

Abstract
We present a 517-gene phylogenetic framework for the breadfruit genus Artocarpus (ca. 70 spp., Moraceae), making use of silica-dried leaves from recent fieldwork and herbarium specimens (some up to 106 years old) to achieve 96% taxon sampling. We explore issues relating to assembly, paralogous loci, partitions, and analysis method to reconstruct a phylogeny that is robust to variation in data and available tools. Although codon partitioning did not result in any substantial topological differences, the inclusion of flanking noncoding sequence in analyses significantly increased the resolution of gene trees. We also found that increasing the size of data sets increased convergence between analysis methods but did not reduce gene-tree conflict. We optimized the HybPiper targeted-enrichment sequence assembly pipeline for short sequences derived from degraded DNA extracted from museum specimens. Although the subgenera of Artocarpus were monophyletic, revision is required at finer scales, particularly with respect to widespread species. We expect our results to provide a basis for further studies in Artocarpus and provide guidelines for future analyses of data sets based on target enrichment data, particularly those using sequences from both fresh and museum material, counseling careful attention to the potential of off-target sequences to improve resolution. [Artocarpus; Moraceae; noncoding sequences; phylogenomics; target enrichment.]
Reduced-representation methods such as target enrichment (HybSeq) have become important tools for phylogenetic studies, enabling high-throughput and cost-effective sequencing of hundreds of loci (Faircloth et al. 2012; Mandel et al. 2014; Weitemier et al. 2014). In this study, we employ HybSeq to investigate the breadfruit genus (Artocarpus J.R.Forst. & G.Forst., Moraceae), analyzing the utility of paralogs, partitioning, noncoding sequences, and herbarium specimens in reconstructing the most data-rich phylogeny of the genus to date.
HybSeq involves hybridizing a randomly sheared sequencing library to bait sequences, typically exons from one or more taxa within or near the target clade. Researchers have employed HybSeq in studies ranging from deep phylogenetics (Prum et al. 2015; Liu et al. 2019) to within-species phylogeography (Villaverde et al. 2018). It is particularly useful for recovering sequences from museum specimens, because target enrichment is suitable for very small DNA fragments and can help overcome the presence of contaminating nonendogenous DNA (Staats et al. 2013; Buerki and Baker 2016; Hart et al. 2016; Brewer et al. 2019). However, making the most of HybSeq data sets, which can comprise hundreds of thousands of characters, requires careful attention to assembly and analysis methods, particularly for degraded DNA from museum specimens. This particularly true because divergent analysis methods can sometimes lead to divergent topologies, all with apparently high statistical support.
The mechanics of HybSeq frequently result in the recovery of nontargeted sequences such as paralogs similar to the target sequences (Hart et al. 2016; Johnson et al. 2016, 2019; Liu et al. 2019) and noncoding sequences flanking the target sequences (e.g. Medina et al. 2019). Both were the case with HybSeq baits we previously developed for Moraceae phylogenetics (Gardner et al. 2016), many of which were represented as paralogous pairs in Artocarpus due to an ancient whole-genome duplication. In almost all cases, they were diverged enough to sort and analyze separately (Johnson et al. 2016). The same targets also typically recovered a several-hundred bp “splash zone” of flanking noncoding sequences (Johnson et al. 2016). The impact of off-target by-catch on phylogenetic reconstruction remains unclear but has the potential to greatly increase the number of phylogenetically informative genes. However, analysis of mixed coding and noncoding sequences can make it difficult to ensure that exons are aligned in frame, particularly when frameshifts are present (Ranwez et al. 2011), hampering partitioning of data sets by codon position. How these issues impact phylogenetic reconstruction remains unclear (Xi et al. 2012; Lanfear et al. 2014).
It is by now well understood that high bootstrap values obtained by concatenating all loci into a supermatrix should not be overinterpreted because near-perfect bootstrap support can mask substantial discordance among gene histories due to incomplete lineage sorting (Kubatko and Degnan 2007; Degnan and Rosenberg 2009; Sayyari and Mirarab 2016). Although there is an increased availability of efficient methods based on the multispecies coalescent model, clear results can be obscured if the underlying gene trees are uninformative (Smith et al. 2015; Sayyari et al. 2017). A major advantage of HybSeq over methods with short, anonymous loci, or large amounts of missing data, is that loci obtained via HybSeq are both long enough to generate single-gene phylogenies and subject to few enough missing taxa per locus for those single-gene phylogenies to be informative. These and other issues are explored below to develop a robust phylogenomic framework that will guide future work in Artocarpus and serve as a model for work in other systems.
Study System
Artocarpus (Fig. 1) contains approximately 70 species of trees with a center of diversity in Borneo and a native range that extends from India to the Solomon Islands (Williams et al. 2017). The genus is best known for important but underutilized crops such as breadfruit (Artocarpus altilis (Parkinson) Fosberg) and jackfruit (Artocarpus heterophyllus Lam.) (Zerega et al. 2010, 2015; Wang et al. 2018;Witherup et al. 2019).
Figure 1.

Diversity of Artocarpus. Subg. Prainea—(A) leaves, (B) syncarp, and (C) immature inflorescences of Artocarpus limpato. Subg. Pseudojaca—(D) leaves and (E) staminate inflorescences of A. fretessii; (F) pistillate inflorescence of A. borneensis; (G) syncarp of A. primackii; (H) syncarp of A. parvus; and (I) staminate (left) and carpellate (right) inflorescences of A. hypargyreus. Subg. Cauliflori—(J) leaves of A. integer; (K–L) staminate inflorescences, (M) syncarps, and (N) carpellate inflorescence of A. heterophyllus. Subg. Artocarpus—(O) leaves and inflorescences of A. altilis; (P) carpellate inflorescence of A. tamaran; (Q) syncarp and (R) carpellate inflorescence of A. odoratissimus; (S) leaves and staminate inflorescence of A. rigidus; (T) syncarps of A. altilis; and (U) staminate inflorescence of A. tamaran.
Artocarpus is monoecious, with spicate to globose staminate (“male”) inflorescences composed of tiny flowers bearing one stamen each. Pistillate (“female”) inflorescences are composed of tightly packed tiny flowers, and in most cases, adjacent flowers are at least partially fused together. Pistillate inflorescences develop into tightly packed accessory fruits composed mainly of fleshy floral tissue, ranging from a few centimeters in diameter in some species to over half a meter long in jackfruit. The tribe Artocarpeae Lam. & DC also includes two smaller Neotropical genera, Batocarpus H.Karst. (3 spp.) and Clarisia Ruiz & Pav. (3 spp.); these always have spicate staminate inflorescences; pistillate flowers may be solitary or condensed into globose heads, but adjacent flowers are never fused.
The most recent complete revision of Artocarpus (Jarrett,1959a, 1959b, 1960) recognized two subgenera, Artocarpus and Pseudojaca Trécul, distinguished by phyllotaxy (leaf arrangement), and the degree of fusion between adjacent pistillate flowers. Since then, several new species have been described (Jarrett 1975; Zhengyi and Xiushi 1989; Kochummen 1998; Berg 2005; Gardner et al. 2020). Berg et al. (2006) revised the Malesian species for the Flora Malesiana, in a few cases combining several taxa into a broadly-circumscribed single species, such as A. altilis (encompassing A. altilis, A. camansi Blanco, A. mariannensis Trécul, A. horridus F.M.Jarrett, A. blancoi Merr., A. pinnatisectus Merr., and A. multifidus F.M.Jarrett); further revisions were proposed for the Flora of Thailand (Berg et al. 2011). Subgenus Pseudojaca was partially revised by Gardner and Zerega (2020), based in part on the analyses presented here. Because a goal of this study is to provide a framework for taxonomic revisions, the nomenclature used here follows Gardner and Zerega (2020) for subgenus Pseodojaca and for the other subgenera follows the narrowest circumscription for each taxon between Jarrett (1959b, 1960), Berg et al. (2006, 2011), and Hart et al. (2016, 2010) (Table 1). The most recent circumscription of Artocarpus recognized four subgenera (Table 1, Fig. 1) and was based on two gene regions and approximately 50% of taxa (Zerega et al. 2010). The subgenera are distinguished by phyllotaxy, the degree of fusion between adjacent pistillate flowers, and the position of inflorescences on the tree: axillary (from a leaf-joint on a small twig) or cauliflorous (from the trunk or a main brank).
Table 1.
A summary of Artocarpus taxonomy following Zerega et al. (2010) at the subgeneric level, and Jarrett (1959–1960) at the section and series level
| Subgenus | Section | Series | Species | Monophyletic |
|---|---|---|---|---|
| Artocarpus | Yes, if A. sepicanus is excluded | |||
| Artocarpus | Yes | |||
| Angusticarpi | Artocarpus lowii, A. montanus*, A. teijsmannii | No. | ||
| Incisifolii | [A. altilis, A. bergii*, A. camansi, A. horridus A. mariannensis], [A. blancoi, A. multifidus, A. pinnatisectus, A. treculianus] | No, but it consists of two monophyletic clades (separated by brackets to the left) defined by geography | ||
| Rugosi | A. corneri*,, A. elasticus, A. jarrettiae*, A. excelsus*, A. kemando, A. maingayi, A. obtusus*, A. scortechinii A. sericicarpus, A. sumatranus, A. tamaran | In most analyses, yes if A. lowii, and A. teijsmannii are included | ||
| Duricarpus | Yes, if A. hirsutus and A. nobilis are included | |||
| Asperifolii | A. brevidpedunculatus, A. chama, A. hispidus, A. hirsutus, A. melinoxylus, A. nobilis, A. odoratissimus, A. rigidus. | Yes, if A. hirsutus and A. nobilis are included, and A. brevipedunculatus is excluded | ||
| Laevifolii | A. anisophyllus, A. lanceifolius, A. sarawakensis* | Yes, if A. sarawakensis and A. brevipedunculatus are included | ||
| Unplaced | A. sepicanus | |||
| Cauliflori | A. annulatus*, A. heterophyllus, A. integer | Yes | ||
| Pseudojaca | Yes, if A. altissimus is excluded. | |||
| Glandulifolium | A. altissimus | Yes | ||
| Pseudojaca | Clavati | A. gongshanensis*, A. hypargyraeus, (A. nanchuanensis), (A. nigrifolius), A. petelotii, A. pithecogallus*, A. styracifolius | Yes, if A. tonkinensis is included. | |
| Peltati |
A. borneensis,
 A. dadah,
A. dadah,
 A. fretessii
A. fretessii
 (including A. albobrunneus), A. fulvicortex, A. glaucus, A. gomezianus, A. griffithii,
(including A. albobrunneus), A. fulvicortex, A. glaucus, A. gomezianus, A. griffithii,
 A. humilis,
A. humilis,
 A. lacucha, A. lamellosus
A. lacucha, A. lamellosus
 (=A. nitidus subsp. nitidus), A. longifolius, A. ovatus, (=A. nitidus subsp. nitidus), A. longifolius, A. ovatus, A. parvus A. parvus (=A. nitidus subsp. lingnanensis), A. primackii,* A. reticulatus, A. rubrosoccatus, A. rubrovenius, A. subrotundifolius, A. thailandicus,* A. tomentosulus, A. tonkinensis, A. vrieseanus, A. xanthocarpus, A. zeylanicus (=A. nitidus subsp. lingnanensis), A. primackii,* A. reticulatus, A. rubrosoccatus, A. rubrovenius, A. subrotundifolius, A. thailandicus,* A. tomentosulus, A. tonkinensis, A. vrieseanus, A. xanthocarpus, A. zeylanicus
|
Yes, if A. tonkinensis is excluded | ||
| Prainea | A. frutescens, A. limpato, A. papuanus, (A. scandens) | Yes | ||
Note: Species marked with an asterisk (*) were described after Jarrett’s revision; we have generally placed them into taxonomic divisions based on the phylogeny presented in this study. Species marked with “n” were previously included in A. nitidus by; those marked with “l” in A. lacucha; and those marked with “g” in A. gomezianus. Species in parentheses were not included in the phylogeny.
The estimated crown age of Artocarpus is approximately 40.07 (29.8–50.81) Ma (Williams et al. 2017), and the genus is associated with an ancient whole-genome duplication (Gardner et al. 2016). The estimated crown age of subgenus Artocarpus is 29.61 Ma (22.33–37.49), whereas it is 18.31 Ma (12.89–24.45) for subgenus Pseudojaca. Widespread interspecific hybridization has not been documented in Artocarpus except between A. altilis (breadfruit) and its Micronesian wild relative A. mariannensis Trécul (Zerega et al. 2005, 2015).
