

Abstract
In non‐small cell lung cancer (NSCLC), activating mutations in the epidermal growth factor receptor (EGFR) induce sensitivity to EGFR tyrosine kinase inhibitors. Despite impressive clinical responses, patients ultimately relapse as a reservoir of drug‐tolerant cells persist, which ultimately leads to acquired resistance mechanisms. We performed an unbiased high‐throughput siRNA screen to identify proteins that abrogate the response of EGFR‐mutant NSCLC to EGFR‐targeted therapy. The deubiquitinase USP13 was a top hit resulting from this screen. Targeting USP13 increases the sensitivity to EGFR inhibition with small molecules in vitro and in vivo. USP13 selectively stabilizes mutant EGFR in a peptidase‐independent manner by counteracting the action of members of the Cbl family of E3 ubiquitin ligases. We conclude that USP13 is a strong mutant EGFR‐specific cotarget that could improve the treatment efficacy of EGFR‐targeted therapies.
Keywords: afatinib, epidermal growth factor receptor, non‐small cell lung cancer, osimertinib, ubiquitin‐specific protease 13
Short abstract
What's new?
Mutations in epidermal growth factor receptor (EGFR) are present in as many as 30 percent of patients with non‐small‐cell lung cancer (NSCLC). EGFR mutations render NSCLC sensitive to EGFR tyrosine kinase inhibitors, though over time many lung tumors become resistant to these therapies. This investigation shows that receptor stabilization and drug resistance in EGFR‐mutant NSCLC is mediated at least in part by the deubiquitinase USP13. Co‐targeting EGFR and USP13 increased NSCLC sensitivity to EGFR inhibitors in vitro and in vivo. The study identifies USP13 targeted therapy as a promising means for overcoming EGFR inhibitor resistance in EGFR‐mutant NSCLC.
Abbreviations
- DUB
ubiquitin‐associated domains
- EGFR
epidermal growth factor receptor
- MAD
mean absolute deviation
- MVP
multivesicular particles
- NSCLC
non‐small cell lung cancer
- UBA
ubiquitin‐binding domain
- USP13
ubiquitin‐specific protease 13
1. INTRODUCTION
Oncogenic epidermal growth factor receptor (EGFR) kinase domain mutations occur in 15% to 30% of non‐small cell lung cancers (NSCLC). 1 EGFR is a transmembrane tyrosine kinase receptor regulating vital cellular functions, including proliferation, cell growth and survival. 2 , 3 The most prevalent mutational hotspots, EGFR E746‐A750 and EGFR L858R, sensitize patients to first‐line EGFR tyrosine kinase inhibitor treatment in NSCLC. 4 , 5 , 6 , 7
EGFR‐targeted therapies result in impressive clinical response, leading to increased overall survival compared to chemotherapy. However, the inescapable limitation of these therapies is acquired resistance caused by the acquisition of secondary genomic alterations leading to disease progression and ultimate fatality. Recent studies show that a period of drug tolerance precedes these genomic alterations in various cancer types. 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 The mechanisms underlying such drug tolerance in EGFR‐mutant NSCLC remain poorly understood. Understanding these mechanisms could lead to the development of therapies that reduce the residual pool of drug‐tolerant cells and the likelihood of the emergence of secondary resistance, leading to improved outcomes.
We aimed to identify molecular mechanisms that reduce the sensitivity to EGFR‐targeted therapy using an unbiased strategy. We performed a high‐throughput siRNA screen with two libraries covering approximately 1500 proteins associated with the ubiquitin and ubiquitin‐like cellular processes with simultaneous inhibition of EGFR with the pan‐HER inhibitor afatinib. We identified the deubiquitinase USP13 as a top‐hit in making EGFR‐mutant lung cancer cells tolerant to afatinib. Accordingly, inhibition of USP13 led to a significant decrease in the survival of lung cancer cells treated with afatinib in vitro and in vivo. USP13 specifically counteracts the downregulation of mutated EGFR by the ubiquitin ligases of the Cbl family. We thus identify USP13 as a novel cotarget to reduce the number of drug‐tolerant cells and hence the pool of cells in which permanent genomic resistance can arise. This opens a window for the improvement of the outcome of these patients.
2. MATERIALS AND METHODS
2.1. Cell culture, transfections and treatments
PC9 (CVCL_B260; Merck), HCC827 (CVCL_2063; ATCC), H1975 (CVCL_1511; ATCC) and BEAS‐2B (CVCL_0168, kindly provided by Prof. Didier Cataldo, Université de Liège, Belgium) cells were cultured in RPMI 1640 (Gibco) supplemented with 10% fetal bovine serum (FBS) (Greiner). HEK293T ((CVCL_0063); ATCC) cells were cultured in Dulbecco's modified Eagle medium (Gibco) supplemented with 10% FBS. All human cell lines were authenticated using STR profiling within the past 3 years. The cell cultures were in a humid incubator at 37°C and 5% CO2 and maintained according to standard procedures. Cell lines were tested for mycoplasma regularly, and experiments were performed on mycoplasma free cells. Lipofectamine RNAiMAX (Thermo) was used for reverse transfection of siRNA and Lipofectamine 2000 (Thermo) was used for transfection of plasmids. siRNA and plasmid constructs are listed in supplementary materials. The concentrations of compounds used in our study (unless otherwise mentioned in the figure legends) were afatinib and osimertinib, 5 nM in PC9, BEAS‐2B and HEK293T cells and 1 nM in HCC827 cells; spautin‐1, 5 μM in PC9 and HEK293T and 10 μM in HCC827.
2.2. High‐throughput siRNA screening targeting ubiquitin(−like) pathways
Two arrayed libraries containing, respectively, 1505 (Qiagen GeneSolutions for human genes custom made) and 1525 (Dharmacon ON‐target plus SMART pool custom made) siRNA pools (composed of 4 siRNAs each pool) targeting different ubiquitin and ubiquitin‐related human mRNA transcripts were employed. Assay plates contained 1 μL of 0.5 μM siRNA per well, and reverse transfection was performed using Lipofectamine RNAiMAX. The final siRNA concentration was 10 nM. PC9 and HCC827 cells were resuspended in RPMI‐1640 supplemented with 10% FBS and seeded onto assay plates (500 and 1000 cells per well, respectively). Cells were incubated for 48 hours and subsequently treated with 5 (PC9) and 1 nM (HCC827) afatinib or DMSO for an additional 48 hours. Cell viability was measured using CellTiter‐Glo (Promega). The experiment was performed in duplicates. Analysis of the viability results was based on a robust Z‐score. 18 The Z‐score = ([mean viability ratio of afatinib and DMSO (siRNA pool)] − [mean viability ratio afatinib and DMSO of the 384 well‐plate])/mean absolute deviation (MAD). The MAD was calculated as follows: MAD = 1.4826 (k is a constant scale factor for normal distribution) × absolute value ([mean viability ratio of afatinib and DMSO (siRNA pool)] − [mean viability ratio afatinib and DMSO of the 384‐well plate]).
2.3. Lentiviral vector production
Lentiviral vectors were produced in HEK293T cells. Briefly, cells were then transfected with either control pLKO.1‐puro or USP13‐targeting pLKO.1‐USP13‐shRNAs vectors (MISSION, Sigma‐Aldrich), GAG, REV and VSV‐G using PEI (Polysciences, Eppelheim, Germany). The supernatants were collected, filtered and supplemented with 10 μg/mL protamine sulfate according to standard procedures. Cells were transduced by incubation with lentiviral vectors‐containing medium for 24 hours.
2.4. Cell viability assays
Cells were seeded in clear bottom 384‐well plates (Greiner Bio‐One) in a total volume of 50 μL per well. At the end‐point analysis, viability was determined by using the ATP‐based luminescence CellTiter‐Glo (Promega). Luminescence output was measured using a Spectramax M3 (Molecular Devices).
2.5. Cell confluence assays
Cell confluence was measured in 384‐ and 96‐well plates using the IncuCyte ZOOM system (Essen BioScience). Phase‐contrast images were taken hourly using the 10× objective during the indicated periods. Data analysis was performed using the IncuCyte ZOOM software.
2.6. Apoptosis assays
2.6.1. Molecular kits
Cells were seeded in white 384‐well plates (1000 cells per well) for viability and caspases 3/7 assays. Cells were transfected, and drugs were added at the indicated concentrations and time‐points. Viability and cleaved‐caspases 3/7 were quantified using the CellTiter‐Glo luminescent kit and the Caspase‐Glo 3/7 kit (Promega). Caspase signals were normalized to the CellTiter‐Glo signals of the same conditions.
