

Abstract
Background & Aims
Non‐alcoholic fatty liver disease (NAFLD) has been associated with multiple metabolic abnormalities. By applying a non‐targeted metabolomics approach, we aimed at investigating whether serum metabolite profile that associates with NAFLD would differ in its association with NAFLD‐related metabolic risk factors.
Methods & Results
A total of 233 subjects (mean ± SD: 48.3 ± 9.3 years old; BMI: 43.1 ± 5.4 kg/m2; 64 male) undergoing bariatric surgery were studied. Of these participants, 164 with liver histology could be classified as normal liver (n = 79), simple steatosis (SS, n = 40) or non‐alcoholic steatohepatitis (NASH, n = 45). Among the identified fasting serum metabolites with higher levels in those with NASH when compared to those with normal phenotype were the aromatic amino acids (AAAs: tryptophan, tyrosine and phenylalanine), the branched‐chain amino acids (BCAAs: leucine and isoleucine), a phosphatidylcholine (PC(16:0/16:1)) and uridine (all FDRp < 0.05). Only tryptophan was significantly higher in those with NASH compared to those with SS (FDRp < 0.05). Only the AAAs tryptophan and tyrosine correlated positively with serum total and LDL cholesterol (FDRp < 0.1), and accordingly, with liver LDLR at mRNA expression level. In addition, tryptophan was the single AA associated with liver DNA methylation of CpG sites known to be differentially methylated in those with NASH.
Conclusions
We found that serum levels of the NASH‐related AAAs and BCAAs demonstrate divergent associations with serum lipids. The specific correlation of tryptophan with LDL‐c may result from the molecular events affecting LDLR mRNA expression and NASH‐associated methylation of genes in the liver.
Keywords: aromatic amino acids, DNA methylation, epigenetics, LDL cholesterol, NAFLD, NASH, non‐targeted metabolomics, tryptophan
Abbreviations
- AAAs
aromatic amino acids
- AAs
amino acids
- BCAAs
branched‐chain amino acids
- BMI
body mass index
- CpG
5'Cytosine—phosphate—Guanine3'
- CPM
count per million
- CVD
cardiovascular disease
- DNL
de novo lipogenesis
- FDR
false discovery rate
- HDL‐c
high density lipoprotein‐ cholesterol
- IQR
interquartile range
- KOBS
kuopio obesity surgery study
- LDL‐c
low density lipoprotein‐ cholesterol
- LDLR
low density lipoprotein receptor
- NAFLD
non‐alcoholic fatty liver disease
- NASH
non‐alcoholic steatohepatitis
- PC(16:0/16:1)
phosphatidylcholine(16:0/16:1)
- PCA
principal component analysis
- SD
standard deviation
- SS
simple steatosis
- T2D
type 2 diabetes
- TMM
trimmed mean of M values
- VLCD
very‐low calorie diet
Key points.
Using serum non‐targeted metabolomics, BCAAs and AAAs were identified to be different among persons with normal liver, SS and NASH; with only tryptophan differing between SS and NASH.
However, these NASH‐associated AAs display divergent association with the serum lipids with the aromatic amino acid, tryptophan correlating strongly with LDL‐c.
Specifically, the correlation of tryptophan with LDLR mRNA expression and NASH‐associated methylation of genes in the liver potentially links LDL‐c to both NASH development and CVD risk.
1. INTRODUCTION
The global epidemic burden of non‐alcoholic steatohepatitis (NASH) is increasing alarmingly. 1 NASH is a chronic progressive hepatic disease often preceded by non‐alcoholic fatty liver disease (NAFLD) and characterized by accumulation of fat, inflammation and possible fibrosis in the liver. 2
Dysregulation of hepatic lipid metabolism starts from the early phases of NAFLD and contributes to several cardiovascular disease risk factors including type 2 diabetes, peripheral insulin resistance and dyslipidemia. 3 , 4 , 5 Although there are many studies highlighting the presence of these risk factors in individuals with NAFLD and NASH, the exact mechanisms behind these risk factors are not fully elucidated. This is also highlighted by the fact that known NAFLD candidate genes have been associated with distinct metabolic profiles. 6 , 7 We have previously proposed that some of these abnormalities could be related to epigenetic modifications, mainly to altered DNA methylation in the liver. 8
Metabolomics approach has proposed various serum or plasma biomarkers for NAFLD. Of those, branched‐chain amino acids (BCAAs) and glutamic acid have been extensively studied. 9 , 10 , 11 Non‐targeted metabolite profiling, an approach that allows hypothesis‐free assessment of a wide spectrum of metabolites resulting from endogenous metabolism, dietary intake, and gut microbial activity, broadens the possibility of novel discoveries related to the pathogenesis of NAFLD. In the present study, we therefore utilized a non‐targeted approach to identify metabolic differences in serum among individuals with normal liver, simple steatosis (SS) and NASH. Furthermore, we investigated whether the serum metabolite profile that associates with NAFLD would differ in its association with NAFLD‐related metabolic risk factors.
2. METHODS
2.1. Study participants
Participants included in this metabolomics study were from the ongoing Kuopio Obesity Surgery Study (KOBS). 12 A total of 233 subjects (mean ± SD: 48.3 ± 9.3 years old; body mass index (BMI): 43.1 ± 5.4 kg/m2; 64 male) undergoing bariatric surgery were included upon the availability of both fasting serum sample and liver histology. Study subjects participated in a 1‐day visit, including an interview on the history of previous diseases and current drug treatment, before the surgery. Fasting blood samples were drawn after 12 hours fasting after 4 weeks of very low‐calorie diet (VLCD). Plasma glucose, insulin and serum lipids were determined as previously described. 12 Written informed consent was obtained from all participants and the study protocol was approved by the Ethics Committee of the Northern Savo Hospital District (54/2005, 104/2008, and 27/2010).
