

Abstract
In this work, we describe the association of a novel homozygous VPS11 variant with adult‐onset generalized dystonia, providing a detailed clinical report and biological evidence of disease mechanism. Vps11 is a subunit of the homotypic fusion and protein sorting (HOPS) complex, which promotes the fusion of late endosomes and autophagosomes with the lysosome. Functional studies on mutated fibroblasts showed marked lysosomal and autophagic abnormalities, which improved after overexpression of the wild type Vps11 protein. In conclusion, a deleterious VPS11 variant, damaging the autophagic and lysosomal pathways, is the probable genetic cause of a novel form of generalized dystonia. ANN NEUROL 2021;89:834–839
Dystonia is a hyperkinetic movement disorder characterized by sustained or intermittent muscle contractions causing abnormal movements and/or postures. If the trunk and at least 2 other sites are involved, dystonia is defined as generalized. 1 Inherited dystonias can be classified as isolated (dystonia is the only motor feature, except for tremor), combined (dystonia is associated with other movement disorders), or complex (dystonia co‐occurs with other neurologic or systemic manifestations). 2
Typically, isolated and combined dystonia have not characteristic features at brain magnetic resonance imaging (MRI). On the other hand, complex dystonia often shows pathognomonic MRI changes. Indeed, complex dystonia is one of the most frequent clinical presentations of neurodegeneration with brain iron accumulation (NBIA), which is a group of genetic disorders displaying progressive iron accumulation in basal ganglia. 2
Vps11 aggregates with other Vps proteins (ie, Vps16, Vps18, Vps33, Vps41, and Vps39) to form the “homotypic fusion and protein sorting complex (HOPS).” The HOPS complex promotes the fusion of late endosomes and autophagosomes with lysosomes. 3 , 4
Homozygous VPS11 mutations were associated with hypomyelinating leukodystrophy 12 (HLD12), characterized by appendicular spasticity, truncal hypotonia, opisthotonic posturing, and seizures. Brain MRIs of affected subjects present a thin corpus callosum and diffused hypomyelination. Two homozygous mutations were described so far (c.2536T>G p.C846G and c.1158_1184del p.L387‐G395del; Fig 1A). 5 , 6 , 7
FIGURE 1.

Genetic analysis, family pedigree, and brain MRI: (A) Vps11 protein domains with the positions of the pathogenic mutations already described and the deleterious variant presented here (underlined). (B) Pedigree of the family under study. Black symbol denotes the affected individual. Grey symbol indicates unknown status. (C) Brain magnetic resonance imaging (MRI) of the subject II.7. Axial T2 (1–3) and coronal FLAIR (4–6) display bilateral T2 hypointensity of substantia nigra, red nucleus, and globus pallidus. Axial FFE (7) shows bilateral pallidal hypointensity. (D) Homozygosity mapping plot displays 2 homozygosity peaks on chromosomes 6 and 11 in the proband. (E) Electropherograms of the VPS11 wild‐type (upper) and the c.136C>T homozygous variant (lower) of the proband (II.7). (F) Alignment of Vps11 protein homologs shows the conservation of the mutated amino acid (Proline 46).
Here, we describe a novel homozygous VPS11 variant probably causative of adult‐onset generalized dystonia. We provide strong evidence of variant deleteriousness and demonstrate its highly damaging impact on the autophagy‐lysosomal pathway.
Materials and Methods
Clinical Data
The subject underwent several neurological examinations, brain MRI, and neurophysiological studies. Blood samples and a skin biopsy were collected. The Ethics Committee of the IRCCS Foundation Ca' Granda Ospedale Maggiore Policlinico (Milan, Italy) approved the study. Written informed consent for publication of clinical details, clinical images, and video recording were obtained from the patient.
Genetic Analysis
The genomic DNA of the proband was analyzed by whole‐exome sequencing (WES) using the Nextera Rapid Capture Exome Library kit (Illumina) on the Illumina NextSeq500 platform. Reads alignment and variant calling/annotation were performed using standard procedures. The candidate VPS11 variant was validated by Sanger sequencing in the affected subject and his relatives. Homozygosity mapping was performed starting from WES data using the Homozygosity Mapper online tool (http://www.homozygositymapper.org/).
