

Abstract
Coal fueled the Industrial Revolution and the global expansion of electrification in the 20th century. In the 21st century, coal use has declined in North America and Europe, but continues to increase in Asia. Coal contains many of the elements of the Periodic Table, in percent-levels or in trace amounts (ppm, ppb). The impact of many of these elements on the environment via air and water discharges from coal-fired plants has been studied with decades of research on their chemical transformations within combustion systems and on their fates upon reintroduction into the environment. The transformations of the trace elements present in coal burned during combustion can be categorized as thermal volatilizations from the coal in the furnace; thermal decomposition of trace element compounds inside the coal; encapsulation inside ash structures through high-temperature vitrification; oxidation of the trace elements with the myriad species contained in flue gas through gas phase (homogeneous) reactions or catalytic (gas-solid) reactions; adsorption and/or reactions with active sites on entrained fly ash particulates contained in the flue gas; and absorption into solutions. These transformations can, in many cases, impact the fraction of these trace elements that are removed by various pollution control devices compared to the fraction released into the environment. The sampling and measurement of trace elements, in the inlet coal, outlet flue gas, aqueous scrubber solutions, and ash matrices, represents a significant challenge. This review focuses on the behavior of trace elements in industrial coal combustion systems with an emphasis on what has been learned over the past century uniquely related to the use of coal in boilers for electricity and heat production. Key accomplishments in measurement, modeling and control of trace element emissions in coal-fired systems are highlighted.
Keywords: Coal, combustion, trace elements, fly ash, mercury, air pollution, coal combustion residuals, rare earth elements
Introduction
Coal’s Importance in World Energy Usage
Coal has powered the world’s industries for centuries, but at a cost: coal contains many elements that affect human health and the health of ecosystems. Over the last 40 years, global coal usage has increased, especially since the beginning of the 21st century, when rising industrial activity in Asia doubled global coal consumption relative to the end of the 20th century. At the same time, coal consumption in Europe, North America, and the former Soviet republics decreased [1]. With current stated policies, coal consumption world-wide is forecast to be flat for the next 10 years and then rising after 2030 through 2050. However, there is the potential for significant reductions if more sustainable policies are adopted, particularly in the Asian countries which are forecast to drive the mid-century increase in consumption [2].
Coal is primarily used for the generation of electricity and commercial heat, with important quantities also used for the production of chemicals, tars, steel, and activated carbons. In 2017, 66.5% of coal worldwide was used for heating and electricity generation, while in the 37 member countries of the Organization for Economic Cooperation and Development (OECD), the proportion was higher at 82.3%. The global share of heat and power generated from coal has held steady at about 40% over the last 40 years, although the share of electricity and heat produced from coal in OECD countries has continued to decrease to 25% in 2018 because of factors such as low-cost natural gas, decreases in the costs for wind and solar electricity generation, and government policies supportive of renewable energy [3].
Coal contains elements, including trace elements (TEs), that affect human health and the health of ecosystems, if released to the environment in specific forms. Knowledge of the impact of TEs released during coal use grew tremendously during the 20th century.
The Impacts of Trace Elements from Coal Combustion on the Environment and Human Health
On a mass basis, the major air pollutants emitted from coal combustion are sulfur dioxide (SO2), nitrogen oxides (NO + NO2 or NOx), and particulate matter (PM), all of which affect human health and the health of ecosystems. When coal is burned, the vast majority of TEs are typically collected in the ash by particulate control systems and isolated from the environment. However, TEs are also emitted to the air (as well as discharged to water bodies) in both the gas phase and as part of the emitted PM. These emissions are typically much smaller in relation to the major pollutants. Nonetheless, the impact of TEs on human health and ecosystems can be significant [4, 5]. Furthermore, care must be taken to adequately isolate and store the collected ash or use it in ways that minimize the migration of toxic elements to the environment.
Very early references to the significance of TEs in coal can be found around the turn of the 20th century when coal was used for drying of certain ingredients, like malted barley, used in beer-making. Arsenic could be volatilized from the coal and subsequently condense on the malt [6]. This led to methods for quantifying the arsenic concentration in coals [7]. Arsenic and other TEs have also long been of concern in the domestic use of coal. For much of the 20th century, coal was used for household cooking and heating with impacts on human health, for example, as documented with respect to Chinese households [4,8,9].
In the 1960s, deposition of mercury from the atmosphere was linked to increases in mercury concentration in aquatic environments and in fish [10]. Anthropogenic releases were suspected as important contributors to mercury in the environment because of the large amount of coal burned for electricity production and other industrial purposes [11]. Billings and Matson [12] were among the first researchers to directly measure gaseous emissions of mercury from a coal-fired power plant. They concluded that 90% of the mercury input to the 700 MWe power plant which they sampled was emitted from the stack to the air. In addition to mercury, other TEs, including radionuclides, were identified as having the potential to impact human and ecological health [13].
Evidence of the impact of TEs from coal combustion on the environment accumulated in the 1960s and 1970s. Measurements were made of certain TEs in air emissions from coal-fired plants [12, 14-17]. More ambitious projects attempted to determine the mass balances of TEs in plants, considering the composition of coal and ash, air emissions, and water discharges [18-24]. Determination of mass balances allowed consideration of the partitioning of TEs among various phases in coal-fired power plants. These efforts have led to a growing understanding of the importance of coal type, minor element concentration, and combustion conditions upon the partitioning of TEs between solid and gaseous phases and upon the mobility of TEs from particles when released to the environment.
Numerous studies document the TE content of size-segregated fly ash starting in the 1970s. In one of the earliest, Natusch and co-workers [14] analyzed size-segregated fly ash samples collected from the particulate control devices and the stacks of coal-fired power plants. For certain trace elements (i.e. As, Cd, Cr, Ni, Pb, Sb, Se, Tl, Zn), the concentration in the fly ash increased with decreasing particle size.
Particles in the range 0.1-1 micron in diameter (i.e., submicron particles) are captured less efficiently than larger particles by particulate control devices [15, 25-27]. These transition-range particles have neither the inertia nor the high diffusivities for efficient removal by impaction or diffusion. It has also been shown that, due to their low settling velocities, the residence time of submicron particles in atmospheric aerosol (ca. 100 h) is an order of magnitude longer than for supermicron particles (10–100 h) [28]. These particles will eventually settle out onto the ground or other surfaces downwind of the combustor. Once deposited, ground moisture and rainfall may dissolve soluble TE-containing compounds out of the ash and carry it into the soil. These compounds may then be adsorbed into plants through roots in the soil, percolate downwards to the water table, or runoff into rivers and lakes. The solubility will depend primarily upon the pH of the soil where the particles deposit, rather than the pH of the ash itself [29,30].
Very small submicron particles (<0.1 micron in diameter), known as the submicron fume or as ultrafine particles, have a very low mass emission but higher number concentrations. However, the residence time of ultrafine particles in an atmosphere aerosol is even longer (ca. 1000 hours) [28]. These particles may present a greater risk of inhalation in human and animal respiratory systems [14,31,32] as they may be deposited more deeply in the lungs if inhaled, and offer large surface area to volume ratios. Mumford and Lewtas reported on the mutagenicity and cytotoxicity of coal fly ash [33]. Gilmour et al. demonstrated that fine (< 2.5 microns) and ultrafine (defined in their study as <0.2 micron) fractions of fly ash from coal combustion generated more pulmonary inflammation in mice than bulk (also known as coarse, >2.5 micron) particles, and that for a subbituminous coal, the ultrafine PM was more toxic than the fine PM [34]. A 2018 modeling study explored the impacts of air quality changes for 20 states in the USA if EPA relaxed air pollution standards for coal-fired power plants from those that were in effect in 2018 to those that were in effect in 2007 [35]. The study concluded that this change would raise atmospheric levels of PM that is less than 2.5 microns in diameter (PM2.5) by 1 to 5 μg/m3 and cause additional premature mortalities in the range of 17-to 39 thousand per year.
Regulation and control of TE air emissions have required increased knowledge of the chemistry of TEs in coal combustion systems and air pollution control devices. Unique chemistry occurs in these systems because of the conditions and/or other elements present in coal combustion systems that would not be present in other systems. The emission of TEs into the air from coal-fired power plants is not the only pathway of concern, although air emissions have received more attention. Discharges of trace elements into water bodies and migration to ground water are also of concern, requiring knowledge of TE partitioning into coal combustion residuals during combustion and post-combustion treatment as well as the mobility of TEs out of these solids.
Coal combustion residuals (CCRs) include bottom ash, fly ash, and solid byproducts from flue gas desulfurization (FGD) units at coal-fired power plants. In the USA, CCRs are considered non-hazardous waste under the Resource, Recovery, and Conservation Act (RCRA); a Final Rule was issued by the U.S. EPA in 2015 on coal ash disposal and remediation of disposal sites [36] In Europe, CCRs are classified as non-hazardous waste and regulated under the Waste Framework Directive (2008/98/EC) [37]. When re-used, CCPs must be registered, subject to the Registration, Evaluation and Authorisation of Chemicals (REACH), Directive 2006/121/EC.
In 2016, 1.2 billion metric tons of CCRs were produced world-wide with an overall utilization rate of 64% [38]. Table 1 shows CCR production and utilization by country or region. If not re-used, CCRs are either stored at the power plant in surface impoundments (bodies of water confined within an enclosure, often containing a mixture of water and ash) or landfills (engineered structures built into or on top of the ground containing dry CCR solids), or sent offsite for landfill disposal. Ash in the environment can weather over time (e.g., in unlined landfills), resulting in concentrations of TEs in groundwater that can, in some cases, exceed local drinking water quality standards [39-42]. Individuals living near coal-fired power plants have increased risks of respiratory and cardiovascular disease, higher infant mortality, and poorer child health, associated with air pollutant emissions and the effects of TEs (including radioactive elements), migrating out of locally stored coal ash [5].
Table 1.
2016 Annual Production and Utilization of CCRs in millions of metric tons [38].
| Country or Region | Produced | Utilized | %Utilized |
|---|---|---|---|
| Australia | 12.3 | 5.4 | 43.5% |
| Asia | |||
| China | 656 | 396 | 70.1% |
| Korea | 10.3 | 8.8 | 85.4% |
| India | 197 | 132 | 67.1% |
| Japan | 12.3 | 12.3 | 99.3% |
| Asia, Other | 18.2 | 12.3 | 67.6% |
| Europe | 140 | ||
| (EU-15) | (40.3) | 38 | 94.3% |
| Middle East & Africa | 32.2 | 3.4 | 10.6% |
| Israel | 1.1 | 1.0 | 90.9% |
| USA | 107.4 | 60.1 | 56.0% |
| Canada | 4.8 | 2.6 | 54.2% |
| Russia | 21.3 | 5.8 | 27.2% |
| Total | 1,222 | 678 | 63.9% |
In the EU and Japan, almost all CCRs are utilized, instead of being disposed (Table 1). In the USA, where approximately half of the CCRs generated are not used, there are about 300 active on-site landfills at coal-fired power plants, which average more than 0.5 km2 (50 hectares) in area and have an average depth of over 12 meters, and more than 700 active on-site surface impoundments, which average more than 0.2 km2 (20 hectares) and have an average depth of 6 meters [43]. In addition to active landfills and impoundments, many more have been closed as coal-fired power plants retire. However, whether actively in use for ash disposal or closed, landfills and impoundments can be a source of TE contamination of surface waters and groundwater.
CCR landfills in the USA must monitor groundwater, control run-off, and have controls for fugitive dust. New landfills must be lined with an impermeable barrier, but many existing landfills are unlined. New and existing surface impoundments have similar requirements for groundwater monitoring. Surface impoundments also have restrictions regarding structural integrity of dams and other containment structures.
Regulations on CCR disposal facilities in the USA were tightened in 2015 [44], in part because of several large breaches of existing CCR ponds, the most notable of which occurred in December, 2008. The breach of a coal ash pond from the Tennessee Valley Authority (TVA) power plant in Kingston, Tennessee flooded more than 1.2 km2 (120 hectares) of land and released over 4.1 million m3 of ash. A survey immediately after the spill showed contamination of surface waters in nearby ponds, but only trace levels were found in the downstream rivers due to dilution [45]. A follow-up study eighteen months after the spill, showed TE concentrations, particularly As, B, Ba, Se and Sr, were below U.S. EPA drinking water limits in river water. Elevated levels were found in ponds and surface water with restricted water exchange and in pore water extracted from the river sediments [46]. Eight years after the spill, the concentrations of trace metals measured in river sediments downstream of the Kingston Plant were compared to concentrations at site located outside the zone of the ash spill. This comparison suggested that up to 80% of the As and 60-90% of the Se in the river downstream of the Kingston Plant were sourced from coal ash [47].
In addition to catastrophic events, like the large-scale rupturing of ash impoundments, CCR impoundments can leak into nearby surface waters or groundwater. For example, Harkness et al. [42] tested groundwater and surface water from 22 unlined coal ash storage impoundments in five states in the southeastern USA. Previous work had shown that boron and strontium isotopes could be used as tracers for the presence of other trace contaminants originating from CCRs [48]. Harkness et al. found isotopic fingerprints for B and Sr characteristic of coal ash at all but one site. Concentrations of CCR contaminants, including sulfate, As, Ca, Fe, Mn, Mo, Se and V, at levels above drinking water levels were observed in 10 out of 24 samples of surface water. Sixty-five groundwater monitoring wells (out of 165) showed high B levels and 49 had high levels of other CCR-contaminants [42].
At many municipal drinking water treatment plants, the disinfection of source water (from rivers and lakes) can produce halogenated organic compounds via the reaction between halides and trace amounts of organic compounds. Some of these disinfection byproducts (DBPs) have been determined to be carcinogenic [49]. Although bromide concentrations in natural waters are generally low, elevated levels of bromide in surface waters have been observed recently and coal-fired power plants have been suggested as one source of these elevated bromide levels [50,51]. In the USA, the implementation of the Mercury and Air Toxics Standards (MATS) in 2014 [52] and the Refined Coal tax credit [53] (both discussed below), motivated some U.S. coal-fired power plants to add bromine to their coal to facilitate Hg removal [54,55]. If those plants also utilized an FGD, an increase in Br concentration was measured in the scrubber effluent. Source apportionment is not always easy or clear-cut, but it is possible that coal-fired power plants have contributed to increases in bromide discharges and DBP formation in municipal drinking water plants downstream of coal-fired power plants.
Review Outline
In summary, TE discharges to the air, soil, and water from coal combustion sources have been shown to have impacts on ecosystems and human health. Devising methods to better control emissions of TEs rests on a clear understanding of the forms of TEs in coal, their transformation during the combustion process, and their behavior in air pollution control devices. This review presents an overview of trace element chemistry in coal combustion systems with an emphasis on what has been learned over the past century uniquely related to the use of coal in boilers for electricity and heat production because these are used world-wide and represent the major pathway for most TEs from coal to be released into the environment for the foreseeable future.
The first section of the review focuses on the combustion system. Before discussing the detailed chemistry of TEs, we explain the concept of modes of occurrence of inorganic elements in coal because the mode of occurrence of a given element can affect its behavior in combustion systems. Semi-volatile elements are those that are partially volatilized from coal during combustion. This category includes all the TEs of environmental concern except mercury. Mercury chemistry differs from the semi-volatile elements (e.g. mercury completely vaporizes in the combustion system, forming different species post combustion with unique properties) and thus is treated separately.
The second section of the review discusses the advances in analytical methods that have allowed greater understanding of the chemistry of TEs and the methods to control TE emissions. Combustion systems are harsh environments and specialized sampling methods had to be developed to collect representative, stable samples from these systems. Measurement techniques were adapted, and new techniques were developed to characterize TE concentrations and forms in coal and coal combustion products. Finally, modeling tools that evolved to predict the behavior of TEs in combustion systems and in air pollution control devices are discussed.
Control of TE emissions to the environment from coal-fired systems was required in the U.S. and other countries as early as the early 21st century. The third section discusses the control of TEs using a variety of air pollution control devices. Emissions into the air of semi-volatile TEs that partitioned to PM are controlled primarily by particulate matter controls on coal-fired systems which represents the vast majority of TE air emissions. However, because of its high volatility and varied speciation in flue gas, mercury emissions were not as easily controlled. Considerable research was carried out in the final decades of the 20th century on mercury control, which is summarized in this section.
Before concluding, we briefly touch on current issues and future concerns regarding TEs in coal combustion systems. No review of such a complex subject can claim to be comprehensive. The authors have endeavored to highlight early, seminal work, which means that more recent work may be omitted or discussed superficially, especially if derived from earlier work.
The Chemistry of Trace Elements in Combustion Systems
The Composition of Trace Elements in Coals
The primary elements in coal are carbon, oxygen, and hydrogen, but coal contains many inorganic elements in minor and trace quantities. The process of transforming organic plant matter to coal, a process known as coalification, is outside the scope of this work, but the reader is referred to, for example, references [56-58] for details. Much work has been published regarding the composition of coals from around the world (see, for example, [59-69]). The U.S. Geological Survey maintains databases on coal quality to which the reader is also directed for more information [67].
The mineral matter in coal comprises crystalline and non-crystalline inorganic compounds in discrete particles as well as inorganic elements dissolved in pore water or adsorbed on organic compounds [70-71]. The origins of mineral matter are the result of various processes that occur from coal formation to extraction. The interested reader can consult reviews by Ward [70,71].
Trace elements are referred to as those elements in coal with a concentration of 100 μg/g (ppmw) or less [73]. Through the 1980s, TEs were not directly analyzed in coal samples, but analyzed in coal ash that was produced by procedures that often involved heating the sample to 750°C-850°C. These high-temperature coal ashing procedures could volatilize elements like Hg or halogens, resulting in inaccurate concentrations when calculated on a whole-coal basis. Since the 1990s, direct methods of analyzing coal for TEs, such as instrumental neutron activation analysis (INAA) provide more accurate measurements of TEs in coal. In a review from the early 21st century, Ketris and Yudovich presented average values of TEs for coals from around the world calculated by the authors based on thousands of analyses of coal and coal ash samples [69]. Results from this review for selected elements are presented in Table 2, separated into brown coal (lignite) and hard (non-lignite) coal. Table 2, while not intended to be comprehensive, provides an idea of the order-of-magnitude concentrations of different TEs.
Table 2.
Average trace element concentrations for selected elements in world coals [69].
| Brown Coal | Hard Coal | |||||
|---|---|---|---|---|---|---|
| Element | No. Samples |
No. Analyses |
Average concentrations (ppmw) |
No. Samples |
No. Analyses |
Average concentrations (ppmw) |
| Ag | 63 | 13,248 | 0.090 ± 0.020 | 96 | 11,799 | 0.100 ± 0.016 |
| As | 66 | 21,092 | 7.6 ± 1.3 | 124 | 22,466 | 9.0 ± 0.7 |
| B | 65 | 5,800 | 56 ± 3 | 103 | 13,834 | 47 ± 3 |
| Be | 80 | 48,001 | 1.2 ± 0.1 | 118 | 114,256 | 2.0 ± 0.1 |
| Br | 27 | 1,061 | 4.4 ± 0.8 | 65 | 6,807 | 6.0 ± 0.8 |
| Cd | 40 | 2,251 | 0.24 ± 0.04 | 75 | 15,079 | 0.20 ± 0.04 |
| Cl | 39 | 1,410 | 120 ± 20 | 83 | 9,981 | 340 ± 40 |
| Co | 95 | 69,660 | 4.2 ± 0.3 | 142 | 125,023 | 6.0 ± 0.2 |
| Cr | 95 | 46,136 | 15 ± 1 | 141 | 64,442 | 17 ± 1 |
| Cs | 40 | 1,808 | 0.98 ± 0.10 | 70 | 11,765 | 1.1 ± 0.12 |
| Cu | 90 | 69,855 | 15 ± 1 | 139 | 75,121 | 16 ± 1 |
| F | 37 | 3,149 | 90 ± 7 | 77 | 11,249 | 82 ± 6 |
| Hg | 48 | 3,623 | 0.10 ± 0.01 | 94 | 34,775 | 0.10 ± 0.01 |
| I | 8 | 52 | 2.3 ± 0.4 | 18 | 264 | 1.5 ± 0.3 |
| Mn | 82 | 23,231 | 100 ± 6 | 134 | 29,521 | 71 ± 5 |
| Mo | 80 | 71,606 | 2.2 ± 0.2 | 131 | 104,901 | 2.1 ± 0.1 |
| Ni | 93 | 70,560 | 9.0 ± 0.9 | 143 | 135,952 | 17 ± 1 |
| Pb | 78 | 67,744 | 6.6 ± 0.4 | 136 | 93,983 | 9.0 ± 0.7 |
| Sb | 43 | 5,220 | 0.84 ± 0.09 | 92 | 13,662 | 1.00 ± 0.09 |
| Se | 39 | 2,323 | 1.0 ± 0.15 | 81 | 16,478 | 1.6 ± 0.1 |
| Tl | 28 | 1,588 | 0.68 ± 0.07 | 49 | 6,105 | 0.58 ± 0.04 |
| V | 97 | 83,964 | 22 ± 2 | 139 | 99,450 | 28 ± 1 |
| Zn | 84 | 78,908 | 18 ± 1 | 137 | 103,924 | 28 ± 2 |
Mode of occurrence of an element in coal refers to the specific compound or compounds, which can be associated with mineral inclusions and exclusions, or with the organic matrix (Figure 1). The mode of occurrence can determine how effectively pre-combustion coal cleaning can reduce the concentration of the element in cleaned coal. Furthermore, the mode of occurrence can affect the behavior of an element in the combustion process. Finkelman summarized what was known in the early 1990s about the modes of occurrence of the 11 TEs identified as hazardous air pollutants in the 1990 U.S. Clean Air Act Amendments (CAAA) [74]. Finkelman acknowledged that elements could be found in multiple modes in a given coal and that the mode of occurrence could vary from coal to coal.
