

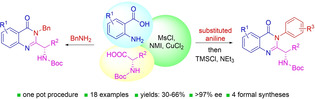
Abstract
Chiral 2‐alkylquinazolinones are key synthetic intermediates, but their preparation in high optical purity is challenging. Thus, a multicomponent procedure integrating anthranilic acids, N‐Boc‐amino acids, and amines in the presence of methanesulfonyl chloride, N‐methylimidazole, and copper(II) chloride was developed to mildly afford N‐Boc‐2‐alkylaminoquinazolin‐4(3H)‐ones with excellent preservation of enantiomeric purity (>99% ee). Copper(II) chloride was essential to retaining enantiopurity, and reaction component structural changes were well tolerated, resulting in an efficient, all‐in‐one procedure that promotes sequential coupling, lactonization, aminolysis, and cyclization in good yields. The method was applied to the rapid assembly of four key intermediates used in the synthesis of high profile quinazolinones, including several PI3K inhibitor drugs.
Keywords: quinazolinone, racemization, copper-mediated, enantiopurity, PI3 kinase
The quinazolinone framework is a synthetically useful intermediate found in a vast number of natural products with a broad spectrum of biological activity. [1] Medicinal chemistry programs centered on this motif have produced several clinical candidates and approved drugs featuring a 2‐alkylamino‐quinazolin‐4(3H)‐one core (Figure 1, red highlight). For example, idelalisib 1 was FDA approved [2] in 2014 as a first‐in‐class selective inhibitor of PI3Kδ for the treatment of some blood cancers. [3] Several quinazolinone‐based PI3K inhibitors[ 3c , 4 ] appeared afterward, such as the antiarthritic triamino‐pyrimidine 2 [5] and the immunomodulating anti‐neoplastic agent, acalisib 3. [6] Ispinesib 4 is a kinesin spindle protein (KSP) inhibitor that is in phase II clinical trials for the treatment of neoplastic diseases. [7] Like ispinesib, quinazolinone 5 features an N‐acylated 2‐alkylaminoquinazolinone, but is valued for its anti‐inflammatory action due to CXCR3 receptor antagonism. [8]
Figure 1.

Selected 2‐alkylaminoquinazolin‐4(3H)‐ones.
Synthetic approaches to the 2‐alkylaminoquinazolin‐4(3H)‐one core frequently converge to N‐Boc protected intermediates 7 and rely on a dehydrative procedure incorporating an anthranilic acid, N‐Boc‐protected amino acid 6 and an aniline in the presence of HOP(OPh)2 or P(OPh)3 in pyridine (Scheme 1).[ 4b , 4c , 4d , 5 , 9 ] Alternatively, isobutyl chloroformate and N‐methylmorpholine (NMM) have been used in place of the phosphite reagents to construct the quinazolinone core,[ 8 , 10 ] while other methods generate anthranilamides via peptide coupling, followed by cyclization using HMDS/I2, [11] TMSCl, [12] or bis(trimethylsilyl)acet‐amide. [13] Preparing anthranil‐amides from 2‐nitrobenzoic acids has also been explored[ 13 , 14 ] as a means to afford Boc‐protected 2‐alkylaminoquinazolin‐4(3H)‐ones 7 with high enantiopurity via Mumm rearrangement, followed by reduction and cyclization. [15] Nonetheless, many of these methods demonstrated one or more limitations such as low overall yield, multistage purification, narrow substrate scope, need for synthetically challenging starting materials, use of sensitizing coupling reagents, [16] or erosion of enantiopurity.
Scheme 1.

General synthetic approaches to N‐Boc‐quinazolinones.
For the purpose of using N‐Boc protected 2‐alkylaminoquinazolin‐4(3H)‐ones as substrates in our quinazolinone rearrangement [17] chemistry leading to benzamidines, we required a method that would avoid these issues and efficiently deliver a variety of these quinazolinones with high enantiopurity. We observed products of variable optical purity with the phosphite protocol that was dependent on the substitution pattern of the anthranilic acid. With the isobutylchloro‐formate/NMM procedure, we obtained low yields of the desired quinazolinones and significant quantities of side products. Alternatively, we found that the coupling of anthranilic acids with anilines mediated by MsCl and N‐methylimidazole (NMI) was promising (Scheme 2). [18] To survey these conditions, we used anthranilic acid 8 a, N‐Boc‐L‐alanine and aniline as substrates and modulated the reagent stoichiometry, solvent, and temperature.