A well-sampled phylogenetic framework for Artocarpus is necessary to inform future taxonomic revision and to clarify relationships within this important genus, in particular the relationships between crop species and their wild relatives, whose conservation is a priority (Castañeda-Álvarez et al. 2016). In this study, we used near-complete (80/83) taxon sampling (at the subspecies level or above) in Artocarpus to reconstruct the most data-rich phylogeny to date for Artocarpus, taking into account the impact of paralogs, codon partitions, noncoding sequences, and analysis method (species tree vs. concatenated supermatrix) on phylogenetic reconstruction in order to develop a truly robust phylogenetic hypothesis. We also used this data set to improve the target capture assembly pipeline HybPiper, which is now optimized for accurately scaffolding small disconnected contigs resulting from degraded DNA. The objectives of the study were to (i) use broad sampling from silica-dried material and herbarium specimens over 100 years old to achieve near-complete taxon sampling for Artocarpus; (ii) test the monophyly of the current taxonomic divisions within Artocarpus to provide a phylogenetic framework for future studies on the taxonomy, conservation, and ecology of the genus; and (iii) examine the impact of paralogs, partitions, and analysis method on phylogenetic reconstruction.
Materials and Methods
A summary of our methods follows. Further details, including protocol modifications for herbarium material and software parameters, can be found in Appendix 1.
Data Accessibility
Raw reads have been deposited in GenBank (BioProject no. PRJNA322184), and alignments and trees have been deposited in the Dryad Data Repository (https://orcid.org/0000-0003-1133-5167). HybPiper and related scripts used in this study are available at https://github.com/mossmatters/HybPiper and https://github.com/mossmatters/phyloscripts.
Taxon Sampling
We sampled all Artocarpus taxa at the subspecies level or above (Jarrett 1959b, 1960, 1975; Zhengyi and Xiushi 1989; Kochummen 1998; Berg 2005; Gardner et al. 2020) and nine taxa of questionable affinities, replicating sampling across geographic or morphological ranges when possible, for a total of 167 ingroup samples belonging to 83 names. Outgroups included one species per genus in the Neotropical Artocarpeae and the sister tribe Moreae. Samples came from field collections preserved in silica gel, botanic gardens, and herbaria (up to 106 years old), totaling 179 samples (Supplementary Table S1 available on dryad at https://doi.org/10.5061/dryad.1rn8pk0pt).
Sample Preparation and Sequencing
DNA extracted from ca. 0.5 cm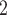 of leaf tissue was quantified on a Qubit fluorometer (Invitrogen, Life Technologies, CA, USA) and assessed on an agarose gel or a High-Sensitivity DNA Assay on a BioAnalyzer 2100 (Agilent). Samples with an average fragment size of
of leaf tissue was quantified on a Qubit fluorometer (Invitrogen, Life Technologies, CA, USA) and assessed on an agarose gel or a High-Sensitivity DNA Assay on a BioAnalyzer 2100 (Agilent). Samples with an average fragment size of  500 bp were sonicated to ca. 550bp using a Covaris M220 (Covaris, Wobum, MA, USA), and libraries were prepared with the Illumina TruSeq Nano HT DNA Library Preparation Kit (Illumina, San Diego, CA, USA) or the KAPA Hyper Prep DNA Library Kit (KAPA, Cape Town, South Africa), using 200 ng of input DNA when possible. Pools of 6–24 libraries were enriched for 333 phylogenetic markers, each with a targeted region between 504 and 2166-bp long (Gardner et al. 2016) with a MYbaits kit (MYcroarray, Ann Arbor, MI, USA) and reamplified with 14 PCR cycles. Sequencing took place on an Illumina MiSeq (
500 bp were sonicated to ca. 550bp using a Covaris M220 (Covaris, Wobum, MA, USA), and libraries were prepared with the Illumina TruSeq Nano HT DNA Library Preparation Kit (Illumina, San Diego, CA, USA) or the KAPA Hyper Prep DNA Library Kit (KAPA, Cape Town, South Africa), using 200 ng of input DNA when possible. Pools of 6–24 libraries were enriched for 333 phylogenetic markers, each with a targeted region between 504 and 2166-bp long (Gardner et al. 2016) with a MYbaits kit (MYcroarray, Ann Arbor, MI, USA) and reamplified with 14 PCR cycles. Sequencing took place on an Illumina MiSeq (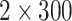 bp, v3) in runs of 30–99 samples.
bp, v3) in runs of 30–99 samples.
Sequence Quality Control and Analyses
In addition to samples prepared for this study, our analyses included reads from all Artocarpus samples from Gardner et al. (2016) as well as the original 333 orthologs from Morus notabilis C.K.Schneid. described by Johnson et al. (2016). Demultiplexed and adapter-trimmed reads were quality trimmed using Trimmomatic 0.39 (Bolger et al. 2014) and assembled with HybPiper 1.2 (Johnson et al. 2016), which represents a compromise between read mapping and de novo assembly and combines local de novo assemblies with scaffolding based on a reference coding sequence (Johnson et al. 2016, 2019). We used the Moraceae reference from Kates et al. (2018), supplemented with additional Artocarpus taxa representing all subgenera. This reference contained the original orthologs from Gardner et al. (2016) in addition to the paralogs identified in Artocarpus by Johnson et al. (2016); paralogs were treated as separate loci and were used for ingroup assemblies only.
HybPiper output used here includes (i) the predicted coding sequence for each target gene (“exon”) and (ii) for the ingroup only, the entire contig assembled for each gene, including noncoding intronic or flanking intergenic sequences (“supercontig”). To reduce bias from sequencing errors, assemblies were masked to remove positions covered by fewer than two reads (Li and Durbin 2009; Li et al. 2009; Quinlan and Hall 2010; Broad Institute 2016). To reduce noise associated with high amounts of missing data, within each gene alignment we removed samples whose exon sequences were less than 150 bp or 20% of the average sequence length for that gene, and samples with fewer than 100 genes after filtering were excluded entirely.
For “exon” sequences, we created in-frame alignments using MACSE 1.02 (Ranwez et al. 2011). For supercontig output, we used MAFFT 7.211 for alignment (–maxiter 1000) (Katoh and Standley 2013). We trimmed alignments to remove columns with  75% gaps using Trimal (Capella-Gutiérrez et al. 2009). Finally, we built gene trees from the exon alignments using FastTree (Price et al. 2009) and visually inspected them for long internal branches to identify alignments containing obvious improperly sorted paralogous sequences; alignments were visually inspected with AliView (Larsson 2014), and 12 genes were discarded, resulting in a final set of 517 genes, including all of the original 333 genes plus 184 paralogs.
75% gaps using Trimal (Capella-Gutiérrez et al. 2009). Finally, we built gene trees from the exon alignments using FastTree (Price et al. 2009) and visually inspected them for long internal branches to identify alignments containing obvious improperly sorted paralogous sequences; alignments were visually inspected with AliView (Larsson 2014), and 12 genes were discarded, resulting in a final set of 517 genes, including all of the original 333 genes plus 184 paralogs.
We used the trimmed alignments to create three data sets:
CDS: exon alignments (in frame), not partitioned by codon position;
Partitioned CDS: exon alignments (in frame), partitioned by codon position; and
CDS+noncoding: supercontig alignments, not partitioned within genes
Each data set was each analyzed with and without paralogs, using the following two methods, for a total of 12 analyses (Table 2): (A) Concatenated supermatrix: all sequences concatenated and partitioned by gene (or by gene and codon, depending on the data set) and analyzed using RAxML 10 (Stamatakis 2006) under the GTRCAT model with 200 rapid bootstrap replicates; (B) Species tree: each gene alignment analyzed using RAxML 10 under GTRCAT with 200 rapid bootstrap replicates. Nodes with  33% support were collapsed using SumTrees 4.3.0 (Sukumaran and Holder 2010), and the resulting trees were used to estimate a species tree with ASTRAL-III 5.5.6 (Mirarab and Warnow 2015), calculating bootstrap (-r, 160) and quartet support (-t 1) for each node (Mirarab and Warnow 2015; Zhang et al. 2017). We used SumTrees to calculate the proportion of gene trees supporting each split; however, quartet support is less sensitive to occasional out-of-place taxa than raw gene-tree support. Attempts to produce a partitioned “supercontig” data set were not successful, because aligning noncoding sequences separately produced unreliable alignments (Appendix 2).
33% support were collapsed using SumTrees 4.3.0 (Sukumaran and Holder 2010), and the resulting trees were used to estimate a species tree with ASTRAL-III 5.5.6 (Mirarab and Warnow 2015), calculating bootstrap (-r, 160) and quartet support (-t 1) for each node (Mirarab and Warnow 2015; Zhang et al. 2017). We used SumTrees to calculate the proportion of gene trees supporting each split; however, quartet support is less sensitive to occasional out-of-place taxa than raw gene-tree support. Attempts to produce a partitioned “supercontig” data set were not successful, because aligning noncoding sequences separately produced unreliable alignments (Appendix 2).
Table 2.
Summary of analyses performed
| Analysis | Data set | Method |
|---|---|---|
| exon.noparalogs* | Coding sequences, without paralogs | Supermatrix, RAxML, GTRCAT, partitioned by gene |
| exon.codon.noparalogs* | Coding sequences, without paralogs | Supermatrix, RAxML, GTRCAT, partitioned by gene and codon position |
| exon* | Coding sequences, with paralogs | Supermatrix, RAxML, GTRCAT, partitioned by gene |
| exon.codon* | Coding sequences, with paralogs | Supermatrix, RAxML, GTRCAT, partitioned by gene and codon position |
| supercontig.noparalogs* | Coding and noncoding sequences, without paralogs | Supermatrix, RAxML, GTRCAT, partitioned by gene |
| supercontig* | Coding and noncoding sequences, with paralogs | Supermatrix, RAxML, GTRCAT, partitioned by gene |
| astral.exon.noparalogs* | Coding sequences, without paralogs | ASTRAL species tree, based on RAxML gene trees estimated under GTRCAT |
| astral.exon.codon.noparalogs* | Coding sequences, without paralogs | ASTRAL species tree, based on RAxML gene trees estimated under GTRCAT and partitioned by codon position |
| astral.exon* | Coding sequences, with paralogs | ASTRAL species tree, based on RAxML gene trees estimated under GTRCAT |
| astral.exon.codon* | Coding sequences, with paralogs | ASTRAL species tree, based on RAxML gene trees estimated under GTRCAT and partitioned by codon position |
| astral.supercontig.noparalogs* | Coding and noncoding sequences, without paralogs | ASTRAL species tree, based on RAxML gene trees estimated under GTRCAT |
| astral.supercontig* | Coding and noncoding sequences, with paralogs | ASTRAL species tree, based on RAxML gene trees estimated under GTRCAT |
| exon.gamma | Coding sequences, with paralogs | Supermatrix, RAxML, GTRGAMMA, partitioned by gene |
| exon.codon.gamma | Coding sequences, with paralogs | Supermatrix, RAxML, GTRGAMMA, partitioned by gene an codon position |
| astral.exon.gamma | Coding sequences, with paralogs | ASTRAL species tree, based on RAxML gene trees estimated under GTRGAMMA |
| astral.exon.codon.gamma | Coding sequences, with paralogs | ASTRAL species tree, based on RAxML gene trees estimated under GTRGAMMA and partitioned by codon position |
| astral.exon.iq | Coding sequences, with paralogs | ASTRAL species tree, based on IQtree gene trees employing the best model for each gene |
| astral.supercontig.iq | Coding and noncoding sequences, with paralogs | ASTRAL species tree, based on IQtree gene trees employing the best model for each gene |
Note: The 12 analyses comprising the “main analysis” are marked with asterisks.
The GTRCAT model was used for the main analyses because it is the generally applicable model recommended for RAxML analyses involving more than 50 taxa (Stamatakis 2006). To test whether substitution models impacted tree inference, we repeated the exon analyses using the GTRGAMMA model, which like GTRCAT allows for rate heterogeneity but is more computationally intensive. We also inferred gene trees using IQTree 2.0 (Minh et al. 2013), with 1000 ultra-fast bootstrap replicates (Hoang et al. 2018). We evaluated substitution models using ModelFinderPlus (Kalyannamoorthy et al. 2017) and calculated maximum-likelihood gene trees using the best fit model selected via the Bayesian information criterion. We then inferred species trees using ASTRAL as described above. Supermatrix analyses took place on the CIPRES Science Gateway (Miller et al. 2010), and all others took place on a cluster at the Chicago Botanic Garden, except for the IQTree analyses, which took place on the Texas Tech High-Performance Computing Cluster. Most processes were run in parallel using GNU Parallel (Tange 2018).
