

Abstract
Aging, the greatest risk factor for Alzheimer's disease (AD), may lead to the accumulation of somatic mutations in neurons. We investigated whether somatic mutations, specifically in longer genes, are implicated in AD etiology. First, we modeled the theoretical likelihood of genes being affected by aging‐induced somatic mutations, dependent on their length. We then tested this model and found that long genes are indeed more affected by somatic mutations and that their expression is more frequently reduced in AD brains. Furthermore, using gene‐set enrichment analysis, we investigated the potential consequences of such long gene disruption. We found that long genes are involved in synaptic adhesion and other synaptic pathways that are predicted to be inhibited in the brains of AD patients. Taken together, our findings indicate that long gene–dependent synaptic impairment may contribute to AD pathogenesis.
Keywords: Alzheimer's disease, DNA damage, long genes, somatic mutations, synaptic adhesion
1. NARRATIVE
Dementia affects 50 million people worldwide, and Alzheimer's disease (AD) is the most common form of dementia, accounting for two‐thirds of all cases. 1 AD is a neurodegenerative disease that is characterized by a decline in memory and cognitive function. Symptoms worsen, become increasingly diverse and more impairing with age, and AD causes much distress for patients and their loved ones. In the best case, current treatments provide some symptom relief and give patients and their families more time to prepare for the inevitably declining disease trajectory. 2 , 3 Thus far, efforts to develop disease‐modifying medications for AD have unfortunately been unsuccessful due to lacking or incomplete knowledge of the (disturbed) biological processes underlying the disease. New insights into AD pathogenesis for better treatment development are therefore urgently needed.
In this article, we propose a new neurobiological mechanism underlying AD, namely, that somatic mutations that accumulate with aging especially affect long genes and lead to decreased long gene expression, which in turn results in disturbed synaptic function. We provide evidence for this hypothesis through analyzing publicly available somatic mutation and gene expression data that were generated and generously provided by other researchers, which therefore did not require any tissue processing from our side. We first give a short overview of the genetic risk factors associated with AD. Subsequently, we introduce and validate a theoretical model of how aging, the most important risk factor for AD, is associated with the accumulation of somatic mutations, particularly in longer genes. Then, we demonstrate that in AD brains, longer genes are more frequently affected by somatic mutations and show a reduced expression, which is predicted to lead to synaptic impairment. Finally, we make suggestions for future research that arise from these insights.
1.1. Inherited risk factors for AD
In a small percentage of AD cases, a clear monogenic cause is present. People carrying rare pathogenic variants (or mutations) in one of three genes—APP, PSEN1, or PSEN2—have a dominantly inherited form of AD with an early age at onset (<65 years). 4 Conversely, late‐onset AD (LOAD, ≥ 65 years) represents the vast majority of AD cases and has a multifactorial etiology: It is caused by the cumulative effect of multiple genetic risk factors combined with lifestyle and environmental factors. The strongest common genetic risk factor for LOAD is the ε4 allele of the apolipoprotein E (APOE) gene (APOEε4). 5 , 6 In addition, an increasing number of inherited common and rare risk variants have been associated with LOAD through so‐called genome‐wide association studies (GWASs) and whole exome/genome sequencing studies, respectively. 7 , 8 , 9 Typically, AD candidate genes and their possible involvement in disease progression have been interpreted in the framework of early onset AD mutations and the main pathological hallmarks of both early and late‐onset AD, that is, the development of extracellular plaques containing amyloid beta (Aβ) and intracellular tangles of hyperphosphorylated tau protein.
RESEARCH IN CONTEXT
Systematic review: The occurrence of somatic mutations with increasing age, the greatest risk factor for Alzheimer's disease (AD), has been hypothesized to occur in the brain and hence contribute to AD pathogenesis. We searched the literature (PubMed) for studies that used sequencing techniques to identify aging‐associated somatic mutations in brain regions and individual neurons of AD patients and healthy controls.
Interpretation: We found that aging‐associated somatic mutations in the brain more often affect longer genes. These long genes show reduced expression in AD brains and encode proteins that are involved in synaptic pathways that are inhibited in AD‐relevant brain regions, especially the hippocampus.
Future directions: To add to our understanding of the effect of long gene disruptions in AD, additional studies are needed in which both RNA sequencing and somatic mutation analysis would be conducted in single neurons from post‐mortem AD hippocampal tissue.
1.2. Aging‐induced somatic mutations in AD
Increasing age is the greatest risk factor for AD, but the (causal) mechanisms through which aging may lead to the development of plaques and tangles and clinical deterioration in patients are incompletely understood. 10 Of interest, somatic mutations—that is non‐inherited genetic variants that only appear in a person's cells (eg, neurons in the brain) throughout his/her lifetime and are not transmitted to future generations—increase with age. 11 In this respect, whole‐genome sequencing of individual neurons from the dentate gyrus, a part of the hippocampus that is the most affected brain region in AD, 12 has recently shown that (healthy) aging of the brain is associated with the accumulation of somatic mutations—that is, somatic single nucleotide variants (sSNVs)—at a more or less linear rate of approximately 40 sSNVs per neuron per year. 13 , 14 This type of DNA damage appears to accumulate in a random manner with increasing age. 11 , 15
If the burden of sSNVs is uniformly scattered at purely random genomic positions, genes spanning longer portions of the genome are expected to accumulate more sSNVs than genes that are smaller in size. Therefore, this age‐related accumulation of sSNVs may disproportionally affect longer genes. Beyond the effects of healthy aging, a higher degree or acceleration of sSNVs can have neuropathological effects. For example, in Cockayne syndrome, a disease associated with brain atrophy and cognitive decline, patients are affected by higher rates of sSNVs due to impaired DNA repair, 14 , 16 and this is especially the case for slow‐ or non‐proliferative cells such as neurons. 17 Although such rare genetic syndromes represent an extreme form of DNA repair dysfunction, they clearly show that genomic maintenance is an active process in neurons. There is substantial evidence for deficiencies in DNA repair in AD as well (reviewed in 18 ). Analysis of sSNVs in the hippocampus has also revealed both clock‐like and oxidative stress–induced signatures, suggesting that there are factors that increase the total mutational burden over and above the typical DNA damage as part of normal aging. 14 , 19 AD‐vulnerable brain regions belong to the most metabolically active regions of the brain. 20 , 21 This high energy demand may make these regions more susceptible to oxidative stress damage as compared to other parts of the brain. 22 Of interest, (long) neuronal genes are known to selectively map to common fragile sites of genomic instability, 23 further increasing their vulnerability to DNA damage.
sSNVs tend to inactivate genes, leading to reduced expression and function of their encoded proteins. 11 , 17 In this respect, it is interesting that, when comparing the hippocampus of old versus young cognitively normal individuals (approximately 80 years vs approximately 20 years old), an overrepresentation of reduced gene expression was reported for longer genes. 24 In turn, this could possibly disrupt the cellular pathways in which these proteins are involved, which may be relevant to AD pathogenesis.