2.2. Liver histology
Liver biopsies were obtained using Trucut needles (Radiplast AB, Uppsala, Sweden) or as a wedge biopsy during elective gastric bypass operations. Overall histological assessment of liver biopsy samples (n = 233) was performed by one pathologist according to the standard criteria, as previously described. 13 Histological diagnosis was divided into three categories: (i) Normal liver without any steatosis, inflammation, ballooning or fibrosis, (ii) Simple steatosis (steatosis > 5%) without evidence of hepatocellular ballooning, inflammation or fibrosis and (iii) NASH. When the subjects were divided into study groups based on the phenotype of their liver histology; 79 had normal liver, 40 had simple steatosis and 45 had NASH (see Table 1). Still, 69 of 233 subjects could not be categorized into these distinct histological phenotypes (for example those with steatosis but also mild fibrosis). However, all participants (n = 233) were included into the correlation analyses (136 with steatosis; 97 with fibrosis; 72 with lobular inflammation; 7 with portal inflammation and 58 with ballooning).
Table 1.
Clinical characteristics and liver histology of study participants according to histological liver phenotype (mean ± SD or median (IQR))
| Normal liver | Simple steatosis | NASH | P a | |
|---|---|---|---|---|
| Total, N (men/women) | 79 (20/59) | 40 (9/31) | 45 (18/27) | .14 |
| Age (years) | 47.4 ± 9.7 | 46.5 ± 8.6 | 49.5 ± 9.6 | .31 |
| BMI (kg/m2) | 42.6 ± 5.5 | 43.3 ± 4.8 | 43.4 ± 5.4 | .61 |
| fS‐Total cholesterol (mmol/L) | 4.2 ± 0.8 | 4.2 ± 0.9 | 4.5 ± 1.1 | .27 |
| fS‐HDL cholesterol (mmol/L) | 1.2 ± 0.3 | 1.1 ± 0.2 | 1.2 ± 0.4 | .27 |
| fS‐LDL cholesterol (mmol/L) | 2.4 ± 0.7 | 2.4 ± 0.9 | 2.5 ± 1.0 | .80 |
| fS‐Triglycerides (mmol/L) | 1.3 (1.0‐2.3) | 1.4 (0.9‐2.0) | 1.6 (1.3‐2.2) | .60 |
| fP‐glucose (mmol/L) | 6.0 ± 1.3* | 6.3 ± 1.9** | 7.3 ± 2.2 | .0005 |
| fS‐insulin (mU/L) | 13.8 (7.8‐18.6)*** | 16.0 (11.0‐23.4) | 20.5 (14.4‐28.8) | .00006 |
| Type 2 diabetes, N(%) | 15 (18.9) | 11 (27.5) | 26 (57.8) | .00003 |
| Lipid lowering medication, N(%) | 22 (28.6) | 9 (22.5) | 20 (44.4) | .07 |
| Glucose lowering medication, N(%) | 14 (17.7) | 11 (25) | 24 (53.3) | .0002 |
| Steatosis grade, N | ||||
| <5% | 79 | 0 | 0 | |
| 5%‐33% | 0 | 32 | 13 | |
| 33%‐66% | 0 | 5 | 19 | |
| >66% | 0 | 3 | 13 | |
| Lobular inflammation, N | 0 | 0 | 45 | |
| Portal inflammation, N | 0 | 0 | 3 | |
| Ballooning, N | 0 | 0 | 37 | |
| Fibrosis, N (stage range) | 0 | 0 | 42 (1‐3) | |
Data are shown as mean ± SD or median (IQR).
fS, fasting serum; fP, fasting plasma; HDL, High density lipoprotein; LDL, Low density lipoprotein; N, number of individuals.
One‐way ANOVA test (continuous variable) or chi‐squared test (categorical variable), after Post hoc Bonferroni correction used for multiple testing.
P < .05 vs NASH,
P < .001 vs NASH,
P < .0001 vs NASH.
2.3. Non‐targeted LC‐MS metabolite profiling analysis
An aliquot (100 µL) of stored (−80°C) fasting serum samples was mixed with 400 µL of acetonitrile (ACN;VWR International, Belgium), and mixed in vortex at maximum speed for 15 seconds, incubated on ice bath for 15 minutes to precipitate the proteins, and centrifuged at 16 000 g, 10 minutes to collect the supernatant. The supernatant was filtered through 0.2 µm PTFE filters in a 96‐well plate. Aliquots of 2 µL were taken from at least half of the plasma samples, mixed in one tube and were used as the quality control sample in the analysis. Additionally, a solvent blank was prepared in the same manner. Small aliquots of the analytical samples were combined to constitute the quality control sample. The samples were analysed with a UHPLC‐qTOF‐MS system (1290 LC, 6540 qTOF‐MS, Agilent Technologies, Waldbronn, Karlsruhe, Germany). The samples were analysed using two different chromatographic techniques, reversed phase (RP) and hydrophilic interaction (hilic) chromatography. Data were acquired in both positive (+) and negative (−) electrospray ionization. The sample tray was kept at 4°C during the analysis. The data acquisition software was MassHunter Acquisition B.04.00 (Agilent Technologies). The quality control and the blank samples were injected after every 12 samples and in the beginning of the analysis. The sample order of the analysis of the samples was randomized. Details on the technical procedures, parameters and data collection are described in the Supplementary Methods.
2.4. Liver RNA expression and DNA methylation
To gain insight into the underlying molecular effects of the amino acids and differential effect on the lipid profile, liver LDL receptor (LDLR) mRNA expression in the same individuals (n = 179; from which 55 individuals (30%) did not have a clean phenotype of normal liver, SS or NASH) obtained from the existing liver RNA sequencing (Illumina HiSeq 2000) data 14 was analysed. Total RNA was extracted and purified using the miRNeasy Mini Kit. RNA sequencing libraries were constructed with the Ribo‐Zero Gold (Illumina) and underwent 50‐nucleotide long paired‐end sequencing on Illumina HiSeq 2500 machine. The reads were aligned against the human reference genome GRCh38 (release 29) using two‐pass STAR RNA‐aligner. 15 On average, 37.5 million (77.4%) read‐pairs were mapped to a unique site of the human genome and were used for further analysis. The Rsubread R package 16 was used to count all reads mapping within exon features. To reduce random variability resulting from low counts, we required at least 80% of samples to have at least 10 reads resulting in 15 243 genes in the final analysis.