Scheme 2.

Exploratory chemistry leading to 12 a formation.
The reactions were initially monitored by NMR and later assessed by TLC and LCMS when purified intermediates were in hand. Analysis of the data revealed that a single equivalent of MsCl and NMI relative to anthranilic acid resulted in a mixture of acid 8 a, amide 9 a and benzoxazinone 10 a. Additional MsCl/NMI (1.2 eq. each) facilitated 10 a formation such that, after the addition of aniline, a mixture of diamide 11 a and 12 a was observed. Heating of 11 a/12 a with TMSCl [12] and NEt3 in dichloromethane resulted in a 47% yield of quinazolinone 12 a and 98% ee (Table 1, entry 1).
Table 1.
Optimization of quinazolinone 12 a formation.[a]
|
| ||||||
|---|---|---|---|---|---|---|
|
entry |
MsCl (equiv.) |
NMI (equiv.) |
CuCl2 (equiv.) |
solvent |
12 a yield (%) |
ee[e] (%) |
|
1 |
1.0[b]+1.2[c] |
1.0[b]+1.2[c] |
– |
DCM |
47 |
98 |
|
2 |
1.0[b]+1.2[c] |
1.0[b]+1.5[c] |
– |
DCM |
57 |
98 |
|
3 |
1.0[b]+1.2[c] |
1.0[b]+2.0[c] |
– |
DCM |
62 |
98 |
|
4 |
2.2[d] |
3.0[d] |
– |
DCM |
66 |
96 |
|
5 |
1.0[b]+1.2[c] |
2.0[b]+1.0[c] |
– |
DCM |
57 |
97 |
|
6 |
1.0[b]+1.2[c] |
1.0[b]+2.0[c] |
– |
DCE |
55 |
93 |
|
7 |
1.0[b]+1.2[c] |
1.0[b]+2.0[c] |
– |
THF |
22 |
91 |
|
8 |
1.0[b]+1.2[c] |
1.0[b]+2.0[c] |
– |
CH3CN |
36 |
93 |
|
9 |
1.0[b]+1.2[c] |
1.0[b]+2.0[c] |
0.25 |
DCM |
63 |
>99 |
|
10 |
1.0[b]+1.2[c] |
1.0[b]+2.0[c] |
0.25 |
DCE |
52 |
98 |
|
11 |
1.0[b]+1.2[c] |
1.0[b]+2.0[c] |
0.50 |
DCE |
45 |
>99 |
|
12 |
1.0[b]+1.2[c] |
1.0[b]+3.0[c] |
0.50 |
DCE |
50 |
>99 |

[a] 1.0 mmol scale in solvent (0.07 M). Reflux refers to the boiling point of the solvent indicated. Yields reflect isolated product.
[b] Equivalency of reagent added in step 1.
[c] Equivalency of reagent added in step 2.
[d] All MsCl and NMI were added in step 1.
[e] Percent ee determined by chiral HPLC comparison of peak AUCs for each enantiomer obtained by this method. MsCl=methanesulfonyl chloride; NMI=N‐methylimidazole; TMSCl=trimethylsilyl chloride; NEt3=triethylamine; DCM=dichloromethane; DCE=1,2‐dichloroethane; THF=tetrahydrofuran; CH3CN=acetonitrile.
