

A large body of evidence has accumulated, demonstrating that there is no biochemical or functional requirement for the use of scFvs in CARs. Instead, highly potent CARs have been constructed based on several engineered non‐immunoglobulin‐based binding scaffolds. This review also includes a critical discussion on the risk of immunogenicity associated with different types of engineered binding domains and humanized or human scFvs.
Keywords: CAR T cells, DARPin, diabody formation, immunogenicity, monobody, nanobody, protein engineering, rcSso7d, scFv clustering, tonic signaling
Abstract
T cells that are genetically engineered to express chimeric antigen receptors (CAR T cells) have shown impressive clinical efficacy against B‐cell malignancies. In contrast to these highly potent CD19‐targeting CAR T cells, many of those directed against other tumor entities and antigens currently suffer from several limitations. For example, it has been demonstrated that many scFvs used as antigen‐binding domains in CARs show some degree of oligomerization, which leads to tonic signaling, T cell exhaustion, and poor performance in vivo. Therefore, in many cases alternatives to scFvs would be beneficial. Fortunately, due to the development of powerful protein engineering technologies, also non‐immunoglobulin‐based scaffolds can be engineered to specifically recognize antigens, thus eliminating the historical dependence on antibody‐based binding domains. Here, we discuss the advantages and disadvantages of such engineered binding scaffolds, in particular with respect to their application in CARs. We review recent studies, collectively showing that there is no functional or biochemical aspect that necessitates the use of scFvs in CARs. Instead, antigen recognition can also be mediated efficiently by engineered binding scaffolds, as well as natural ligands or receptors fused to the CAR backbone. Finally, we critically discuss the risk of immunogenicity and show that the extent of nonhuman amino acid stretches in engineered scaffolds—even in those based on nonhuman proteins—is more similar to humanized scFvs than might be anticipated. Together, we expect that engineered binding scaffolds and natural ligands and receptors will be increasingly used for the design of CAR T cells.
Abbreviations
- CAR
chimeric antigen receptor
- scFv
single‐chain variable fragment
- DARPin
designed ankyrin repeat protein
- FN3
10th type III domain of human fibronectin
- TanCAR
tandem CAR
- EGFR
epidermal growth factor receptor
- HER2
human epidermal growth factor receptor 2
- hRBP4
human retinol‐binding protein 4
Introduction
Immunotherapy for the treatment of cancer has experienced a breakthrough in the last decade. One of the most promising approaches in this field is CAR T cell therapy, that is, the adoptive transfer of T cells genetically engineered to express chimeric antigen receptors (CARs) [1, 2]. The standard CAR molecule combines an extracellular antigen‐binding domain with intracellular signaling domains, which activate the T cell in response to antigen recognition. The most commonly used CARs consist of an extracellular single‐chain variable fragment (scFv) that is directed against a tumor‐associated antigen and usually derived from murine, humanized or human antibody sequences, followed by a hinge or spacer region, a transmembrane domain and intracellular signaling domains derived from a costimulatory receptor (usually CD28 or 4‐1BB) and from CD3ζ (Fig. 1A). Very briefly, the manufacturing of CAR T cells involves isolation of T cells from the patients' blood, transduction with a lentivirus or retrovirus carrying the CAR gene, ex vivo expansion, and infusion of the final CAR T cell product into the patient (Fig. 1B). The remarkable clinical responses of patients treated with CAR T cells targeting the antigen CD19 expressed on B cell acute lymphoblastic leukemia (B‐ALL) and B cell lymphoma led to FDA approval of two CD19‐targeting CAR T cell products in 2017: Kymriah (tisagenlecleucel) and Yescarta (axicabtagene ciloleucel) [3]. Despite the highly promising clinical outcomes achieved by these products, CAR T cells frequently cause adverse events such as neurotoxicity and the release of large amounts of cytokines, resulting in a condition termed cytokine release syndrome [4, 5]. Furthermore, in contrast to the treatment of B cell malignancies, such high potency of CAR T cells has rarely been observed with solid tumors. Thus, there is still considerable room for improvement of this relatively young therapeutic approach. Since the CAR molecule is assembled from several domains, each of these components may be independently optimized to yield CAR T cells with improved therapeutic properties [6]. Moreover, the right components need to be matched to yield an efficient CAR molecule. For example, depending on the targeted epitope (proximal or distal to the membrane), different spacer lengths between the antigen‐targeting domain and the transmembrane domain may be required to yield an optimal distance between the T cell and the target cell membranes [7, 8, 9, 10].
Fig. 1.
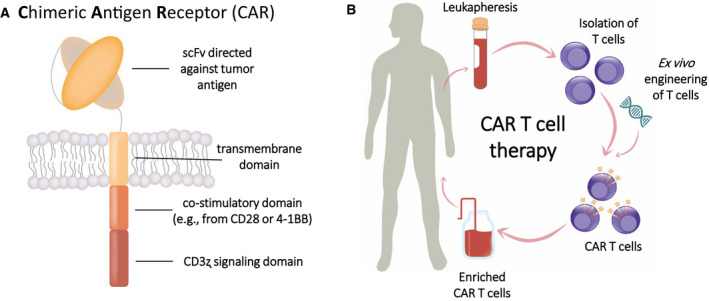
(A) Structure of a standard second‐generation CAR, consisting of an scFv targeting a tumor‐associated antigen, a transmembrane domain, and intracellular signaling domains derived from a costimulatory receptor and CD3ζ. (B) Basic scheme of CAR T cell therapy. Leukocytes are harvested by leukapheresis from the patient, followed by isolation of T cells and genetic engineering to induce CAR expression. After expansion, enriched CAR T cells are administered to the patient.
In this review, we focus on the antigen‐binding domain of CAR molecules. We discuss potential limitations of scFvs and advantages of engineered binding scaffolds as new options for CARs. We give an overview of the most commonly used binding scaffolds based on either nonhuman or human proteins and highlight those which have already been used within a CAR. Finally, we critically discuss the potential immunogenicity of engineered binding scaffolds in comparison with that of humanized and 'fully human' antibody fragments.
Limitations of scFvs as antigen recognition domains on CARs
In the vast majority of CARs currently under preclinical and clinical investigation, scFvs are used as the antigen‐targeting domain. Indeed, scFvs can be very attractive because of their small size and the possibility to engineer them to bind virtually any target molecule accessible on the cell surface. In addition, antibodies are already available for most antigens, facilitating the rapid and inexpensive generation of scFvs [11]. However, the major disadvantage of scFvs is their dependency on correct pairing between the linked VH and VL domains. This domain architecture entails the risk of uncontrolled heterodimerization between two scFv molecules. That is, instead of forming a correct VH‐VL pair within an scFv molecule, a VH can also pair with the VL of a neighboring molecule and vice versa, thereby forming so‐called diabodies [12] (Fig. 2). Depending on the length of the peptide linker, even complexes of three or four cross‐paired scFvs can be formed [13] (Fig. 2). This tendency of scFvs to oligomerize is well established in the protein engineering field and has been demonstrated not only for isolated soluble scFvs [12, 13, 14], but also for scFvs fused to a crystallizable fragment (Fc) [15] and, importantly, for scFvs integrated into CARs ([16] and [17]). This phenomenon can be explained by the lack of the natural structural support from the CH1‐CL interface that is normally part of a full‐length antibody and/or by partial unfolding at the VH‐VL interface, ultimately resulting in domain swapping [18]. Alternatively, mispairing may simply be a result of a stochastic process during protein folding, where a given VH may randomly pair either with its intramolecular VL partner or with a neighboring VL from another molecule. Apart from mispairing, scFvs have extended hydrophobic patches that are normally not solvent‐exposed in a full‐length antibody, potentially causing aggregation in the absence of domain swapping [18, 19].
Fig. 2.

Schematic representation of a conventional scFv and its oligomeric states, which are formed by all scFvs to various extents, depending on linker length and the specific scFv variant, among other factors. These oligomerization effects may occur both in a soluble format and when scFvs are integrated into CARs.
When incorporated into CARs, oligomerization of scFvs and subsequent CAR clustering can be detrimental since it can lead to antigen‐independent constitutive signaling of the CARs, which is referred to as tonic signaling [16, 18, 20]. Long and colleagues observed such tonic signaling in CAR T cells harboring an scFv against the disialoganglioside GD2, which subsequently led to T cell exhaustion and thereby poor in vivo performance of the GD2‐specific CAR [16]. This effect was further accompanied by lower CAR T cell expansion in vitro and increased T cell apoptosis. Importantly, the authors additionally investigated CARs containing four other scFvs, of which three induced tonic CAR signaling and T cell exhaustion at varying degrees: two different CARs recognizing CD22 (scFvs HA22 and m971) and one CAR directed against HER2 (scFv 4D5). Only the clinically used and highly potent CD19‐directed CAR (scFv FMC63) did not show constitutive activation and T cell exhaustion, which may at least partially explain the success of CAR T cells based on this scFv in the clinics. Taken together, these studies highlight that tonic signaling can be observed at varying degrees with many commonly used scFvs and can impact the performance of CAR T cell therapy.