Figure 1.

Forms of occurrence of inorganic elements in coal.
Figure 1 illustrates the three primary categories of TE association in coal. Much of the inorganic material was associated with the original sediment layers which formed the organic structure of the coal. This material is usually organically bound in the final coal product. Larger portions of inorganic material in the original sediment layers may not be incorporated into the organic structure but become mineral inclusions. That is, the organic matrix surrounds the mineral phase. Other exclusions of inorganic material were mixed into the coal during geological events after the coal seam was formed or from non-coal overburden resulting from mining operations. Excluded minerals also include inclusions that are liberated from the carbon matrix during grinding [75].
Most minor inorganic elements are found in discrete minerals, but in certain coals Na, Ca, K, and Mg are associated with the organic matrix. Inorganic TEs are also found primarily in discrete minerals, either in specific mineral phases (e.g., Zn in sphalerite) or substituted in other minerals (e.g., Hg in pyrite). In certain coals Cr, Hg, Mn, Sb, Se, and halogens can be wholly or partially associated with the organic matrix [70-71,76].
Behavior of Trace Elements due to Coal Combustion
Measurement of TE emissions from coal-fired stacks preceded an understanding of how those elements were distributed in air emissions and, in some cases, in what form [14,15,17,77,78]. As discussed in more detail below, certain elements can vaporize during combustion and, in some instances, recondense (homogeneously or heterogeneously) and/or adsorb on or react with ash surfaces, which results in enrichment of the elements in the suspended ash (i.e., fly ash). Small particles offer relatively larger surface areas for these vapor-solid mechanisms to occur. Trace elements can be grouped according to the volatility of the element and its most likely molecular or atomic forms of occurrence in the combustion system as well as by observed partitioning between gas-phase, fly ash, and residual ash in coal-fired power plants [79-83]. Figure 2 depicts this classification of selected TEs into three groups:
Figure 2.

Classification of trace elements by observed behavior in coal combustion systems (adapted from [81]).
Group 1, completely volatilized during combustion with little enrichment in fly ash;
Group 2, partially volatilized during combustion, enriched in fly ash, and depleted in bottom ash;
Group 3, minimal vaporization during combustion with equal distribution between bottom ash and fly ash.
Certain elements showed intermediate behavior between groups that was not predicted from theoretical considerations of boiling points [80], leading to more work to elucidate the chemistry of transformation taking place in the coal boiler and air pollution control devices (APCDs). For example, the high boiling points of B2O3 and B (1800°C and 3297°C, respectively) would suggest that boron is minimally volatilized, contrary to observations in combustion systems.
Early models from the 1990s for predicting the air emission of TEs from coal combustion systems used emission factors that were derived from field observations. An emission factor represents the fraction of the input of a given TE that is emitted from the stack. Szpunar [84] published emission factors for the hazardous air pollutants (HAPs) as defined in the CAAA in the USA derived from field data on emissions. At the time these models were being developed, most coal-fired power plants had a particulate matter (PM) control device as the last control element before the stack. Due to their low pressure drop and corresponding low required fan power requirements, electrostatic precipitators (ESPs) were the most common PM control devices. More sophisticated emission-factor models took into account the distribution of a particular element in the fly ash as a function of particle size and the efficiency of PM removal in ESPs, baghouses or venturi scrubbers as a function of particle size [85,86]. While these models provided a means for estimating emissions, they did not shed light on the underlying chemical reactions taking place within coal combustion systems.
Helble [86] compared emission factors for individual elements with the collection efficiency of PM control devices at coal-fired boilers, noting that for most TEs, the emission factor of the element was approximately equal to the penetration efficiency of the PM control device with a few notable exceptions: mercury, selenium and, in some plants, arsenic. Helble proposed that certain TEs, such as selenium and arsenic, might be more strongly associated with the submicron particulate matter, which was known to be collected less efficiently than supermicron particles in PM control devices [25]. Mercury, on the other hand, was shown to be emitted primarily in the vapor phase [84].
Early work by Natusch and co-workers [14] demonstrated that certain TEs had an increasing concentration in emitted fly ash with decreasing particle size. The grouping of TEs by their potential volatility in the combustion zone [79-82], previously diagrammed in Figure 2, was consistent with the observations of Natusch et al [14], Helble [86], and others.
Unraveling the chemistry of TEs in the combustion and post combustion zones has been an active area of research for more than forty years [30,87-90]. The schematic diagram in Figure 3 (which has been drawn in various forms by many researchers, but was first published by Haynes et al. in 1982 [91]) illustrates the major pathways for inorganic elements in coal combustion systems, from the combustion zone to the inlet of particulate control devices. There are several important “chemical reactors” embedded within the overall process that determine the fate of TEs.
Figure 3.

Pathways for trace elements in coal combustion systems.
Semi-Volatile Trace Elements
A fraction of all TEs vaporize during the combustion process, with the degree of vaporization dependent upon the element’s volatility, its form of occurrence in the coal, and matrix effects from surrounding matter that may inhibit or facilitate vaporization [92]. Understanding the extent of vaporization as a function of combustion conditions (e.g., local temperature, local oxygen concentration) was the focus of much early research. Meserole and Chow [85] derived regression equations for vaporization of specific TEs during combustion based on field data. Yan and co-workers calculated vaporization of individual elements in the combustion zone using thermodynamic equilibrium [93]. More sophisticated single-particle models for vaporization of individual elements around a single burning coal particle were developed, which took into account the effects of local temperature and oxygen concentration [94-97] and the associations of certain elements with the mineral matter [98].
Predicting vaporization of TEs is a first step in understanding their fate in coal combustion systems. In addition to Hg, semi-volatile elements (As, Sb, Se) vaporize in the combustion zone almost entirely [99-102]. More refractory elements (e.g. Al, Fe) may partially vaporize depending on temperatures, local environments, modes of occurrence, and volatility. These elements often nucleate first, and then grow by coagulation and condensation as temperatures fall and partial pressures of increasingly volatile species become supersaturated [103-105]. Nucleation, coagulation, and condensation in the combustion and post-combustion regions produce submicron particles, while release of included material, char fragmentation, and condensation yields fine fragment and coarse particles. Submicron particles offer relatively larger surface areas for condensation processes promoting preferential enrichment. Extremely high number concentrations and high collision frequencies cause coagulating nuclei to grow quickly into an accumulation mode (more commonly known as the submicron fume). Together, these processes result in three size modes of fly ash particles centered at approximately 0.4, 2 and 20 μm, corresponding to accumulation, fine fragmentation, and coarse modes [106-110].
Models were developed to predict the size distribution of fly ash in the combustion zone and the partitioning of semi-volatile TEs to the fly ash [111-113]. Lockwood et al [112] concluded that “heterogeneous condensation of the metal vapours alone is insufficient to explain fully the fate of metal vapours. Surface reaction and gas phase chemical reaction should also be considered, but the necessary chemical kinetic data are currently lacking."
The submicron particles in the combustion and post combustion zone provide a relatively large surface area for chemical reaction. It has been known for some time that the composition of the submicron aerosol is different from that of the supermicron particles [ 14, 114, 115]. The latter have an overall composition similar to that of the bulk average ash in the parent coal, although the composition of individual supermicron particles is not uniform [116,117]. The composition of submicron particles, in contrast, is often enriched in certain major elements (Na, Ca, Fe), minor elements (S, Cl), and TEs (As, Se, Sb, Cd) relative to the bulk ash composition [118]. However, such enrichment in submicron ash has been shown to be dependent on the type of coal or, more specifically, the mode of occurrence of the element in the coal [92,119]. Furthermore, the degree of enrichment of As and Se in submicron particles was shown to depend on the concentrations of iron and calcium in the fine fly ash and the amount of SO2 present in the flue gas, leading to the hypothesis that, for many coals, the enrichment was the result of a surface chemical reaction between As or Se with Fe or Ca, with interference from a competitive reaction of Fe or Ca oxides with SO2 [120]. Models for the reaction of individual fly ash particles with gaseous arsenic [121] and gaseous selenium [122] as a function of temperature, fly ash size and fly ash composition were subsequently developed to provide more insight into mechanisms in actual combustion systems.
Mercury Chemistry
The chemistry of mercury in coal combustion systems has been an active area of study, because of the early documentation of Hg accumulation in the environment from anthropogenic activities, as discussed in the Introduction, which resulted in regulations to limit Hg emissions from incinerators, municipal waste combustors, and coal-combustion sources. Regulations have often been developed to reduce emissions by requiring application of control technologies.
Unlike the other TEs emitted from the stacks of coal combustion systems, Hg is not effectively controlled by PM control devices like ESPs and baghouses. Therefore, considerable research and development work was required to devise effective control strategies for Hg emissions. Control technology could not have been reliably and cost-effectively implemented without a sound scientific understanding of Hg chemistry. In this section, the focus is on Hg chemistry in the combustion and post-combustions zones, while a subsequent section will address how the science was applied to Hg control technologies.
Figure 4 provides an overview of Hg chemistry, which, as will be noted below, is not as simple as the figure would indicate. In the high-temperature regions of coal-fired boilers, Hg primarily exists as the gaseous elemental form (Hg0). As the flue gas cools in the post combustion zone, Hg0 may be converted to gaseous oxidized forms (Hg2+) or partition to particulate-bound mercury (Hgp). As noted above, methods for measurement of mercury in flue gas can distinguish between Hg0 and Hg2+, but cannot identify specific oxidized mercury compounds. In the presence of halogens, gas-phase equilibrium conditions favor the formation of Hg2+ at the temperatures of APCDs [123, 124]. However, the oxidation of Hg0 is kinetically limited by gaseous (homogeneous) and gas-solid (heterogeneous) reaction rates. As a result, mercury enters the APCDs as a mixture of Hgp, Hg2+, and Hg0. Oxidized forms (HgCl2, etc.) have a high solubility in water, whereas Hg0 is only sparingly soluble [125]; thus, oxidized forms of mercury are much easier to remove from the flue gas in flue gas desulfurization processes, as will be discussed in later sections. The chemical form (elemental or oxidized) and partitioning (gas-phase or particulate phase) of Hg affects the ability to control Hg emissions.
Figure 4.

Overview of mercury chemistry in flue gas.
The extent of conversion of Hg0 to Hg2+ and Hgp depends on the flue gas composition, the amount and properties of fly ash, and the flue gas quench (i.e. cool down) rate. Thermodynamic arguments suggest that hot flue gases containing Hg and halogens (like Cl) should contain a high proportion of gaseous oxidized mercury (e.g., HgCl2) as first noted in works on Hg behavior in incinerators [126,127]. An early survey of Hg emissions from coal-fired power plants in the USA showed a correlation between the amount of Cl in the coal and the fraction of oxidized mercury in the flue gas [128]. However, the concentration of oxidized mercury in flue gas was often found to be considerably lower than expected from simple equilibrium considerations, as first noted in work on waste incinerators. Widmer and co-workers postulated that the rate at which the flue gas cools post combustion (the quench rate) was a factor in the gas phase oxidation of Hg0 to HgCl2 and subsequently performed bench-scale experiments to demonstrate the effect of quench rate on oxidation [129]. Quench rate affects the oxidation of mercury by chlorine-containing species, because the major gas-phase pathway for the formation of Hg2+ is via reaction with atomic chlorine, which is kinetically limited at the typical quench rates of commercial combustion systems [130,131]. That is, boiler quench rates are often too high to maintain high atomic Cl concentrations which limits HgCl2 formation.
The halogen content of coals varies widely. For example, in a detailed study of coals from the USA [195], Cl concentrations ranged from 10 to 5,000 μg/g. Low-rank coals, such as lignite, have lower Cl contents than bituminous coals, as the average values in Table 2 illustrate. Bromine concentrations are typically 2 to 4% of the Cl concentrations, also shown in Table 2 and Reference [132]. Iodine concentrations are lower than bromine concentrations, although with fewer samples available, it is hard to generalize.
Laboratory testing of homogeneous oxidation (Figure 5) showed that, compared to chlorine, bromine was much more effective at oxidizing mercury at typical commercial combustion system quench rates [133]. Full-scale testing of the effects of different halogens on the degree of Hg oxidation at the APCD inlet demonstrated that Cl was less effective than Br or I in terms of mercury oxidizing efficiency (on an equal weight basis) [134].
Figure 5.

Homogeneous oxidation of mercury by halogens in a laboratory combustor as a function of initial halogen concentration [133].
Early work on gas-phase kinetic modeling using power plant conditions in the late 1990s and early 2000s provided considerable insight into the effects of quench rate, halogen speciation, and other constituents in the gas such as H2O and NO, for example [ 130, 131, 135-137]. After extensive work on gas-phase oxidation kinetics, it became clear that gas-phase models generally underpredicted the degree of mercury oxidation in full-scale boilers at the inlet to the air pollution control devices (approximately 150°C). These results suggested that homogeneous (gas-phase) oxidation was not the only pathway for Hg oxidation. Coal flue gas contains fly ash, containing both metal oxides and unburned carbon, both of which have been shown to be effective, in some circumstances, both for heterogenous oxidation of mercury and for adsorption of oxidized Hg.
Different metals and metal oxides have been shown to be effective oxidation catalysts for Hg in coal flue gas [138]. Metal oxides in fly ash, principally forms of iron oxide, catalyze the oxidation of Hg0 to Hg2+ in the presence of halogens [139-143]. Carbon surfaces, such as the surfaces of unburned carbon in fly ash or activated carbon, have also been shown to act as oxidation catalysts for Hg [141, 144].
Mercury adsorption on carbon is one method to capture Hg from the gas phase. The unburned carbon in fly ash, which can sometime be present in significant concentrations, can adsorb oxidized Hg in the flue gas. Hower et al. [145] observed a correlation between Hg content and carbon content of bituminous fly ash samples collected from ESP hoppers. Senior and Johnson [146] showed a correlation between Hg removal across full-scale ESPs and the carbon content of the fly ash in coal-fired power plants burning bituminous coals, which had between 1.5 and 67 wt.% carbon in ash. These field studies suggested that Hg was adsorbed onto the carbon in fly ash and subsequently removed with the carbon in the particulate control device. More detailed laboratory experiments confirmed the ability of unburned carbon in fly ash to adsorb Hg [141, 147, 148]. This result led to the development of technologies that use activated carbon adsorption as a pollution control method, as discussed in the Trace Elements in Air Pollution Control Devices section of this paper.
Methods
Elucidating chemical mechanisms in a complex environment would not be possible without a variety of analytical methods, some of which were developed specifically to understand TEs behaviors in coal combustion systems. In this section, advancements in sampling procedures, analytical methods, and modeling are discussed.
Sampling Procedures
Before chemical analysis can commence, a stable and representative sample must be obtained. Collecting samples from combustion systems and air pollution control devices presents challenges. The gas phase, whether in the combustion or post-combustion zone, is hot (60 to 1500°C) and wet (8-18 vol% moisture). Phase changes and varying dew points add to the sampling complexity. Flue gas often contains corrosive acid gases (e.g., SO2, SO3, HCl) and a considerable burden of particulate matter as illustrated in Table 3. We note that Table 3 is a broad representation, but allows the reader to envision some of the important species that can interact with TEs such as oxygen, sulfur oxides, nitrogen oxides, halogen species such as hydrogen chloride, and entrained particles.
Table 3.
Flue gas composition at boiler exit estimated from combustion calculations; fly ash concentrations from [25].
| Specie | Units | Range |
|---|---|---|
| CO2 | vol% | 13-16% |
| O2 | vol% | 3-4% |
| H2O | vol% | 8-12% |
| N2 | vol% | Balance, ~73% |
| Fly ash | g/Nm3, dry | 5-20 |
| HCl | mg/Nm3, dry | 1-200 |
| SO2 | mg/Nm3, dry | 400-10,000 |
| SO3 | mg/Nm3, dry | 2-100 |
| NOx* | mg/Nm3, dry | 200-1,000 |
| CO | mg/Nm3, dry | 20-1,000 |
| Total Hg | μg/Nm3, dry | 5-30 |
NO + NO2, mg/m3 calculated as at NO2
Sampling Trace Elements in Flue Gas
Certain TEs in coal combustion flue gas can exist in both gaseous and solid forms along the flue gas path from the combustion zone to the stack, notably As, Hg, Se, and Sb. Quantifying TE concentrations in flue gas requires dedicated sampling systems that could extract a representative sample of the hot gas for analysis. Starting in the 1970s, methods for sampling flue gas for TEs were based on wet chemical methods (i.e., methods designed to capture the elements of interest in solutions for analysis) [12]. An example is U.S. EPA Method 29 [149], illustrated in Figure 6. EPA Method 29 was designed to measure the solid particulate and gaseous emissions of Hg and 16 other TEs (Sb, As, Ba, Be, Cd, Cr, Co, Cu, Pb, Mn, Ni, P, Se, Ag, Tl, Zn). Because of the presence of particles in the flue gas, the method uses a probe inserted into the flue gas stream. The sample gas is drawn through the probe at a nearly identical “isokinetic” velocity to that of the bulk gas. By matching sample and duct gas velocities, streamlines remain straight, ensuring a representative sample of particles. A heated filter removes particles in the sample train. The probe itself is glass-lined so that the acid gases in the sample do not contact metal surfaces. The probe, filter, and sample lines are heated to prevent condensation of water. Following the filter, the sample gas is bubbled through a series of solutions in impingers intended to absorb key chemicals of interest out of the gas. Following sampling, particles collected on the filter and from dried probe rinses are typically weighed, and digested in strong acids for wet chemical analysis.
Figure 6.

U.S. EPA Method 29 for measurement of TEs in flue gas, adapted from [149].
There are practical limitations to the impinger-based methods. The methods often require long sampling times (2-6 hours) in order to obtain TE concentrations above analytical detection limits. Further, if the intent is to absorb chemicals with widely different properties, a complex sample train composed of relatively large amounts of glassware and tubing is required. Furthermore, the glass impingers contain strongly oxidizing and acidic reagents requiring labor-intensive sample recovery and analytical procedures for quantification. Alternate measurement methods have been developed for Hg that are faster and less expensive, including sorbent traps [150] and continuous emission monitors [151]. All methods must still have appropriate sample conditioning systems to remove particles and prevent interference from moisture and acid gases. Continuous emission monitors for gaseous Hg have been widely adopted at coal-fired power plants, which typically use specialized probes that dilute the sample gas (to prevent condensation of moisture) and separate particles from the sample gas, thus delivering a low-moisture and largely particle-free sample to the measurement apparatus.
Size-Segregated Particulate Sampling in Flue Gas
The sampling methods discussed so far either collect all the particulate matter in the flue gas on a filter or discard most of the particulate matter. However, the ability to collect a size-segregated sample of particulate matter for chemical analysis was critical to understanding the interactions between TEs in the gas phase and ash in the combustion and post-combustion environments. This was made possible through the development of cascade impactors, particularly those operated at very low (sub-atmospheric) pressures. The current state of the technology, low-pressure impactors can segregate the PM into 13 size-segregated samples with up to seven different submicron size categories in quantities sufficient for chemical analysis. It should be noted that while cascade impactors are used in most instances, cascade cyclone systems are also available for particle classification and collection. Often both approaches utilize after filters to collect the smallest size fraction of particles.
Impactors and cyclones utilize particle inertia to separate particles from gas. A cascade impactor is an inertial separation device consisting of size-dependent PM collection stages. In each stage the inlet particle-laden gas stream is forced to make a turn around an obstruction before passing through a specifically sized nozzle. In general, the nozzle sizes get smaller and smaller as the gas travels through the impactor. The stage may consist of a single jet (Figure 7a) or multiple jets (Figure 7b). As the gas makes the turn around the obstruction, particles with a mass that corresponds to a size larger than the design size for the stage will have too much momentum to make the turn and enter the nozzle and will impact the collection plate surrounding the nozzle and be removed from the gas. The correspondence between particle mass and particle size is known as the aerodynamic diameter (note: physical and aerodynamic diameters are identical only for spheres of unit density, 1 g/cm3). Smaller particles will travel with the gas to the next smaller sized stage meaning that smaller and smaller particles are captured on each subsequent stage.
Figure 7.

Cross-sectional schematic of cascade impactor stages with (a) single jet or (b) multiple jets.
Extraction of particles from gases by means of inertial separation and impaction has been used since the 19th century (see Marple [152] for a detailed history of the impactor). The cascade impactor, first developed by May [153], allowed particles to be collected and segregated by size. May’s impactor used four stages, each with a single jet, to collect particles in the range of 50 micron down to 1.5 micron in aerodynamic diameter. The Battelle impactor, developed in the 1950s, used six single-jet stages to collect particles down to 0.5 micron in aerodynamic diameter [154]. Andersen [155] developed a six-stage impactor with multiple jets per stage, which allowed the collection of larger samples. In the late 1960s, cascade impactors started to be used to sample directly from ducts and stacks of industrial processes, including coal-fired boilers. These impactors, such as the University of Washington impactor [156], were made of stainless steel to allow direct insertion into hot flue gases. Hering and co-workers [157] improved the cut-off diameter of collection by designing an eight-stage impactor with a sonic orifice in between stages four and five, thus allowing the lower stages to operate at reduced (sub-atmospheric) pressure. The lowest stage collected particles down to 0.05 micron in aerodynamic diameter. While Hering and co-workers used a single jet per stage for their low-pressure impactor, Berner and co-workers [158] developed a multi-jet low pressure impactor that could collect particles down to 0.08 micron in aerodynamic diameter. Hillamo and Kauppinen [159] demonstrated a version of the Berner impactor that could collect particles as small as 0.032 micron in aerodynamic diameter on the lowest stage.