With this result in hand, a systematic study of reaction parameters was undertaken to optimize the process. Firstly, we found it was advantageous to run the reaction without isolation of any intermediates, hence we optimized the process as a one‐pot procedure. The addition of molecular sieves in step 1 was necessary to avoid hydrolysis of benzoxazinone 10 a (see supplementary Table S1). Increasing the equivalency of NMI in step 2 improved the yield of 12 a without eroding the enantiomeric purity (Table 1, entries 2–3); however, adding additional quantities of NMI beyond 1.0 equivalent in step 1 reduced the ee value of the product (entries 4–5, Table 1). Exchange of solvent from dichloromethane to dichloroethane, THF, or acetonitrile with the other reaction parameters of entry 3 were inferior with respect to both yield and optical purity (entries 6–8). Changes to the temperature in steps 1 or 2, or altering the equivalency of aniline (step 3) or TMSCl/NEt3 (step 4) did not improve the enantiomeric purity of the product (Table S1, entries 8–11). As some racemization was still occurring, we took note of reports showing that this issue in mixed anhydride type peptide couplings could be mitigated with the addition of CuCl2. [19] The addition of CuCl2 (0.25 equiv.) in DCM afforded quinazolinone 12 a in 63% yield and >99% ee (Table 1, entry 9). Switch of solvent to DCE resulted in a slight loss in enantiomeric purity and yield, though increasing the CuCl2 equivalency and increasing the amount of NMI in step 2 restored the enantiopurity to >99% (entry 12). This became important later for some substrates in our scope that required heating at a higher temperature than what could be accomplished in DCM. Notably, we revisited the isobutyl chloroformate/NMM conditions after seeing the copper(II) chloride effect, but only trace amounts of quinazolinone 12 a were observed when these reagents were switched out for the MsCl/NMI system in our new protocol.
Moving forward with our one‐pot protocol, we surveyed a collection of substituted anthranilic acids, anilines or benzylamine, and α‐substituted, N‐Boc protected amino acids and their associated effects on yield and enantiomeric purity (Table 2). Substitution of the anthranilic acid core revealed a 15% better yield for a C5‐trifluoromethyl analog 12 d compared to a C5‐methoxy derivative 12 c, presumably due to increased susceptibility of the former case to attack by a weakly nucleophilic aniline or stabilization of negative charge in the intermediates. Nonetheless, anthranilic acids 8 a–e were effective substrates, delivering quinazolinones 12 a–e in reasonable yields and excellent enantiomeric purity in DCM. When a C6‐fluorine atom was present on the anthranilic acid, the corresponding diamide was not completely converted to quinazolinone 12 f in step 4; however, switching to the alternative DCE conditions (Table 1, entry 12) permitted a higher reaction temperature and afforded 12 f in 30% yield and 98% ee. The relative low yield of 12 f may be due its instability, as C6‐halogenated quinazolinones have been reported to be problematic. [4a] Nonetheless, the use of DCE/CuCl2 was beneficial for a few other substrates, especially those that employed deactivated anilines or bulky N‐Boc protected amino acids, such as those leading to 12 j, 12 l and 12 m. For example, quinazolinone 12 l was obtained in 41% yield and 95% ee using DCM and 0.25 equiv. of CuCl2 but in DCE and with 0.50 equiv. of CuCl2, 12 l was obtained in the same yield but with an improved 99% ee.
Table 2.
Method substrate scope, yields and enantiomeric purity of N‐Boc‐2‐alkylaminoquinazolinones.[a]
|
|

[a] Reactions run on 1.0 mmol scale/0.07 M. Performed in DCM and with CuCl2 (0.25 equiv.) unless otherwise noted; reflux refers to DCM boiling point unless DCE was used. Yields reflect isolated product. Percent ee determined by chiral HPLC comparison of peak AUCs for each enantiomer obtained by this method.
[b] Used DCE/CuCl2 (0.50 equiv.); reflux refers to DCE boiling point.
[c] Used BnNH2 (4.0 equiv.) and step 4 was unnecessary.
Good yields and excellent ee values were observed for a variety of anilines and benzyl amine when paired with N‐Boc‐ alanine (12 g–12 k). Notably, for 12 k in which benzylamine was used in place of an aniline, fewer equivalents of the amine were required (4.0 equiv.) and the cyclization step employing TMSCl/NEt3 was unnecessary, resulting in direct formation of 12 k in 61% yield and >99% ee. This may be due to the greater nucleophilicity of benzylamine compared to the use of anilines. The procedure also tolerated the incorporation of more bulky amino acid side chains such as those in valine, methionine and cyclopropylglycine, as shown by the preparation of quinazolinones 12 m–o.
To assess if the protocol was beneficial in the synthesis of previously reported N‐Boc‐2‐alkylamino‐quinazolinones, we approached the formal syntheses of four quinazolinone drugs or candidates for which the requisite N‐Boc‐quinazolinones were known. Quinazolinone 12 p, a key idelalisib 1 synthetic intermediate,[ 3a , 14 , 15 ] had been reportedly prepared via a phosphite‐mediated strategy, though the yield and enantiopurity was not reported (Table 3, entry 1).