In line with the functional data discussed above, recent experiments in our laboratories provided direct biochemical evidence that scFvs indeed induce CAR clustering on the T cell surface [17]. Briefly, we showed that CARs based on strictly monomeric backbones were dimerized/oligomerized by a low‐affinity version of the HER2‐directed scFv 4D5, resulting in multivalent antigen recognition and thereby avidity‐based activation of this low‐affinity CAR. Of note, co‐expression of two separate membrane‐anchored constructs comprising the VH and VL of 4D5, respectively, resulted in a functional CAR, but eliminated the clustering‐induced avidity effects. Thus, these data strongly suggest that the removal of the linker between the VH and VL prevented diabody formation, CAR clustering, and avidity‐based activation of the otherwise monomeric low‐affinity CARs.
Potential mispairing problems are expected to be even more pronounced in so‐called tandem CARs (TanCARs), in which two scFvs are expressed in tandem (i.e., fused to each other) to yield a bispecific CAR (Fig. 3A) [21, 22, 23]. Such TanCARs have mostly been constructed with the goal to design CARs with OR gate function. That is, those TanCARs recognize target cells expressing antigen A, or antigen B or both, thereby strongly reducing the risk of resistance due to antigen loss. While the construction of TanCARs containing two scFvs is definitely feasible, extensive optimization may be required to avoid mispairing between different scFvs (either those within a TanCAR or between neighboring TanCAR molecules). For example, the generation of a CD19‐CD22 bispecific CAR required a loop configuration, in which the CD22 scFv is expressed in between the VH and VL of the CD19 scFv (CD19VL‐CD22VH‐CD22VL‐CD19VH; Fig. 3B) [24, 25, 26]. Although this creative design was highly successful, resulting in a potent CAR that is currently being evaluated in clinical trials, it nevertheless demonstrates that the construction of TanCARs based on two scFvs is a particularly challenging task. In contrast, the construction of a TanCAR based on two engineered single‐domain binding scaffolds is more straightforward, since this strategy precludes undesirable domain swapping and oligomerization (Fig. 3C).
Fig. 3.

Schemes of (A) a tandem CAR based on two scFvs, (B) a tandem 'loop' CAR having a special configuration of the CD19 and CD22 scFv (CD19 VL‐CD22 VH‐CD22 VL‐CD19 VH), and (C) a tandem CAR based on two engineered binding scaffolds.
The potential of engineered binding scaffolds as alternatives to scFvs in CARs
Specific interaction of the CAR molecule with the target antigen and subsequent induction of T cell effector functions such as cytokine secretion and cytotoxicity is an essential prerequisite for effective CAR T cell therapy. An ideal antigen‐binding domain of a CAR molecule should meet the following requirements: (i) It should recognize its target molecule with high affinity and specificity, (ii) it should be stable and well expressed by primary human T cells, (iii) it should not induce CAR clustering and tonic signaling, and (iv) it should be as close as possible to human germline‐encoded proteins to minimize the risk of immunogenicity (immunogenicity will be extensively discussed below). While scFvs typically fulfill the first two requirements, many scFvs suffer from their tendency to induce tonic CAR signaling [16], as discussed above.
Of note, the development of various display technologies, such as phage display [27, 28], yeast display [29, 30, 31], and ribosome display [32, 33], allows the generation of artificial binding sites also in non‐immunoglobulin‐based protein scaffolds. In other words, the emergence of these protein engineering technologies eliminated the historical dependency on antibody‐derived constructs for antigen recognition. This resulted in the generation of many different types of engineered binding scaffolds, which can be considered as non‐antibody‐based alternatives to scFvs. Engineered binding scaffolds are typically developed by randomly mutating a certain surface area on a stable protein domain, followed by the selection of suitable binders from the resulting library by using one of the display methods mentioned above. Numerous different engineered binding scaffolds have been described in the literature (reviewed in Ref. [34, 35]). They are derived from various organisms (human, bacteria, archaea, plants, or even artificially designed proteins), cover a broad variety in their architecture (i.e., α‐helical or β‐sheet proteins, or combinations thereof), vary in their sizes, and comprise completely different structural elements on which the engineered binding surface is located (e.g., flexible loop regions, or rigid structures based on α‐helices or β‐sheets) (Fig. 4A,B). In some scaffolds, the randomized positions in the loop regions resemble the complementarity‐determining regions (CDRs) within antibodies (Fig. 4C) with an overall similar global fold, as exemplarily illustrated for monobodies (Fig. 4A). Notably, alternative binding scaffolds harbor several advantages: (i) They are typically highly stable and well‐expressed; (ii) many scaffolds do not contain any disulfide bonds (in contrast to scFvs), which makes them applicable in the reducing environment present in the cytoplasm [36]; and (iii) they are usually composed of only one protein domain. The latter is the most important advantage of engineered binding scaffolds with respect to their application in CARs, since the single‐domain architecture precludes any mispairing issues as is observed with scFvs. As mentioned above, numerous engineered binding scaffolds have been described in the last two decades. Since the focus of this review is directed toward the application of those engineered proteins in CAR T cells, only a representative selection of binding scaffolds (particularly those that have been used for the generation of CARs) will be discussed below and presented in Fig. 4. For a more comprehensive overview of engineered binding scaffolds, we refer the reader to other reviews [37, 38, 39].
Fig. 4.

(A) Structures of the most commonly used engineered binding scaffolds derived from the Protein Data Bank (PDB) (DARPin: 4JB8 [107], affibody: 2KZI [108], nanobody: 5F1K [109], Sso7d: 1SSO [110], monobody/adnectin 3QWQ [111], and anticalin: 1L6M [112]). Randomized positions within the respective scaffolds are highlighted in red. (B) Overview of the key features of depicted binding scaffolds. (C) Complementarity‐determining regions (CDRs) in an scFv. The VL domain is colored in light gray, the VH domain in dark gray, and the respective CDRs are colored in dark and light red for the VL and VH domains, respectively. The depicted scFv is directed against IL‐15 (PDB: 2XQB). All graphics were generated using pymol.
Examples for binding scaffolds based on nonhuman proteins
Designed ankyrin repeat proteins
Among the best‐known alternative scaffolds are designed ankyrin repeat proteins (DARPins). DARPins comprise several structurally similar repeats that are based on a consensus sequence derived from several natural ankyrin repeat proteins [40, 41]. Depending on the number of repeats, DARPins are approximately 14–17 kDa in size. The engineered binding surface is composed of both surface residues on α‐helices and loop residues (Fig. 4A). DARPins have been selected for binding to a wide range of targets and can exhibit high affinities in the picomolar range while at the same time providing the advantage of high thermostability [42, 43, 44]. Further, these compactly folded scaffolds can even be fused to each other without impairing folding, stability, or specificity. As an example, this allowed the generation of bivalent DARPin fusions targeting two different subdomains on HER2, thereby leading to enhanced receptor inactivation [45]. Another multispecific DARPin targeting VEGF and hepatocyte growth factor (HGF), while also binding to human serum albumin (HSA) for increased half‐life [46], is currently tested in combination with a tyrosine kinase inhibitor in a clinical trial for patients with non‐small cell lung cancer (NCT03418532).
Affibodies
Affibodies are extremely small (approx. 6–7 kDa) protein scaffolds derived from the Z‐domain of Staphylococcus aureus protein A (Fig. 4A). In contrast to antibodies, whose binding surface is based on loop regions, the engineered binding site on affibodies is located on rigid α‐helices [34, 38, 47]. Affibodies have been selected against various targets and HER2‐ and IL‐17A‐specific affibodies are currently in clinical testing (NCT03655353 and NCT03591887, respectively) [34, 39, 48, 49].
Sso7d and Sac7d
Sso7d is another minimalist binding scaffold with a size of only 7 kDa (Fig. 4A). This DNA‐binding protein derived from the hyperthermophilic archaeon Sulfolobus solfataricus is free of cysteines and extremely thermostable (T m of 99°C [50]). In addition to Sso7d, its close homolog Sac7d (derived from Sulfolobus acidocaldarius) has also been used for the selection of highly stable recognition domains [51]. Sso7d and Sac7d have been engineered to bind to completely different types of target molecules, including a small organic molecule (fluorescein), a peptide fragment (derived from β‐catenin) [52], and various proteins such as human Fc [53], among many others.