1.3. Long genes are more frequently affected in AD
We built a theoretical model that predicts the likelihood of a gene being affected by at least one sSNV based on its length and the age of an individual. Our model estimated that an average‐sized gene has a 1% chance of acquiring at least one mutation by age 65 (ie, the age threshold for a LOAD diagnosis). In contrast, the likelihood for the longest gene in the genome, CNTNAP2, to be affected by an sSNV by age 65 is markedly higher, at 60%. We tested our model using publicly available post‐mortem brain sSNV data from three studies. This confirmed our predictions: The 272 longest genes in the genome (ie, genes with a log size of more than two standard deviations above the mean) were overrepresented among the genes affected by sSNVs in all three data sets, and the length of genes with sSNVs was longer than average in all three studies.
As indicated above, sSNVs are likely to lead to reduced expression (and function) of the affected genes. Therefore, we tested if, compared to healthy individuals, this reduced expression of longer genes can be observed more frequently in (the brains of) AD patients. We confirmed that long genes were indeed much more likely than shorter genes to show reduced expression in AD brains. This abnormal expression pattern was found in six brain regions commonly affected in AD (temporal cortex, superior temporal gyrus, parahippocampal gyrus, inferior frontal gyrus, dorsolateral prefrontal cortex, and especially hippocampus). In contrast, in two other brain regions that are more resilient to AD (frontal pole and cerebellum), longer genes were not more likely to have reduced expression. Furthermore, to show that the observed reduced gene expression is not due to neuronal loss resulting from AD itself, we analyzed data of differential gene expression between AD patients and controls in individual neurons from the entorhinal cortex, a brain area in the vicinity of the hippocampus that is (also) among the first to be affected in AD. We found that longer genes are more likely to show reduced expression in individual inhibitory neurons from this brain region, but this effect was not seen in excitatory neurons.
1.4. Long genes encode proteins involved in synaptic pathways
Long genes are more likely to have brain‐related functions and to be expressed in the brain. 25 To further explore the implications of long gene susceptibility to sSNVs, we performed pathway analysis to investigate which biological processes and molecular networks were enriched in the set of 272 longest genes in the genome. This indicated that long genes are involved in multiple synaptic functions such as synaptic organization, adhesion, transmission, and plasticity. We found that several of these synaptic pathways were also enriched within the differential gene expression data from the eight AD‐related brain regions. Furthermore, based on the direction of gene expression, the “synaptogenesis signaling” pathway was predicted to be inhibited in three brain regions (temporal cortex, parahippocampal gyrus, and hippocampus), whereas four additional synaptic function‐related pathways were inhibited in the hippocampus. Moreover, our finding of reduced expression of longer genes in inhibitory but not excitatory neurons of the entorhinal cortex (see above) fits very well with a recent paper that identified a diminished synaptic inhibitory‐excitatory balance in mouse entorhinal cortex as an early AD marker, preceding Aβ plaque formation. 26
1.5. Synaptic impairment in AD
Converging evidence from the literature suggests that synaptic impairment may be critical in AD development and progression. For instance, many of the AD GWAS genes encode proteins with essential roles in synaptic function and adhesion. 27 The relevance of impaired synaptic processes in AD is further corroborated by the fact that synaptic loss is the strongest neuropathological correlate of cognitive decline in AD. 28 , 29 , 30 Of interest, genes that have been associated through GWASs with AD‐related brain phenotypes—for example, hippocampal volume and cerebrospinal fluid levels of phosphorylated tau—are also enriched for processes such as synaptic plasticity. 31 , 32 In addition, the familial AD genes APP and PSEN1/2 have roles in synaptogenesis, synaptic adhesion, and neurotransmission. 27 Moreover, LRP1B is encoded by one of the genome's longest genes and is highly expressed in AD‐vulnerable brain regions. 33 , 34 This protein serves as a receptor of apoE—with APOE ε4 being the strongest common genetic risk factor for LOAD (see above)—and it interacts with both APP and the postsynaptic scaffolding protein PSD95, 35 , 36 implying a role in regulating synaptic function. In this way, the very long gene LRP1B may link early and late‐onset AD through the effect of its encoded protein on synaptic signaling.
Finally, several very long genes that we found to be both affected by sSNVs and downregulated in the hippocampus of AD patients encode proteins with important roles in synaptic function. For example, CNTNAP2, the longest human gene (see above), encodes a neuronal adhesion molecule and has been further implicated in AD etiology through the latest meta‐analytic GWAS. 8 Reduced CNTNAP2 expression levels have also been observed in the AD hippocampus. 37 Other examples of proteins encoded by very long genes that are both affected by sSNVs and downregulated in the AD hippocampus include PTPRT, a regulator of synaptogenesis, 38 and RIMS2, a modulator of neurotransmitter release. 39 In addition, the SLC4A10 40 and RYR2 41 proteins are involved in synaptic plasticity.
1.6. Limitations
Our study has two main limitations. First, the sSNVs that we analyzed to test our model were derived from two studies that examined bulk hippocampal tissue and not single neurons. Therefore, it is possible that some of these sSNVs occurred in brain cells other than neurons or as the result of developmental mosaicism, and follow‐up studies are needed that specifically investigate sSNVs at the level of single neurons in AD (see below). Second, because post‐mortem tissue is necessarily collected late in the disease course, the observed reduced expression of long genes in AD‐vulnerable brain regions could be a consequence of synaptic loss resulting from the AD pathology itself — due to neuronal atrophy — rather than being the underlying cause of it. 42 However, in support of our findings, we also observed that the expression of long genes is reduced in single inhibitory neurons of AD patients.