Table 3.
Comparison of yield and %ee using various methods to generate N‐Boc‐quinazolinone 12 p.
|
| |||||
|---|---|---|---|---|---|
|
entry |
8 f, X |
method |
ref |
12 p yield (%)[a] |
12 p ee (%)[b] |
|
1 |
NH2 |
P(OPh)3/pyridine aniline, 70 °C, 8 h |
9c |
not reported |
|
|
2 |
NH2 |
P(OPh)3/pyridine aniline, 70 °C, 8 h[c] |
9c[d] |
30 |
94 |
|
3 |
NO2 |
SOCl2 coupling, Mumm rearrangement, reduction/cyclization |
15 |
36 |
97–98[e] |
|
4 |
NH2 |
See Table 2, one pot, DCE, 1 mmol scale [c] |
this work |
33 |
>99 |
|
5 |
NH2 |
See Table 2, one pot, DCE, 10 mmol scale |
this work |
43 |
>99 |

[a] Isolated yields.
[b] Percent ee determined by chiral HPLC comparison of peak AUCs for each enantiomer.
[c] Reactions run on 1.0 mmol scale.
[d] Procedure from ref. 9c for 12 p was reproduced in our lab and resulting yields and %ee of 12 p are reported (see SI).
[e] Ref. 15 reported a 98% ee in the main manuscript but 97% ee in the SI for 12 p.
Using their published [9c] protocol, we generated 12 p in 30% yield and 94% ee (entry 2). The generation of quinazolinone 12 p by Mumm rearrangement of an imidoyl chloride, followed by reduction and cyclization was reported to occur in 36% yield and 97–98% ee. [15] Using anthranilic acid 8 f (X=NH2), our one‐pot, copper(II) chloride‐mediated strategy afforded 12 p in 33% yield and 99% ee on a 1 mmol scale, and in 43% yield on a 10 mmol scale without loss of enantiomeric purity (>99% ee, entry 5).
The high stereofidelity of the method was further demonstrated in the formal synthesis of acalisib 3 (Scheme 3). Using our protocol in DCM, anthranilic acid 8 g was converted into quinazolinone 12 q (56% yield, >99% ee) which was deprotected with TFA to afford amine 13 a in 88% yield and >99% ee. The transformation of 13 a to acalisib 3 has been reported, [6a] though the yields and enantiopurity of 13 a prepared by that method were not provided. Nonetheless, the final enantiopurity of acalisib 3 was reported as >99% ee after proceeding through a four‐step protocol. We converge to the same intermediate 13 a using the one‐pot procedure with a reasonable 49% yield and excellent enantiopurity.
Scheme 3.

Formal syntheses of acalisib 3, ispinesib 4 and quinazolinone 5 using the one‐pot procedure on a 1.0 mmol scale (0.07 M). Yields reflect isolated product and %ee was determined by chiral HPLC comparison of peak AUCs.
For both the formal synthesis of ispinesib 4 and that of CXCR3 antagonist 5, isobutyl chloroformate and N‐methylmorpholine were combined with the necessary anthranilic acid and aniline to generate N‐Boc quinazolinones 12 r and 12 s, respectively (Scheme 3). Using our protocol in DCE, the reaction of anthranilic acid 8 e, N‐Boc‐D‐valine, and benzylamine proceeded without the need of step 4 and expeditiously afforded 12 r, the key intermediate in the synthesis of ispinesib 4, with 98% ee and in a 41% yield.[ 7 , 10 ] Quinazolinone 12 s, which was used to prepare [8] CXCR3 antagonist 5 and was generated using isobutyl chloroformate/NMM in 30% yield over 4 steps (no reported %ee), was instead assembled using our one‐pot protocol in DCM in 66% yield and >99% ee. Ultimately, these four key quinazolinone intermediates (12 p, 12 r, 12 s, and 13 a) were successfully generated using the one‐pot protocol and resulted in excellent enantiomeric purity and yields that are reasonable when considering the number of steps otherwise required to make them. These formal syntheses, taken together with the quinazolinone collection shown in Table 2, underscore the utility of this method in preserving the enantiopurity of the desired quinazolinone products.