In sampling from combustion flue gas, the isokinetically collected sample gas is often diluted with a dry, inert gas like nitrogen to remain above the dew point, preventing condensation of moisture on the stages (particularly at the low pressures of the stages that collect the smallest particles), and to freeze any subsequent reaction or agglomeration in the gas during sampling. Certain coals (e.g., bituminous) produce a higher fraction of large (>10 μm) particles, which can quickly overload the initial stages of the impactor. When this occurs, subsequent particles will no longer stick to the stage plate surface, but will bounce off and through the nozzle onto the next (smaller sized) impactor stage. This effect will bias the particle size distribution measurement and may cause particles from different PM formation modes to blend together, which would contaminate chemical measurements. A cyclone pre-separator is often used upstream of the cascade impactor to remove the largest particles, if good resolution of submicron particles is desired. Impaction surfaces may also be treated with thin coatings of inert materials (typically high-purity grease) to minimize particle bounce effects. The stages (and the pre-separator catch) are subsequently weighed to generate a distribution of particle mass as a function of aerodynamic diameter.
Development of the ability to collect representative samples of size-segregated fly ash and gaseous TEs from flue gas for subsequent analysis enabled the understanding of chemical mechanisms. The next section discusses analytical methods applied to such samples to quantify concentration and speciation of TEs.
Analytical Methods
Trace Elements in Coal and Ash
Considerable work was done in the 1980s and 1990s on methods for identifying the modes of occurrence of TEs in coal and coal byproducts. Knowing the form of the TE in the coal (the starting point) and the form of the TE in the gas phase or solid phase post combustion (the end point), led to greater understanding of the physical and chemical mechanisms controlling TE transformations in combustion environments. In some cases, analytical methods were developed or adapted specifically for samples from coal combustion systems.
Several factors led to the generation of useful information on mode of occurrence. Large collaborative programs, in which multiple researchers analyzed the same coals, took place in the 1990s [160,161]. In addition to traditional mineralogical analysis, several highly specialized methods were used to good effect. Selective leaching (also known as chemical fractionation) provided element-specific, quantitative information on associations of TEs with specific mineral phases [161-164]. X-ray absorption fine structure spectroscopy (XAFS) used specially tuned x-ray energy to probe the local structure, including bond distance and electronic states, of a specific element. Much use was made of XAFS and X-ray absorption near edge spectroscopy (XANES) to understand the speciation of elements in coal [165-167]. Trace element microanalyzers such as laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) and ion microprobes such as the Sensitive High-Resolution Ion Microprobe with Reversed Geometry (SHRIMP-RG) were used to probe the distribution of TEs in individual mineral inclusions, which clearly demonstrated associations with specific mineral phases [168].
Specialized analytical methods were also developed and applied to fly ash and carbonaceous sorbents to better understand the fate of TEs in the combustion or post-combustion zones. Batch wet chemistry methods developed to digest coal ash, while time-consuming, allowed comprehensive analysis by ICP-MS, inductively coupled plasma atomic emission spectrometry (ICP-AES), graphite furnace atomic emission spectrometry (GFAAS), and neutron activation analysis (NAA) even when the sample size was on the order of milligrams, micrograms, or nanograms of material. Both XAFS and XANES were used to study speciation of many different elements in fly ash, including Hg, As, Cr, and Se [169-174]. Analysis of leachates from fly ash by ion chromatography also yielded speciation information [175-177]. Surface chemical analysis of fly ash composition has been carried out by such methods as scanning electron microscopy/energy dispersive spectroscopy (SEM/EDS), LA-ICP-MS [178], and X-ray photoelectron spectroscopy (XPS) [179-183].
Laboratory testing of the leaching of TEs from CCRs has been used to predict the potential for leaching in the environment. Laboratory work in the 1980s was conducted using column leaching and batch methods [184-187]. The speciation of certain elements like As and Se was also measured in leachate to better understand the impact and environmental fate of these elements [185, 188, 189]. Laboratory evaluations of leaching were improved to better represent the varied environments in which CCRs may reside. The Leaching Environmental Assessment Framework developed by U.S. EPA is a set of laboratory protocols that provide flexibility in test methods to simulate key environmental conditions affecting leaching [190, 191].
Utilizing the specialized sampling methods discussed above, the size distribution of coal combustion aerosols has been measured by optical, electrical or inertial means in laboratory combustors and at coal-fired boilers (see, for example, [106,192]). As discussed in subsequent sections, knowledge of the number and mass distribution of the fly ash, either in-process or in-stack, provides valuable information on the physical mechanisms of ash formation and environmental fate. However, to understand the chemical mechanisms leading to speciation and emissions of TEs, chemical analysis of size-segregated fly ash was necessary.
Early work on fly ash from a coal-fired power plant by Davison et al. [114] analyzed fly ash from the hopper of a PM collection device, which had been mechanically sieved, and in-stack samples collected using a cascade impactor. Trace elements in the size-classified samples were analyzed by different methods, depending on the specific element, including DC arc emission spectrometry, atomic absorption spectrometry (AAS), X-ray fluorescence spectrometry, and spark source mass spectrometry. Other analysis methods have been used for chemical analysis of individual impactor stages, including instrumental NAA [193] and GFAAS [20, 107]. Seames and co-workers [194] modified U.S. EPA's Toxicity Characterization Leaching Protocol (TCLP) Method 1310 to allow the testing of inorganic compounds contained on cascade impactor stages. The modified method was used to explore the solubility of four TEs from five different types of coal fly ash as a function of particle size, which provided information on element speciation in ash.
Wet Chemistry Methods for Mercury Quantification
Mercury emissions from incinerators became a concern in the 1980s [195]. In the USA, EPA’s Method 101A (“Hg in sewage sludge incinerators”) was first promulgated in 1982 as a method for measuring both particulate-bound and gaseous mercury. This was an impinger-based, wet chemistry method for measuring total mercury in incinerator stacks. Because incinerator feed streams often contain Cl, theoretical considerations suggested that Hg in air emissions from incinerators would be in an oxidized form, i.e., HgCl2 [196]. Methods were developed in the 1980s to measure elemental and total mercury in incinerator air emissions to allow discrimination between elemental and oxidized forms of mercury [197].
In the 1990s, wet chemistry and batch or discrete sampling methods (e.g., EPA Method 101A, EPA Method 29) were accepted methods for measuring total gaseous and particulate mercury from coal combustion sources. The Ontario Hydro method was subsequently developed as a wet-chemical method intended to measure elemental and oxidized gaseous mercury as well as particulate-phase mercury, later codified as ASTM Method D-6784 [198].
Wet chemical methods for measuring Hg proved cumbersome and expensive. Typically, three replicate measurements were made, each six hours in duration. The sampling train (see Figure 6) was complex, carrying out the measurement required trained personnel, and results from an off-site laboratory were ready in one to two weeks. Such methods hampered research and development efforts to understand Hg chemistry and to develop Hg control technologies for coal combustion systems.
Mercury Sorbent Traps
To produce less expensive and faster measurement of Hg in flue gas, dry batch methods (also known as sorbent trap methods) were developed. Sorbent trap methods involved a probe inserted into the flue gas duct, as with the wet chemistry methods, but the probe itself contained glass tubes filled with solid sorbents that trapped Hg as the sample gas was pulled through them [68]. The sample time of sorbent trap methods can vary from 30 minutes to a week. On-site analysis of the sorbent in the traps was introduced to provide rapid turnaround for the results. Specialized sorbent traps for distinguishing between gaseous oxidized and gaseous elemental mercury were also developed [199] as well as sorbent traps specific to gas-phase As and Se [200].
Continuous Emission Monitors for Mercury Measurement
While sorbent traps provided faster measurement than wet chemical methods, the discrete nature of the sampling process could not produce the continuous measurement needed to dynamically control Hg emissions in certain control technology applications. Continuous emission monitoring (CEM) systems for Hg measurement in flue gas face similar sampling challenges as discrete methods in that a hot, particle-laden gas containing water and acid gases must be sampled. The most frequently used detection methods for continuous measurement of Hg concentration in combustion gases are AAS and atomic fluorescence spectroscopy (AFS). To use either detection method, Hg must be present in the gaseous elemental form. Because gaseous mercury can be in the elemental form or in the oxidized form, reductants are used to convert all the mercury in the gas to the elemental form so that it could be detected. The detection methods are susceptible to interference or performance degradation from acid gases present in combustion flue gas (HCl, SOx, etc.).
Early models of CEMs used liquid-filled impingers in the sampling systems to convert oxidized mercury in part of the sample to elemental mercury, while measuring only elemental mercury in another part of the sample gas. This allowed CEMs to measure both total gaseous mercury and elemental gaseous mercury in flue gas while removing acid gases. More recently, conversion to elemental mercury in the CEM is done in the gas phase, either thermally or catalytically. Dilution of the sample gas is used as a way to greatly reduce the interference of acid gases on the measurement. Newer CEMs can also measure total or elemental mercury, and have proved to be rugged and reliable enough for permanent installation as compliance monitors. Details can be found in [151].
Significant advances in Hg measurement technology in combustion systems have occurred since early efforts in the 1970s. Without such advances, the study of Hg chemistry in combustion systems as well as the development of effective control technology would have been severely hampered. Furthermore, the promulgated emission limits are based on measurements in coal-fired utility boilers, and compliance with those limits depends on reliable, continuous stack measurements of Hg.
Modeling and Computational Methods
New computational methods were also required to understand the complex chemistry of TEs in coal combustion flue gas and in air pollution control devices. This section highlights the tools that contributed to the many models developed to understand and to predict TE behavior. Later sections discuss some of the modeling efforts in context.
Thermodynamic equilibrium models have been used to define the limits of possible behavior of TEs in both oxidizing and reducing conditions typical of coal combustion or gasification environments [123, 124]. These equilibrium models rest upon early U.S. National Institutes of Standards and Technology and Joint U.S. Army, Navy, and Air Force (NIST-JANAF) databases from the 1970s with updated and expanded thermodynamic data [201,202]. Such models provide insight into the chemical forms of occurrence that are possible for each TE, but cannot predict the actual presence of any specific specie, and therefore must be used with care. With the notable exception of Hg, the molecular forms of gas-phase TEs are primarily formed in the highly dynamic environment surrounding the TE’s original ash or coal particle location. As discussed in more detail in subsequent sections, gas-phase mercury oxidation is kinetically limited, even at higher temperatures, because of low concentrations of Hg and kinetic limitations on halogen speciation. Gas-phase mercury can approach equilibrium distributions in the presence of certain catalysts (e.g., SCR catalysts). Post combustion, the flue gases are subject to rapid cooling as heat is extracted to superheat steam, which means the assumption of equilibrium is not valid.
Powerful computational tools have been developed which can accurately predict key properties of molecules that are useful for understanding chemical reactions. These developments have come about because of dramatic increases in computer speed and memory and because of the design of efficient quantum chemical algorithms. The goal of ab initio quantum computations is to provide an accurate approximation of the energy states of atoms and molecules. Chemical properties are obtained from derivatives of the energy with respect to external parameters. Many properties can be computed by these methods, including (but not limited to):
Bond energies and bond lengths;
Reaction energies;
Vibrational frequencies;
Reaction pathways and mechanisms.
The functions used to describe atomic orbitals are known as basis functions and collectively form a basis set. Larger basis sets give a better approximation of the atomic orbitals, but they require more computational power. Many basis sets are available, depending on the nature of the atoms and the bonds between atoms. Choosing the correct basis set and using the appropriate level of theory are crucial for obtaining an accurate description of atoms and hence their properties.
Starting in the late 1990s, detailed kinetic mechanisms were developed to predict the gas-phase oxidation of mercury in coal combustion systems. Individual rates constants were developed using ab initio quantum computations, first with a focus on Cl species [130,137,203-208], then extended to bromine [208-212] and iodine species [212-214]. The interaction of Hg and surfaces has also been modeled using quantum computational methods. Carbonaceous surfaces, which might represent unburned carbon in fly ash or activated carbon sorbents, have been the subject of a number of studies (for example, [183, 215-219]). Metal and metal oxide surfaces, which might represent metal oxides in fly ash, metal oxides in SCR catalysts, or noble metals, have also been modeled (for example, [220-223]). While most of the quantum chemical modeling has applied to Hg chemistry, these methods have also been applied to selenium and arsenic species (for example, [224-225]).
Quantum computational chemistry does not replace experimental measurements, but can be used to explore new or unknown chemistry. This has proved useful for certain TEs such as mercury, selenium, and arsenic in coal combustion systems.
Trace Elements in Air Pollution Control Devices
In this section, we discuss some of the ways that APCDs affect trace elements. Trace element chemistry starts in the combustion zone, but doesn’t always end when flue gas leaves the boiler and enters the various APCDs. In some cases, years of research were needed to unravel the complexities.
There is no standard set of APCDs, but the most commonly used APCDs on the newest generation of coal-fired power plants will be assumed here. For more detailed information, the reader is referred to [226]. Figure 8 depicts a common configuration of the current generation of coal-fired power plants that are being built around the world. These plants use pulverized coal, that is, coal that has been ground to about 90 microns average size. The coal is pneumatically conveyed to the boiler through distribution piping and then injected into the combustion zone using specially designed burners. Coal particles experience the high-temperature combustion zone (~1700°C), generating a mixture of gaseous and solid combustion products (flue gas). A portion of the solid ash components (bottom ash) settle within and are removed from the boiler, and the remaining portion (fly ash) are carried by the gas flow downstream. The combustion zone contains heat transfer surfaces in which water is boiled to make steam, then, post combustion, the steam is superheated by means of more heat transfer surface, which cools the flue gas to 350 to 400°C. After passing through the air heater, a gas-gas heater exchanger to preheat the combustion air, the flue gas is further cooled to 130 to 200°C.
Figure 8.

Modern pulverized coal-fired power plant, from coal pile to stack with common air pollution control devices: selective catalytic reduction (SCR), electrostatic precipitator (ESP) or baghouse, wet gas desulfurization (FGD) scrubber.
As the gas cools, it passes through a series of APCDs — that function as a series of chemical reactor/PM separator units — in order to reduce the concentrations of certain regulated pollutants. However, the APCDs also often transform some of the minor and trace elements in the flue gas and entrained ash as it passes through them. Temperatures and residence times will vary from one boiler to another, depending on the fuel characteristics, operating conditions, and specific design of APCDs. Figure 9 provides a representative time-temperature history.
Figure 9.

Representative flue gas time-temperature history for pulverized coal-fired boiler (refer to text for definitions of acronyms).
Selective Catalytic Reduction
Selective Catalytic Reduction (SCR) is commonly used to reduce NO and NO2 in the flue gas. Arsenic and mercury have significant interactions with SCR catalysts, which are discussed here. SCR processes typically use a V2O5 catalyst to promote the reaction of ammonia and NO at a gas temperature of approximately 370°C. For NO, the global reaction is written as:
A similar global reaction can be written for NO2, although at SCR temperatures almost all the NOx consists of NO. The detailed mechanism [2272] is initiated by the adsorption of an ammonia molecule on a vanadia (V5+-OH) site. The ammonia molecule is activated by reacting with a V=O redox site, which is converted to (V4+-OH). The activated ammonia reacts with gaseous (or weakly adsorbed) NO to produce N2 and H2O. In a final step, O2 oxidizes V4+-OH back to V=O.
The catalyst consists of porous ceramic material impregnated with V2O5 and supported on closely spaced plates or extruded in a honeycomb configuration. Gaseous ammonia reagent is mixed into the flue gas at the inlet to the SCR reactor. In addition to reducing NOx, SCR catalysts interact with gaseous or particulate-bound TEs in the flue gas.
Catalyst Deactivation by Arsenic
Ideally, the catalyst would retain its initial activity for NOx reduction indefinitely, although catalyst manufacturers typically assume a certain loss of catalyst activity as a function of time when specifying a catalyst for specific operating conditions. Over time the activity of an SCR catalyst declines either from physical means (e.g., blockage of pores by fly ash, erosion) or by chemical reactions between the active sites and flue gas constituents (e.g., As, Na, K) [228]. Sometimes this decline is steeper than predicted, because the plant operating conditions or the fuel properties were not as originally assumed.
SCRs were first used on coal-fired boilers in Japan and Germany, which burned coals with relatively low As contents, and thus little impact of As on SCR operation was observed except in specific power plant configurations. Certain power plants in Germany recirculated fly ash back into the boiler and experienced accelerated deactivation of the SCR catalyst [229]. Reinjecting fly ash back into the combustion zone causes As from the ash to vaporize, thus increasing the concentration of gaseous arsenic (As2O3) in the flue gas. Gaseous arsenic reacts with vanadium sites in SCR catalysts, which reduces the catalytic activity for NOx reduction. Fly ash recirculation at a power plant in Germany was estimated to increase the total As concentration in the flue gas by 6.5 times [229].
As SCRs began to be widely deployed in the USA, excessive deactivation from As was noted at certain plants, either because of fly ash re-injection at cyclone-fired boilers [228] or because the coal in use had a very low calcium content in the ash [230]. As discussed above, gaseous arsenic reacts with calcium compounds in the fly ash. If the coal calcium content is too low relative to the As content of the coal, gaseous arsenic oxide will be present in the SCR reactor at high enough concentrations to cause accelerated deactivation of the catalyst.
Based on thermodynamic considerations, arsenic is assumed to be gaseous As2O3 or the dimer As4O6 at the temperature of the SCR. Catalyst deactivation by arsenic oxide is thought to occur by blocking micropores in the catalyst, which prevents NO and NH3 from reaching active sites, and by chemical reaction with vanadium sites. One approach to improving performance under high-arsenic conditions is to reformulate the catalyst, both chemically and physically [231]. Another approach is to add limestone (primarily CaCO3) to the boiler [228-232]. In the furnace, limestone decomposes to CaO, which reacts with gaseous arsenic oxide upstream of the SCR to form solid Ca3(AsO4)2, which can then be removed in the downstream particulate control device [232]. Other power plants use blends of higher calcium-containing coals with the original coal to obtain the same effect.
Figure 10 illustrates the effect of adding limestone to reduce the gas-phase As concentration in three coal-fired combustion systems that used fly ash re-injection, and thus had high arsenic oxide concentrations in the gas [228]. The figure shows gaseous arsenic concentrations measured at the inlet of the SCR, that is, at operating temperatures in the range of 350°C to 400°C. The addition of limestone reduced the concentrations of vapor-phase arsenic by about two orders of magnitude. Knowledge of the chemistry of arsenic in coal combustion systems supported a successful strategy for mitigating the effects of arsenic on SCR catalysts.
Figure 10.

Measured arsenic concentration at SCR inlet (in μg/Nm3) as a function of coal arsenic content. Data of Pritchard et al. [228] shown for boilers with 100% fly ash recycle, with and without limestone injection for three different plants.
Catalytic Oxidation of Mercury
Gaseous oxidized mercury is more easily removed from flue gas than gaseous elemental mercury, particularly in FGD units. Thus, maximizing oxidized mercury in the flue gas is an effective strategy for control of emissions in APCDs. Commercial SCR catalysts used at coal-fired power plants for NOx control are good mercury oxidation catalysts. Measurements of mercury speciation after full-scale SCRs showed that substantial oxidation of elemental mercury can take place across the catalyst [233, 234]. This has been observed in bench-scale experiments (see, for example, [235-241]) either using simulated flue gas or in slipstream reactors using actual flue gas [242,243].
In the SCR, V2O5 catalyst adsorbs Hg0, where it reacts with HCl to form HgCl2. A reaction scheme was proposed by Madsen [241] as follows:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
Steps (1) through (5) represent a Mars-Maesson redox mechanism for the catalytic oxidation of elemental mercury. Vanadium is a transition metal that can be found in several different oxidation states, which facilitates the oxidation of elemental mercury. In well-controlled laboratory experiments, the SCR catalyst has been shown to reduce gaseous oxidized mercury to Hg0(g) [241, 244], but this has not been observed in full-scale plants. Most of the research in this area has been carried out using Cl as the halogen, but bromine and iodine have also been shown to promote Hg oxidation across SCR catalysts [240, 244, 245].
In the SCR elemental mercury must compete with ammonia for access to catalytic sites, and the ammonia concentration at the SCR inlet is several orders of magnitude greater than the Hg concentration in the gas. It has been observed that the presence of ammonia in the flue gas inhibits the oxidation of elemental mercury [242] and can also cause oxidized Hg to be reduced to elemental Hg [241, 244]. In SCR reactors, ammonia concentration decreases inside the catalyst from the inlet to the outlet as it reacts with NOx. Thus, Hg oxidation increases along the flue gas path within the SCR reactor. Models of mercury oxidation across SCR catalyst have incorporated these effects, as well as the importance of the pore structure of the catalysts on mercury oxidation [246-248].
While V2O5 is the active agent in most SCR catalysts at coal-fired power plants, the catalysts contain other metal oxides as substrates (Al2O3, TiO2) or modifying additives (WO3, MoO3). These other metal oxide components of the catalyst can also affect Hg oxidation [232].
Particulate Control Devices
All modern coal-fired boilers have PM control devices, which can, in some cases, affect the behavior of certain TEs. Two primary PM control devices are used in coal-fired boilers: ESPs and fabric filters. ESPs remove particles by first charging the particles with an electric discharge and then subjecting the charged particles to an electric field, which induces the charged particles to move to a collection surface. Once the particles are collected, there is limited interaction between the gas and the particles. Helble [86] surveyed reported emissions of TEs from U.S. power plants (all equipped with ESPs) sampled in the 1990s. With the exception of As, Se, and Hg, the emission rates of TEs could be modeled adequately using the rate of emissions of particulate matter from the ESP.