Recently, Traxlmayr et al. generated libraries based on a charge‐reduced version of Sso7d (reduced charge Sso7d, rcSso7d) to diminish unspecific interactions with mammalian cell surfaces resulting from excess positive charges on this DNA‐binding protein [50]. The resulting rcSso7d libraries were selected for binding to various antigens, such as human epidermal growth factor receptor (EGFR) [50] and for specific recognition of an oncogenic, mutated variant of human K‐Ras [36]. Recently, rcSso7d was also engineered to recognize human retinol‐binding protein 4 (hRBP4) in a small molecule‐dependent manner [54]. This ultimately yielded a molecular switch that enabled the functional control of CAR T cells by administration of an orally available small molecule, as will be further discussed below.
Nanobodies
Camelids are among the very few animals which express antibodies composed of only a heavy chain. As a consequence, the binding sites in those camelid antibodies only comprise a single domain, that is, the variable domain of the heavy chain (VHH). Separate expression of these VHH domains yields single‐domain antibody fragments (~15 kDa) that are commonly referred to as nanobodies (Fig. 4A) [55]. Because the light chain is missing in nanobodies, they do not require additional folding steps as is required for scFvs, where the VH and VL need to assemble. Thus, while nanobodies are antibody‐derived, they possess the critical advantage of a single‐domain architecture, similar to most non‐antibody‐derived scaffold proteins.
Another benefit of nanobodies is their ability to reach some epitopes which are not accessible to conventional antibodies [56]. A good example for such a binding mode is nanobodies selected against the glycoprotein 140 of human immunodeficiency virus (HIV), which have been shown to neutralize the majority of known HIV strains [57]. The authors speculated that the longer CDR3 loop can bind to a neutralizing epitope, which normally is inaccessible to conventional antibody formats. Currently, several bispecific nanobodies are investigated in clinical trials (NCT03384745, NCT03468426, and NCT03972150). In early 2019, the first nanobody termed caplacizumab was approved by the FDA. Caplacizumab is a bivalent agent comprising two identical humanized nanobodies targeting von Willebrand factor, a key protein for blood coagulation. This protein is not only the first nanobody to be ever approved, but it is also the first therapy for patients with acquired thrombotic thrombocytopenic purpura, which is a rare disease characterized by excessive blood clotting [58].
Examples for binding scaffolds based on human proteins
Monobodies/Adnectins
Monobodies—also termed adnectins—are small (10 kDa) and cysteine‐free scaffolds based on the 10th type III domain of human fibronectin (FN3) [59, 60]. They possess a β‐sandwich structure and an engineered binding surface that is typically based on three solvent‐exposed loops, resembling the CDR loops of antibodies (Fig. 4A) [59, 60, 61]. In an alternative library design, the binding site has been engineered on the 'side' of the FN3 domain, that is, including a β‐sheet and two‐loop regions (termed 'side‐and‐loop library'), thereby forming a concave binding surface [61].
Due to the human origin and high stability of the FN3 domain (T m of 86°C [62]), monobodies have evolved as some of the most commonly used non‐antibody‐based scaffolds. Among the many monobodies generated so far, CT‐322 is among the most advanced products. It targets human VEGFR‐2, is attached to polyethylene glycol (PEG) to increase its half‐life, and was already tested in a phase II study for patients with recurrent glioblastoma which resulted in a partial response in at least one patient [63]. Alternatively, due to the lack of disulfide bonds (as is the case for most other binding scaffolds), monobodies have also been used for blocking various intracellular molecules and signaling pathways [64, 65].
Anticalins
Anticalins are engineered scaffolds based on the lipocalin family, of which 15 members have been described in humans [66]. Those proteins are approximately 20 kDa in size and have a characteristic eight‐stranded β‐barrel fold, which binds to small hydrophobic ligands such as fatty acids and vitamins [67, 68]. The four structurally flexible loops at the entrance of the binding pocket can be engineered to yield antigen‐specific anticalins (Fig. 4A) [69]. This has been used to generate anticalins directed against many different antigens for a broad spectrum of applications, involving therapeutic approaches and in vivo imaging as well as diagnostics [70]. Importantly, anticalins targeting VEGF‐A, PCSK9, hepcidin, IL‐4‐Rα, and a bispecific product targeting HER2 and the costimulatory receptor 4‐1BB have entered the clinical development for the treatment of a diverse set of diseases [71].
Examples for engineered binding scaffolds used as antigen recognition domains in CARs
Despite the availability of a range of different binding scaffolds, the vast majority of CARs has been constructed based on scFvs. Nevertheless, over the last years several studies have accumulated, in which alternative binding scaffolds were successfully used as antigen recognition domains in CARs.
One example is a CAR in which a HER2‐specific DARPin was integrated into a CAR backbone of the second‐generation comprising intracellular signaling domains derived from CD28 and CD3ζ. This DARPinCAR could be expressed in T cells at levels comparable to that of a CAR based on a HER2‐specific scFv and mediated lysis of various HER2‐expressing human and murine tumor cell lines [72].
In another study, two HER2‐targeting DARPins were integrated into CARs comprising signaling domains derived from CD28, 4‐1BB, and CD3ζ [73]. In in vitro assays, the resulting DARPinCARs showed slightly higher capacities to induce T cell effector functions than an scFv‐based control CAR. Surprisingly, when tested in a mouse model, only one out of two DARPinCARs and the scFv‐CAR showed efficient tumor clearance. Interestingly, both the functional DARPinCAR and the scFv‐CAR target the same membrane distal epitope on HER2 while the other DARPin was shown to bind to the membrane‐proximal domain IV of HER2, suggesting that the location of the epitope plays an important role independent of the used binding domain [73].
De Munter et al. designed a bispecific CAR targeting both HER2 and CD20 by linking two llama‐derived nanobodies. Both bispecific and monospecific nanobody‐based CARs (termed nanoCARs) were expressed at levels comparable to that of a monospecific scFv‐based CAR. In addition, target cells engineered to express either HER2 or CD20, as well as those expressing both antigens, were efficiently lysed by the respective nanoCAR T cells [74]. This study is a good example, where the use of single‐domain binding scaffolds avoided the problem of mispairing—even though two antigen‐binding modules were expressed in series in one CAR molecule.
Xie et al. [75] generated nanobody‐based CAR T cells to target the tumor microenvironment. They selected nanobodies specific for either programmed death‐ligand 1 (PD‐L1) or the fibronectin splice variant EIIIB, an immune checkpoint and a marker of tumor extracellular matrix, respectively. Both the PD‐L1‐ and the EIIIB‐specific nanoCAR T cells efficiently delayed tumor growth in a B16 melanoma model.
Apart from DARPins and nanobodies, also monobodies have been applied for the design of CAR molecules. Monobodies, which had previously been engineered to bind to EGFR with low nanomolar affinities [76], were incorporated into CARs harboring two costimulatory domains. In general, the expression levels of monobody‐based CARs in primary T cells were comparable to that of an scFv‐based control CAR [77]. The EGFR‐specific monobody showed slightly lower affinity than the scFv derived from the EGFR‐specific clinical antibody cetuximab. The authors argued that this difference in affinity could be used for better discrimination of EGFR‐high‐ vs. EGFR‐low‐expressing cells, since it is known that high‐affinity CAR T cells recognize also healthy tissue expressing low antigen levels. Indeed, target cells with low levels of EGFR were still recognized by the scFv‐based CAR T cells while being spared by the monobody‐CAR T cells [77]. In contrast, target cells expressing high levels of EGFR were efficiently lysed by both types of CARs.
In a recent study, we engineered two completely different binding scaffolds (rcSso7d and monobodies) to recognize hRBP4 only when loaded with an orally available small molecule drug called A1120. That is, selected binders bound to the drug‐loaded conformation of hRBP4 with high affinity while exhibiting approximately 500‐fold lower affinity to hRBP4 in the absence of this small molecule [54]. This yielded a molecular ON switch, in which a protein–protein interaction can be turned on with an orally available small molecule drug. By incorporating one of these engineered molecular switches into a CAR, the assembly of a functional CAR molecule could be regulated by administration of the drug A1120. This ultimately enabled the functional control of primary human CAR T cells in vitro, as demonstrated by regulated cytokine secretion and cytotoxic activity.
Finally, in another study from our group rcSso7d‐based binders against EGFR and affibody‐based binders against HER2 were used to analyze the relationship between CAR dimerization, affinity of the antigen‐binding domain, and CAR T cell activation. This analysis was only enabled by the availability of those monomeric single‐domain binding scaffolds. In contrast, when we used an scFv directed against HER2, it was not possible to investigate the effect mediated by CAR dimerization, since dimerization/oligomerization was already induced by the scFv in an uncontrolled manner, as discussed above [17].