Last, several control experiments were carried out to determine the point at which racemization occurred (Scheme 4). The conversion of anthranilic acid 8 a to benzoxazinone 10 a in DCE without CuCl2 was arrested after step 2 (Scheme 4, panel 1). Isolation and characterization revealed that 10 a was obtained in an 86% yield, >99% ee, and showed no racemization occurring in the first two steps.
Scheme 4.
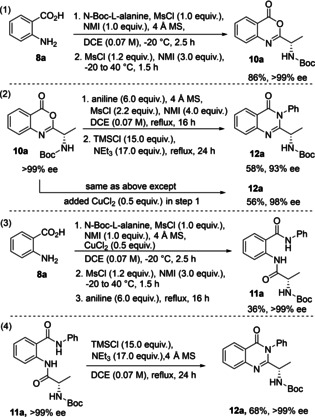
Control experiments revealing need for CuCl2.
Treatment of isolated benzoxazinone 10 a (>99% ee) with aniline in DCE with and without copper (II) chloride showed erosion of enantiomeric purity in the absence of the additive (Scheme 4, panel 2). Formation of diamide 11 a from acid 8 a did not reveal racemization, and isolation of the ring closing reaction with diamide 11 a and TMSCl/NEt3 showed no loss of enantiopurity in the absence of copper(II) chloride to form 12 a (panel 4). Collectively, these results point to the sensitivity of benzoxazinone 10 a and the diamide forming reaction (step 3, Scheme 2) as the source of loss of enantiomeric purity and the point at which CuCl2 is needed to prevent it.
Given that metal‐coordination with the equivalent nitrogen atom in quinazolinones has been characterized, [20] we reasoned that the CuCl2 may coordinate the benzoxazinone core nitrogen atom in 10 a, as shown in Scheme 5 (intermediate A). Reaction between mesyl chloride and NMI forms HCl which can further activate the benzoxazinone carbonyl toward nucleophilic addition of the aniline, followed by elimination to form ring‐opened intermediate C. Loss of the amide‐nitrogen coordinated copper species can ultimately afford vis‐amide 11 a. Alternatively, when copper(II) chloride was not present, we proposed that the stereocenter may be compromised by an alternative pathway en route to the formation of the bis‐amide intermediate, in accordance with the results shown in Scheme 4. We took note of a report [4a] from Patel and co‐workers who, while generating quinazolinone PI3K inhibitors, proposed a mechanism by which the quinazolinones formed a nitrilium ion that ultimately led to ring opening and bis‐amide formation. In the case of the benzoxazinone 10 a, we propose that a structurally similar nitrilium ion intermediate may form by virtue of the uncoordinated imine‐like nitrogen atom that fragments the activated ring to generate nitrilium ion E in the absence of copper(II) chloride. In the key step leading to racemization, loss of the acidic propargylic‐like proton may generate a benzenamine F that could be intercepted by an intramolecular 6‐endo‐dig cyclization involving the nearby carboxylic acid group to afford benzoxazinylidene G. Attack of the aniline on the carbonyl of G would be expected to afford an intermediate H that, upon ring opening and subsequent tautomerization, would generate racemized bis‐amide 11 a’. This rationale accounts for the observed racemization leading to 11 a’ in the absence of CuCl2 and the stabilization of the stereocenter when the reagent is used. Further, this mechanistic proposal underscores the importance of using the non‐basic reagent, N‐methylimidazole, as part of the amide coupling. As noted earlier, adding CuCl2 to the previously reported isobutyl chloroformate/NMM conditions failed to improve reaction yield or preserve enantiopurity. In light of the proposed mechanism, this made sense as the N‐methyl morpholine reagent would be expected to preferentially coordinate the copper(II) chloride over the substrate 10 a.
Scheme 5.

Proposed role of CuCl2 in attenuating racemization.