In a fabric filter, all the gas passes through a porous material, which collects the ash in a thin layer (or filter cake). After sufficient ash accumulates, the filter cake is removed. A key difference between ESPs and fabric filters is that in a fabric filter, the flue gas is in intimate contact with fly ash removed from previously flue gas until the ash is removed, which can be many hours. In this regard, fabric filters are similar to fixed bed reactors, and there can be physical or chemical interaction between the flue gas and the ash, which does not occur in ESPs.
In 2010, pilot-scale work was carried out at the U.S. EPA’s multi-pollutant control research facility [249]. Several different coals were burned and metal emissions (Sb, As, Be, Cd, Cr, Co, Pb, Mn, Hg, Ni, Se) were measured after a fabric filter or after an ESP. The removal efficiencies (coal to stack) of all of the non-Hg TEs tested (except Se) were consistently similar to the removal of the bulk PM across the control device, whether ESP or fabric filter.
Full-scale data on TE emissions from coal-fired power plants was obtained by the U.S. EPA Information Collection Request (ICR) program in 2010 [250]. Owners/operators of a limited number of coal- fired electric utility steam generating units in the USA were required to conduct stack measurements for selected hazardous air pollutants. In total, 214 coal-fired boilers were sampled; the boilers selected had the newest PM control devices, mostly fabric filters. Each of these plants was required to test the stack for Sb, As, Be, Cd, Cr, Co, Pb, Mn, Hg, Ni, Se, PM (total filterable, fine – dry and wet), O2, CO2, and moisture. The composition of the coal used during the stack test was also measured. Because TE emissions were measured at the stack, the emission rates of the TEs measured reflect the effects of all the APCDs. As summarized in [251], approximately half the boilers in the ICR dataset had no SO2 control, and this subset provides information on the effectiveness of PM control devices for removing TEs.
The removal of TEs (from coal to stack) for the boilers in the ICR dataset with only PM control devices is shown in Figure 11 in which data from multiple boilers is averaged. With the exception of Hg and Se, the TEs had high rates of removal in both fabric filters and ESPs. Mercury and selenium had lower rates of removal than the other TEs. Furthermore, rates of removal were lower on average in boilers that had ESPs for PM control as compared to those with fabric filters. Table 4 and Table 5 provide more details of the removals of Hg and Se from the dataset, broken out by PM control device and the rank of coal burned (bituminous or subbituminous). A few of the boilers used activated carbon injection (ACI) upstream of the PM control device, as noted.
Figure 11.
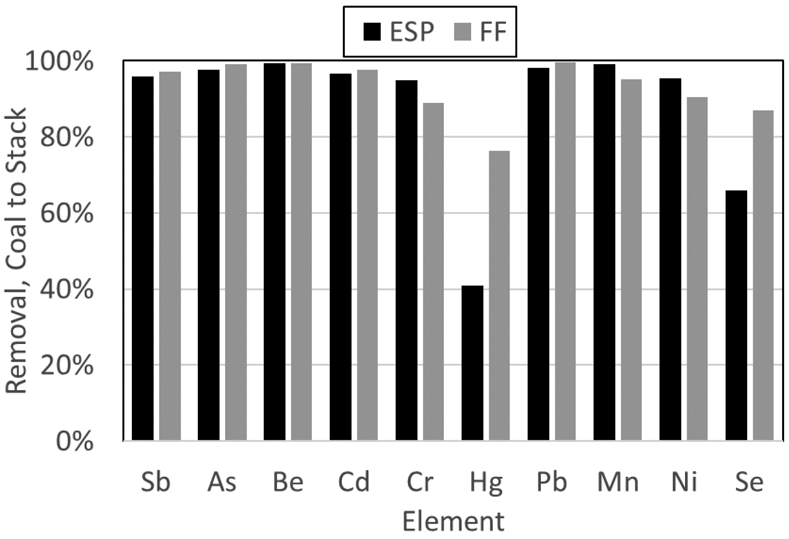
Average removal of metals from coal to stack in 2010 ICR dataset for boilers with only PM control [251].
Table 4.
Mercury removal, coal to stack, in 2010 ICR dataset for boilers with only PM control [251]. CESP = cold-side ESP; ACI = activated carbon injection; FF = fabric filter.
| Bituminous- fired |
Subbituminous-fired | |||
|---|---|---|---|---|
| PM Control |
Number of Boilers |
Average Hg Removal % |
Number of Boilers |
Average Hg Removal % |
| CESP | 24 | 37% | 16 | 52% |
| CESP/ACI | -- | -- | 7 | 58% |
| FF | 18 | 82% | 6 | 43% |
| FF/ACI | 2 | 91% | 6 | 94% |
Table 5.
Selenium removal, coal to stack, in 2010 ICR dataset for boilers with only PM control [251]. CESP = cold-side ESP; ACI = activated carbon injection; FF = fabric filter.
| Bituminous-fired | Subbituminous-fired | |||
|---|---|---|---|---|
| PM Control |
Number of Boilers |
Average Se Removal % |
Number of Boilers |
Average Se Removal % |
| CESP | 24 | 45% | 20 | 88% |
| CESP/ACI | 1 | 36% | 7 | 81% |
| FF | 19 | 82% | 5 | 94% |
| FF/ACI | 1 | 99% | 5 | 90% |
Table 4 shows that for bituminous coals, fabric filters were more effective at removing Hg than ESPs. Bituminous fly ash can contain high concentrations of unburned carbon, which can act as a sorbent for Hg, while subbituminous and lignite fly ash typically have very lower concentrations of unburned carbon [146]. This is particularly evident in fabric filters, which have a long contact time between flue gas and fly ash.
Previous work shows that bituminous-fired boilers with ESPs had low rates of selenium capture [122]. Sulfur competes with selenium for calcium on the surface of fly ash, and more selenium can be removed by the fly ash in the boiler at temperatures of 400-600°C if the SO2 concentration in the flue gas is lower [119, 120]. The ICR dataset (Table 5) showed that, as expected, bituminous-fired boilers with ESPs had low rates of Se removal. However, bituminous-fired boilers with fabric filters had much higher rates of Se removal than those with ESPs, because of low-temperature sorption of Se on the fly ash in the fabric filter. However, the lower sulfur contents and higher calcium contents of subbituminous coals resulted in higher removal of Se at subbituminous-fired boilers, as compared to bituminous-fired boilers for similar PM control devices.
Particulate matter control devices remove most of the TEs of environmental concern at rates that are similar to the removal rate of PM with the exception of Hg and Se. These two elements are more volatile than other TEs and, as discussed previously, interact chemically with flue gas constituents, including fly ash. Fabric filters, which have better contact between the flue gas and the fly ash, are more efficient at removing these elements than ESPs.
Activated Carbon Injection for Mercury Emission Control
Powdered activated carbon (PAC) can be injected directly into the flue gas as a means to control Hg emissions. The PAC is then collected along with the fly ash in the PM control device. Early applications of PAC for Hg control were in incinerators (see, for example, [252]). PAC worked well in municipal solid waste (MSW) incinerators, because the high concentration of Cl in the feed material resulted in almost all Hg in an oxidized form, which has proven easier to adsorb on activated carbon than the elemental form. In addition, the concentrations of Hg in untreated incinerator flue gas are often two orders of magnitude greater than in untreated coal-derived flue gas. The higher concentrations of Hg in incinerator flue gas also facilitate removal by activated carbon.
The PAC sorbents that had worked well in MSW incinerators did not always work well in coal-fired power plants, because in many cases Hg is in both oxidized and elemental forms, depending on, among other things, the Cl content of the coal. Figure 12 compares the effectiveness of PAC injection for Hg removal at an MSW incinerator [252] versus a coal-fired power plant [253]. Both plants injected the same type of PAC and had similar air pollution control equipment: a dry scrubber followed by a fabric filter. The coal-fired plant burned a coal with a very low Cl content, which resulted in very little oxidized mercury in the flue gas. As the figure suggests, adapting a control technology that had worked on incinerators at coal-fired power plants was not simple.
Figure 12.

Mercury removal as a function of PAC injection rate in mg per actual cubic meter of flue gas at MSW plant [252] and coal-fired power plant [253].
The need to find effective Hg control for coal-fired power plants drove considerable fundamental research into the mechanisms of the interaction between Hg and other gases with carbon surfaces starting in the 1990s. Fixed-bed reactors have been widely used in the laboratory for these studies, in which mixtures of gases containing Hg0 or HgCl2 are passed through a temperature-controlled bed of PAC. Mercury concentration (and sometimes speciation) is measured at the outlet of the fixed bed to determine when the carbon is saturated with Hg, also known as the breakthrough point. Early work focused on fixed-bed experiments using inert carrier gas (Ar or N2), for example, [254-257]. Research then shifted to using carrier gas that more closely simulated flue gas (see, for example, [258-261]).
Uptake of Hg0 or HgCl2 on activated carbon in a nitrogen atmosphere is via physical adsorption and demonstrates decreased sorption as temperature increases [255,256,260]. However, in the presence of other gaseous species typically found in coal flue gas (see Table 3), the capacity of PAC to adsorb Hg0 can increase dramatically. Fixed-bed experiments often used a baseline gas containing O2, CO2, H2O and N2 with Hg0. Activated carbon captured little Hg0 in the baseline gas [258,259,261]. Adding the acid gases HCl, SO2, NO or NO2 individually to the baseline gas increases Hg0 sorption, which suggests a chemisorption mechanism. In one set of experiments, when both NO2 and SO2 were present in the baseline gas, the Hg sorption capacity of the sorbent was low and the elemental Hg was oxidized to a gaseous oxidized form, suggesting oxidation on the surface without sorption [261]. It was hypothesized [262] that NO2 (and to a lesser extent O2) could oxidize Hg bound to the carbon surface and the oxidized mercury could subsequently react with halogen compounds. However, NO2 could also oxidize SO2 to sulfate on the surface, which could prevent the surface site from oxidizing Hg or could cause oxidized mercury to be displaced (desorbed into the gas phase). Thus, oxidation of mercury by activated carbon continues to take place, even after the sorbent has achieved the equilibrium adsorption capacity for Hg. The adsorption of H2SO4 (the low-temperature form of SO3 in flue gas) also “poisons” activated carbon with respect to Hg sorption by a similar mechanism [263,264].
Direct evidence of Hg bonding on carbon surfaces is difficult to obtain. Li et al. [265] exposed activated carbons in a fixed bed to gas containing elemental mercury in nitrogen and then used XAFS to probe the Hg bonding on the surface of exposed carbons. The authors concluded that Hg bonding on the carbon surface was associated with oxygen (as oxygen functional groups). Experiments using simulated flue gas mixtures in fixed-bed experiments have shown more complex results. Huggins et al. [169] examined different activated carbons exposed to simulated flue gas in a fixed-bed reactor. XAFS results of Hg bonding showed that for carbons with either iodine or sulfur added to the carbon surface, Hg formed a complex with the added element (I or S).
Undoped activated carbon showed more variation in Hg bonding, depending on whether HCl or HgCl2 was present in the simulated flue gas. Hutson and co-workers used XAS and XPS to probe Hg bonding on different activated carbons exposed to simulated flue gas in a fixed-bed reactor [182]. Even though Hg was present in the gas in the elemental form, on the surface, Hg was consistently observed to be in an oxidized form. A virgin activated carbon and a chlorine-impregnated activated carbon appeared to contain Hg bound to chlorinated sites and possibly to sulfate species, whereas for bromine-impregnated carbon, Hg was bound primarily to bromine. Wilcox et al. [183] used XPS to characterize the binding of elemental mercury to brominated and non-brominated activated carbons. XPS results were consistent with elemental mercury being oxidized by bromine on the surface of brominated carbon, although surface oxygen groups could not be ruled out as playing a role in oxidation.
Activated carbons consist of regions of planar carbon in fused aromatic rings, also known as graphene layers. There are two different types of carbon sites in graphene layers: basal and edge sites. The edge sites (also called zigzag sites) have higher densities of unpaired electrons, as compared to basal sites, which makes these sites more active in chemisorption leading to the formation of stable surface compounds [266]. Olson and co-workers proposed a detailed mechanism for Hg sorption and oxidation that was centered on carbon edge sites [262,267]. Other researchers proposed that oxygen complexes on the surface of the carbon, possibly carbonyl or lactone groups, were active sites for sorption of elemental mercury [265,268,269].
Lizzio and DeBarr [270] studied adsorption and oxidation of SO2 on activated carbon surfaces experimentally. They observed that SO2 adsorption capacity was suppressed when more oxygen was adsorbed on the carbon surface, leading them to conclude that oxygen reduced the number of what they termed “free” sites on the carbon surface. These carbon sites were hypothesized to be responsible for the adsorption of SO2 and its subsequent oxidation. Their work has parallels to elemental mercury oxidation and adsorption, because “free” reactive carbon sites (also called zigzag sites or basic carbon sites) have been identified as important for Hg adsorption and oxidation [262] and because adsorption/oxidation of SO2 has been shown to directly compete with Hg adsorption/oxidation on activated carbon [181]. Theoretical calculations on Hg binding to carbon edge sites in graphene structures showed that the Hg-C bond formed was strongest when oxygen was the nearest neighbor to Hg on the surface [183]. Thus, zigzag carbon sites appear to be critical to the adsorption and oxidation of elemental mercury on carbon, but surface oxygen may play a role in the chemisorption process.
Flue Gas Desulfurization Chemistry
FGD units are used to remove SO2 and other acid gases from the flue gas. These reactors contact an alkaline material with the gas to form sulfates, primarily, and minor products such as halides. The most common type of scrubber, the wet FGD, contacts the flue gas with an aqueous slurry of limestone (although lime (CaO) or caustic soda (NaOH) are also used), either by spraying the slurry into the gas or by bubbling the gas through the slurry. Calcium sulfite and/or sulfate forms and precipitates out of the slurry. Complex aqueous chemistry takes place in wet FGD systems, affecting the fate of certain TEs, as discussed below.
Mercury Chemistry in Wet FGDs
Removal of Hg across wet FGDs is largely a function of the fraction of Hg2+ in the incoming gas [128]. If the flue gas contains mostly oxidized mercury and assuming favorable chemistry in the scrubber (discussed below), wet FGDs can remove up to 95% of the incoming Hg. In scrubbers using lime or limestone reagents (the most commonly deployed), the solid product can be calcium sulfate (synthetic gypsum, CaSO4·2H2O), calcium sulfite (CaSO3), or a mixture of the two compounds. The liquid stream is typically discharged, with or without some level of water treatment. In zero-liquid-discharge plants, the liquid is sent to an evaporation pond or recycled. The Hg captured from the gas phase can, therefore, be incorporated into the solids or into the liquid discharge.
The oxidation-reduction (redox) potential in the scrubber solution strongly influences the fate of Hg in wet FGDs [271]. In scrubbers with low redox potential (inhibited oxidation systems), Hg is predominantly precipitated out of solution. In scrubbers with high redox potential (forced oxidation scrubbers), Hg remains mostly dissolved in solution. Blythe and co-workers [272] took slurry samples from full-scale wet FGDs and measured Hg in the solid and liquid fractions separately, observing that for forced oxidation scrubbers, the fraction of Hg in the solid phase decreased with increasing redox potential.
In some cases, wet FGDs do not appear to capture gas-phase Hg efficiently even when most of the incoming gaseous Hg is oxidized. Initial observations that some wet FGDs can actually emit more elemental mercury than enters [273,274] spurred research efforts into the aqueous chemistry of Hg in wet FGDs. Much was already known on the subject based on early work on Hg chemistry in aqueous solutions containing sulfur species in the atmosphere [275-278]. Since the 1990s, aqueous Hg chemistry in environments characteristic of wet limestone scrubbers in coal-fired power plants has been studied extensively, for example, [279-282].
The main pathways for aqueous Hg chemistry in FGD scrubbers as proposed by Currie et al. [281] are shown in Figure 13, which illustrates mechanisms involving chloride, although as discussed below, these apply to other halogens as well. Gas-phase oxidized mercury is very soluble in aqueous solutions and can decompose to form chloride complexes, mixed chloride-sulfite complexes or sulfites. Equilibrium favors the formation of Hg(SO3)2−2 in the presence of excess sulfite. Certain mercuric–sulfite complexes can decompose to form elemental mercury. Because of the low solubility of elemental mercury in water [252], scrubbers may emit elemental mercury if too much of the oxidized mercury in solution is reduced.
Figure 13.
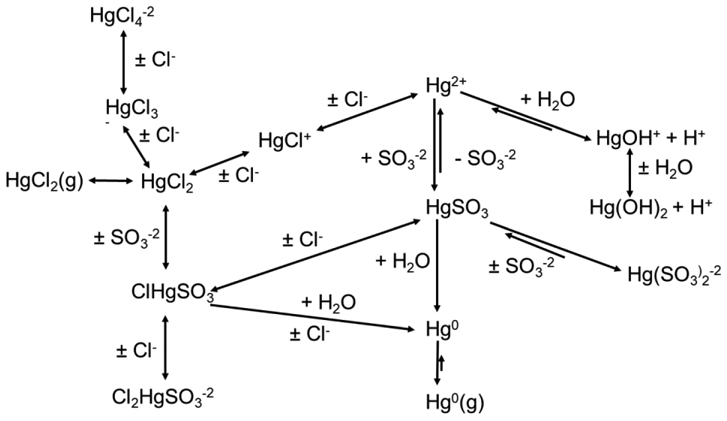
Aqueous pathways for mercury in wet FGD scrubbers adapted from [281].
Early work on aqueous Hg chemistry assumed that the main halogen in the scrubber was Cl. Recent work has focused on the effect of different halogens on Hg in FGDs. Dissolved Hg2+ can form complexes with multiple ligands. The stability of dissolved HgX2 compounds increases as Cl < Br < I and Hg can complex with multiple halogen ligands [283]:
| (6) |
As can be seen in Figure 13, the presence of halides in the scrubber could reduce the reduction of dissolved Hg2+ to Hg0, by formation of Hg-halide complexes. The solubility of HgX2 compounds is HgCl2 > HgBr2 > HgI2 [284]. If sufficient iodide is present in a simulated scrubber solution, re-emission of oxidized mercury (presumably as HgI2) has been observed [283, 285]. Thus, wet FGD scrubbers are effective at removing oxidized gas-phase mercury, but the aqueous chemistry in the scrubber can result in re-emission of some of the absorbed Hg.
Selenium Chemistry in Wet FGDs
Gaseous SeO2 that is not chemisorbed on fly ash and removed in the PM control device, enters the wet FGD scrubber in the gas phase or as Se partitioned on PM with the latter primarily being fine fragment and submicron particles that were not collected in (or condensed after) the PM control device. At the inlet to a wet FGD, the gas contacts the aqueous scrubbing solution and is quenched from ~150°C to ~55°C, resulting in additional condensation (homogeneous or heterogeneous) of some vapor-phase Se to form new particulate Se. Full-scale testing of Se gas-solid partitioning across a wet FGD scrubber at a coal-fired power plant showed evidence of gas-to-particle conversion of Se across the scrubber, based on the dependence of Se concentration on particle diameter downstream of the scrubber and on thermodynamic calculations [286]. Once in the scrubber solution, Se can partition to the liquid phase or to the solid phase [287-289]. If in the former, Se will be discharged from the scrubber in the waste water.
The U.S. EPA, under authority of the Clean Water Act, promulgated effluent guidelines for steam electric power plants that specified effluent limits for Se, As, Hg, and nitrogen in wastewater discharged from wet FGD scrubbers [290]. A survey of Se concentrations and speciation in 18 different wet FGDs in 2016 found that all the scrubbers had Se concentrations higher than the proposed effluent limitation guidelines for discharge of FGD wastewater [288]. Therefore, treatment of the FGD wastewater to remove TEs may be required.
The speciation of Se in scrubbers is complex and has significant implications for downstream wastewater control processes. The most common option for treating FGD wastewater is physical-chemical precipitation in which chemicals are added to the wastewater to remove suspended solids and dissolved metals as precipitates. Physio-chemical treatment removes most TEs of concern, including dissolved selenite (Se[IV]), but does not remove dissolved selenate (Se[VI]). Secondary treatment, typically anoxic/anaerobic bioreactors, may be required to remove selenate [291]. Understanding the chemistry of Se in wet FGDs is necessary to design an effective wastewater treatment system.
Speciation of Se in the scrubber varies with pH and redox state, which are determined by operating conditions. This is illustrated by Figure 14, which shows an equilibrium calculation for Se in the form of a Pourbaix (Eh-pH) diagram. Typical values of pH and redox potential for forced oxidation and inhibited oxidation scrubbers are shown. Selenite is dominant in inhibited oxidation FGD systems while selenate is most prevalent for forced oxidation FGD systems [288]. Metals such as manganese can catalyze the oxidation of selenite to selenate in FGDs [292]. Transition metals such as iron and manganese enter the scrubber in trace amounts in the limestone reagent or in fly ash particles not captured in the PM control device.
Figure 14.

Pourbaix diagram for10−10 M Se in water, adapted from [288].
Putting Chemistry to Work: Hg Control
After decades of deliberation, research, and field demonstrations, the U.S. EPA promulgated the MATS rule in 2012, with technical corrections issued in 2016 [52], which applied to coal- and oil-fired electricity generators starting in 2015. The MATS rule falls under the 1990 CAAA, which regulates hazardous air pollutants. The rule sets numerical limits on emissions of Hg and selected TEs (Sb, As, Be, Cd, Cr, Co, Pb, Mn, Hg, Ni, and Se) as well as certain acid gases. With the exception of Hg, sources had the option of complying with the limits for TEs by controlling PM emissions as a surrogate, because as discussed above most of the TEs are removed in the PM control device. The MATS rule set numerical emission limits on Hg that effectively required 80-90% removal of Hg, based on the input coal Hg.
Beginning in 2011, another government rule in the US resulted in more modest Hg emission reductions from coal-fired power plants. The U.S. Internal Revenue Code (IRC) section 45 provides a production tax credit to facilities using a modified coal (also known as refined coal) such that use of the refined coal provides a minimum of 20% reduction in NOx emissions rates and a minimum of 40% reduction in either Hg or SO2 emissions rates [53]. These reduction in air pollutants are typically verified through laboratory tests unrelated to actual plant operations. Almost all plants employing refined coal choose the option to reduce Hg emissions, in addition to NOx emissions. Coal from a mine on its way to be burned by a power plant is treated (usually on-site at the plant) at a facility that may dry the coal (if it is lignite) and add halogen-containing compounds (often calcium bromide, CaBr2) together with additives to achieve NOx reduction [293]. In 2018, refined coal made up about 20% of the coal (116 million metric tons) used in the power sector [294].
Removal of Particulate-bound Hg
There are currently only two commercialized pathways to remove Hg from the flue gas of coal-fired boilers: adsorption of Hg on surfaces (e.g., fly ash, sorbents) with subsequent removal by the PM control equipment or absorption of oxidized Hg species in scrubbers. Methods for controlling of Hg emissions from coal-fired boilers were developed based on the fundamental knowledge of the chemistry of Hg, as discussed above.
Mercury can be adsorbed on the unburned carbon in fly ash or on specially designed sorbents, which are typically injected into the flue gas in powdered form and then subsequently captured in the PM control device. Adsorption of gaseous oxidized mercury proceeds readily at temperatures below 150°C to 175°C. However, in the case of flue gas containing primarily elemental mercury, specialty sorbents had to be developed.
For flue gas containing predominantly Hg0 with relatively low concentrations of halogen species, activated carbon is not an effective Hg sorbent. Increasing the concentration of halogen species in the flue gas is one method of increasing sorption of Hg0. Another method is to impregnate the carbon surface with halogens. Ghorishi and co-workers [295] added Cl to a commercial activated carbon and observed significantly greater removal of Hg0 than observed in the virgin carbon in a bench-scale entrained-flow reactor in which the carbon was suspended in a flowing gas at temperature for a short time. Bromine-impregnated carbons were made commercially in the early 2000s, either by contacting virgin activated carbon with a bromine-containing gas [296] or by spraying the carbon with an aqueous Br salt solution. Iodine-impregnated activated carbon performed better than bromine-impregnated activated carbon on an equal mass basis [297], but the iodine-impregnated carbon was more expensive than the brominated carbon. Brominated carbons were adopted widely for Hg emissions control, because of reasonably good performance and lower cost..
Field testing of activated carbon injection took place in the late 1990s and 2000s at a variety of plants, which burned many different coals and had different APCDs. Flue gas with low halogen concentrations was an initial challenge, but impregnating the activated carbon with halogens or adding additional halogens to the fuel improved the performance of activated carbon. In some power plants, there were adequate concentrations of halogen in the flue gas, but activated carbon was not effective. These were plants burning high-sulfur coal, in which a certain portion of the sulfur is converted to SO3 in the boiler (typically on the order of 1%) [298]. Plants with SCRs convert an additional 1-2% of the sulfur to SO3. As noted above, the presence of SO3 “poisons” activated carbon, preventing it from adsorbing Hg. However, activated carbon formulations were developed that were more resistant to the effects of SO3 on Hg adsorption [299].
Removal of Gaseous Oxidized Hg
In order to achieve 80-90% Hg removal using flue gas desulfurization scrubbers, at least 90% of the Hg entering the scrubber should be oxidized. Mercury can be oxidized either homogeneously or heterogeneously along the flue gas path from boiler to air pollution control device. The presence of halogen species is critical for both oxidation pathways. In some cases, the coal being burned has a low native halogen content, which can be supplemented by adding additional halogen (Cl, Br, I) to the coal with the oxidizing potential varying as I > Br > Cl. Note that the oxidizing potential of the halogens is in reverse order to the relative concentrations of these halogens in coal, which is why halogens are sometimes added to coal.
In theory, blending a high-chlorine coal with a low-chlorine coal will increase the amount of Cl in the flue gas, but this doesn't always provide enough Hg control and hasn't been widely adopted as a strategy. As noted above, on an equivalent molar basis, Br is more effective than Cl for oxidation of elemental mercury in coal-fired boilers. Bromine also proved to be more cost-effective than iodine in many cases. Work carried out by Vosteen and co-workers in Europe on the effect of Br addition on Hg emissions in incinerators spurred numerous investigations on the impact of Br addition to coal combustion systems [54]. Numerous trials of Br addition to coal-fired power plants were carried out in U.S. plants, beginning in the early 2000s, for example, [300].
In the USA, the popularity of the IRC section 45 production tax credit [53] combined with the promulgation of the MATS rule [52] resulted in widespread adoption of Br addition to coal at power plants. There were unintended consequences of Br addition, which did not immediately come to light. A study of the effects of Br addition (including direct Br addition to coal and injection of brominated PAC) concluded, based on surveys of coal-fired power plants, that corrosion in the air heater was the major observed impact. The most commonly reported corrosion problems in power plants were when Br was added to the fuel at concentrations greater than 100 μg Br/g coal (ppmw), but only in boilers firing subbituminous coals or subbituminous-bituminous blends. The high moisture content of the flue gas associated with subbituminous coal firing resulted in high acid dew points on the cold metal surfaces of the air heater, which resulted in corrosion especially for plants operating under conditions of very low ambient air temperatures [55,301]. Because of the differences in dew point temperatures for HCl vs. HBr, high HCl contents in the flue gas do not cause air heater corrosion.
A further unintended consequence of adding Br to the fuel was the effect on Se partitioning: adding Br can shift Se from fly ash to the gas phase and thence into the wet FGD. For example, testing at a subbituminous-coal fired boiler with SCR, ESP and wet FGD showed 110% increase in gas-phase Se at the wet FGD inlet by increasing the Br in the coal to 30 ppmw Br addition compared to the sub-ppmw baseline level [302].
As discussed above, SCR catalysts were effective at oxidizing elemental mercury in the presence of sufficient halogen in the flue gas. Coal-fired plants that had both and SCRs and FGD scrubbers could achieve high levels of overall Hg removal, provided sufficient halogen was present in the coal or added to the boiler and if re-emission of gas-phase Hg in the scrubber was minimized.
Additives to wet FGDs have been used to reduce the re-emission of Hg. Most such additives are organic or inorganic compounds that contain a sulfide or thiol group, which will react with dissolved ionic Hg to form very stable compounds and/or precipitate out of the liquid. Some of these additives are used in industrial wastewater treatment systems [303]. Another approach has been to add activated carbon to the scrubber liquid, which will adsorb ionic Hg and will be easily separated from the calcium sulfate solids so as not to contaminate the byproduct [304]. In some cases, re-emission of Hg can be minimized by optimizing scrubber operating conditions. Some of the FGD parameters that could be optimized are as follows [305].
Oxidation-reduction potential (ORP);
Iron content in the limestone together with process conditions to increase the precipitation of iron fines (which can adsorb Hg);
Halide concentrations in the scrubber liquid (Cl concentrations controlled by operation conditions, while Br and I are sometimes added);
Fraction of suspended solids in the scrubber slurry (which affects concentration of fines, controlling surface area for adsorption).
Another method to enhance removal of gaseous Hg is to oxidize the elemental mercury using a Pd or Au based catalyst with subsequent adsorption [306-308]. The URS method developed by Blythe and co-workers [306] employs a fixed catalyst bed while the method developed by Seames et al. coats the catalyst on the clean side of a fabric filter [307,308].
Hg Control in U.S. Coal-Fired Power Plants
The U.S. MATS rule required 80-90% reduction in Hg air emissions, relative to the Hg input in the fuel. Coal-fired plants adopted different strategies, depending on the fuel being burned and on the APCDs in use. When the MATS rule was implemented, the two most common strategies were using FGD scrubbers, often together with additional oxidizing agents and/or SCR, and using activated carbon injection. Have these strategies been successful? A recent estimate of Hg emissions from coal-fired power plants in the USA, from 1993 to 2017 shows how chemistry was put to work [309]. Figure 15 shows a reduction in Hg air emissions of 60% from 1993 to 2017. The overall reduction in Hg emissions was only 31%, however, because Hg in liquid and solid waste increased as a result of capturing more particulate-bound Hg and more Hg in FGD scrubbers.
Figure 15.

Change in Hg emissions from U.S. coal-fired power plants, 1993-2017 [309].
Current Trends and Future Challenges
This review focuses on research work in the late 20th century and early 2000s on trace element behavior in pulverized coal combustion systems and APCDs. Today, research on trace element behavior in coal-based systems continues around the world. In this section, we briefly mention topics of current research in the area of trace elements in coal, with the understanding that an in-depth examination of these topics is beyond the scope of this review.
Much of the early work on trace element behavior was concentrated on pulverized coal combustion systems as the most widely deployed systems for commercial heating and electricity generation in the 20th century. There has been a significant increase in the commercial application of coal-fired fluidized bed boilers. Circulating fluidized bed (CFB) boilers have been increasingly deployed in the early 21st century in Asia, particularly in China. Trace element behavior in these systems is a subject of current interest.
Oxy-combustion systems developed and tested in the 2000s have attracted interest as a way to efficiently combust coal while capturing CO2 emissions. Traditionally, coal combustion systems have used the naturally occurring concentration of O2 in air as the oxidation source, with the inert N2 acting as a heat sink and flue gas diluent. Oxy-combustion systems, whether based on pulverized coal combustion or fluidized bed combustion, use increased concentrations of O2 (typically 30 to 100 vol%) as the oxidizer in order to produce a flue gas with increased CO2 concentration to facilitate its capture and sequestration. The O2-enriched oxidizer is usually combined with recycled flue gas to control peak flame temperatures by using the inert CO2 in the flue gas as a heat sink, thus avoiding damage to combustor boiler tubes and refractory materials. Recycling flue gas back into the combustion zone, as in oxy-combustion, results in higher concentrations of TEs in the flue gas. Differences in the combustion conditions between pulverized coal systems and fluidized bed systems as well as differences between combustion in air and combustion in oxygen and CO2 change the behavior of TEs, and has resulted in considerable on-going research. For oxy-coal combustion, CO2 capture and processing involve new sour gas, cryogenic, and high-pressure separations that will be incompatible with particulate phase species, requiring very high levels of PM control. Mercury-induced corrosion of aluminum heat exchangers in CO2 compression equipment is of particular concern.
Gasification of coal is a process by which coal reacts with steam and oxygen at high temperature and in an oxygen-deficient atmosphere to produce syngas, which is a mixture of carbon monoxide, hydrogen, water, and other reduced-phase hydrocarbons. This syngas can be burned similarly to natural gas for the generation of electricity, or used to make liquid chemicals, fuels, or hydrogen. Integrated gasification combined cycle (IGCC) systems are not widely deployed for electricity generation. However, gasification is used to produce chemicals and fuels from coal, notably in China. Hydrogen production from coal gasification with CO2 capture and storage (CCS) has become an area of research interest in the 2010s as a method for reducing the content of petroleum-derived compounds in transportation and industrial processes. Transformations of trace elements in gasification processes have been studied since the 1980s. Renewed interest in gasification and new applications of the process means continued interest in the behavior of trace elements in gasification systems.
Interest in reducing the use of fossil carbon fuel sources in the electricity and industrial sectors has led to a renewed interest in biomass as a fuel. In some cases, biomass is co-fired with coal. Blends of biomass and coal result in novel chemical pathways for trace elements. Biomass generally has lower concentrations of sulfur and higher concentrations of alkali elements and chlorine as compared to coal. When coal and biomass are blended together in combustion or gasification systems, the chemistry of trace elements as well as the formation of particulate matter can be significantly different than in cases when the fuels are utilized alone.
Much of the previous research on the trace and minor elements in coal and coal byproducts have been driven by environmental concerns. A current area of active research is in the extraction of rare earth elements (REEs), which comprise the lanthanide elements, as well as scandium and yttrium, from coal and coal by-products produced domestically and worldwide. These elements are widely used in high-technology products such as catalysts, cell phones, hard drives, hybrid engines, lasers, fluorescent lamps, batteries, magnets, medical devices, and televisions. The possible recovery of rare earth and other critical elements from abundant coal and by-products represents a paradigm shift for coal and may provide the economic incentives needed to pursue more aggressive environmental mitigation strategies.
Conclusions
Tremendous progress was achieved in the latter half of the 20th century and early 21st century in the field of trace element chemistry in coal combustion systems. Fundamental chemical mechanisms were devised for semi-volatile elements and for Hg, leading to effect control strategies for emissions of TEs to air and water. These advances were supported by new methods for characterizing TEs in coal and ash, for sampling flue gas, for making size-segregated particulate matter measurements, and for modeling the complex chemistry of TEs in combustion systems. Regulation and control of TE emissions was made possible by increased knowledge of the chemistry of TEs in combustion systems and air pollution control devices. In some parts of the world, the use of coal for generating heat and electricity has been reduced, which reduces the air emissions of TEs. However, coal will continue to be used for heat and electricity production for the foreseeable future. Methods for utilizing coal may evolve to be cleaner and more efficient, which will require new discoveries regarding TE chemistry. Finally, the impacts of TEs from coal usage will be felt in the environment long after coal usage ceases from the legacy impacts of TEs mobilized out of coal ash and coal combustion residuals.
Acknowledgements
The authors would like to thank the five anonymous reviewers of the manuscript who provided valuable comments and suggestions that improved the manuscript. In addition to the journal reviewers, the authors are grateful to Drs. C. Andrew Miller, Chun-Wai Lee, and Rebecca Dodder (U.S. EPA) for their comments and suggestions. The research described in this article has been reviewed by the U.S. EPA Center for Environmental Measurement and Modeling and approved for publication. The contents of this article should not be construed to represent the policy of the U.S. Government or any agency thereof. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof.
References
- (1).British Petroleum, BP Statistical Review of World Energy; British Petroleum: London, June, 2019, 2019; p 64. [Google Scholar]
- (2).U.S. Energy Information Agency, International Energy Outlook 2019 with projections to 2050; U.S. Energy Information Agency: Washington, D.C., September 24, 2019, 2019; p 85. [Google Scholar]
- (3).International Energy Agency, Coal Information 2019, https://www.iea.org/reports/coal-information-2019 (2020-01-30, Date last accessed). [Google Scholar]
- (4).Finkelman R; Belkin HE; Zheng B, Health impacts of domestic coal use in China. Proc. Natl. Acad. Sci 1999, 96, 3427–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kravchenko J; Lyerly HK, The impact of coal-powered electrical plants and coal ash impoundments on the health of residential communities. North Carolina Medical Journal 2018, 79 (5), 289–300. [DOI] [PubMed] [Google Scholar]
- (6).Smyth WP, Poisoning of English Beer and Food. The Sanitarian 1904, 52 (415), 515. [Google Scholar]
- (7).Ault RG, Determination of Arsenic in Malting Coal. Journal of the Institute of Brewing 1961, 67 (1), 14–28. [Google Scholar]
- (8).Chapman RS; Mumford JL; He X; Harris DB; Yang R; Jiang W, Assessing indoor air pollution exposure and lung cancer risk in Xuan Wei, China. Journal of the American College of Toxicology 1989, 8 (5), 941–948. [Google Scholar]
- (9).Mumford JL; Chapman R; Harris DB; He XZ; Cao SR; Xian YL; Li XM, Indoor air exposure to coal and wood combustion emissions associated with a high lung cancer rate in Xuan Wei, China. Environment International 1989, 15 (1-6), 315–320. [Google Scholar]
- (10).Johnels A; Westermark T; Berg W; Persson P; Sjöstrand B, Pike (Esox lucius L.) and some other aquatic organisms in Sweden as indicators of mercury contamination in the environment. Oikos 1967, 18, 323–333. [Google Scholar]
- (11).Joensuu OI, Fossil Fuels as a Source of Mercury Pollution. Science 1971, 72, 1027–1028. [DOI] [PubMed] [Google Scholar]
- (12).Billings CE; Matson WR, Mercury Emissions from Coal Combustion. Science 1972, 176, 1232–1233. [DOI] [PubMed] [Google Scholar]
- (13).Van Hook R, Potential health and environmental effects of trace elements and radionuclides from increased coal utilization. Environmental Health Perspectives 1979, 33, 227–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Natusch D; Wallace J; Evans C, Toxic trace elements: preferential concentration in respirable particles. Science 1974, 183 (4121), 202–204. [DOI] [PubMed] [Google Scholar]
- (15).Germani MS; Zoller WH, Vapor Phase Concentrations of Arsenic, Selenium, Bromine, Iodine, and Mercury in the Stack of a Coal-Fired Power Plant. Environ Sci Technol 1988, 22, 1079–1085. [DOI] [PubMed] [Google Scholar]
- (16).Andren AW; Klein DH; Talmi Y, Selenium in coal-fired steam plant emissions. Environmental Science & Technology 1975, 9 (9), 856–858. [Google Scholar]
- (17).Littlejohn RF, Emission of trace elements from coal-fired industrial boilers. A survey of relevant literature. International Journal of Energy Research 1984, 8 (4), 375–386. [Google Scholar]
- (18).Diehl RC; Hattman EA; Schultz H; Haren RJ, Fate of trace mercury In the combustion of coal. U.S. Department of the Interior. Bureau of Mines: Pittsburgh, PA, 1972; Vol. 54. [Google Scholar]
- (19).Bolton N; Carter J; Emery J; Feldman C; Fulkerson W; Hulett L; Lyon W, Trace element mass balance around a coal-fired steam plant. In Trace Elements In Fuel, Babu SP, Ed. American Chemical Society: Washington, DC, 1975; Vol. 141, pp 175–187. [Google Scholar]
- (20).Kaakinen JW; Jorden RM; Lawasani MH; West RE, Trace element behavior in coal-fired power plant. Environmental Science & Technology 1975, 9 (9), 862–869. [Google Scholar]
- (21).Schultz H; Hattman EA; Booher WB, Fate of some trace elements during coal pretreatment and combustion. Am. Chem. Soc., Div. Fuel Chem., Prepr 1976, 18 (4). [Google Scholar]
- (22).Roffman HK; Kary RE; Hudgins T In Ecological distribution of trace elements emitted by coal-burning power generating units employing scrubbers and electrostatic precipitators, Papers presented before the fourth Symposium on Coal Utilization: NCA/BCR Coal Conference and Expo IV, October 18-19-20, 1977, Louisville, Kentucky, National Coal Association: 1977; p 192. [Google Scholar]
- (23).Meij R; Janssen LHJM; van der Kooij J, Air pollutant emissions from coal-fired power stations. KEMA: Arnhem, The Netherlands, 1986; Vol. 4. [Google Scholar]
- (24).Meij R Tracking trace elements at a coal-fired power plant equipped with a wet flue-gas desulphurisation facility; KEMA: Arnhem, The Netherlands, 1989. [Google Scholar]
- (25).Ylätalo SI; Hautanen J, Electrostatic precipitator penetration function for pulverized coal combustion. Aerosol Science and Technology 1998, 29 (1), 17–30. [Google Scholar]
- (26).Clarke LB, The Behavior of Trace Elements During Coal Combustion and Gasification: An Overview. In Managing Hazardous Air Pollutants: State of the Art, Chow W; Connor KK, Eds. Lewis Publishers: Boca Raton, FL, 1993; pp 358–370. [Google Scholar]
- (27).Shendrikar A; Ensor D; Cowen S; Woffinden G; McElroy M, Size-dependent penetration of trace elements through a utility baghouse. Atmospheric Environment 1983, 17 (8), 1411–1421. [Google Scholar]
- (28).Esmen NA; Corn M, Residence time of particles in urban air. Atmospheric Environment 1971, 5 (8), 571–578. [DOI] [PubMed] [Google Scholar]
- (29).Kauppinen EI; Pakkanen TA, Coal combustion aerosols: a field study. Environmental Science & Technology 1990, 24 (12), 1811–1818. [Google Scholar]
- (30).Swaine DJ, Trace elements in coal and their dispersal during combustion. Fuel Processing Technology 1994, 39 (1-3), 121–137. [Google Scholar]
- (31).Seaton A; Godden D; MacNee W; Donaldson K, Particulate air pollution and acute health effects. The Lancet 1995, 345, 176–178. [DOI] [PubMed] [Google Scholar]
- (32).U.S. EPA, Particulate Matter Research Needs for Human Health Risk Assessment to Support Future Reviews of the National Ambient Air Quality Standards for Particulate Matter; EPA/600/R97/132F; National Center for Environmental Assessment Research Triangle Park, NC, 1998. [Google Scholar]
- (33).Mumford JL; Lewtas J, Mutagenicity and cytotoxicity of coal fly ash from fluidized-bed and conventional combustion. Journal of Toxicology and Environmental Health, Part A Current Issues 1982, 10 (4-5), 565–586. [DOI] [PubMed] [Google Scholar]
- (34).Gilmour MI; O'Connor S; Dick CA; Miller CA; Linak WP, Differential pulmonary inflammation and in vitro cytotoxicity of size-fractionated fly ash particles from pulverized coal combustion. Journal of the Air & Waste Management Association 2004, 54 (3), 286–295. [DOI] [PubMed] [Google Scholar]
- (35).Thomson VE; Huelsman K; Ong D, Coal-fired power plant regulatory rollback in the United States: Implications for local and regional public health. Energy Policy 2018, 123, 558–568. [Google Scholar]
- (36).Final Rule, Hazardous and Solid Waste Management System; Disposal of Coal Combustion Residuals from Electric Utilities, 80 Federal Register 21,302, 2015. https://www.federalregister.gov/documents/2015/04/17/2015-00257/hazardous-and-solid-waste-management-system-disposal-of-coal-combustion-residuals-from-electric (2020-08-31, date of last access)
- (37).Waste Framework Directive, Directive 2008/98/EC of the European Parliament. https://ec.europa.eu/environment/waste/legislation/a.htm (2020-08-31, date of last access).