To summarize the overall bootstrap support of each tree with a single statistic, we calculated “percent resolution,” which represents the proportion of bipartitions with  50% bootstrap support (Kates et al. 2018). We visualized trees using FigTree 1.4.3 (Rambaut 2016) and analyzed and compared trees in R 3.5.1 (R Core Development Team 2008) using ape 5.2 (Paradis et al. 2004), phytools 0.6-60 (Revell 2012), Lattice 0.20-38 (Sarkar 2008), and Phangorn 2.4.0 (Schliep 2011). Analyses included analyses of differences in topologies and pairwise Robison-Foulds (RF) distances (the sum of disagreeing bipartitions) for all trees.
50% bootstrap support (Kates et al. 2018). We visualized trees using FigTree 1.4.3 (Rambaut 2016) and analyzed and compared trees in R 3.5.1 (R Core Development Team 2008) using ape 5.2 (Paradis et al. 2004), phytools 0.6-60 (Revell 2012), Lattice 0.20-38 (Sarkar 2008), and Phangorn 2.4.0 (Schliep 2011). Analyses included analyses of differences in topologies and pairwise Robison-Foulds (RF) distances (the sum of disagreeing bipartitions) for all trees.
Results
Sequencing and Assembly
Of the 179 sequenced accessions, 164 resulted in assemblies with at least 25 genes (Supplementary Fig. S1 and Table S1 available on dryad), including all attempted taxa except for Artocarpus nigrifolius C.Y.Wu and A. nanchuanensis, C.Y.Wu, two species closely allied to A. hypargyreus Hance, which was assembled, and A. scandens Miq. sensu Jarrett, considered conspecific with A. frutescens Becc. by Berg et al. (2006), which was also assembled. Less successful samples generally had few reads and may have been out-competed by other samples during hybridization, reamplification, or both. Fewer reads were also associated with shorter assembled sequences (Supplementary Fig. S1 available on dryad). Only samples with at least 100 genes were used for phylogenetic analyses, resulting in the loss of five additional samples and one taxon, Artocarpus reticulatus Miq. Adding the Morus notabilis sequences resulted in a final data set of 160 samples representing 80 out of 83 named Artocarpus taxa at the subspecies/variety level or above (96%) and nine taxa of uncertain affinity.
Overall, samples collected more recently showed improved sequencing results (Supplementary Fig. S2 available on dryad), primarily because the majority of samples collected since 2000 were dried on silica gel. Whether a sample was dried on silica gel was significantly associated with increased gene length as a percentage of average length (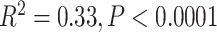 ) and to a lesser extent with the total number of genes recovered (
) and to a lesser extent with the total number of genes recovered (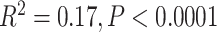 ). All 16 unsuccessful (
). All 16 unsuccessful (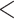 25 genes) assemblies were taken from herbarium sheets (collected between 1917 and 1997), rather than silica-dried material. Among 67 successfully assembled herbarium samples, younger age was associated with increased gene length, although the model was a poor fit (
25 genes) assemblies were taken from herbarium sheets (collected between 1917 and 1997), rather than silica-dried material. Among 67 successfully assembled herbarium samples, younger age was associated with increased gene length, although the model was a poor fit (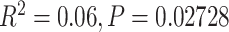 ), but not with an increase in the number of genes recovered (
), but not with an increase in the number of genes recovered (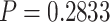 ) (Supplementary Fig. S2 available on dryad). By the same token, we observed a decrease in average DNA fragment size in older samples (Supplementary Fig. S3 available on dryad). Lowering the maximum assembly k-mer values for herbarium samples with under 400 genes increased recovery by an average of 20 genes.
) (Supplementary Fig. S2 available on dryad). By the same token, we observed a decrease in average DNA fragment size in older samples (Supplementary Fig. S3 available on dryad). Lowering the maximum assembly k-mer values for herbarium samples with under 400 genes increased recovery by an average of 20 genes.
Gene recovery was high; the average sample (of the 160 passing the final filter) had sequences for 448/517 genes (87%). The median assembled gene had 2 exons (mean: 3.4; 25–75%: 2–3). In the final filtered data set of 333 genes, the average gene had exon sequences for 151/160 samples (94%, range 57–160, median 154) and noncoding sequences for 130 (81%, range 49–151, median 131). For the 184 paralogs, the average gene in the final filtered data set had exon sequences for 117/160 samples (73%, range 32–148, median 126) and intron sequences for 101 (63%, range 30–132, median 110) (Supplementary Table S2 available on dryad). The supermatrix of trimmed exon alignments for the primary 333 genes contained 407,310 characters; and the full set of 517 exon alignments, including 184 paralogs, contained 569,796 characters. The supermatrix of 333 trimmed supercontig alignments contained 813,504 characters, and the full set of 517 genes contained 1,181,279 characters. The full set of exon alignments had 21% gaps or undetermined characters, whereas the full set of supercontig alignments was 36.87% gaps or undetermined characters.
Phylogenetic Disagreement
A strict consensus of the 12 phylogenetic trees under GTRCAT (henceforth: “main analysis”) had 100/159 (63%) nodes resolved (mean RF distance 53), revealing agreement in backbone relationships between the major subgenera but substantial disagreement at shallower nodes (Fig. 2). The six ASTRAL phylogenies differed little from one another, whereas supermatrix analyses had somewhat greater divergence (Fig. 3, Supplementary Table S3 available on dryad).
Figure 2.

Strict consensus of all 12 main-analysis trees (excluding only those analyses in which exons and introns were aligned separately, for reasons discussed in the text). Infrageneric taxa are labeled according to Jarrett’s (1959b, 1960) taxonomic divisions, as modified by Zerega et al. (2010) and this study. Recently described taxa that were split from older taxa recognized by Jarrett are classified according to Jarrett’s species concepts. Labels to the right of the tree denote major nonmonophyletic taxonomic divisions.
Figure 3.

Plot showing the first two axes of a PCA analysis of Robinson-Foulds (RF) distances between all 12 main analyses.
Partitions and model selection
In exon data sets, partitioning by codon position (Fig. 2, Supplementary Figs. S5–S8 available on dryad) had little impact on final topology, with only a single within-species rearrangement (RF 4), but in the ASTRAL analysis, partitioning by codon position caused Artocarpus sepicanus 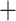 A. altissimus to form a grade rather than a clade, as in all other analyses (RF 12). The choice of model (GTRCAT vs. GTRGAMMA) also produced only minor changes (Supplementary Figs. S9–S12 available on dryad). For the two sets of gene trees estimated using IQtree, generally simpler models than GTRCAT or GTRGAMMA were selected; nevertheless, the resulting ASTRAL tree showed only minor changes at shallow depths (Supplementary Figs. S13 and S14 available on dryad).
A. altissimus to form a grade rather than a clade, as in all other analyses (RF 12). The choice of model (GTRCAT vs. GTRGAMMA) also produced only minor changes (Supplementary Figs. S9–S12 available on dryad). For the two sets of gene trees estimated using IQtree, generally simpler models than GTRCAT or GTRGAMMA were selected; nevertheless, the resulting ASTRAL tree showed only minor changes at shallow depths (Supplementary Figs. S13 and S14 available on dryad).
Paralogs
Addition of paralogs led to slightly more disagreement (Fig. 2, Supplementary Figs. S15–S18 and Table S3 available on dryad). In the exon data set, changes to the positions of Artocarpus parvus Gagnep. ( A. nitidus Trécul subp. lingnanensis (Merr.) F.M.Jarrett) and A. gomezianus Wall. ex Tréc. affected the backbone of ser. Peltati F.M.Jarrett, subg. Pseudojaca, in the supermatrix analysis (RF 58); in the ASTRAL analysis, there were fewer rearrangements, mainly in the same clade (RF 20). However, disagreement was reduced when noncoding sequences were included (supermatrix RF 22; ASTRAL RF 8).
A. nitidus Trécul subp. lingnanensis (Merr.) F.M.Jarrett) and A. gomezianus Wall. ex Tréc. affected the backbone of ser. Peltati F.M.Jarrett, subg. Pseudojaca, in the supermatrix analysis (RF 58); in the ASTRAL analysis, there were fewer rearrangements, mainly in the same clade (RF 20). However, disagreement was reduced when noncoding sequences were included (supermatrix RF 22; ASTRAL RF 8).
Introns
Inclusion of noncoding sequences (Fig. 2, Supplementary Figs. S19–S22 and Table S3 available on dryad) led to similar amounts of disagreement, with rearrangements at the series level in subg. Pseudojaca and subg. Artocarpus. Disagreement was greater in the supermatrix analyses (no paralogs) (RF 62) than in ASTRAL analyses (RF 36). Addition of paralogs reduced disagreement in both cases (supermatrix RF 38; ASTRAL RF 26).
Analysis
The greatest differences among the 12 trees were between ASTRAL and supermatrix trees (Figs. 2, 4, Supplementary Figs. S23–S25 and Table S3 available on dryad), with a mean RF distance between the six supermatrix trees and six ASTRAL trees of 78. Again, addition of noncoding regions or paralogs reduced disagreement between supermatrix and ASTRAL analyses; average RF distance for exons/no-paralogs was 85, exons 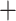 paralogs 77, supercontig/no-paralogs 71, and supercontigs + paralogs 70. Agreement was higher among ASTRAL trees (mean RF 21, 138/159 nodes in agreement) than among supermatrix trees (mean RF 48, 116/159 nodes in agreement). Differences (RF 66) between ASTRAL and supermatrix analyses for the full data set (supercontigs
paralogs 77, supercontig/no-paralogs 71, and supercontigs + paralogs 70. Agreement was higher among ASTRAL trees (mean RF 21, 138/159 nodes in agreement) than among supermatrix trees (mean RF 48, 116/159 nodes in agreement). Differences (RF 66) between ASTRAL and supermatrix analyses for the full data set (supercontigs 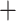 paralogs for all genes) at the species level can be ascribed to mostly minor repositionings within subclades involving two outgroup taxa (Bagassa guianensis Aubl. and Batocarpus orinoceros H. Karst.) and 14 ingroup taxa (Fig. 4).
paralogs for all genes) at the species level can be ascribed to mostly minor repositionings within subclades involving two outgroup taxa (Bagassa guianensis Aubl. and Batocarpus orinoceros H. Karst.) and 14 ingroup taxa (Fig. 4).
Figure 4.

Comparison between the full-data set (supercontigs for all genes) supermatrix and ASTRAL trees, with disagreeing branches highlighted, showing moderate disagreement at shallow phylogenetic depths but complete agreement at deeper nodes. Left: maximum-likelihood tree based on all supercontigs, partitioned by gene, including all paralogs; all branch lengths are proportional to mean substitutions per site. Right: ASTRAL tree based on all supercontigs; internal branch lengths are proportional to coalescent units; terminal branch lengths were arbitrarily assigned to improve visualization. Pie charts at nodes represent the proportion of gene trees supporting each split, and numbers represent bootstrap support.
Phylogenetic Resolution
Percent resolution based on bootstrap values was 90–95% for all supermatrix trees in the main analysis and did not differ materially between analyses. Among ASTRAL trees, resolution was between 84% and 97% for all analyses. By slight margins, the best-resolved trees for both supermatrix and ASTRAL analyses were those based on the largest data set (Supplementary Table S3 available on dryad). Resolution based on quartet support for ASTRAL trees was between 57% and 60%, reflecting substantial gene-tree discordance (Supplementary Table S4 available on dryad, Fig. 4). For resolution measured by gene-tree support (percentage of nodes supported by at least half of the 517 gene trees), scores ranged from 17% to 24%. In general, analyses including paralogs had reduced gene-tree support, and trees based on supercontigs with no paralogs had the highest scores (24% for both ASTRAL and supermatrix).