Cell Cultures and Plasmid Transfection
Skin fibroblasts of the patients and 3 controls were cultured in Dulbecco's Modified Eagle's Medium (DMEM) high glucose supplemented with 15% fetal bovine serum (Euroclone), 1% penicillin/streptomycin (Sigma‐Aldrich), and 1% of Amphotericin B (Sigma‐Aldrich). Patient fibroblasts were transfected through lipofectamine with a wild‐type VPS11 untagged plasmid (HG21081‐UT; Sinobiological).
Protein Blotting and Biochemical Studies
Western blot analyses were performed in triplicate on whole‐protein lysates from cultured fibroblasts using the following primary antibodies: Vps11 (HPA039020; Sigma‐Aldrich); actin (A2066; Sigma‐Aldrich), lysosome‐associated membrane glycoprotein 1 (LAMP1; ab25630; Abcam), β‐glucocerebrosidase (GBA; EPR5143 [3]; Abcam), α‐glucosidase (GAA; 02D05; Genzyme), acid ceramidase (ASAH1; sc‐28486; Santa Cruz Biotechnology), p62 (MABN130, Sigma‐Aldrich), LC3A/B (ab58610, Abcam), and Beclin‐1 (D40C5, Cell Signaling Technology). Enzyme activities of lysosomal and plasma membrane glycohydrolases were determined in total cell lysates using already published methods. 8 A 2‐tailed Student's t test was performed to assess the statistical significance of the protein amount and lysosomal activities differences in the patient compared to the fibroblasts of 3 controls.
Electron Microscopy
Approximately 5 × 106 fibroblasts were incubated with the fixing solution (glutaraldehyde 3%). Samples were dehydrated through graded alcohols, infiltrated, and embedded in Spurr's resin. Ultrathin sections (60 nm) were cut with Ultrotome Nova (LKB). Observations were performed using an EM 109 transmission electron microscope (Zeiss).
Results
Clinical Features
The family of the proband lived in a very small and geographically isolated village of Southern Italy for many generations. Family members considered the distant consanguinity of the parents to be very plausible (I.1 and I.2). The father of the proband died at the age of 42 because of a cerebral hemorrhage; the mother died at the age of 51 because of the complications of asthma due to bacterial pneumonia. Two of proband's brothers (II.2 and II.6) died within the first year of life for unknown reasons. All the siblings, which had by far passed their brother's age at onset, were reported to be healthy. All available siblings were neurologically examined and none of them displayed any sign of dystonia or neurological involvement (Fig 1B).
The proband was born at term after an uneventful pregnancy and had normal psychomotor development. From the age of 30 years, the proband developed progressive dystonic postures, initially affecting the left upper limb. In the following 5 years, dystonia progressively became generalized, involving the trunk, limbs, neck, and larynx. Moreover, progressive dysarthria and dysphagia appeared.
The proband reached our clinic at 40 years of age presenting with severe generalized dystonia affecting the trunk, face, neck (torticollis), and limbs (left > right), accompanied by moderate dysphagia and complete anarthria. Moderate lower limb hyperreflexia was also observed (Video). The proband did not display any evident major cognitive impairment; however, due to the limitations related to complete anarthria and severe motor impairment, an in‐depth neuropsychological assessment was not performed.
Brain MRI displayed mild brain atrophy with more conspicuous involvement of the basal ganglia. Interestingly, substantia nigra, red nucleus, and globus pallidus presented a marked symmetrical hypointensity in T2‐weighted sequences. The T2*‐weighted sequence showed symmetrical bipallidal hypointensity, which may be suggestive of iron accumulation (Fig 1C). Neurophysiological studies were unremarkable.
Therapeutic challenges with levodopa and anticholinergics did not bring any benefit. Botulinum toxin injections brought a slight improvement of the dystonic posture of the neck and left limbs. At the age of 50, the patient underwent bilateral deep brain stimulation of the internal globus pallidus (GPi DBS). No significant improvement of the dystonic features was observed after DBS (Video).
Genetic Analysis
The suspected consanguinity of the parents suggested a homozygous mutation as the cause of the disease. WES was performed on the proband. No pathogenic mutations were found in known disease genes for inherited dystonia and NBIA. A filtering analysis for rare (allele frequency < 0.1%) homozygous variants with protein impact was performed. Only the c.136C>T, p.P46S variant in VPS11 gene was identified. Rare biallelic variants were identified either in obvious noncandidate genes or were carried also by healthy siblings. The VPS11 variant lies on chromosome 11 in 1 of the 2 homozygous regions identified by homozygosity mapping (GRCh37: Chr 6: 30993880–31238851 and Chr11: 117985545–123454978; Fig 1E). Sanger sequencing confirmed the variant to be homozygous in the proband (II.7) and heterozygous in subjects II.3, II.4, and II.8 (Fig 1B, E).