- (38).Harris D; Heidrich C; Feuerborn J Global aspects on Coal Combustion Products, 2019. https://www.coaltrans.com/insights/article/global-aspects-on-coal-combustion-products (2020-08-31, date of last access) [Google Scholar]
- (39).Lee S; Spears DA, The long-term weathering of PFA and implications for groundwater pollution. Quarterly Journal of Engineering Geology and Hydrogeology 1995, 28 (Supplement 1), S1–S15. [Google Scholar]
- (40).Lee S; Spears DA, Potential contamination of groundwater by pulverized fuel ash. Geological Society, London, Special Publications; 1998, 128 (1), 51–61. [Google Scholar]
- (41).Choi S-K; Lee S; Song Y-K; Moon H-S, Leaching characteristics of selected Korean fly ashes and its implications for the groundwater composition near the ash disposal mound. Fuel 2002, 81 (8), 1083–1090. [Google Scholar]
- (42).Harkness JS; Sulkin B; Vengosh A, Evidence for coal ash ponds leaking in the southeastern United States. Environmental Science & Technology 2016, 50 (12), 6583–6592. [DOI] [PubMed] [Google Scholar]
- (43).U.S. EPA. Frequent Questions about the 2015 Coal Ash Disposal Rule. November 2, 2018. https://www.epa.gOv/coalash/frequent-questions-about-2015-coal-ash-disposal-rule#main-content (07 July 2020, date last accessed)
- (44).Hazardous and Solid Waste Management System; Disposal of Coal Combustion Residuals From Electric Utilities: Final Rule; United States Environmental Protection Agency: Washington, DC, 2015. [Google Scholar]
- (45).Ruhl L; Vengosh A; Dwyer GS; Hsu-Kim H; Deonarine A; Bergin M; Kravchenko J, Survey of the potential environmental and health impacts in the immediate aftermath of the coal ash spill in Kingston, Tennessee. Environmental Science & Technology 2009, 43 (16), 6326–6333. [DOI] [PubMed] [Google Scholar]
- (46).Ruhl L; Vengosh A; Dwyer GS; Hsu-Kim H; Deonarine A, Environmental impacts of the coal ash spill in Kingston, Tennessee: an 18-month survey. Environmental Science & Technology 2010, 44 (24), 9272–9278. [DOI] [PubMed] [Google Scholar]
- (47).Ramsey AB; Faiia AM; Szynkiewicz A, Eight years after the coal ash spill–Fate of trace metals in the contaminated river sediments near Kingston, eastern Tennessee. Applied Geochemistry 2019, 104, 158–167. [Google Scholar]
- (48).Lin R; Howard BH; Roth EA; Bank TL; Granite EJ; Soong Y, Enrichment of rare earth elements from coal and coal by-products by physical separations. Fuel 2017, 200, 506–520. [Google Scholar]
- (49).Richardson SD; Plewa MJ; Wagner ED; Schoeny R; DeMarini DM, Occurrence, genotoxicity, and carcinogenicity of regulated and emerging disinfection by-products in drinking water: A review and roadmap for research. Mutat. Res 2007, 636, 178–242. [DOI] [PubMed] [Google Scholar]
- (50).VanBriesen J, Potential drinking water effects of bromide discharges from coal-fired electric power plants. EPA NPDES Comments 2014, 1–38. [Google Scholar]
- (51).Good KD; VanBriesen JM, Coal-fired power plant wet flue gas desulfurization bromide discharges to U.S. watersheds and their contributions to drinking water sources. Environmental Science & Technology 2018, 53 (1), 213–223. [DOI] [PubMed] [Google Scholar]
- (52).U.S. EPA, National Emission Standards for Hazardous Air Pollutants From Coal- and Oil-Fired Electric Utility Steam Generating Units and Standards of Performance for Fossil-Fuel-Fired Electric Utility, Industrial-Commercial-Institutional, and Small Industrial-Commercial-Institutional Steam Generating Units; Final Rule; 40 CFR Parts 60 and 63, Federal Register, Vol. 81, No. 66, April 6, 2016. [Google Scholar]
- (53).U.S. Internal Revenue Service. Credit for Renewable Electricity Production and Refined Coal Production, and Publication of Inflation Adjustment Factor and Reference Prices for Calendar Year 2019. Federal Register 2019, 84(109), 26508–26509. [Google Scholar]
- (54).Vosteen BW; Kanefke R; Koser H, Bromine-enhanced mercury abatement from combustion flue gases-recent industrial applications and laboratory research. VGB Powertech 2006, 86 (3), 70–75. [Google Scholar]
- (55).Dombrowski K; Aramasick K; Srinivasan N Balance of Plant Impacts of Bromine for Mercury Control. In Air Quality IX Conference, Arlington, VA, October 2013. [Google Scholar]
- (56).Schobert HH, The Geochemistry of Coal I. The Classification and Origin of Coal. J. Chem. Edu 1989, 66 (3), 242–244. [Google Scholar]
- (57).Schobert HH, The Geochemistry of Coal II. The Components of Coal. J. Chem. Edu 1989, 66 (4), 290–293. [Google Scholar]
- (58).Schweinfurth SP Coal: A complex natural resource an overview of factors affecting coal quality and use in the United States; Circular 1143; U.S. Geological Survey Reston, VA, 2002; 37 pp. [Google Scholar]
- (59).Gluskoter HJ; Ruch RR; Miller WG; Cahill RA; Dreher GB; Kuhn JK Trace Elements in Coal: Occurrence and Distribution; Circular 499; Illinois State Geological Society: Urbana, IL, 1977; p 154. [Google Scholar]
- (60).Hardesty DR; Pohl JH Combustion of pulverized coals: an assessment of research needs. I. Background, justification, and mineral matter; SAND-78-8804; Sandia National Laboratory: Livermore, CA, 1979. [Google Scholar]
- (61).Lu X; Zeng H; Xu T; Yan R, Chemometric studies of distribution of trace elements in seven Chinese coals. Fuel 1995, 74 (9), 1382–1386. [Google Scholar]
- (62).Dai S; Ren D; Chou C-L; Finkelman RB; Seredin VV; Zhou Y, Geochemistry of trace elements in Chinese coals: A review of abundances, genetic types, impacts on human health, and industrial utilization. International Journal of Coal Geology 2012, 94, 3–21. [Google Scholar]
- (63).Riley KW; French DH; Farrell OP; Wood RA; Huggins FE, Modes of occurrence of trace and minor elements in some Australian coals. International Journal of Coal Geology 2012, 94, 214–224. [Google Scholar]
- (64).Shah P; Strezov V; Nelson PF, Speciation of chromium in Australian coals and combustion products. Fuel 2012, 102, 1–8. [Google Scholar]
- (65).Tian HZ; Lu L; Hao JM; Gao JJ; Cheng K; Liu KY; Qiu PP; Zhu CY, A Review of Key Hazardous Trace Elements in Chinese Coals: Abundance, Occurrence, Behavior during Coal Combustion and Their Environmental Impacts. Energy & Fuels 2013, 27 (2), 601–614. [Google Scholar]
- (66).Verma SK; Masto RE; Gautam S; Choudhury DP; Ram LC; Maiti SK; Maity S, Investigations on PAHs and trace elements in coal and its combustion residues from a power plant. Fuel 2015, 162, 138–147. [Google Scholar]
- (67).U.S. Geological Survey, World Coal Quality Inventory, https://www.usgs.gov/centers/eersc/science/world-coal-quality-inventory-data?qt-science_center_objects=0#qt-science_center_objects (accessed February 11, 2020). [Google Scholar]
- (68).Swaine DJ, Trace Elements in Coal. Butterworths: London, 1990; 278 pp. [Google Scholar]
- (69).Ketris MP; Yudovich YE, Estimations of Clarkes for Carbonaceous biolithes: World averages for trace element contents in black shales and coals. International Journal of Coal Geology 2009, 78 (2), 135–148. [Google Scholar]
- (70).Ward CR, Analysis and significance of mineral matter in coal seams. International Journal of Coal Geology 2002, 50 (1-4), 135–168. [Google Scholar]
- (71).Ward CR, Analysis, origin and significance of mineral matter in coal: An updated review. International Journal of Coal Geology 2016, 165, 1–27. [Google Scholar]
- (72).Dai S; Hower JC; Finkelman RB; Graham IT; French D; Ward CR; Eskenazy G; Wei Q; Zhao L, Organic associations of non-mineral elements in coal: A review. International Journal of Coal Geology 2020, 218, 103347. [Google Scholar]
- (73).Raask E, The Mode of Occurrence and Concentration of Trace Elements in Coal. Prog Energy Combust Sci 1985, 11, 97–118. [Google Scholar]
- (74).Finkelman RB, Modes of occurrence of potentially hazardous elements in coal: levels of confidence. Fuel Processing Technology 1994, 39 (1-3), 21–34. [Google Scholar]
- (75).Raask E, Mineral impurities in coal combustion: behavior, problems, and remedial measures. Hemisphere Publishing Corporation: New York, 1985. [Google Scholar]
- (76).Xu M; Yan R; Zheng C; Qiao Y; Han J; Sheng C, Status of trace element emission in a coal combustion process: a review. Fuel Processing Technology 2003, 85 (2-3), 215–237. [Google Scholar]
- (77).U.S. EPA. Study of Hazardous Air Pollutant Emissions from Electric Utility Steam Generating Units -- Final Report to Congress, EPA-453/R-98-004a, February 1998, 502 p.
- (78).Electric Utility Trace Substances Synthesis Report; EPRI TR-104614; Electric Power Research Institute: Palo Alto, CA, 1994. [Google Scholar]
- (79).Smith IM Trace elements from coal combustion: emissions; IEACR-01; International Energy Agency: London, 1987; p 87. [Google Scholar]
- (80).Clarke LB; Sloss LL, Trace elements: emissions from coal combustion and gasification. IEA Coal Research: London, 1992; Vol. 49. [Google Scholar]
- (81).Ratafia-Brown JA, Overview of trace element partitioning in flames and furnaces of utility coal-fired boilers. Fuel Processing Technology 1994, 39 (1-3), 139–157. [Google Scholar]
- (82).Meij R, Trace element behavior in coal-fired power plants. Fuel Processing Technology 1994, 39 (1-3), 199–217. [Google Scholar]
- (83).Meij R, The distribution of trace elements during the combustion of coal. In Environmental Aspects of Trace Elements in Coal. Energy & Environment, S. DJ; G. F, Eds. Springer: Dordrecht, The Netherlands, 1995; pp 111–127. [Google Scholar]
- (84).Szpunar CB Air Toxic Emissions from Combustion of Coal: Identifying and Quantifying Hazardous Air Pollutants from U.S. Coals; Argonne National Laboratory: Argonne, Illinois, 1992. [Google Scholar]
- (85).Meserole FB; Chow W, Controlling trace species in the utility industry. In Managing Hazardous Air Pollutants: State of the Art, Chow W; Connor KK, Eds. Lewis Publishers: Boca Raton, FL, 1993; pp 371–379. [Google Scholar]
- (86).Helble JJ, A model for the air emissions of trace metallic elements from coal combustors equipped with electrostatic precipitators. Fuel Processing Technology 2000, 63, 125–147. [Google Scholar]
- (87).Smith RD, The trace element chemistry of coal during combustion and the emissions from coal-fired plants. Progress in Energy and Combustion Science 1980, 6 (1), 53–119. [Google Scholar]
- (88).Damle A; Ensor D; Ranade M, Coal combustion aerosol formation mechanisms: a review. Aerosol Science and Technology 1981, 1 (1), 119–133. [Google Scholar]
- (89).Danihelka P; Volná Z; Jones JM; Williams A, Emission of trace toxic metals during pulverized fuel combustion of Czech coals. International Journal of Energy Research 2003, 27 (13), 1181–1203. [Google Scholar]
- (90).Zhang J; Zhao Y; Ding F; Zeng H; Zheng C, Preliminary study of trace element emissions and control during coal combustion. Frontiers of Energy and Power Engineering in China 2007, 1 (3), 273–279. [Google Scholar]
- (91).Haynes BS; Neville M; Quann RJ; Sarofim AF, Factors Governing the Surface Enrichment of Fly Ash and Volatile Trace Species. J. Colloid Interface Sci 1982, 87 (1), 266–278. [Google Scholar]
- (92).Raeva AA; Klykov OV; Kozliak EI; Pierce DT; Seames WS, In Situ Evaluation of Inorganic Matrix Effects on the Partitioning of Three Trace Elements (As, Sb, Se) at the Outset of Coal Combustion. Energy & Fuels 2011, 25 (10), 4290–4298. [Google Scholar]
- (93).Yan R; Gauthier D; Flamant G, Volatility and chemistry of trace elements in a coal combustor. Fuel 2001, 80 (15), 2217–2226. [Google Scholar]
- (94).Quann RJ; Sarofim AF In Vaporization of refractory oxides during pulverized coal combustion, Symposium (International) on Combustion, Elsevier: 1982; pp 1429–1440. [Google Scholar]
- (95).Quann RJ; Neville M; Janghorbani M; Mims CA; Sarofim AF, Mineral matter and trace-element vaporization in a laboratory-pulverized coal combustion system. Environmental Science & Technology 1982, 16 (11), 776–781. [DOI] [PubMed] [Google Scholar]
- (96).Senior CL; Flagan RC, Ash Vaporization and Condensation During Combustion of a Suspended Coal Particle. Aerosol Science and Technology 1982, 1, 371–383. [Google Scholar]
- (97).Lee CM; Davis KA; Heap MP; Eddings E; Sarofim A, Modeling the vaporization of ash constituents in a coal-fired boiler. Proceedings of the Combustion Institute 2000, 28, 2375–2382. [Google Scholar]
- (98).James DW; Krishnamoorthy G; Benson SA; Seames WS, Modeling trace element partitioning during coal combustion. Fuel Processing Technology 2014, 126, 284–297. [Google Scholar]
- (99).Senior CL; Bool III LE; Morency JR, Laboratory Study of Trace Element Vaporization from Combustion of Pulverized Coal. Fuel Processing Technology 2000, 63 (2-3), 109–124. [Google Scholar]
- (100).Zeng T; Sarofim AF; Senior CL, Vaporization of Arsenic, Selenium and Antimony during Coal Combustion. Combustion and Flame 2001, 126, 1714–1724. [Google Scholar]
- (101).Yan R; Gauthier D; Flamant G; Peraudeau G, Fate of Selenium in Coal Combustion: Volatilization and Speciation in Flue Gas. Environ Sci Technol 2001, 35, 1406–1410. [DOI] [PubMed] [Google Scholar]
- (102).Noda N; Ito S, The release and behavior of mercury, selenium, and boron in coal combustion. Powder Technology 2008, 180 (1-2), 227–231. [Google Scholar]
- (103).Flagan RC; Taylor DD Laboratory studies of submicron particles from coal combustion, In Symposium (International) on Combustion, Elsevier: 1981; pp 1227–1237. [Google Scholar]
- (104).Senior CL; Flagan RC, Laboratory Studies of Sub-Micron Aerosol Formation from Pulverized Coal Combustion Using Model Compounds. Aerosol Science and Technology 1983, 2, 287. [Google Scholar]
- (105).Neville MJ; McCarthy JF, Size fractionation of submicrometer coal combustion aerosol for chemical analysis. Atmospheric Environment 1983, 17 (12), 2599–2604. [Google Scholar]
- (106).McElroy M; Carr R; Ensor D; Markowski G, Size distribution of fine particles from coal combustion. Science 1982, 215 (4528), 13–19. [DOI] [PubMed] [Google Scholar]
- (107).Linak WP; Miller CA; Seames WS; Wendt JO; Ishinomori T; Endo Y; Miyamae S, On trimodal particle size distributions in fly ash from pulverized-coal combustion. Proceedings of the Combustion Institute 2002, 29 (1), 441–447. [Google Scholar]
- (108).Seames WS, An initial study of the fine fragmentation fly ash particle mode generated during pulverized coal combustion. Fuel Processing Technology 2003, 81 (2), 109–125. [Google Scholar]
- (109).Yu D; Xu M; Yao H; Sui J; Liu X; Yu Y; Cao Q, Use of elemental size distributions in identifying particle formation modes. Proceedings of the Combustion Institute 2007, 31, 1921–1928. [Google Scholar]
- (110).Liu H; Wang Y; Wendt JOL, Particle Size Distributions of Fly Ash Arising from Vaporized Components of Coal Combustion: A Comparison of Theory and Experiment. Energy & Fuels 2018, 32, 4300–4307. [Google Scholar]
- (111).Yousif S; Lockwood F; Abbas T Modeling of toxic metal emissions from solid fuel combustors, Proceedings of the Symposium (International) on Combustion, Elsevier: 1998; pp 1647–1654. [Google Scholar]
- (112).Lockwood F; Yousif S, A model for the particulate matter enrichment with toxic metals in solid fuel flames. Fuel Processing Technology 2000, 65, 439–457. [Google Scholar]
- (113).Han J; Xu M; Yao H; Furuuchi M; Sakano T; Kim HJ, Simulating the transformation of heavy metals during coal or sewage sludge combustion. Journal of Environmental Science and Health Part A 2007, 42 (2), 217–224. [DOI] [PubMed] [Google Scholar]
- (114).Davison RL; Natusch DF; Wallace JR; Evans CA Jr, Trace elements in fly ash. Dependence of concentration on particle size. Environmental Science & Technology 1974, 8 (13), 1107–1113. [Google Scholar]
- (115).Markowski GR; Filby R, Trace element concentration as a function of particle size in fly ash from a pulverized coal utility boiler. Environmental Science & Technology 1985, 19 (9), 796–804. [DOI] [PubMed] [Google Scholar]
- (116).Helble JJ; Srinivasachar S; Boni AA, Factors influencing the transformation of minerals during pulverized coal combustion. Progress In Energy and Combustion Science 1990, 16 (4), 267–279. [Google Scholar]
- (117).Yan L; Gupta R; Wall T, Fragmentation behavior of pyrite and calcite during high-temperature processing and mathematical simulation. Energy & Fuels 2001, 15 (2), 389–394. [Google Scholar]
- (118).Senior CL; Bool III LE; Srinivasachar S; Pease BR; Porle K, Pilot Scale Study of Trace Element Vaporization and Condensation during Combustion of Sub-Bituminous Coal. Fuel Processing Technology 2000, 63 (2-3), 149–166. [Google Scholar]
- (119).Seames WS; Wendt JOL, Partitioning of arsenic, selenium, and cadmium during the combustion of Pittsburgh and Illinois #6 coals in a self-sustained combustor. Fuel Processing Technology 2000, 63, 179–196. [Google Scholar]
- (120).Seames WS; Wendt JOL, Regimes of association of arsenic and selenium during pulverized coal combustion. Proceedings of the Combustion Institute 2007, 31 (2), 2839–2846. [Google Scholar]
- (121).Senior CL; Lignell DO; Sarofim AF; Mehta A, Modeling arsenic partitioning in coal-fired power plants. Combustion and Flame 2006, 147 (3), 209–221. [Google Scholar]
- (122).Senior C; Van Otten B; Wendt JOL; Sarofim A, Modeling the behavior of selenium in Pulverized-Coal Combustion systems. Combustion and Flame 2010, 157 (11), 2095–2105. [Google Scholar]
- (123).Linak WP; Wendt JO, Trace metal transformation mechanisms during coal combustion. Fuel Processing Technology 1994, 39 (1-3), 173–198. [Google Scholar]
- (124).Frandsen F; Dam-Johansen K; Rasmussen P, Trace elements from combustion and gasification of coal—an equilibrium approach. Progress In Energy and Combustion Science 1994, 20 (2), 115–138. [Google Scholar]
- (125).Clever HL; Johnson SA; Derrick ME, The solubility of mercury and some sparingly soluble mercury salts in water and aqueous electrolyte solutions. Journal of Physical and Chemical Reference Data 1985, 14 (3), 631–680. [Google Scholar]
- (126).Bergström JG, Mercury behaviour in flue gases. In Incineration of Municipal Waste, Dean RB, Ed. Academic Press: London, 1988; pp 55–62. [Google Scholar]
- (127).Hall B; Schager P; Lindqvist O, Chemical reactions of mercury in combustion flue gases. Water Air & Soil Pollution 1991, 56 (1), 3–14. [Google Scholar]
- (128).Afonso RF; Senior CL Assessment of Mercury Emissions from Full Scale Power Plants. In Proceedings of the EPRI-EPA-DOE-AWMA Mega Symposium and Mercury Conference; Air & Waste Management Association: Pittsburgh, 2001. [Google Scholar]
- (129).Widmer NC; Cole JA; Seeker WR; Gaspar JA, Practical Limitation of Mercury Speciation in Simulated Municipal Waste Incinerator Flue Gas. Combustion Science and Technology 1998, 134 (1), 315–326. [Google Scholar]
- (130).Sliger RN; Kramlich JC; Marinov NM, Towards the development of a chemical kinetic model for the homogeneous oxidation of mercury by chlorine species. Fuel Processing Technology 2000, 65, 423–438. [Google Scholar]
- (131).Senior CL; Sarofim AF; Zeng T; Helble JJ; Mamani-Paco R, Gas-phase transformations of mercury in coal-fired power plants. Fuel Processing Technology 2000, 63, 197–213. [Google Scholar]
- (132).Kolker A; Quick JC; Senior CL; Belkin HE Mercury and halogens in coal--Their role in determining mercury emissions from coal combustion; Fact Sheet 2012-3122; U.S. Geological Survey: Reston, Virginia, 2012. [Google Scholar]
- (133).Van Otten B; Buitrago PA; Senior CL; Silcox GD, Gas-Phase Oxidation of Mercury by Bromine and Chlorine in Flue Gas. Energy & Fuels 2011, 25 (8), 3530–3536. [Google Scholar]
- (134).Richardson CF; Dombrowski K; Chang R, Mercury control evaluation of halogen injection into coal-fired furnaces. In Proceedings of the 9th Electric Utilities Environmental Conference, Tucson, AZ, 2006. [Google Scholar]
- (135).Niksa S; Helble JJ; Fujiwara N, Kinetic Modeling of Homogeneous Mercury Oxidation: The Importance of NO and H2O in Predicting Oxidation in Coal-Derived Systems. Environ Sci Technol 2001, 35, 3701–3706. [DOI] [PubMed] [Google Scholar]
- (136).Edwards JR; Srivastava RK; Kilgroe JD, A Study of Gas-Phase Mercury Speciation Using Detailed Chemical Kinetics. J Air Waste Manag Assoc 2001, 51, 869–877. [DOI] [PubMed] [Google Scholar]
- (137).Li L-C; Deng P; Tian A-M; Xu M-H; Zheng C-G; Wong N-B, A study on the reaction mechanism and kinetic of mercury oxidation by chlorine species. Journal of Molecular Structure: THEOCHEM 2003, 625 (1-3), 277–281. [Google Scholar]
- (138).Presto AA; Granite EJ, Survey of catalysts for oxidation of mercury in flue gas. Environmental Science & Technology 2006, 40 (18), 5601–5609. [DOI] [PubMed] [Google Scholar]
- (139).Ghorishi SB Fundamentals of mercury speciation and control in coal-fired boilers; EPA-600/R-98-014; U.S. EPA: February 1998. [Google Scholar]
- (140).Galbreath KC; Zygarlicke CJ; Olson ES; Pavlish JH; Toman DL, Evaluating mercury transformation mechanisms in a laboratory-scale combustion system. Science of the Total Environment 2000, 261 (1-3), 149–155. [DOI] [PubMed] [Google Scholar]
- (141).Dunham GE; DeWall RA; Senior CL, Fixed-bed studies of the interactions between mercury and coal combustion fly ash. Fuel Processing Technology 2003, 82 (2-3), 197–213. [Google Scholar]
- (142).Norton GA; Yang H; Brown RC; Laudal DL; Dunham GE; Erjavec J, Heterogeneous oxidation of mercury in simulated post combustion conditions. Fuel 2003, 82 (2), 107–116. [Google Scholar]
- (143).Ghorishi SB; Lee CW; Jozewicz WS; Kilgroe JD, Effects of fly ash transition metal content and flue gas HCl/SO2 ratio on mercury speciation in waste combustion. Environmental Engineering Science 2005, 22 (2), 221–231. [Google Scholar]
- (144).Pavlish JH; Sondreal EA; Mann MD; Olson ES; Galbreath KC; Laudal DL; Benson SA, Status review of mercury control options for coal-fired power plants. Fuel Processing Technology 2003, 82 (2-3), 89–165. [Google Scholar]
- (145).Hower JC; Trimble AS; Eble CF; Palmer CA; Kolker A, Characterization of fly ash from low-sulfur and high-sulfur coal sources: partitioning of carbon and trace elements with particle size. Energy Sources 1999, 21 (6), 511–525. [Google Scholar]
- (146).Senior CL; Johnson SA, Impact of Carbon-in-Ash on Mercury Removal across Particulate Control Devices in Coal-Fired Power Plants. Energy & Fuels 2005, 19, 859–863. [Google Scholar]
- (147).Serre SD; Silcox GD, Adsorption of elemental mercury on the residual carbon in coal fly ash. Industrial & Engineering Chemistry Research 2000, 39 (6), 1723–1730. [Google Scholar]
- (148).López-Antón MA; Díaz-Somoano M; Martínez-Tarazona MR, Retention of elemental mercury in fly ashes in different atmospheres. Energy & Fuels 2007, 21 (1), 99–103. [Google Scholar]
- (149).U.S. EPA, Air Emission Measurement Center (EMC). https://www.epa.gov/emc (accessed February 12, 2020). [Google Scholar]
- (150).Senior CL, Batch Methods for Mercury Monitoring. In Mercury Control: for Coal-Derived Gas Streams, Granite EJ; Pennline HW; Senior CL, Eds. Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2015; pp 91–107. [Google Scholar]
- (151).Laudal DL, Continuous Mercury Monitors for Fossil Fuel-Fired Utilities. In Mercury Control: for Coal-Derived Gas Streams, Granite EJ; Pennline HW; Senior CL, Eds. Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2015; pp 71–89. [Google Scholar]
- (152).Marple VA, History of impactors—the first 110 years. Aerosol Science and Technology 2004, 38 (3), 247–292. [Google Scholar]
- (153).May KR, The cascade impactor: an instrument for sampling coarse aerosols. Journal of Scientific Instruments 1945, 22 (10), 187. [Google Scholar]
- (154).Mitchell RI; Pilcher JM, Improved cascade impactor for measuring aerosol particle sizes. Industrial & Engineering Chemistry 1959, 51 (9), 1039–1042. [Google Scholar]
- (155).Andersen AA, New sampler for the collection, sizing, and enumeration of viable airborne particles. Journal of Bacteriology 1958, 76 (5), 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Pilat MJ; Ensor DS; Bosch JC, Source test cascade impactor. Atmospheric Environment 1970, 4 (6), 671–679. [Google Scholar]
- (157).Hering SV; Flagan RC; Friedlander SK, Design and evaluation of new low-pressure impactor. I. Environmental Science & Technology 1978, 12 (6), 667–673. [Google Scholar]
- (158).Berner A; Lürzer C; Pohl F; Preining O; Wagner P, The size distribution of the urban aerosol in Vienna. Science of the Total Environment 1979, 13 (3), 245–261. [Google Scholar]
- (159).Hillamo RE; Kauppinen EI, On the performance of the Berner low pressure impactor. Aerosol Science and Technology 1991, 14 (1), 33–47. [Google Scholar]
- (160).Davidson RM Modes of occurrence of trace elements in coal: results from an international collaborative programme; CCC/CD-36; IEA Coal Research: London, 2000. [Google Scholar]
- (161).Senior CL; Zeng T; Che J; Ames MR; Sarofim AF; Olmez I; Huggins FE; Shah N; Huffman GP; Kolker A; Mroczkowski S; Palmer C; Finkelman R, Distribution of Trace Elements in Selected Pulverized Coals as a Function of Particle Size and Density. Fuel Processing Technology 2000, 63 (2-3), 215–241. [Google Scholar]
- (162).Palmer C; Mroczkowski S; Finkelman R; Crowley SS; Bullock JS, The use of sequential leaching to quantify the modes of occurrence of elements in coal. In 15th Pittsburgh Coal Conference, Pittsburgh, PA, 1998. [Google Scholar]
- (163).Gentzis T; Goodarzi F, Chemical Fractionation of Trace Elements in Coal and Coal Ash. Energy Sources 1999, 21 (3), 233–256. [Google Scholar]
- (164).Hoffart A; Seames W; Kozliak E; Riedinger S; Francini J; Carlson C, A two-step acid mercury removal process for pulverized coal. Fuel 2006, 85 (9), 1166–1173. [Google Scholar]
- (165).Huffman GP; Huggins FE; Shah N; Zhao J, Speciation of arsenic and chromium in coal by XAFS spectroscopy. Fuel Processing Technology 1994, 39, 47–62. [Google Scholar]
- (166).Huggins FE; Huffman G, Modes of occurrence of trace elements in coal from XAFS spectroscopy. International Journal of Coal Geology 1996, 32 (1-4), 31–53. [Google Scholar]
- (167).Huggins FE; Huffman GP; Kolker A; Mroczkowski S; Palmer C; Finkelman R, Combined Application of XAFS Spectroscopy and Sequential Leaching for Determination of Arsenic Speciation in Coal. Energy & Fuels 2002, 16, 1167–1172. [Google Scholar]
- (168).Kolker A; Wooden J; Persing H; Zielinski R, Stanford-USGS SHRIMP-RG ion microprobe: a new approach to determining the distribution of trace elements in coal. ACS Division of Fuel Chemistry, Preprints 2000, 45 (3), 542–546. [Google Scholar]
- (169).Huggins FE; Huffman GP; Dunham GE; Senior CL, XAFS Examination of Mercury Sorption on Three Activated Carbons. Energy & Fuels 1999, 13, 114–121. [Google Scholar]
- (170).Shoji T; Huggins FE; Huffman GP, XAFS Spectroscopy Analysis of Selected Elements in Fine Particulate Matter Derived from Coal Combustion. Energy & Fuels 2002, 16, 325–329. [Google Scholar]
- (171).Huggins FE; Yap N; Huffman GP; Senior CL, XAFS Characterization of Mercury Captured from Combustion Gases on Sorbents at Low Temperatures. Fuel Processing Technology 2003, 82, 167–196. [Google Scholar]
- (172).Huggins FE; Huffman GP; Linak WP; Miller CA, Quantifying Hazardous Species in Particulate Matter Derived from Fossil-Fuel Combustion. Environ Sci Technol 2004, 38, 1836–1842. [DOI] [PubMed] [Google Scholar]
- (173).Huggins FE; Senior CL; Chu P; Ladwig K; Huffman GP, Selenium and arsenic speciation in fly ash from full-scale coal-burning utility plants. Environ Sci Technol 2007, 41 (9), 3284–3289. [DOI] [PubMed] [Google Scholar]
- (174).Jiao F; Wijaya N; Zhang L; Ninomiya Y; Hocking R, Synchrotron-based XANES speciation of chromium in the oxy-fuel fly ash collected from lab-scale drop-tube furnace. Environ Sci Technol 2011, 45 (15), 6640–6. [DOI] [PubMed] [Google Scholar]
- (175).Niss ND; Schabron JF; Brown TH, Determination of Selenium Species in Coal Fly Ash Extracts. Environ Sci Technol 1993, 27, 827–829. [Google Scholar]
- (176).Jackson BP; Miller WP, Arsenic and selenium speciation in coal fly ash extracts by ion chromatography-inductively coupled plasma mass spectrometry. J Anal Atom Spectrom 1998, 13, 1107–1112. [Google Scholar]
- (177).Narukawa T; Takatsu A; Chiba K; Riley KW; French DH, Investigation on chemical species of arsenic, selenium and antimony in fly ash from coal fuel thermal power stations. J Environ Monit 2005, 7 (12), 1342–8. [DOI] [PubMed] [Google Scholar]
- (178).Piispanen MH; Arvilommi SA; Broeck B. V. d.; Nuutinen LH; Tiainen MS; Perämäki PJ; Laitinen RS, A Comparative Study of Fly ash Characterization by LA-ICP-MS and SEM-EDS†. Energy & Fuels 2009, 23 (7), 3451–3456. [Google Scholar]
- (179).Papirer E; Lacroix R; Donnet J-B; Nanse G; Fioux P, XPS Study of the halogenation of carbon black-part 1. Bromination. Carbon 1994, 32 (7), 1341–1358. [Google Scholar]
- (180).Papirer E; Lacroix R; Donnet J-B; Nansé G; Fioux P, XPS study of the halogenation of carbon black—Part 2. Chlorination. Carbon 1995, 33 (1), 63–72. [Google Scholar]
- (181).Olson ES; Crocker CR; Benson SA; Pavlish JH; Holmes MJ, Surface compositions of carbon sorbents exposed to simulated low-rank coal flue gases. J. Air & Waste Manage. Assoc 2005, 55 (6), 747–754. [DOI] [PubMed] [Google Scholar]
- (182).Hutson ND; Attwood BC; Scheckel KG, XAS and XPS characterization of mercury binding on brominated activated carbon. Environ. Sci. & Tech 2007, 41 (5), 1747–1752. [DOI] [PubMed] [Google Scholar]
- (183).Wilcox J; Sasmaz E; Kirchofer A; Lee S-S, Heterogeneous mercury reaction chemistry on activated carbon. Journal of the Air & Waste Management Association 2011, 61 (4), 418–426. [DOI] [PubMed] [Google Scholar]
- (184).Van der Sloot HA; Wijkstra J; van Dalen A; Das HA; Slanina J; Dekkers JJ; Wals GD Leaching of trace elements from coal solid waste; Netherlands Energy Research Foundation: The Netherlands, 1982. [Google Scholar]
- (185).Wadge A; Hutton M, The leachability and chemical speciation of selected trace elements in fly ash from coal combustion and refuse incineration. Environmental Pollution 1987, 48 (2), 85–99. [DOI] [PubMed] [Google Scholar]
- (186).de Groot GJ; Wijkstra J; Hoede D; van der Sloot HA, Leaching characteristics of selected elements from coal fly ash as a function of the acidity of the contact solution and the liquid/solid ratio. In Environmental aspects of stabilization and solidification of hazardous and radioactive wastes, ASTM International: 1989. [Google Scholar]
- (187).Hjelmar O, Leachate from land disposal of coal fly ash. Waste Management & Research 1990, 8 (6), 429–449. [Google Scholar]
- (188).Wang T; Wang J; Tang Y; Shi H; Ladwig K, Leaching Characteristics of Arsenic and Selenium from Coal Fly Ash: Role of Calcium. Energy & Fuels 2009, 23, 2959–2966. [Google Scholar]
- (189).Luo Y; Giammar DE; Huhmann BL; Catalano JG, Speciation of Selenium, Arsenic, and Zinc in Class C Fly Ash. Energy & Fuels 2011, 25 (7), 2980–2987. [Google Scholar]
- (190).Thorneloe S; Kosson DS; Sanchez F; Garrabrants A; Helms G, Evaluating the Fate of Metals in Air Pollution Control Residues from Coal-Fired Power Plants. Environ Sci Technol 2010, 44, 7351–7356. [DOI] [PubMed] [Google Scholar]
- (191).U.S. EPA. Leaching Environmental Assessment Framework (LEAF). https://www.epa.gov/hw-sw846/leaching-environmental-assessment-framework-leaf-methods-and-guidance#LEAF%20Methods (03 July 2020, date last accessed).
- (192).Taylor DD; Flagan RC, The influence of combustor operation on fine particles from coal combustion. Aerosol Science and Technology 1981, 1 (1), 103–117. [Google Scholar]
- (193).Bool III LE; Helble JJ, A laboratory study of the partitioning of trace elements during pulverized coal combustion. Energy & Fuels 1995, 9 (5), 880–887. [Google Scholar]
- (194).Seames WS; Sooroshian J; Wendt JO, Assessing the solubility of inorganic compounds from size-segregated coal fly ash aerosol impactor samples. Journal of Aerosol Science 2002, 33 (1), 77–90. [Google Scholar]
- (195).Westergard B, Mercury from Hogdalen incineration plant in Stockholm, 1972-1985. Waste Management & Research 1986, 4 (1), 21–21. [Google Scholar]
- (196).Hall B; Lindqvist O; Ljungström E, Mercury chemistry in simulated flue gases related to waste incineration conditions. Environmental Science & Technology 1990, 24 (1), 108–111. [Google Scholar]
- (197).Metzger M; Braun H, In-situ mercury speciation in flue gas by liquid and solid sorption systems. Chemosphere 1987, 16 (4), 821–832. [Google Scholar]
- (198).ASTM International, D 6784: Standard Test Method for Elemental, Oxidized, Particle-Bound and Total Mercury in Flue Gas Generated from Coal-Fired Stationary Sources (Ontario Hydro Method). 2002. [Google Scholar]
- (199).Prestbo EM; Bloom NS, Mercury Speciation Adsorption (MESA) Method Intercomparison Results in Combustion Flue Gas. In 11th Annual Pittsburgh Coal Conference, Pittsburgh, PA, 1994. [Google Scholar]
- (200).Schneider AP; Cook PM; Cross JA; Mertz AM; Motovilov D; Ng-Feng A; Moyer WP; Sipershteyn A; Tobin EM; Siperstein J, Innovative Measurement Technologies: Hg, SO3, NH3, HCl, HBr, As, Se. In Power Plant Pollutant and Effluent Control MEGA Symposium: Best Practices and Future Trends, Baltimore, MD, 2018. [Google Scholar]
- (201).Frandsen FJ; Helble JJ; Erickson TA; Kuhnel V; Linak WP Prediction of trace element partitioning in utility boilers, In EPRI-DOE-EPA Combined Utility Air Pollutant Control Symposium: the Mega Symposium, Washington, DC, August 25-26, 1997; Offen G; Ruth L; Lachapelle D, Eds. Electric Power Research Institute: Washington, DC, 1997. [Google Scholar]
- (202).Stull DR; Prophet H JANAF Thermochemical Tables, Second Edition; NBS 37; U.S. Government Printing Office: Washington, D.C., 1971. [Google Scholar]
- (203).Wilcox JL; Robles J; Marsden DCJ; Blowers P, Theoretically Predicted Rate Constants for Mercury Oxidation by Hydrogen Chloride in Coal Combustion Flue Gases. Environ Sci Technol 2003, 37, 4199–4204. [DOI] [PubMed] [Google Scholar]
- (204).Xu M; Qiao Y; Zheng C; Li L; Liu J, Modeling of homogeneous mercury speciation using detailed chemical kinetics. Combustion and Flame 2003, 132 (1-2), 208–218. [Google Scholar]
- (205).Zheng C; Liu J; Liu Z; Xu M; Liu Y, Kinetic mechanism studies on reactions of mercury and oxidizing species in coal combustion. Fuel 2005, 84 (10), 1215–1220. [Google Scholar]
- (206).Wilcox J, A kinetic investigation of high-temperature mercury oxidation by chlorine. The Journal of Physical Chemistry A 2009, 113 (24), 6633–6639. [DOI] [PubMed] [Google Scholar]
- (207).Liu J; Qu W; Yuan J; Wang S; Qiu J; Zheng C, Theoretical studies of properties and reactions involving mercury species present in combustion flue gases. Energy & Fuels 2010, 24 (1), 117–122. [Google Scholar]
- (208).Balabanov NC; Peterson KA, Mercury and Reactive Halogens: The Thermochemistry of Hg + {Cl2, Br2, BrCl, ClO, and BrO}. J Phys Chem A 2003, 107, 7465–7470. [Google Scholar]
- (209).Goodsite ME; Plane J; Skov H, A theoretical study of the oxidation of Hg0 to HgBr2 in the troposphere. Environmental science & technology 2004, 38 (6), 1772–1776. [DOI] [PubMed] [Google Scholar]
- (210).Balabanov NB; Shepler BC; Peterson KA, Accurate global potential energy surface and reaction dynamics for the ground state of HgBr2. J Phys Chem A 2005, 109 (39), 8765–8773. [DOI] [PubMed] [Google Scholar]
- (211).Wilcox J; Okano T, Ab initio-based Mercury Oxidation Kinetics via Bromine at Postcombustion Flue Gas Conditions. Energy & Fuels 2011, 25 (4), 1348–1356. [Google Scholar]
- (212).Shepler BC; Balabanov NC; Peterson KA, Ab Initio Thermochemistry Involving Heavy Atoms: An Investigation of the Reactions of Hg + IX (X = I, Br, Cl, O). J Phys Chem A 2005, 109, 10363–10372. [DOI] [PubMed] [Google Scholar]
- (213).Auzmendi-Murua I; Castillo A; Bozzelli JW, Mercury oxidation via chlorine, bromine, and iodine under atmospheric conditions: thermochemistry and kinetics. J Phys Chem A 2014, 118 (16), 2959–75. [DOI] [PubMed] [Google Scholar]
- (214).Auzmendi-Murua I; Bozzelli JW, Gas Phase Mercury Oxidation by Halogens (Cl, Br, I) in Combustion Effluents: Influence of Operating Conditions. Energy & Fuels 2016, 30 (1), 603–615. [Google Scholar]
- (215).Padak B; Brunetti M; Lewis A; Wilcox J, Mercury binding on activated carbon. Environmental Progress 2006, 25 (4), 319–326. [Google Scholar]
- (216).Liu J; Cheney MA; Wu F; Li M, Effects of chemical functional groups on elemental mercury adsorption on carbonaceous surfaces. Journal of Hazardous Materials 2011, 186 (1), 108–113. [DOI] [PubMed] [Google Scholar]
- (217).Li J; Maroto-Valer MM, Computational and experimental studies of mercury adsorption on unburned carbon present in fly ash. Carbon 2012, 50 (5), 1913–1924. [Google Scholar]
- (218).Liu J; Qu W; Joo SW; Zheng C, Effect of SO2 on mercury binding on carbonaceous surfaces. Chemical Engineering Journal 2012, 184, 163–167. [Google Scholar]
- (219).Liu J; Qu W; Zheng C, Theoretical studies of mercury–bromine species adsorption mechanism on carbonaceous surface. Proceedings of the Combustion Institute 2013, 34 (2), 2811–2819. [Google Scholar]
- (220).Aboud S; Sasmaz E; Wilcox J, Mercury adsorption on PdAu, PdAg and PdCu alloys. Main Group Chemistry 2008, 7 (3), 205–215. [Google Scholar]
- (221).Guo P; Guo X; Zheng C, Roles of γ-Fe2O3 in fly ash for mercury removal: Results of density functional theory study. Applied Surface Science 2010, 256 (23), 6991–6996. [Google Scholar]
- (222).Suarez Negreira A; Wilcox J, DFT study of Hg oxidation across vanadia-titania SCR catalyst under flue gas conditions. The Journal of Physical Chemistry C 2013, 117 (4), 1761–1772. [Google Scholar]