Taken together, these studies underline the versatility of engineered binding scaffolds for the design of CARs. Thus, there is no functional requirement that necessitates the use of scFvs as antigen‐binding domains in CARs. Instead, the suitability of an antigen recognition domain will be dictated by its recognized epitope, its affinity and specificity, its tendency to dimerize/oligomerize, and its expression rate when fused to a CAR backbone. In some cases, the optimal choice will be an scFv, whereas in other instances an engineered binding scaffold might be more suitable.
Potential immunogenicity
Apart from the biochemical and functional properties of engineered binding scaffolds, their potential immunogenicity is another important factor that needs to be considered. Especially for binding scaffolds derived from nonhuman proteins, the risk of being recognized by the patient's immune system is a potential drawback. Indeed, it was already observed in early clinical studies that the administration of CAR T cells can induce both a humoral and cellular immune response, which leads to blockade and limited persistence of the CAR T cells [78]. Interestingly, patients, who relapsed with antigen‐positive tumor cells after treatment with anti‐CD19 CAR T cells based on a murine scFv, could be successfully treated with CAR T cells based on either a humanized anti‐CD19 scFv [79] or a 'fully human' anti‐CD22 scFv. This observation suggests that the rejection of the CAR T cells based on the murine scFv could be overcome by using scFvs that are closer to human germline sequences. Therefore, the CAR T cell field is moving toward the use of humanized or 'fully human' scFvs [25, 79, 80].
However, it is important to note that every engineered binding domain—even those derived from human sequences—is not 'fully human'. That is, in order to generate an antigen‐binding site, a number of surface positions need to be mutated. Even our immune system modifies the sequences of the variable domains of antibodies through junctional diversity [81] and somatic hypermutation [82] in order to achieve the high affinities that are typical for antibodies. Thus, apart from assembling the antibody genes from germline‐encoded V, D, and J segments, B cells undergo molecular processes to introduce additional diversity, thereby deviating from the germline sequence.
The risk of T cell‐mediated immunogenicity of a given polypeptide sequence depends on many factors, including protein/peptide processing efficiency, altered processing by the immune proteasome, recognition by a T cell receptor (TCR), and—most importantly—whether the resulting peptide is efficiently displayed on any of the major histocompatibility complex (MHC) alleles expressed in a given patient (or a patient population) [83, 84]. Several assays have been developed to predict—or at least estimate—the risk of immunogenicity, including in silico tools, in vitro models with peripheral blood mononuclear cells, and in vivo models using transgenic mice or nonhuman primates [84]. In case a certain sequence stretch in a protein is predicted to be potentially immunogenic, it is possible to reduce the immunogenicity of this epitope by inserting mutations. For example, the number of predicted T cell epitopes in green fluorescent protein (GFP) and Pseudomonas exotoxin A has been successfully reduced without compromising protein function [85, 86]. However, while those prediction models may help to reduce the risk of immunogenicity of lead candidates, a definitive answer on the immunogenicity of an engineered therapeutic protein will only be obtained upon testing in a reasonably sized patient population (covering a representative set of MHC molecules) [84].
As already mentioned, in addition to T cell‐mediated immune responses, B cell‐mediated immunity may also play a role. For example, potent humoral immune responses were observed in an early clinical trial with repeated administration of T cells expressing a first‐generation CAR based on a murine scFv directed against carbonic anhydrase IX (CAIX) [78]. In this trial, the humoral responses were anti‐idiotypic in nature and neutralized the function of the CAIX‐specific CAR T cells. Notably, the occurrence of anti‐idiotypic antibodies in the blood coincided with the inability to detect circulating CAR T cells by anti‐idiotype antibody‐based flow cytometric analysis, whereas qPCR analysis of CAR DNA still showed their presence. In another study, repeated infusion of CAR T cells electroporated with mRNA coding for a CAR based on a murine scFv against human mesothelin resulted in anaphylaxis, likely caused by IgE antibodies directed against the CAR [87].
Given the lack of perfectly reliable models for the prediction of immunogenicity, it is generally desired to design therapeutic proteins that are as close to human germline as possible. For example, a chimeric antibody, in which the entire variable domains are of nonhuman origin, is considered more likely to be immunogenic than a humanized antibody which only comprises nonhuman CDRs and possibly some nonhuman framework positions. However, caution is needed, because the word 'humanized' is somewhat misleading, since it implies that the resulting monoclonal antibodies (mAbs) are human and therefore not distinguishable from an endogenous human protein. This is clearly not the case, given the nonhuman origin of their CDR loops. In addition, usually some framework mutations are needed to maintain binding and/or stability of the resulting humanized mAbs [88, 89].
Although the number of nonhuman amino acid positions is clearly not the only determinant of immunogenicity, we aimed at roughly comparing this deviation from human sequences in different antigen recognition domains. Therefore, we determined the number of amino acid positions differing from human germline‐encoded genes for the following antigen‐binding formats (Fig. 5): (i) scFvs derived from 'humanized' blockbuster antibodies (being defined here by worldwide sales of at least US$ 1 billion in 2016 [90]); (ii) scFvs derived from 'human' blockbuster antibodies; (iii) nanobodies based on camelid VHH domains; (iv) monobodies based on the human FN3 domain; (v) anticalins based on human lipocalins; (vi) binders based on Sso7d or rcSso7d derived from S. solfataricus; and (vii) affibodies based on the Z‐domain of protein A from S. aureus. As expected, 'human' scFvs are closest to human germline sequences (Fig. 5). More surprisingly, engineered monobodies and anticalins tend to contain fewer nonhuman amino acid positions than humanized scFvs (Fig. 5). It should be noted that for the humanized and human scFvs (as well as for nanobodies) these numbers are probably underestimating the deviations from germline, due to four factors: First, we used the DomainGapAlign tool from IMGT (www.imgt.org), which was also recommended for alignment to human antibody germline sequences by the WHO INN Expert Group in April 2015 [88]. However, due to the short lengths of the D segments and additional junctional diversity and/or somatic hypermutation, it was not possible to assign those D segments to the CDR‐H3s. As a consequence, parts of the usually considerably mutated CDR‐H3 sequences were excluded from the analysis (Fig. S1), thereby reducing the number of nonhuman amino acid positions. Second, it is known that each individual only carries a fraction of the known germline antibody gene segments (e.g., approximately 50 out of 250 known V segments [91]). As a consequence, aligning an antibody against the full set of germline gene segments in the IMGT database will yield a closest match, which might not be present in a given patient. In other words, the closest V segment in that patient might differ even more from the given antibody sequence, which would further increase the number of nonhuman positions in that scFv. Third, it also needs to be taken into consideration that for proteins based on human germline genes (such as antibodies or monobodies), scattered mutations will result in novel nonhuman epitopes, even if these stretches only contain a certain percentage of mutated residues. As an example, a 12‐amino acid peptide containing six mutated residues does not look like a human peptide any more. Thus, while the entire peptide will be seen as 'nonhuman', the alignment only defines six mutated amino acid positions, thus again underestimating the extent of nonhuman sequence stretches. Fourth, the linkers between VH and VL in scFvs were also excluded from the analysis. Thus, the data for humanized and human scFvs and for nanobodies in Fig. 5 should be interpreted as conservative estimates.
Fig. 5.
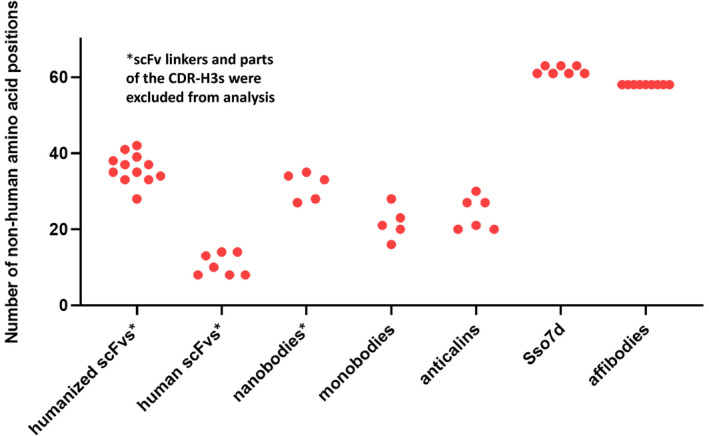
Analysis of the number of nonhuman amino acid positions in human and humanized scFvs as compared to engineered binding scaffolds based on human or nonhuman protein domains. To determine the deviation from human germline sequences for humanized and human scFvs, the VH and VL sequences of humanized and human blockbuster antibodies were obtained from the DrugBank (https://www.drugbank.ca/) and subsequently aligned to human antibody germline genes using the DomainGapAlign tool from IMGT (www.imgt.org). An alignment is exemplarily shown for trastuzumab in Fig. S1. Since the D germline segments could not be unambiguously assigned to the CDR‐H3 sequences, parts of the CDR‐H3s were omitted from analysis. Therefore, the actual number of nonhuman amino acid positions will be slightly higher for human and humanized scFvs, as well as for nanobodies, which were also aligned using the DomainGapAlign tool. To determine the number of nonhuman amino acid positions in engineered binding scaffolds, monobodies and anticalins were aligned against the original human proteins (FN3 domain and lipocalins, respectively), whereas Sso7d and affibodies were assumed to be 100% nonhuman. Since the charge‐reduced version of Sso7d (rcSso7d) is shortened by 2 amino acids, the amount of nonhuman positions is either 61 (rcSso7d‐based binders) or 63 (Sso7d‐based mutants). All names and sequences of analyzed proteins are depicted in Table S1.