More detailed analysis of differences between species trees based on the exon and supercontig data sets revealed that even if final species trees had similar resolution, supercontig trees were based on more information because the gene trees were significantly more informative. Inclusion of noncoding sequences significantly increased the number of splits with over 30% support (mean of +8). Because nodes under 30% were collapsed for species tree estimation, the species tree in the supercontig data set was based on 9% more splits across the 517 gene trees (total of 51,307) than the species tree in the exon data set (total 47,067). These patterns persisted in no-paralog data sets (mean increase in nodes over 30%: 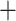 9; overall difference in splits for 333 collapsed trees: 36,373 vs. 33,404 or 9%). Because addition of noncoding sequences also increased agreement between supermatrix and ASTRAL analyses (see above), this suggests at least some disagreement between supermatrix and species-tree analyses arises not only from incomplete lineage sorting but also from lack of resolution at the gene-tree level, something that has also been observed at deeper phylogenetic scales (Pease et al. 2018). Although we used low bootstrap support as an indicator of poor resolution in single-locus gene trees, we again caution against over-interpreting high bootstrap values, particularly in multilocus trees.
9; overall difference in splits for 333 collapsed trees: 36,373 vs. 33,404 or 9%). Because addition of noncoding sequences also increased agreement between supermatrix and ASTRAL analyses (see above), this suggests at least some disagreement between supermatrix and species-tree analyses arises not only from incomplete lineage sorting but also from lack of resolution at the gene-tree level, something that has also been observed at deeper phylogenetic scales (Pease et al. 2018). Although we used low bootstrap support as an indicator of poor resolution in single-locus gene trees, we again caution against over-interpreting high bootstrap values, particularly in multilocus trees.
Phylogenetic Relationships
The genus Artocarpus was monophyletic in all 12 main analyses, as were subgenera Cauliflori (F.M.Jarrett) Zerega and Prainea (King) Zerega, Supardi & Motley (Table 1). Subgenus Artocarpus was monophyletic excluding A. sepicanus Diels, and subgenus Pseudojaca was monophyletic excluding A. altissimus (Miq.) J.J.Sm. In 10/12 analyses, A. sepicanus and A. altissimus formed a clade sister to subgenera Cauliflori and Artocarpus; however, in codon-partitioned ASTRAL analyses, they formed a grade in the same position (Supplementary Figs. S8 and S10 available on dryad). The backbone phylogeny was otherwise identical in all 12 trees: subgenus Prainea was sister to all other Artocarpus, which comprised a grade in this order: subgenus Pseudojaca, A. sepicanus  A. altissimus (usually), followed by subgenus Cauliflori
A. altissimus (usually), followed by subgenus Cauliflori  subgenus Artocarpus. Apart from the monophyly of the genus, which was supported by 61% of gene trees in the complete data set (supercontig, all genes), subgeneric relationships had much less support at the gene-tree level. The position of subg. Prainea was supported by 28% of gene trees; subg. Pseudojaca by 7%, and subgenera Artocarpus/Cauliflori by 4%. Quartet support was substantially higher (Fig. 4).
subgenus Artocarpus. Apart from the monophyly of the genus, which was supported by 61% of gene trees in the complete data set (supercontig, all genes), subgeneric relationships had much less support at the gene-tree level. The position of subg. Prainea was supported by 28% of gene trees; subg. Pseudojaca by 7%, and subgenera Artocarpus/Cauliflori by 4%. Quartet support was substantially higher (Fig. 4).
Within subgenus Artocarpus, both of Jarrett’s sections were monophyletic (excepting A. sepicanus, A. hirsutus Lam., and A. nobilis Thwaites, which she considered anomalous and did not place in sections), but none of the five series were monophyletic. However, series Rugosi F.M.Jarrett, characterized by rugose staminate inflorescences, was “nearly monophyletic” in most analyses, requiring only the inclusion of three nonrugose species (Artocarpus teijsmannii Miq., A. lowii King, and A. excelsus F.M.Jarrett). Members of series Incisifolii F.M.Jarrett, characterized by incised adult leaves, formed two nonsister clades, one in the Philippines and one ranging from Indonesia to Oceania. Within subgenus Pseudojaca, section Pseudojaca was monophyletic (excluding A. altissimus), as was series Clavati F.M.Jarrett—characterized by clavate interfloral bracts. Series Peltati F.M.Jarrett—characterized by peltate interfloral bracts—would be monophyletic if A. tonkinensis A.Chev. were excluded, the latter species being sister to series Clavati in all main analyses.
Most species (for which we included at least two samples) were monophyletic, but several were not monophyletic in any analysis, including Artocarpus treculianus Elmer, A. sarawakensis F.M. Jarrett, A. lanceifolius Roxb., A. rigidus Blume, and A. teijsmannii Miq. Berg’s and Jarrett’s broad concept of A. nitidus (including A. borneensis Merr., A. griffithii (King) Merr., A. humilis Becc., A. lamellosus Blanco, A. parvus, A. vrieseanus Miq. var. subsessilis F.M.Jarrett, and A. xanthocarpus Merr.) and A. lacucha Roxb. ex Buch.-Ham. (including A. dadah Miq., A. fretessii Teijm. & Binn. ex Hassk., A. lacucha, A. ovatus Blanco, A. vrieseanus Miq. var. refractus (Becc.) F.M.Jarrett and A. vrieseanus Miq. var. papillosus F.M.Jarrett) were also not monophyletic. The type of Artocarpus teijsmannii Miq. ssp. subglabrus C.C. Berg was sister to A. sepicanus in all analyses, whereas ssp. teijsmannii was within subgenus Artocarpus, series Rugosi.
The neotropical Artocarpeae formed a clade sister to Artocarpus in all 12 trees. Although Batocarpus was monophyletic in all supermatrix analyses, neither Batocarpus nor Clarisia was monophyletic in any ASTRAL tree.
Discussion
Taxon Sampling
Although other studies have successfully applied target enrichment to recover sequences from herbarium and museum material (Guschanski et al. 2013; Hart et al. 2016), to our knowledge, this is among the first to use herbarium collections to achieve near-complete taxon sampling in a tropical plant genus of this size (ca. 70 spp.). The ability to successfully sequence herbarium material was indispensable for this study. For 34 of 90 (38%) ingroup taxa in the final analyses (including subspecies and the nine individuals of uncertain affinities), we did not have access to any fresh or silica-dried material and relied exclusively on herbarium specimens. In some cases, the only readily available samples were approximately 100 years old (e.g. A. treculianus sensu stricto (coll. 1910–1911: 369–370 genes recovered after filtering), A. nigrescens Elmer (coll. 1919: 431 genes), and A. pinnatisectus (type coll. 1913: 425 genes)). Although old samples had a lower success rate than silica-dried material, and sample degradation contributed to shorter assembled contigs, age alone was not significantly associated with recovery of fewer loci. Instead, the number of reads obtained was the most important factor in determining the number of loci recovered (Supplementary Fig. S2 available on dryad). We hope these results encourage others to aim for complete taxon sampling with minimally destructive sampling from natural history collections when newly collected material is not available, so long as identifications can be confirmed by taxonomic experts. We note that during the course of this study, we corrected a substantial number of misidentifications.
Although fieldwork remains among the most important aspects for systematic biology studies, phylogenetic reconstruction can benefit dramatically from incorporation of DNA from museum specimens. In this study, we successfully sequenced several DNA extractions from museum specimens that had been unusable for Sanger sequencing because PCR amplification failed (presumably due to small fragment size) (Zerega et al. 2010; Williams et al. 2017). The ability to achieve near-complete taxon sampling from museum material will open new opportunities for phylogeny-based analyses of clades with species that are difficult to collect, rare, or extinct, but present in herbarium collections. Our results suggest that near-complete taxon sampling can improve consistency between analyses, resulting in more reliable phylogenies. A previous study (Kates et al. 2018) using a smaller data set of 22 Artocarpus species, found substantial disagreement between analyses in the backbone phylogeny of Artocarpus. Here, all 12 main analyses recovered almost identical backbones, disagreeing occasionally regarding positions of A. altissimus and A. sepicanus. Others have likewise found that missing taxa can substantially impact phylogenetic reconstructions (de la Torre-Bárcena et al. 2009). Robust taxon sampling also has serious implications for biodiversity conservation. Artocarpus treculianus is listed as Vulnerable by IUCN (World Conservation Monitoring Centre 1998). Due to availability of sequences from century-old herbarium sheets, we now know that this species is not monophyletic and that the synonymized A. nigrescens Elmer should probably be reinstated. Splitting a Vulnerable species into two will result, at the very least, in two Vulnerable species, but narrower circumscriptions may also increase the threat level. Availability of material from collections has also revealed new species including A. bergii E.M. Gardner, Zerega & Arifiani (Gardner et al., in press), a close ally of breadfruit from the Maluku Islands and A. montanus E.M. Gardner & Zerega (Gardner et al. 2020), a montane species endemic to Vietnam.
Impact of Various Analysis Methods
Analyzing data in different ways can help produce more robust phylogenetic hypotheses by revealing which relationships are independent of analysis method. Of the variants we tested, codon partitioning had the smallest impact, resulting in no major topological changes except for the relationship of A. sepicanus and A. altissimus. This is not surprising, as RAxML’s GTRCAT model provides for rate heterogeneity even absent explicit partitioning (Stamatakis 2006). The other comparisons revealed more disagreement, mostly at shallow phylogenetic depths. However, in all cases, disagreement decreased if additional sequences (paralogs or noncoding) were added to a data set. This suggests more data can lead to a certain amount of convergence in analyses, even though simply adding more data to a supermatrix may not improve the accuracy of the resulting species tree (Degnan and Rosenberg 2009).
Based on these results, we conclude that for our study, greater benefit resulted from analyzing more data, in particular noncoding sequences, than from partitioning by codon position, and the same may be true for analyses at similar phylogenetic scales, particularly when methods provide for rate heterogeneity. Moreover, the results of attempts to produce a codon-partitioned supercontig data set suggest that overpartitioning may bias analyses, particularly in the presence of missing data; the ASTRAL analyses of that data set, which effectively had sub-partitions because each gene tree containing three partitions was estimated separately, was more congruent with the main analyses (Appendix 2).
We therefore recommend that when possible, flanking noncoding sequences be included in analyses. The benefits of gene trees with fewer polytomies, and thus more reliable species trees, likely outweigh any minimal advantage gained in partitioning by codon position, at least for a data set like ours. In light of increased congruence between analyses as our data set was enlarged, we suggest using as many loci and as much flanking noncoding sequence as is available, with the caveat to exercise caution with regard to taxa with excessive missing data. The cutoffs we used,  20% of the average sequence length and
20% of the average sequence length and  20% of loci, might be made more stringent, as some inter-analysis disagreement appeared to center around samples with more missing data. We also note that the paralogs in Artocarpus likely result from an ancient whole-genome duplication (Gardner et al. 2016; Williams et al. 2017) and were thus easy to separate for use as additional loci; this may not be the case for paralogs of more recent origin, which should be approached with caution.
20% of loci, might be made more stringent, as some inter-analysis disagreement appeared to center around samples with more missing data. We also note that the paralogs in Artocarpus likely result from an ancient whole-genome duplication (Gardner et al. 2016; Williams et al. 2017) and were thus easy to separate for use as additional loci; this may not be the case for paralogs of more recent origin, which should be approached with caution.
Although adding or extending loci may reduce disagreement between analyses, it may not always increase phylogenetic resolution. A handful of genes may have insufficient informative characters to resolve a phylogeny, and resolution may increase as loci are added, but with hundreds of genes, lack of informative characters is not the problem. Here, consistently high bootstrap values masked substantial gene-tree discordance, which actually increased when paralogous loci were added. Other phylogenomic studies have also found high rates of gene-tree discordance (Degnan and Rosenberg 2009; Wickett et al. 2014; Copetti et al. 2017; Pease et al. 2018; Liu et al. 2019). Because gene-tree discordance can result from biological processes such as incomplete lineage sorting or ancient hybridization, it may reflect a lack of phylogenetic resolution, but rather a biological reality that cannot be accurately represented by a single bifurcating tree. Nonetheless, just as bootstrap support can convey a misleading sense of certainty, support measured by the rate of gene-tree support can exaggerate uncertainty. For example, if a gene tree generally supports a clade, but has one out-of-place taxon, perhaps due to an incomplete or erroneous sequence, that gene tree will not be counted as supporting the clade in question. Support measured as the proportion of gene-tree quartets supporting each node, not the frequency of the exact clade being tested, may provide a more realistic measure of support (Sayyari and Mirarab 2016); in our analyses, they were generally lower than bootstrap values but substantially higher than gene-tree support.
