

Abstract
Objective
To evaluate the efficacy and safety of E6011, a humanized IgG2 monoclonal antibody against human fractalkine (FKN), in a phase II, double‐blind, placebo‐controlled study in rheumatoid arthritis (RA) patients.
Methods
Patients with moderate‐to‐severe RA who had an inadequate response to methotrexate were randomly assigned to a placebo group or to E6011 100‐mg, 200‐mg, or 400/200‐mg groups at a 2:1:2:2 ratio. During the 24‐week period, patients received the study drug subcutaneously at weeks 0, 1, and 2 and then once every 2 weeks. The primary end point was the American College of Rheumatology 20% improvement criteria (ACR20) response rate at week 12.
Results
Study drugs were administered to 190 patients (placebo, n = 54; E6011 100 mg, n = 28; E6011 200 mg, n = 54; E6011 400/200 mg, n = 54), and 169 patients completed treatment. A significant difference from placebo was not found in ACR20 response rates at week 12 (37.0% [placebo], 39.3% [100 mg], 48.1% [200 mg], and 46.3% [400/200 mg], using nonresponder imputation). As a secondary end point, ACR20 response rate in the 200‐mg and 400/200‐mg groups attained statistical significance at week 24 (35.2% [placebo], 39.3% [100 mg], 53.7% [200 mg], and 57.4% [400/200 mg]). Subsequent exploratory subgroup analysis revealed greater efficacy of E6011, particularly in patients with a higher baseline proportion of CD16+ monocytes; ACR20 response rates in this patient subgroup at week 24 were 30.0% (placebo), 46.7% (100 mg), 57.7% (200 mg), and 69.6% (400/200 mg). E6011 administered for 24 weeks was well tolerated.
Conclusion
This is the first evidence that E6011, a novel cell trafficking inhibitor targeting the FKN–CX3CR1 interaction, is modestly effective with 24 weeks of treatment in RA patients, although the primary end point was not met.
INTRODUCTION
Rheumatoid arthritis (RA) is an autoimmune disease characterized by persistent synovitis and systemic inflammation, ultimately resulting in joint damage, disability, decreased quality of life, and other comorbidities, when insufficiently treated. Disease‐modifying antirheumatic drugs (DMARDs) are key therapeutic agents. They include conventional synthetic DMARDs, of which methotrexate (MTX) is the anchor drug, as well as biologic and targeted synthetic DMARDs targeting tumor necrosis factor (TNF), interleukin‐6 (IL‐6) receptor, T cell costimulation, B cells (CD20), and JAKs. Recent guidelines for the management of RA recommend rapid attainment of sustained remission or low disease activity in each patient (1, 2, 3, 4). However, ~50–70% of patients fail to achieve remission or maintain low disease activity, even when they initially respond well to current therapies (5, 6).
Fractalkine (FKN) is a membrane‐bound CX3C chemokine that possesses a chemokine/mucin hybrid structure and transmembrane domain (7, 8). FKN is expressed on vascular endothelial cells (ECs), and its unique structure gives it 2 functional forms: an adhesion molecule when present in its membrane‐bound form and a chemoattractant in its soluble form after shedding by metalloproteases (9). Expression of FKN is up‐regulated on vascular ECs at inflamed lesions, such as RA synovia (10, 11). Notably, both forms of FKN are recognized by its receptor, CX3CR1, which is expressed on monocyte/macrophages and cytotoxic effector lymphocytes, including natural killer cells and cytotoxic T cells (7, 12, 13). Among monocytes, CX3CR1 is highly expressed on CD16+ monocytes, which are known to be increased in RA (14, 15). CD16+ monocytes adhere to vascular endothelium via FKN–CX3CR1 interactions, where they produce large amounts of proinflammatory cytokines (e.g., TNF and IL‐6) and chemokines that recruit other types of immune cells to areas where CD16+ monocytes are located (16, 17). This results in augmented inflammatory reactions in affected synovia (18). CX3CR1 is also expressed on terminally differentiated CD4+ and CD8+ T cells, which are increased in the peripheral blood of RA patients. These cells preferentially produce interferon‐γ, TNF, granzyme A, and perforin (11), ultimately contributing to tissue damage, which is indicative of the role the FKN–CX3CR1 axis plays in RA pathophysiology.