2.6.2. Flow cytometry
Cells were seeded in 24‐well plates (50,000 cells per well). Drugs were added 24 hours postseeding and incubated for the indicated periods. Cells were detached and resuspended in Annexin‐V‐binding buffer (BD Pharmingen) and stained with Annexin‐V‐APC and 7‐AAD (BD Pharmingen). The samples were subsequently analyzed with a BD Canto flow cytometer.
2.7. Cell cycle assays
Cells were cultured in 24‐ or 6‐well plates and subsequently detached via trypsinization. Cells were washed and resuspended in 500 μL cold phosphate‐buffered saline (PBS). Cells were then fixed using 70% ice‐cold ethanol and then washed with ice‐cold PBS. A total of 106 cells were used per analysis and stained with 5 μL 7‐AAD in 100 μL flow cytometry buffer (PBS containing 1% bovine serum albumin (BSA) and 0.05% sodium azide). After 10 minutes, 200 μL of flow cytometry buffer was added, and the 7‐AAD signal in the cells in suspension was measured via flow cytometry (BD Canto).
2.8. qRT‐PCR
Total mRNA extraction was extracted using the Nucleospin RNA plus kit (Macherey‐Nagel). First‐strand cDNA was synthesized using SuperScript II Reverse Transcriptase (Life Technologies). Target and reference genes were quantified in duplicates on a LightCycler480 (Roche) using SYBR Green I Master (Roche). The mean CT values of the target genes were normalized to the geometric mean of two housekeeping genes (SDHA and TBP). Primers are listed in supplementary materials.
2.9. Western blotting
Cells were lysed in a standard Triton X‐100 lysis buffer containing 25 mM Tris‐HCl (pH = 7.5), 150 mM NaCl, 1 mM EDTA, 1% Triton X‐100, supplemented with protease and phosphatase inhibitors. Lysates were cleared by centrifugation, and protein concentrations were determined by the Bradford protein assay kit (Bio‐Rad). Equal amounts of total proteins were loaded per lane on 10% polyacrylamide gels, and proteins were separated by electrophoresis and blotted on nitrocellulose membranes. Membranes were blocked with 5% non‐fat milk dissolved in dissolved in tris‐buffered saline ‐ tween 20 (TBS‐T) and stained overnight with the primary antibody (listed in supplementary materials) and subsequently labeled with near‐infrared fluorescent secondary antibodies (LI‐COR). Membranes were developed using the LI‐COR Odyssey FC system. Protein quantifications and image analyses were performed using the Image Studio Software (LI‐COR).
2.10. Immunoprecipitation
Cells were lysed in Triton X‐100 lysis buffer. Equal amounts of total proteins were used for every immunoprecipitation condition. Samples were incubated with 1 μg of an antibody recognizing the protein of interest and 10 μL of Protein G Sepharose 4 Fast Flow antibody purification resin slurry (GE Healthcare). After 2 hours incubation, beads were washed three times with cold PBS and subsequently boiled in Western blotting loading buffer. Immunoprecipitation and whole cell lysate samples were then processed by Western blotting.
2.11. Confocal imaging
Cells were fixed with 4% paraformaldehyde and blocked with 1% BSA in PBS‐T (PBS + 0.1% Tween‐20) and finally washed twice with PBS‐T. Cells were stained with primary antibodies (mouse EGFR and rabbit USP13) for 1 hour and with secondary fluorescently labeled antibodies and washed three times with PBS‐T. Hoechst 33342 was used for nuclear counterstaining. Images were acquired using a ZEISS LSM710 NLO confocal microscope using the ZEN 2009 software (Carl Zeiss).
2.12. Mouse xenograft model
Female Swiss nu/nu homozygous mice (4 weeks old; Crl:NU‐Foxn1nu; strain code: 088‐Homozygous) were purchased from Charles River Laboratories. PC9 cells were stably transduced with pLKO.1‐puro (empty vectors), pLKO.1‐USP13‐shRNA 1 or 2, and selected using 3 μg/mL puromycin. The transduced PC9 cells were resuspended in cold 50% Matrigel in RPMI 1640 and injected subcutaneously into the mice's right flank (106 cells per mouse). For each transduced cell line, 20 mice were injected. Tumor volumes were measured using a Vernier caliper and calculated using the formula: Volume = Length × Width × [(Length + Width)/2] x π/6. Every 3 days, tumor volumes and mice body weights were determined. At an average tumor volume of 100 mm3, mice were assigned to a control (vehicle) or an afatinib (5 mg/kg) group; each group consisted of 10 mice with an equal average in tumor volumes. Mice were treated daily with 5 mg/kg afatinib or vehicle (DMSO) dissolved in 0.4% Tween‐80 and 1% methylcellulose via oral gavage. The experiment was terminated before tumor volumes reached 1000 mm3. All the procedures related to animal handling, care and treatments were performed according to the guidelines approved by the University of Gent Animal Care and Use Ethical Committee.
2.13. Sensitivity and toxicity index
Calculation of the sensitivity (assessed in the cancer cells) and toxicity (assessed in the normal epithelial cells) indexes was as follows: Sensitivity index = (Viability NT siRNA plus Afatinib/Viability NT)/(Viability siRNA x plus afatinib/viability siRNA x); Toxicity index = Viability NT siRNA/Viability siRNA x, in which x refers to every gene included in the siRNA screen.
2.14. Statistical analysis
Statistics for the cell viability and apoptosis data were performed with the one‐way analysis of variance (ANOVA) (using the Tukey post hoc test) to compare the means of three independent experiments measuring viability and apoptosis in vitro. The sample size for the in vivo experiment (mouse tumor xenografts) was calculated using the power analysis software Gpower 3.1. Input values were based on the in vitro results obtained in Figure 2. The paired two‐way ANOVA (using the Tukey post hoc test) was performed to compare the means of the tumor volumes and over the duration of the experiment. The means correspond to five mice per condition. The unpaired, two‐tailed t‐test with Welsh's correlation was used to compare the means of the tumor weights for the control shRNA plus afatinib and the USP13 shRNA B plus afatinib. The means correspond to five mice per condition. Western blots are representative images of experiments that were independently repeated at least two times and on several occasions more than three times. qRT‐PCR results were obtained from at least three independent repeats. Flow cytometry experiments were repeated at least two times (cell cycle) or at least four times (apoptosis). Error bars represent the SEM unless otherwise specified.
FIGURE 2.