In summary, we have devised an efficient means of assembling N‐Boc‐protected 2‐alkylaminoquinazolin‐4(3H)‐ones which are valuable intermediates in synthetic processes leading to bioactive natural product derivatives, promising lead compounds and marketed drugs. Faced with a need to generate an array of these intermediates reliably and with high enantiomeric purity, we developed a MsCl/NMI/CuCl2 mediated protocol that incorporates commercially available, substituted anthranilic acids, N‐Boc‐amino acids, and amines. Notably, the peptide coupling, lactonization, aminolysis, and cyclization occurs in one‐pot without the need for isolation of intermediates and affords products in modest to good yields and excellent enantiopurities. Moreover, when benzylamine was employed in place of the aniline component, diamide cyclization occurred spontaneously, thus obviating the need for the TMSCl/NEt3 step. The utility and enantiopurity‐preserving feature of the transformation, including the Boc‐deprotection step leading to 13 a that retained the enantiopurity with high fidelity, was further demonstrated by the rapid formal syntheses of quinazolinone‐based drugs, idelalisib 1, acalisib 3, and ispinesib 4 and bioactive quinazolinone 5.
Experimental Section
General Procedures A (DCM/0.25 eq. CuCl2) and B (DCE/0.50 eq. CuCl2) for the Synthesis of Quinazolinones 12
Procedure A (DCM/0.25 eq. CuCl2). To the mixture of Boc‐L or D amino acid (1.00 mmol), anhydrous CuCl2 (34 mg, 0.25 mmol) and 4 Å MS (140 mg) in anhydrous DCM (15 mL) under N2 and cooled to −20 °C was added dropwise N‐methylimidazole (NMI) (80 μL, 1.00 mmol) and MsCl (77 μL, 1.00 mmol). After being stirred at −20 °C for 1 h, anthranilic acid or substituted anthranilic acid 8 (1.00 mmol) was added into the reaction mixture and stirred at −20 °C for 1.5 h. Then, MsCl (93 μL, 1.20 mmol) and NMI (160 μL, 2.00 mmol) was added dropwise into the reaction mixture at −20 °C and slowly heated to 40 °C in parallel synthesizer heating mantle for 1.5 h. After being cooled to rt, aniline or substituted aniline (6.00 mmol) was added dropwise into the reaction mixture and heated to reflux in a parallel synthesizer heating mantle for 16 h. Subsequently, triethylamine (2.37 mL, 17.00 mmol) and TMSCl (1.91 mL, 15.00 mmol) was added dropwise into the reaction mixture at 0 °C and slowly heated to reflux in Parallel synthesizer heating mantle for 24 h. The reaction mixture was diluted with DCM (100 mL), washed with 1 M HCl, saturated aqueous Na2CO3, and brine in turn, dried over Na2SO4, and filtered, then the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography to afford 12.
Procedure B (DCE/0.50 eq. CuCl2). To the mixture of Boc‐L or D amino acid (1.00 mmol), anhydrous CuCl2 (68 mg, 0.50 mmol) and 4 Å MS (140 mg) in anhydrous DCE (15 mL) under N2 cooled to −20 °C was added dropwise NMI (80 μL, 1.00 mmol) and MsCl (77 μL, 1.00 mmol). After being stirred at −20 °C for 1 h, anthranilic acid or substituted anthranilic acid 8 (1.00 mmol) (137 mg, 1.00 mmol) was added into the reaction mixture and stirred at −20 °C for 1.5 h. Then, MsCl (93 μL, 1.20 mmol) and NMI (240 μL, 3.00 mmol) was added dropwise into the reaction mixture at −20 °C and slowly heated to 40 °C in Parallel synthesizer heating mantle for 1.5 h. After being cooled to rt, aniline or substituted aniline (6.00 mmol) was added dropwise into the reaction mixture and heated to reflux in Parallel synthesizer heating mantle for 16 h. Subsequently, triethylamine (2.37 mL, 17.00 mmol) and TMSCl (1.91 mL, 15.00 mmol) was added dropwise into the reaction mixture at 0 °C and slowly heated to reflux in Parallel synthesizer heating mantle for 24 h. The reaction mixture was diluted with DCM (100 mL), washed with 1 M HCl, saturated aqueous Na2CO3, and brine in turn, dried over Na2SO4, and filtered, then the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography to afford 12.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
J.E.G. gratefully acknowledges funding from the National Institutes of Health (R01AI118814 and U19AI142762). We thank the UW‐Madison Medicinal Chemistry Center and the Analytical Instrumentation Center in the UW‐Madison School of Pharmacy for use of instrumentation related to chiral HPLC, NMR, HRMS, and polarimetry.