Previously, we investigated the safety and efficacy of E6011, a humanized IgG2 monoclonal antibody against human FKN, in a phase I/II, open‐label, multiple‐ascending‐dose study in patients with active RA. E6011 was found to be safe and showed efficacy signals in these patients (19). We further evaluated E6011 in a phase II, double‐blind, placebo‐controlled study to confirm its efficacy, safety, and dose‐response relationship in patients with moderately to severely active RA who had an inadequate response to MTX. Herein, we present the results of the 24‐week treatment phase (double‐blind portion) of this clinical trial. This is the first study to reveal the clinical benefit of blocking the FKN–CX3CR1 axis for treatment of RA.
PATIENTS AND METHODS
Study design
This multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group comparison study was performed to evaluate the efficacy and safety of 3 dosages of E6011, compared with placebo, in RA patients in Japan (ClinicalTrials.gov identifier: NCT02960438). The following 4 treatment groups were selected for the study: E6011 100‐mg group, E6011 200‐mg group, E6011 400/200‐mg group, and placebo group. The study consisted of screening, observation, treatment (double‐blind), extension (open‐label), and follow‐up phases. Screening assessments were performed within 42 days prior to treatment initiation. The protocol was approved by the institutional review board of each study institution. This study was conducted in accordance with the standard operating procedures of the sponsor, which were designed to ensure adherence with the Declaration of Helsinki and Good Clinical Practice.
Patients
Japanese patients with active RA (ages 18–74 years) who were diagnosed according to the 1987 American College of Rheumatology (ACR) classification criteria (20) or the 2010 ACR/European League Against Rheumatism criteria (21) were screened for eligibility. Inclusion criteria included the following: tender joint count of ≥6 (of 68 joints), swollen joint count of ≥6 (of 66 joints), and C‐reactive protein (CRP) level of ≥0.6 mg/dl, or erythrocyte sedimentation rate of ≥28 mm/hour after receiving MTX (6–16 mg/week) for ≥12 weeks before trial entry. Patients were excluded if they had previously been treated with biologics and discontinued treatment due to inadequate response or had a history of biologic treatment for RA within 12 weeks prior to the study. All participants provided written informed consent before participation.
Randomization and blinding
Patients who met the eligibility criteria during the screening and observation phase were randomly allocated to the placebo, 100‐mg, 200‐mg, and 400/200‐mg groups at a 2:1:2:2 ratio. This dynamic allocation (minimization method) was performed using the following factors: CRP level at the screening phase, disease duration, and history of biologic treatment. Randomization was performed centrally using an interactive web response system (IWRS). The individual responsible for randomization generated the list of randomized drug numbers. At screening, the investigator or designee accessed the IWRS to register patient information. The independent enrollment center confirmed the eligibility of the patient, assigned each patient to a treatment group using a dynamic allocation algorithm, and provided the drug number to the investigator via email. Upon completing all planned assessments at the observation phase, the investigator prescribed the study drug for the eligible patient based on the drug number specified by the independent enrollment center. Therefore, in a double‐blind manner, patients received either E6011 or placebo at weeks 0, 1, and 2, and then once every 2 weeks until week 22.
Procedures
During the treatment phase (24 weeks), patients were subcutaneously injected with either E6011 or placebo at weeks 0, 1, and 2, and then once every 2 weeks until week 22 in a double‐blind manner. In the E6011 100‐mg, E6011 200‐mg, and placebo groups, patients received the study drug (100 mg, 200 mg, or placebo, respectively) at weeks 0, 1, and 2, and then once every 2 weeks. In the E6011 400/200‐mg group, patients received 400 mg at weeks 0, 1, 2, 4, 6, 8, and 10 and then 200 mg once every 2 weeks. In our previous phase I/II study (19), 400 mg of E6011 sufficiently improved clinical symptoms. However, in the present study, administration of the 400‐mg dose required subcutaneous injection at 4 sites with 100 mg/ml of study drug. Therefore, 400‐mg administrations were limited to 10 weeks (for the primary end point) to reduce the burden on patients, and from week 12, patients received 200‐mg subcutaneous administrations (injection at 2 sites).
Patients who completed evaluations at week 24 of the treatment phase entered the extension phase. The extension phase lasted until 104 weeks after the start of study treatment, and patients received open‐label E6011 200 mg every 2 weeks until week 102. If patients completed or discontinued the study, a follow‐up visit was conducted 28 days after completion or discontinuation of the study, and a follow‐up visit or telephone interview was conducted 70 days after the last dosing. Here, we present the results of the 24‐week treatment phase (double‐blind portion) of this clinical trial.
Assessments
Efficacy
The primary end point was ACR 20% improvement criteria (ACR20) response rate at week 12. Major secondary end points were rates of ACR20 response at week 24, rates of ACR50 and ACR70 responses at weeks 12 and 24, and improvements in individual ACR components (number of tender joints, number of swollen joints, patient’s and physician’s global assessments, Health Assessment Questionnaire (22), and CRP level) over 24 weeks. Other secondary end points included change in Disease Activity Score in 28 joints using the CRP level (DAS28‐CRP) (23) and the Clinical Disease Activity Index (CDAI) (24) over 24 weeks.
Biomarker
Peripheral blood samples were used to measure CD16+ monocytes at baseline and at weeks 2, 4, 8, 12, and 24. Whole blood was lysed with BD Pharm Lyse (BD Biosciences) and then incubated with Fc Receptor Blocking Reagent (Miltenyi Biotec). Blocked samples were incubated with Alexa Fluor 647–conjugated anti‐human CD14 (BioLegend) and fluorescein isothiocyanate–conjugated anti‐human CD16 (Abcam) for 30 minutes on ice and analyzed using a FACSCanto II apparatus (BD Biosciences). The percentage of CD16+ cells in total monocytes was calculated with a sequential gating strategy using FlowJo (BD Biosciences).
Safety
Safety was evaluated based on adverse events (AEs), clinical laboratory parameters, vital signs, standard 12‐lead electrocardiogram (ECG) results, chest radiographs, neurologic findings, and CD4+ blood cell counts. AEs were coded using the Medical Dictionary for Regulatory Activities, version 20.1. Severity of AEs was graded on a 5‐point scale according to the Common Terminology Criteria for Adverse Events (CTCAE; version 4.0).
Statistical analysis
The primary end point was analyzed using a logistic regression model with CRP level at baseline, RA disease duration, and history of treatment with biologics as covariates for comparison between the placebo group and either the E6011 200‐mg or E6011 400/200‐mg group. The overall significance level was defined as α = 0.025 (1‐sided). The Benjamini‐Hochberg method was used to control the overall Type I error rate.
Sample size was conservatively calculated at a 1‐sided significance level of α = 0.0125 (α = 0.025/2) considering multiplicity. The ACR20 response rate at week 12 was expected to be 30% in the placebo group and ≥60% in both the E6011 200‐mg and 400/200‐mg groups. Sample sizes of 50 for the 200‐mg, 400/200‐mg, and placebo groups had 91% power to detect a difference in response rate of 35% between the placebo group and each E6011 group and 79% power to detect a difference in response rate of 30% based on a chi‐square test.
Multiplicity adjustment was not considered for secondary efficacy analyses. For ACR20 (excluding week 12), ACR50, and ACR70 response rates, analyses similar to those for the primary end point were conducted. Each component of the ACR response criteria, DAS28‐CRP, and CDAI, and any changes from baseline, were summarized at each visit, according to treatment group. Changes from baseline were also analyzed using analysis of covariance with baseline value, CRP level at baseline, RA disease duration, and prior biologic treatment as covariates. The significance level for comparisons between the placebo group and each E6011 treatment group (100‐mg, 200‐mg, or 400/200‐mg) was defined as α = 0.05 (2‐sided). The ACR20 and ACR50 response rates at week 24 were also analyzed in subgroups according to the baseline proportion of CD16+ monocytes.
The efficacy analysis set was the group of randomized patients who received the study drug and had ≥1 evaluable postdose primary efficacy data set available. The safety analysis set was the group of patients who received ≥1 dose of the study drug and had ≥1 evaluable postdose safety data set available. For efficacy analyses, the approach used to handle missing data for the ACR response criteria was the nonresponder imputation (NRI) method, and for continuous variables, the last observation carried forward method was used. For safety analysis, AEs that emerged during the 24‐week treatment phase were evaluated.
RESULTS
Patient disposition and baseline characteristics
Between November 10, 2016 and September 7, 2017, 194 patients were randomly allocated to each treatment group. After randomization, 4 of 194 patients discontinued the study before starting treatment with the study drug, because they failed to meet entry criteria. All treated patients (n = 190) were included in the efficacy and safety analyses. Of the 190 patients who received ≥1 dose of the study drug (placebo, n = 54; E6011 100 mg, n = 28; E6011 200 mg, n = 54; 400/200 mg, n = 54), 169 completed the planned treatment regimen, while 21 prematurely discontinued treatment within the 24‐week double‐blind period. All patients were included in each analysis. The number of patients who discontinued treatment was similar between the placebo and E6011 treatment groups (Figure 1).
Figure 1.
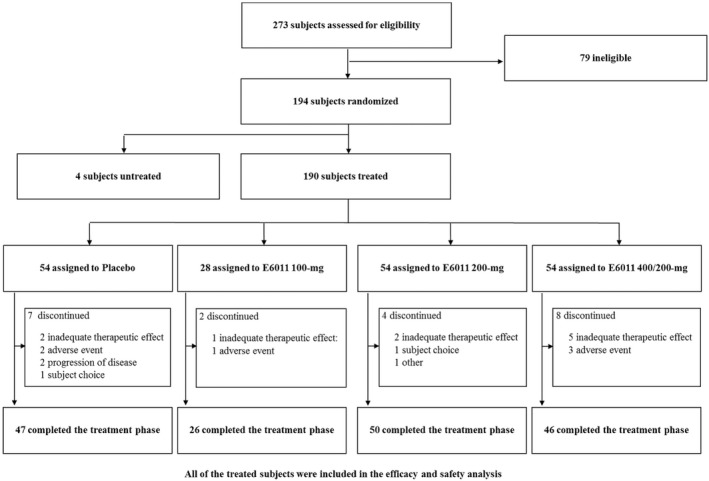
Patient disposition during the 24‐week double‐blind period.
Baseline demographic characteristics were similar among the 4 treatment groups (Table 1). Most patients (78.9%) were female, and the median age was 56.0 years. The mean ± disease duration was 7.1 ± 6.85 years. Approximately 23% of patients (43 of 190) had previously received biologic. The mean ± SD dose of MTX was 9.9 ± 2.84 mg/week, the mean ± SD baseline CRP level was 1.30 ± 1.49 mg/dl, and the mean ± SD baseline tender joint count (of 68 joints) and swollen joint count (of 66 joints) were 15.3 ± 7.90 and 12.6 ± 5.98, respectively. The proportion of oral glucocorticoid use in the E6011 400/200‐mg treatment group was numerically higher, although the difference was not significant, compared with the other groups. Mean baseline scores for clinical measures were comparable across treatment groups.
Table 1.
Patient baseline demographics and laboratory data*
|
Placebo group (n = 54) |
E6011 100‐mg group (n = 28) |
E6011 200‐mg group (n = 54) |
E6011 400/200‐mg group (n = 54) |
|
|---|---|---|---|---|
| Age, years | 57.6 ± 9.86 | 56.5 ± 10.4 | 56.5 ± 10.4 | 55.2 ± 9.13 |
| Sex, no. (%) | ||||
| Male | 9 (16.7) | 6 (21.4) | 10 (18.5) | 15 (27.8) |
| Female | 45 (83.3) | 22 (78.6) | 44 (81.5) | 39 (72.2) |
| Weight, kg | 54.7 ± 11.3 | 55.4 ± 11.0 | 57.9 ± 14.4 | 55.7 ± 11.2 |
| RA duration, years | 6.9 ± 7.35 | 6.4 ± 5.46 | 7.1 ± 6.58 | 7.6 ± 7.38 |
| Prior biologic use, no. (%) | 12 (22.2) | 7 (25.0) | 12 (22.2) | 12 (22.2) |
| MTX dose, mg/week | 9.6 ± 2.20 | 9.9 ± 3.22 | 10.1 ± 2.97 | 10.1 ± 3.10 |
| Oral glucocorticoids | ||||
| Yes, no. (%) | 23 (42.6) | 11 (39.3) | 24 (44.4) | 31 (57.4) |
| Dose, mg/day† | 3.65 ± 2.17 | 4.82 ± 2.33 | 4.15 ± 2.34 | 3.54 ± 2.13 |
| RF‐positive, no. (%)‡ | 45 (83.3) | 23 (82.1) | 46 (85.2) | 44 (81.5) |
| Anti‐CCP–positive, no. (%)§ | 51 (94.4) | 26 (92.9) | 46 (85.2) | 45 (83.3) |
| TJC (of 68 joints) | 13.7 ± 6.81 | 14.1 ± 7.24 | 16.3 ± 7.15 | 16.6 ± 9.61 |
| SJC (of 66 joints) | 12.7 ± 6.81 | 11.3 ± 5.27 | 12.4 ± 4.89 | 13.5 ± 6.41 |
| CRP at screening, mg/dl | 1.25 ± 1.04 | 1.38 ± 1.60 | 1.60 ± 3.33 | 1.34 ± 1.53 |
| CRP at baseline, mg/dl | 1.50 ± 1.63 | 1.44 ± 1.87 | 1.08 ± 0.84 | 1.24 ± 1.63 |
| DAS28‐CRP | 5.04 ± 0.88 | 4.99 ± 1.01 | 5.08 ± 0.73 | 5.20 ± 0.93 |
Except where indicated otherwise, values are the mean ± SD. RA = rheumatoid arthritis; MTX = methotrexate; RF = rheumatoid factor; anti‐CCP = anti–cyclic citrullinated peptide; TJC = tender joint count; SJC = swollen joint count; DAS28‐CRP = Disease Activity Score in 28 joints using C‐reactive protein.
Concomitant dose at baseline is shown in prednisolone equivalent.
Positivity defined as >15 IU/ml.
Positivity defined as ≥4.5 units/ml.
ACR20 response rates at week 12 (using NRI) were 37.0% (placebo), 39.3% (100 mg), 48.1% (200 mg), and 46.3% (400/200 mg). Although the rates were higher in the 200‐mg and 400/200‐mg groups compared with the placebo group, statistical significance was not reached (P = 0.188 for the 200‐mg and 400/200‐mg groups, using the logistic regression model with the Benjamini‐Hochberg method) (Figure 2A). Therefore, although this study did not meet the primary end point, it met multiple secondary end points. At week 24, ACR20 response rates were 35.2% (placebo), 39.3% (100 mg), 53.7% (200 mg), and 57.4% (400/200 mg), and the response rates in the E6011 200‐mg and 400/200‐mg groups were significantly higher than in the placebo group (P = 0.023 for the 200‐mg group and P = 0.010 for the 400/200‐mg group, using the logistic regression model with the Benjamini‐Hochberg method) (Figure 2B). ACR50 response rates at weeks 12 and 24 (using NRI) were 14.8% and 16.7% (placebo), 10.7% and 17.9% (100 mg), 25.9% and 25.9% (200 mg), and 18.5% and 27.8% (400/200 mg), respectively. ACR70 response rates at weeks 12 and 24 (using NRI) were 3.7% and 5.6% (placebo), 3.6% and 14.3% (100 mg), 9.3% and 11.1% (200 mg), and 7.4% and 13.0% (400/200 mg), respectively (Figures 2A and B).
Figure 2.
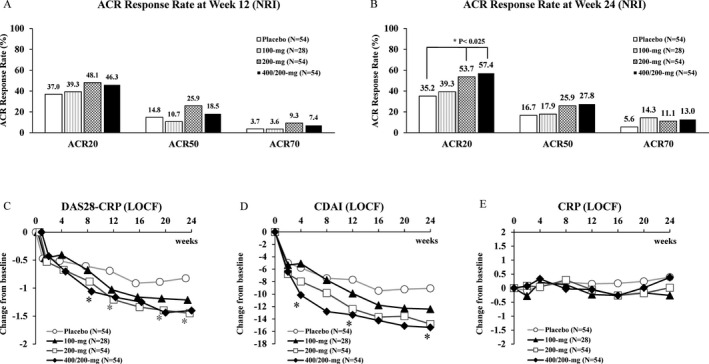
A and B, American College of Rheumatology 20% improvement criteria (ACR20), ACR50, and ACR70 response rate at weeks 12 (A) and 24 (B) (using nonresponder imputation [NRI]). C–E, Mean change from baseline in the Disease Activity Score in 28 joints using the C‐reactive protein level (DAS28‐CRP) (C), the Clinical Disease Activity Index (CDAI) (D), and the CRP level (E), according to E6011 dose (using the last observation carried forward [LOCF] approach). * = P < 0.05, versus placebo, in C and D.
The DAS28‐CRP and CDAI decreased sequentially from baseline after treatment with E6011. Decreases in the DAS28‐CRP and CDAI were statistically significant between the placebo group and the 200‐mg or 400/200‐mg group as early as week 8 (for DAS28‐CRP; Figure 2C) or week 4 (for CDAI; Figure 2D). In contrast, any apparent reduction in CRP level was not observed to be associated with E6011 treatment within the double‐blind period (Figure 2E).
Levels of CD16+ monocytes, which highly express CX3CR1, in whole monocytes were sequentially measured. Baseline levels ranged broadly from 1.60% to 42.1%, and the median value derived from all patients was 10.35% (data not shown). Median values in each group at baseline were comparable (9.43% [placebo], 12.80% [100 mg], 11.15% [200 mg], and 10.80% [400/200 mg]). For further exploratory analyses, patients were divided into 2 groups by taking the baseline median yielded from all patients (10.35%) and applying it to CD16+ monocyte–low and CD16+ monocyte–high populations. In the population with low CD16+ monocytes, there was no trend in terms of ACR20 response at week 24 (43.3% [placebo], 20.0% [100 mg], 54.5% [200 mg], and 45.5% [400/200 mg]) (Figure 3). The response in the CD16+ monocyte–high population showed a marked dose‐dependent increase in ACR20 response rate (30.0% [placebo], 46.7% [100 mg], 57.7% [200 mg], and 69.6% [400/200 mg]). These results were also confirmed in the ACR50 responses at week 24 (20.0% [placebo], 10.0% [100 mg], 13.6% [200 mg], and 13.6% [400/200 mg] in the CD16 monocyte–low population, and 15.0% [placebo], 26.7% [100 mg], 34.6% [200 mg], and 39.1% [400/200 mg] in the CD16+ monocyte–high population) (Figure 3).
Figure 3.
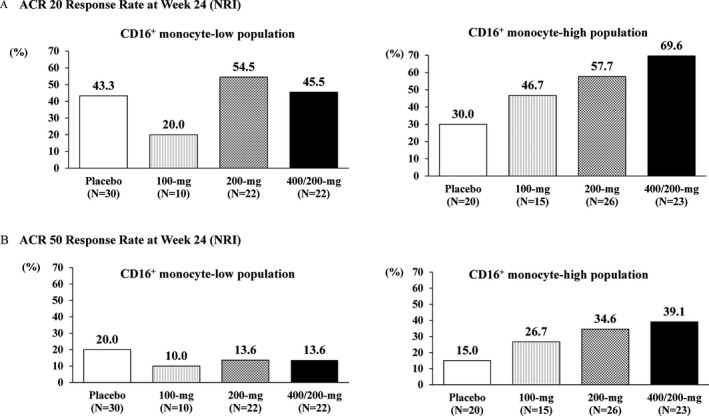
Subgroup analysis by baseline proportion of CD16+ monocytes. ACR20 response rates (A) and ACR50 response rates (B) at week 24 in populations with a low or high proportion of CD16+ monocytes are shown. See Figure 2 for definitions.
After initiation of treatment, CD16+ monocyte levels in total monocytes decreased significantly at week 2 in all E6011 groups, and reductions were sustained throughout the treatment period without dose dependency (Figure 4). AEs and treatment‐related AEs occurred more frequently in the E6011 treatment groups than in the placebo group (AEs, 63.0% in the placebo group and 73.5% in the E6011 groups; treatment‐related AEs, 22.2% in the placebo group and 39.7% in the E6011 groups) (Table 2). A dose response was found in the incidence of AEs (63.0% [placebo], 67.9% [100 mg], 70.4% [200 mg], and 79.6% [400/200 mg]), but not in the incidence of treatment‐related AEs (22.2% [placebo], 46.4% [100 mg], 33.3% [200 mg], and 42.6% [400/200 mg]).
Figure 4.
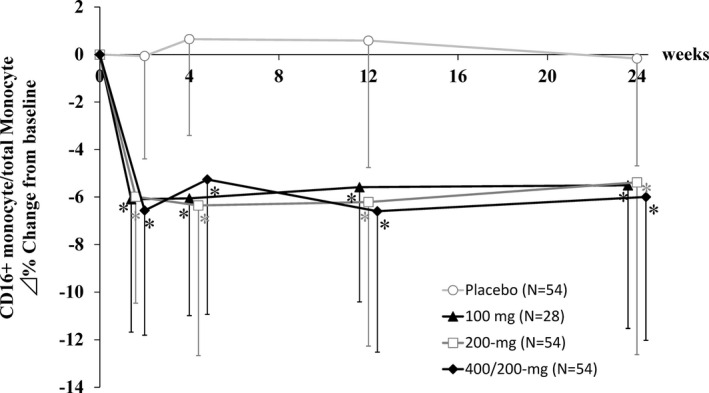
Changes in the proportion of CD16+ monocytes after E6011 treatment. Symbols and lines show the mean ± SD. * = P < 0.001 versus placebo.
Table 2.
AEs and laboratory data*
|
Placebo group (n = 54) |
E6011 100‐mg group (n = 28) |
E6011 200‐mg group (n = 54) |
E6011 400/200‐mg group (n = 54) |
E6011 total (n = 136) |
|
|---|---|---|---|---|---|
| All AEs | 34 (63.0) | 19 (67.9) | 38 (70.4) | 43 (79.6) | 100 (73.5) |
| Treatment‐related AEs | 12 (22.2) | 13 (46.4) | 18 (33.3) | 23 (42.6) | 54 (39.7) |
| AE maximum grade | |||||
| Grade 1 | 7 (13.0) | 4 (14.3) | 10 (18.5) | 9 (16.7) | 23 (16.9) |
| Grade 2 | 25 (46.3) | 13 (46.4) | 25 (46.3) | 32 (59.3) | 70 (51.5) |
| Grade 3 | 2 (3.7) | 1 (3.6) | 3 (5.6) | 0 | 4 (2.9) |
| Grade 4 | 0 | 1 (3.6) | 0 | 2 (3.7) | 3 (2.2) |
| Grade 5 | 0 | 0 | 0 | 0 | 0 |
| Serious AEs | 2 (3.7) | 1 (3.6) | 2 (3.7) | 3 (5.6) | 6 (4.4) |
| Death | 0 | 0 | 0 | 0 | 0 |
| AEs leading to withdrawal | 2 (3.7) | 1 (3.6) | 0 | 2 (3.7) | 3 (2.2) |
| AEs that occurred in ≥5% of patients in any group | |||||
| Nasopharyngitis | 16 (29.6) | 7 (25.0) | 10 (18.5) | 18 (33.3) | 35 (25.7) |
| URI | 2 (3.7) | 2 (7.1) | 4 (7.4) | 2 (3.7) | 8 (5.9) |
| Stomatitis | 1 (1.9) | 0 | 2 (3.7) | 5 (9.3) | 7 (5.1) |
| Bronchitis | 1 (1.9) | 2 (7.1) | 1 (1.9) | 3 (5.6) | 6 (4.4) |
| Back pain | 1 (1.9) | 1 (3.6) | 3 (5.6) | 2 (3.7) | 6 (4.4) |
| Pharyngitis | 2 (3.7) | 0 | 3 (5.6) | 2 (3.7) | 5 (3.7) |
| Dental caries | 0 | 0 | 0 | 3 (5.6) | 3 (2.2) |
| Headache | 3 (5.6) | 0 | 0 | 1 (1.9) | 1 (0.7) |
| Laboratory data | |||||
| Hemoglobin, gm/liter | −2.0 ± 9.7 | −1.6 ± 7.2 | 0.5 ± 10.3 | −3.3 ± 10.4 | −1.4 ± 9.9 |
| Lymphocytes, 109/liter | −0.05 ± 0.61 | 0.08 ± 0.38 | −0.01 ± 0.34 | −0.06 ± 0.57 | −0.01 ± 0.45 |
| Neutrophils, 109/liter | −0.09 ± 2.02 | −0.40 ± 1.42 | −0.34 ± 1.49 | −0.39 ± 2.22 | −0.37 ± 1.79 |
| ALT, units/liter | 0.6 ± 12.0 | 3.9 ± 26.5 | 3.4 ± 12.8 | 0.3 ± 14.1 | 2.3 ± 16.9 |
| Creatinine, μmoles/liter | 0.7 ± 5.7 | 1.9 ± 6.1 | 0.7 ± 5.2 | 2.0 ± 21.3 | 1.4 ± 14.0 |
| HDL cholesterol, mmoles/liter | −0.04 ± 0.28 | 0.05 ± 0.28 | 0.02 ± 0.24 | 0.05 ± 0.28 | 0.04 ± 0.26 |
| LDL cholesterol, mmoles/liter | 0.01 ± 0.44 | −0.05 ± 0.40 | −0.03 ± 0.55 | 0.03 ± 0.44 | −0.01 ± 0.48 |
| Creatine kinase, IU/liter | 5.4 ± 25.3 | 7.2 ± 26.4 | 2.1 ± 34.8 | 18.4 ± 74.0 | 9.6 ± 53.1 |
Adverse event (AE) values are the number (%) of patients. Laboratory data are the mean ± SD change from baseline at week 24 (using last observation carried forward). AEs emerged until week 24. However, for patients who discontinued the study drug during the treatment phase, AEs emerged until 70 days after the last intended dose. A patient with ≥2 AEs in the same preferred term was counted only once for that preferred term. MedDRA version 20.1 was used. URI = upper respiratory tract infection; ALT = alanine aminotransferase; HDL = high‐density lipoprotein; LDL = low‐density lipoprotein.