Taxonomic Considerations
Our results provide a phylogenetic framework for a taxonomic revision of Artocarpus, currently in progress (Table 1). A summary of taxonomic implications is discussed here, and more details with regard to characters can be found in the Appendix. The subgeneric divisions made by Jarrett (1959a,1959b, 1960) and Zerega et al. (2010) can be maintained with minor modifications to account for the anomalous A. sepicanus and A. altissimus, which in 10/12 main analyses formed a clade. It is curious that these species should be closely allied (having different leaf phyllotaxy and differences in degree of perianth tissue fusion of adjacent pistillate flowers—the defining characters of the subgenera). The disagreement as to their affinity in the codon-partitioned ASTRAL analyses warrants further investigation, raising the possibility that the apparent affinity may be due to long-branch attraction (Roch et al. 2019). The only apparent morphological affinity between them is bifid styles, a moraceous plesiomorphy (Clement and Weiblen 2009), present occasionally in subgenus Artocarpus but unique to A. altissimus in subgenus Pseudojaca.
In addition, the phylogeny supports the broad outlines of Jarrett’s (1959b, 1960) sections, validating her careful morphological and anatomical studies, which built on those of Renner (1907). The sections within subgenus Artocarpus might be maintained with the inclusion of A. hirsutus and A. nobilis in section Duricarpus F.M.Jarrett–an affinity noted by Jarrett (1959b) and Berg et al. (2006). Jarrett noted that those species had characters intermediate between sections Artoarpus and Duriarpus, and indeed, their positions in all main analyses were sister to most of the rest of section Duricarpus.
At the series level within subgenus Artocarpus, a wholesale reconsideration is probably necessary, especially in series Angusticarpi F.M.Jarrett, which never formed a consistent clade or grade. Artocarpus teijsmannii subsp. subglabrus, which differs from A. sepicanus only in petiole characters, appears to be conspecific with the latter. Of special interest in the clade containing A. altilis (breadfruit) are putative new species that are wild relatives of breadfruit (A. bergii, endemic to the Maluku Islands and one accession of uncertain affinity also originating in Maluku and cultivated in the Bogor Botanical Gardens (cf. camansi). The status of Artocarpus horridus F.M.Jarrett is unclear; one accession fell in its expected place together with other samples from the Moluccas, but the position of the other, sister to the entire clade, must be treated with caution, as that sample had among the highest proportions of missing data. Substantial discordance between analyses in the breadfruit clade may reflect hybridization, which has been observed between A. altilis and A. mariannensis and may warrant further investigation in the clade as a whole.
Within subgenus Pseudojaca, to the extent we included multiple accessions per species, our results mostly supported Jarrett’s (1960) revision. The series were largely monophyletic, with the exception of the position of A. tonkinensis (with peltate interfloral bracts) nested within the clade distinguished by clavate interfloral bracts. The ancestral state for interfloral bracts is likely peltate (Clement and Weiblen 2009), so Artocarpus tonkinensis may simply represent a plesiomorphic taxon sister to a derived clade. At the species levels, some taxonomic changes from Berg et al. (2006) are necessary. Those proposed for subgenus Pseudojaca are outlined in Gardner and Zerega (2020) and already reflected in the nomenclature used here. For example, as Williams et al. (2017) found, the five taxa sunk into Artocarpus lacucha by Berg et al. (2006) (A. dadah Miq., A. ovatus Blanco, A. fretesii, A. vrieseanus var. refractus, and A. vrieseanus var. subsessilis) do not belong together. Additionally, the subspecies of A. nitidus ( A. lamellosus) do not form a clade, nor do the subspecies of A. gomezianus; these have been revised accordingly (Gardner and Zerega 2020). However, the varieties of A. vrieseanus sensu Jarrett (1960) form a clade. Discordance between analyses within Pseudojaca may reflect ancient hybridization or the young age of this clade relative to the rest of Artocarpus (Williams et al. 2017), but so far no evidence of widespread hybridization between extant species has come to light; this is another area warranting further investigation.
A. lamellosus) do not form a clade, nor do the subspecies of A. gomezianus; these have been revised accordingly (Gardner and Zerega 2020). However, the varieties of A. vrieseanus sensu Jarrett (1960) form a clade. Discordance between analyses within Pseudojaca may reflect ancient hybridization or the young age of this clade relative to the rest of Artocarpus (Williams et al. 2017), but so far no evidence of widespread hybridization between extant species has come to light; this is another area warranting further investigation.
The Chinese species described since Jarrett’s (1960) revision all belong to Series Clavati. Our sampling did not include A. nanchuanensis, but this species is morphologically similar to A. hypargyreus, and subsequent sequencing after the main analyses were complete confirmed the affinity (Gardner and Zerega 2020). We were unable to successfully sequence A. nigrifolius, but an examination of the type suggests that it is conspecific with A. hypargyreus.
Pending a complete revision, we propose the following adjustments to achieve monophyletic sections: Artocarpus hirsutus and A. nobilis are transferred to sect. Duricarpus, and A. teijsmannii subsp. subglabrus is reduced to the synonomy of A. sepicanus.
Conclusion
We provide a robust phylogenetic framework for Artocarpus, making use of herbarium specimens up to 106 years old to supplement our own collections and achieve near-complete taxon sampling, demonstrating the value of even very old natural history collections in improving phylogenetic studies. Our results will inform future evolutionary and systematic studies of this important group of plants. More generally, the results may guide future analyses of HybSeq data sets, particularly those combining fresh with museum material, by counseling careful attention to data set construction and analysis method to produce the most informative phylogenetic hypotheses.
The increasing availability of phylogenomic data sets has dramatically changed the practice of revisionary systematics. Data sets containing hundreds or thousands of loci produce trees with extremely high statistical support, apparently providing ironclad frameworks for making taxonomic decisions. However, apparent high support for relationships may often be an artifact of the massive number of characters available for phylogenetic inference, masking real uncertainties, revealed only by employing a variety of analytical methods. By the same token, focusing on exclusively conserved coding regions—an inherent feature of some reference-based assembly methods—can result in unnecessarily uninformative gene trees, leading to poor support at the species-tree level. Using a data set with near-complete taxon sampling, we demonstrated that decisions made in how to conduct analyses can substantially affect phylogenetic reconstruction, resulting in discordant phylogenies, each with high statistical support. Employing multiple analytical methods can help separate truly robust phylogenetic relationships from those that only appear to be well-supported but are inconsistent across analyses. Although codon partitioning and model choice did not substantially alter our phylogeny, inclusion of flanking noncoding sequences in analyses significantly increased the number of informative splits at the gene-tree level, resulting ultimately in more robust species trees. In general, increasing the size of data sets, through inclusion of paralogous genes, increased convergence between analysis methods without reducing gene-tree conflict. This likely resulted from biological, not analytical processes; for this reason, we prefer quartet-based scoring methods as the most informative ways of determining support for species trees.
Supplementary Material
Acknowledgments
A study such as this would not have been possible without the careful foundational work of the late F.M. Jarrett and the late C.C. Berg. We thank Postar Miun, Jeisin Jumian, Markus Gubilil, Aloysius Laim, Brono Saludin, Jegong anak Suka, Salang anak Nyegang, Jugah anak Tagi, Wan Nuur Fatiha Wan Zakaria, and Harto for assistance in the field; the Pritzker Laboratory for Molecular Systematics at the Field Museum of Natural History (K. Feldheim) for the use of sequencing facilities; J. Fant, E. Williams, H. Noble, R. Overson, and B. Cooper for assistance in the lab; four anonymous reviewers for helpful comments on the manuscript; Sabah Biodiversity Centre, Chief Conservator of Forests and Deputy Chief Conservator of Forests (Research & Development), Sabah Forestry Department for permission to conduct field research in Sabah (permit nos. JKI4/MBS.r000-2/2(128), JKM/MBS.1000-2/2 (175), and JKM/MBS.1000-2/2 JLD.4 (150); the Sarawak Forest Department for permission to conduct field research in Sarawak (permit no. NCCD.907.4.4(JLD.13)-195 and park permit no. 135/2016); the Sabah Agriculture Department (Au Wai Fong, Jain Linton, Jabi Tananak) and the National Tropical Botanical Garden for access to living collections; and the following herbaria for access to collections for examination and/or sampling: BM, BKF, BO, CAL, CANB, F, IBSC, K, KEP, FTBG, HAST, KUN, L, MIN, MO, NY, P, PNH, SAN, SING, SAR, SNP, U, US.
Appendix 1 – Methods in detail
Taxon Sampling
We sampled all Artocarpus taxa at the subspecies level or above recognized by Jarrett (1959b, 1960), Berg et al. (2006), and Kochummen (1998), all three obsolete species that Jarrett (1959b) sunk into Artocarpus treculianus Elmer, and all of the new species described by Wu and Chang (1989), for a total of 83 named Artocarpus taxa. We also sampled nine taxa of questionable affinities. We replicated samples across geographic or morphological ranges when possible, for a total of 167 ingroup samples. As outgroups, we sampled one member of each genus in the Neotropical Artocarpeae (Batocarpus and Clarisia) and the sister tribe Moreae (Morus L., Streblus Lour., Milicia Sim., Trophis P. Browne, Bagassa Aubl., and Sorocea A. St.-Hil.). We obtained samples from our own field collections preserved in silica gel (from Malaysia, Thailand, Hong Kong, Bangladesh, and India, and from botanic gardens in Indonesia, Malaysia, and Hawai’i, USA) and from herbarium specimens up to 106 years old (from the following herbaria: BM, BO, CHIC, E, F, HAST, HK, K, KUN, L, MO, NY, KEP, S, SAN, SNP, US). In total, we included 179 samples (Supplementary Table S1 available on dryad).