X. Li, J. E. Golden, Adv. Synth. Catal. 2021, 363, 1638.
References
- 1.
- 1a. Resende D. I. S. P., Boonpothong P., Sousa E., Kijjoa A., Pinto M. M. M., Nat. Prod. Rep. 2019, 36, 7–34; [DOI] [PubMed] [Google Scholar]
- 1b. Khan I., Ibrar A., Ahmed W., Saeed A., Eur. J. Med. Chem. 2015, 90, 124–169; [DOI] [PubMed] [Google Scholar]
- 1c. Kshirsagar U. A., Org. Biomol. Chem. 2015, 13, 9336–9352; [DOI] [PubMed] [Google Scholar]
- 1d. Demeunynck M., Baussanne I., Curr. Med. Chem. 2013, 20, 794–814; [PubMed] [Google Scholar]
- 1e. Michael J. P., Nat. Prod. Rep. 2003, 20, 476–493. [DOI] [PubMed] [Google Scholar]
- 2. Markham A., Drugs 2014, 74, 1701–1707. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a.K. W. Fowler, D. Huang, E. A. Kesicki, H. C. Ooi, A. R. Oliver, F. Ruan, J. Treiberg, (Icos Corporation), PCT Int. Appl. WO 2005/113556 A1, 2005;
- 3b. Garces A. E., Stocks M. J., J. Med. Chem. 2019, 62, 4815–4850; [DOI] [PubMed] [Google Scholar]
- 3c. Perry M. W. D., Abdulai R., Mogemark M., Petersen J., Thomas M. J., Valastro B., Westin Eriksson A., J. Med. Chem. 2019, 62, 4783–4814. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Patel L., Chandrasekhar J., Evarts J., Forseth K., Haran A. C., Ip C., Kashishian A., Kim M., Koditek D., Koppenol S., Lad L., Lepist E.-I., McGrath M. E., Perreault S., Puri K. D., Villaseñor A. G., Somoza J. R., Steiner B. H., Therrien J., Treiberg J., Phillips G., J. Med. Chem. 2016, 59, 9228–9242; [DOI] [PubMed] [Google Scholar]
- 4b. Perreault S., Chandrasekhar J., Cui Z.-H., Evarts J., Hao J., Kaplan J. A., Kashishian A., Keegan K. S., Kenney T., Koditek D., Lad L., Lepist E.-I., McGrath M. E., Patel L., Phillips B., Therrien J., Treiberg J., Yahiaoui A., Phillips G., J. Med. Chem. 2017, 60, 1555–1567; [DOI] [PubMed] [Google Scholar]
- 4c. Wei M., Zhang X., Wang X., Song Z., Ding J., Meng L.-H., Zhang A., Eur. J. Med. Chem. 2017, 125, 1156–1171; [DOI] [PubMed] [Google Scholar]
- 4d. Thakur A., Tawa G. J., Henderson M. J., Danchik C., Liu S., Shah P., Wang A. Q., Dunn G., Kabir M., Padilha E. C., Xu X., Simeonov A., Kharbanda S., Stone R., Grewal G., J. Med. Chem. 2020, 63, 4256–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Patel L., Chandrasekhar J., Evarts J., Haran A. C., Ip C., Kaplan J. A., Kim M., Koditek D., Lad L., Lepist E.-I., McGrath M. E., Novikov N., Perreault S., Puri K. D., Somoza J. R., Steiner B. H., Stevens K. L., Therrien J., Treiberg J., Villaseñor A. G., Yeung A., Phillips G., J. Med. Chem. 2016, 59, 3532–3548. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a.K. D. Puri, J. B. Evarts, B. Lannutti, N. A. Giese, (Calistoga Pharmaceuticals Inc.), PCT Int. Appl. WO 2010/123931 A1, 2010;
- 6b. Kater A. A. P., Tonino S. H., Spiering M., Chamuleau M. E. D., Liu R., Adewoye A. H., Gao J., Dreiling L., Xin Y., Doorduijn J. K., Kersten M. J., Blood Cancer J. 2018, 8, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sorbera L., Bolos J., Serradell N., Bayes M., Drugs Future 2006, 31, 778–787. [Google Scholar]
- 8. Johnson M., Li A.-R., Liu J., Fu Z., Zhu L., Miao S., Wang X., Xu Q., Huang A., Marcus A., Xu F., Ebsworth K., Sablan E., Danao J., Kumer J., Dairaghi D., Lawrence C., Sullivan T., Tonn G., Schall T., Collins T., Medina J., Bioorg. Med. Chem. Lett. 2007, 17, 3339–3343. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Yamazaki N., Higashi F., Tetrahedron Lett. 1972, 13, 5047–5050; [Google Scholar]
- 9b. Rabilloud G., Sillion B., J. Heterocycl. Chem. 1980, 17, 1065–1068; [Google Scholar]
- 9c.A. Boruah, S. Hosahalli, S. K. Panigrahi, (Aurigene Discovery Technologies Limited), PCT Int. Appl. WO 2014/106800 A2, 2014.