1.7. Conclusions and directions for future research
We found that, through aging, longer genes are more frequently affected by sSNVs in the hippocampus, the most affected brain region in AD. In addition, long genes show reduced expression in multiple brain regions of AD patients, including the hippocampus. Furthermore, we showed that many of the longest genes in the genome code for proteins that are involved in synaptic adhesion and function. Based on expression data, these synaptic pathways were also predicted to be inhibited in the AD hippocampus and other AD‐vulnerable brain regions that are important for memory and cognition. Taken together, our findings provide novel insights in how aging‐induced DNA damage may promote AD pathogenesis through having a negative effect on synaptic function.
As for future research, we propose three main avenues to pursue. First, studies are needed that conduct concurrent RNA and DNA sequencing (eg, as in 43 ) of single neurons and possibly other cell types from brain tissue samples of AD patients and non‐demented controls. In this way, the putative causal relationship between specific sSNVs in (long) genes and their reduced expression could be corroborated. Second, studies should be conducted that are aimed at further unraveling the links between sSNVs in specific genes, synaptic impairment, and AD pathology. With regard to the latter, studies in animal or cellular models that manipulate the functions of specific long genes can be instrumental in elucidating the molecular chain of events following sSNVs. Third, animal models could be used to investigate earlier disease stages, and this to confirm that the observed reduction in (synaptic) gene expression and function is driving the AD pathology rather than being the result of end stage disease.
2. CONSOLIDATED RESULTS AND STUDY DESIGN
2.1. Model for sSNV likelihood through aging: effect of gene length
The human genome contains 20,535 unique protein‐coding genes that vary greatly in size (data retrieved from Ensembl Biomart [GENCODE v19, GRCH37p13] 44 ). The distribution of human gene length has a long tail encompassing extremely long genes in the mega base pair (bp) range (Figure 1A). After log‐transformation, 272 genes have a log bp size of more than two standard deviations above the mean and are designated as the set of “very long” genes (Figure 1B and Supplementary Table 1).
FIGURE 1.

Longer genes have an increased likelihood to be affected by sSNVs. (A) The length distribution of human genes has a long tail that extends toward a group of extremely long genes of 1‐2 mega base pairs (272 very long genes are indicated with open circles (see below) and gene length in base pairs (bp). Gene length information was retrieved from Ensembl Biomart (GENCODE v19, GRCh37p13). (B) Gene length follows a log‐normal distribution with parameters μ = 4.35 (22.5 kb) and σ = 0.68 (dashed line). The outlier bin near 1 kb represents the large family of olfactory receptors that have gone through extreme evolutionary expansion. The 272 genes that are indicated by the open circles in 1B and in the shaded gray area under the curve in 1C show the subgroup of very long genes (genes with gene length > μ+2σ) that were used for the enrichment analyses in this study. (C) Binomial probability model for gene conservation over time in which somatic mutations (sSNVs) take place at a fixed and uniform rate across the genome, age in years (y). An average‐sized gene mostly survives the mutational burden of aging, with only ≈1% of its copies being affected by somatic mutations in a 65‐year‐old subject. For longer genes, however, ≈60% of copies are expected to have been affected by at least one sSNV between the sixth and seventh decades of life. (D) sSNVs occur more often in longer genes (Kolmogorov‐Smirnov test: P < 1.0 × 10−4). Gene length distributions for genes having potential pathogenic sSNVs from the studies by Park et al. (Red, 208 genes), Ivashko‐Pachima et al. (Blue, 499 genes), Lodato et al. (Green, 175 genes), and all human protein‐coding genes (Black, 20535 genes) are shown. Circles following the same color code plotted below density graph represent individual gene lengths. Box plots visualize the median with flanking lower and upper hinges (corresponding to the 25th and 75th percentiles), and the whiskers represent the 95% confidence interval
Assuming that the ≈40 sSNVs that accumulate yearly in neurons 13 , 14 occur at random positions in their genomes (6.4 billion base pairs), the probability per year, per nucleotide of acquiring an sSNV (ω) is 6.2 × 10−9. Hence, we modeled the chance for an sSNV occurring in a gene with the binomial equation , in which a is subject age in years, ω is the per‐nucleotide probability of an sSNV per year, and is the number of DNA nucleotides forming the transcribed region of the gene of interest (ie, the gene length). For an average‐sized gene (22.5 kbp, based on log distribution, see below), our model estimates that this mutational rate would result in ≈0.9% of gene copies acquiring at least one sSNV by the age of 65 (Figure 1C). The model further predicts that longer genes are more likely to be affected by sSNVs. For instance, 65 years of the same mutational rate is expected to affect 60.5% of the copies of the longest human gene, CNTNAP2 (Figure 1C).
We then tested whether sSNVs are more likely to affect longer genes by comparing the lengths of genes affected by sSNVs in the hippocampus to the gene length distribution of all protein‐coding genes that we retrieved from Ensembl Biomart (see above), using Kolmogorov‐Smirnoff tests. 45 We retrieved human hippocampal sSNV data from three recent publications: Lodato et al., 14 Park et al., 19 and Ivashko‐Pachima et al. 46 (see “Detailed Methods and Results” and Table 4). As predicted by our model, the length of sSNV‐harboring genes is longer than average (P = 1.0 × 10−4) (Figure 1D). Using hypergeometric tests, 47 we found that the set of 272 very long genes is enriched in the single cell sSNVs from the Lodato et al. study (4.3‐fold increase, P = 9.6 × 10−5), and in the hippocampal sSNVs from the studies by Park et al. and Ivashko‐Pachima et al. (3.3‐fold increase, P = 1.41 × 10−3, and 2.4‐fold increase, P = 6.90 × 10−4, respectively).
TABLE 4.
Data resource information for data used in this article
| Type of data | Brain region | Details | Original paper |
|---|---|---|---|
| sSNV | Dentate gyrus/prefrontal cortex | NIH NeuroBioBank; WGS of single isolated neuronal nuclei | Lodato et al. 14 |
| sSNV | Hippocampus | Netherlands Brain Bank and Human Brain and Spinal Fluid Resource Center; WES of laser capture micro dissected hippocampal formations | Park et al. 19 |
| sSNV | Hippocampus | Banner Sun Health Research Institute; RNA‐seq based mutation analysis (from GSE67333) | Ivashko‐Pachima et al. 46 |
| RNAseq | Cerebellum | Mayo clinic (AMP‐AD); bulk tissue | Allen et al. 50 |
| RNAseq | Temporal cortex | Mayo clinic (AMP‐AD); bulk tissue | Allen et al. 50 |
| RNAseq | Frontal pole (BA10) | Mount Sinai/JJ Peters VA Medical Center Brain Ban (AMP‐AD); bulk tissue | Wang et al. 51 |
| RNAseq | Superior temporal gyrus (BA22) | Mount Sinai/JJ Peters VA Medical Center Brain Ban (AMP‐AD); bulk tissue | Wang et al. 51 |
| RNAseq | Parahippocampal gyrus (BA36) | Mount Sinai/JJ Peters VA Medical Center Brain Ban (AMP‐AD); bulk tissue | Wang et al. 51 |
| RNAseq | Inferior frontal gyrus (BA44) | Mount Sinai/JJ Peters VA Medical Center Brain Ban (AMP‐AD); bulk tissue | Wang et al. 51 |
| RNAseq | Dorsolateral prefrontal cortex | Mount Sinai/JJ Peters VA Medical Center Brain Ban (AMP‐AD); bulk tissue | Mostafavi et al. 52 |
| RNAseq | Hippocampus | Netherlands Brain Bank; bulk tissue | Van Rooij et al. 53 |
| RNAseq | Entorhinal Cortex | Victorian Brain bank; single‐nucleus RNA sequencing | Grubman et al. 54 |
NOTE. Abbreviations: AMP‐AD, Accelerating Medicines Partnership Alzheimer's Disease Project; BA, Brodmann area; RNAseq, RNA sequencing; sSNV, somatic single nucleotide variant; WES, whole exome sequencing; WGS, whole genome sequencing.
2.2. Transcriptomic data analyses
We extracted differentially expressed genes, that is, genes that show significantly increased or decreased expression when comparing AD patients to non‐demented controls, from previously published RNA sequencing data resources for nine brain regions (cerebellum, temporal cortex, frontal pole, superior temporal gyrus, parahippocampal gyrus, inferior frontal gyrus, dorsolateral prefrontal cortex [DLPFC], hippocampus, and [single cell data from the] entorhinal cortex) (see “Detailed Methods and Results”). The transcriptomic data were filtered for protein‐coding genes and binned in 50 consecutive groups based on transcribed gene length. We compared the number of genes showing AD‐associated decreased expression in each bin with that of the total gene pool using hypergeometric tests. 47 These analyses showed a sharp increase in downregulated genes at the far end of the gene length distribution (top 2%) in the temporal cortex (P = 1.2 × 10−8), superior temporal gyrus (Brodmann Area [BA]22) (P = 1.6 × 10−6), parahippocampal gyrus (BA36) (P = 1.3 × 10−11), inferior frontal gyrus (BA44) (P = 3.1 × 10−14), DLPFC (P = 5.0 × 10−5), and hippocampus (P = 1.1 × 10−16) (Figure 2). This effect was not observed in the cerebellum and the frontal pole (BA10), regions that are known to be more resilient to AD 10 and can be considered negative controls (Figure 2). The analysis of single neuron transcriptome data from the entorhinal cortex revealed that genes in the top 2% bin of gene length showed reduced expression in inhibitory neurons of (P = 8.2 × 10−9) but not in excitatory neurons (Figure 3). Additional hypergeometric tests showed that the set of 272 very long genes was overrepresented within the significantly differentially expressed genes in the temporal cortex (P = 1.22 × 10−5), BA22 (P = 1.74 × 10−2), BA36 (P = 2.64 × 10−8), and hippocampus (P = 6.47 × 10−8). In contrast, we observed fewer differentially expressed very long genes in the cerebellum and DLPFC (P = 2.03 × 10−5 and P = 2.01 × 10−2) (Table 1). Eighteen of the 19 long genes affected by sSNVs from the Park et al. and Ivashko‐Pachima et al. studies for which RNA transcripts were detected showed decreased expression in the AD hippocampus (Table 2).
FIGURE 2.
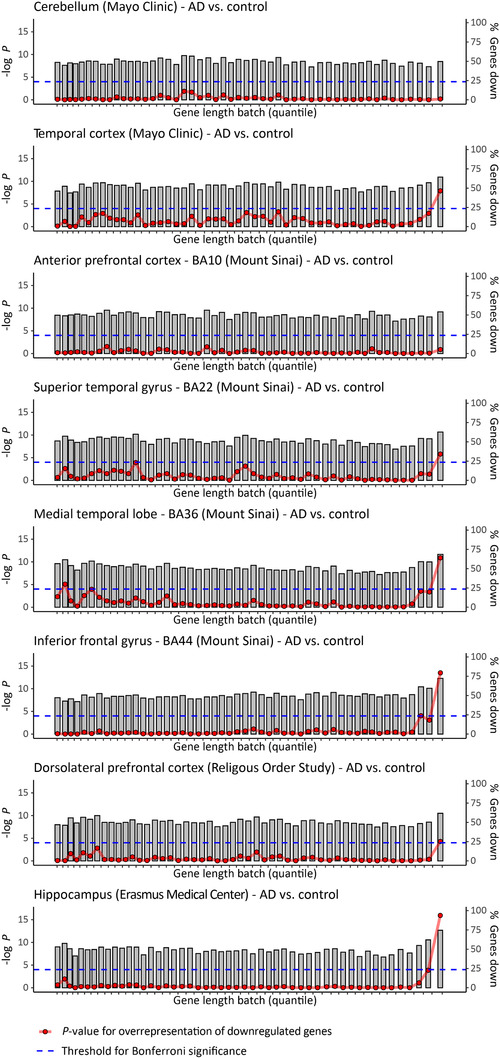
Long genes are significantly downregulated in AD‐relevant brain regions. Plots show differentially expressed genes, that is, genes that show significantly increased or decreased expression when comparing AD patients to non‐demented controls, from previously published RNA sequencing studies (Table 4). Protein‐coding genes are binned in 50 consecutive groups (gray bars), based on transcribed gene length. We compared the number of genes showing either increased or decreased expression in each bin (height of gray bar) with that of the total gene pool using hypergeometric tests (red circles, Bonferroni threshold for significance is indicated with dashed blue line)
FIGURE 3.
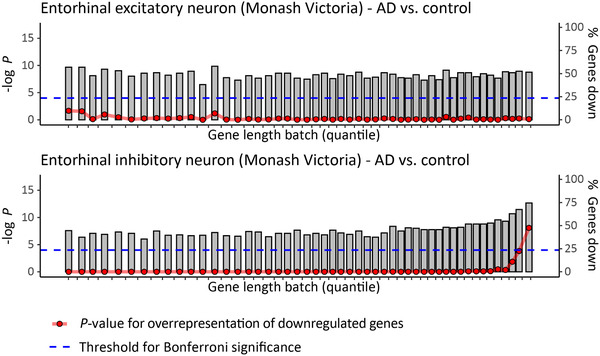
Long genes are significantly downregulated in inhibitory neurons of the entorhinal cortex. Plots show differentially expressed genes, that is, genes that show significantly increased or decreased expression when comparing AD patients to non‐demented controls, in single inhibitory or excitatory neurons from the entorhinal cortex (Table 4). Protein‐coding genes are binned in 50 consecutive groups (gray bars), based on transcribed gene length. We compared the number of genes showing either increased or decreased expression in each bin (height of gray bar) with that of the total gene pool using hypergeometric tests (red circles, Bonferroni threshold for significance is indicated with dashed blue line)
TABLE 1.
Over‐ and underrepresentation of very long genes in genes differentially expressed in the brain of AD patients
| Brain region | Number of genes detected (very long) | Number of differentially expressed genes (very long) | Over‐/underrepresentation |
|---|---|---|---|
| Cerebellum | 14291 (258) | 5128 (63) | ‐1.47 (P = 2.03 × 10‐5) |
| Temporal cortex | 14292 (258) | 6129 (143) | 1.29 (P = 1.22 × 10‐5) |
| Frontal pole (BA10) | 13788 (263) | 334 (5) | ‐ 1.27 (P = 1.51 × 10‐1) |
| Superior temporal gyrus (BA22) | 13789 (263) | 688 (20) | 1.52 (P = 1.74 × 10‐2) |
| Parahippocampal gyrus (BA36) | 13789 (263) | 4814 (134) | 1.46 (P = 2.64 × 10‐8) |
| Inferior frontal gyrus (BA44) | 13789 (263) | 151 (3) | 1.04 (P = 2.27 × 10‐1) |
| Dorsolateral prefrontal cortex | 13512 (250) | 1647 (22) | ‐1.39 (P = 2.10 × 10‐2) |
| Hippocampus | 14533 (250) | 7411 (156) | 1.22 (P = 6.47 × 10‐5) |
NOTE. A hypergeometric test was performed to generate the P‐ values of over‐and underrepresentation.
Abbreviation: AD, Alzheimer's disease.
TABLE 2.
Genes that are affected by sSNVs in the hippocampus 19 , 46 and differentially expressed in the AD hippocampus 53
| sSNV study | Decreased mRNA expression in the AD hippocampus | Increased mRNA expression in the AD hippocampus |
|---|---|---|
| Park et al. 19 | CAMTA1, CNTNAP2, CSMD2, NRXN1, PTPRT | NAV2 |
| Ivashko‐Pachima et al. 46 | ANK2, DCC, FAT3, GRIK2, HS6ST3, KALRN, MYT1L, NELL1, RIMS2, RYR2, SLC4A10, TENM2, TENM3 | – |
Abbreviations: AD, Alzheimer's disease; sSNV, somatic single nucleotide variant.
2.3. Enrichment analyses
We used Ingenuity Pathway Analysis (IPA) to test for enrichment of canonical pathways within our predefined set of very long genes, using a false discovery rate (FDR) correction for multiple testing. 48 Five of the 10 most enriched pathways in the very long gene set are directly related to synaptic function, that is, “synaptogenesis signaling” (P = 3.47 × 10−6), “synaptic long‐term depression”(P = 1.02 × 10−5), “CREB signaling in neurons” (P = 2.51 × 10−4) (which are the three most significantly enriched pathways), “synaptic long‐term potentiation” (P = 1.05 × 10−3), and “glutamate receptor signaling” (P = 2.57 × 10−3). With the Panther classification system 49 we assessed enrichment of gene ontology (GO) terms within the same set of genes, with Fisher exact test and applying FDR correction. In keeping with the IPA results, the GO term analysis revealed that the set of very long genes is enriched for multiple synaptic functions such as synaptic organization, adhesion, transmission, and plasticity. The full results of the enrichment analyses of the 272 longest genes are provided in the supplement (Supplementary Table 2).
We performed IPA canonical pathway enrichment analyses on the tissue level transcriptomic data (all differentially expressed protein‐coding genes with corrected P < 0.05) to predict whether the pathways that were enriched in the set of very long genes were activated or inhibited in AD brains. The “synaptogenesis signaling” pathway was predicted to be inhibited within three brain regions in which very long genes were overrepresented within the differentially expressed genes, that is, temporal cortex (P = 1.15 × 10−6; Z = ‐3.05), BA36 (P = 6.17 × 10−6; Z = ‐3.61), and hippocampus (P = 3.47 × 10−6; Z = ‐5.70). All four other synaptic function–related pathways that were enriched within the set of very long genes—that is, “synaptic long‐term depression,” “CREB signaling in neurons,” “synaptic long‐term potentiation,” and “glutamate receptor signaling”—were also predicted to be inhibited based on the differentially expressed genes in the hippocampus (P = 1.20 × 10−8; Z = ‐2.28, P = 5.01 × 10−14; Z = ‐2.91, P = 3.89 × 10−10; Z = ‐3.21, and P = 3.09 × 10−7; Z = ‐4.36) (Table 3 and Supplementary Table 3).