The gene level count values were then normalized using a trimmed‐mean of M‐values (TMM), converted to count per million (CPM) and were logarithmically (log2) transformed using edgeR. 17 The technical factors were obtained from STAR RNA‐aligner (summary of mapping statistics) and from Picard 18 (quality metrics). Principal component analysis (PCA) was used to identify effects of confounding factors on the transcriptome. The following technical factors were included in the analysis: uniquely aligned reads % and 3′ bias. For DNA methylation studies, DNA from human liver biopsies was extracted using DNeasy blood and tissue kit (QIAGEN) as per the standard protocol provided by the manufacturer. The extracted DNA concentration and purity were measured followed by analysis of DNA methylation in liver using Infinium HumanMethylation 450 bead chip (Illumina), as described previously. 8 For the correlation analysis between liver DNA methylation and serum non‐targeted metabolomics, 58 participants (normal liver, 24; simple steatosis (SS), 19 and NASH, 14; in addition to one with only mild fibrosis) with both measurements available were used.
2.5. Statistical analysis and selection of metabolites to be identified
Data are presented as mean ± SD or median (interquartile range: IQR). In histological analyses, we primarily focused on subjects, who could be characterized into three study groups (normal liver, simple steatosis (SS) and NASH; n = 164), but when conducting correlation analyses with individual variables all study subjects were included (n = 233, see Methods).
For the non‐targeted metabolomics analyses, after data normalization, differential features across study groups were identified using a linear model fit separately for each feature using R version 3.5.3 (R Core Team, 2017, Vienna). The feature level was used as the dependent variable, and the group as the independent variable. The significance of the difference between the groups was tested using a type III F‐test. Results were adjusted for multiple comparisons using Benjamini‐Hochberg false discovery rate (FDR). FDR adjusted P < .05 were considered significant. After this filtering procedure, the differential metabolites were identified based on the MS/MS spectral comparison with pure standard compounds, or via search of the candidate compounds in the databases including the Human Metabolome database, METLIN, ChemSpider and SciFinder, and the results were verified with the MS/MS spectral features included in the in‐house database based on pure chemical compounds, public databases or from data reported in earlier publications. We also tested the significant findings for pairwise differences using pairwise t‐tests based on estimated marginal means as implemented in the ‘emmeans’ R‐package version 1.2., and further corrected for multiple testing using FDR. 19
For the metabolites that remained significant after the adjustments for multiple testing we further investigated their correlation with variables of interest (BMI, fasting plasma glucose, serum insulin and serum lipid levels, liver histology, LDLR expression and DNA methylation). For that, we calculated cross‐sectional correlations using Pearson's test for variables with normal distribution or for those natural‐log‐transformed to obtain normal distribution. For testing correlations with categorical liver histology data (steatosis grade, lobular inflammation, and fibrosis stage) Spearman's rank test were used. The results were adjusted for multiple comparisons using Benjamini‐Hochberg FDR for clinical variables, liver histology, liver mRNA expression and NASH‐associated DNA methylation independently. A two‐sided FDR P < .1 was considered statistically significant for all the correlation analyses performed, which were conducted with the SPSS version 25 program (IBM Inc, Armonk, NY).
3. RESULTS
3.1. Metabolites associated with NASH
The clinical characteristics of the participants in this study according to liver histology groups are shown in Table 1. While there were no significant differences in age, BMI or serum lipids between the study groups, individuals with normal liver had lower levels of fasting glucose (P < .05) and insulin (P < .0001) as compared to individuals with SS and NASH (all P < .05) Accordingly, in the normal liver group there were less participants with type 2 diabetes (T2D) and those using glucose lowering treatment compared to group with NASH (P < .0001).
From a total of 362 metabolic features nominally associated with NASH, we focused on the identified metabolites that remained associated with NASH at FDRp < 0.05 (linear regression model after correction for multiple testing, Table S1). Still, among the unidentified features significantly associated with NASH, we observed that the majority of them (six of 10) were lipids (Table S1).
The most prominent differences were found in amino acids (AAs), among them the aromatic AAs (AAAs) tryptophan, tyrosine and phenylalanine and BCAAs leucine and isoleucine (Figure 1). Serum levels for all these AAs were higher in individuals with NASH compared to those with normal liver (FDRp < 0.001). Tyrosine, phenylalanine, leucine and isoleucine were also increased in individuals with SS compared to those with normal liver (FDRp < 0.05). Tryptophan was the only AA significantly higher in those with NASH when compared to those with SS (FDRp < 0.05, Figure 1).
Figure 1.
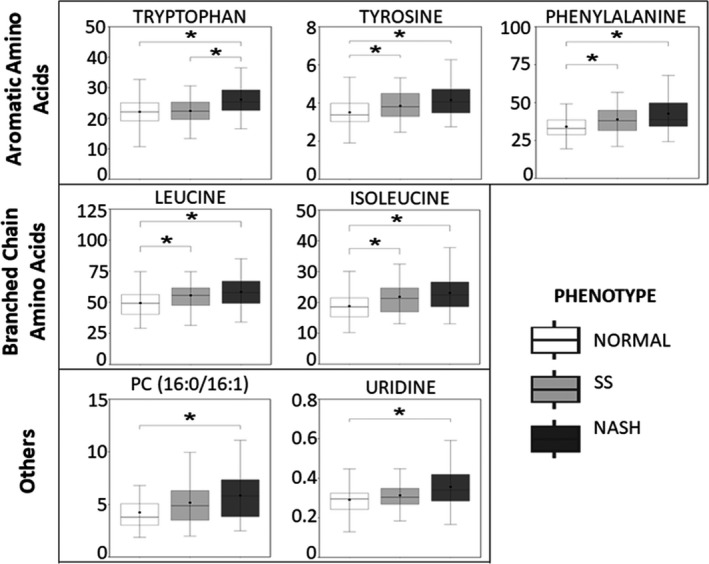
Top identified metabolites differing between participants with normal liver, simple steatosis (SS) and non‐alcoholic steatohepatitis (NASH). Each box plot represents the median peak area for individual metabolite as the centre line and whiskers for 1.5*IQR; mean is plotted as a dot. Significance between groups is shown by asterisk. *Pairwise test, FDRp < 0.05. Y‐axis represents MS peak area ×105
From metabolites other than AAs, phosphatidylcholine (PC(16:0/16:1)) and uridine were also higher in those with NASH when compared with individuals with normal liver. However, levels of these metabolites were not different between NASH and SS groups or between SS and normal liver groups (Figure 1). All the associations of NASH with the identified and non‐identified metabolic features remained significant (P < .05) after adjusting for age, sex and type 2 diabetes in the linear regression models (Table S1). Further adjustments for the use of either lipid or glucose lowering medication also did not change the results (Table S1).