The p.P46S VPS11 variant is novel and absent from public databases (ie, 1000G and gnomAD). The Proline at position 46 is highly conserved in mammalian orthologues (Fig 1F) and is localized in the Vps11 N‐terminal β‐propeller domain (see Fig 1A), which is known to be essential for protein–protein interactions. 9 Several in silico tools predict the VPS11 variant to be deleterious (Polyphen 2: Possibly Damaging 0.495; MutationTaster: Disease Causing 0.999; CADD‐PHRED: Deleterious 23.4). Moreover, the Pro > Ser substitution is nonconservative and the peculiar properties of Proline residues (eg, their rigid conformation with low flexibility) are frequently important to stabilize protein structure. For these reasons, VPS11 was selected as the candidate etiological gene.
Functional Studies
Immunoblot studies on total lysates from cultured fibroblasts showed a slight but statistically significant increase of Vps11 protein amount in mutated cells compared with controls. In addition, patient fibroblasts showed a striking increase in autophagic proteins p62, LC3A, and LC3B, with normal Beclin‐1 levels, and augmented lysosomal proteins and hydrolases (LAMP1, β‐glucocerebrosidase, acid α‐glucosidase, and acid ceramidase), suggesting a significant accumulation of the late‐endosomal and lysosomal components (Fig 2A).
FIGURE 2.
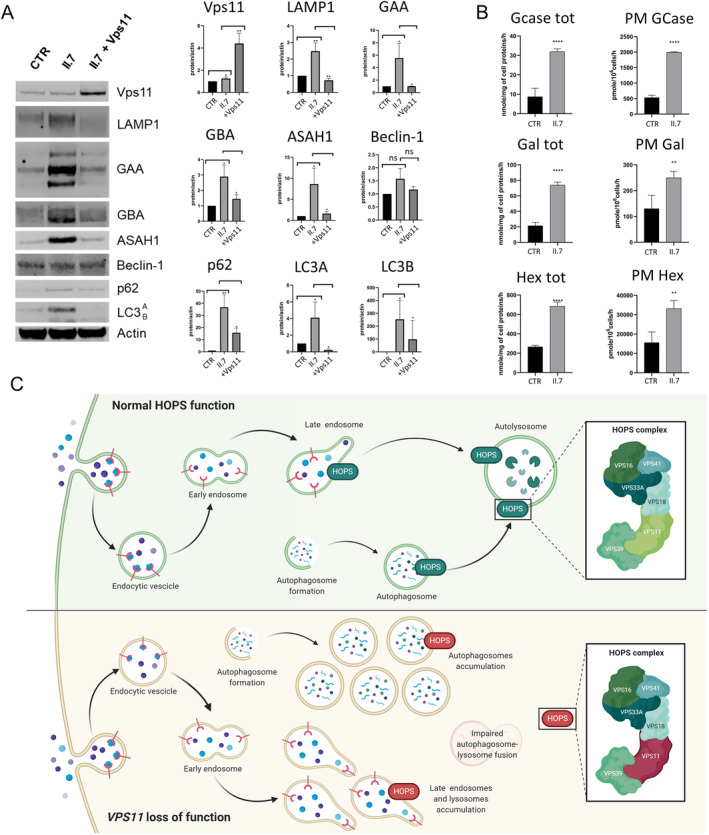
Immunoblot of autophagic and lysosomal proteins and lysosomal enzymatic activities: (A) Immunoblots of fibroblast lysates display a statistically significant increase of autophagic and lysosomal proteins in proband cells (II.7) compared with controls (CTR). The overexpression of wild‐type Vps11 protein in patient fibroblasts (II.7 + Vps11) ameliorates the abnormal phenotype. * = p < 0.05, ** = p < 0.02. (B) Lysosomal and plasmatic membrane enzymatic activities in patient fibroblasts (II.7) are significantly higher than in controls (CTR). GCase = β‐glucocerebrosidase, Gal = β‐galactosidase, Hex = β‐hexosaminidase, PM = plasma membrane, ** = p < 0.01, **** = p < 0.0001. (C) Cartoon model of the pathological autophagic and lysosomal functions associated with the identified VPS11 deleterious variant. The homozygous VPS11 loss‐of‐function variant causes a dysfunction of the homotypic fusion and protein sorting (HOPS) complex leading to a defect of the fusion of lysosomes with autophagosomes. Consequently, the accumulation of abnormal lysosomal and autophagic vesicles develops. Adapted from the template “Mutation of HOPS Complex Subunits”, by BioRender.com (2020), and from Steel D et al Ann Neurol 2020.