Again, we would like to emphasize that the number of nonhuman amino acid positions in a protein is not expected to perfectly correlate with its immunogenic potential. Nevertheless, it can be expected that there is some trend. This was also the underlying assumption in the therapeutic antibody field, which moved from fully murine hybridoma antibodies to chimeric mAbs and finally to humanized and 'fully human' mAbs. Most importantly, the data in Fig. 5 clearly demonstrate that judging the risk of immunogenicity of a therapeutic protein solely based on its human vs. nonhuman origin would be an oversimplification and in many cases incorrect. For example, nanobodies derived from camelids tend to contain a comparable or even slightly lower number of nonhuman amino acid positions than humanized scFvs, which can be explained by the fact that—despite having more nonhuman framework positions—they only contain three CDR loops and are only approximately half the size of an scFv. Even binding scaffolds based on nonhuman proteins such as affibodies or Sso7d‐based binders only show slightly elevated levels of such nonhuman positions compared with humanized scFvs. Taking into consideration the expected underestimation of the deviation of scFvs from germline (as discussed above), the difference between humanized scFvs and engineered nonhuman binding scaffolds will be even smaller.
Finally, it should also be noted that in addition to foreign sequences within protein domains also the junctions between different CAR domains unavoidably create non‐native peptide stretches that may induce an immune response as has been observed, for example, with the oncogenic fusion protein Bcr‐Abl [92, 93].
Natural receptors or ligands as alternatives to scFvs on CARs
Besides engineered binding scaffolds and scFvs, natural ligands and receptors can also be used as alternative binding moieties in CARs. They harbor several advantages compared with engineered protein scaffolds: (i) They naturally occur in the human body, thereby minimizing the risk of potential immunogenicity; (ii) many of them can bind multiple targets, which broadens the range of applications and prevents escape due to antigen downregulation; and (iii) some of their ligands are upregulated upon stress conditions and can thereby often be found in the tumor microenvironment.
Natural killer group 2 member D‐CARs
One example for such a natural receptor is natural killer group 2 member D (NKG2D), a type II transmembrane protein present on various immune cells, such as natural killer (NK) cells, CD8+ T cells, and γδ T cells [94]. NKG2D binds to eight known ligands (MICA, MICB, and ULBP1 to ULBP6) which are present on a wide range of tumors of various categories, including carcinomas, sarcomas, leukemias, lymphomas, and multiple myelomas [95]. Notably, NKG2D ligands are not only expressed by tumor tissue, but can also be found on immunosuppressive cells such as regulatory T cells (Tregs) and myeloid‐derived suppressor cells (MDSCs) [96]. Given these expression patterns of NKG2D ligands, the extracellular domain of NKG2D is a promising candidate for an antigen recognition domain on a CAR. First reports of an NKG2D‐based CAR demonstrated efficacy in murine models and in vitro activity against human tumor cell lines [97, 98]. Lehner and colleagues developed an NKG2D‐CAR with a reengineered extracellular domain by fusing the C terminus of NKG2D in a type I membrane protein orientation to generate a second‐generation CAR with integrated CD28 costimulation [99]. Subsequently, NKG2D was fused to CAR backbones with 4‐1BB costimulation and—for expression of the CAR in NK cells—also to DAP12 [100, 101]. Meanwhile, NKG2D CAR T cells and also NKG2D CAR NK cells have already been tested in several clinical phase I studies [101, 102, 103]. Overall, the administration of NKG2D‐CAR cells in absence of lymphodepleting conditioning was well‐tolerated without significant side effects. Notably, despite limited persistence, promising clinical responses were observed in patients with AML and colorectal cancer [101, 103].
Lymphocyte function‐associated antigen 1‐CARs
Another antigen that is commonly overexpressed by tumors is intercellular adhesion molecule 1 (ICAM1). It can be found on carcinomas but is also expressed at low level on endothelial and immune cells [104]. In previous studies, Park and colleagues have affinity‐matured the natural ligand, lymphocyte function‐associated antigen 1 (LFA‐1) by directed evolution to achieve several variants covering a huge affinity range (K D 1 nm–1 mm) [105]. When these LFA‐1 variants with different affinities to ICAM1 were incorporated into a CAR comprising two costimulatory domains (CD28 and 4‐1BB), the authors found that both antigen density and affinity to ICAM1 were directly proportional to CAR T cell activity. Interestingly, CAR T cells incorporating high‐affinity binders not only killed tumor cells, but also induced systemic toxicity in a mouse model. In contrast, lower‐affinity CARs in the micromolar range could delay tumor growth without any observed toxicity. This study elegantly shows that natural ligands or receptors can even be affinity‐tuned by introducing a limited number of mutations (in this case only 1–3 mutations, depending on the LFA‐1 variant). The availability of recognition domains in a broad affinity range may be a critical advantage, allowing the investigator to choose the optimal affinity to yield highly efficacious CAR T cells, while only causing limited systemic toxicity.
Interleukin 13 (IL‐13) mutein‐CARs
An attractive strategy to treat glioblastoma involves CAR T cells recognizing the IL‐13‐receptor α 2 (IL‐13Rα2), which is commonly overexpressed in brain tumors. In these CARs, antigen recognition is mediated by mutated versions of the antigen's natural ligand, that is, IL‐13 muteins. A recent case study reported a complete clinical response lasting for more than 7 months in a glioblastoma patient treated with repeated infusions of IL‐13 mutein CAR T cells [106]. Together, IL‐13 muteins represent a further example of natural ligands, which have been engineered through introduction of a limited number of mutations to achieve a therapeutically optimal affinity.
Conclusion
Due to the availability of monoclonal antibodies against virtually any antigen, scFvs represent a convenient option for the design of CARs. However, many scFvs have been shown to trigger tonic CAR signaling and T cell exhaustion [16, 18, 20], presumably caused by scFv clustering. As extensively discussed in this review, there is no biochemical or functional requirement for the use of scFvs, as demonstrated by multiple studies using engineered binding scaffolds or natural ligands or receptors for the construction of CARs. Therefore, we anticipate that these alternative antigen recognition domains will be increasingly used in the CAR field, hopefully overcoming some of the current challenges in CAR T cell therapy.
Conflicts of interest
C.U.Z., B.S., M.L., and M.W.T. have filed patent applications related to the technologies described in this manuscript. The other authors declare no conflict of interest.
Author contributions
JMT and STR performed sequencing alignments. CUZ and MWT analyzed the sequencing alignments to yield mutation counts. CUZ, BS, ML, and MWT wrote the manuscript. All authors read and edited the manuscript.
Supporting information
Fig. S1. Representative VH and VL alignments. VH and VL sequences of human and humanized antibodies were obtained from the DrugBank (https://www.drugbank.ca/) and aligned against human antibody germline sequences using the DomainGapAlign tool from IMGT (www.imgt.org).
Table S1. Amino acid sequences of VH and VL domains derived from human and humanized antibodies and engineered binding scaffolds used for the analysis of the number of non‐human amino acid positions in Figure 5.
Acknowledgements
This work is supported by the Austrian Science Fund (FWF Project W1224—Doctoral Program on Biomolecular Technology of Proteins—BioToP), the Federal Ministry for Digital and Economic Affairs of Austria, and the National Foundation for Research, Technology and Development of Austria to the Christian Doppler Research Association (Christian Doppler Laboratory for Next Generation CAR T Cells) and private donations to the St. Anna Children's Cancer Research Institute (Vienna, Austria).
Contributor Information
Manfred Lehner, Email: manfred.lehner@ccri.at.
Michael W. Traxlmayr, Email: michael.traxlmayr@boku.ac.at.