3.2. AAAs, but not BCAAs, are related to serum total and LDL cholesterol (LDL‐c)
We next correlated the serum metabolite levels of the identified features with basic clinical characteristics (Figure 2). As expected, all the AAs that significantly associated with NASH correlated positively with fasting levels of insulin and triglycerides (FDRp < 0.1, Figure 2). Based on the selection process, all these top ranking identified metabolites were positively correlated with histological steatosis and inflammation (Figure 2). However, only tyrosine and tryptophan of the AAAs, but not the BCAAs, correlated positively with both total and LDL‐c (FDRp < 0.1, Figure 2). None of the NASH‐associated AAs correlated with BMI or glucose levels (FDRp > 0.1). Both PC(16:0/16:1) and uridine correlated positively with fasting serum levels of total cholesterol and triglycerides (FDRp < 0.01, Figure 2). After excluding those participants who were not categorized into having either normal liver, SS or NASH, the correlations remained essentially the same between each of the metabolites and the respective clinical characteristics (Table S2).
Figure 2.
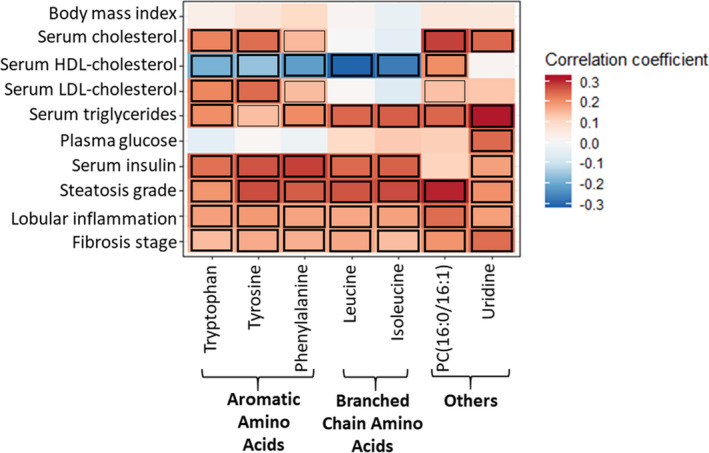
Heatmap representing the correlations between the clinical variables (row wise) and the top identified metabolites significantly associated with NASH (column wise). For all the clinical variables, Pearson's test was used except for the categorical histological variables (Steatosis grade, grade of lobular inflammation, and fibrosis stage) where Spearman's correlation test was used. For serum triglycerides and insulin log transformed values have been used. Colour of the cells indicates strength of a relationship (r). The cells marked with black borders in the heatmap demonstrate significant correlations (thick‐FDRp < 0.05; thin‐FDRp < 0.1). Abbreviations: HDL‐high density lipoprotein; LDL‐low density lipoprotein
3.3. Liver LDLR mRNA expression associated strongly with the AAAs tryptophan and tyrosine
Elevated LDL‐c levels in NAFLD have previously been associated with downregulation of the LDLR. 20 To gain insight into the possible molecular effects underlying the differential lipid profile associated with AAAs, we correlated liver LDLR mRNA expression with the AA levels. We observed that only the levels of tyrosine and tryptophan were significantly (FDRp < 0.1) and negatively correlated with liver LDLR mRNA expression (Figure 3). These correlations remained significant even after adjusting for the use of statin medication (56 of 179 individuals were statin users), which is known to directly alter LDLR expression. 21
Figure 3.
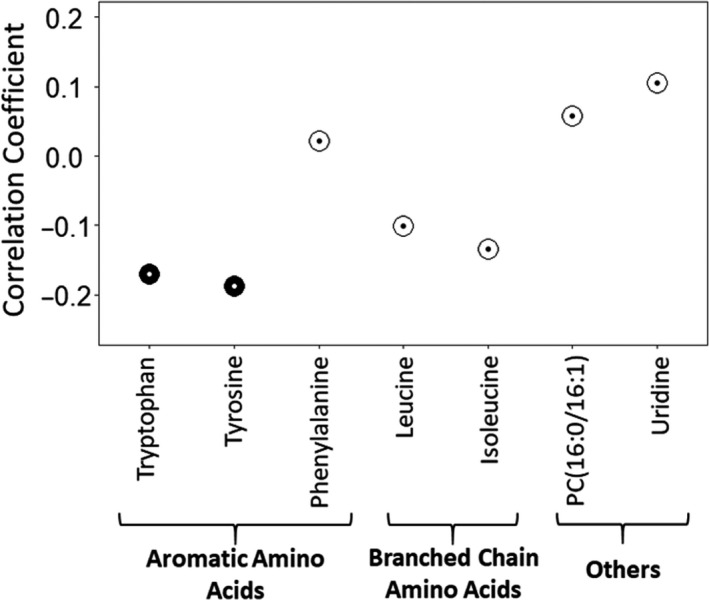
Pearson correlation coefficients between LDLR mRNA gene expression (based on RNA sequencing) and top identified metabolites significantly associated with NASH (n = 179). Each dot corresponds to the correlation coefficient for individual metabolite. The colour of the circle represents significance of the correlation: black (FDRp < 0.1); white (FDRp > 0.1)
To further explore the mechanism behind altered LDLR expression, we plotted correlations of tryptophan and tyrosine with average DNA methylation at CpG sites mapping to LDLR. Only tryptophan (r = 0.304, P = .021), but not tyrosine (r = 0.165, P = .217), correlated with average methylation at CpG sites annotated to this gene.