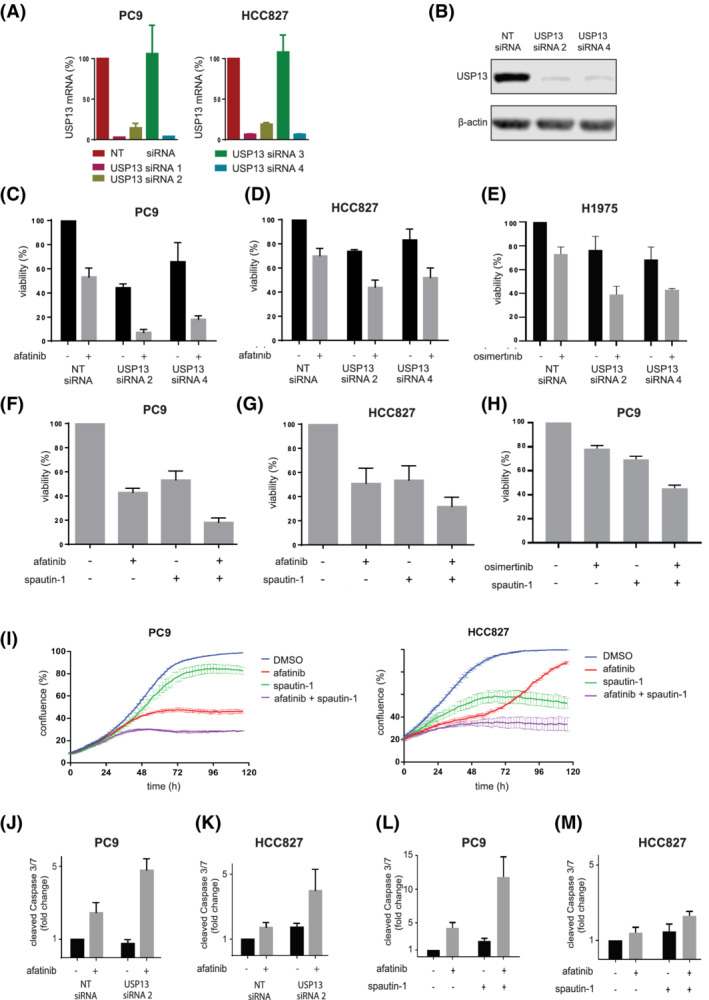
USP13 targeting increases sensitivity for EGFR inhibition through apoptosis. A, Quantitative RT‐PCR for USP13 mRNA in PC9 and HCC827 reverse‐transfected with non‐targeting (NT) and USP13 siRNAs 1‐4. Analyses were performed 24 hours posttransfection. Values were normalized to NT siRNA condition. Bars represent mean ± SEM. B, USP13 expression levels in PC9 reverse‐transfected with NT and USP13 siRNAs 2 and 4 for 72 hours. β‐Actin is a loading control. C,D, Cell viability analysis of PC9 (C) and HCC827 (D) cells reverse‐transfected with indicated siRNAs and treated with afatinib or vehicle (DMSO) 48 hours posttransfection. Cell viability was measured 96 hour posttransfection. Results were normalized to the NT siRNA DMSO control. Bars represent mean viability (%) ± SEM. E, Cell viability analysis of H1975 reverse‐transfected with indicated siRNAs and treated with osimertinib or vehicle (DMSO) 48 hour posttransfection. Cell viability was measured 96 hours posttransfection. Bars represent mean viability (%) ± SEM. F,G, Cell viability analysis of PC9 (F) and HCC827 (G) treated with afatinib, spautin‐1 or their combination for 72 hours. Bars represent mean ± SEM. H, Cell viability analysis of PC9 treated with osimertinib, spautin‐1 or their combination for 72 hours. I, Confluence analysis of PC9 and HCC827 treated with afatinib, spautin‐1 or their combination for 120 hours. Lines represent lowess curve through actual data points, the SEM is shown by bars per data point. Confluence was determined every hour. Graph is representative of three independent experiments. J,K, Caspases 3/7 cleavage analysis in PC9 (J) and HCC827 (K) cells reverse‐transfected with indicated siRNAs and treated with afatinib or vehicle (DMSO) 48‐hour posttransfection. Caspases 3/7 cleavage was measured 96‐hour posttransfection and normalized to the cell viability results for the same experiment. Results were normalized to non‐targeting (NT) siRNA DMSO control. Bars represent the mean ± SEM. L,M, As in (J,K) but with cells treated with afatinib, spautin‐1 or their combination [Color figure can be viewed at wileyonlinelibrary.com]
3. RESULTS
3.1. Ubiquitin system‐wide high‐throughput siRNA screen reveals specific mediators of EGFR TKI drug tolerance
To identify proteins that affect drug tolerance for EGFR TKIs, we performed a broad ubiquitin and ubiquitin‐like siRNA screen in treatment‐naive EGFR‐mutant NSCLC cells exposed to the pan‐HER/EGFR TKI afatinib. To enhance the likelihood of finding cell‐context‐independent mechanisms, we performed the screen in parallel using two human lung adenocarcinoma cell lines, PC9 and HCC827. These cell lines are the two most frequently studied, gold‐standard cell lines representing treatment‐naive EGFR mutant NSCLCs. We chose the pan‐HER inhibitor afatinib as it has the lowest IC50 values for the inhibition of EGFR ΔE746‐A750 and EGFR L858R among the clinically approved inhibitors (including osimertinib) and to increase the likelihood of identifying mechanisms that have a pan‐HER significance. 19 The afatinib concentrations were based on titration experiments of clinically achievable doses (Figure 1A and Figure S1).
FIGURE 1.
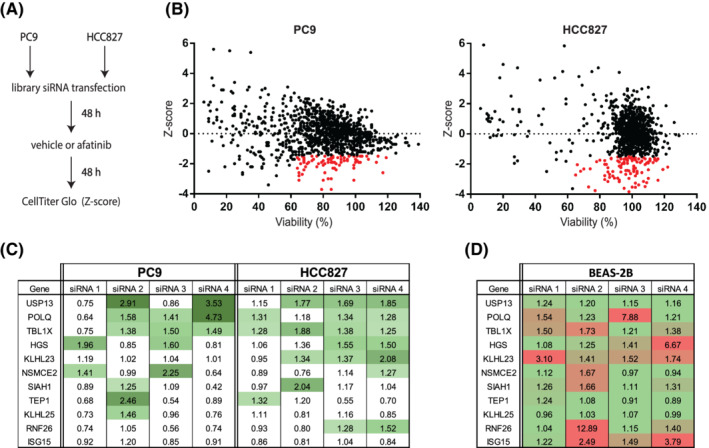
Ubiquitin system‐wide siRNA screen identifies targets of intrinsic insensitivity to afatinib. A, Screen methodology. Cells were reverse‐transfected in 384 well plates with pools of four siRNAs per target and treated with DMSO (vehicle) or afatinib 48 hours posttransfection. Cell viability was measured 96 hours posttransfection. B, Screen results. Z‐scores represent the relative viability differences of DMSO vs afatinib‐treated cells. Negative scores represent stronger effects on cell viability caused by afatinib compared to the median effects of the drug. Red dots indicate siRNA pools that did not decrease viability below 65% (DMSO) and had a Z‐score of at least −1.5. C, Sensitivity index for the individual siRNAs, which were common hits in PC9 and HCC827. Green intensity increasing indicates higher sensitivity indexes. D, Toxicity index for different siRNAs in normal lung epithelial BEAS‐2B cells. Green and red indicate low and high toxicity values, respectively [Color figure can be viewed at wileyonlinelibrary.com]
Candidate proteins were selected based on their capacity to increase afatinib sensitivity upon protein knockdown (Z‐scores ≤ −1.5) without strongly reducing cell viability in the absence of afatinib (viability >65%). With these criteria, we identified 75 and 121 siRNA pools in PC9 and HCC827, respectively (Figure 1B and Table S1).
From these two datasets, we continued to validate the 11 siRNA pools effective in both cell lines. We repeated the viability assays using the individual siRNAs from each pool (Figure S2) and calculated a sensitivity index corresponding to the effect of each siRNA on the sensitivity of the cells to afatinib under each condition. A sensitivity factor > 1.25 was the threshold to determine that a specific siRNA sufficiently increased the effect of afatinib. In the four final top hits (USP13, POLQ, TBL1X and HGS), we found at least two different siRNAs that sensitized NSCLC cells to afatinib (Figure 1C). To gain insight into the potential effect on non‐malignant epithelial cells and exclude EGFR mutation‐independent effects, we assessed the knockdown of the top candidates in the normal lung epithelial (EGFR wild‐type) BEAS‐2B cell line (Figure S3). In this case, we calculated a toxicity index for each siRNA by calculating the ratio between the effect of the individual siRNAs and the non‐targeting control siRNA (Figure 1D). Among the four top candidates, USP13 showed little toxicity in the BEAS‐2B model. At the same time, TBL1X, HGS and POLQ knockdown resulted in higher toxicity indexes. Overall, knockdown of USP13 displayed the most potent afatinib‐sensitizing effect on the viability of PC9 and HCC827 cells (Figure 1C), with a minimal impact on the viability of BEAS‐2B cells (Figure 1D).
3.2. USP13 cotargeting decreases drug tolerance and sensitizes EGFR‐mutant NSCLC to EGFR inhibition
We identified USP13 as the most potent candidate target to reduce tolerance and increase afatinib sensitivity in EGFR mutant NSCLC. We confirmed the knockdown efficiency by qRT‐PCR using the same USP13 siRNAs employed in the screen. We found that three out of four downregulated USP13 in both PC9 and HCC827 cells (Figure 2A). The knockdown of USP13 was confirmed by Western blotting in PC9 cells (Figure 2B).
The downregulation of USP13 in PC9 cells decreased their viability to 44% to 66%, while afatinib reduced it to 53%. When combined, cell viability decreased to 7% to 18% (Figure 2C). Similar results were obtained in HCC827 cells (Figure 2D), confirming that USP13 knockdown strongly enhances afatinib in treatment‐naive lung cancer cell lines. We subsequently tested whether USP13 inhibition could sensitize cells harboring the EGFR L858R/T790M double resistance mutation. We treated H1975 cells with USP13 siRNA and the EGFR inhibitor osimertinib. The knockdown of USP13 also led to a substantial cell viability reduction upon osimertinib treatment (Figure 2E).