References
- 1. June CH & Sadelain M (2018) Chimeric antigen receptor therapy. N Engl J Med 379, 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Salter AI, Pont MJ & Riddell SR (2018) Chimeric antigen receptor‐modified T cells: CD19 and the road beyond. Blood 131, 2621–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. The Lancet . (2017) CAR T‐cells: an exciting frontier in cancer therapy. Lancet 390, 1006. https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(17)32395-4/fulltext [DOI] [PubMed] [Google Scholar]
- 4. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, Komanduri KV, Lin Y, Jain N, Daver N et al. (2018) Chimeric antigen receptor T‐cell therapy – assessment and management of toxicities. Nat Rev Clin Oncol 15, 47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez‐Cuyar LF, Yeung C, Liles WC, Wurfel M, Lopez JA et al. (2017) Endothelial activation and blood‐brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR‐T cells. Cancer Discov 7, 1404–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guedan S, Calderon H, Posey AD Jr & Maus MV (2019) Engineering and design of chimeric antigen receptors. Mol Ther Methods Clin Dev 12, 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, Raubitschek AA, Forman SJ & Press OW (2008) Antigen sensitivity of CD22‐specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol 180, 7028–7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O'Neill A, Irlam J, Chester KA, Kemshead JT, Shaw DM et al. (2005) The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother 28, 203–211. [DOI] [PubMed] [Google Scholar]
- 9. Hudecek M, Lupo‐Stanghellini M‐T, Kosasih PL, Sommermeyer D, Jensen MC, Rader C & Riddell SR (2013) Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1‐specific chimeric antigen receptor T cells. Clin Cancer Res 19, 3153–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hudecek M, Sommermeyer D, Kosasih PL, Silva‐Benedict A, Liu L, Rader C, Jensen MC & Riddell SR (2015) The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res 3, 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lerner RA (2016) Combinatorial antibody libraries: new advances, new immunological insights. Nat Rev Immunol 16, 498–508. [DOI] [PubMed] [Google Scholar]
- 12. Todorovska A, Roovers RC, Dolezal O, Kortt AA, Hoogenboom HR & Hudson PJ (2001) Design and application of diabodies triabodies and tetrabodies. J Immunol Methods 248, 47–66. [DOI] [PubMed] [Google Scholar]
- 13. Gil D & Schrum AG (2013) Strategies to stabilize compact folding and minimize aggregation of antibody‐based fragments. Adv Biosci Biotechnol 4, 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wörn AP & Plückthun A(2001) Stability engineering of antibody single chain Fv fragments. J Mol Biol 305, 898–1010. [DOI] [PubMed] [Google Scholar]
- 15. Wu AM, Tan GJ, Sherman MA, Clarke P, Olafsen T, Forman SJ & Raubitschek AA (2001) Multimerization of a chimeric anti‐CD20 single‐chain Fv‐Fc fusion protein is mediated through variable domain exchange. Protein Eng 14, 1025–1033. [DOI] [PubMed] [Google Scholar]
- 16. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR et al. (2015) 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 21, 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Salzer B, Schueller CM, Zajc CU, Peters T, Schoeber MA, Kovacic B, Buri MC, Lobner E, Dushek O, Huppa JB et al. (2020) Engineering AvidCARs for combinatorial antigen recognition and reversible control of CAR function. Nature Communications 11, 10.1038/s41467-020-17970-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ajina A & Maher J (2018) Strategies to address chimeric antigen receptor tonic signaling. Mol Cancer Ther 17, 1795–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nieda L, Honegger A, Krebber C & Plückthun A (1997) Disrupting hydrophobic patches at the variable constant domain interface‐ improved in vivo folding and physical characterization of an engineered scfv fragment. Protein Eng 10, 435–444. [DOI] [PubMed] [Google Scholar]
- 20. Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, Kawalekar OU, Guedan S, McGettigan SE, Posey AD Jr et al. (2015) Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res 3, 356–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A et al. (2015) Human Epidermal Growth Factor Receptor 2 (HER2) –specific chimeric antigen receptor–modified t cells for the immunotherapy of HER2‐positive sarcoma. J Clin Oncol 33, 1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, Corder A, Schonfeld K, Koch J, Dotti G et al. (2013) TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids 2, e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zah E, Lin MY, Silva‐Benedict A, Jensen MC & Chen YY (2016) T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res 4, 498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Labanieh L, Majzner RG & Mackall CL (2018) Programming CAR‐T cells to kill cancer. Nat Biomed Eng 2, 377–391. [DOI] [PubMed] [Google Scholar]
- 25. Fry TJ, Shah NN, Orentas RJ, Stetler‐Stevenson M, Yuan CM, Ramakrishna S, Wolters P, Martin S, Delbrook C, Yates B et al. (2018) CD22‐targeted CAR T cells induce remission in B‐ALL that is naive or resistant to CD19‐targeted CAR immunotherapy. Nat Med 24, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qin H, Ramakrishna S, Nguyen S, Fountaine TJ, Ponduri A, Stetler‐Stevenson M, Yuan CM, Haso W, Shern JF, Shah NN & Fry TJ (2018) Preclinical development of bivalent chimeric antigen receptors targeting both CD19 and CD22. Mol Therapy Oncol 11, 127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Winter G, Griffiths AD, Hawkins RE & Hoogenboom HR (1994) Making antibodies by phage display technology. Annu Rev Immunol 12, 433–455. [DOI] [PubMed] [Google Scholar]
- 28. Hoogenboom HR & Chames P (2000) Natural and designer binding sites made by phage display technology. Immunol Today 21, 371–378. [DOI] [PubMed] [Google Scholar]
- 29. Cherf GM & Cochran JR (2015) Applications of yeast surface display for protein engineering. Methods Mol Biol 1319, 155–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gai SA & Wittrup KD (2007) Yeast surface display for protein engineering and characterization. Curr Opin Struct Biol 17, 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Angelini A, Chen TF, de Picciotto S, Yang NJ, Tzeng A, Santos MS, Van Deventer JA, Traxlmayr MW & Wittrup KD (2015) Protein engineering and selection using yeast surface display. Methods Mol Biol 1319, 3–36. [DOI] [PubMed] [Google Scholar]
- 32. He M & Khan F (2005) Ribosome display: next‐generation display technologies for production of antibodies in vitro. Expert Rev Proteom 2, 421–430. [DOI] [PubMed] [Google Scholar]
- 33. Plückthun A In: Ribosome Display and Related Technologies (Douthwaite J., Jackson R. eds) . Methods in Molecular Biology (Methods and Protocols), vol 805. Springer, New York, NY. 10.1007/978-1-61779-379-0_1 [DOI] [Google Scholar]
- 34. Owens B (2017) The rise of non‐antibody based scaffolds. Nat Biotechnol 35, 602–603. [DOI] [PubMed] [Google Scholar]
- 35. Skrlec K, Strukelj B & Berlec A (2015) Non‐immunoglobulin scaffolds: a focus on their targets. Trends Biotechnol 33, 408–418. [DOI] [PubMed] [Google Scholar]
- 36. Kauke MJ, Traxlmayr MW, Parker JA, Kiefer JD, Knihtila R, McGee J, Verdine G, Mattos C & Wittrup KD (2017) An engineered protein antagonist of K‐Ras/B‐Raf interaction. Sci Rep 7, 5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Konning D & Kolmar H (2018) Beyond antibody engineering: directed evolution of alternative binding scaffolds and enzymes using yeast surface display. Microb Cell Fact 17, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Simeon R & Chen Z (2018) In vitro‐engineered non‐antibody protein therapeutics. Protein Cell 9, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vazquez‐Lombardi R, Phan TG, Zimmermann C, Lowe D, Jermutus L & Christ D (2015) Challenges and opportunities for non‐antibody scaffold drugs. Drug Discov Today 20, 1271–1283. [DOI] [PubMed] [Google Scholar]
- 40. Boersma YL & Pluckthun A (2011) DARPins and other repeat protein scaffolds: advances in engineering and applications. Curr Opin Biotechnol 22, 849–857. [DOI] [PubMed] [Google Scholar]
- 41. Jost CP & Plückthun A(2014) DARPins other alternative scaffolds and bispecific IgGs. Curr Opin Chem Biol 27, 102–112. [DOI] [PubMed] [Google Scholar]
- 42. Pluckthun A (2015) Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu Rev Pharmacol Toxicol 55, 489–511. [DOI] [PubMed] [Google Scholar]
- 43. Binz HK, Amstutz P, Kohl A, Stumpp MT, Briand C, Forrer P, Grutter MG & Pluckthun A (2004) High‐affinity binders selected from designed ankyrin repeat protein libraries. Nat Biotechnol 22, 575–582. [DOI] [PubMed] [Google Scholar]
- 44. Kohl A, Binz HK, Forrer P, Stumpp MT, Pluckthun A & Grutter MG (2003) Designed to be stable: crystal structure of a consensus ankyrin repeat protein. Proc Natl Acad Sci U S A 100, 1700–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jost C, Schilling J, Tamaskovic R, Schwill M, Honegger A & Plückthun A (2013) Structural basis for eliciting a cytotoxic effect in HER2‐overexpressing cancer cells via binding to the extracellular domain of HER2. Structure 21, 1979–1991. [DOI] [PubMed] [Google Scholar]
- 46. Binz HK, Bakker TR, Phillips DJ, Cornelius A, Zitt C, Gottler T, Sigrist G, Fiedler U, Ekawardhani S, Dolado I et al. (2017) Design and characterization of MP0250, a tri‐specific anti‐HGF/anti‐VEGF DARPin(R) drug candidate. MAbs 9, 1262–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Richards DA (2018) Exploring alternative antibody scaffolds: antibody fragments and antibody mimics for targeted drug delivery. Drug Discov Today Technol 30, 35–46. [DOI] [PubMed] [Google Scholar]
- 48. Sorensen J, Velikyan I, Sandberg D, Wennborg A, Feldwisch J, Tolmachev V, Orlova A, Sandstrom M, Lubberink M, Olofsson H et al. (2016) Measuring HER2‐receptor expression in metastatic breast cancer using [68Ga]ABY‐025 affibody PET/CT. Theranostics 6, 262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Frejd FY & Kim KT (2017) Affibody molecules as engineered protein drugs. Exp Mol Med 49, e306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Traxlmayr MW, Kiefer JD, Srinivas RR, Lobner E, Tisdale AW, Mehta NK, Yang NJ, Tidor B & Wittrup KD (2016) Strong enrichment of aromatic residues in binding sites from a charge‐neutralized hyperthermostable Sso7d scaffold library. J Biol Chem 291, 22496–22508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mouratou B, Schaeffer F, Guilvout I, Tello‐Manigne D, Pugsley AP, Alzari PM & Pecorari F (2007) Remodeling a DNA‐binding protein as a specific in vivo inhibitor of bacterial secretin PulD. Proc Natl Acad Sci U S A. 104, 17983–17988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gera N, Hussain M, Wright RC & Rao BM (2011) Highly stable binding proteins derived from the hyperthermophilic Sso7d scaffold. J Mol Biol 409, 601–616. [DOI] [PubMed] [Google Scholar]
- 53. Gera N, Hill AB, White DP, Carbonell RG & Rao BM (2012) Design of pH sensitive binding proteins from the hyperthermophilic Sso7d scaffold. PLoS One 7, e48928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zajc CU, Dobersberger M, Schaffner I, Mlynek G, Puhringer D, Salzer B, Djinovic‐Carugo K, Steinberger P, De Sousa Linhares A, Yang NJ et al. (2020) A conformation‐specific ON‐switch for controlling CAR T cells with an orally available drug. Proc Natl Acad Sci U S A 117, 14926–14935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jank L, Pinto‐Espinoza C, Duan Y, Koch‐Nolte F, Magnus T & Rissiek B (2019) Current approaches and future perspectives for nanobodies in stroke diagnostic and therapy. Antibodies 8, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ingram JR, Schmidt FI & Ploegh HL (2018) Exploiting nanobodies´ singular traits. Ann Rev Immunol 36, 1–21. [DOI] [PubMed] [Google Scholar]
- 57. Koch K, Kalusche S, Torres JL, Stanfield RL, Danquah W, Khazanehdari K, von Briesen H, Geertsma ER, Wilson IA, Wernery U et al. (2017) Selection of nanobodies with broad neutralizing potential against primary HIV‐1 strains using soluble subtype C gp140 envelope trimers. Sci Rep 7, 8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Morrison C (2019) Nanobody approval gives domain antibodies a boost. Nat Rev Drug Dis 18, 485–487. [DOI] [PubMed] [Google Scholar]
- 59. Koide AB, Bailey CW, Huang X & Koide S(1998) The Fn3 domain as a scaffold for novel binding proteins. J Mol Biol 284, 1142–1151. [DOI] [PubMed] [Google Scholar]
- 60. Koide S, Koide A & Lipovsek D (2012) Target‐binding proteins based on the 10th human fibronectin type III domain. Methods Enzymol 503, 135–156. [DOI] [PubMed] [Google Scholar]
- 61. Koide A, Wojcik J, Gilbreth RN, Hoey RJ & Koide S (2012) Teaching an old scaffold new tricks: monobodies constructed using alternative surfaces of the FN3 scaffold. J Mol Biol 415, 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hackel BJ, Kapila A & Wittrup KD (2008) Picomolar affinity fibronectin domains engineered utilizing loop length diversity, recursive mutagenesis, and loop shuffling. J Mol Biol 381, 1238–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schiff D, Kesari S, de Groot J, Mikkelsen T, Drappatz J, Coyle T, Fichtel L, Silver B, Walters I & Reardon D (2015) Phase 2 study of CT‐322, a targeted biologic inhibitor of VEGFR‐2 based on a domain of human fibronectin, in recurrent glioblastoma. Invest New Drugs 33, 247–253. [DOI] [PubMed] [Google Scholar]
- 64. Wojcik J, Lamontanara AJ, Grabe G, Koide A, Akin L, Gerig B, Hantschel O & Koide S (2016) Allosteric inhibition of Bcr‐Abl kinase by high affinity monobody inhibitors directed to the Src Homology 2 (SH2)‐kinase interface. J Biol Chem 291, 8836–8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schmit NE, Neopane K & Hantschel O (2019) Targeted protein degradation through cytosolic delivery of monobody binders using bacterial toxins. ACS Chem Biol 14, 916–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schiefner A & Skerra A (2015) The Menagerie of human lipocalins: a natural protein scaffold for molecular recognition of physiological compounds. Account Chem Res 48, 976–985. [DOI] [PubMed] [Google Scholar]
- 67. Lakshmi B, Mishra M, Srinivasan N & Archunan G (2015) Structure‐based phylogenetic analysis of the lipocalin superfamily. PLoS One 10, e0135507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Breustedt DA, Schonfeld DL & Skerra A (2006) Comparative ligand‐binding analysis of ten human lipocalins. Biochim Biophys Acta 1764, 161–173. [DOI] [PubMed] [Google Scholar]
- 69. Gebauer M & Skerra A (2019) Engineering of binding functions into proteins. Curr Opin Biotechnol 60, 230–241. [DOI] [PubMed] [Google Scholar]
- 70. Richter A, Eggenstein E & Skerra A (2014) Anticalins: exploiting a non‐Ig scaffold with hypervariable loops for the engineering of binding proteins. FEBS Lett 588, 213–218. [DOI] [PubMed] [Google Scholar]
- 71. Rothe C & Skerra A (2018) Anticalin proteins as therapeutic agents in human diseases. BioDrugs 32, 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hammill JA, VanSeggelen H, Helsen CW, Denisova GF, Evelegh C, Tantalo DG, Bassett JD & Bramson JL (2015) Designed ankyrin repeat proteins are effective targeting elements for chimeric antigen receptors. J Immunother Cancer 3, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Siegler E, Li S, Kim YJ & Wang P (2017) Designed Ankyrin repeat proteins as Her2 targeting domains in chimeric antigen receptor‐engineered T cells. Hum Gene Ther 28, 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. De Munter S, Ingels J, Goetgeluk G, Bonte S, Pille M, Weening K, Kerre T, Abken H & Vandekerckhove B (2018) Nanobody based dual specific CARs. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xie YJ, Dougan M, Jailkhani N, Ingram J, Fang T, Kummer L, Momin N, Pishesha N, Rickelt S, Hynes RO & Ploegh H (2019) Nanobody‐based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc Natl Acad Sci U S A 116, 7624–7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Emanuel SL, Engle LJ, Chao G, Zhu RR, Cao C, Lin Z, Yamniuk AP, Hosbach J, Brown J, Fitzpatrick E et al. (2011) A fibronectin scaffold approach to bispecific inhibitors of epidermal growth factor receptor and insulin‐like growth factor‐I receptor. MAbs 3, 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Han X, Cinay GE, Zhao Y, Guo Y, Zhang X & Wang P (2017) Adnectin‐based design of chimeric antigen receptor for T cell engineering. Mol Ther 25, 2466–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lamers CH, Willemsen R, van Elzakker P, van Steenbergen‐Langeveld S, Broertjes M, Oosterwijk‐Wakka J, Oosterwijk E, Sleijfer S, Debets R & Gratama JW (2011) Immune responses to transgene and retroviral vector in patients treated with ex vivo‐engineered T cells. Blood 117, 72–82. [DOI] [PubMed] [Google Scholar]
- 79. Maude SL, Barrett DM, Rheingold SR, Aplenc R, Teachey DT, Callahan C, Baniewicz D, White C, Talekar MK, Shaw PA et al. (2016) Efficacy of humanized CD19‐targeted Chimeric Antigen Receptor (CAR)‐modified T cells in children and young adults with relapsed/refractory acute lymphoblastic leukemia. Blood 128, 217–217.27207794 [Google Scholar]
- 80. Haso W, Lee DW, Shah NN, Stetler‐Stevenson M, Yuan CM, Pastan IH, Dimitrov DS, Morgan RA, FitzGerald DJ, Barrett DM et al. (2013) Anti‐CD22‐chimeric antigen receptors targeting B‐cell precursor acute lymphoblastic leukemia. Blood 121, 1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Meek K (1990) Analysis of junctional diversity during B lymphocyte development. Science 250, 820. [DOI] [PubMed] [Google Scholar]
- 82. Di Noia JM & Neuberger MS (2007) Molecular mechanisms of antibody somatic hypermutation. Annual Rev Biochem 76, 1–22. [DOI] [PubMed] [Google Scholar]
- 83. Baker M, Reynolds HM, Lumicisi B & Bryson CJ (2010) Immunogenicity of protein therapeutics: the key causes, consequences and challenges. Self/Nonself 1, 314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Brinks V, Weinbuch D, Baker M, Dean Y, Stas P, Kostense S, Rup B & Jiskoot W (2013) Preclinical models used for immunogenicity prediction of therapeutic proteins. Pharm Res 30, 1719–1728. [DOI] [PubMed] [Google Scholar]
- 85. King C, Garza EN, Mazor R, Linehan JL, Pastan I, Pepper M & Baker D (2014) Removing T‐cell epitopes with computational protein design. Proc Natl Acad Sci U S A 111, 8577–8582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mazor R, Eberle JA, Hu X, Vassall AN, Onda M, Beers R, Lee EC, Kreitman RJ, Lee B, Baker D et al. (2014) Recombinant immunotoxin for cancer treatment with low immunogenicity by identification and silencing of human T‐cell epitopes. Proc Natl Acad Sci U S A 111, 8571–8576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, Zhao Y, Kalos M & June CH (2013) T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res 1, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Jones TD, Carter PJ, Pluckthun A, Vasquez M, Holgate RG, Hotzel I, Popplewell AG, Parren PW, Enzelberger M, Rademaker HJ et al. (2016) The INNs and outs of antibody nonproprietary names. MAbs 8, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wang F, Sen S, Zhang Y, Ahmad I, Zhu X, Wilson IA, Smider VV, Magliery TJ & Schultz PG (2013) Somatic hypermutation maintains antibody thermodynamic stability during affinity maturation. Proc Nat Acad Sci 110, 4261. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 90. Carter PJ & Lazar GA (2018) Next generation antibody drugs: pursuit of the 'high‐hanging fruit'. Nat Rev Drug Discov 17, 197–223. [DOI] [PubMed] [Google Scholar]
- 91. Ralph DK & Matsen FAIV (2019) Per‐sample immunoglobulin germline inference from B cell receptor deep sequencing data. PLOS Computat Biol 15, e1007133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bocchia M, Korontsvit T, Xu Q, Mackinnon S, Yang SY, Sette A & Scheinberg DA (1996) Specific human cellular immunity to bcr‐abl oncogene‐derived peptides. Blood 87, 3587–3592. [PubMed] [Google Scholar]
- 93. ten Bosch GJ, Toornvliet AC, Friede T, Melief CJ & Leeksma OC (1995) Recognition of peptides corresponding to the joining region of p210BCR‐ABL protein by human T cells. Leukemia 9, 1344–1348. [PubMed] [Google Scholar]
- 94. Raulet DH (2003) Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol 3, 781–790. [DOI] [PubMed] [Google Scholar]
- 95. Spear P, Wu M‐R, Sentman M‐L & Sentman CL (2013) NKG2D ligands as therapeutic targets. Cancer Immun 13. [PMC free article] [PubMed] [Google Scholar]
- 96. Sentman CL & Meehan KR (2014) NKG2D CARs as cell therapy for cancer. Cancer J 20, 156–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zhang T, Lemoi BA & Sentman CL (2005) Chimeric NK‐receptor‐bearing T cells mediate antitumor immunotherapy. Blood 106, 1544–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zhang T, Barber A & Sentman CL (2006) Generation of antitumor responses by genetic modification of primary human T cells with a chimeric NKG2D receptor. Cancer Res 66, 5927–5933. [DOI] [PubMed] [Google Scholar]
- 99. Lehner M, Gotz G, Proff J, Schaft N, Dorrie J, Full F, Ensser A, Muller YA, Cerwenka A, Abken H et al. (2012) Redirecting T cells to Ewing's sarcoma family of tumors by a chimeric NKG2D receptor expressed by lentiviral transduction or mRNA transfection. PLoS One 7, e31210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Song D‐G, Ye Q, Santoro S, Fang C, Best A & Powell DJ Jr (2013) Chimeric NKG2D CAR‐expressing T cell‐mediated attack of human ovarian cancer is enhanced by histone deacetylase inhibition. Hum Gene Therapy 24, 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, Jin Q, Su L, Liu X, Wang K et al. (2019) Adoptive transfer of NKG2D CAR mRNA‐engineered natural killer cells in colorectal cancer patients. Mol Ther 27, 1114–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Baumeister SH, Murad J, Werner L, Daley H, Trebeden‐Negre H, Gicobi JK, Schmucker A, Reder J, Sentman CL, Gilham DE et al. (2019) Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol Res 7, 100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sallman DA, Brayer J, Sagatys EM, Lonez C, Breman E, Agaigué S, Verma B, Gilham DE, Lehmann FF & Davila ML (2018) NKG2D CAR therapy induced remission in a AML patient. Haematologica 103, 424–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Figenschau SL, Knutsen E, Urbarova I, Fenton C, Elston B, Perander M, Mortensen ES & Fenton KA (2018) ICAM1 expression is induced by proinflammatory cytokines and associated with TLS formation in aggressive breast cancer subtypes. Scientific Rep 8, 11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Park S, Shevlin E, Vedvyas Y, Zaman M, Park S, Hsu YS, Min IM & Jin MM (2017) Micromolar affinity CAR T cells to ICAM‐1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep 7, 14366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J et al. (2016) Regression of glioblastoma after chimeric antigen receptor T‐cell therapy. N Engl J Med 375, 2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Seeger MA, Zbinden R, Flütsch A, Gutte PGM, Engeler S, Roschitzki‐Voser H & Grütter MG (2013) Design, construction, and characterization of a second‐generation DARP in library with reduced hydrophobicity. Protein Sci 22, 1239–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Eigenbrot C, Ultsch M, Dubnovitsky A, Abrahmsén L & Härd T (2010) Structural basis for high‐affinity HER2 receptor binding by an engineered protein. Proc Nat Acad Sci U S A 107, 15039–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Li T, Qi S, Unger M, Hou YN, Deng QW, Liu J, Lam CMC, Wang XW, Xin D, Zhang P et al. (2016) Immuno‐targeting the multifunctional CD38 using nanobody. Sci Rep 6, 27055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Baumann H, Knapp S, Lundbäck T, Ladenstein R & Härd T (1994) Solution structure and DNA‐binding properties of a thermostable protein from the archaeon Sulfolobus solfataricus. Nat Struct Biol 1, 808–819. [DOI] [PubMed] [Google Scholar]
- 111. Ramamurthy V, Krystek Stanley R, Bush A, Wei A, Emanuel Stuart L, Das Gupta R, Janjua A, Cheng L, Murdock M, Abramczyk B et al. (2012) Structures of Adnectin/protein complexes reveal an expanded binding footprint. Structure 20, 259–269. [DOI] [PubMed] [Google Scholar]
- 112. Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN & Strong RK (2002) The Neutrophil Lipocalin NGAL is a bacteriostatic agent that interferes with siderophore‐mediated iron acquisition. Mol Cell 10, 1033–1043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Representative VH and VL alignments. VH and VL sequences of human and humanized antibodies were obtained from the DrugBank (https://www.drugbank.ca/) and aligned against human antibody germline sequences using the DomainGapAlign tool from IMGT (www.imgt.org).
Table S1. Amino acid sequences of VH and VL domains derived from human and humanized antibodies and engineered binding scaffolds used for the analysis of the number of non‐human amino acid positions in Figure 5.