–607. [DOI] [PubMed] [Google Scholar]
- 135.Gordon WR, Arnett KL, Blacklow SC. The molecular logic of Notch signaling–a structural and biochemical perspective. J Cell Sci. 2008;121:3109–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zeng F, Harris RC. Epidermal growth factor, from gene organization to bedside. Semin Cell Dev Biol. 2014;28:2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Harris RC, Chung E, Coffey RJ. EGF receptor ligands. Exp Cell Res. 2003;284:2–13. [DOI] [PubMed] [Google Scholar]
- 138.Dreux AC, Lamb DJ, Modjtahedi H, Ferns GA. The epidermal growth factor receptors and their family of ligands: their putative role in atherogenesis. Atherosclerosis. 2006;186:38–53. [DOI] [PubMed] [Google Scholar]
- 139.Heath WF, Merrifield RB. A synthetic approach to structure-function relationships in the murine epidermal growth factor molecule. Proc Natl Acad Sci U S A. 1986;83:6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lin HH, Stacey M, Saxby C, et al. Molecular analysis of the epidermal growth factor-like short consensus repeat domain-mediated protein-protein interactions: dissection of the CD97-CD55 complex. J Biol Chem. 2001;276:24160–24169. [DOI] [PubMed] [Google Scholar]
- 141.Montelione GT, Wüthrich K, Burgess AW, et al. Solution structure of murine epidermal growth factor determined by NMR spectroscopy and refined by energy minimization with restraints. Biochemistry. 1992;31:236–249. [DOI] [PubMed] [Google Scholar]
- 142.Rehder D The role of vanadium in biology. Metallomics. 2015;7:730–742. [DOI] [PubMed] [Google Scholar]
- 143.Michibata H, Iwata Y, Hirata J. Isolation of highly acidic and vanadium-containing blood cells from among several types of blood cell from ascidiidae species by density-gradient centrifugation. J Exp Zool. 1991;257:306–313. [Google Scholar]
- 144.Fukui K, Ueki T, Ohya H, Michibata H. Vanadium-binding protein in a vanadium-rich ascidian ascidia sydneiensis samea: CW and pulsed EPR studies. J Am Chem Soc. 2003;125:6352–6353. [DOI] [PubMed] [Google Scholar]
- 145.Ueki T, Kawakami N, Toshishige M, Matsuo K, Gekko K, Michibata H. Characterization of vanadium-binding sites of the vanadium-binding protein Vanabin2 by site-directed mutagenesis. Biochim Biophys Acta. 2009;1790:1327–1333. [DOI] [PubMed] [Google Scholar]
- 146.Kawakami N, Ueki T, Matsuo K, Gekko K, Michibata H. Selective metal binding by Vanabin2 from the vanadium-rich ascidian, Ascidia sydneiensis samea. Biochim Biophys Acta. 2006;1760:1096–1101. [DOI] [PubMed] [Google Scholar]
- 147.Hamada T, Asanuma M, Ueki T, et al. Solution structure of Vanabin2, a Vanadium(IV)-binding protein from the vanadium-rich ascidian ascidia sydneiensis samea. J Am Chem Soc. 2005;127:4216–4222. [DOI] [PubMed] [Google Scholar]
- 148.Bhopatkar AA, Ghag G, Wolf LM, Dean DN, Moss MA, Rangachari V. Cysteine-rich granulin-3 rapidly promotes amyloid-beta fibrils in both redox states. Biochem J. 2019;476:859–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ghag G, Wolf LM, Reed RG, et al. Fully reduced granulin-B is intrinsically disordered and displays concentration-dependent dynamics. Protein Eng Des Sel. 2016;29:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ghag G, Holler CJ, Taylor G, Kukar TL, Uversky VN, Rangachari V. Disulfide bonds and disorder in granulin-3: An unusual handshake between structural stability and plasticity. Protein Sci. 2017;26:1759–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Holler CJ, Taylor G, Deng Q, Kukar T. Intracellular proteolysis of progranulin generates stable, lysosomal granulins that are haploinsufficient in patients with frontotemporal dementia caused by GRN mutations. eNeuro. 2017;4:ENEURO 0100–0117.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Palfree RG, Bennett HP, Bateman A. The evolution of the secreted regulatory protein progranulin. PLoS One. 2015;10:e0133749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hrabal R, Chen Z, James S, Bennett HP, Ni F. The hairpin stack fold, a novel protein architecture for a new family of protein growth factors. Nat Struct Biol. 1996;3:747–752. [DOI] [PubMed] [Google Scholar]
- 154.Tolkatchev D, Malik S, Vinogradova A, et al. Structure dissection of human progranulin identifies well-folded granulin/epithelin modules with unique functional activities. Protein Sci. 2008;17:711–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hamer DH. Metallothionein. Annu Rev Biochem. 1986;55:913–951. [DOI] [PubMed] [Google Scholar]
- 156.Kaegi JHR, Schaeffer A. Biochemistry of metallothionein. Biochemistry. 1988;27: 8509–8515. [DOI] [PubMed] [Google Scholar]
- 157.Moffatt P, Denizeau F. Metallothionein in physiological and physiopathological processes. Drug Metab Rev. 1997;29:261–307. [DOI] [PubMed] [Google Scholar]
- 158.Acharya C, Blindauer CA. Unexpected interactions of the cyanobacterial metallothionein SmtA with uranium. Inorg Chem. 2016;55:1505–1515. [DOI] [PubMed] [Google Scholar]
- 159.Bell SG, Vallee BL. The metallothionein/thionein system: an oxidoreductive metabolic zinc link. Chembiochem. 2009;10:55–62. [DOI] [PubMed] [Google Scholar]
- 160.Rodriguez-Menendez S, Garcia M, Fernandez B, et al. The zinc-metallothionein redox system reduces oxidative stress in retinal pigment epithelial cells. Nutrients. 2018;10:1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Maret W Redox biochemistry of mammalian metallothioneins. JBIC J Biol Inorg Chem. 2011;16:1079–1086. [DOI] [PubMed] [Google Scholar]
- 162.Sabolic I, Breljak D, Skarica M, Herak-Kramberger CM. Role of metallothionein in cadmium traffic and toxicity in kidneys and other mammalian organs. Biometals. 2010;23:897–926. [DOI] [PubMed] [Google Scholar]
- 163.Martinho A, Goncalves I, Cardoso I, et al. Human metallothioneins 2 and 3 differentially affect amyloid-beta binding by transthyretin. FEBS J. 2010;277:3427–3436. [DOI] [PubMed] [Google Scholar]
- 164.Goncalves I, Quintela T, Baltazar G, Almeida MR, Saraiva MJ, Santos CR. Transthyretin interacts with metallothionein 2. Biochemistry. 2008;47:2244–2251. [DOI] [PubMed] [Google Scholar]
- 165.Coyle P, Philcox JC, Carey LC, Rofe AM. Metallothionein: the multipurpose protein. Cell Mol Life Sci. 2002;59:627–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Roschitzki B, Vašák M. A distinct Cu4-thiolate cluster of human metallothionein-3 is located in the N-terminal domain. JBIC J Biol Inorg Chem. 2002;7:611–616. [DOI] [PubMed] [Google Scholar]
- 167.Messerle BA, Schäffer A, Vasák M, Kägi JH, Wüthrich K. Three-dimensional structure of human [113Cd7]metallothionein-2 in solution determined by nuclear magnetic resonance spectroscopy. J Mol Biol. 1990;214:765–779. [DOI] [PubMed] [Google Scholar]
- 168.Zeyer KA, Reinhardt DP. Engineered mutations in fibrillin-1 leading to Marfan syndrome act at the protein, cellular and organismal levels. Mutat Res Rev Mutat Res. 2015;765:7–18. [DOI] [PubMed] [Google Scholar]
- 169.Handford PA. Fibrillin-1, a calcium binding protein of extracellular matrix. Biochim Biophys Acta Biomembr Mol Cell Res. 2000;1498:84–90. [DOI] [PubMed] [Google Scholar]
- 170.Wang D, Horton JR, Zheng Y, Blumenthal RM, Zhang X, Cheng X. Role for first zinc finger of WT1 in DNA sequence specificity: Denys-Drash syndrome-associated WT1 mutant in ZF1 enhances affinity for a subset of WT1 binding sites. Nucleic Acids Res. 2018;46:3864–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Borel F, Barilla KC, Hamilton TB, Iskandar M, Romaniuk PJ. Effects of Denys-Drash syndrome point mutations on the DNA binding activity of the Wilms’ tumor suppressor protein WT1. Biochemistry. 1996;35:12070–12076. [DOI] [PubMed] [Google Scholar]