The combination of spautin‐1, a chemical inhibitor of USP13 20 and afatinib decreased the viability significantly in PC9 and HCC827 cells compared to the respective single‐agent treatments (Figures 2F,G). To evaluate whether these observations could also apply to other EGFR inhibitors, we treated PC9 and H1975 cells with spautin‐1 and osimertinib. We found the same combined effect suggesting that the USP13 inhibition also decreases the tolerance to other EGFR inhibitors than afatinib (Figure 2H and Figure S4).
We further confirmed these findings using a confluence‐based life cell imaging technique in which PC9 and HCC827 cells treated with afatinib, spautin‐1 or both were monitored for 120 hours. After 72 hours, the combination of afatinib and spautin‐1 decreased the confluence levels (Figure 2I), comparable to our previous findings (Figure 2F,G). The effects of the combination of afatinib and spautin‐1 escalated in the first 48‐72 hours and were subsequently maintained (Figure 2I). Afatinib treatment of PC9 and HCC827 cells stably expressing USP13 shRNA caused more potent growth suppression than the nontargeting (NT) shRNA control cell line treated with afatinib (Figure S4), supporting the specificity of the effects seen with spautin‐1.
3.3. USP13 suppression induces apoptosis upon EGFR inhibition of mutant NSCLC cells
USP13 downregulation or inhibition severely compromised the viability of PC9 and HCC827 cells when cotreated with afatinib. This effect was not observed in the lung epithelial BEAS‐2B cell line (Figure S4), suggesting specificity for lung cancer cells carrying an EGFR mutation. We then analyzed the effect of USP13 levels/activity modulation on the cell cycle and apoptosis rate in NSCLC cells treated with afatinib. USP13 silencing/inhibition resulted in a modest accumulation of cells in G1/G0. However, it did not significantly increase the G1/G0 cell cycle arrest caused by EGFR inhibition (Figure S5).
Afatinib increased 2.4‐ and 1.7‐times the apoptosis rate in PC9 and HCC827 cells transfected with control siRNA (Figures 2J,K), while USP13 siRNA led to a 4.1‐ and 3.9‐fold increase in apoptosis when combined with afatinib in PC9 and HCC827 (Figure 2J,K). Spautin‐1 also led to a modest but reproducible induction of apoptosis in PC9 (Figure 2L) and HCC827 (Figure 2M) cells. Still, the combination of spautin‐1 and afatinib increased caspases 3/7 cleavage by 12‐ and 2.5‐fold in PC9 and HCC827 cells, respectively, compared to untreated cells. We confirmed this substantial increase in caspase cleavage/activation by flow cytometry (Figure S5).
3.4. A positive feedback loop between EGFR and USP13
USP13 is a protein involved in the deubiquitination and stabilization of multiple substrates. To investigate the mechanism of action underlying the strong effect of USP13 and EGFR cotargeting, we investigated whether USP13 could affect the protein expression of EGFR. First, we overexpressed USP13 in PC9 cells and observed a significant increase in the total protein levels of EGFR (Figure 3A). We excluded that USP13 regulates EGFR at the transcript level, further establishing that the effect is mediated via a posttranslational mechanism (Figure 3B).
FIGURE 3.
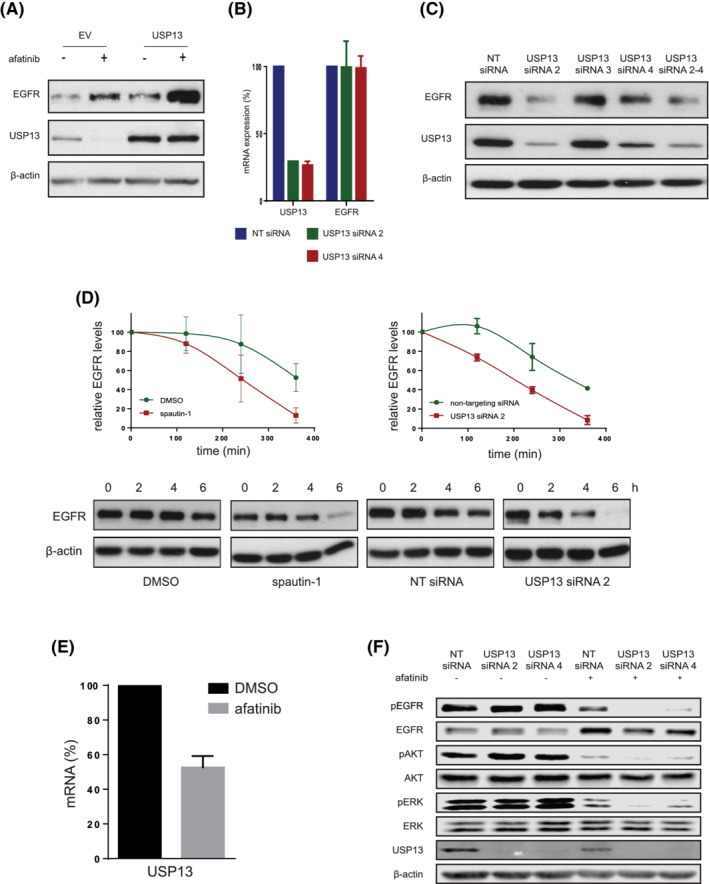
Positive feedback loop between EGFR and USP13. A, USP13 and EGFR levels were determined in PC9 cells transfected with either empty vector or pEF‐FLAG‐USP13, and treated with afatinib or DMSO (−) 24 hours posttransfection. Cells were lysed 48 hours posttransfection. β‐Actin was used as a loading control. B, USP13 and EGFR mRNA levels in PC9 cells reverse‐transfected with NT or USP13 siRNAs 2 and 4. Cells were lysed 24 hours posttransfection. C, As in (A) using PC9 cells reverse‐transfected with non‐targeting (NT) or USP13 siRNAs 2‐4. Cells were lysed 96 hours posttransfection. β‐Actin was used as the loading control. D, EGFR degradation rates were determined in the presence of cycloheximide (CHX) (40 μg/mL, 1 hour pretreatment). PC9 cells were also treated with spautin‐1 or DMSO for 72 hours (left graph), or NT or USP13 siRNA B (right graph) for 96 hours. EGFR and β‐actin levels were quantified, and EGFR values normalized to the β‐actin (loading control) values. Graphs show mean ± SEM. E, USP13 mRNA levels were determined using qRT‐PCR in PC9 cells 24 hours posttreatment with afatinib or DMSO. F, Levels of indicated proteins were determined in reverse‐transfected PC9 cells with NT or USP13 siRNAs 2 and 4. Cells were treated with afatinib or DMSO (as indicated) for 48 hours and lysed 96 hours posttransfection. β‐Actin was used as the loading control [Color figure can be viewed at wileyonlinelibrary.com]
USP13 downregulation with siRNA decreased the total protein levels of EGFR, indicating a clear connection between USP13 expression and EGFR protein levels (Figure 3C). Moreover, USP13 inhibition and silencing lead to a decreased half‐life of the mutant EGFR protein in NSCLC cells, indicating that USP13 likely stabilizes EGFR (Figure 3D).
On the other hand, we found that EGFR inhibition downregulated the endogenous levels of USP13 (Figure 3A). EGFR appears to regulate transcription of USP13, as afatinib treatment resulted in a significant reduction of USP13 mRNA and protein levels (Figure 3A,E,F). Inhibition of the USP13‐mediated stabilization of EGFR combined with EGFR inhibition led to further dampening of ERK1/2 and Akt activation, compared to EGFR inhibition alone. In addition, the combined effect of USP13 downregulation by EGFR inhibition and USP13 siRNA also increased the negative effect on EGFR stability (Figure 3F). Taken together, these data indicate a feedback loop between USP13 and EGFR.
3.5. USP13 inhibits the ubiquitin‐mediated degradation of mutant EGFR
To understand the interplay between USP13 and mutant EGFR proteins, we first analyzed their respective subcellular localization. In PC9 cells, most of the EGFR was in the cytosol, presumably in endosomes, as previously reported. 21 Only some localized at the plasma membrane (Figure 4A). USP13 was diffusely found in the nucleus and the cytosol, but also present in some areas of the plasma membrane, where it colocalized with EGFR (Figure 4A, insets).
FIGURE 4.