- (223).Ling L; Fan M; Wang B; Zhang R, Application of computational chemistry in understanding the mechanisms of mercury removal technologies: a review. Energy & Environmental Science 2015, 8 (11), 3109–3133. [Google Scholar]
- (224).Monahan-Pendergast M; Przybylek M; Lindblad M; Wilcox J, Theoretical predictions of arsenic and selenium species under atmospheric conditions. Atmospheric Environment 2008, 42 (10), 2349–2357. [Google Scholar]
- (225).Zhang S; Hu X; Lu Q; Zhang J, Density Functional Theory Study of Arsenic and Selenium Adsorption on the CaO (001) Surface. Energy & Fuels 2011, 25 (7), 2932–2938. [Google Scholar]
- (226).Steam - its generation and use. 42nd ed.; Babcock & Wilcox: Charlotte, North Carolina, 2015. [Google Scholar]
- (227).Topsøe N; Topsøe H; Dumesic J, Vanadia/titania catalysts for selective catalytic reduction (SCR) of nitric-oxide by ammonia: I. Combined temperature-programmed in-situ FTIR and on-line mass-spectroscopy studies. Journal of Catalysis 1995, 151 (1), 226–240. [Google Scholar]
- (228).Pritchard S; DiFrancesco C; Kaneko S; Kobayashi N; Suyama K; Lida K Optimizing SCR catalyst design and performance for coal-fired boilers, In Proceedings of the EPA/EPRI Symposium on Stationary Combination NOx Control, 1995. [Google Scholar]
- (229).Gutberlet H, Influence of furnace type on poisoning of DENOX catalysts by arsenic. VGB Kraftwerkstechnik 1988, 68 (3), 264–271. [Google Scholar]
- (230).Staudt JE; Engelmeyer T; Weston WH; Sigling R, The impact of arsenic on coal fired power plants equipped with SCR. In Proceedings of ICAC Forum, Houston, TX, 2002. [Google Scholar]
- (231).Morita I; Hirano M; Bielawski GT, Development and Commercial Operating Experience of SCR DeNOx Catalysts for Wet-Bottom Coal-Fired Boilers. In Proceedings of Power-Gen International, Orlando, FL, 1998. [Google Scholar]
- (232).Ake T; Erickson C; Medeiros W; Hutcheson L; Barger M; Rutherford S, Limestone injection for protection of SCR catalyst. In Proceedings of the EPA-EPRI-NETL-AWMA Combined Power Plant Air Pollutant Control Mega Symposium, Washington, DC, 2003. [Google Scholar]
- (233).Gutberlet H; Schlueter A; Licata A, SCR Impacts on Mercury Emissions from Coal-Fired Boilers In Electric Power Research Institute SCR Workshop, Memphis, TN, 2000. [Google Scholar]
- (234).Laudal D Effect of Selective Catalytic Reduction on Mercury, 2002 Field Studies Update; Product ID 100558; EPRI: Palo Alto, CA, 2002. [Google Scholar]
- (235).Hocquel M; Unterberger S; Hein KRG, Effects of Mercury on SCR-catalysts. In Conference on Selective Catalytic Reduction (SCR) and Selective Non-Catalytic Reduction (SNCR) for NOx Control, Pittsburgh, PA, 2002. [Google Scholar]
- (236).Lee CW; Srivastava RK; Ghorishi SB; Hastings TW; Stevens FM, Investigation of Selective Catalytic Reduction Impact on Mercury Speciation under Simulated NOx Emissions Control Conditions. J Air Waste Manag Assoc 2004, 54, 1560–1566. [DOI] [PubMed] [Google Scholar]
- (237).Zhuang Y; Laumb J; Liggett R; Holmes M; Pavlish J, Impacts of acid gases on mercury oxidation across SCR catalyst. Fuel Processing Technology 2007, 88 (10), 929–934. [Google Scholar]
- (238).Eom Y; Jeon SH; Ngo TA; Kim J; Lee TG, Heterogeneous mercury reaction on a selective catalytic reduction (SCR) catalyst. Catalysis Letters 2008, 121 (3-4), 219–225. [Google Scholar]
- (239).Lee CW; Serre SD; Zhao Y; Lee SJ; Hastings TW, Mercury oxidation promoted by a selective catalytic reduction catalyst under simulated powder river basin coal combustion conditions. Journal of the Air & Waste Management Association 2008, 58 (4), 484–493. [DOI] [PubMed] [Google Scholar]
- (240).Kamata H; Ueno S-I; Naito T; Yukimura A, Mercury oxidation over the V2O5 (WO3)/TiO2 commercial SCR catalyst. Industrial & Engineering Chemistry Research 2008, 47 (21), 8136–8141. [Google Scholar]
- (241).Madsen K Mercury Oxidation over Selective Catalytic Reduction (SCR) Catalysts. Ph.D. Thesis. Danish Technical University, Lyngby, Denmark, 2011. [Google Scholar]
- (242).Richardson CF; Machalek T; Miller S; Dene C; Chang R, Effect of NOx Control Processes on Mercury Speciation in Utility Flue Gas. J Air Waste Manag Assoc 2002, 52, 941–947. [DOI] [PubMed] [Google Scholar]
- (243).Cao Y; Gao Z; Zhu J; Wang Q; Huang Y; Chiu C; Parker B; Chu P; Pan W-P, Impacts of halogen additions on mercury oxidation, in a slipstream selective catalyst reduction (SCR), reactor when burning sub-bituminous coal. Environmental Science & Technology 2008, 42 (1), 256–261. [DOI] [PubMed] [Google Scholar]
- (244).Stolle R; Koeser H; Gutberlet H, Oxidation and reduction of mercury by SCR DeNOx catalysts under flue gas conditions in coal fired power plants. Applied Catalysis B: Environmental 2014, 144, 486–497. [Google Scholar]
- (245).Vosteen BW; Straube S; Koeser H, Mercury Sorption and Mercury Oxidation by Chlorine and Bromine at SCR DeNOx Catalyst (Part A: Oxidation). In 9th Annual EUEC, Tucson, Arizona, 2006. [Google Scholar]
- (246).Niksa S; Fujiwara N, A predictive mechanism for mercury oxidation on selective catalytic reduction catalysts under coal-derived flue gas. Journal of the Air & Waste Management Association 2005, 55 (12), 1866–1875. [DOI] [PubMed] [Google Scholar]
- (247).Senior CL, Oxidation of Mercury across Selective Catalytic Reduction Catalysts in Coal-Fired Power Plants. J. Air & Waste Manage. Assoc 2006, 56, 23–31. [DOI] [PubMed] [Google Scholar]
- (248).Beretta A; Usberti N; Lietti L; Forzatti P; Di Blasi M; Morandi A; La Marca C, Modeling of the SCR reactor for coal-fired power plants: Impact of NH3 inhibition on Hg0 oxidation. Chemical Engineering Journal 2014, 257, 170–183. [Google Scholar]
- (249).Singer CF Surrogacy Testing in the MPCRF, Prepared for U.S. EPA; Arcadis: Durham, NC, March 30, 2011. [Google Scholar]
- (250).U.S. EPA. Mercury and Air Toxics Standards for Utilities – Technology Transfer Network. https://www3.epa.gov/airtoxics/utility/utilitypg.html (2020-08-26, date of last access)
- (251).Senior CL; Sjostrom S Behavior of Mercury and Selenium in Coal-Fired Boilers and Air Pollution Control Devices: A Review of Recent Full-Scale Data. In Air & Waste Management Association 104th Annual Conference and Exhibition, Orlando, FL, June 21-24, 2011. [Google Scholar]
- (252).White DM; Kelly WE; Stucky MJ Field Test of Carbon Injection for Mercury Control Camden County Municipal Waste Combustor; EPA-600/R-93-181; U.S. EPA: Research Triangle Park, NC, September 1993. [Google Scholar]
- (253).Sjostrom S Evaluation of Sorbent Injection for Mercury Control Topical Report for: Basin Electric Power Cooperative's Laramie River Station; U.S. DOE Cooperative Agreement No. DE-FC26-03NT41986, Topical Report No. 41986R11; ADA-ES, Inc.: Littleton, CO, 2006. [Google Scholar]
- (254).Krishnan SV; Gullett BK; Jozewicz WS, Sorption of Elemental Mercury by Activated Carbons. Environ. Sci. Technol 1994, 26, 1506–1512. [DOI] [PubMed] [Google Scholar]
- (255).Karatza D; Lancia A; Musmarra D; Pepe F, Adsorption of metallic mercury on activated carbon. In Symposium (International) on Combustion, Elsevier: 1996; Vol. 26, pp 2439–2445. [Google Scholar]
- (256).Karatza D; Lancia A; Musmarra D; Pepe F; Volpicelli G, Kinetics of adsorption of mercuric chloride vapors on sulfur impregnated activated carbon. Combustion Science and Technology 1996, 112 (1), 163–174. [Google Scholar]
- (257).Granite EJ; Pennline HW; Hargis RA, Novel Sorbents for Mercury Removal from Flue Gas. Industrial & Engineering Chemistry Research 2000, 39, 1020–1029. [Google Scholar]
- (258).Young BC; Musich MA, Screening of carbon-based sorbents for the removal of elemental mercury from simulated combustion flue gas. Prepr. Pap.-Am. Chem. Soc., Div. Fuel Chem 1995, 40 (4), 833–837. [Google Scholar]
- (259).Ghorishi B; Gullett BK, Sorption of mercury species by activated carbons and calcium-based sorbents: effect of temperature, mercury concentration and acid gases. Waste Management & Research 1998, 16 (6), 582–593. [Google Scholar]
- (260).Carey TR; Hargrove OW; Richardson CF; Chang R; Meserole FB, Factors affecting mercury control in utility flue gas using activated carbon. Journal of the Air & Waste Management Association 1998, 48 (12), 1166–1174. [DOI] [PubMed] [Google Scholar]
- (261).Miller SJ; Dunham GE; Olson ES; Brown TD, Mercury sorbent development for coal-fired boilers. In Proceedings of the Conference on Air Quality: Mercury, Trace Elements, and Particulate Matter, Crystal City, VA, 1998. [Google Scholar]
- (262).Olson E; Laumb J; Benson S; Dunham G; Sharma R; Mibeck B; Miller S; Holmes M; Pavlish J, Chemical mechanisms in mercury emission control technologies. Journal de Physique IV France 2003, 107, 979–982. [Google Scholar]
- (263).Morris EA; Morita K; Jia CQ, Understanding the effects of sulfur on mercury capture from coal-fired utility flue gases. Journal of Sulfur Chemistry 2010, 31 (5), 457–475. [Google Scholar]
- (264).Morris EA; Kirk DW; Jia CQ; Morita K, Roles of sulfuric acid in elemental mercury removal by activated carbon and sulfur-impregnated activated carbon. Environmental Science & Technology 2012, 46 (14), 7905–7912. [DOI] [PubMed] [Google Scholar]
- (265).Li YH; Lee CW; Gullett BK, The effect of activated carbon surface moisture on low temperature mercury adsorption. Carbon 2002, 40 (1), 65–72. [Google Scholar]
- (266).Bandosz TJ; Ania CO, Surface chemistry of activated carbons and its characterization. In Activated Carbon Surfaces in Environmental Remediation, Bandosz TJ, Ed. Elsevier, Ltd.: Oxford, UK, 2006. [Google Scholar]
- (267).Olson ES; Azenkeng A; Laumb JD; Jensen RR; Benson SA; Hoffmann MR, New developments in the theory and modeling of mercury oxidation and binding on activated carbons in flue gas. Fuel Processing Technology 2009, 90 (11), 1360–1363. [Google Scholar]
- (268).Li YH; Lee CW; Gullett BK, Importance of activated carbon's oxygen surface functional groups on elemental mercury adsorption. Fuel 2003, 82 (4), 451–457. [Google Scholar]
- (269).Zhang B; Xu P; Qiu Y; Yu Q; Ma J; Wu H; Luo G; Xu M; Yao H, Increasing oxygen functional groups of activated carbon with non-thermal plasma to enhance mercury removal efficiency for flue gases. Chemical Engineering Journal 2015, 263, 1–8. [Google Scholar]
- (270).Lizzio AA; DeBarr JA, Mechanism of SO2 removal by carbon. Energy & Fuels 1997, 11 (2), 284–291. [Google Scholar]
- (271).Gutberlet H; Boehm GM, The influence of induced oxidation on the operation of wet FGD systems. In Proceedings of Air Quality V, Arlington, VA, 2005. [Google Scholar]
- (272).Blythe GM; Richardson MK; Steen WA; Dene CE; Nolan PS, Investigation of mercury control by wet FGD systems. In Proceedings of Air Quality VIII, Arlington, VA, 2011. [Google Scholar]
- (273).Farthing GA Advanced Emissions Control Development Program–Phase I Final Report, Contract: DE-FC22-94PC94251; Babcock & Wilcox Corporation: Barberton, OH, 1996. [Google Scholar]
- (274).Amrhein GT; Kudlac GA; Madden DM, Full-scale testing of mercury control for wet FGD systems. In Proceedings of the 27th International Coal Utilization and Fuel Systems Conference, Clearwater, FL, 2002; Vol. 2, p 20. [Google Scholar]
- (275).Munthe J; Xiao ZF; Lindqvist O, The aqueous reduction of divalent mercury by sulfite. Water Air & Soil Pollution 1991, 56 (1), 621–630. [Google Scholar]
- (276).Van Loon L; Mader E; Scott SL, Reduction of the aqueous mercuric ion by sulfite: UV spectrum of HgSO3 and its intramolecular redox reaction. The Journal of Physical Chemistry A 2000, 104 (8), 1621–1626. [Google Scholar]
- (277).Van Loon LL; Mader EA; Scott SL, Sulfite stabilization and reduction of the aqueous mercuric ion: kinetic determination of sequential formation constants. The Journal of Physical Chemistry A 2001, 105 (13), 3190–3195. [Google Scholar]
- (278).Ryaboshapko A; Bullock R; Ebinghaus R; Ilyin I; Lohman K; Munthe J; Petersen G; Seigneur C; Wängberg I, Comparison of mercury chemistry models. Atmospheric Environment 2002, 36 (24), 3881–3898. [Google Scholar]
- (279).Chang JCS; Ghorishi SB, Simulation and evaluation of elemental mercury concentration increase in flue gas across a wet scrubber. Environmental Science & Technology 2003, 37 (24), 5763–5766. [DOI] [PubMed] [Google Scholar]
- (280).Blythe G; DeBerry D; Marsh B; Paradis J; Range J; Rhudy R, Bench-scale evaluation of the fate of mercury in wet FGD systems. In Proceedings of the EPRI-EPA-DOE-AWMA Mega Symposium, Washington, DC, 2004. [Google Scholar]
- (281).Currie J; DeBerry DW; Blythe GM; Pletcher S; Rhudy R, Bench-scale kinetics study of mercury reactions in FGD liquors. In Proceedings of the DOE NETL Mercury Control Technology Conference, Pittsburgh, PA, 2006. [Google Scholar]
- (282).Dίaz-Somoano M; Unterberger S; Hein KRG, Mercury emission control in coal-fired plants: The role of wet scrubbers. Fuel processing Technology 2007, 88 (3), 259–263. [Google Scholar]
- (283).Heidel B; Klein B, Reemission of elemental mercury and mercury halides in wet flue gas desulfurization. International Journal of Coal Geology 2017, 170, 28–34. [Google Scholar]
- (284).Bittig M; Pieper B, A basic equation for mercury(II) absorption. In Proceedings of VGB Workshop Mercury Control, Berlin, Germany, 2016. [Google Scholar]
- (285).Masoomi I; Heidel B; Schmid MO; Scheffknecht G, Effect of additives on mercury partitioning in wet limestone flue gas desulfurization. Clean Energy 2020, 4 (2), 132–141. [Google Scholar]
- (286).Senior CL; Tyree CA; Meeks ND; Acharya C; McCain JD; Cushing KM, Selenium Partitioning and Removal Across a Wet FGD Scrubber at a Coal-Fired Power Plant. Environ Sci Technol 2015, 49 (24), 14376–82. [DOI] [PubMed] [Google Scholar]
- (287).Impact of Wet FGD Design and Operations on Selenium Speciation: 2010 Update. EPRI, Palo Alto, CA: 2010) 1019870. [Google Scholar]
- (288).Allen JO; Ferens-Foulet CK; Acharya C, Effluent Trace Metals Survey and Related Plant Operations at 18 Flagship FGD Units. In Power Plant Pollutant Control and Carbon Management "MEGA" Symposium, Baltimore, MD, 2016. [Google Scholar]
- (289).Córdoba P, Partitioning and speciation of selenium in wet limestone flue gas desulphurisation systems: A review. Fuel 2017, 202, 184–195. [Google Scholar]
- (290).40 CFR Part 423, Effluent Limitations Guidelines and Standards for the Steam Electric Power Generating Point Source Category; Final Rule, https://ecfr.federalregister.gov/current/title-40/chapter-I/subchapter-N/part-423 (2020-09-07, date of last access) [Google Scholar]
- (291).U.S. EPA, Technical Development Document for the Effluent Limitation Guidelines and Standards for the Steam Electric Power Generating Point Source Category; EPA-821-R-15-007; U.S. EPA: Washington, D.C., 2015. [Google Scholar]
- (292).Akiho H; Ito S; Matsuda H; Yoshioka T, Elucidation of the mechanism of reaction between S2O8(2−), Selenite and Mn2+ in aqueous solution and limestone-gypsum FGD liquor. Environ Sci Technol 2013, 47 (19), 11311–7. [DOI] [PubMed] [Google Scholar]
- (293).Abbott M; Gadgil M; Wasikowski S; Kotch G; Owens T; Watson M, Advances in Refined-Coal Technology for Emissions Reduction. In Power Plant Pollutant Control "MEGA" Symposium, Baltimore, MD, 2014. [Google Scholar]
- (294).Prest BC; Krupnick A How Clean is “Refined Coal”? An Empirical Assessment of a Billion-Dollar Tax Credit, Report 19-05. Resources for the Future, November 2019. https://media.rff.org/documents/Refined_Coal_Report_rev.pdf (19 June, 2020 date last accessed). [Google Scholar]
- (295).Ghorishi SB; Keeney RM; Serre SD; Gullett BK; Jozewicz WS, Development of a Cl-impregnated activated carbon for entrained-flow capture of elemental mercury. Environmental Science & Technology 2002, 36 (20), 4454–4459. [DOI] [PubMed] [Google Scholar]
- (296).Nelson SG Sorbents and Methods for the Removal of Mercury from Combustion gases. US 6,953,494, October 11, 2005.
- (297).Sjostrom S; Ebner T; Slye R; Chang R; Strohfus M; Pelerine J; Smokey S, Full-scale evaluation of mercury control at Great River Energy’s Stanton Generating Station using injected sorbents and a spray dryer/baghouse. In Air Quality III Conference, Arlington, VA, 2002. [Google Scholar]
- (298).Campbell T; Sjostrom S; Dillon M; Brignac P; O’Palko A; Chang R; Raichle P; Auclair A; Keyes H., Mercury Control with Activated Carbon: Results from Plants with High SO3. In Proceedings of DOE-EPRI-U.S. EPA-A& WMA Combined Power Plant Air Pollutant Control Symposium–The Mega Symposium, Baltimore, MD, 2008. [Google Scholar]
- (299).Pollack NR; Hayden RA Sorbent formulation for removal of mercury from flue gas. US 8,216,535, 2012.
- (300).Sjostrom S; Starns T; Amrhein J; Bustard J; Durham M; Penrod W; Linville C; O’Palko A; Chang R, Full-scale evaluation of mercury control by injecting activated carbon upstream of a spray dryer and fabric filter. In Joint EPRI DOE EPA Combined Utility Air Pollution Control Symposium, The Mega Symposium, Washington, DC, 2004. [Google Scholar]
- (301).Dombrowski K; Srinivasan N Bromine Balance of Plant Study. In Reinhold NOx Conference, Salt Lake City, UT, February 18-19, 2013. [Google Scholar]
- (302).Gadgil M Multi Pollution Control using Mitagent™ Combustion Additive. In 2013 APC Round Table & Exposition, St. Louis, MO, July 8-19, 2013. [Google Scholar]
- (303).Tarabocchia J; Peldszuz R, Mercury separation from flue gas and scrub water with Trimercapto-s-triazine (TMT). In DOE-EPRI-U.S. EPA-A&WMA Combined Power Plant Air Pollutant Control Symposium, Washington, DC, 2003. [Google Scholar]
- (304).Elliott P; Riethmann T, Testing of STEAG’s Mercury Control Technology at a US Midwest Utility. In Air Quality VIII Conference, Crystal City, VA, 2011. [Google Scholar]
- (305).Blythe GM, Mercury Capture in Wet Flue Gas Desulfurization Systems. In Mercury Control: for Coal-Derived Gas Streams, Granite EJ; Pennline HW; Senior CL, Eds. Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2015; pp 261–275. [Google Scholar]
- (306).Blythe GM; Dombrowski K; Machalek T; Richardson CF; Richardson MK Pilot testing of mercury oxidation catalysts for upstream of wet FGD systems, Final report; DOE Cooperative Agreement No: DE-FC26-01NT41185; URS Corporation: Austin, TX, 2006. [Google Scholar]
- (307).Hrdlicka JA; Seames WS; Mann MD; Muggli DS; Horabik CA, Mercury oxidation in flue gas using gold and palladium catalysts on fabric filters. Environmental Science & Technology 2008, 42 (17), 6677–6682. [DOI] [PubMed] [Google Scholar]
- (308).Seames W; Mann MD; Muggli DS Mercury oxidation of flue gas using catalytic barrier filters. US 7,618,603, 2009.
- (309).Gingerich DB; Zhao Y; Mauter MS, Environmentally significant shifts in trace element emissions from coal plants complying with the 1990 Clean Air Act Amendments. Energy Policy 2019, 132, 1206–1215. [Google Scholar]