Sample Preparation and Sequencing
We sampled approximately 0.5 cm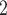 of dried leaf from each sample for DNA extraction. For herbarium specimens, we sampled from a fragment packet when feasible and when it was clear that the material in the fragment packet originated from the specimen on the sheet (something that cannot always be assumed with very old specimens). DNA was extracted using one of three methods; (i) the Qiagen DNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol; (ii) the MoBio PowerPlant Pro DNA Kit, (MoBio Laboratories, Carlsbad, CA, USA); or (iii) a modified CTAB protocol (Doyle and Doyle 1987). For kit extractions, the protocols were modified for herbarium material by extending initial incubation times (Williams et al. 2017) and adding an additional 200
of dried leaf from each sample for DNA extraction. For herbarium specimens, we sampled from a fragment packet when feasible and when it was clear that the material in the fragment packet originated from the specimen on the sheet (something that cannot always be assumed with very old specimens). DNA was extracted using one of three methods; (i) the Qiagen DNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol; (ii) the MoBio PowerPlant Pro DNA Kit, (MoBio Laboratories, Carlsbad, CA, USA); or (iii) a modified CTAB protocol (Doyle and Doyle 1987). For kit extractions, the protocols were modified for herbarium material by extending initial incubation times (Williams et al. 2017) and adding an additional 200 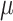 L of ethanol to the column-binding step. CTAB extractions of herbarium specimens, which often had high but impure DNA yields, were cleaned using a 1:1.8:5 ratio of sample, SPRI beads, and isopropanol, the latter added to prevent the loss of small fragments (Lee 2014). For herbarium specimens, we sometimes combined two or more separate extractions in order to accumulate enough DNA for library preparation. We assessed degradation of DNA from herbarium specimens using either an agarose gel or a High-Sensitivity DNA Assay on a BioAnalyzer 2100 (Agilent) and did not sonicate samples whose average fragment size was less than 500 bp. The remaining DNA samples were sonicated to a mean insert size of 550 bp using a Covaris M220 (Covaris, Wobum, MA, USA). Libraries were prepared with either the Illumina TruSeq Nano HT DNA Library Preparation Kit (Illumina, San Diego, CA, USA) or the KAPA Hyper Prep DNA Library Kit following the manufacturer’s protocol, except that reactions were performed in one-third volumes to save reagent costs. We used 200 ng of input DNA when possible; for some samples, input was as low as 10 ng. For herbarium samples with degraded DNA, we usually did not perform size selection, unless there were some fragments that were above 550 bp. We also diluted the adapters from 15 to 7.5
L of ethanol to the column-binding step. CTAB extractions of herbarium specimens, which often had high but impure DNA yields, were cleaned using a 1:1.8:5 ratio of sample, SPRI beads, and isopropanol, the latter added to prevent the loss of small fragments (Lee 2014). For herbarium specimens, we sometimes combined two or more separate extractions in order to accumulate enough DNA for library preparation. We assessed degradation of DNA from herbarium specimens using either an agarose gel or a High-Sensitivity DNA Assay on a BioAnalyzer 2100 (Agilent) and did not sonicate samples whose average fragment size was less than 500 bp. The remaining DNA samples were sonicated to a mean insert size of 550 bp using a Covaris M220 (Covaris, Wobum, MA, USA). Libraries were prepared with either the Illumina TruSeq Nano HT DNA Library Preparation Kit (Illumina, San Diego, CA, USA) or the KAPA Hyper Prep DNA Library Kit following the manufacturer’s protocol, except that reactions were performed in one-third volumes to save reagent costs. We used 200 ng of input DNA when possible; for some samples, input was as low as 10 ng. For herbarium samples with degraded DNA, we usually did not perform size selection, unless there were some fragments that were above 550 bp. We also diluted the adapters from 15 to 7.5 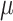 M, and usually performed only a single SPRI bead cleanup between adapter ligation and PCR amplification. Many of these libraries contained substantial amounts of adapter dimer, so we adjusted the post-PCR SPRI bead cleanup ratio to 0.8
M, and usually performed only a single SPRI bead cleanup between adapter ligation and PCR amplification. Many of these libraries contained substantial amounts of adapter dimer, so we adjusted the post-PCR SPRI bead cleanup ratio to 0.8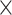 . Libraries were enriched for 333 phylogenetic markers (Gardner et al. 2016) with a MYbaits kit (MYcroarray, Ann Arbor, MI, USA) following the MYbaits manufacturer’s protocol (version 3). Hybridization took place in pools of 6–24 libraries; within each pool, we used equal amounts of all libraries (20–100 ng, as available), and tried to avoid pooling samples with dramatically different phylogenetic distances to the bait sequences (Morus and Artocarpus), as closer taxa can out-compete multiplexed distant taxa in hybridization reactions, as we previously found when pooling Dorstenia L. and Parartocarpus Baill. with Artocarpus (Johnson et al. 2016). We reamplified enriched libraries with 14 PCR cycles using the conditions specified in the manufacturer’s protocol. In some cases, adapter dimer remained even after hybridization; in those cases, we removed it either using a 0.7
. Libraries were enriched for 333 phylogenetic markers (Gardner et al. 2016) with a MYbaits kit (MYcroarray, Ann Arbor, MI, USA) following the MYbaits manufacturer’s protocol (version 3). Hybridization took place in pools of 6–24 libraries; within each pool, we used equal amounts of all libraries (20–100 ng, as available), and tried to avoid pooling samples with dramatically different phylogenetic distances to the bait sequences (Morus and Artocarpus), as closer taxa can out-compete multiplexed distant taxa in hybridization reactions, as we previously found when pooling Dorstenia L. and Parartocarpus Baill. with Artocarpus (Johnson et al. 2016). We reamplified enriched libraries with 14 PCR cycles using the conditions specified in the manufacturer’s protocol. In some cases, adapter dimer remained even after hybridization; in those cases, we removed it either using a 0.7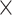 SPRI bead cleanup or, in cases where the library fragments were very short (ca. 200 bp, compared with 144 bp for the dimer), by size-selecting the final pools to
SPRI bead cleanup or, in cases where the library fragments were very short (ca. 200 bp, compared with 144 bp for the dimer), by size-selecting the final pools to 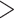 180 bp on a BluePippin size-selector using a 2% agarose gel cassette (Sage Science, Beverley, MA, USA). Pools of enriched libraries were sequenced on an Illumina MiSeq (600 cycle, version 3 chemistry) alongside samples for other studies in three multiplexed runs each containing 30–99 samples.
180 bp on a BluePippin size-selector using a 2% agarose gel cassette (Sage Science, Beverley, MA, USA). Pools of enriched libraries were sequenced on an Illumina MiSeq (600 cycle, version 3 chemistry) alongside samples for other studies in three multiplexed runs each containing 30–99 samples.
Sequence Quality Control and Analyses
Demultiplexing and adapter trimming took place automatically through Illumina BaseSpace (basespace.illumina.com). All reads have been deposited in GenBank (BioProject no. PRJNA322184). Raw reads were quality trimmed using Trimmomatic (Bolger et al. 2014), with a quality cutoff of 20 in a 4-bp sliding window, discarding any reads trimmed to under 30 bp. In addition to the samples sequenced for this study, reads used for assemblies included all Artocarpus samples sequenced in Johnson et al. (2016) (available under the same BioProject number). Common methods for target capture assembly include mapping reads to a reference (Weitemier et al. 2014; Hart et al. 2016) and de novo assemblies (Mandel et al. 2014; Faircloth 2015), but both have drawbacks. Read mapping can result in lost data, particularly indels and noncoding regions, unless a close reference is available. On the other hand, de novo assemblies can also result in lost data if loci cannot be assembled into single scaffolds. A compromise approach, implemented in HybPiper, is to combine local de novo assemblies—which may result in many small contigs per locus—with scaffolding based on a reference coding sequence, which need not be closely related; a reference with less than 30% sequence, typically within the same family or order, will usually suffice (Johnson et al. 2016, 2019). The resulting assemblies thus cover the maximum available portion of each locus, notwithstanding the existence of long gaps, and also make use of all available on-target reads, including introns, not simply those that can be aligned to a reference.
We assembled sequences using HybPiper 1.2, which represented an update of the original pipeline optimized for short reads from highly fragmented DNA from museum specimens. HybPiper’s guided assembly method uses the reference to scaffold localized de novo assemblies. This is particularly advantageous when dealing with very short reads from degraded DNA, because for those samples, reads covering a single exon may assemble into more than one contig. In those cases, HybPiper uses the reference to scaffold and concatenate multiple contigs into a “supercontig” containing the gene of interest as well as any flanking noncoding sequences (Johnson et al. 2016). The new version of HybPiper is optimized to accurately handle many small contigs covering a single gene, deduplicating overlaps and outputting high-confidence predicted coding sequences even in the presence of many gaps caused by fragmentary local assemblies. HybPiper as well as all related scripts used in this study are available at https://github.com/mossmatters/HybPiper and https://github.com/mossmatters/phyloscripts. We generated a new HybPiper reference for this study, using reads from all four subgenera of Artocarpus. Target-enriched reads from A. camansi Blanco (the same individual used for whole-genome sequencing in the original marker development [Gardner et al. 2016], Artocarpus limpato Miq., Artocarpus heterophyllus, and A. lacucha (the latter three from reads sequenced in Johnson et al. 2016) were assembled de novo using SPAdes (Bankevich et al. 2012), and genes were predicted using Augustus (Keller et al. 2011), with Arabidopsis Hehyn. as the reference. Predicted genes were annotated using a BLASTn search seeded with the HybPiper target file of 333 phylogenetic marker genes from Johnson et al. (2016). Paralogs were annotated as follows: genes covering at least 75% of the primary ortholog (labeled “p0” and matching the original targeted A. camansi sequence) were labeled as “paralogs” (“p1,” “p2,” etc.). Genes covering less than 75% of the primary ortholog (labeled “e0”) were labeled as “extras” (“e1,” “e2,” etc.), denoting uncertainty as to whether they are paralogs or merely genes with a shared domain. To avoid the assembly of chimeric paralogs, we did not use the original orthologs to scaffold multiple contigs into single genes; all annotated paralogs were from de novo assembled contigs. Single-copy genes were labeled as “single” in the new reference. We used this new 4-taxon reference to guide all ingroup assemblies, and we used the original set of Morus notabilis targets (Johnson et al. 2016) to guide all outgroup assemblies.
We set the per-gene coverage cutoff to 8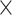 , except for certain low-read samples where gene recovery was improved by lowering the coverage cutoff to 4
, except for certain low-read samples where gene recovery was improved by lowering the coverage cutoff to 4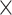 (10 samples) or 2
(10 samples) or 2 (18 samples). HybPiper relies on SPAdes for local de novo assemblies. SPAdes creates several assemblies with different k-mer values, with the maximum estimated from the reads (up to 127 bp), and then merges them into a final assembly. For herbarium samples that initially recovered fewer than 400 genes, we reran HybPiper, manually setting the maximum k-mer values for assembly to 55 instead of allowing SPAdes to automatically set it. To extract noncoding sequences and annotate gene features along assembled contigs, we used the HybPiper script “intronerate.py.” We assessed target recovery success using the get_seq_lengths.py and gene_recovery_heatmap.r scripts from HybPiper.
(18 samples). HybPiper relies on SPAdes for local de novo assemblies. SPAdes creates several assemblies with different k-mer values, with the maximum estimated from the reads (up to 127 bp), and then merges them into a final assembly. For herbarium samples that initially recovered fewer than 400 genes, we reran HybPiper, manually setting the maximum k-mer values for assembly to 55 instead of allowing SPAdes to automatically set it. To extract noncoding sequences and annotate gene features along assembled contigs, we used the HybPiper script “intronerate.py.” We assessed target recovery success using the get_seq_lengths.py and gene_recovery_heatmap.r scripts from HybPiper.
To mask low-coverage regions likely to contain sequencing errors, we mapped each sample’s reads to its HybPiper supercontigs using BWA (Li and Durbin 2009), removed PCR duplicates using Picard (Broad Institute 2016), and calculated the depth at each position with Samtools (Li et al. 2009). Using BedTools (Quinlan and Hall 2010), we then hard-masked all positions covered by less than two unique reads. We then used the masked supercontigs and the HybPiper gene annotation files to generate masked versions of the standard HybPiper outputs (using intron_exon_extractor.py): (i) the predicted coding sequence for each target gene (“exon”); (ii) the entire contig assembled for each gene (“supercontig”); and (iii) the predicted noncoding sequences for each gene (“noncoding,” including introns, UTRs, and intergenic sequences).
To the HybPiper output, we added the original orthologs (CDS only) identified in Morus notabilis (Gardner et al. 2016). Because paralogs were only assembled for ingroup samples (due to an Artocarpus-specific whole-genome duplication [Gardner et al. 2016]), we added the corresponding “p0” or “e0” from Morus to each paralog alignment to serve as an outgroup.
We filtered each set of sequences as follows. For “exon” sequences, we subtracted masked bases (Ns) and removed sequences less than 150 bp and sequences covering less than 20% of the average sequence length for that gene. For “supercontig” sequences, we removed sequences whose corresponding “exon” sequences had been removed. Samples with less than 100 genes remaining after filtering were excluded from the main analyses.
Alignment and trimming then proceeded as follows. For “exon” output, after removing the genes and sequences identified during the filtering stage, we created in-frame alignments using MACSE (Ranwez et al. 2011). For “supercontig” output, we used MAFFT for alignment (–maxiter 1000) (Katoh and Standley 2013). We trimmed all alignments to remove all columns with  75% gaps using Trimal (Capella-Gutiérrez et al. 2009).
75% gaps using Trimal (Capella-Gutiérrez et al. 2009).
To quickly inspect gene trees for artifacts, we built gene trees from the trimmed “exon” alignments using FastTree (Price et al. 2009) and visually inspected the gene trees for outlier long branches within the ingroup to identify alignments containing improperly sorted paralogous sequences. In some cases, we visually inspected alignments using AliView (Larsson 2014). We discarded a small number of genes whose alignments contained paralogous sequences, for a final set of 517 genes, including all of the original 333 genes.
We used the trimmed alignments to create three sets of gene alignment data sets:
CDS: “exon” alignments, not partitioned by codon position;
Partitioned CDS: 333 “exon” alignments, partitioned by codon position; and
Supercontig: “supercontig” alignments, not partitioned within genes
We also attempted to create a codon-partitioned supercontig alignment by separately aligning “exon” and “intron” sequences and then concatenating them, resulting in three partitions per gene. However, this data set differed substantially from the supercontig data set, resulting in substantially differing (and nonsensical) topologies even when the partitions were removed; samples with a high proportion of very short or missing noncoding sequences clustered together, perhaps because aligning very short noncoding sequences without longer coding sequences to anchor them produced unreliable alignments. We therefore did not include the partitioned exon+intron data set in the main analyses (discussed further in Appendix 2).
To investigate whether including both copies of a paralogous locus impacted phylogenetic reconstruction, we created versions of each data set with and without paralogs. We analyzed each of these six data sets using the following two methods, for a total of 12 analyses: (A) Concatenated supermatrix: all genes were concatenated into a supermatrix, with each gene partitioned separately (i.e. 1 or 3 partitions per gene, depending on the data set) and analyzed using RAxML 10 (Stamatakis 2006) under GTR+CAT model with 200 rapid bootstrap replicates, rooted with the Moreae outgroups; (B) Species tree: each gene alignment was analyzed using RAxML 10 under the GTR+CAT model with 200 rapid bootstrap replicates, rooted with the Moreae outgroups. Nodes with  33% support were collapsed into polytomies using SumTrees (Sukumaran and Holder 2010), and the resulting trees were used to estimate a species tree with ASTRAL-III (Mirarab and Warnow 2015). We estimated node support with multilocus bootstrapping (-r, 160 bootstrap replicates) and by calculating the proportion of quartet trees that support each node (-t 1) (Mirarab and Warnow 2015; Zhang et al. 2017). For the final trees, we also used SumTrees to calculate the proportion of gene trees supporting each split. Quartet support is directly related to the method ASTRAL uses for estimating species trees—decomposing gene trees into quartets (Mirarab and Warnow 2015); it is also less sensitive to occasional out-of-place taxa than raw gene-tree support.
33% support were collapsed into polytomies using SumTrees (Sukumaran and Holder 2010), and the resulting trees were used to estimate a species tree with ASTRAL-III (Mirarab and Warnow 2015). We estimated node support with multilocus bootstrapping (-r, 160 bootstrap replicates) and by calculating the proportion of quartet trees that support each node (-t 1) (Mirarab and Warnow 2015; Zhang et al. 2017). For the final trees, we also used SumTrees to calculate the proportion of gene trees supporting each split. Quartet support is directly related to the method ASTRAL uses for estimating species trees—decomposing gene trees into quartets (Mirarab and Warnow 2015); it is also less sensitive to occasional out-of-place taxa than raw gene-tree support.
Because all RAxML analyses were conducted using the GTRCAT model, we also repeated the analyses of the CDS data sets using the GTRGAMMA model to investigate the robustness of the recovered topologies to slight model differences.
To summarize the overall bootstrap support of each tree with a single statistic, we calculated “percent resolution” as the number of bipartitions with  50% bootstrap support divided by the total number of bipartitions and represents the proportion of nodes that one might consider resolved (Kates et al. 2018). We visualized trees using FigTree (Rambaut 2016) and the APE package in R (Paradis et al. 2004). To compare trees, we used the phytools package in R (Revell 2012) to plot a consensus tree and to calculate a RF distance matrix for all trees. The RF distance between tree
50% bootstrap support divided by the total number of bipartitions and represents the proportion of nodes that one might consider resolved (Kates et al. 2018). We visualized trees using FigTree (Rambaut 2016) and the APE package in R (Paradis et al. 2004). To compare trees, we used the phytools package in R (Revell 2012) to plot a consensus tree and to calculate a RF distance matrix for all trees. The RF distance between tree  and tree
and tree  equals the number of bipartitions unique to
equals the number of bipartitions unique to  plus the number of bipartitions unique to
plus the number of bipartitions unique to  . We visualized the first two principal components of the matrix using the Lattice package in R (Sarkar 2008). In addition, we conducted pairwise topology comparisons using the “phylo.diff” function from the Phangorn package in R (Schliep 2011) and an updated version of “cophylo” from phytools (github.com/liamrevell/phytools/). All statistical analyses took place in R (R Core Development Team 2008).
. We visualized the first two principal components of the matrix using the Lattice package in R (Sarkar 2008). In addition, we conducted pairwise topology comparisons using the “phylo.diff” function from the Phangorn package in R (Schliep 2011) and an updated version of “cophylo” from phytools (github.com/liamrevell/phytools/). All statistical analyses took place in R (R Core Development Team 2008).
Supermatrix analyses took place on the CIPRES Science Gateway (Miller et al. 2010). All other analyses took place on a computing cluster at the Chicago Botanic Garden, and almost all processes were run in parallel using GNU Parallel (Tange 2018). Alignments and trees have been deposited in the Dryad Data Repository (accession no. TBA).
Appendix 2 – Partitioned exon  noncoding data set
noncoding data set
Results
The partitioned supermatrix analyses in which exons and introns were aligned separately were extremely divergent, particularly in the supermatrix analyses. The mean RF distance to any other tree for the partitioned supercontig trees was 138 (180 for the supermatrix trees and 96 for the ASTRAL trees). Likewise, the strict consensus of all 16 trees had only 48/159 nodes resolved. Re-running the supermatrix analysis with partitions by gene only did not improve concordance (mean RF 176) (Supplementary Fig. S8 available on dryad). The supermatrix trees for which introns and exons were aligned separately all contained a unique, taxonomically nonsensical clade of 38 samples, nested either within subgenus Pseudojaca or subgenus Artocarpus, characterized by increased missing data. A high proportion of missing “intron” sequences ( 50%) appead to be the best predictor for membership in the nonsense clade; all members also had below-average “intron” sequence lengths, although sequence length seemed somewhat less correlated membership in the clade. Pruning tips with missing “intron” sequences dramatically reduced the divergence of the trees in question from other trees (Supplementary Fig. S26 available on dryad). The ASTRAL trees inferred from exons and introns aligned separately were not as severely divergent as the supermatrix trees and did not contain the same nonsense clade (Supplementary Fig. S27 available on dryad).
50%) appead to be the best predictor for membership in the nonsense clade; all members also had below-average “intron” sequence lengths, although sequence length seemed somewhat less correlated membership in the clade. Pruning tips with missing “intron” sequences dramatically reduced the divergence of the trees in question from other trees (Supplementary Fig. S26 available on dryad). The ASTRAL trees inferred from exons and introns aligned separately were not as severely divergent as the supermatrix trees and did not contain the same nonsense clade (Supplementary Fig. S27 available on dryad).
Discussion
The divergent and questionable topologies of the supermatrix analyses for which introns and exons were aligned separately seems to relate at least in part to the alignment method, as removing the codon partitions did not improve the concordance of the supermatrix analysis. It is likely that intronic sequences, especially incomplete ones from samples with fewer or shorter reads, do not align well absent exons to anchor the alignments. Thus, even if missing data do not bias analyses per se (de la Torre-Bárcena et al. 2009), it may in effect create phylogenetically misleading artifacts due to improper alignment or lack of sufficient characters (Rubin et al. 2012). However, partitioning the introns separately seems to have amplified this problem. Because RAxML does not provide for sub-partitions, each gene’s first two codons, third codon, and introns were treated as independent, unlinked partitions in the supermatrix analyses. By contrast, the ASTRAL analyses of the same data set, which effectively had sub-partitions because each gene tree containing three partitions was estimated separately, were much less divergent, suggesting that overpartitioning can bias phylogenetic analyses.
Supplementary Material
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.1rn8pk0pt
Author Contributions
E.M.G. obtained funding, did lab work, and led the writing of the manuscript; all authors contributed to the manuscript. M.G.J. developed the updates to HybPiper. E.M.G. and M.G.J. conducted the bioinformatic analyses. E.M.G. and N.J.C.Z. did fieldwork and herbarium work. J.T.P. facilitated and participated in fieldwork in Sabah. A.S.A.P. facilitated and participated in fieldwork in Sarawak. D.A. did herbarium work in Bogor. Sahromi facilitated and participated in fieldwork in Bogor. N.J.W. obtained funding and supervised the bioinformatic aspects of the project. N.J.C.Z. obtained funding and supervised the overall project.
Funding
This work was supported by the United States National Science Foundation (DEB award numbers 0919119 to N.J.C.Z., 1501373 to N.J.C.Z. and E.M.G., 1342873 and 1239992 to N.J.W., and DBI award number 1711391 to E.M.G.); the Northwestern University Plant Biology and Conservation Program; The Initiative for Sustainability and Energy at Northwestern University; the Garden Club of America; the American Society of Plant Taxonomists; a Systematics Research Fund grant from the Linnean Society and the Systematics Association; the Botanical Society of America; and Texas Tech College of Arts and Sciences.
References
- Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D., Pyshkin A. V., Sirotkin A. V., Vyahhi N., Tesler G., Alekseyev M.A., Pevzner P.A.. 2012. SPAdes: a new genome assembly algorithm and Its applications to single-cell sequencing. J. Comput. Biol. 19:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg C.C. 2005. Flora Malesiana precursor for the treatment of Moraceae 8: other genera than Ficus. Blumea. 50:535–550. [Google Scholar]
- Berg C.C., Pattharahirantricin N., Chantarasuwan B., Santisuk T., Larsen K.. 2011. Flora of Thailand, Vol. 10, Pt. 4: Cecropiaceae and Moraceae. Forest Herbarium, Royal Forest Department. [Google Scholar]
- Berg C.C., Corner E.J.H., Jarrett F.M.. 2006. Moraceae, genera other than Ficus. Flora Malesiana ser. I, vol. 17(1). Foundation for Flora Malesiana. [Google Scholar]
- Bolger A.M., Lohse M., Usadel B.. 2014. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer G.E., Clarkson J.J., Maurin O., Zuntini A.R., Barber V., Bellot S., Biggs N., Cowan R.S., Davies N.M.J., Dodsworth S., Edwards S.L., Eiserhardt W.L., Epitawalage N., Frisby S., Grall A., Kersey P.J., Pokorny L., Leitch I.J., Forest F., Baker W.J.. 2019. Factors affecting targeted sequencing of 353 nuclear genes from herbarium specimens spanning the diversity of angiosperms. Front. Plant Sci. 10:1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad Institute. 2016. Picard tools. Available from: https://broadinstitute.github.io/picard/. [Google Scholar]
- Buerki S., Baker W.J.. 2016. Collections-based research in the genomic era. Biol. J. Linn. Soc. 117:5–10. [Google Scholar]
- Capella-Gutiérrez S., Silla-Martínez J.M., Gabaldón T.. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 25:1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castañeda-Álvarez N.P., Khoury C.K., Achicanoy H.A., Bernau V., Dempewolf H., Eastwood R.J., Guarino L., Harker R.H., Jarvis A., Maxted N., Müller J. V., Ramirez-Villegas J., Sosa C.C., Struik P.C., Vincent H., Toll J.. 2016. Global conservation priorities for crop wild relatives. Nat. Plants. 2:16022. [DOI] [PubMed] [Google Scholar]
- Clement W.L., Weiblen G.D.. 2009. Morphological evolution in the mulberry family (Moraceae). Syst. Bot. 34:530–552. [Google Scholar]
- Copetti D., Búrquez A., Bustamante E., Charboneau J.L.M., Childs K.L., Eguiarte L.E., Lee S., Liu T.L., McMahon M.M., Whiteman N.K., Wing R.A., Wojciechowski M.F., Sanderson M.J.. 2017. Extensive gene tree discordance and hemiplasy shaped the genomes of North American columnar cacti. Proc. Natl. Acad. Sci. USA. 114:12003–12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Bárcena J.E., Kolokotronis S.-O., Lee E.K., Stevenson D.W., Brenner E.D., Katari M.S., Coruzzi G.M., DeSalle R.. 2009. The impact of outgroup choice and missing data on major seed plant phylogenetics using genome-wide EST data. PLoS One. 4:e5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan J.H., Rosenberg N.A.. 2009. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24:332–340. [DOI] [PubMed] [Google Scholar]
- Doyle J., Doyle J.. 1987. Genomic plant DNA preparation from fresh tissue-CTAB method. Phytochem. Bull. 19:11–15. [Google Scholar]
- Faircloth B.C. 2015. PHYLUCE is a software package for the analysis of conserved genomic loci. Bioinformatics. 32:786–788. [DOI] [PubMed] [Google Scholar]
- Faircloth B.C., McCormack J.E., Crawford N.G., Harvey M.G., Brumfield R.T., Glenn T.C.. 2012. Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Syst. Biol. 61:717–726. [DOI] [PubMed] [Google Scholar]
- Gardner E.M., Arifiani D., Zerega N.J.C. in press. Artocarpus bergii (Moraceae): a new species in the breadfruit clade from the Moluccas. Syst. Bot. [Google Scholar]
- Gardner E.M., Chaveerach A., Sudmoon R., Zerega N.J.C.. 2020. Two new species of Artocarpus (Moraceae) from Thailand and Vietnam Phytotaxa. 453:265–274. [Google Scholar]
- Gardner E.M., Johnson M.G., Ragone D., Wickett N.J., Zerega N.J.C.. 2016. Low-coverage, whole-genome sequencing of Artocarpus camansi (Moraceae) for phylogenetic marker development and gene discovery. Appl. Plant Sci. 4:1600017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner E.M., Zerega N.J.C.. 2020. Taxonomic updates to Artocarpus subgenus Pseudojaca (Moraceae), with a particular focus on the taxa in Singapore. Gard. Bull. Singapore. 72. [Google Scholar]
- Guschanski K., Krause J., Sawyer S., Valente L.M., Bailey S., Finstermeier K., Sabin R., Gilissen E., Sonet G., Nagy Z.T., Lenglet G., Mayer F., Savolainen V.. 2013. Next-generation museomics disentangles one of the largest primate radiations. Syst. Biol. 62:539–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart M.L., Forrest L.L., Nicholls J.A., Kidner C.A.. 2016. Retrieval of hundreds of nuclear loci from herbarium specimens. Taxon. 65:1081–1092. [Google Scholar]
- Hoang D.T., Chernomor O., Von Haeseler A., Minh B.Q., Vinh L.S.. 2018. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35:518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett F.M. 1959a. Studies in Artocarpus and allied genera, I. General Considerations. J. Arnold Arbor. 40:1–29. [Google Scholar]
- Jarrett F.M. 1959b. Studies in Artocarpus and allied genera III. A revision of Artocarpus subgenus Artocarpus. J. Arnold Arbor. 40:113–155, 298,–326, 327–368. [Google Scholar]
- Jarrett F.M. 1960. Studies in Artocarpus and allied genera, IV. A revision of Artocarpus subgenus Pseudojaca. J. Arnold Arbor. 41:73–109, 111–140. [Google Scholar]
- Jarrett F.M. 1975. Four new Artocarpus species from Indo-Malesia (Moraceae). Blumea. 22:409–410. [Google Scholar]
- Johnson M.G., Gardner E.M., Liu Y., Medina R., Goffinet B., Shaw A.J., Zerega N.J.C., Wickett N.J.. 2016. HybPiper: extracting coding sequence and introns for phylogenetics from high-throughput sequencing reads using target enrichment. Appl. Plant Sci. 4:1600016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M.G., Pokorny L., Dodsworth S., Botigué L.R., Cowan R.S., Devault A., Eiserhardt W.L., Epitawalage N., Forest F., Kim J.T., Leebens-Mack J.H., Leitch I.J., Maurin O., Soltis D.E., Soltis P.S., Wong G.K., Baker W.J., Wickett N.J.. 2019. A universal probe set for targeted sequencing of 353 nuclear genes from any flowering plant designed using k-medoids clustering. Syst. Biol. 68:594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy S., Minh B.Q., Wong T.K.F., Von Haeseler A., Jermiin L.S.. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 14:587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kates H.R., Johnson M.G., Gardner E.M., Zerega N.J.C., Wickett N.J.. 2018. Allele phasing has minimal impact on phylogenetic reconstruction from targeted nuclear gene sequences in a case study of Artocarpus. Am. J. Bot. 105:404–416. [DOI] [PubMed] [Google Scholar]
- Katoh K., Standley D.M.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30:772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller O., Kollmar M., Stanke M., Waack S.. 2011. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics. 27:757–63. [DOI] [PubMed] [Google Scholar]
- Kochummen K.M. 1998. New species and varieties of Moraceae from Malaysia. Gard. Bull. Singapore. 50:197–219. [Google Scholar]
- Kubatko L.S., Degnan J.H.. 2007. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 56:17–24. [DOI] [PubMed] [Google Scholar]
- Lanfear R., Calcott B., Kainer D., Mayer C., Stamatakis A.. 2014. Selecting optimal partitioning schemes for phylogenomic datasets. BMC Evol. Biol. 14:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson A. 2014. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics. 30:3276–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R.. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. 2009. The sequence alignment/map format and SAMtools. Bioinformatics. 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Johnson M.G., Cox C.J., Medina R., Devos N., Vanderpoorten A., Hedenäs L., Bell N.E., Shevock J.R., Aguero B., Quandt D., Wickett N.J., Shaw A.J., Goffinet B.. 2019. Resolution of the ordinal phylogeny of mosses using targeted exons from organellar and nuclear genomes. Nat. Commun. 10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B.N. 2014. Solid Phase Reverse Immobilization (SPRI) Bead Technology for Micro RNA Clean Up using the Agencourt RNAClean XP Kit. Beckman Coulter Life Sciences. [Google Scholar]
- Mandel J.R., Dikow R.B., Funk V. a., Masalia R.R., Staton S.E., Kozik A., Michelmore R.W., Rieseberg L.H., Burke J.M.. 2014. A target enrichment method for gathering phylogenetic information from hundreds of loci: an example from the compositae. Appl. Plant Sci. 2:1300085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina R., Johnson M.G., Liu Y., Wickett N.J., Shaw A.J., Goffinet B.. 2019. Phylogenomic delineation of Physcomitrium (Bryophyta: Funariaceae) based on targeted sequencing of nuclear exons and their flanking regions rejects the retention of Physcomitrella, Physcomitridium and Aphanorrhegma. J. Syst. Evol. 57: 404–417. [Google Scholar]
- Miller M.A., Pfeiffer W., Schwartz T.. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. 2010 Gateway Computing Environment Workshop. GCE 2010. IEEE. [Google Scholar]
- Minh B.Q., Nguyen M.A.T., Von Haeseler A.. 2013. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30:1188–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab S., Warnow T.. 2015. ASTRAL-II: coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics. 31:i44–i52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E., Claude J., Strimmer K.. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 20:289–290. [DOI] [PubMed] [Google Scholar]
- Pease J.B., Brown J.W., Walker J.F., Hinchliff C.E., Smith S.A.. 2018. Quartet Sampling distinguishes lack of support from conflicting support in the green plant tree of life. Am. J. Bot. 105:385–403. [DOI] [PubMed] [Google Scholar]
- Price M.N., Dehal P.S., Arkin A.P.. 2009. Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26:1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prum R.O., Berv J.S., Dornburg A., Field D.J., Townsend J.P., Lemmon E.M., Lemmon A.R.. 2015. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature. 526:569–573. [DOI] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M.. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Development Team. 2008. R: a language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing. [Google Scholar]
- Rambaut A. 2016. FigTree v1.4.3. Institute of Evolutionary Biology, University of Ediburgh. Available from: http://tree.bio.ed.ac.uk/software/figtree/. [Google Scholar]
- Ranwez V., Harispe S., Delsuc F., Douzery E.J.P.. 2011. MACSE: Multiple alignment of coding SEquences accounting for frameshifts and stop codons. PLoS One. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revell L.J. 2012. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3:217–223. [Google Scholar]
- Roch S., Nute M., Warnow T.. 2019. Long-branch attraction in species tree estimation: inconsistency of partitioned likelihood and topology-based summary methods. Syst. Biol. 68:281–297. [DOI] [PubMed] [Google Scholar]
- Rubin B.E.R., Ree R.H., Moreau C.S.. 2012. Inferring phylogenies from RAD sequence data. PLoS One. 7:e33394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar D. 2008. Lattice: multivariate data visualization with r. New York (NY): Springer. [Google Scholar]
- Sayyari E., Mirarab S.. 2016. Fast coalescent-based computation of local branch support from quartet frequencies. Mol. Biol. Evol. 33:1654–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayyari E., Whitfield J.B., Mirarab S.. 2017. Fragmentary gene sequences negatively impact gene tree and species tree reconstruction. Mol. Biol. Evol. 34:3279–3291. [DOI] [PubMed] [Google Scholar]
- Schliep K.P. 2011. phangorn: phylogenetic analysis in R. Bioinformatics. 27:592–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S.A., Moore M.J., Brown J.W., Yang Y.. 2015. Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evol. Biol. 15:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staats M., Erkens R.H.J., van de Vossenberg B., Wieringa J.J., Kraaijeveld K., Stielow B., Geml J., Richardson J.E., Bakker F.T.. 2013. Genomic treasure troves: complete genome sequencing of herbarium and insect museum specimens. PLoS One. 8:e69189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 22:2688–2690. [DOI] [PubMed] [Google Scholar]
- Sukumaran J., Holder M.T.. 2010. DendroPy: a Python library for phylogenetic computing. Bioinformatics. 26:1569–1571. [DOI] [PubMed] [Google Scholar]
- Tange O. 2018. GNU Parallel 2018. Available from: 10.5281/zenodo.1146014. [DOI] [Google Scholar]
- Villaverde T., Pokorny L., Olsson S., Rincón-Barrado M., Johnson M.G., Gardner E.M., Wickett N.J., Molero J., Riina R., Sanmartín I.. 2018. Bridging the micro- and macroevolutionary levels in phylogenomics: Hyb-Seq solves relationships from populations to species and above. New Phytol. 220:636–650. [DOI] [PubMed] [Google Scholar]
- Wang M.M.H., Gardner E.M., Chung R.C.K., Chew M.Y., Milan A.R., Pereira J.T., Zerega N.J.C.. 2018. Origin and diversity of an underutilized fruit tree crop, cempedak (Artocarpus integer, Moraceae). Am. J. Bot. 105:898–914. [DOI] [PubMed] [Google Scholar]
- Weitemier K., Straub S.C.K., Cronn R.C., Fishbein M., Schmickl R., McDonnell A., Liston A.. 2014. Hyb-Seq: Combining Target Enrichment and Genome Skimming for Plant Phylogenomics. Appl. Plant Sci. 2:1400042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickett N.J., Mirarab S., Nguyen N., Warnow T., Carpenter E., Matasci N., Ayyampalayam S., Barker M.S., Burleigh J.G., Gitzendanner M.A., Ruhfel B.R., Wafula E., Der J.P., Graham S.W., Mathews S., Melkonian M., Soltis D.E., Soltis P.S., Miles N.W., Rothfels C.J., Pokorny L., Shaw A.J., DeGironimo L., Stevenson D.W., Surek B., Villarreal J.C., Roure B., Philippe H., DePamphilis C.W., Chen T., Deyholos M.K., Baucom R.S., Kutchan T.M., Augustin M.M., Wang J., Zhang Y., Tian Z., Yan Z., Wu X., Sun X., Wong G.K.-S., Leebens-Mack J.. 2014. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. USA. 111:E4859–E4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams E.W., Gardner E.M., Harris R., Chaveerach A., Pereira J.T., Zerega N.J.C.. 2017. Out of Borneo: biogeography, phylogeny, and divergence date estimates of Artocarpus (Moraceae). Ann. Bot. 119:611–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witherup C., Zuberi M.I., Hossain S., Zerega N.J.C.. 2019. Genetic diversity of Bangladeshi jackfruit (Artocarpus heterophyllus) over time and across seedling sources. Econ. Bot. 73:233–248. [Google Scholar]
- World Conservation Monitoring Centre. 1998. Artocarpus treculianus. Available from: 10.2305/IUCN.UK.1998.RLTS.T33246A9771111.en. [DOI] [Google Scholar]
- Wu C.Y., Chang S.S.. 1989. Taxa nova nonnulla Moracearum Sinensium. Acta Bot. Yunnanica. 11:24–34. [Google Scholar]
- Xi Z., Ruhfel B.R., Schaefer H., Amorim A.M., Sugumaran M., Wurdack K.J., Endress P.K., Matthews M.L., Stevens P.F., Mathews S., Davis C.C.. 2012. Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. Proc. Natl. Acad. Sci. USA. 109:17519–17524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerega N.J.C., Nur Supardi M.N., Motley T.J.. 2010. Phylogeny and recircumscription of Artocarpeae (Moraceae) with a focus on Artocarpus. Syst. Bot. 35:766–782. [Google Scholar]
- Zerega N.J.C., Ragone D., Motley T.. 2005. Systematics and species limits of breadfruit (Artocarpus, Moraceae). Syst. Bot. 30:603–615. [Google Scholar]
- Zerega N.J.C., Wiesner-Hanks T., Ragone D., Irish B., Scheffler B., Simpson S., Zee F.. 2015. Diversity in the breadfruit complex (Artocarpus, Moraceae): genetic characterization of critical germplasm. Tree Genet. Genomes. 11:1–26. [Google Scholar]
- Zhang C., Sayyari E., Mirarab S.. 2017. ASTRAL-III: Increased scalability and impacts of contracting low support branches BT—comparative genomics: 15th International Workshop, RECOMB CG 2017, Barcelona, Spain, October 4-6, 2017, Proceedings. In: Meidanis J., Nakhleh L., editors. Cham: Springer International Publishing. p. 53–75. [Google Scholar]
- Zhengyi W., Xiushi Z.. 1989. Taxa nova nonnulla Moracearum Sinensium. Acta Bot. Yunnanica. 11:24–34. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw reads have been deposited in GenBank (BioProject no. PRJNA322184), and alignments and trees have been deposited in the Dryad Data Repository (https://orcid.org/0000-0003-1133-5167). HybPiper and related scripts used in this study are available at https://github.com/mossmatters/HybPiper and https://github.com/mossmatters/phyloscripts.