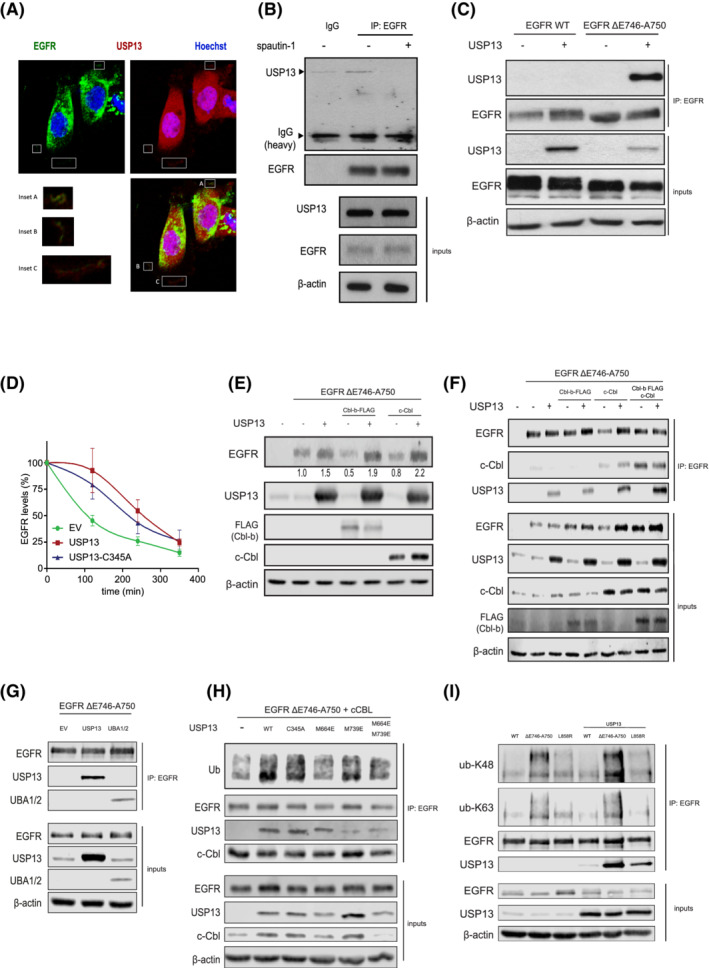
USP13‐EGFR interaction relies on c‐Cbl and the UBA domains of USP13. A, Confocal images of PC9 cells displaying localizations of USP13 (red), EGFR (green) and DNA (Hoechst, blue). Insets show colocalization of USP13 and EGFR. B, Immunoprecipitation (IP) of endogenous EGFR from PC9 cells treated with DMSO or spautin‐1 for 24 hours was analyzed by Western blotting as indicated. Inputs correspond to 5% of the sample used for IP. C, IP of EGFR from HEK293T cells overexpressing EGFR wild‐type (WT) or ΔE746‐A750 mutant and USP13. IP was performed 24 hours posttransfection. Samples were treated as in (B). D, EGFR degradation rates were determined by cycloheximide‐chase experiments and Western blotting. HEK293T cells were transfected with EGFR ΔE746‐A750 mutant and empty vector (EV), USP13 wild‐type or C345A mutant. Graph shows mean ± SEM. E, HEK293T cells were transfected with EGFR‐ΔE746‐A750, Cbl‐b, c‐Cbl and/or USP13. Cells were lysed 24 hours posttransfection and analyzed by Western blotting. F, IP of EGFR ΔE746‐A750 mutant from HEK293T cells also overexpressing Cbl‐b, c‐Cbl and/or USP13. Samples were treated as in E. G, As in (F) using HEK293T cells overexpressing USP13 full‐length or its UBA1/2 domains. H, As in (F) using HEK293T cells overexpressing USP13 wild‐type (WT) or mutants in its catalytic site (C345A), UBA1 (M664E) or UBA2 (M739E) domains (as indicated). I, IP of EGFR wild type, or mutants ΔE746‐A750 or L858R from HEK293T cells in the absence or presence of overexpressed USP13. IP product was immunoblotted using ubiquitin chain specifics K48 or K63 antibodies as indicated [Color figure can be viewed at wileyonlinelibrary.com]
We found that endogenous USP13 coimmunoprecipitated with endogenous mutant EGFR (Figure 4B). Treatment with spautin‐1 abolished the interaction (Figure 4B). We then recapitulated the interaction between exogenous USP13 and EGFR in HEK293T cells. The interaction of wild‐type EGFR with USP13 was hardly detectable, whereas USP13 coimmunoprecipitated with the EGFR ΔE746‐A750 and L858R mutants (Figure 4C,I), indicating that USP13 associates much more tightly with mutant than wild‐type EGFR.
We then compared the half‐life of the EGFR ΔE746‐A750 mutant in HEK293T cells treated with cycloheximide and overexpressing wild‐type USP13 or its catalytical inactive C345A mutant, 22 (Figure 4D). Surprisingly, both versions of USP13 extended the half‐life of mutant EGFR, indicating that the stabilizing effect does not require the isopeptidase activity of USP13.
The E3 ubiquitin ligases c‐Cbl and Cbl‐b control endosomal sorting and degradation of EGFR within lysosomes. 23 In HEK293T cells expressing EGFR‐ΔE740‐A746, USP13 antagonized the destabilization of mutant EGFR mediated by c‐Cbl or Cbl‐b (Figure 4E). c‐Cbl enhanced the interaction between EGFR‐ΔE740‐A746 and USP13, while Cbl‐b increased the binding of both c‐Cbl and USP13 to mutant EGFR (Figure 4F).
These results suggest that c‐Cbl controls the binding between USP13 and mutant EGFR, and Cbl‐b modulates c‐Cbl binding and subsequent recruitment of USP13 to mutant EGFR.
USP13 harbors two functional ubiquitin‐associated domains (UBA). The UBA domains of USP13 have been reported to preferentially bind K63‐linked ubiquitin chains, 24 which are known to be involved in the degradation of EGFR. 24 , 25 Therefore, we considered the possibility that USP13 binds to ubiquitinated mutant EGFR via its UBA domains and, by doing so, prevents EGFR endosomal sorting and lysosomal degradation. We found that the isolated UBA1 + 2 domains of USP13 were sufficient to mediate the interaction with mutant EGFR in cells (Figure 4G). By employing the USP13‐C345A mutant or versions mutated in UBA1, UBA2 or both, we established that the UBA domains' integrity is necessary for USP13 to bind to mutant EGFR (Figure 4H). The USP13 wild type and C345A mutant were more efficient than versions mutated in their UBA domains to bind to and maintain the ubiquitination of mutant EGFR (Figure 4H). Thus, USP13 is likely to stabilize ubiquitinated mutant EGFR. We then analyzed the nature of the ubiquitin chains found on mutant EGFR and found that the EGFR‐ΔE746‐A750 mutant carried more ubiquitin chains of K48 and K63 types than wild‐type EGFR (Figure 4I). The fraction of mutant EGFR bound to USP13 was also enriched in those two types of ubiquitin chains (Figure 4I). We hypothesize that USP13 stabilizes mutant EGFR at the level of its endosomal sorting and/or lysosomal degradation, which has been associated with K48 and K63 ubiquitination.
Overall, we showed that USP13 abrogates sorting/degradation of ubiquitinated mutant EGFR, independent of its catalytic activity but requiring its UBA domains that are known to bind to K63‐type ubiquitin chains preferentially. 24
3.6. In vivo USP13 downregulation sensitizes EGFR mutant NSCLC tumors to afatinib
We set up a mouse xenograft tumor model using PC9 cells transduced with lentiviral vectors containing USP13 shRNAs or control shRNA. Three different groups of 20 female Swissnu/nu mice each were injected subcutaneously with 106 cells. At a tumor volume of 100 mm3, each group was divided into two subgroups and treated daily (for 21 days) by oral gavage with either afatinib or vehicle control. On Day 7, one mouse of each subgroup was sacrificed to check the USP13 knockdown efficiency by Western blot. We found that both USP13 shRNAs reduced USP13 expression in vivo compared to the control shRNA although with different efficiencies (Figure 5A). The most efficient USP13 shRNA also resulted in a decrease in the levels of total and phosphorylated (ie, active) EGFR, which correlated with a reduction in the activation of Akt and ERK1/2 (Figure 5A). The downregulation of USP13 also increased active caspase 3, indicating tumor apoptosis (Figure 5A). We measured the volumes of the tumors every 3 days and found that the USP13 shRNAs alone did not significantly inhibit tumor growth. Afatinib alone induced a robust tumor growth arrest, while the combination of afatinib with the most efficient USP13 shRNA led to an almost complete disappearance of the tumors (Figure 5B,C). The body weight of the mice was not affected by our treatments (Figure 5D). We found a decreased cell density and pronounced hyalinization in H&E‐stained tumor samples having reduced USP13 levels, especially in combination with afatinib. Those effects were proportionate to the USP13 knockdown efficiency (Figure 5E,F). Tumors in the control group were actively cycling, with 70% of Ki‐67 positivity. Treatment with afatinib alone diminished the number of proliferating cells to approximately 36%, while USP13 shRNA only slightly decreased cell proliferation. Afatinib, combined with the most efficient USP13 knockdown, caused a near‐complete proliferation arrest in the tumors (Figures 5G,H). Overall, these data indicate that targeting of USP13 strongly potentiates the anticancer effects of afatinib in vivo.