TABLE 3.
Canonical pathway enrichment analysis for differentially expressed genes in brain regions of AD patients
| Pathway | Brain region | |||||||
|---|---|---|---|---|---|---|---|---|
| Cerebellum | Temporal cortex | Frontal pole (BA10) | Superior temporal gyrus (BA22) | Parahippocampal gyrus (BA36) | Inferior frontal gyrus (BA44) | DLPFC | Hippocampus | |
| Synaptogenesis Signaling Pathway |
P = 3.98 × 10‐2 Z‐score = 0.816 |
P = 1.15 × 10‐6 Z‐score = ‐3.051 | – |
P = 4.90 × 10‐4 Z‐score = ‐1.706 |
P = 6.17 × 10‐6 Z‐score = ‐3.606 |
– |
P = 7.95 × 10‐1 Z‐score = 0.426 |
P = 3.47 × 10‐6 Z‐score = ‐5.692 |
| Synaptic Long‐Term Depression |
P = 1.85 × 10‐1 Z‐score = ND |
P = 1.00 × 10‐3 Z‐score = ‐0.707 |
– | – |
P = 1.82 × 10‐5 Z‐score = ‐1.265 |
– |
P = 7.35 × 10‐1 Z‐score = 0.258 |
P = 1.20 × 10‐8 Z‐score = ‐2.770 |
| CREB Signaling in Neurons |
P = 1.32 × 10‐1 Z‐score = ND |
P = 2.19 × 10‐2 Z‐score = ‐1.633 |
– | – |
P = 8.91 × 10‐4 Z‐score = ‐1.890 |
– |
P = 6.08 × 10‐1 Z‐score = 0 |
P = 5.01 × 10‐14 Z‐score = ‐2.907 |
| Synaptic Long‐Term Potentiation |
P = 1.64 × 10‐1 Z‐score = ND |
P = 1.95 × 10‐2 Z‐score = ‐1.342 |
– | – |
P = 3.02 × 10‐3 Z‐score = ‐1.633 |
– |
P = 4.86 × 10‐1 Z‐score = ‐0.277 |
P = 3.89 × 10‐10 Z‐score = ‐3.212 |
| Glutamate Receptor Signaling |
P = 2.84 × 10‐1 Z‐score = ND |
P = 4.68 × 10‐2 Z‐score = ND |
– | – |
P = 9.12 × 10‐3 Z‐score = ND |
– |
P = 7.95 × 10‐1 Z‐score = ND |
P = 3.09 × 10‐7 Z‐score = ‐4.359 |
NOTE. Results are shown for the five most significantly enriched synaptic pathways among the 272 longest genes in the genome (Supplementary Table 1). Significant P‐values (P < 0.05) and Z‐scores (Z ≤ ‐2 or Z ≥ 2) are indicated in bold.
Abbreviations: AD, Alzheimer's disease; BA, Brodmann area; DLPFC, dorsolateral prefrontal cortex; ND, Not determined.
3. DETAILED METHODS AND RESULTS
To test our model and hypothesis, we used publicly available resources to obtain data of sSNVs in the hippocampus and differential gene expression in AD brain regions (Table 4).
3.1. sSNV data sets
Genes affected by exonic sSNVs in both people with early onset neurodegeneration due to genetic disorders of DNA repair and healthy controls were obtained from the whole‐genome sequencing study at single‐neuron level by Lodato et al. 14 Genes affected by sSNVs in both AD patients and controls were retrieved from the studies at whole‐tissue level—more specifically, the hippocampus—by Park et al. 19 and Ivashko‐Pachima et al. 46
3.2. RNA sequencing data sets
Furthermore, uniformly processed RNA sequencing (RNA‐seq) data (weighted linear model based on diagnosis) from seven brain areas (cerebellum, temporal cortex, frontal pole [Brodmann Area (BA) 10], two subregions of the temporal cortex [superior temporal gyrus (BA22) and parahippocampal gyrus (BA36)], inferior frontal gyrus [BA44], and dorsolateral prefrontal cortex [DLPFC]) was obtained from the AMP‐AD knowledge portal on the Synapse platform (syn2580853). 50 , 51 , 52 On the AMP‐AD knowledge portal, the data, analysis results, analytical methodology, and research tools generated by multiple consortia are made available with support of the National Institute on Aging's Alzheimer's Disease Translational Research Program. To study the hippocampus, an additional RNA‐seq data set—that is, data of differential mRNA expression between AD patients and controls—from the Netherlands brain bank study was used. 53
To show that the effects we observed are not due to changes in brain cell composition, we used a single‐cell RNA‐seq data set, that is, data of differential mRNA expression between AD patients and controls, in individual neurons from the entorhinal cortex. 54 Grubman et al. used the single‐cell transcriptomic profiles of these entorhinal neurons to classify neurons into inhibitory and excitatory cells, using the RCA (Reference Component Analysis) method. 55
CONFLICT OF INTEREST
Geert Poelmans is director of DrugTarget ID, Ltd. (The Netherlands). All other authors declare no conflicts of interest.
Supporting information
Supplementary information
Supplementary information
Supplementary information
ACKNOWLEDGMENTS
The results published here are in whole or in part based on data obtained from the AMP‐AD Knowledge Portal (https://doi.org/10.7303/syn2580853). Study data were provided by the following sources: The Mayo Clinic Alzheimers Disease Genetic Studies, led by Dr. Nilufer Taner and Dr. Steven G. Younkin, Mayo Clinic, Jacksonville, FL using samples from the Mayo Clinic Study of Aging, the Mayo Clinic Alzheimers Disease Research Center, and the Mayo Clinic Brain Bank. Data collection was supported through funding by NIA grants P50 AG016574, R01 AG032990, U01 AG046139, R01 AG018023, U01 AG006576, U01 AG006786, R01 AG025711, R01 AG017216, R01 AG003949, National Institute of Neurological Disorders and Stroke (NINDS) grant R01 NS080820, CurePSP Foundation, and support from Mayo Foundation. Study data include samples collected through the Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona. The Brain and Body Donation Program is supported by the NINDS (U24 NS072026 National Brain and Tissue Resource for Parkinsons Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimers Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimers Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05‐901, and 1001 to the Arizona Parkinson's Disease Consortium), and the Michael J. Fox Foundation for Parkinsons Research. Data generated from post‐mortem brain tissue collected through the Mount Sinai VA Medical Center Brain Bank and were provided by Dr. Eric Schadt from Mount Sinai School of Medicine. Data were provided by the Rush Alzheimer's Disease Center, Rush University Medical Center, Chicago. Data collection was supported through funding by NIA grants P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36836, U01AG32984, and U01AG46152, the Illinois Department of Public Health, and the Translational Genomics Research Institute.
Soheili‐Nezhad S, van der Linden RJ, Olde Rikkert M, Sprooten E, Poelmans G. Long genes are more frequently affected by somatic mutations and show reduced expression in Alzheimer's disease: Implications for disease etiology. Alzheimer's Dement. 2021;17:489–499. 10.1002/alz.12211
Funding information: The research presented in this paper was supported by the Dutch charity foundation ”Stichting Devon.” In addition, ES is supported by a Hypatia Tenure Track Grant (Radboudumc), Christine Mohrmann Fellowship (Radboud University), and a National Alliance for Research on Schizophrenia & Depression (NARSAD) Young Investigator Grant (Brain and Behavior Research Foundation, ID: 25034).
REFERENCES
- 1. Patterson C. World Alzheimer Report 2018: The State of the Art of Dementia Research: New Frontiers. London, UK: Alzheimer's Disease International (ADI); 2018. [Google Scholar]
- 2. Livingston G, Huntley J, Sommerlad A, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396(10248):413‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yiannopoulou KG, Papageorgiou SG. Current and future treatments in Alzheimer disease: an update. J Cent Nerv Syst Dis. 2020;12:1179573520907397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bekris LM, Yu CE, Bird TD, Tsuang DW. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23(4):213‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van der Lee SJ, Wolters FJ, Ikram MK, et al. The effect of APOE and other common genetic variants on the onset of Alzheimer's disease and dementia: a community‐based cohort study. Lancet Neurol. 2018;17(5):434‐444. [DOI] [PubMed] [Google Scholar]
- 6. Genin E, Hannequin D, Wallon D, et al. APOE and Alzheimer disease: a major gene with semi‐dominant inheritance. Mol Psychiatry. 2011;16(9):903‐907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45(12):1452‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jansen IE, Savage JE, Watanabe K, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51(3):404‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bellenguez C, Charbonnier C, Grenier‐Boley B, et al. Contribution to Alzheimer's disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol Aging. 2017;59:220. e1‐e9. [DOI] [PubMed] [Google Scholar]
- 10. Thomas KR, Bangen KJ, Weigand AJ, et al. Objective subtle cognitive difficulties predict future amyloid accumulation and neurodegeneration. Neurology. 2020;94(4):e397‐e406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kennedy SR, Loeb LA, Herr AJ. Somatic mutations in aging, cancer and neurodegeneration. Mech Ageing Dev. 2012;133(4):118‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mrdjen D, Fox EJ, Bukhari SA, Montine KS, Bendall SC, Montine TJ. The basis of cellular and regional vulnerability in Alzheimer's disease. Acta Neuropathol. 2019;138(5):729‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hoang ML, Kinde I, Tomasetti C, et al. Genome‐wide quantification of rare somatic mutations in normal human tissues using massively parallel sequencing. Proc Natl Acad Sci U S A. 2016;113(35):9846‐9851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lodato MA, Rodin RE, Bohrson CL, et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science. 2018;359(6375):555‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lodato MA, Walsh CA. Genome aging: somatic mutation in the brain links age‐related decline with disease and nominates pathogenic mechanisms. Hum Mol Genet. 2019;28(R2):R197‐R206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baez S, Couto B, Herrera E, et al. Tracking the cognitive, social, and neuroanatomical profile in early neurodegeneration: type III Cockayne syndrome. Front Aging Neurosci. 2013;5:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 2014;15(7):465‐481. [DOI] [PubMed] [Google Scholar]
- 18. Hou Y, Song H, Croteau DL, Akbari M, Bohr VA. Genome instability in Alzheimer disease. Mech Ageing Dev. 2017;161(Pt A):83‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park JS, Lee J, Jung ES, et al. Brain somatic mutations observed in Alzheimer's disease associated with aging and dysregulation of tau phosphorylation. Nat Commun. 2019;10(1):3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greicius MD, Srivastava G, Reiss AL, Menon V. Default‐mode network activity distinguishes Alzheimer's disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci U S A. 2004;101(13):4637‐4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhou Y, Wang K, Liu Y, Song M, Song SW, Jiang T. Spontaneous brain activity observed with functional magnetic resonance imaging as a potential biomarker in neuropsychiatric disorders. Cogn Neurodyn. 2010;4(4):275‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Frisard M, Ravussin E. Energy metabolism and oxidative stress: impact on the metabolic syndrome and the aging process. Endocrine. 2006;29(1):27‐32. [DOI] [PubMed] [Google Scholar]
- 23. Smith DI, Zhu Y, McAvoy S, Kuhn R. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett. 2006;232(1):48‐57. [DOI] [PubMed] [Google Scholar]
- 24. Vermeij WP, Dolle ME, Reiling E, et al. Restricted diet delays accelerated ageing and genomic stress in DNA‐repair‐deficient mice. Nature. 2016;537(7620):427‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Raychaudhuri S, Korn JM, McCarroll SA, et al. Accurately assessing the risk of schizophrenia conferred by rare copy‐number variation affecting genes with brain function. PLoS Genet. 2010;6(9):e1001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Petrache AL, Rajulawalla A, Shi A, et al. Aberrant excitatory‐inhibitory synaptic mechanisms in entorhinal cortex microcircuits during the pathogenesis of Alzheimer's disease. Cereb Cortex. 2019;29(4):1834‐1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dourlen P, Kilinc D, Malmanche N, Chapuis J, Lambert JC. The new genetic landscape of Alzheimer's disease: from amyloid cascade to genetically driven synaptic failure hypothesis? Acta Neuropathol. 2019;138(2):221‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298(5594):789‐791. [DOI] [PubMed] [Google Scholar]
- 29. Forner S, Baglietto‐Vargas D, Martini AC, Trujillo‐Estrada L, LaFerla FM. Synaptic impairment in Alzheimer's disease: a dysregulated symphony. Trends Neurosci. 2017;40(6):347‐357. [DOI] [PubMed] [Google Scholar]
- 30. Li K, Wei Q, Liu FF, et al. Synaptic dysfunction in Alzheimer's disease: abeta, tau, and epigenetic alterations. Mol Neurobiol. 2018;55(4):3021‐3032. [DOI] [PubMed] [Google Scholar]
- 31. Chung J, Wang X, Maruyama T, et al. Genome‐wide association study of Alzheimer's disease endophenotypes at prediagnosis stages. Alzheimers Dement. 2018;14(5):623‐633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Soheili‐Nezhad S, Jahanshad N, Guelfi S, et al. Imaging genomics discovery of a new risk variant for Alzheimer's disease in the postsynaptic SHARPIN gene. Hum Brain Mapp. 2020;41(13):3737‐3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haas J, Beer AG, Widschwendter P, et al. LRP1b shows restricted expression in human tissues and binds to several extracellular ligands, including fibrinogen and apoE‐carrying lipoproteins. Atherosclerosis. 2011;216(2):342‐347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hawrylycz MJ, Lein ES, Guillozet‐Bongaarts AL, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489(7416):391‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cam JA, Zerbinatti CV, Knisely JM, Hecimovic S, Li Y, Bu G. The low densitylipoprotein receptor‐related protein 1B retains beta‐amyloid precursor protein at the cell surface and reduces amyloid‐beta peptide production. J Biol Chem. 2004;279(28):29639‐29646. [DOI] [PubMed] [Google Scholar]
- 36. Marschang P, Brich J, Weeber EJ, et al. Normal development and fertility of knockout mice lacking the tumor suppressor gene LRP1b suggest functional compensation by LRP1. Mol Cell Biol. 2004;24(9):3782‐3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van Abel D, Michel O, Veerhuis R, Jacobs M, van Dijk M, Oudejans CB. Direct downregulation of CNTNAP2 by STOX1A is associated with Alzheimer's disease. J Alzheimers Dis. 2012;31(4):793‐800. [DOI] [PubMed] [Google Scholar]
- 38. Lee JR. Protein tyrosine phosphatase PTPRT as a regulator of synaptic formation and neuronal development. BMB Rep. 2015;48(5):249‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kaeser PS, Deng L, Fan M, Sudhof TC. RIM genes differentially contribute to organizing presynaptic release sites. Proc Natl Acad Sci U S A. 2012;109(29):11830‐11835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sinning A, Liebmann L, Hubner CA. Disruption of Slc4a10 augments neuronal excitability and modulates synaptic short‐term plasticity. Front Cell Neurosci. 2015;9:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. More J, Galusso N, Veloso P, et al. N‐Acetylcysteine prevents the spatial memory deficits and the Redox‐dependent RyR2 decrease displayed by an Alzheimer's disease rat model. Front Aging Neurosci. 2018;10:399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barbash S, Sakmar TP. Length‐dependent gene misexpression is associated with Alzheimer's disease progression. Sci Rep. 2017;7(1):190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kong SL, Li H, Tai JA, et al. Concurrent single‐cell RNA and targeted DNA sequencing on an automated platform for comeasurement of genomic and transcriptomic signatures. Clin Chem. 2019;65(2):272‐281. [DOI] [PubMed] [Google Scholar]
- 44. Cunningham F, Achuthan P, Akanni W, et al. Ensembl 2019. Nucleic Acids Res. 2019;47(D1):D745‐D51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grishkevich V, Yanai I. Gene length and expression level shape genomic novelties. Genome Res. 2014;24(9):1497‐1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ivashko‐Pachima Y, Hadar A, Grigg I, et al. Discovery of autism/intellectual disability somatic mutations in Alzheimer's brains: mutated ADNP cytoskeletal impairments and repair as a case study. Mol Psychiatry. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fisher RA. Statistical methods for research workers. In: Kotz S, Johnson NL, eds. Breakthroughs in Statistics: Methodology and Distribution. New York, NY: Springer New York; 1992:66‐70. [Google Scholar]
- 48. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57(1):289‐300. [Google Scholar]
- 49. Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. PANTHER version 14: more genomes, a new PANTHER GO‐slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019;47(D1):D419‐D26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Allen M, Carrasquillo MM, Funk C, et al. Human whole genome genotype and transcriptome data for Alzheimer's and other neurodegenerative diseases. Sci Data. 2016;3:160089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang M, Beckmann ND, Roussos P, et al. The Mount Sinai cohort of large‐scale genomic, transcriptomic and proteomic data in Alzheimer's disease. Sci Data. 2018;5:180185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mostafavi S, Gaiteri C, Sullivan SE, et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer's disease. Nat Neurosci. 2018;21(6):811‐819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. van Rooij JGJ, Meeter LHH, Melhem S, et al. Hippocampal transcriptome profiling combined with protein‐protein interaction analysis elucidates Alzheimer's disease pathways and genes. Neurobiol Aging. 2019;74:225‐233. [DOI] [PubMed] [Google Scholar]
- 54. Grubman A, Chew G, Ouyang JF, et al. A single‐cell atlas of entorhinal cortex from individuals with Alzheimer's disease reveals cell‐type‐specific gene expression regulation. Nat Neurosci. 2019;22(12):2087‐2097. [DOI] [PubMed] [Google Scholar]
- 55. Lake BB, Ai R, Kaeser GE, et al. Neuronal subtypes and diversity revealed by single‐nucleus RNA sequencing of the human brain. Science. 2016;352(6293):1586‐1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information
Supplementary information
Supplementary information