3.4. Tryptophan strongly correlates with liver methylation of CpG sites related to NASH
We have previously shown that NASH associates with differential DNA methylation in the human liver. 8 To explore whether this could link to divergent associations between AAs and NASH‐related metabolic phenotypes, we investigated the association of the AA levels with DNA methylation of all CpG sites from the Illumina array. In this approach we observed that all the AAs were nominally correlated (P < .05) with DNA methylation of a high number of CpG sites (Figure S1A). However, when we analysed correlations only with the previously published NASH‐associated methylated sites (FDRp < 0.001) 8 tryptophan differed from the other AAs because it correlated with larger number of CpG sites associated with NASH (P < .05; 1319 of 1605 CpG's) (Figure S1B). Of 1319 CpG's, 1287 remains significant even at FDRp < 0.1. The tryptophan‐specific association with DNA methylation is highlighted also when plotting correlations between the methylation levels of the top 30 NASH‐associated CpG sites with the levels of the AAs (Figure 4A). The names of the corresponding mapped genes related to these NASH‐related CpG sites are shown in Figure 4B. We next checked the correlations between methylation of NASH‐associated CpG sites and the tryptophan indole derivatives; indole propionic acid and indole acetic acid that we identified in the participant's serum. 22 As illustrated in Figure S2, only tryptophan correlated strongly with most of the respective CpG sites that were associated with NASH and not the indole derivatives.
Figure 4.
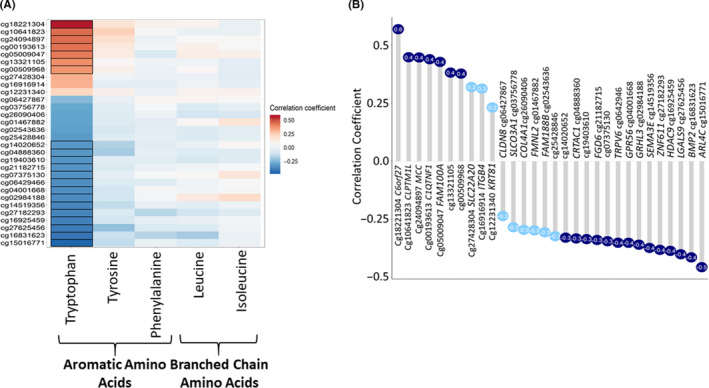
DNA methylation and tryptophan. (A) Correlation heatmap for NASH‐associated amino acids in serum with the top 30 NASH‐associated liver CpG sites. 8 Colour indicates strength of relationship (correlation coefficient): blue colour‐negative, red colour‐positive. Significant correlations (FDRp < 0.1) highlighted with black borders. (B) Lollipop plot representing the correlations of tryptophan with top 30 NASH‐associated CpG sites with corresponding genes mapped close to the CpG sites. Dark blue circles FDRp < 0.1 and light blue FDRp > 0.1
4. DISCUSSION AND CONCLUSIONS
In the current study, we applied non‐targeted metabolomics profiling of fasting serum of obese individuals with and without histologically proven NASH. We identified the AAAs tryptophan, tyrosine and phenylalanine along with the BCAAs leucine and isoleucine, in addition to PC(16:0/16:1) and uridine, as NASH‐associated metabolites. Although all these metabolites correlated with fasting insulin, only the AAAs tryptophan and tyrosine were positively correlated with fasting LDL‐c indicating specific pathways related to these AAs. Interestingly, LDLR liver mRNA expression levels correlated with the levels of these AAs, indicating a plausible link with the LDL dysregulation in NASH. Furthermore, only tryptophan correlated with both DNA methylation of LDLR and other NASH‐specific DNA methylation sites in the liver, suggesting that links between tryptophan and liver DNA methylation may explain, at least in part, the divergent associations of NASH‐related metabolites with metabolic outcomes.
Our observation of higher AA levels in serum of subjects with NASH and SS than in subjects with normal livers (Figure 1) are in line with earlier reported associations between AAs and NAFLD. 23 , 24 Both AAAs and BCAAs are found to be increased in individuals with NAFLD and NASH, and similar to our results, many of them (such as leucine, isoleucine and tyrosine) have been correlated with insulin resistance and steatosis. 25 , 26 BCAAs and tyrosine have also been linked with hepatic fat accumulation in early stages of NAFLD, independently of insulin resistance. 24 It has also been suggested that higher rate of whole‐body protein turnover and impaired AA catabolism occurring as a late event in the progression of steatosis to NASH 23 may be the cause of higher levels of all these AAs in the circulation. 26 , 27 Furthermore, altered gut permeability present in NAFLD and NASH may drive the diet‐ or gut‐derived metabolites (eg the AAs) readily into the systemic circulation resulting in their elevated levels. 28
Especially AAAs as a group have been associated with the prevalence of liver diseases. 29 , 30 One possible explanation for increased AAAs in serum may be the overall impaired hepatic metabolism of AAAs because of lipid accumulation in the liver. 24 , 31 In line with this, we found serum levels of the AAAs tryptophan, phenylalanine and tyrosine to be elevated in those with NASH compared to those with normal liver (Figure 1). Earlier, phenylalanine, the substrate for tyrosine in the body, has been found to be elevated in obesity, NAFLD and NASH. 23 , 25 , 32 , 33 On the other hand, serum tyrosine levels has also been associated with insulin resistance and with fibrotic staging of NASH. 34 Interestingly, we found specifically tryptophan levels to be higher in serum of individuals with NASH than in those with either SS or normal livers, but not between those with SS and normal livers as shown for the other AAs. This highlights the possible role of tryptophan specifically to the histological findings characterizing NASH, such as fibrosis or liver inflammation.
The most interesting novel finding in our study is the specific correlations of tryptophan and tyrosine with LDL and total cholesterol (Figure 2). Their levels in serum correlated with liver expression of LDLR suggesting a potential mechanism for elevated LDL cholesterol (Figure 3). Downregulation of hepatic LDLR mRNA in subjects with NAFLD has been suggested to contribute to increased amount of LDL cholesterol in the circulation. 20 Taken together, these results indicate a probable crosstalk between tryptophan, tyrosine and the LDL biology. Therefore, our findings suggest that tryptophan may contribute to both CVD risk and NASH by modifying LDL biology.
Interestingly, only serum tryptophan levels were associated with the average liver methylation of LDLR specific CpG sites suggesting that this AA could be acting at the epigenetic level by modifying DNA methylation. 35 , 36 Furthermore, after exploring correlations between identified NASH‐associated metabolites and methylation of CpG sites related to NASH, we observed that only tryptophan correlated significantly with the previously shown top 30 NASH‐specific DNA‐methylation sites 8 (Figure 4A,B). This specific link between tryptophan and DNA methylation could be related to the structure of tryptophan, which differs from the other related AAAs tyrosine and phenylalanine by the presence of an indole moiety. 37 We could not find any correlations of NASH‐associated CpG sites with the levels of the other identified indole derivatives, indole propionic acid and indole acetic acid. However, the possibility of regulation of DNA methylation by indole moieties still remains because indole derivatives are known to be active DNA methylation modulators. 38
Besides being positively associated with NASH in the present study, serum levels of the BCAAs leucine and isoleucine are strongly associated with the HDL‐c and fasting insulin levels (Figure 2). This has also been reported before 39 , 40 suggesting a link between HDL‐c and mechanisms that lead to insulin resistance. 33 , 39 Regarding BCAAs in NASH, disrupted crosstalk between BCAAs and the liver tri‐carboxylic acid cycle have been reported previously suggesting a contribution to dysfunctional mitochondrial metabolism in NAFLD. 41 Our results thus support the earlier findings that elevated levels of BCAAs could contribute to insulin resistance and development of NASH. 40 , 42
Another interesting finding was that among the lipid features associated with NASH, the levels of PC(16:0/16:1) were higher in individuals with NASH than in the ones with SS and normal livers. This lipid has not been previously linked to NAFLD, but proposed as a biomarker for hepatocellular carcinoma. 43 Lipids synthesized mostly from carbohydrates in the liver by de novo lipogenesis (DNL) are potential sources contributing to fatty liver. In fact, fasting levels of PC(16:0/16:1) seem to reflect both the increase in DNL and dietary carbohydrate‐fat ratio. 44 This goes in line with the fact that carbohydrate restriction has emerged, along with other measures, an effective dietary intervention for NAFLD, 45 and that short‐term carbohydrate restriction results in improved liver lipid metabolism and decreased PC(16:0/16:1) in plasma. 46 , 47 Therefore, the identification of higher PC(16:0/16:1) levels in NASH in our study could be reflecting both the advanced stage of liver disease already present in NASH as well as the imbalance in glucose and lipid metabolism in these patients. Thus, PC(16:0/16:1) could be a potential marker to be further investigated as a modifiable factor in the prevention of NAFLD/NASH.
Finally, we also found serum uridine levels to be higher in individuals with NASH compared to those with normal liver. Uridine has been established as an intrinsic link between pyrimidine metabolism and liver lipid accumulation. 48 Long‐term feeding of uridine has been shown to induce glucose intolerance and severe hepatic lipid accumulation in mice. 49 In humans, the evidence relating uridine with fatty liver is scarce, and limited to specific situations such as to the carbohydrate‐restricted diet in obese patients with NAFLD. 47
The current study is of cross‐sectional nature and thus limits our conclusions related to causation. Furthermore, because our study subjects were morbidly obese, we also cannot generalize the results to normal weight subjects. Our patients in the study were on a VLCD for 4 weeks before the surgery. Therefore, we cannot conclude that the results would be the same without preceding VLCD. However, we had a range of liver phenotypes from normal liver to NASH, based on liver histology, and therefore could explore factors related to NAFLD and related metabolic phenotypes within an obese population, known to be at high risk for NAFLD. Even though we had limited sample size for the analyses where we included liver gene expression and DNA methylation data, the results provided us a clear information about the possible mechanism linking NASH‐associated metabolites with metabolic outcomes. However, the availability of data about intrahepatic metabolite content would have provided us with better understanding about the mechanism linking NASH‐associated metabolites with metabolic outcomes at tissue‐level. Although through this technology we were able to bring new hits associated with NASH, such as PC(16:0/16:1), some of the lipid features derived from non‐targeted metabolomics that were altered in NASH are yet to be identified.
In conclusion, serum levels of the NASH‐related AAAs and BCAAs demonstrate divergent associations with serum lipids. Despite similar associations with steatosis and fasting insulin, the differences in the association with serum lipids, especially total and LDL cholesterol, suggest AA‐specific pathways. Although the correlation of tryptophan with LDL metabolism and hepatic LDLR mRNA expression suggest that LDL metabolism could mediate the association of tryptophan with NASH, we acknowledge that also other AA correlated with LDL‐c, but not so strongly with NASH. Thus, the unique strong correlation of tryptophan with methylation of NASH‐related CpG sites suggests a novel mechanism linking tryptophan with LDL‐c, CVD risk and development of NASH.
5. ETHICS APPROVAL STATEMENT AND PATIENT CONSENT STATEMENT
Written informed consent was obtained from all participants and the study protocol was approved by the Ethics Committee of the Northern Savo Hospital District (54/2005, 104/2008, and 27/2010) and were in accordance with the Helsinki Declaration.
CONFLICT OF INTEREST
The authors have no relevant conflict of interest to disclose.
AUTHOR CONTRIBUTIONS
VDd.M. and RS researched data, collected the clinical data and wrote the manuscript; VM researched and collected the data; AK, and KH analysed and interpreted the metabolomics data; DK, ZM, and PP provided and analysed the RNA sequencing data; CL, EN and AP were responsible for the DNA methylation data and reviewed the manuscript; JP designed the study, collected the clinical data and reviewed the manuscript. All the authors have critically read, reviewed and approved the final version of the manuscript.
Funding information
Kuopio Obesity Surgery Study (PI JP) was supported by the Finnish Diabetes Research Foundation, Kuopio University Hospital Project grant (EVO/VTR grants 2005‐2019), the Academy of Finland grant (Contract no. 138006), the Finnish Cultural Foundation and the University of Eastern Finland Spearhead Funding. RS was supported by GenomMed Doctoral Programme, co‐funded by Horizon 2020 Framework Programme of the European Union (Marie Skłodowska Curie grant agreement no. 740264). DK was supported by the Academy of Finland (Contract no. 316458). PP and ZM were supported by the National Institutes of Health (NIH) grants HL‐095056, HL‐28481 and U01 DK105561.
Supporting information
Supplementary Material
ACKNOWLEDGEMENT
The authors acknowledge CSC–IT Center for Science, Finland, for computational resources.
de Mello VD, Sehgal R, Männistö V, et al. Serum aromatic and branched‐chain amino acids associated with NASH demonstrate divergent associations with serum lipids. Liver Int.2021;41:754–763. 10.1111/liv.14743
Vanessa D. de Mello and Ratika Sehgal share the first authorship.
Handling editor: Stefano Romeo
REFERENCES
- 1. Povsic M, Wong OY, Perry R, Bottomley J. A structured literature review of the epidemiology and disease burden of non‐alcoholic steatohepatitis (NASH). Adv Ther. 2019;36(7):1574‐1594. 10.1007/s12325-019-00960-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liu W, Baker RD, Bhatia T, Zhu L, Baker SS. Pathogenesis of nonalcoholic steatohepatitis. Cell Mol Life Sci. 2016;73(10):1969‐1987. 10.1007/s00018-016-2161-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gastaldelli A, Cusi K. From NASH to diabetes and from diabetes to NASH: mechanisms and treatment options. JHEP Rep. 2019;1(4):312‐328. 10.1016/j.jhepr.2019.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wong CR, Lim JK. The association between nonalcoholic fatty liver disease and cardiovascular disease outcomes. Clin Liver Dis. 2018;12(2):39‐44. 10.1002/cld.721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Francque SM, van der Graaff D, Kwanten WJ. Non‐alcoholic fatty liver disease and cardiovascular risk: pathophysiological mechanisms and implications. J Hepatol. 2016;65(2):425‐443. 10.1016/j.jhep.2016.04.005 [DOI] [PubMed] [Google Scholar]
- 6. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461‐1465. 10.1038/ng.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dongiovanni P, Meroni M, Baselli G, et al. PCSK7 gene variation bridges atherogenic dyslipidemia with hepatic inflammation in NAFLD patients. J Lipid Res. 2019;60(6):1144‐1153. 10.1194/jlr.p090449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Mello VD, Matte A, Perfilyev A, et al. Human liver epigenetic alterations in non‐alcoholic steatohepatitis are related to insulin action. Epigenetics. 2017;12(4):287‐295. 10.1080/15592294.2017.1294305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pirola CJ, Sookoian S. Multiomics biomarkers for the prediction of nonalcoholic fatty liver disease severity. World J Gastroenterol. 2018;24(15):1601‐1615. 10.3748/wjg.v24.i15.1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Camps J, Joven J. Metabolite profiling can change health‐care delivery to obese patients with fatty liver disease: the search for biomarkers. Clin Chem Lab Med. 2017;55(4):501‐506. 10.1515/cclm-2016-0762 [DOI] [PubMed] [Google Scholar]
- 11. Gitto S, Schepis F, Andreone P, Villa E. Study of the serum metabolomic profile in nonalcoholic fatty liver disease: research and clinical perspectives. Metabolites. 2018;8(1):17. 10.3390/metabo8010017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pihlajamäki J, Grönlund S, Simonen M, et al. Cholesterol absorption decreases after roux‐en‐Y gastric bypass but not after gastric banding. Metabolism. 2010;59(6):866‐872. 10.1016/j.metabol.2009.10.004 [DOI] [PubMed] [Google Scholar]
- 13. Männistö VT, Simonen M, Soininen P, et al. Lipoprotein subclass metabolism in nonalcoholic steatohepatitis. J Lipid Res. 2014;55(12):2676‐2684. 10.1194/jlr.P054387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Benhammou JN, Ko A, Alvarez M, et al. Novel lipid long intervening noncoding RNA, oligodendrocyte Maturation‐Associated long intergenic noncoding RNA, regulates the liver steatosis gene Stearoyl‐Coenzyme A desaturase as an enhancer RNA. Hepatol Comm. 2019;3(10):1356‐1372. 10.1002/hep4.1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Comput Appl Biosci. 2013;29(1):15‐21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liao Y, Smyth GK, Shi W. The R package rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47(8):e47. 10.1093/nar/gkz114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA‐seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40(10):4288‐4297. 10.1093/nar/gks042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Broad Institute Picard Tools . Picard toolkit. [Accessed 16 December 2019]. Available from http://broadinstitute.github.io/picard. Updated 2019
- 19. Russell L, Henrik S, Jonathon L, Paul B, Maxime H. Emmeans: estimated marginal means, aka least‐squares means. R package version 1.4.6. Available from https://CRAN.R‐project.org/package=emmeans. https://github.com/rvlenth/emmeans. Updated 2020
- 20. Min H‐K, Kapoor A, Fuchs M, et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012;15(5):665‐674. 10.1016/j.cmet.2012.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu Q, Chen Y, Xu C. Statins and new‐onset diabetes mellitus: LDL receptor may provide a key link. Front Pharmacol. 2017;8:372. 10.3389/fphar.2017.00372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roager HM, Licht TR. Microbial tryptophan catabolites in health and disease. Nat Commun. 2018;9(1):3294‐3310. 10.1038/s41467-018-05470-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kalhan SC, Guo L, Edmison J, et al. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metab Clin Exp. 2011;60(3):404‐413. 10.1016/j.metabol.2010.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cheng S, Wiklund P, Autio R, et al. Adipose tissue dysfunction and altered systemic amino acid metabolism are associated with non‐alcoholic fatty liver disease. PLoS One. 2015;10(10):e0138889. 10.1371/journal.pone.0138889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gaggini M, Carli F, Rosso C, et al. Altered amino acid concentrations in NAFLD: impact of obesity and insulin resistance. Hepatology. 2018;67(1):145‐158. 10.1002/hep.29465 [DOI] [PubMed] [Google Scholar]
- 26. Goffredo M, Santoro N, Tricò D, et al. A branched‐chain amino acid‐related metabolic signature characterizes obese adolescents with non‐alcoholic fatty liver disease. Nutrients. 2017;9(7):642. 10.3390/nu9070642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Newgard C. Interplay between lipids and branched‐chain amino acids in development of insulin resistance. Cell Metab. 2012;15(5):606‐614. 10.1016/j.cmet.2012.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ray K. Leaky guts: intestinal permeability and NASH Nature reviews . Gastroenterol Hepatol. 2015;12(3):123. 10.1038/nrgastro.2015.15 [DOI] [PubMed] [Google Scholar]
- 29. Siomkajło M, Rybka J, Mierzchała‐Pasierb M, et al. Specific plasma amino acid disturbances associated with metabolic syndrome. Endocrine. 2017;58(3):553‐562. 10.1007/s12020-017-1460-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yoo HJ, Jung KJ, Kim M, et al. Liver cirrhosis patients who had normal liver function before liver cirrhosis development have the altered metabolic profiles before the disease occurrence compared to healthy controls. Front Physiol. 2019;10:1421. 10.3389/fphys.2019.01421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dejong CHC, van de Poll MCG, Soeters PB, Jalan R, Damink O, Steven WM. Aromatic amino acid metabolism during liver failure. J Nutr. 2007;137(6):1579S‐1585S. 10.1093/jn/137.6.1579s [DOI] [PubMed] [Google Scholar]
- 32. Swierczynski J, Sledzinski T, Slominska E, Smolenski R, Sledzinski Z. Serum phenylalanine concentration as a marker of liver function in obese patients before and after bariatric surgery. Obes Surg. 2008;19(7):883‐889. 10.1007/s11695-008-9521-z [DOI] [PubMed] [Google Scholar]
- 33. Newgard CB, An J, Bain JR, et al. A branched‐chain amino acid‐related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9(6):565‐566. 10.1016/j.cmet.2009.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miwa Kawanaka M, Nishino K, Oka T, et al. Tyrosine levels are associated with insulin resistance in patients with nonalcoholic fatty liver disease. Hepatic Med Evid Res. 2015;7:29‐35. 10.2147/HMER.S79100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Murphy SK, Yang H, Moylan CA, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology. 2013;145(5):1076‐1087. 10.1053/j.gastro.2013.07.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ahrens M, Ammerpohl O, von Schönfels W, et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease‐specific and remodeling signatures after bariatric surgery. Cell Metab. 2013;18(2):296‐302. 10.1016/j.cmet.2013.07.004 [DOI] [PubMed] [Google Scholar]
- 37. Palego L, Betti L, Rossi A, Giannaccini G. Tryptophan biochemistry: structural, nutritional, metabolic, and medical aspects in humans. J Amino Acids. 2016;2016:1‐13. 10.1155/2016/8952520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eider SSL, Dorleta OA, Del Carmen MMM, et al. Indole derivatives, pharmaceutical compositions containig such indoles and their use as DNA methylation modulators. Patent US2015336889A1. 2015.
- 39. Yang P, Hu W, Fu Z, et al. The positive association of branched‐chain amino acids and metabolic dyslipidemia in chinese han population. Lipids Health Dis. 2016;15(1):120. 10.1186/s12944-016-0291-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kujala UM, Peltonen M, Laine MK, et al. Branched‐chain amino acid levels are related with surrogates of disturbed lipid metabolism among older men. Front Med. 2016;3:57. 10.3389/fmed.2016.00057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sunny NE, Kalavalapalli S, Bril F, et al. Cross‐talk between branched‐chain amino acids and hepatic mitochondria is compromised in nonalcoholic fatty liver disease. Am J Physiol Endocrinol Metab. 2015;309(4):E311‐E319. 10.1152/ajpendo.00161.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Satapati S, Kucejova B, Duarte JAG, et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J Clin Investig. 2015;125(12):4447‐4462. 10.1172/jci82204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cotte AK, Cottet V, Aires V, et al. Phospholipid profiles and hepatocellular carcinoma risk and prognosis in cirrhotic patients. Oncotarget. 2019;10(22):2161‐2172. 10.18632/oncotarget.26738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Inoue M, Senoo N, Sato T, et al. Effects of the dietary carbohydrate–fat ratio on plasma phosphatidylcholine profiles in human and mouse. J Nutr Biochem. 2017;50:83‐94. 10.1016/j.jnutbio.2017.08.018 [DOI] [PubMed] [Google Scholar]
- 45. Rinella ME, Sanyal AJ. Management of NAFLD: A stage‐based approach. Nat Rev Gastroenterol Hepatol. 2016;13(4):196‐205. 10.1038/nrgastro.2016.3 [DOI] [PubMed] [Google Scholar]
- 46. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Investig. 2005;115(5):1343‐1351. 10.1172/JCI200523621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mardinoglu A, Wu H, Bjornson E, et al. An integrated understanding of the rapid metabolic benefits of a carbohydrate‐restricted diet on hepatic steatosis in humans. Cell Metab. 2018;27(3):559‐571.e5. 10.1016/j.cmet.2018.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Le TT, Ziemba A, Urasaki Y, Hayes E, Brotman S, Pizzorno G. Disruption of uridine homeostasis links liver pyrimidine metabolism to lipid accumulation. J Lipid Res. 2013;54(4):1044‐1057. 10.1194/jlr.m034249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Urasaki Y, Pizzorno G, Le TT. Chronic uridine administration induces fatty liver and pre‐diabetic conditions in mice. PLoS One. 2016;11(1):e0146994. 10.1371/journal.pone.0146994 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material