- 10.G. Bergnes, E. Ha, G. Yiannikouros, P. Kalaritis, B. E. Yonce, K. A. Welday, (Cytokinetics, Inc.), PCT Int. Appl. WO 2003/070701 A2, 2003.
- 11. Kshirsagar U. A., Mhaske S. B., Argade N. P., Tetrahedron Lett. 2007, 48, 3243–3246. [Google Scholar]
- 12.J. B. Evarts, B. Lannutti, H. Webb, (Gilead Calistoga Llc), PCT Int. Appl. WO 2013/082540 A1, 2013.
- 13.D. Xi, T. Wang, X. Feng, S. Wu, T. Zhang, L. Wang, (Sunshine Lake Pharma Co., Ltd., Calitor Sciences, Llc), PCT Int. Appl. WO 2016/14960 A1, 2016.
- 14. Mekala N., Buddepu S. R., Dehury S. K., Moturu K. M. V. R., Indukuri S. K. V., Vasireddi U. R., Parimi A. R., RSC Adv. 2018, 8, 15863–15869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhichkin P., Kesicki E., Treiberg J., Bourdon L., Ronsheim M., Ooi H. C., White S., Judkins A., Fairfax D., Org. Lett. 2007, 9, 1415–1418. [DOI] [PubMed] [Google Scholar]
- 16. McKnelly K. J., Sokol W., Nowick J. S., J. Org. Chem. 2020, 85, 1764–1768. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Jaffett V. A., Nerurkar A., Cao X., Guzei I. A., Golden J. E., Org. Biomol. Chem. 2019, 17, 3118–3128; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Schroeder C. E., Neuenswander S. A., Yao T., Aubé J., Golden J. E., Org. Biomol. Chem. 2016, 14, 3950–3955; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17c. Schroeder C. E., Yao T., Sotsky J., Smith R. A., Roy S., Chu Y.-K., Guo H., Tower N. A., Noah J. W., McKellip S., Sosa M., Rasmussen L., Smith L. H., White E. L., Aubé J., Jonsson C. B., Chung D., Golden J. E., J. Med. Chem. 2014, 57, 8608–8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Mao L., Wang Z., Li Y., Han X., Zhou W., Synlett 2011, 2011, 129–133; [Google Scholar]
- 18b. Li B., Zhang J., Xu Y., Yang X., Li L., Tetrahedron Lett. 2017, 58, 2374–2377; [Google Scholar]
- 18c. Gangarapu N. R., Reddy E. K., Sajith A. M., Yellappa S., Chandrasekhar K. B., ChemistrySelect 2017, 2, 7706–7710. [Google Scholar]
- 19.
- 19a. Miyazawa T., Donkai T., Yamada T., Kuwata S., Int. J. Pept. Protein Res. 1992, 40, 49–53; [DOI] [PubMed] [Google Scholar]
- 19b. Miyazawa T., Otomatsu T., Yamada T., Kuwata S., Tetrahedron Lett. 1984, 25, 771–772; [Google Scholar]
- 19c. Han S.-Y., Kim Y.-A., Tetrahedron 2004, 60, 2447–2467. [Google Scholar]
- 20. Aly M. M., Mohamed Y. A., El-Bayouki K. A. M., Basyouni W. M., Abbas S. Y., Eur. J. Med. Chem. 2010, 45, 3365–3373. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary