

Abstract
The consequences of the Cretaceous–Paleogene (K–Pg) boundary (KPB) mass extinction for the evolution of plant diversity remain poorly understood, even though evolutionary turnover of plant lineages at the KPB is central to understanding assembly of the Cenozoic biota. The apparent concentration of whole genome duplication (WGD) events around the KPB may have played a role in survival and subsequent diversification of plant lineages. To gain new insights into the origins of Cenozoic biodiversity, we examine the origin and early evolution of the globally diverse legume family (Leguminosae or Fabaceae). Legumes are ecologically (co-)dominant across many vegetation types, and the fossil record suggests that they rose to such prominence after the KPB in parallel with several well-studied animal clades including Placentalia and Neoaves. Furthermore, multiple WGD events are hypothesized to have occurred early in legume evolution. Using a recently inferred phylogenomic framework, we investigate the placement of WGDs during early legume evolution using gene tree reconciliation methods, gene count data and phylogenetic supernetwork reconstruction. Using 20 fossil calibrations we estimate a revised timeline of legume evolution based on 36 nuclear genes selected as informative and evolving in an approximately clock-like fashion. To establish the timing of WGDs we also date duplication nodes in gene trees. Results suggest either a pan-legume WGD event on the stem lineage of the family, or an allopolyploid event involving (some of) the earliest lineages within the crown group, with additional nested WGDs subtending subfamilies Papilionoideae and Detarioideae. Gene tree reconciliation methods that do not account for allopolyploidy may be misleading in inferring an earlier WGD event at the time of divergence of the two parental lineages of the polyploid, suggesting that the allopolyploid scenario is more likely. We show that the crown age of the legumes dates to the Maastrichtian or early Paleocene and that, apart from the Detarioideae WGD, paleopolyploidy occurred close to the KPB. We conclude that the early evolution of the legumes followed a complex history, in which multiple auto- and/or allopolyploidy events coincided with rapid diversification and in association with the mass extinction event at the KPB, ultimately underpinning the evolutionary success of the Leguminosae in the Cenozoic. [Allopolyploidy; Cretaceous–Paleogene (K–Pg) boundary; Fabaceae, Leguminosae; paleopolyploidy; phylogenomics; whole genome duplication events]
The Cretaceous–Paleogene boundary (KPB) at 66 Ma, is defined by the mass extinction event that resulted in major turnover in the earth’s biota, including the extinction of non-avian dinosaurs (Lyson et al. 2019). The KPB event determined in significant part the composition of the modern biota, because many lineages that were successful in the wake of the mass extinction event remained abundant and diverse throughout the Cenozoic until the present. Well-known examples of successful post-KPB lineages are the mammals and birds, both inconspicuous elements of the Cretaceous fauna, while their core clades Placentalia and Neoaves became some of the most prominent and diverse groups of vertebrate fauna across the Cenozoic (Claramunt and Cracraft 2015; Phillips 2015). Plants were also severely affected by the KPB (McElwain and Punyasena 2007), with a clear shift in floristic composition evident from major turnover of dominant species and loss of diversity indicated by a 57–78% drop in macrofossil species richness across boundary-spanning fossil sites in North America (Wilf and Johnson 2004) and disappearance of 15–30% of pollen and spore species in palynological assemblages in North America and New Zealand (Vajda and Bercovici 2014). In addition, consecutive global spikes in spores of fungi and ferns in the palynological record (Vajda et al. 2001; Barreda et al. 2012) are consistent with sudden KPB ecosystem collapse and a recovery period characterized by low diversity vegetation dominated by ferns. Although the KPB is not considered a major extinction event for plants, with no plant families apparently lost (McElwain and Punyasena 2007; Cascales-Miñana and Cleal 2014), a sudden increase in net diversification rate in the Paleocene has been inferred from paleobotanical data (Silvestro et al. 2015), suggesting increased origination following the KPB.
Macroevolutionary dynamics of plant clades across the KPB have received less attention than prominent vertebrate clades, even though plants are the main primary producers and structural components of terrestrial ecosystems. Therefore, the diversification of the Cenozoic biota cannot be fully understood without understanding the effect of the KPB on evolutionary turnover of plant diversity. A potentially important aspect of plant evolution during this period is the apparent concentration of whole genome duplication (WGD) events around the KPB (Fawcett et al. 2009; Vanneste et al. 2014; Lohaus and Van de Peer 2016; but see Cai et al. 2019). This is explained by the idea that polyploid lineages had enhanced survival and establishment across the KPB (Lohaus and Van de Peer 2016) and greater potential to rapidly diversify thereafter compared to diploids (Levin and Soltis 2018). Recent work is revealing the prevalence and significance of WGDs in shaping the evolution of the flowering plants (Wendel 2015; Soltis et al. 2016; Yang et al. 2018; Cai et al. 2019; Conover et al. 2019). Determining the phylogenetic placements and timing of WGDs is a central issue in plant evolution, but remains challenging, with often conflicting lines of evidence, such that many WGDs and their phylogenetic positions remain putative and poorly understood (e.g., Conover et al. 2019).
We examine the role of the KPB in shaping Cenozoic plant diversity by investigating the origin and early evolution of the legume family, including the placement and timing of WGDs. The legume family (Leguminosae or Fabaceae), perhaps more than any other plant clade, appears to parallel the example of Placentalia and Neoaves. No clearly identifiable legume fossils predate the KPB (Herendeen and Dilcher 1992)—the oldest unequivocal legume fossil is 65.35 Ma (Lyson et al. 2019)—but the family was already abundant and diverse in the earliest modern type rainforests in the late Paleocene (Wing et al. 2009; Herrera et al. 2019). The oldest fossils clearly referable to (stem groups of) subfamilies are from close to the Paleocene–Eocene Thermal Maximum (PETM)—morphotype # CJ76 of c. 58 Ma (Wing et al. 2009) can be referred to Caesalpinioideae and Barnebyanthus buchananensis of c. 56 Ma to Papilionoideae (Crepet and Herendeen 1992)—and legumes are ubiquitous in Eocene, Oligocene, and Neogene floras (Herendeen and Dilcher 1992). Legumes range from gigantic rainforest canopy trees and lianas, to shrubs, herbs, geoxyles, and (semi-)aquatics, arguably presenting the most spectacular evolutionary and ecological radiation of any angiosperm family (McKey 1994). Legumes occur nearly everywhere except for Antarctica and exert considerable ecological dominance globally, especially in tropical rainforests, savannas, and dry forests of the Americas, Africa, and Australia as well as forming one of the most prominent components of the global (temperate) herbaceous flora. The characteristic “pod” or “legume” fruit provides a unique diagnostic synapomorphy for the clade, which contains many important crop species cultivated for their seeds and fruits (e.g., beans, (chick)peas, lentils, peanuts), and legumes are also well-known for their ability to fix atmospheric nitrogen via symbiosis with bacteria in root nodules which is shared by the majority of legume species. The six main lineages of legumes, recently recognized as subfamilies (LPWG 2017), apparently diverged nearly simultaneously (Koenen et al. 2020), mirroring Placentalia (Teeling and Hedges 2013), and Neoaves (Suh et al. 2015; Suh 2016).
The apparent rapid diversification of the legumes soon after the KPB, and the occurrence of multiple WGDs during their early evolution (Cannon et al. 2015; Stai et al. 2019), make the family an excellent model to investigate the association of WGDs with the KPB. However, there is uncertainty about how many WGDs were involved in the early evolution of legumes and their phylogenetic placements. Several taxa in subfamily Papilionoideae have been shown to share a WGD (Mudge et al. 2005; Cannon et al. 2006), that was subsequently shown to subtend the subfamily as a whole and is not shared with other subfamilies, in which three additional and independent WGDs were hypothesized (Cannon et al. 2015). More recently, WGDs were hypothesized to have occurred independently early in the evolution of each subfamily (except Duparquetioideae, for which there are no nuclear genomic or cytological data) based in part on haploid chromosome numbers, with the WGD in Cercidoideae excluding the genus Cercis, the sister group to the rest of that subfamily (Stai et al. 2019). While Stai et al. (2019) presented convincing evidence that Cercis lacks a polyploid history, their assertion that the genus retained ancestral genomic features including a haploid chromosome number of 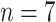 , was partly based on its phylogenetic position (as an “early-diverging” lineage), and lacked any explicit reconstruction of chromosomal evolution (Mayrose et al. 2009). However, the phylogenetic positions of Cercis and Cercidoideae alone cannot establish that these taxa retained ancestral traits (Crisp and Cook 2005), while recent analyses of genome-scale nuclear gene data placed Cercidoideae as the sister group of Detarioideae (Koenen et al. 2020), not as sister to the rest of the legumes as suggested by Stai et al. (2019). Furthermore, haploid chromosome numbers of 6–8 are also found in subfamilies Detarioideae, Caesalpinioideae, and commonly in Papilionoideae, even though paleopolyploidy in Detarioideae and Papilionoideae is well established (Cannon et al. 2015; Ren et al. 2019). Moreover, rather than the five independent WGDs proposed by Stai et al. (2019), alternative explanations of a single WGD shared across all legumes, or, given the likely non-polyploidy of Cercis, one or more WGDs shared across multiple subfamilies, would be more parsimonious. These alternative hypotheses remain to be tested using a representative set of gene trees with adequate taxon sampling.
, was partly based on its phylogenetic position (as an “early-diverging” lineage), and lacked any explicit reconstruction of chromosomal evolution (Mayrose et al. 2009). However, the phylogenetic positions of Cercis and Cercidoideae alone cannot establish that these taxa retained ancestral traits (Crisp and Cook 2005), while recent analyses of genome-scale nuclear gene data placed Cercidoideae as the sister group of Detarioideae (Koenen et al. 2020), not as sister to the rest of the legumes as suggested by Stai et al. (2019). Furthermore, haploid chromosome numbers of 6–8 are also found in subfamilies Detarioideae, Caesalpinioideae, and commonly in Papilionoideae, even though paleopolyploidy in Detarioideae and Papilionoideae is well established (Cannon et al. 2015; Ren et al. 2019). Moreover, rather than the five independent WGDs proposed by Stai et al. (2019), alternative explanations of a single WGD shared across all legumes, or, given the likely non-polyploidy of Cercis, one or more WGDs shared across multiple subfamilies, would be more parsimonious. These alternative hypotheses remain to be tested using a representative set of gene trees with adequate taxon sampling.
Uncertainty also surrounds the age of the legume family. While legumes are not known with certainty from any Cretaceous fossil site, the family has a long stem lineage dating to c. 80–100 Ma (Wang et al. 2009; Magallón et al. 2015), which means that the timing of the initial radiation of the family and legume WGDs relative to the KPB are uncertain. In Placentalia and Neoaves, divergence time estimates also remain contentious; some molecular divergence time estimates suggest that these clades originated and diversified well before the KPB, implying that many lineages of both clades survived the end-Cretaceous event (Cooper and Penny 1997; Meredith et al. 2011; Jetz et al. 2012). However, like legumes, both groups first appear in the Paleocene fossil record. A phylogenetic study of mammals combining molecular sequence data and morphological characters for extant and fossil taxa, found only a single placental ancestor crossing the KPB (O’Leary et al. 2013; but see Springer et al. 2013; dos Reis et al. 2014). Others have argued that diversification of Placentalia followed a “soft explosive” model, with a few lineages crossing the KPB followed by rapid ordinal level Paleocene radiation (Phillips 2015; Phillips and Fruciano 2018). Recent time-calibrated phylogenies for birds showed the age of Neoaves to also be close to the KPB (Jarvis et al. 2014; Claramunt and Cracraft 2015; Prum et al. 2015), with rapid post-KPB divergence represented by a hard polytomy (Suh 2016). For legumes, it is similarly unlikely that the modern subfamilies have Cretaceous crown ages. These clades, especially Papilionoideae, Caesalpinioideae, and Detarioideae, appear to have rapidly diversified following their origins, which would imply mass survival of many legume lineages across the KPB. Furthermore, diversification of the six legume subfamilies appears to have occurred rapidly (Lavin et al. 2005), indeed nearly simultaneously (Koenen et al. 2020), with long stem branches subtending each subfamily. Therefore, two hypotheses seem plausible: 1) legumes have a Cretaceous crown age and subfamily stem lineages diverged prior to the KPB, while subfamily crown radiations occurred (shortly) after the KPB, corresponding to a “soft explosive” model or 2) a single legume ancestor crossed the KPB and rapidly diversified into six lineages in the wake of the mass extinction event, corresponding to a “hard explosive” model, with the subfamily radiations associated with the PETM and/or Eocene climatic optimum. Current molecular crown age estimates for legumes range from c. 59 to 64 Ma (Lavin et al. 2005; Bruneau et al. 2008; Simon et al. 2009). These studies, however, lacked extensive sampling of outgroup taxa relying instead on fixing the legume stem age, thereby compromising the ability to estimate the crown age. Furthermore, these studies used chloroplast sequences, whose evolutionary rates are known to vary strongly across legumes (Lavin et al. 2005; Koenen et al. 2020). Nuclear gene data are likely better suited for estimating divergence times (Christin et al. 2014).
In this study, we evaluate the number of WGDs during early legume evolution and assess whether any of them are shared across multiple subfamilies. We use gene tree reconciliation methods to identify the most likely placement of WGDs among the earliest divergences within the legumes (i.e., those before the diversification of the subfamily crown groups; hereafter referred to as the “backbone”) and test their placement with a probabilistic method using gene count data. We also evaluate the possibility of allopolyploidy involving one or more lineages with phylogenetic supernetwork reconstruction and gene tree reconciliation with multilabeled (MUL) trees. In addition, we evaluate whether the origin of legumes and WGDs are closely associated with the KPB by inferring a new legume chronogram based on 36 informative and relatively clock-like nuclear genes and 20 fossil calibration points, and by assessing the timing of duplication nodes in gene trees.
Materials and Methods
Gene Tree Inference
We used sets of homolog clusters generated prior to extracting orthologs for species tree inference using the Yang and Smith (2014) pipeline, derived from genomes and transcriptomes of representatives of five of the six legume subfamilies and an extensive eudicot outgroup (Supplementary Table S1 available on Dryad at http://dx.doi.org/10.5061/dryad.zkh18936s) assembled by Koenen et al. (2020). We do not include the monospecific subfamily Duparquetioideae for which large-scale nuclear genomic data are presently unavailable. These homolog clusters include multiple sequences per taxon representing paralogs for non-terminal gene duplications; duplications restricted to a terminal taxon are not included. Amino acid sequences of these clusters were aligned with MAFFT v. 7.187 (Katoh and Standley 2013) using the G-INSi algorithm. To avoid having multiple fragments of paralog copies present, which could inflate the number of gene duplications, sites with  5% missing data were removed with BMGE (Criscuolo and Gribaldo 2010) after which all sequences with more than 75% gaps were removed. These data removal steps also eliminated clusters with significant missing data. Tree estimation was repeated on these clusters, using RAxML v. 8.2 (Stamatakis 2014) with the WAG + G model and 100 rapid bootstrap replicates.
5% missing data were removed with BMGE (Criscuolo and Gribaldo 2010) after which all sequences with more than 75% gaps were removed. These data removal steps also eliminated clusters with significant missing data. Tree estimation was repeated on these clusters, using RAxML v. 8.2 (Stamatakis 2014) with the WAG + G model and 100 rapid bootstrap replicates.
Mapping of Gene Duplications
From the homolog trees, we extracted rooted clades as input gene trees for gene duplication mapping analysis with Phyparts (Smith et al. 2015). This method counts for each node the number of gene trees in which at least two descendent taxa are represented by at least two paralogous sequences. Aquilegia and Papaver were used as the outgroup to root and extract the paralog clades. Phyparts was run with and without a 50% bootstrap cutoff.
In addition, we performed gene tree reconciliation with a model of gene duplication and loss (horizontal transfers not considered) using Notung v 2.9 (Stolzer et al. 2012) on the rosid portion of the species tree. Because Notung accounts for incomplete lineage sorting (ILS) when using non-binary trees (i.e., trees with polytomies), we introduced six polytomies for poorly supported, short internodes in the species tree (at the base of Fabales and within Caesalpinioideae and Papilionoideae). Additionally, an analysis was run with two additional polytomies within the legume backbone, since ILS likely occurred among the first divergences in the family (Koenen et al. 2020). All other internodes within the legume family are considered to be well-supported (Koenen et al. 2020), suggesting that ILS will have less impact on these. Input gene trees were extracted from homolog clusters as for the Phyparts analysis, but with all non-rosid taxa as the outgroup, such that the older Pentapetalae hexaploidization is not included. First, we used the –rearrange option in Notung with an 80% bootstrap threshold to rearrange poorly supported branches in gene trees according to relationships found in the species tree. This has the drawback that in the case of missing data or duplicate gene loss, some genuine gene duplications with lower support are reconciled to a more inclusive clade. However, without this rearrangement step, many more gene duplications are inferred across all nodes, presumably in part caused by gene tree estimation errors. Next, we ran the reconciliation analysis in –phylogenomics mode and analyzed the number of inferred duplications on each node, setting the cost of duplications at 1.5 (the default), and gene losses at 0.1 to avoid a strong influence of missing data from transcriptomes on reconciliation scores. We explored other settings but the results did not change significantly.
Testing Placements of WGDs Using Gene Count Data
We used the WGDgc package in R (Rabier et al. 2014) to test the placements of WGDs hypothesized by Phyparts and Notung. This probabilistic method models background gene duplication and loss rates using a birth and death process, while adding WGDs on specific branches of the species tree. Birth–death and duplicate gene retention rates for WGDs are estimated with maximum likelihood and the overall likelihood is compared across different configurations of WGDs on the species tree. We extracted gene count data from the rosid gene trees used in the Notung analysis, after removing several transcriptome accessions with relatively high levels of missing data. Furthermore, to use the “oneInBothClades” conditional likelihood option, Eucalyptus grandis and Punica granatum were removed to ensure there are two large clades at the root, the nitrogen-fixing clade of angiosperms (consisting of Cucurbitales, Rosales, Fagales, and Fabales) and a clade consisting of the remaining sampled rosid orders. Accordingly, count data were filtered to remove all gene families that did not have at least one copy in both main clades at the root. Additionally, we removed all gene families that did not have at least one copy in each of the five sampled legume subfamilies to reduce possible negative impacts of missing data on the inferences. Analyses were run with different models with two, three or four WGDs within legumes. The WGD shared by Salix purpurea and Populus trichocarpa is additionally modeled in all analyses. Likelihood ratio tests (LRTs) were used to compare the most likely (nested) models with different numbers of WGDs.  values for the LRTs at different confidence levels are given in Rabier et al. (2014).
values for the LRTs at different confidence levels are given in Rabier et al. (2014).
Gene Tree Reconciliation with Allopolyploidy
To visualize potential reticulation, we redrew the filtered supernetwork (Whitfield et al. 2008) of Koenen et al. (2020) with the Convex Hull method in SplitsTree4 (Huson and Bryant 2005). Potential branches in the species tree that could be involved in allopolyploidy were identified for analysis with GRAMPA (Gregg et al. 2017). Because GRAMPA cannot infer multiple WGDs, we generated a filtered gene tree set excluding duplications associated with previously identified independent WGDs in Detarioideae and Papilionoideae so that these do not influence the reconciliation scores. To do this, we used the gene trees generated for the WGDgc analysis and reduced Cercidoideae, Detarioideae, and Papilionoideae to single accessions (Bauhinia tomentosa, Anthonotha fragrans, and Medicago truncatula, respectively), collapsing all duplications that are particular to these subfamilies. An independent autopolyploidy event is not well established for Caesalpinioideae even though this subfamily showed a polyploid signal in Ks plots (Cannon et al. 2015). Therefore, we retained the transcriptomes of Albizia julibrissin, Entada abyssinica, Inga spectabilis, and Microlobius foetidus since they were well-represented in gene trees. In this way, we test whether polyploidy in Caesalpinioideae is likely derived from independent autopolyploidy or allopolyploidy, or instead from an earlier WGD shared with other subfamilies. For this analysis, gene trees with <50% average bootstrap support were excluded.
Divergence Time Analyses
The 20 fossils used to calibrate molecular clock analyses on the species tree are listed in Table 1 and discussed in detail in Supplementary Appendix S1 available on Dryad.
Table 1.
Fossil calibrations used in the divergence time analyses.
Calibration
|
Definition | Fossil | Age (Ma) |
|---|---|---|---|
| Eudicots | |||
| 26 | CG eudicots | Tricolpate pollen; England and Gabon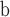
|
126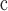
|
| 27 | CG Ranunculales |
Teixeiraea lusitanica – flower; Portugal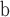
|
113 |
| 38 | CG Pentapetalae | Pentamerous flower with distinct calyx and corolla; USA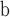
|
100 |
| 48 | SG Ericales |
Pentapetalum trifasciculandricus—flowers; USA
|
89.8 |
| 94 | SG Myrtaceae | “Flower number 3” from the Table Nunatak Formation, Antarctica
|
83.6 |
| 105 | SG Brassicales |
Dressiantha bicarpelata—flowers; USA
|
89.8 |
| 112 | CG Rosaceae |
Prunus wutuensis—fruits; China
|
49.4 |
| 116 | SG Cannabaceae |
Aphananthe cretacea and Gironniera gonnensis—fruits; Germany
|
66 |
| 122 | SG Juglandaceae |
Polyptera manningi—fruits; USA
|
64.4 |
| 133 | SG Populus |
Populus wilmattae—leaves, infructescences and fruits; USA
|
37.8 |
| X14 | SG Fagales |
Protofagacea allonensis—flowers; USA
|
83.6 |
| Legumes | |||
| A | SG Leguminosae |
Paracacioxylon frenguellii—wood with vestured pits; Argentina
|
63.5 |
| C | SG Cercis |
Cercis parvifolia—leaves and C. herbmeyeri—fruits; USA
|
36 |
C
|
SG Bauhinia | cf. Bauhinia—simple leaf with bilobed lamina; Tanzania
|
46 |
| F | SG Resin-producing clade |
Hymenaea mexicana—vegetativeand floral remains in amber; Mexico
|
22.5 |
| G | SG Detarioideae |
Aulacoxylon sparnacense—wood and amber; France
|
53 |
G
|
SG Resin-producing clade | Same as G | 53 |
H
|
CG Amherstieae |
Aphanocalyx singidaensis—bifoliolate leaves; Tanzania
|
46 |
| I2 | SG Styphnolobium/Cladrastis |
Styphnolobium and Cladrastis—leaves and fruits; USA
|
37.8 |
| M2 | SG Robinioid clade |
Robinia zirkelii—wood; USA
|
33.9 |
| Q | SG Acacieae/Ingeae | Flattened polyads with 16 pollen grains; Brazil, Colombia, Cameroon and Egypt
|
33.9 |
| Q2 | SG Acacia s.s. | Polyads with pseudocolpi; Australia
|
23 |
| Z | SG Caesalpinioideae | Bipinnate leaves; Colombia
|
58 |
See Supplementary Appendix 1 available on Dryad for detailed discussion of these fossil calibrations.
1 available on Dryad for detailed discussion of these fossil calibrations.
CG  Crown group; SG
Crown group; SG  Stem group; Ma
Stem group; Ma  Million years ago.
Million years ago.
 Numbers 26, 27, 38, 48, 94, 105, 112, 116, 122 and 133 refer to calibrations from Magallón et al. (2015) as listed in their Supplementary Information Methods S1; letters A, D, F, G, I2, M2, and Q refer to calibrations from Bruneau et al. (2008) and/or Simon et al. (2009).
Numbers 26, 27, 38, 48, 94, 105, 112, 116, 122 and 133 refer to calibrations from Magallón et al. (2015) as listed in their Supplementary Information Methods S1; letters A, D, F, G, I2, M2, and Q refer to calibrations from Bruneau et al. (2008) and/or Simon et al. (2009).
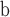 Magallón et al. (2015) and references therein.
Magallón et al. (2015) and references therein.
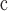 Prior set as normal with standard deviation of 1.0, and truncated between minimum and maximum bounds of 113 and 136 Ma, respectively.
Prior set as normal with standard deviation of 1.0, and truncated between minimum and maximum bounds of 113 and 136 Ma, respectively.
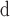 Xing et al. (2014) and reference therein.
Xing et al. (2014) and reference therein.
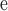
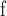 Note that the new fossil discovered by Lyson et al. (2019) at c. 65.35 Ma is slightly older than the fossil listed here and is currently the oldest known fossil evidence of SG Leguminosae; however, since the currently used fossil does not constrain this node because of the long stem lineage of the family, substituting this calibration with the new Lyson et al. (2019) fossil would not influcence our results.
Note that the new fossil discovered by Lyson et al. (2019) at c. 65.35 Ma is slightly older than the fossil listed here and is currently the oldest known fossil evidence of SG Leguminosae; however, since the currently used fossil does not constrain this node because of the long stem lineage of the family, substituting this calibration with the new Lyson et al. (2019) fossil would not influcence our results.
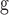
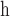 Alternative prior 1 as used in FLC analysis with eight local clocks.
Alternative prior 1 as used in FLC analysis with eight local clocks.
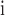
 De Franceschi and De Ploëg (2003).
De Franceschi and De Ploëg (2003).
 Lavin et al. (2003) and references therein.
Lavin et al. (2003) and references therein.
 Simon et al. (2009): Supplementary Information and references therein.
Simon et al. (2009): Supplementary Information and references therein.
Using SortaDate (Smith et al. 2018), we analyzed the 1103 gene trees from Koenen et al. (2020) to estimate total tree length (a proxy for sequence variation or informativeness), root-to-tip variance (a proxy for clock-likeness) and compatibility of bipartitions with the ML tree inferred using the full data set (the RAxML tree inferred with the LG4X model). We selected the best genes for dating based on arbitrary cutoff values: i) total tree length greater than 5, ii) root-to-tip variance less than 0.005, and iii) at least 10% of bipartitions compatible with the ML tree. This yielded 36 genes, which were concatenated with an aligned length of 14,462 amino acid sites. We also used the “pxlstr” program of the Phyx package (Brown et al. 2017) to calculate taxon-specific root-to-tip lengths from the ML tree, after pruning Ranunculales, on which the tree was rooted. These values were used to define local clocks. Arabidopsis thaliana, Linum usitatissimum, and Polygala lutea were removed because of much higher root-to-tip lengths relative to their closest relatives. Panax ginseng was also removed because of a low root-to-tip length relative to other sampled asterids, leaving a total of 72 taxa.
We used BEAST v.1.8.4 (Drummond et al. 2012) with various clock models to estimate divergence times based on the alignment of the selected 36 genes and the 20 fossil calibrations (Supplementary Appendix S1 available on Dryad). Analyses were run with the LG + G model of amino acid substitution using a birth–death tree prior, and the ML tree to fix the topology. Fossil calibrations were set as uniform priors between minimum ages specified in Table 1 and a maximum age of 126 Ma (oldest fossil evidence of eudicots) as listed in Supplementary Table S2 available on Dryad, with the exception of the root node, for which we used a normal prior at 126 Ma with a standard deviation of 1.0, truncated to minimum and maximum ages of 113 Ma (the Aptian–Albian boundary) and 136 Ma (the oldest crown angiosperm fossil, see Magallón et al. 2015). We ran analyses under the uncorrelated lognormal (UCLN), strict, random (RLC), and three different fixed local clock (FLC) models (Supplementary Appendix S1 available on Dryad).
Analyses sampling from the prior (without data) were run for 100 million generations, the strict clock, FLC3 and FLC6 analyses were run for 25 million generations and all other clock analyses for 50 million generations, confirming convergence with Tracer v1.7.1 (Rambaut et al. 2018). For the non-prior analyses, the first 10% of the total number of generations was discarded as burn-in before summarizing median branch lengths and substitution rates with TreeAnnotator from the BEAST package.
To infer ages of gene duplication nodes, we made four new subsets of gene trees for time-scaling. The first includes all gene trees for which duplications were mapped on the collapsed legume backbone by Notung, but including only well-sampled taxa (see Supplementary Table S1 available on Dryad), and all other rosids as outgroup taxa. The other three sets were obtained by taking sequences of all non-legume taxa in the nitrogen-fixing clade of angiosperms as outgroup alongside sequences of selected, well-sampled accessions for each of the subfamilies Caesalpinioideae, Detarioideae, and Papilionoideae, creating separate sets of gene trees for each of these subfamilies. We chose these three subfamilies because they are well-sampled and their paleopolyploidy is well established. In this way, we could assess if the WGD events in different subfamilies occurred at different times or whether they are coincident as expected for shared WGDs, although this in itself does not constitute evidence for shared events. For Detarioideae all four sampled transcriptomes were included, for Caesalpinioideae we included only those of Entada abyssinica, Microlobius foetidus, Albizia julibrissin, and Inga spectabilis, and for Papilionoideae the genomes of Medicago truncatula, Glycine max, Phaseolus vulgaris, and Arachis ipaensis were included. For each set, sequences were realigned and new gene trees were inferred with RAxML, using the PROTGAMMAAUTO model. The resulting trees were rooted with Notung with respect to the species tree relationships. For the family-wide trees we further tested whether all legume sequences formed a clade to make sure no gene duplications predating the divergence of legumes (e.g., from the Pentapetalae gamma event) were included. For each subfamily gene tree set, we ran a phyparts analysis and all gene trees with duplications mapping to the crown node of the subfamily were selected. All gene trees in the family-wide and subfamily-specific sets were individually time-scaled using penalized likelihood (Sanderson 2002) in the R package ape (function “chronos”) (Paradis et al. 2004; Paradis 2013). Based on simulations, it was shown that although the correlated clock model estimates more accurate substitution rates, the strict clock estimates more accurate branch lengths (Paradis 2013). Since our purpose is to estimate ages, not rates, we used the strict clock in these analyses, and set the smoothing parameter to 1 as done by Paradis (2013). The root age was set at 110 Ma for the family-wide gene tree set and to 105 Ma for the subfamily-specific gene tree sets based on crown age estimates for rosids and the nitrogen-fixing clade of angiosperms from time-scaling analyses on the species tree (Supplementary Figs. S6–S13 available on Dryad). After time-calibration, ages of duplication nodes were extracted and histograms and density plots of these were made in R.
Results
The removal of sites with  5% missing data and fragmentary sequences from the 9282 homolog clusters generated by Koenen et al. (2020), led to the removal of 640 clusters with large amounts of missing data. From trees inferred from the remaining 8642 homologs, we extracted different sets of rooted gene trees for analysis: i) 8038 trees for the Phyparts analyses that include all sampled taxa except Ranunculales which were used for rooting, ii) 8324 trees including only rosid taxa for the Notung and WGDgc analyses, and iii) 4371 pruned trees with only taxa from the nitrogen-fixing clade of angiosperms, including four Caesalpinioideae species and one species from each remaining subfamily, and average BS
5% missing data and fragmentary sequences from the 9282 homolog clusters generated by Koenen et al. (2020), led to the removal of 640 clusters with large amounts of missing data. From trees inferred from the remaining 8642 homologs, we extracted different sets of rooted gene trees for analysis: i) 8038 trees for the Phyparts analyses that include all sampled taxa except Ranunculales which were used for rooting, ii) 8324 trees including only rosid taxa for the Notung and WGDgc analyses, and iii) 4371 pruned trees with only taxa from the nitrogen-fixing clade of angiosperms, including four Caesalpinioideae species and one species from each remaining subfamily, and average BS  50%, for the GRAMPA analysis. Exemplar gene trees are included in Supplementary Figure S1 available on Dryad, showing evidence of several gene duplications within legumes. These also show that due to differential gene loss, the patterns in individual gene trees are not always clear and general patterns can only be inferred from analyzing large numbers of gene trees. Because of the way these homolog sets were assembled, duplications restricted to terminal lineages are not included, therefore testing for WGDs postulated by Stai et al. (2019) specific to Dialioideae and within Cercidoideae (excluding Cercis), is not possible with this data set. For time-calibrating the species tree, 36 informative and relatively clock-like genes were selected from the 1103 orthologs of Koenen et al. (2020). To estimate the timing of gene duplication nodes, we analyzed 863 gene trees extracted from the Notung analysis including taxa from multiple subfamilies and 246, 250, and 272 trees including only Caesalpinioideae, Detarioideae, and Papilionoideae, respectively. Supplementary Table S1 available on Dryad gives an overview of accessions included per analysis, and numbers of trees and sequences included per taxon. Alignments, gene trees, and gene count data are included in Supplementary Data S1–S7 available on Dryad.
50%, for the GRAMPA analysis. Exemplar gene trees are included in Supplementary Figure S1 available on Dryad, showing evidence of several gene duplications within legumes. These also show that due to differential gene loss, the patterns in individual gene trees are not always clear and general patterns can only be inferred from analyzing large numbers of gene trees. Because of the way these homolog sets were assembled, duplications restricted to terminal lineages are not included, therefore testing for WGDs postulated by Stai et al. (2019) specific to Dialioideae and within Cercidoideae (excluding Cercis), is not possible with this data set. For time-calibrating the species tree, 36 informative and relatively clock-like genes were selected from the 1103 orthologs of Koenen et al. (2020). To estimate the timing of gene duplication nodes, we analyzed 863 gene trees extracted from the Notung analysis including taxa from multiple subfamilies and 246, 250, and 272 trees including only Caesalpinioideae, Detarioideae, and Papilionoideae, respectively. Supplementary Table S1 available on Dryad gives an overview of accessions included per analysis, and numbers of trees and sequences included per taxon. Alignments, gene trees, and gene count data are included in Supplementary Data S1–S7 available on Dryad.
Inferring Phylogenetic Locations of WGDs
In the Phyparts analysis, we find significantly elevated numbers of gene duplications at several nodes where WGDs were previously hypothesized to have occurred, including the Salix/Populus clade (Tuskan et al. 2006) and one consistent with the known gamma hexaploidization subtending Pentapetalae (Jiao et al. 2012) (Fig. 1a and Supplementary Fig. S2 available on Dryad). For Pentapetalae, many homologs show more than one gene duplication at that node, with nearly twice as many duplications (1901) as the number of homologs with duplications (1105), as expected for two consecutive rounds of WGD. Some of these duplications may also stem from older events, since missing data and/or gene loss for the three non-Pentapetalae taxa in our data set could mean that we do not find duplicates of older WGDs in these taxa. Within legumes, high numbers of gene duplications at particular nodes suggest that there were three early WGD events, one located on the stem lineage of the family and one each on the stem lineage of subfamilies Papilionoideae and Detarioideae (Fig. 1a and Supplementary Fig. S2 available on Dryad). When applying a bootstrap filter to the homolog trees ( 50% support), numbers of duplications are considerably lower, but the pattern is the same (Fig. 1a and Supplementary Fig. S2 available on Dryad). At the root of the family, the number of gene duplications drops from 1646 to 99 when applying this bootstrap filter, in line with the difficulty of resolving the deepest dichotomies of the legume phylogeny (Koenen et al. 2020). Notably, for the legume crown node we also find evidence for a significant fraction of homologs showing more than one gene duplication, with 1646 duplications from only 1229 homologs mapping to that node. This could suggest multiple rounds of WGD (e.g., Supplementary Fig. S1e,f available on Dryad), although some of these can be attributed to duplications in both paralog copies of genes duplicated at the Pentapetalae gamma event, and for many others support values across gene trees are low. For other hypothesized WGDs, numbers of homologs with more than one duplication are much lower, suggesting they involved a single round of polyploidization. Using gene tree reconciliation with Notung, we found similar results (Fig. 1b and Supplementary Figs. S3 and S4 available on Dryad), although here the Pentapetalae node was not included. However, numbers of duplications particular to Detarioideae are higher than in the Phyparts analysis. The opposite is true for Papilionoideae, where Notung finds higher numbers of gene duplications on the node uniting Caesalpinioideae and Papilionoideae, and on several nodes within Papilionoideae relative to the Phyparts results.
50% support), numbers of duplications are considerably lower, but the pattern is the same (Fig. 1a and Supplementary Fig. S2 available on Dryad). At the root of the family, the number of gene duplications drops from 1646 to 99 when applying this bootstrap filter, in line with the difficulty of resolving the deepest dichotomies of the legume phylogeny (Koenen et al. 2020). Notably, for the legume crown node we also find evidence for a significant fraction of homologs showing more than one gene duplication, with 1646 duplications from only 1229 homologs mapping to that node. This could suggest multiple rounds of WGD (e.g., Supplementary Fig. S1e,f available on Dryad), although some of these can be attributed to duplications in both paralog copies of genes duplicated at the Pentapetalae gamma event, and for many others support values across gene trees are low. For other hypothesized WGDs, numbers of homologs with more than one duplication are much lower, suggesting they involved a single round of polyploidization. Using gene tree reconciliation with Notung, we found similar results (Fig. 1b and Supplementary Figs. S3 and S4 available on Dryad), although here the Pentapetalae node was not included. However, numbers of duplications particular to Detarioideae are higher than in the Phyparts analysis. The opposite is true for Papilionoideae, where Notung finds higher numbers of gene duplications on the node uniting Caesalpinioideae and Papilionoideae, and on several nodes within Papilionoideae relative to the Phyparts results.
Figure 1.

Numbers of gene duplications mapped over the species tree. a) Results from a phyparts analysis on the species tree topology of Koenen et al. (2020) and b) results from a Notung analysis on the rosids portion of the same tree. Relative sizes of circles on nodes indicate the number of duplications as per the legend. Actual numbers are indicated for nodes with relatively high numbers of duplications, in a) the two numbers are derived from ML topologies without and with a bootstrap filter of 50%, respectively.
The likely phylogenetic locations of WGDs based on mapping of gene duplications were further tested with WGDgc (Rabier et al. 2014), using gene count data harvested from the rosid gene tree set. The best-scoring model with two WGDs has one WGD specific to Detarioideae and one shared by Papilionoideae and Caesalpinioideae (Fig. 2a). This model received a higher likelihood than a model with two WGDs specific to Detarioideae and Papilionoideae (Fig. 2d), or other models with two WGDs. When adding a third Papilionoideae-specific WGD, the LRT score of 25.76 suggests that this three-WGD model is significantly better at the 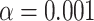 confidence level (
confidence level (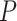 value
value 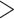 9.550, see Rabier et al. 2014) (Fig. 2b). Other models with three WGDs received lower likelihood scores (Fig. 2e). The second best-scoring three-WGD model is that with independent WGDs in Caesalpinioideae, Detarioideae, and Papilionoideae corresponding to the results of Cannon et al. (2015) and Stai et al. (2019). Adding a fourth WGD on the legume crown node (Fig. 2c) further improves the likelihood, but the LRT score of 7.94 is only significant at a lower confidence level of
9.550, see Rabier et al. 2014) (Fig. 2b). Other models with three WGDs received lower likelihood scores (Fig. 2e). The second best-scoring three-WGD model is that with independent WGDs in Caesalpinioideae, Detarioideae, and Papilionoideae corresponding to the results of Cannon et al. (2015) and Stai et al. (2019). Adding a fourth WGD on the legume crown node (Fig. 2c) further improves the likelihood, but the LRT score of 7.94 is only significant at a lower confidence level of 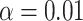 (
(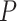 value
value 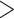 5.412, see Rabier et al. 2014). Alternative placement of a fourth WGD within legumes (Fig. 2f) has a lower likelihood than placing it on the legume crown node and received an LRT score of 1.16 which is not significant even at
5.412, see Rabier et al. 2014). Alternative placement of a fourth WGD within legumes (Fig. 2f) has a lower likelihood than placing it on the legume crown node and received an LRT score of 1.16 which is not significant even at 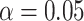 (
(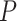 value
value 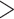 2.706, see Rabier et al. 2014).
2.706, see Rabier et al. 2014).
Figure 2.

Possible placements of legume WGD events on the species tree and their log-likelihoods based on the gene count method implemented in WGDgc. Top row: Models with the highest likelihood scores for a) two WGDs, b) three WGDs, and c) four WGDs, with LRT scores indicated above the arrows between each panel. Bottom row: d) The second most likely model with two WGDs, e) The three next most likely models with three WGDs, from left to right: the model corresponding to results from Cannon et al. (2015) and Stai et al. (2019); an alternative model to b) with a shared WGD for Caesalpinioideae, Dialioideae, and Papilionoideae; and the model with a pan-legume WGD as suggested by the Phyparts and Notung analyses (Fig. 1), f) The second most likely model with four WGDs. The WGD subtending Populus and Salix in the outgroup taxa is not shown but was included in all analyses. Caes  Caesalpinioideae, Cerc
Caesalpinioideae, Cerc  Cercidoideae, Detar
Cercidoideae, Detar  Detarioideae, Dial
Detarioideae, Dial  Dialioideae and Pap
Dialioideae and Pap  Papilionoideae. Circles represent WGDs, the numbers above them indicate the estimated duplicate gene retention rates.
Papilionoideae. Circles represent WGDs, the numbers above them indicate the estimated duplicate gene retention rates.
Distinguishing between Auto- and Allopolyploidy along the Legume Backbone
An allopolyploid event along the legume backbone could provide an alternative explanation for the high numbers of gene duplications mapping to the legume crown node. Only one or a few subfamilies need to be derived from such an event for duplicate gene copies to map to the legume crown node if the parental lineages of the polyploid diverged at the base of the family. Under this scenario, no pan-legume WGD would be inferred and the subfamilies could each be subtended by independent WGDs and be ancestrally non-polyploid as suggested by Cannon et al. (2015) and Stai et al. (2019). Alternatively, a WGD could be shared across two or more subfamilies. In the filtered supernetwork, complex tangles of “boxed” relationships coincide with the putative placements of WGDs inferred with Phyparts, Notung, and WGDgc: at the bases of Papilionoideae, Detarioideae, and the family as a whole (Fig. 3). This suggests that at least three WGDs occurred early in the evolution of the legumes, one of which occurred along the backbone before or among the first divergences in the family. For most subfamilies, however, there is little reticulation involving the root edges, except in Caesalpinioideae, suggesting that (at least) this subfamily could have resulted from an allopolyploid event.
Figure 3.

A filtered supernetwork drawn with the Convex Hull algorithm shows tangles of gene tree relationships at the bases of the legumes, and subfamilies Detarioideae and Papilionoideae, that correspond to WGDs, as well as possible reticulation at the base of Caesalpinioideae. The filtered supernetwork was inferred from the 1103 1-to-1 ortholog gene tree set, and only bipartitions that received more than 80% bootstrap support in gene tree analyses were included. Edge lengths and colors are by their weight, a measure of prevalence of the bipartition that the edge represents among the gene trees. Ellipses with dashed outlines indicate increased complexity at putative locations of WGDs.
GRAMPA identified eight MUL trees representing allopolyploid events (Fig. 4a–f), that had lower (better) reconciliation scores than the singly labeled species tree (Fig. 4g). MUL trees with just autopolyploidy (Fig. 4h,i) received higher (worse) scores. The two best-scoring MUL trees (Fig. 4a) included an allopolyploid event involving Cercidoideae or Detarioideae as the second parental lineage for the clade combining the other three sampled subfamilies. The same second parental lineages are implied in the fourth and fifth best-scoring trees, for the Caesalpinioideae + Papilionoideae clade (Fig. 4c). Given that strong gene tree conflict was observed among the orthologs analyzed by Koenen et al. (2020), these MUL trees may receive better scores due to ILS and/or gene tree estimation errors. The only low scoring MUL tree with an independent allopolyploid event restricted to Caesalpinioideae (Fig. 4f) scored only slightly better than the singly labeled tree (Fig. 4g). The remaining low scoring MUL trees involve a shared allopolyploidy event for Caesalpinioideae and Papilionoideae (Fig. 4b,e) or one in which it is shared with Dialioideae (Fig. 4d). The lowest scoring of these involves an allopolyploid event subtending Caesalpinioideae + Papilionoideae with the second parental lineage stemming from a divergence that occurred before the first legume dichotomy in the species tree (Fig. 4b), in line with the high number of duplications mapped onto the legume crown node in the Phyparts and Notung analyses (Fig. 1). An allopolyploid event shared by Caesalpinioideae and Papilionoideae is also in line with the high likelihood of a WGD on the node uniting these subfamilies obtained with WGDgc (Fig. 2).
Figure 4.

Hypotheses involving allopolyploidy derived from GRAMPA and their reconciliation scores compared to hypotheses involving only autopolyploidy. (a)–(f) All eight allopolyploid hypotheses that gave lower (better) reconcilation scores than (g), which represents the null hypothesis with no allopolyploidy. Hypotheses involving an additional autopolyploid event in Caesalpinioideae (h), or at the legume crown node (i), lead to higher (worse) reconciliation scores. Large circles indicate putative allo- or autopolyploidy events accounted for in the analysis (as per the legend), small circles indicate autopolyploid events in Papilionoideae and Detarioideae that were not taken into account and removed from the input gene trees prior to the analysis. Solid lines represent the species tree topology; dashed lines connect to the putative second parental lineage of the allopolyploid, with hypothetical extinct lineages indicated with a 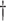 . Caes
. Caes  Caesalpinioideae, Cerc
Caesalpinioideae, Cerc  Cercidoideae, Detar
Cercidoideae, Detar  Detarioideae, Dial
Detarioideae, Dial  Dialioideae and Pap
Dialioideae and Pap  Papilionoideae.
Papilionoideae.
Divergence Time Estimation
The oldest definitive fossil evidence of crown group legumes is from the Late Paleocene, consisting of bipinnate leaves from c. 58 Ma (Wing et al. 2009; Herrera et al. 2019) and papilionoid-like flowers from c. 56 Ma (Crepet and Herendeen 1992), representing Caesalpinioideae and Papilionoideae, respectively. The older fossil woods with vestured pits, from the Early Paleocene of Patagonia (Brea et al. 2008) and the Middle Paleocene of Mali (Crawley 1988), could represent stem relatives of the family (vestured pits are found in Papilionoideae, Caesalpinioideae, and Detarioideae, so this is likely an ancestral legume trait). Similarly, early Paleocene (65.35 Ma) fossil fruits and leaflets from Colorado (described after our analyses were complete; Lyson et al. 2019) also represent ancestral legume characters and cannot be placed to subfamily. Therefore, based on fossil evidence, c. 58 Ma can be considered the minimum age of the legume crown node. Molecular age estimates (95% Highest Posterior Density (HPD) intervals) for the crown node range from 65.47–86.45 Ma to 73.46–81.18 Ma under the UCLN and RLC models, respectively, to minima and maxima between 64.63 and 68.85 Ma under various FLC models (Supplementary Table S3 available on Dryad), the latter suggesting a close association of initial legume diversification with the KPB (Fig. 5). Time-scaled trees for all clock analyses, annotated with 95% HPD intervals, are in Supplementary Figures S6–S13 available on Dryad; 95% HPD intervals for selected nodes are listed in Supplementary Table S3 available on Dryad.
Figure 5.

The origin of the legumes is closely associated with the KPB. a) Chronogram estimated with eight fixed local clocks (FLC8 model) in BEAST, with the clock partitions indicated by colored branches, from an alignment of 36 genes selected as both clock-like and highly informative and hence well-suited for dating analyses. Blue shading represents 500 post-burn-in trees (“densitree” plot) indicating posterior distributions of node ages. Yellow stars indicate putative legume WGD events, the placement of a putative allopolyploid event is equivocal and is indicated by two stars labeled with question marks (one on the stem lineage of the family and one on the stem lineage of Caesalpinioideae because the time-scaling analysis of gene duplications presented in (c) is based on this subfamily). Labeled circles indicate placements and ages of fossil calibrations listed in Table 1. Note that fossil A is placed on the legume stem node but postdates the median crown age estimates for the family and is therefore not plotted on the legume stem lineage (similar for fossils 27 and 38). b) Prior and posterior distributions for the crown age of legumes under different clock models, as indicated in the legend. c) Density plots of age estimates for duplication nodes in gene trees, for all duplications that mapped onto the legume crown node in the Notung analysis in gray and for duplications in the three well-sampled subfamilies Papilionoideae, Caesalpinioideae, and Detarioideae as indicated in the legend.
Placement of Eocene fossils of Detarioideae and Cercidoideae within the crown groups of those clades (Bruneau et al. 2008; Simon et al. 2009; Estrella et al. 2017), yields older crown age estimates for these clades. However, with these calibrations (alternative prior 1, Supplementary Table S2 available on Dryad), a 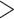 10-fold higher substitution rate along the stem lineages of these two subfamilies relative to the rates within both crown clades is inferred (c.
10-fold higher substitution rate along the stem lineages of these two subfamilies relative to the rates within both crown clades is inferred (c. 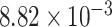 vs.
vs. 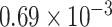 substitutions site
substitutions site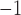 myr
myr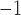 , with identical rates estimated independently for Cercidoideae and Detarioideae; Supplementary Fig. S14a available on Dryad). This rate is also nearly five times higher than the mean rate across the tree as a whole (
, with identical rates estimated independently for Cercidoideae and Detarioideae; Supplementary Fig. S14a available on Dryad). This rate is also nearly five times higher than the mean rate across the tree as a whole (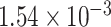 substitutions site
substitutions site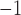 myr
myr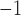 ), while the crown clades of these two subfamilies have estimated rates about half those of the mean. Analyses with the same clock partitioning but calibrated with Late Eocene Cercis fossils and Mexican amber (Hymenaea) as the oldest crown group evidence for Cercidoideae and Detarioideae, respectively, do not infer such strong substitution rate shifts, with all clock partitions estimated to have substitution rates ranging from
), while the crown clades of these two subfamilies have estimated rates about half those of the mean. Analyses with the same clock partitioning but calibrated with Late Eocene Cercis fossils and Mexican amber (Hymenaea) as the oldest crown group evidence for Cercidoideae and Detarioideae, respectively, do not infer such strong substitution rate shifts, with all clock partitions estimated to have substitution rates ranging from 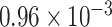 to
to 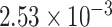 substitutions site
substitutions site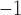 myr
myr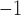 (Supplementary Fig. S14b available on Dryad). Either way, different placements of these fossils have little effect on the crown age estimates for the family in the FLC analyses (Supplementary Table S3, Figs. S11, S12, and S15h–j available on Dryad).
(Supplementary Fig. S14b available on Dryad). Either way, different placements of these fossils have little effect on the crown age estimates for the family in the FLC analyses (Supplementary Table S3, Figs. S11, S12, and S15h–j available on Dryad).
Age estimates for duplication nodes show that (at least) Caesalpinioideae and Papilionoideae are derived from one or more WGDs that occurred close to the KPB (Fig. 5c and Supplementary Fig. S16 available on Dryad). The WGD specific to Detarioideae appears to be more recent, in the Eocene (Fig. 5c and Supplementary Fig. S16 available on Dryad). The duplication nodes corresponding to the legume backbone inferred from the Notung analysis are likely a mixture of Detarioideae WGD duplications and older legume WGDs. This is surprising since it implies that Detarioideae paralogs do not always form sister clades in the gene trees, which could be caused by gene tree estimation errors or an allopolyploid origin for that subfamily. The large spread of ages for the duplication nodes (Fig. 5c) may be attributed to substitution rate variation across genes, which, in the absence of fossil calibrations, is unaccounted for. However, we note that in the case of allopolyploidy, the estimated ages of duplication nodes reflect the divergence time of the two parental lineages rather than the allopolyploid event itself, thereby overestimating the age of polyploidy.
Discussion
In this study, we investigate possible links between WGDs, lack of phylogenetic resolution surrounding the earliest rapid successive divergences within the Leguminosae (Koenen et al. 2020) and the mass extinction event at the KPB. The key findings are that many gene duplications are reconciled on the crown node of the legumes (Fig. 1) suggesting a WGD event shared by all subfamilies, while gene count data support shared paleopolyploidy of Caesalpinioideae and Papilionoideae (Fig. 2). These contrasting results can be reconciled by the inference of an allopolyploidization event shared by two or more subfamilies (Figs. 3 and 4). Furthermore, we show that this event and a further independent WGD restricted to Papilionoideae, as well as the rapid initial diversification of the family, probably coincided with the major biotic turnover associated with the mass extinction event at the KPB (Fig. 5). In combination, this series of events has resulted in considerable phylogenomic complexity which likely contributes to the difficulty of resolving deep-branching relationships among the legume subfamilies (Koenen et al. 2020). These insights, from one of the most evolutionarily successful post-KPB plant clades, suggest that the KPB was a pivotal moment for the origins of Cenozoic flowering plant diversity.
Paleopolyploidy in the Leguminosae
Our analyses provide evidence for at least three WGD events early in the evolution of legumes, one before or among the first divergences in the family, plus independent WGDs subtending subfamilies Detarioideae and Papilionoideae. Our results suggest two hypotheses for the oldest WGD event: i) it is placed on the stem lineage, representing a pan-legume WGD or ii) it involved allopolyploidy between two lineages derived from the first divergence within the family. The first hypothesis is supported by results from the Phyparts and Notung analyses (Fig. 1), while the WGDgc analysis only rejects a pan-legume WGD with the highest confidence interval in the LRT (Fig. 2). The second hypothesis is supported by the GRAMPA analysis (Fig. 4). Under the second hypothesis, duplicated genes would be reconciled onto the crown node of the family when using methods not accounting for allopolyploidy (Fig. 1). While this makes a pan-legume WGD less likely, all results show at least one WGD among the first divergences of the family (Figs. 1–4) shared across more than one subfamily, rather than restricted to a single subfamily. We show that it is unlikely that an independent WGD occurred in Caesalpinioideae (Figs. 1 and 2), including in the case of allopolyploidy (Fig. 4). Most evidence instead suggests that Caesalpinioideae and Papilionoideae, perhaps together with Dialioideae, share a WGD (Figs. 1b, 2a–c, and 4a–e), and that this was likely an allopolyploid event (Fig. 4a–e). This implies that subfamily Papilionoideae as a whole underwent two successive rounds of WGD, which is overwhelmingly supported by the gene count method (Fig. 2b), with even some modest support for three rounds of WGD (Fig. 2c), but with lower confidence.
It is possible that missing data due to inclusion of transcriptome data, rather than fully sampled genomes, influenced our analyses. In particular, for Dialioideae, where only a single transcriptome is sampled, it remains uncertain whether Dialioideae shares a WGD with Caesalpinioideae and Papilionoideae, or not. The gene count method is likely to be particularly sensitive to missing data, as it does not take gene tree topology into account, thereby potentially erroneously favoring a WGD shared by the better-sampled Caesalpinioideae and Papilionoideae rather than a pan-legume WGD (Fig. 2a,b). Missing data could also affect identification of which parental lineages were involved in an ancient allopolyploid event and which subfamilies are derived from it. However, given that GRAMPA takes gene tree topology into account, the inference that allopolyploidy is more likely than autopolyploidy is likely robust, and moreover, none of the other results reject allopolyploidy.
Apart from including more fully sequenced genomes, denser taxon sampling is also necessary to resolve the number and placement of WGDs with higher precision, accuracy and confidence. In particular, it will be desirable to include Poeppigia and Baudouinia or Eligmocarpus to span the first two divergences of Dialioideae (Zimmerman et al. 2017) and determine if a putative Dialioideae WGD was shared by all members of that subfamily, as well as Duparquetia orchidacea, the sole member of Duparquetioideae, for which nuclear genomic and cytogenetic data are lacking, its phylogenetic placement is based solely on chloroplast data (Koenen et al. 2020) and any potential history of polyploidy remains unknown.
Our results contrast with those of Cannon et al. (2015) and Stai et al. (2019) who suggested that all WGDs are restricted to individual subfamilies. The hypothesis of a pan-legume WGD contrasts most strongly with their hypothesis of four or five independent WGDs each confined to a single subfamily. An allopolyploid event shared across two or three subfamilies that excludes at least Cercidoideae and Detarioideae is more in line with the idea that Cercis has not undergone a WGD since the origin of the legumes (Stai et al. 2019). However, none of our results support a separate WGD restricted to Caesalpinioideae (which is well-sampled in our data sets) as inferred by Cannon et al. (2015), as well as in the analysis of WGDs across Viridiplantae by the One Thousand Plant Transcriptomes Initiative (2019). While the former study relied on 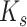 plots for inference of this particular WGD, the latter also used a Multi-tAxon Paleopolyploidy Search (MAPS) analysis of gene trees (Li et al. 2015). However, these analyses were performed for a total of 244 putative WGDs across the green plant phylogeny, using a standardized approach and including only six to eight taxa in each MAPS analysis (three ingroup and three outgroup taxa for the analysis of the putative Caesalpinioideae WGD) and without the sort of extensive gene tree filtering we performed here. Reanalysis of the One Thousand Plant Transcriptomes Initiative (2019) gene trees with Notung and Phyparts suggests that their data also do not support a Caesalpinioideae-specific WGD (Supplementary Appendix S2 available on Dryad).
plots for inference of this particular WGD, the latter also used a Multi-tAxon Paleopolyploidy Search (MAPS) analysis of gene trees (Li et al. 2015). However, these analyses were performed for a total of 244 putative WGDs across the green plant phylogeny, using a standardized approach and including only six to eight taxa in each MAPS analysis (three ingroup and three outgroup taxa for the analysis of the putative Caesalpinioideae WGD) and without the sort of extensive gene tree filtering we performed here. Reanalysis of the One Thousand Plant Transcriptomes Initiative (2019) gene trees with Notung and Phyparts suggests that their data also do not support a Caesalpinioideae-specific WGD (Supplementary Appendix S2 available on Dryad).
Estimating the Timeline of Legume Evolution
Our analyses suggest that the legume crown age dates back to the Maastrichtian or Early Paleocene, potentially within 1 or 2 million years before or after the KPB (Fig. 5, Supplementary Figs. S6–S13, Table S3 available on Dryad), although such high precision is unwarranted due to the idiosyncrasies of the molecular clock. These results update those of Lavin et al. (2005), Bruneau et al. (2008), and Simon et al. (2009) and provide the first age estimates for legumes based on nuclear genomic data. The FLC analyses (i.e., assuming 3, 6, or 8 different clade-specific substitution rates) even suggest that potentially only a single legume ancestor crossed the KPB giving rise to the six subfamilies during the early Paleocene, conforming to a “hard explosive” model. However, across the different analyses, part of the posterior density of crown age estimates spans the late Maastrichtian (Fig. 5), suggesting a “soft explosive” model, with the six subfamily lineages diverging in the Late Cretaceous, crossing the KPB, and giving rise to the modern subfamily crown groups in the Cenozoic. These different explosive models have been used to describe the origin and early diversification of placental mammals (Phillips 2015; Fig. 1). For birds, the timing of diversification relative to the KPB has also been controversial (Ksepka and Phillips 2015), but it now appears likely that Neoaves underwent explosive radiation from a single ancestor that crossed the KPB (Suh 2016). Apart from legumes, Placentalia, and Neoaves, also frogs (Feng et al. 2017), fishes (Alfaro et al. 2018), multiple lineages in Menispermaceae (Wang et al. 2012) and lichen-forming fungi (Huang et al. 2019) apparently all diversified rapidly following the KPB, suggesting this is a common pattern across organismal groups. We present here, to our knowledge, the first example of a major plant clade whose origin and initial diversification appears to be closely linked to the KPB (although we note that e.g., Rubiaceae (Antonelli et al. 2009) and Meliaceae (Koenen et al. 2015) have crown age estimates close to the KPB, but this does not appear to correlate with rapid initial diversification). Thus, even if extinction was less severe for plants than for animals at the KPB, the Paleocene was nevertheless a time of major origination of lineages across biota, and other examples of KPB-related accelerated plant diversification from larger angiosperm timetrees can be expected.
The FLC and strict clock models produce similar age estimates, but the RLC and UCLN models, which relax the clock assumption more, yield older divergence time estimates. By allowing independent substitution rates on all branches, the RLC and UCLN models are potentially overfitting the data to attempt to satisfy the marginal prior on node ages (Brown and Smith 2017). As inferred from analyses run without data, the marginal prior constructed across all nodes can be considered “pseudo-data” (Brown and Smith 2017) that are derived from interactions among the node calibration priors (based on fossil ages) and with the branching process prior (constant birth–death model in our case), and should therefore not overly inform node ages. FLC and strict clock models lend greater weight to the molecular data and can overrule marginal prior distributions on divergence times (Supplementary Fig. S15 available on Dryad) whilst still respecting hard maximum and minimum bounds of fossil constraints on calibrated nodes, as suggested by our results. It is also clear from running analyses without data, that the marginal age prior on the (uncalibrated) legume crown node is poorly informed, with the 95% HPD interval between 80.03 and 109.70 Ma (Fig. 5b and Supplementary Table S3 available on Dryad), the minimum of which is much older than the oldest legume fossils, presumably caused by overly conservative maximum bounds on calibrated nodes (Phillips 2015). UCLN and RLC analyses also inferred relatively high substitution rates for some deep branches in the outgroup during the Lower Cretaceous, relative to more derived and terminal branches (Supplementary Figs. S6 and S8 available on Dryad), presumably to satisfy the poorly informed marginal priors. Phillips (2015) suggested that setting less conservative maxima on priors could remedy this problem, but our analysis with such prior settings shows little effect (Supplementary Figs. S7 and S16k available on Dryad), with some of the deepest branches still showing much higher substitution rates. Since there is no evidence for, nor any reason to assume that substitution rates along those branches should be elevated relative to terminal branches, we conclude that this is caused by overfitting rate heterogeneity across branches under the influence of the marginal prior. Furthermore, the RLC analyses fitted c. 45 local clocks across the phylogeny, a high number relative to the 142 branches in the tree (implying a separate clock for every  3 branches on average), which is also indicative of overfitting. This could also be seen as evidence that the data are not the product of clock-like evolution, but it becomes difficult to estimate how much the clock deviates if the marginal prior on node ages is too influential. FLC analyses provide a more pragmatic approach by defining local clocks based on root-to-tip length distributions across clades and pruning outlier taxa (see Methods and Supplementary Fig. S5 available on Dryad). This approach largely accounts for the violation of the molecular clock but does not relax the clock such that the marginal prior on node ages is given excessive weight relative to the molecular signal. Furthermore, because the genes we selected are reasonably clock-like and highly informative, it is desirable that these data inform the node ages with sufficient weight. One drawback of using this approach is that the large amount of sequence data combined with the FLC model, results in unrealistically precise estimates.
3 branches on average), which is also indicative of overfitting. This could also be seen as evidence that the data are not the product of clock-like evolution, but it becomes difficult to estimate how much the clock deviates if the marginal prior on node ages is too influential. FLC analyses provide a more pragmatic approach by defining local clocks based on root-to-tip length distributions across clades and pruning outlier taxa (see Methods and Supplementary Fig. S5 available on Dryad). This approach largely accounts for the violation of the molecular clock but does not relax the clock such that the marginal prior on node ages is given excessive weight relative to the molecular signal. Furthermore, because the genes we selected are reasonably clock-like and highly informative, it is desirable that these data inform the node ages with sufficient weight. One drawback of using this approach is that the large amount of sequence data combined with the FLC model, results in unrealistically precise estimates.
Polyploidy (Senchina et al. 2003) as well as the KPB itself (Berv and Field 2018), have been implicated as potentially causing transient substitution rate increases, raising the possibility that substitution rates during early legume evolution could have deviated temporarily but markedly from the "background" rate of Cretaceous rosids. This would render ages inferred for the first few dichotomies and those of the subfamilies less certain. The age estimates inferred for these nodes rely on the assumption that the substitution rate did not vary significantly within clock partitions, and most importantly within the rosid partition which includes most of the backbone of the family and the stem lineage subtending it. The WGD events along the legume backbone and subtending subfamilies Papilionoideae and Detarioideae could have affected substitution rates along those branches. By selecting for smaller stature and shorter generation times and reducing population sizes (Berv and Field 2018), the KPB could additionally have prompted increased rates along some or all subfamily stem lineages, and, in the case of “hard” explosive diversification after the KPB, perhaps also along the legume stem lineage. A third factor that could influence node age estimates involving the first few legume divergences is extensive gene tree incongruence (Koenen et al. 2020), including among some of the 36 genes used for time-scaling. Divergence time analyses accommodate this incongruence within a single topology, meaning that additional substitutions are inferred for conflicting gene trees, which can inflate branch lengths between rapid speciation events (Mendes and Hahn 2016). Taken together, these three factors could mean that the time frame for early legume evolution appears too long in our results, with (some of the) subfamily ages likely being slightly older than estimated here, and divergence of the subfamilies happening nearly simultaneously (Koenen et al. 2020), rather than spanning the c. 3–5 million years inferred here (Fig. 5a and Supplementary Figs. S6–S13 available on Dryad). On the other hand, the time frame over which successive speciation events cause ILS depends primarily on the asymptotic effective population sizes (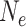 ) of the daughter species and their mean generation times, which can both be high for woody perennials, the most likely ancestral habit of Leguminosae. Reciprocal monophyly of sequences sampled from two species becomes highly likely when the number of generations since speciation is substantially larger than
) of the daughter species and their mean generation times, which can both be high for woody perennials, the most likely ancestral habit of Leguminosae. Reciprocal monophyly of sequences sampled from two species becomes highly likely when the number of generations since speciation is substantially larger than 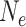 (Rosenberg 2003), which could require millions of years if
(Rosenberg 2003), which could require millions of years if 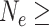 10,000 and the generation time
10,000 and the generation time 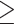 100 years. Substantial ILS (c. 30% gene trees deviating from the species tree) is well documented among genera Homo, Pan, and Gorilla (Scally et al. 2012) despite the 4 million years separating the two speciation events. Similar observations in plant groups with long generation times and moderately large
100 years. Substantial ILS (c. 30% gene trees deviating from the species tree) is well documented among genera Homo, Pan, and Gorilla (Scally et al. 2012) despite the 4 million years separating the two speciation events. Similar observations in plant groups with long generation times and moderately large 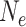 (Copetti et al. 2017; Chen et al. 2019) suggests this is also common in long-lived woody plants. Hence, the substantial gene tree conflict for the main legume lineages (Koenen et al. 2020) could be due to ILS assuming that successive speciation events occurred within a few millions of years, as inferred here (Fig. 5a and Supplementary Figs. S6–S13 available on Dryad).
(Copetti et al. 2017; Chen et al. 2019) suggests this is also common in long-lived woody plants. Hence, the substantial gene tree conflict for the main legume lineages (Koenen et al. 2020) could be due to ILS assuming that successive speciation events occurred within a few millions of years, as inferred here (Fig. 5a and Supplementary Figs. S6–S13 available on Dryad).
The placement of Cercidoideae and Detarioideae fossils within the stem or crown groups of these subfamilies, and hence the timing of their origins, remains uncertain (Supplementary Appendix S1 available on Dryad). Nevertheless, the new timeline for legume evolution presented here confirms the rapid diversification of legume lineages during the early Cenozoic as inferred by Lavin et al. (2005). While stem age estimates of each subfamily are remarkably close to each other, crown age estimates are strikingly different (Supplementary Table S3 available on Dryad). Caesalpinioideae are found to have the oldest crown age (late Paleocene), followed by Papilionoideae with a crown age in the Early Eocene. Overall, the subfamily age estimates suggest that early diversification of the legume subfamilies coincided with Paleocene biotic recovery, the Eocene climatic optima and Oligocene turnover in response to global cooling.
Angiosperm WGDs have been suggested to be non-randomly distributed through time and significantly clustered around the KPB (Fawcett et al. 2009; Vanneste et al. 2014; Lohaus and Van de Peer 2016). We show that two of the early legume WGDs are also temporally close to the KPB (Fig. 5), lending further support to the idea that polyploid survival and establishment were enhanced at or soon after the KPB with its associated rapid turnover of lineages (Lohaus and Van de Peer 2016; Levin and Soltis 2018). Polyploidy could have helped ancestral legumes and other plant lineages to both survive the mass extinction event and rapidly diversify owing to differential gene loss and other processes of diploidization (Adams and Wendel 2005; Dodsworth et al. 2016). On the other hand, many paleopolyploidy events significantly pre- and postdate the KPB and more extensive sampling of recently diversified groups may reveal a weaker pattern of KPB clustering, or a pattern of WGDs associated with episodes of rapid global change more generally (Cai et al. 2019; Levin 2020). Nevertheless, the timings of two WGDs as well as the initial diversification of the legumes close to the KPB (Fig. 5) are in line with the boundary being a pivotal moment in the evolutionary history of life on earth, selecting for polyploid lineages in plants (Lohaus and Van de Peer 2016) and leading to biotic turnover which initiated rapid diversification of lineages that would become dominant throughout the Cenozoic (Phillips 2015; Claramunt and Cracraft 2015; this study). Furthermore, the prevalence of WGDs across the plant tree of life (e.g., Wendel 2015; Soltis et al. 2016; Yang et al. 2018; Cai et al. 2019; Conover et al. 2019; One Thousand Plant Transcriptomes Initiative 2019), potentially in association with rapid environmental change more generally (Cai et al. 2019), as well as in relation to the diversification of several large clades (e.g., Jiao et al. 2012; Barker et al. 2016; this study), further emphasizes just how prevalent and important polyploidization has been for plant evolution.
The Added Complications of Paleopolyploidy on Evolutionary Inferences in Deep Time
Alongside rapid diversification and consequent lack of phylogenetic signal (Koenen et al. 2020), WGD events are also likely to contribute to the difficulties of resolving the deep nodes in Papilionoideae (Cardoso et al. 2012, 2013), Detarioideae (Estrella et al. 2018), and Leguminosae (Koenen et al. 2020). WGDs themselves may have promoted increased lineage diversification rates resulting in short internodes and ILS. If the polyploidy event happened some time before the first legume divergences, or in the case of allopolyploidy, divergence of gene copies happened prior to lineage splitting and orthology detection should be easier. However, if the polyploidy event happened immediately before rapid cladogenesis, a potentially large fraction of paralogous gene copies would not have diverged at this point, making orthology detection challenging. In either case, paralogous or homoeologous gene copies will have been differentially lost, pseudogenized or sub- or neo-functionalized, further complicating correct orthology detection (Wendel 2015; Cheng et al. 2018). Together with ILS, this could explain the large fraction of gene trees supporting alternative topologies at the root of the legumes (Koenen et al. 2020). An allopolyploid event involving two or more early legume lineages (Fig. 4) offers an alternative explanation for gene tree discordance, but discriminating between these alternatives is not straightforward. It is notable that other large plant clades, such as Pentapetalae (Zeng et al. 2017), Asteraceae (Barker et al. 2016; Huang et al. 2016), Brassicaceae (Couvreur et al. 2010; Huang et al. 2015), and Malvaceae (Conover et al. 2019), also show lack of resolution in clades subtended by WGDs similar to that revealed here for the legume family and subfamilies Papilionoideae and Detarioideae. This suggests that the association of polyploidy with rapid divergence, lack of phylogenetic signal, and gene tree conflict, is a common feature in the evolution of angiosperms and origination of major plant clades.
A large number of homolog clusters do not show gene duplications along the legume backbone or within any of the subfamilies, suggesting that loss of paralog copies is widespread, as observed for ancient WGDs more generally (Adams and Wendel 2005; Dehal and Boore 2005; Brunet et al. 2006; Scannell et al. 2007; Tiley et al. 2016). If many of those losses occurred along the stem lineages of the six subfamilies after their divergence, different paralog copies could have been retained in different lineages, adding to gene tree conflict. Loss of paralog copies along subfamily stem lineages will also complicate distinguishing whether a gene duplication corresponds to a WGD shared among two or more subfamilies, or a subfamily-specific nested WGD. Lack of support in homolog trees showing gene duplications further complicates this issue, making it extremely challenging to accurately reconstruct phylogenetic relationships and the history of WGDs. Given these difficulties, sampling a wider range of complete genomes will be important, since with transcriptome data it is unknown whether duplicate gene copies are lost or simply not expressed in tissues from which RNA was extracted. Furthermore, increased taxon sampling will counteract negative impacts of missing data, because some duplicate gene copies may have been lost in species sampled here, but not necessarily across the whole clade or subfamily which those species represent. Despite all these complications, our analyses allow us to reject some hypotheses such as an independent WGD subtending Caesalpinioideae, and to formulate a new hypothesis involving ancient allopolyploidy, potentially reconciling the large number of gene duplications inferred at the root of the legumes (Fig. 1) with the presumed non-polyploid history of Cercis within the legumes (Stai et al. 2019).
However, this hypothesis may well be an approximation of the full complexity of genome evolution and polyploidy that occurred in legumes in association with the KPB. These WGD events occurred c. 66 Ma and much evidence has been obscured by subsequent genome reorganization and loss of the large majority of duplicate gene copies. These issues limit the degree of complexity that can be reconstructed for such ancient events compared to more recently evolved polyploidy. For instance, many angiosperm polyploid complexes are known to have involved recurrent allo- and autopolyploidy yielding extremely complex genomic relationships and variable ploidy levels, for example, such as in the well-studied perennial soybean polyploid complex (e.g., Doyle et al. 2004). If a similar polyploid complex gave rise to the six major legume lineages, these could have had different ploidy levels with differing ancestries of subgenomes in cases of allopolyploidy.
Concluding Remarks
We show that the early evolution of the legumes followed a complex scenario with multiple nested auto- and/or allopolyploidy events, and rapid divergence of the six main lineages against the background of a mass extinction event that involved major turnover in the Earth’s biota and biomes. WGD likely contributed to the survival and evolutionary diversification of the legumes in the wake of the KPB, and to the rise to ecological dominance of legumes in early Cenozoic tropical forests. At the same time, these events make it difficult to reconstruct early legume evolutionary history, including evolutionary relationships, divergence times and the phylogenetic locations of WGD events themselves. The similarities between the origins of the legumes and those of other major Cenozoic clades such as mammals and birds are striking. All three of these prominent Cenozoic clades show recalcitrant basal polytomies and parallel trajectories of rapid early divergence closely associated with the KPB, further emphasizing the importance of the KPB mass extinction event and the earth system succession that followed in its aftermath (Hull 2015) in shaping the modern biota.
Acknowledgments
We thank the S3IT of the University of Zurich for the use of the ScienceCloud computational infrastructure and Robin van Velzen, Steven Cannon, Pascal-Antoine Christin, and two anonymous reviewers for constructive feedback that greatly improved the manuscript.
Supplementary Material
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.zkh18936s.
Funding
This work was supported by the Swiss National Science Foundation (Grants 31003A_135522 and 31003A_182453 to C.E.H.); the Department of Systematic & Evolutionary Botany, University of Zurich; the Natural Sciences and Engineering Research Council of Canada (Grant to A.B.), the U.K. National Environment Research Council (Grant NE/I027797/1 to R.T.P.), and the Fonds de la Recherche Scientifique of Belgium (Grant J.0292.17 to O.H.).
References
- Adams K.L., Wendel J.F.. 2005. Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 8(2):135–141. [DOI] [PubMed] [Google Scholar]
- Alfaro M.E., Faircloth B.C., Harrington R.C., Sorenson L., Friedman M., Thacker C.E., Oliveros C.H., Èerný D., Near T.J.. 2018. Explosive diversification of marine fishes at the Cretaceous–Palaeogene boundary. Nat. Ecol. Evol. 2:688–696. [DOI] [PubMed] [Google Scholar]
- Antonelli A., Nylander J.A., Persson C., Sanmartín I.. 2009. Tracing the impact of the Andean uplift on Neotropical plant evolution. Proc. Natl. Acad. Sci. USA 106:9749-9754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker M.S., Li Z., Kidder T.I., Reardon C.R., Lai Z., Oliveira L.O., Scascitelli M., Rieseberg L.H.. 2016. Most Compositae (Asteraceae) are descendants of a paleohexaploid and all share a paleotetraploid ancestor with the Calyceraceae. Am. J. Bot. 103:1203–1211. [DOI] [PubMed] [Google Scholar]
- Barreda V.D., Cúneo N.R., Wilf P., Currano E.D., Scasso R.A., Brinkhuis H.. 2012. Cretaceous/Paleogene floral turnover in Patagonia: drop in diversity, low extinction, and a Classopollis Spike. PLoS One 7(12):e52455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berv J.S., Field D.J.. 2018. Genomic signature of an Avian Lilliput Effect across the K–Pg extinction. Syst. Biol. 67(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brea M., Zamuner A.B., Matheos S.D., Iglesias A., Zucol A.F.. 2008. Fossil wood of the Mimosoideae from the early Paleocene of Patagonia, Argentina. Alcheringa. 32:427–441. [Google Scholar]
- Brown J.W., Smith S.A.. 2017. The past sure is tense: on interpreting phylogenetic divergence time estimates. Syst. Biol. 67:340–353. [DOI] [PubMed] [Google Scholar]
- Brown J.W., Walker J.F., Smith S.A.. 2017. Phyx: phylogenetic tools for unix. Bioinformatics 33:1886–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau A., Mercure M., Lewis G.P., Herendeen P.S.. 2008. Phylogenetic patterns and diversification in the caesalpinioid legumes. Botany 86:697–718. [Google Scholar]
- Brunet F.G., Crollius H.R., Paris M., Aury J.M., Gibert P., Jaillon O., Laudet V., Robinson-Rechavi M.. 2006. Gene loss and evolutionary rates following whole-genome duplication in teleost fishes. Mol. Biol. Evol. 23(9):1808–1816. [DOI] [PubMed] [Google Scholar]
- Cai, L., Xi, Z., Amorim, A.M., Sugumaran, M., Rest, J.S., Liu, L., Davis, C.C.. 2019. Widespread ancient whole-genome duplications in Malpighiales coincide with Eocene global climatic upheaval. New Phytol. 221: 565-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon S.B., McKain M.R., Harkess A., Nelson M.N., Dash S., Deyholos M.K., Peng Y., Joyce B., Stewart Jr C.N., Rolf M., Kutchan T.. 2015. Multiple polyploidy events in the early radiation of nodulating and non-nodulating legumes. Mol. Biol. Evol. 32(1):193–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon S.B., Sterc L., Rombauts S., Sato S., Cheung F., Gouzy J., Wang X., Mudge J., Vasdewani J., Schiex T., Spannagl M.. 2006. Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc. Natl. Acad. Sci. USA 103:14959–14964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso D., de Queiroz L.P., Pennington R.T., de Lima H.C., Fonty E., Wojciechowski M.F., Lavin M.. 2012. Revisiting the phylogeny of papilionoid legumes: New insights from comprehensively sampled early-branching lineages. Am. J. Bot. 99:1991–2013. [DOI] [PubMed] [Google Scholar]
- Cardoso D., Pennington R.T., de Queiroz L.P., Boatwright J.S., van Wyk B.-E., Wojciechowski M.F., Lavin M.. 2013. Reconstructing the deep-branching relationships of the papilionoid legumes. S. Afr. J. Bot. 89:58–75. [Google Scholar]
- Cascales-Miñana B., Cleal C.J.. 2014. The plant fossil record reflects just two great extinction events. Terra Nova 26:195–200. [Google Scholar]
- Chen J., Li L., Milesi P., Jansson G., Berlin M., Karlsson B., Aleksic J., Vendramin G.G., Lascoux M.. 2019. Genomic data provide new insights on the demographic history and the extent of recent material transfers in Norway spruce. Evol. Appl. 12:1539-1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F., Wu J., Cai X., Liang, J., Freeling M., Wang X.. 2018. Gene retention, fractionation and subgenome differences in polyploid plants. Nat. Plants 4: 258-268. [DOI] [PubMed] [Google Scholar]
- Christin P.-A., Spriggs E., Osborne C.P., Strömberg C.A.E., Salamin N., Edwards E.J.. 2014. Molecular dating, evolutionary rates, and the age of the grasses. Syst. Biol. 63:153–165. [DOI] [PubMed] [Google Scholar]
- Claramunt S., Cracraft J.. 2015. A new time tree reveals Earth history’s imprint on the evolution of modern birds. Sci. Adv. 1(11):e1501005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover J.L., Karimi N., Stenz N., Ané C., Grover C.E., Skema, C., Tate J.A., Wolff K., Logan S.A., Wendel J.F., Baum D.A.. 2019. A Malvaceae mystery: a mallow maelstrom of genome multiplications and maybe misleading methods? J Integrative Pl. Biol. 61: 12-31. [DOI] [PubMed] [Google Scholar]
- Cooper A., Penny D.. 1997. Mass survival of birds across the Cretaceous-Tertiary Boundary: molecular evidence. Science 275:1109–1113. [DOI] [PubMed] [Google Scholar]
- Copetti D., Búrquez A., Bustamante E., Charboneau J.L., Childs K.L., Eguiarte L.E., Lee S., Liu T.L., McMahon M.M., Whiteman N.K., Wing R.A.. 2017. Extensive gene tree discordance and hemiplasy shaped the genomes of North American columnar cacti. Proc. Natl. Acad. Sci. USA 114:12003-12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couvreur T.L.P., Franzke A., Al-Shehbaz I.A., Bakker F.T., Koch M.A., Mummenhoff K.. 2010. Molecular phylogenetics, temporal diversification, and principles of evolution in the mustard family (Brassicaceae). Mol. Biol. Evol. 27:55–71. [DOI] [PubMed] [Google Scholar]
- Crawley M. 1988. Palaeocene wood from the Republic of Mali. Bull. Br. Mus. (Nat. Hist.) Geol. 44:3–14. [Google Scholar]
- Crepet W.L., Herendeen PS.. 1992. Papilionoid flowers from the early Eocene of southeastern North America. In: Herendeen P.S., Dilcher D.L., editors. Advances in legume systematics part 4: The fossil record. Richmond, UK: Royal Botanic Gardens, Kew. p. 43–55. [Google Scholar]
- Criscuolo A., Gribaldo S.. 2010. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 10:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisp M.D., Cook L.G.. 2005. Do early branching lineages signify ancestral traits? Trends Ecol. Evol. 20: 122-128. [DOI] [PubMed] [Google Scholar]
- De Franceschi D., De Ploëg G.. 2003. Origine de l’ambre des faciés sparnaciens (Èocéne infèrieur) du Bassin de Paris: le bois de l’ambre producteur. Geodiversitas. 25:633–647. [Google Scholar]
- de la Estrella M., Forest F., Klitgård B., Lewis G.P., Mackinder B.A., de Queiroz L.P., Bruneau A.. 2018. A new phylogeny-based tribal classification of subfamily Detarioideae, an early branching clade of florally diverse tropical arborescent legumes. Sci. Rep. 8(1):6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Estrella M., Forest F., Wieringa J.J., Fougère-Danezan M., Bruneau A.. 2017. Insights on the evolutionary origin of Detarioideae, a clade of ecologically dominant tropical African trees. New Phytol. 214(4):1722–1735. [DOI] [PubMed] [Google Scholar]
- Dehal P., Boore J.L.. 2005. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 3(10):e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodsworth S, Chase M.W., Leitch A.R.. 2016. Is post-polyploidization diploidization the key to the evolutionary success of angiosperms? Bot. J. Linn. Soc. 180(1):1–5. [Google Scholar]
- dos Reis M., Donoghue P.C.J., Yang Z.. 2014. Neither phylogenomic nor palaeontological data support a Palaeogene origin of placental mammals. Biol. Lett. 10:20131003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle J.J., Doyle J.L., Rauscher J.T., Brown A.H.D.. 2004. Evolution of the perennial soybean polyploid complex (Glycine subgenus Glycine): a study of contrasts. Biol. J. Linn. Soc. 82(4):583-597. [Google Scholar]
- Drummond A.J., Suchard M.A., Xie D., Rambaut A.. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29:1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett J.A., Maere S., Van de Peer Y.. 2009. Plants with double genomes might have had a better chance to survive the Cretaceous – Tertiary extinction event. Proc. Natl. Acad. Sci. USA 106:5737–5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y.-J., Blackburn D.C., Liang D., Hillis D.M., Wake D.B., Cannatella D.C., Zhang P.. 2017. Phylogenomics reveals rapid, simultaneous diversification of three major clades of Gondwanan frogs at the Cretaceous–Paleogene boundary. Proc. Natl. Acad. Sci. USA 114(29):E5864–E5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herendeen, P.S. 1992. The fossil history of the Leguminosae from the Eocene of southeastern North America. In: Herendeen P.S., Dilcher D.L., editors. Advances in legume systematics part 4: The fossil record. Richmond, UK: Royal Botanic Gardens, Kew. p. 85–160. [Google Scholar]
- Herendeen P.S., Jacobs B.F.. 2000. Fossil legumes from the Middle Eocene (46.0 Ma) Mahenge Flora of Singida, Tanzania. Am. J. Bot. 87:1358–1366. [PubMed] [Google Scholar]
- Gregg W.T., Ather S.H., Hahn M.W.. 2017. Gene-tree reconciliation with MUL-trees to resolve polyploidy events. Syst. Biol. 66(6):1007-1018. [DOI] [PubMed] [Google Scholar]
- Herendeen P.S., Dilcher D.L.. 1992. Advances in legume systematics part 4. The fossil record. Richmond, UK: Royal Botanic Gardens, Kew. [Google Scholar]
- Herrera F., Carvalho M.R., Wing S.L., Jaramillo C., Herendeen P.S.. 2019. Middle to Late Paleocene Leguminosae fruits and leaves from Colombia. Aust. Syst. Bot. 32:385-408. [Google Scholar]
- Huang C.-H., Sun R., Hu Y., Zeng L., Zhang N., Cai L., Zhang Q., Koch M.A., Al-Shehbaz I., Edger P.P., Pires J.C., Tan D.-Y., Zhong Y., Ma H.. 2015. Resolution of Brassicaceae phylogeny using nuclear genes uncovers nested radiations and supports convergent morphological evolution. Mol. Biol. Evol. 33:394–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.-H., Zang C., Liu M., Hu Y., Gao T., Qi J., Ma H.. 2016. Multiple polyploidization events across Asteraceae with two nested events in the early history revealed by nuclear phylogenomics. Mol. Biol. Evol. 33:2820–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.P., Kraichak E., Leavitt S.D., Nelsen M.P., Lumbsch H.T.. 2019. Accelerated diversifications in three diverse families of morphologically complex lichen-forming fungi link to major historical events. Sci. Rep. 9:1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull P. 2015. Life in the aftermath of mass extinctions. Curr. Biol. 25:R941–R952. [DOI] [PubMed] [Google Scholar]
- Huson D.H., Bryant D.. 2005. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23(2):254-267. [DOI] [PubMed] [Google Scholar]
- Jacobs B.F., Herendeen P.S.. 2004. Eocene dry climate and woodland vegetation in tropical Africa reconstructed from fossil leaves from northern Tanzania. Palaeogeogr. Palaeocl. 213:115–123. [Google Scholar]
- Jarvis E.D., Mirarab S., Aberer A.J., Li B., Houde P., Li C., Ho S.Y., Faircloth B.C., Nabholz B., Howard J.T., Suh A.. 2014. Whole genome analyses resolve the early branches in the tree of life of modern birds. Science 346:1320–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetz W., Thomas G.H., Joy J.B., Hartmann K., Mooers A.O.. 2012. The global diversity of birds in space and time. Nature 491(7424):444–448. [DOI] [PubMed] [Google Scholar]
- Jia H., Manchester S.R.. 2014. Fossil Leaves and Fruits of Cercis L. (Leguminosae) from the Eocene of Western North America. Int. J. Plant Sci. 175:601–612. [Google Scholar]
- Jiao Y., Leebens-Mack J., Ayyampalayam S., Bowers J.E., McKain M.R., McNeal J., Rolf M., Ruzicka D.R., Wafula E., Wickett N.J., Wu X., Zhang Y., Wang J., Zhang Y., Carpenter E.J., Deyholos M.K., Kutchan T.M., Chanderbali A.S., Soltis P.S., Stevenson D.W., McCombie R., Pires J.C., Wong G.K.-S., Soltis D.E., DePamphilis C.W.. 2012. A genome triplication associated with early diversification of the core eudicots. Genome Biol. 13(1):R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller G. 2014. Deccan volcanism, the Chicxulub impact, and the end-Cretaceous mass extinction: coincidence? Cause and effect? In: Keller G., Kerr A.C., editors. Volcanism, impacts, and mass extinctions: causes and effects, vol. 505. Boulder (CO): Geological Society of America. p. 57–89. [Google Scholar]
- Koenen E.J., Clarkson J.J., Pennington T.D., Chatrou L.W.. 2015. Recently evolved diversity and convergent radiations of rainforest mahoganies (Meliaceae) shed new light on the origins of rainforest hyperdiversity. New Phytol. 207:327-339. [DOI] [PubMed] [Google Scholar]
- Koenen E.J.M., Ojeda D.I., Steeves R., Migliore J., Bakker F.T., Wieringa J.J., Kidner C., Hardy O.J., Pennington R.T., Bruneau A., Hughes C.E.. 2020. Large-scale genomic sequence data resolve the deepest divergences in the legume phylogeny and support a near-simultaneous evolutionary origin of all six subfamilies. New Phytol. 225:1355-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksepka D.T., Phillips M.J.. 2015. Avian diversification patterns across the K–Pg boundary: influence of calibrations, datasets, and model misspecification. Ann. Mo. Bot. Gard. 100(4):300–328. [Google Scholar]
- Lavin M., Herendeen P.S., Wojciechowski M.F.. 2005. Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the tertiary. Syst. Biol. 54:575–594. [DOI] [PubMed] [Google Scholar]
- Lavin, M., Wojciechowski, M.F., Gasson, P., Hughes, C. and Wheeler, E.. 2003. Phylogeny of robinioid legumes (Fabaceae) revisited: Coursetia and Gliricidia recircumscribed, and a biogeographical appraisal of the Caribbean endemics. Syst. Bot. 28:387–409. [Google Scholar]
- Levin D.A., Soltis D.E.. 2018. Factors promoting polyploid persistence and diversification and limiting diploid speciation during the K–Pg interlude. Curr. Opin. Plant Biol. 42:1–7. [DOI] [PubMed] [Google Scholar]
- Levin, D.A. 2020. Has the polyploid wave ebbed? Front. Plant Sci. 11: 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohaus R., Van de Peer Y.. 2016. Of dups and dinos: evolution at the K/Pg boundary. Curr. Opin. Plant Biol. 30:62–69. [DOI] [PubMed] [Google Scholar]
- LPWG (Legume Phylogeny Working Group). 2017. A new subfamily classification of the Leguminosae based on a taxonomically comprehensive phylogeny. Taxon 66:44–77. [Google Scholar]
- Li, Z., Baniaga, A.E., Sessa, E.B., Scascitelli, M., Graham, S.W., Rieseberg, L.H., Barker, M.S.. 2015. Early genome duplications in conifers and other seed plants. Sci. Adv. 1(10), p.e1501084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyson, T.R., Miller, I.M., Bercovici, A.D., Weissenburger, K., Fuentes, A.J., Clyde, W.C., Hagadorn, J.W., Butrim, M.J., Johnson, K.R., Fleming, R.F., Barclay, R.S.. 2019. Exceptional continental record of biotic recovery after the Cretaceous–Paleogene mass extinction. Science 366: 977-983. [DOI] [PubMed] [Google Scholar]
- Magallón S., Gómez-Acevedo S., Sánchez-Reyes L.L., Hernández-Hernández T.. 2015. A metacalibrated time-tree documents the early rise of flowering plant phylogenetic diversity. New Phytol. 207:437–453. [DOI] [PubMed] [Google Scholar]
- Mayrose I., Barker M.S., Otto S.P.. 2009. Probabilistic models of chromosome number evolution and the inference of polyploidy. Syst. Biol. 59(2):132-144. [DOI] [PubMed] [Google Scholar]
- McElwain J.C., Punyasena S.W.. 2007. Mass extinction events and the plant fossil record. Trends Ecol. Evol. 22:548–557. [DOI] [PubMed] [Google Scholar]
- McKey D. 1994. Legumes and nitrogen: The evolutionary ecology of a nitrogen-demanding lifestyle. In: Sprent J.I., McKey D., editors. Advances in legume systematics part 5. The nitrogen factor. Richmond, UK: Royal Botanic Gardens, Kew. p. 211–228. [Google Scholar]
- Mendes F.K., Hahn M.W.. 2016. Gene tree discordance causes apparent substitution rate variation. Syst. Biol. 65(4):711–721. [DOI] [PubMed] [Google Scholar]
- Meredith R.W., Janecka J.E., Gatesy J., Ryder O.A., Fisher C.A., Teeling E.C., Goodbla A., Eizirik E., Simão TL., Stadler T., Rabosky D.L.. 2011. Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification. Science. 334(6055):521–524. [DOI] [PubMed] [Google Scholar]
- Miller J.T., Murphy D.J., Ho S.Y.W., Cantrill D.J., Seigler D.. 2013. Comparative dating of Acacia: combining fossils and multiple phylogenies to infer ages of clades with poor fossil records. Aust. J. Bot. 61:436–445. [Google Scholar]
- Mudge J., Cannon S.B., Kalo P., Oldroyd G.E.D., Roe B.A., Town C.D., Young N.D.. 2005. Highly syntenic regions in the genomes of soybean, Medicago truncatula, and Arabidopsis thaliana. BMC Plant Biol. 5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary M.A., Bloch J.I., Flynn J.J., Gaudin T.J., Giallombardo A., Giannini N.P., Goldberg S.L., Kraatz B.P., Luo Z.X., Meng J., Ni X.. 2013. The placental mammal ancestor and the post–K-Pg radiation of placentals. Science 339(6120):662–667. [DOI] [PubMed] [Google Scholar]
- One Thousand Plant Transcriptomes Initiative, 2019. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574: 679-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E., Claude J., Strimmer K.. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20(2):289-290. [DOI] [PubMed] [Google Scholar]
- Paradis, E. 2013. Molecular dating of phylogenies by likelihood methods: a comparison of models and a new information criterion. Mol. Phylogenet. Evol. 67(2):436-444. [DOI] [PubMed] [Google Scholar]
- Phillips M.J. 2015. Geomolecular dating and the origin of placental mammals. Syst. Biol. 65(3):546–557. [DOI] [PubMed] [Google Scholar]
- Phillips M.J., Fruciano C.. 2018. The soft explosive model of placental mammal evolution. BMC Evol. Biol. 18:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poinar Jr G.O., Brown A.E.. 2002. Hymenaea mexicana sp. nov. (Leguminosae: Caesalpinioideae) from Mexican amber indicates Old World connections. Bot. J. Linn. Soc. 139:125–132. [Google Scholar]
- Prum R.O., Berv J.S., Dornburg A., Field D.J., Townsend J.P., Lemmon E.M., Lemmon A.R.. 2015. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 526:569–573. [DOI] [PubMed] [Google Scholar]
- Rabier C.E., Ta T., Ané C.. 2014. Detecting and locating whole genome duplications on a phylogeny: a probabilistic approach. Mol. Biol. Evol. 31(3):750-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A., Drummond A.J., Xie D., Baele G., Suchard M.A.. 2018. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67(5):901–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren L., Huang W., Cannon S.B.. 2019. Reconstruction of ancestral genome reveals chromosome evolution history for selected legume species. New Phytol. 223: 2090-2103. [DOI] [PubMed] [Google Scholar]
- Rosenberg N.A. 2003. The shapes of neutral gene genealogies in two species: probabilities of monophyly, paraphyly, and polyphyly in a coalescent model. Evolution 57(7):1465-1477. [DOI] [PubMed] [Google Scholar]
- Sanderson M.J. 2002. Estimating absolute rates of molecular evolution and divergence times: a penalized likelihood approach. Mol. Biol. Evol. 19(1):101-109. [DOI] [PubMed] [Google Scholar]
- Scally A., Dutheil J.Y., Hillier L.W., Jordan G.E., Goodhead I., Herrero J., Hobolth A., Lappalainen T., Mailund T., Marques-Bonet T., McCarthy S.. 2012. Insights into hominid evolution from the gorilla genome sequence. Nature 483(7388):169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannell D.R., Frank A.C., Conant G.C., Byrne K.P., Woolfit M., Wolfe K.H.. 2007. Independent sorting-out of thousands of duplicated gene pairs in two yeast species descended from a whole-genome duplication. Proc. Natl. Acad. Sci. USA 104(20):8397–8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senchina D.S., Alvarez I., Cronn R.C., Liu B., Rong J., Noyes R.D., Paterson A.H., Wing R.A., Wilkins T.A., Wendel J.F.. 2003. Rate variation among nuclear genes and the age of polyploidy in Gossypium. Mol. Biol. Evol. 20(4):633–643. [DOI] [PubMed] [Google Scholar]
- Silvestro D., Cascales-Miñana B., Bacon C.D., Antonelli A.. 2015. Revisiting the origin and diversification of vascular plants through a comprehensive Bayesian analysis of the fossil record. New Phytol. 207(2):425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M.F., Grether R., de Queiroz L.P., Skema C., Pennington R.T., Hughes C.E.. 2009. Recent assembly of the Cerrado, a Neotropical plant diversity hotspot, by in situ evolution of adaptations to fire. Proc. Natl. Acad. Sci. USA 106:20359–20364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S.A., Brown J.W., Walker J.F.. 2018. So many genes, so little time: a practical approach to divergence-time estimation in the genomic era. PLoS One 13(5):e0197433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S.A., Moore M.J., Brown J.W., Yang Y.. 2015. Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evol. Biol. 15:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis D.E., Visger C.J., Marchant D.B., Soltis P.S.. 2016. Polyploidy: pitfalls and paths to a paradigm. Am. J. Bot. 103:1146–1166. [DOI] [PubMed] [Google Scholar]
- Springer M.S., Meredith R.W., Teeling E.C., Murphy W.J.. 2013. Technical comment on “The placental mammal ancestor and the post–K-Pg radiation of placentals”. Science 341:613. [DOI] [PubMed] [Google Scholar]
- Stai J.S., Yadav A., Sinou C., Bruneau A., Doyle J.J., Fernández-Baca D., Cannon S.B.. 2019. Cercis: a non-polyploid genomic relic within the generally polyploid legume family. Front. Plant Sci. 10:345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML Version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolzer M., Lai H., Xu M., Sathaye D., Vernot B., Durand D.. 2012. Inferring duplications, losses, transfers and incomplete lineage sorting with nonbinary species trees. Bioinformatics 28(18):i409-i415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh A. 2016. The phylogenomic forest of bird trees contains a hard polytomy at the root of Neoaves. Zool. Scr. 45:50–62. [Google Scholar]
- Suh A., Smeds L., Ellegren H.. 2015. The dynamics of incomplete lineage sorting across the ancient adaptive radiation of neoavian birds. PLoS Biol. 13(8):e1002224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling E.C., Hedges S.B.. 2013. Making the impossible possible: rooting the tree of placental mammals. Mol. Biol. Evol. 30:1999–2000. [DOI] [PubMed] [Google Scholar]
- Tiley G.P., Ané C., Burleigh J.G.. 2016. Evaluating and characterizing ancient whole-genome duplications in plants with gene count data. Genome Biol. Evol. 8(4):1023-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuskan G.A., Difazio S., Jansson S., Bohlmann J., Grigoriev I., Hellsten U., Putnam N., Ralph S., Rombauts S., Salamov A., Schein J.. 2006. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science. 313(5793):1596–1604. [DOI] [PubMed] [Google Scholar]
- Vajda V., Bercovici A.. 2014. The global vegetation pattern across the Cretaceous–Paleogene mass extinction interval: A template for other extinction events. Global Planet Change 122:29–49. [Google Scholar]
- Vajda V., Raine J.I., Hollis C.J.. 2001. Indication of global deforestation at the Creataceous-Tertiary boundary by New Zealand fern spike. Science 294:1700–1702. [DOI] [PubMed] [Google Scholar]
- Vanneste K., Baele G., Maere S., Van de Peer Y.. 2014. Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the Cretaceous–Paleogene boundary. Genome Res. 24(8):1334–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Moore M.J., Soltis P.S., Bell C.D., Brockington S.F., Alexandre R., Davis C.C., Latvis M., Manchester S.R., Soltis D.E.. 2009. Rosid radiation and the rapid rise of angiosperm-dominated forests. Proc. Natl. Acad. Sci. USA 106(10):3853–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Ortiz R.D.C., Jacques F.M., Xiang X.G., Li H.L., Lin L., Li R.Q., Liu Y., Soltis P.S., Soltis D.E., Chen Z.D.. 2012. Menispermaceae and the diversification of tropical rainforests near the Cretaceous–Paleogene boundary. New Phytol. 195:470-478. [DOI] [PubMed] [Google Scholar]
- Wendel J.F. 2015. The wondrous cycles of polyploidy in plants. Am. J. Bot. 102:1753–1756. [DOI] [PubMed] [Google Scholar]
- Whitfield J., Cameron S.A., Huson D., Steel M.. 2008. Filtered Z-closure supernetworks for extracting and visualizing recurrent signal from incongruent gene trees. Syst. Biol. 57:939–947. [DOI] [PubMed] [Google Scholar]
- Wilf P., Johnson K.R.. 2004. Land plant extinction at the end of the Cretaceous: a quantitative analysis of the North Dakota megafloral record. Paleobiology 30:347–368. [Google Scholar]
- Wing S.L., Herrera F., Jaramillo C.A., Gómez-Navarro C., Wilf P., Labandeira C.C.. 2009. Late Paleocene fossils from the Cerrejón Formation, Colombia, are the earliest record of Neotropical rainforest. Proc. Natl. Acad. Sci. USA 106:18627–18632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y.X., Onstein R.E., Carter R.J., Stadler T., Linder H.P.. 2014. Fossils and a large molecular phylogeny show that the evolution of species richness, generic diversity and turnover rates are disconnected. Evolution 68:2821–2832. [DOI] [PubMed] [Google Scholar]
- Yang Y., Moore M.J., Brockington S.F., Mikenas J., Olivieri J., Walker J.F., Smith S.A.. 2018. Improved transcriptome sampling pinpoints 26 ancient and more recent polyploidy events within Caryophyllales, including two allopolyploidy events. New Phytol. 217:855–870. [DOI] [PubMed] [Google Scholar]
- Yang Y., Smith S.A.. 2014. Orthology inference in nonmodel organisms using transcriptomes and low-coverage genomes: improving accuracy and matrix occupancy for phylogenomics. Mol. Biol. Evol. 31:3081–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L., Zhang N., Zhang Q., Endress P.K., Huang J., Ma H.. 2017. Resolution of deep eudicot phylogeny and their temporal diversification using nuclear genes from transcriptomic and genomic datasets. New Phytol. 214:1338–1354. [DOI] [PubMed] [Google Scholar]
- Zimmerman E., Herendeen P.S., Lewis G.P., Bruneau A.. 2017. Floral evolution and phylogeny of the Dialioideae, a diverse subfamily of tropical legumes. Am. J. Bot. 104(7):1019-1041. [DOI] [PubMed] [Google Scholar]