FIGURE 5.
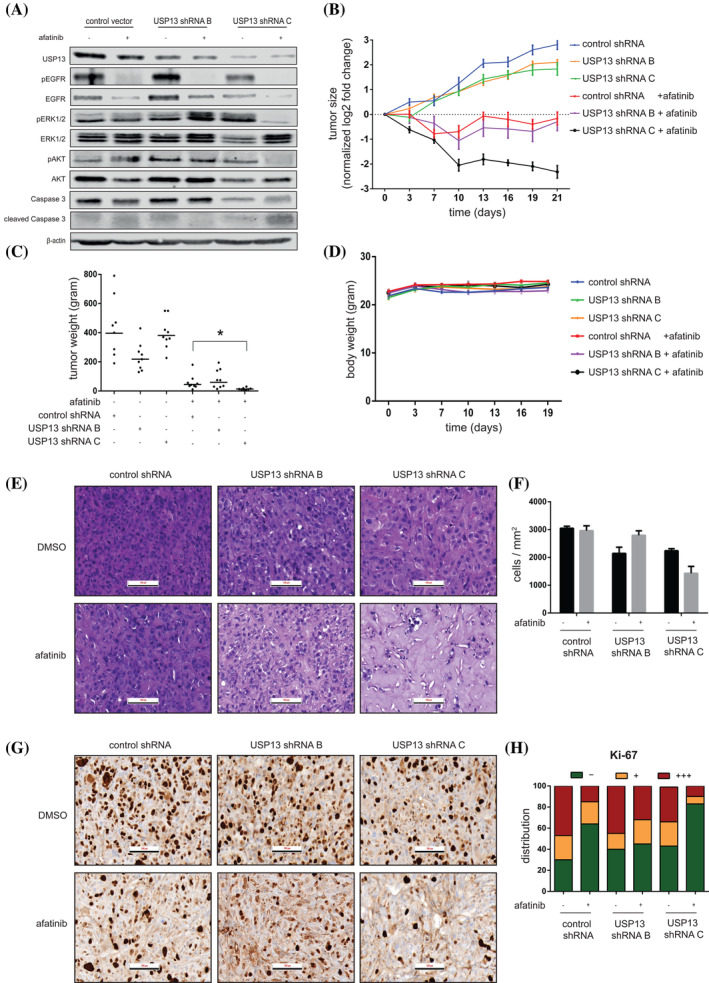
USP13 knockdown enhances the anticancer effects of afatinib in a human NSCLC mouse xenograft model. A, Xenograft tumors from genetically engineered PC9 cells (as indicated) were harvested and lysed after 7 days of treatment (control or afatinib) and levels of indicated proteins were analyzed by Western blotting. B, Growth of the indicated tumors during 21 days. Day 0 represents the start of afatinib treatment. Data points represent the mean ± SEM of a minimum of eight mice per group. C, Weight of the indicated tumors after resection after 21 days of treatment. Dots represent the mass (mg) of individual tumors. Horizontal lines show mean ± SEM. D, Weight of the mice carrying the indicated tumors over time. Day 0 represents the start of afatinib treatment. Data points represent the mean ± SEM of a minimum of eight mice per group. E, Representative H&E staining of xenograft tumor tissues produced by the respective genetically engineered PC9 cells. Scale bar indicates 100 μm. F, Quantification of the number of nuclei per mm2 found in (E). Bars represent the mean ± SEM of a minimum of three mice per group. G, Representative Ki‐67 staining of tumor tissues obtained from the indicated PC9 cells. Scale bar indicates 100 μm. H, Quantification of the percentage of absent (−), weakly positive (+) or strongly positive (+++) Ki‐67 nuclear staining in the tumors from (G). Bars show the relative group distribution per condition [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
EGFR‐targeted therapies have drastically improved patients' outcomes with advanced EGFR mutant lung cancer, but acquired resistance consistently leads to ultimate treatment failure, disease progression and patient demise. Early functional drug tolerance to the therapy leads to the accumulation of a pool of persisting cells that, over time, can gain novel resistance mutations. 16 , 17 Drug tolerance may be facilitated by feedback activation of survival pathways, 16 , 26 , 27 , 28 altered epigenetic states 29 , 30 or regulation by the microenvironment. 31 , 32 Several mechanisms of constitutive primary and secondary resistance have been identified in lung cancer, 33 but the molecular mechanisms underlying early drug tolerance to EGFR‐TKI remain unknown.
To identify such mechanisms, we performed an unbiased siRNA screen targeting the ubiquitin and ubiquitin‐like systems. The ubiquitin system can virtually regulate all cellular physiological and pathological signaling and has recently emerged as a clinically relevant and druggable pathway. 34
Using this method, we have identified USP13 as a prominent regulator of EGFR‐TKI drug tolerance in EGFR‐mutant NSCLC. Targeting of USP13 by si/shRNA‐mediated silencing or by the small‐molecule inhibitor spautin‐1 sensitizes EGFR‐mutant NSCLC cells to EGFR inhibition in vitro and in vivo. Although several studies have investigated E3 ubiquitin ligases (ie, c‐Cbl and Cbl‐b) and deubiquitination in the regulation of EGFR, 23 , 35 , 36 the uncovering of an interplay between the ubiquitin system and the therapeutic targeting of mutant EGFR with TKIs is novel. We demonstrate that USP13 targeting leads to an increased sensitivity toward EGFR inhibition by both osimertinib and afatinib in NSCLC harboring the most common EGFR mutations (ΔE746‐A750 and L858R/T790M) while sparing EGFR wild‐type cells and tumors. This observation is highly relevant since it could predict a potential increased therapeutic ratio when applied in vivo and clinically. Although both osimertinib and afatinib were included in our study, a comparison between these compounds combined with USP13 was not made as these compounds differ in IC50 and biological activity. 37 USP13 is likely to stabilize mutant EGFR in a ubiquitinated state, and inhibition of USP13 destabilizes mutant EGFR and abrogates signaling of mutant EGFR in lung cancer cells. Cotargeting of EGFR and USP13 led to a robust increase in treatment efficacy both in vitro and in vivo, mainly due to a strong induction of apoptosis rather than enhancing the cell cycle arrest caused by afatinib alone. 38 , 39
Given that USP13 targeting caused EGFR destabilization, some of the effects may be independent of the EGFR kinase activity as EGFR can suppress cell death in manners that are independent of its kinase activity. 40 , 41 Moreover, these findings are in line with our previous work that showed that siRNA‐mediated EGFR downregulation could increase the biological effect of EGFR TKIs (including afatinib) compared to single‐agent drug targeting in EGFR mutant NSCLC. 42
EGFR downregulation is regulated by several adaptor proteins, including the E3 ubiquitin ligases c‐Cbl and Cbl‐b. 23 They ubiquitinate EGFR, provoking its internalization into early endosomes and sorting into multivesicular particles (MVPs). EGFR is then either degraded in lysosomes or recycled back to the cell membrane. 43 , 44 Deubiquitinating enzymes also control EGFR turnover. One study reported that up to 15 DUBs are directly or indirectly implicated in EGFR regulation. 45 Among those, USP8/UBPY, AMSH, USP9x, USP2a and Cezanne‐1/OTUD7B have been characterized more in detail but only in the context of wild‐type EGFR. 45 , 46 , 47 , 48 Overall, those deubiquitinases exert multiple effects depending on (a) their specific substrate(s) (ie, EGFR itself or EGFR adaptor proteins); (b) the targeted ubiquitin chains (ie, K48, K63 or others); and (c) their spatiotemporal effects during the EGFR degradation process.
We found that USP13 inhibits mutant EGFR degradation by a process that leads to the accumulation of ubiquitinated (K48 and K63) EGFR, which is bound to c‐Cbl and USP13. We also report that the interaction between USP13 and mutant EGFR is more robust than the wild‐type EGFR. The association is dependent on the presence of c‐Cbl, as we observed that increased expression of c‐Cbl enhanced USP13‐mutant EGFR interaction. Of note, USP13 did not interfere with the ubiquitin ligase activity of c‐Cbl/Cbl‐b since modulation of USP13 levels/activity did not change the ubiquitination status of EGFR. The general mechanism of action of USP13 remains poorly understood, as both isopeptidase activity‐dependent (eg, Skp2 49 ) and ‐independent (eg, Siah‐2 22 ) effects have been described. Since ubiquitinated (K48 and K63) EGFR accumulated in the presence of USP13, we propose that the control of EGFR stability by USP13 does not require deubiquitination of the receptor. Also, we show that a catalytically inactive form of USP13 (C345A mutant) is still capable of stabilizing mutant EGFR. The isopeptidase‐independent mechanism of action proposed so far for USP13 relies on its UBA domains, which bind to ubiquitinated substrates, possibly inducing structural changes that stabilize them. 22 Coincidentally, K63‐linked ubiquitin chains are required for EGFR sorting and degradation. 25 This type of chains has also been reported as the preferred substrates for the USP13 UBA domains. 24 We found that the UBA domains of USP13 were required for the binding to and stabilizing EGFR, as mutated UBA domains in USP13 resulted in a loss of USP13‐EGFR interaction and of EGFR stabilization. We show that the C‐terminal part of USP13 (containing the UBA domains) is sufficient to bind to mutant EGFR. We thus propose a model in which USP13 recognizes mutant EGFR, K63‐ubiquitinated by c‐Cbl via its UBA domains; USP13 binding could then shield EGFR, which may even be further ubiquitinated at K48, preventing its interaction with the downstream endosomal/lysosomal machinery. Of note, since spautin‐1 was able to abolish the interaction between USP13 and mutant EGFR, it is conceivable that spautin‐1 binds to or somehow affects the function of the USP13 UBA domains.
The extensive morphological transformation observed in vivo in the EGFR mutant tumors in which USP13 and EGFR were targeted, including massive hyalinization, is reminiscent of the effects seen in c‐KIT‐mutant gastrointestinal stromal tumors treated with imatinib. 50 Coincidentally, it was recently found that spautin‐1 reduced the expression of GLUT1 in prostate cancer cells, 51 which is under the control of EGFR. It is important to note that USP10/13 were not involved in that context, which agrees with the selectivity of the binding of USP13 to mutant as opposed to wild‐type EGFR in our study. No other molecular mechanism was put forward to explain the effect of spautin‐1 on the EGFR signaling in prostate cancer cells. 51 . Imatinib also determines the localization of GLUT1. 52 Possibly, GLUT1 could be the conduit to the similar in vivo morphological changes displayed by the tumors in our study treated with combined USP13 shRNA and EGFR inhibition.
Currently, spautin‐1 is the only described USP13 inhibitor. Although this compound is effective in combination with EGFR inhibitors, little is known of its toxicological profile and stability in vivo. The few studies in which spautin‐1 was used in vivo focused on biological effects but reported little or no toxicological parameters. 53 , 54 Hence, further studies are required to determine whether spautin‐1 is a suitable compound for in vivo use and clinical studies. Our study underlines the need for clinically applicable USP13 inhibitors.
Another recent study gives additional support for further developing the therapeutic targeting of USP13 in lung cancer in addition to the combined targeting with mutant EGFR we have discovered. Wu et al demonstrate that amplification of USP13 in some NSCLC leads to enhanced AKT and MAPK signaling that sustains cancer progression. 55 Based on these and our data, we postulate that USP13 expression may serve as a predictive biomarker for the response to EGFR‐targeted therapies. Further studies are required to correlate USP13 expression in EGFR mutant NSCLC with the clinical outcome of patients treated with EGFR TKIs.
In summary, our study unveils a critical role for USP13 in early drug tolerance to the EGFR inhibitors afatinib and osimertinib by controlling the turnover of mutant EGFR. Combined targeting of USP13 and EGFR strongly reduces the viability of EGFR mutant lung cancer cells in vitro and in vivo while sparing non‐malignant (EGFR wild‐type) lung epithelial cells. Thus, we have identified USP13 as an essential mediator of the innate therapy insensitivity in the context of EGFR‐mutant lung cancer treated with EGFR TKIs. The EGFR mutant‐specific effects suggest that USP13 is an excellent candidate for further clinical exploration combined with EGFR inhibition.
CONFLICT OF INTEREST
The authors declare no potential conflicts of interest.
ETHICS STATEMENT
All the procedures related to animal handling, care and treatments were approved by the University of Gent Animal Care and Use Ethical Committee.
Supporting information
Appendix S1: Supporting Information
Table S1 Analysis of the siRNA screen data
ACKNOWLEDGEMENTS
We thank Prof. Karine Breckpot and Prof. Cleo Goyvaerts (VUB) for assistance regarding the use of the lentiviral core facility; Prof. Luc Bouwens and Eddy Himpen (VUB) for assistance regarding the use of the confocal microscope; and Prof. Luc Leyns (VUB) for access to his laboratory equipment.
JDG received a grant from the Cancer Plan 29‐39 Belgium. GJG had grants from FWO (G0C7514N), BELSPO Interuniversity Attraction Poles (IAP‐P7‐07), VUB (starting ZAP credit) and Innoviris (BB2B program). GJG and JDG are cofinanced by an Interdisciplinary Research Program (IRP) for Excellence on Lung Cancer Research (VUB) and Wetenschappelijk Fonds Willy Gepts (WFWG) (UZ Brussel/VUB). PAB thanks funding from the CCSG grant (NCI Cancer Center Support Grant P30 CA030199) for the siRNA screen. PG and CE are PhD Fellows of the FWO and were also funded by the Emmanuel van der Schueren scholarship from Kom op Tegen Kanker. AN is an FWO postdoctoral fellow.
Giron P, Eggermont C, Noeparast A, et al. Targeting USP13‐mediated drug tolerance increases the efficacy of EGFR inhibition of mutant EGFR in non‐small cell lung cancer. Int. J. Cancer. 2021;148:2579–2593. 10.1002/ijc.33404
Funding information Belgian Federal Science Policy Office, Grant/Award Number: IAP‐P7‐07; Cancer Plan 29‐39 Belgium; Fonds Wetenschappelijk Onderzoek, Grant/Award Number: G0C7514N; Innoviris, Grant/Award Number: BB2B program; Kom op tegen Kanker; NCI Cancer Center Support Grant, Grant/Award Number: P30 CA030199; UZ Brussel/VUB (Willy Gepts fund); Vrije Universiteit Brussel
Contributor Information
Gustavo J. Gutierrez, Email: ggutierr@vub.be.
Jacques De Grève, Email: jacques.degreve@uzbrussel.be.
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article. Additional data will be made available from the corresponding authors upon reasonable request.
REFERENCES
- 1. Midha A, Dearden S, McCormack R. EGFR mutation incidence in non‐small‐cell lung cancer of adenocarcinoma histology: a systematic review and global map by ethnicity (mutMapII). Am J Cancer Res. 2015;5:2892‐2911. [PMC free article] [PubMed] [Google Scholar]
- 2. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169‐181. [DOI] [PubMed] [Google Scholar]
- 3. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non‐small cell lung cancer. Nature. 2018;553:446‐454. [DOI] [PubMed] [Google Scholar]
- 4. Soria J‐C, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR ‐mutated advanced non–small‐cell lung cancer. N Engl J Med. 2018;378:113‐125. [DOI] [PubMed] [Google Scholar]
- 5. Mitsudomi T, Kobayashi Y. Afatinib in lung cancer harboring EGFR mutation in the LUX‐lung trials: six plus three is greater than seven? Transl Lung Cancer Res. 2016;5:446‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small‐cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129‐2139. [DOI] [PubMed] [Google Scholar]
- 7. Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung, cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497‐1500. [DOI] [PubMed] [Google Scholar]
- 8. Woolston A, Khan K, Spain G, et al. Genomic and transcriptomic determinants of therapy resistance and immune landscape evolution during anti‐EGFR treatment in colorectal cancer. Cancer Cell. 2019;36:35.e9‐50.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bell CC, Fennell KA, Chan YC, et al. Targeting enhancer switching overcomes non‐genetic drug resistance in acute myeloid leukaemia. Nat Commun. 2019;10:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hugo W, Zaretsky JM, Sun L, et al. Genomic and transcriptomic features of response to anti‐PD‐1 therapy in metastatic melanoma. Cell. 2016;165:35‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Knoechel B, Roderick JE, Williamson KE, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46:364‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mehta A, Kim YJ, Robert L, et al. Immunotherapy resistance by Inflammation‐induced dedifferentiation. CANCER Discov. 2018;8:935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shlush LI, Mitchell A, Heisler L, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature. 2017;547:104‐108. [DOI] [PubMed] [Google Scholar]
- 14. Shaffer SM, Dunagin MC, Torborg SR, et al. Rare cell variability and drug‐induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546:431‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fong CY, Gilan O, Lam EYN, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015;525:538‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hata AN, Niederst MJ, Archibald HL, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22:262‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramirez M, Rajaram S, Steininger RJ, et al. Diverse drug‐resistance mechanisms can emerge from drug‐tolerant cancer persister cells. Nat Commun. 2016;7:10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Birmingham A, Selfors LM, Forster T, et al. Statistical methods for analysis of high‐throughput RNA interference screens. Nat Methods. 2009;6:569‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shintouo CM, Shey RA, Nebangwa DN, et al. In silico design and validation of ovmane1, a chimeric antigen for human onchocerciasis diagnosis. Pathogens. 2020;9:1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu J, Xia H, Kim M, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chung B, Raja SM, Clubb RJ, et al. Aberrant trafficking of NSCLC‐associated EGFR mutants through the endocytic recycling pathway promotes interaction with Src@. BMC Cell Biol. 2009;10:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Scortegagna M, Subtil T, Qi J, et al. USP13 enzyme regulates Siah2 ligase stability and activity via noncatalytic ubiquitin‐binding domains. J Biol Chem. 2011;286:27333‐27341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pennock S, Wang Z. A tale of two Cbls: interplay of c‐Cbl and Cbl‐b in epidermal growth factor receptor downregulation. Mol Cell Biol. 2008;28:3020‐3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Y‐H, Zhou C‐J, Zhou Z‐R, Song A‐X, Hu H‐Y. Domain analysis reveals that a deubiquitinating enzyme USP13 performs non‐activating catalysis for Lys63‐linked polyubiquitin. PLos One. 2011;6(12):e29362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang F, Zeng X, Kim W, et al. Lysine 63‐linked polyubiquitination is required for EGF receptor degradation. Proc Natl Acad Sci. 2013;110:15722‐15727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug resistance via feedback activation of stat3 in oncogene‐addicted cancer cells. Cancer Cell. 2014;26:207‐221. [DOI] [PubMed] [Google Scholar]
- 27. Raaby Gammelgaard K, Vad‐Nielsen J, Simone Clement M, et al. Up‐regulated FGFR1 expression as a mediator of intrinsic TKI resistance in EGFR‐mutated NSCLC. Transl Oncol. 2019;12:432‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shah KN, Bhatt R, Rotow J, et al. Aurora kinase A drives the evolution of resistance to third‐generation EGFR inhibitors in lung cancer. Nat Med. 2019;25:111‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sharma SV, Lee DY, Li B, et al. A chromatin‐mediated reversible drug‐tolerant state in cancer cell subpopulations. Cell. 2010;141:69‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Byers LA, Diao L, Wang J, et al. An epithelial‐mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19:279‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wilson TR, Fridlyand J, Yan Y, et al. Widespread potential for growth‐factor‐driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505‐509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hirata E, Girotti MR, Viros A, et al. Intravital imaging reveals how BRAF inhibition generates drug‐tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell. 2015;27:574‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meador CB, Hata AN. Acquired resistance to targeted therapies in NSCLC: updates and evolving insights. Pharmacol Ther. 2020;210:107522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fan Q, Wang Q, Cai R, Yuan H, Xu M. The ubiquitin system: orchestrating cellular signals in non‐small‐cell lung cancer. Cell Mol Biol Lett. 2020;25:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Clague MJ, Urbé S. Endocytosis: the DUB version. Trends Cell Biol. 2006;16:551‐559. [DOI] [PubMed] [Google Scholar]
- 36. Ray P, Tan YS, Somnay V, et al. Differential protein stability of EGFR mutants determines responsiveness to tyrosine kinase inhibitors. Oncotarget. 2016;7:68597‐68613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hirano T, Yasuda H, Tani T, et al. In vitro modeling to determine mutation specificity of EGFR tyrosine kinase inhibitors against clinically relevant EGFR mutants in non‐small‐cell lung cancer. Oncotarget. 2015;6:38789‐38803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu C‐H, Chou C‐C, Tu H‐F, et al. Antibody‐assisted target identification reveals afatinib, an EGFR covalent inhibitor, down‐regulating ribonucleotide reductase. Oncotarget. 2018;9:21512‐21529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Modjtahedi H, Cho BC, Michel MC, Solca F. A comprehensive review of the preclinical efficacy profile of the ErbB family blocker afatinib in cancer. Naunyn Schmiedebergs Arch Pharmacol. 2014;387:505‐521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tan X, Thapa N, Sun Y, Anderson RAA. A kinase‐independent role for EGF receptor in autophagy initiation. Cell. 2015;160:145‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weihua Z, Tsan R, Huang W‐C, et al. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell. 2008;13:385‐393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen G, Kronenberger P, Teugels E, Umelo IA, De Grève J. Targeting the epidermal growth factor receptor in non‐small cell lung cancer cells: the effect of combining RNA interference with tyrosine kinase inhibitors or cetuximab. BMC Med. 2012;10:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang F, Kirkpatrick D, Jiang X, Gygi S, Sorkin A. Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol Cell. 2006;21:737‐748. [DOI] [PubMed] [Google Scholar]
- 44. Tomas A, Futter CE, Eden ER. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 2014;24:26‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Savio MG, Wollscheid N, Cavallaro E, et al. USP9X controls EGFR fate by deubiquitinating the endocytic adaptor Eps15. Curr Biol. 2016;26:173‐183. [DOI] [PubMed] [Google Scholar]
- 46. Liu Z, Zanata SM, Kim J, et al. The ubiquitin‐specific protease USP2a prevents endocytosis‐mediated EGFR degradation. Oncogene. 2013;32:1660‐1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pareja F, Ferraro DA, Rubin C, et al. Deubiquitination of EGFR by Cezanne‐1 contributes to cancer progression. Oncogene. 2012;31:4599‐4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alwan HAJ, Van Leeuwen JEM. UBPY‐mediated epidermal growth factor receptor (EGFR) deubiquitination promotes EGFR degradation. J Biol Chem. 2007;282:1658‐1669. [DOI] [PubMed] [Google Scholar]
- 49. Chen M, Gutierrez GJ, Ronai ZA. Ubiquitin‐recognition protein Ufd1 couples the endoplasmic reticulum (ER) stress response to cell cycle control. Proc Natl Acad Sci U S A. 2011;108:9119‐9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vassos N, Agaimy A, Schlabrakowski A, Hohenberger W, Schneider‐Stock R, Croner RS. An unusual and potentially misleading phenotypic change in a primary gastrointestinal stromal tumour (GIST) under imatinib mesylate therapy. Virchows Arch. 2011;458:363‐369. [DOI] [PubMed] [Google Scholar]
- 51. Liao Y, Guo Z, Xia X, et al. Inhibition of EGFR signaling with Spautin‐1 represents a novel therapeutics for prostate cancer. J Exp Clin Cancer Res. 2019;38:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Prenen H, Stefan C, Landuyt B, et al. Imatinib mesylate inhibits glucose uptake in gastrointestinal stromal tumor cells by downregulation of the glucose transporters recruitment to the plasma membrane. Am J Biochem Biotechnol. 2005;1:95‐102. [Google Scholar]
- 53. Xiao J, Feng X, Huang X‐Y, et al. Spautin‐1 ameliorates acute pancreatitis via inhibiting impaired autophagy and alleviating calcium overload. Mol Med. 2016;22:643‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Horie R, Nakamura O, Yamagami Y, et al. Apoptosis and antitumor effects induced by the combination of an mTOR inhibitor and an autophagy inhibitor in human osteosarcoma MG63 cells. Int J Oncol. 2016;48:37‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu Y, Zhang Y, Liu C, et al. Amplification of USP13 drives non‐small cell lung cancer progression mediated by AKT/MAPK signaling. Biomed Pharmacother. 2019;114:108831. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Table S1 Analysis of the siRNA screen data
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article. Additional data will be made available from the corresponding authors upon reasonable request.