Transient overexpression of wild‐type Vps11 protein in patient fibroblasts significantly improved the pathological phenotype at the protein level, demonstrating the deleterious effect of the Vps11 p.P46S variant (see Fig 2A).
Lysosomal enzymatic activities in patient fibroblasts (ie, β‐glucocerebrosidase, β‐galactosidase, and β‐hexosaminidase) were 3 to 4 times higher than in controls. The plasma membrane enzymatic activities were also significantly increased, of about 2 to 3 times compared with controls (see Fig 2B).
Electron microscopy of the patient's fibroblasts showed large clear vacuolar structures, which were completely absent in control fibroblasts (Fig 3). This result appears to be consistent with a morphological alteration of the autophagic and/or endo‐lysosomal compartments, in line with the results of protein studies.
FIGURE 3.

Electron microscopy analysis: Patient fibroblasts (D, E, F) show large clear vacuolar structures in the cytoplasm (black arrows), which are absent in control fibroblasts (A, B, C), derived from 3 healthy subjects. This abnormality is consistent with an alteration of the autophagic and/or endo‐lysosomal compartments.
Discussion
The identification of a VPS11 deleterious variant in a patient with adult‐onset generalized dystonia and neurodegeneration confirms the important role of the lysosomal and autophagic pathways in the pathogenesis of neurodegenerative disorders. 7
In a previous study, knockdown of VPS11 gene in HeLa cells caused an increase of autophagy markers (ie, p62, LC3A, and LC3B). Moreover, the accumulation of immature autophagosomes and the reduction of autolysosomes were observed. These data suggest that VPS11 knockdown severely impairs autophagy flux. 6 In the present study, patient fibroblasts presented an overexpression of autophagic proteins p62 and LC3B without alteration of Beclin‐1 levels, suggesting an accumulation of autophagosomes without autophagy induction. Moreover, an increased amount and activity of lysosomal hydrolases suggested the accumulation of late endosomes and/or lysosomes. In addition, considering the increased plasma membrane lysosomal activities, these stored vesicles are probably partly exocytosed. The transfection of patient‐derived fibroblasts with the wild‐type VPS11 plasmid and consequent Vps11 overexpression ameliorated the pathological phenotype at the protein level, suggesting a pathogenic role of the identified VPS11 variant. These results are in line with previous VPS11 knockdown models, indicating that the p.P46S variant is probably a loss‐of‐function mutation (Fig 2C).
Hörtnagel et al conducted a microscope analysis on tissue samples derived from subjects affected with HLD12 carrying a homozygous VPS11 mutation (p.C846G). In these patients, electron microscopy on skin biopsies showed several intra‐cellular clear vacuoles. 7 In our study, electron microscopy analysis on the patient's fibroblasts showed a strikingly consistent result, suggesting that the cytoplasmic accumulation of clear vacuoles, consistent with an alteration of the endo‐lysosomal compartment, is the pathological hallmark of VPS11‐related disease.
Very interestingly, mutations of VPS16 and VPS41 genes were recently found to cause complex dystonia, often associated with brain abnormalities compatible with brain iron accumulation. 10 , 11 , 12 Vps16 and Vps41 associate with Vps11 to form the HOPS complex. Therefore, our work supports the hypothesis that the disruption of the HOPS normal function is crucial in the pathogenesis of this group of complex dystonias.
Our findings suggest that VPS11 mutations are associated with at least 2 different phenotypes: the infantile‐onset hypomyelinating leukodystrophy 12 (HLD12) and the later onset VPS11‐associated neurodegeneration. The identification of additional cases will help to disentangle the genotype–phenotype correlation in VPS11‐related disorders. Phenotypic heterogeneity associated with mutations of a single gene is not a novelty in the field of inherited dystonias; for example, PLA2G6 mutations can cause both Infantile Neuroaxonal Dystrophy (INAD) and the later onset PLA2G6‐related dystonia‐parkinsonism.
Intriguingly, lysosomal impairment was already associated with brain iron accumulation, such as in the case of WDR45 and ATP13A2 gene mutations, which are directly involved in autophagic and lysosomal functions, respectively. Moreover, lysosomes and autophagy play a central role in the cellular recycling of iron from aged organelles, such as damaged mitochondria. 13 One can speculate that the abnormality of lysosomal‐autophagic function described here probably adversely affects the handling of intracellular iron and could contribute to the process of neurodegeneration with brain iron accumulation. However, more studies are needed to comprehend the link between lysosomal‐autophagic dysfunction and neurodegeneration in VPS11‐associated neurodegeneration. In addition, neuropathological evidence would be necessary to definitively establish the nature of MRI abnormalities of this disorder.
In conclusion, this work strongly suggests the association of a recessive deleterious variant of the VPS11 gene with a novel form of generalized dystonia and provides in vitro evidence for a crucial role of the lysosomal‐autophagic pathway in the pathogenesis of this neurodegenerative disorder.
Author Contributions
E.M. and A.D.F. contributed to the conception and design of the study. E.M., F.C., L.B., S.S., L.S., F.B., G.F., M.M., M.G., C.E., M.A., and S.C. contributed to the acquisition and analysis of data. E.M., S.D., G.P.C., N.B., and A.D.F. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
The authors declared no conflict of interest.
Supporting information
Video S1. Supporting information
Acknowledgments
The authors thank all the family members for the collaboration in this study and the Associazione "Centro Dino Ferrari" for its support.
References
- 1. Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord 2013;28:863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Klein C, Lohmann K, Marras C, et al. GeneReviews®: hereditary dystonia overview. Seattle, WA: National Center for Biotechnology Information, 1993. [Google Scholar]
- 3. Peplowska K, Markgraf DF, Ostrowicz CW, et al. The CORVET tethering complex interacts with the yeast Rab5 homolog Vps21 and is involved in endo‐lysosomal biogenesis. Dev Cell 2007;12:739–750. [DOI] [PubMed] [Google Scholar]
- 4. Ostrowicz CW, Bröcker C, Ahnert F, et al. Defined subunit arrangement and Rab interactions are required for functionality of the HOPS tethering complex. Traffic 2010;11:1334–1346. [DOI] [PubMed] [Google Scholar]
- 5. Hörtnagel K, Krägeloh‐Mann I, Bornemann A, et al. The second report of a new hypomyelinating disease due to a defect in the VPS11 gene discloses a massive lysosomal involvement. J Inherit Metab Dis 2016;39:849–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang J, Lachance V, Schaffner A, et al. A founder mutation in VPS11 causes an autosomal recessive Leukoencephalopathy linked to Autophagic defects. PLoS Genet 2016;12:e1005848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Edvardson S, Gerhard F, Jalas C, et al. Hypomyelination and developmental delay associated with VPS11 mutation in Ashkenazi‐Jewish patients. J Med Genet 2015;52:749–753. [DOI] [PubMed] [Google Scholar]
- 8. Aureli M, Loberto N, Chigorno V, et al. Remodeling of sphingolipids by plasma membrane associated enzymes. Neurochem Res 2011;36:1636–1644. [DOI] [PubMed] [Google Scholar]
- 9. Behrmann H, Lürick A, Kuhlee A, et al. Structural identification of the Vps18 β‐propeller reveals a critical role in the HOPS complex stability and function. J Biol Chem 2014;289:33503–33512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van der Welle REN, Jobling R, Burns C, et al. VPS41 recessive mutation causes ataxia and dystonia with retinal dystrophy and mental retardation by inhibiting HOPS function and mTORC1 signaling. bioRxiv 2019. [Google Scholar]
- 11. Steel D, Zech M, Zhao C, et al. Loss‐of‐function variants in HOPS complex genes VPS16 and VPS41 cause early‐onset dystonia associated with lysosomal abnormalities. Ann Neurol 2020;88:867–877. 10.1002/ana.25879. [DOI] [PubMed] [Google Scholar]
- 12. Cai X, Chen X, Wu S, et al. Homozygous mutation of VPS16 gene is responsible for an autosomal recessive adolescent‐onset primary dystonia. Sci Rep 2016;6:25834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kurz T, Eaton JW, Brunk UT. The role of lysosomes in iron metabolism and recycling. Int J Biochem Cell Biol 2011;43:1686–1697. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Supporting information