The incidence of grade 3 CTCAEs and grade 4 AEs and serious AEs that led to treatment discontinuation or dose interruptions was similar between the placebo group and E6011 treatment groups, and no apparent dose response was observed. AEs that occurred in ≥5% of patients in any E6011 treatment group included nasopharyngitis, upper respiratory tract infection, stomatitis, bronchitis, back pain, pharyngitis, and dental caries. Among the AEs that occurred in ≥5% of patients in any E6011 group, the following AEs occurred at a rate of ≥2‐fold that observed in patients in the placebo group: stomatitis (1.9% in the placebo group versus 5.1% in E6011 groups), bronchitis (1.9% versus 4.4%), back pain (1.9% versus 4.4%), and dental caries (0% versus 2.2%). No clinically meaningful changes were observed in laboratory data or other safety assessments, such as standard 12‐lead ECG results, chest radiographs, neurologic findings, or CD4+ blood cell counts.
DISCUSSION
E6011 is a novel investigational drug used to neutralize FKN, which is highly expressed in inflamed lesions and suppresses immune cell accumulation at affected lesions. This is the first multicenter, randomized, double‐blind, placebo‐controlled study to evaluate the efficacy and safety of E6011 for up to 24 weeks in patients with active RA who had an inadequate response to MTX.
With E6011, there was a trend toward increasing ACR20 response rate at week 12, the primary end point, although this was not statistically significant (Figure 2A). Statistical significance was attained at week 24, which was the secondary end point, even though the response rate was lower than we expected and lower than that of existing marketed biologics for RA (Figure 2B). Regarding other secondary end points, ACR50 response rates were numerically higher in the E6011 groups than in the placebo group at week 24 (Figure 2B), although this was not statistically significant. CRP level was not reduced in correlation with symptomatic improvement during the 24 weeks (Figure 2E). The CDAI, which does not include CRP level, reached statistical significance earlier, at week 4, in the 400/200‐mg group (Figure 2D). Such discrepancy in the clinical measures used may support the notion of a distinct mode of action of E6011, which suppresses cell migration from the circulation and accumulation in inflamed tissue but does not directly neutralize cytokines. The ACR response rate that is widely utilized as an end point in clinical studies may not accommodate evaluation of the cell trafficking inhibitor, E6011. Further studies are required to investigate the end point that sufficiently reflects the local effect of E6011.
For biomarker analysis, we focused on CD16+ monocytes because of their importance in RA pathophysiology and high expression of the FKN receptor, CX3CR1. Although there was no relationship between background features of the disease and the proportion of CD16+ monocytes (data not shown), we conducted a subsequent exploratory analysis as to whether baseline levels of these cells were related to the response to E6011. As shown in Figure 3, CD16+ monocyte–high populations tended to respond better than CD16+ monocyte–low populations, while there were some variations in data on ACR20 response. While a more obvious dose‐response tendency for ACR50 response was observed in CD16+ monocyte–high populations, these results should be interpreted carefully, as this subgroup analysis was ad hoc and exploratory and was immature for providing statistically convincing data because of its small sample size. Although this subsequent exploratory analysis had limitations, it provided some indication that E6011 may represent a potential treatment option for RA patients, especially with a precision medicine approach considering the baseline proportion of CD16+ monocytes.
After initiation of E6011 treatment, CD16+ monocyte levels were found to decrease quickly by week 2, without any dose response (Figure 4). No relationship was found between the magnitude of CD16+ monocyte level reduction and clinical response (data not shown). The FKN–CX3CR1 interaction is known to elicit signals to promote the survival of human monocytes through activation of phosphoinositide 3‐kinase (25). It is believed that E6011 may inhibit monocyte survival, although FKN–CX3CR1 may not be a central or unique axis for monocyte survival or maintenance because of the lack of dose dependency observed. Decreased magnitude of CD16+ monocytes cannot pharmacodynamically reflect the dose of E6011 administered. Therefore, the importance of CD16+ monocytes in predicting response or as a pharmacodynamic biomarker for E6011 remains elusive and should be further explored.
Regarding the safety profile of E6011, the incidence of AEs showed modest dose dependency. Notably, nasopharyngitis, stomatitis, bronchitis, back pain, and dental caries occurred more frequently with higher doses of E6011, although these AEs were either mild or moderate (grade 1 or 2). Moreover, given that these AEs were the most common ones reported in other clinical trials, and that some of them represent potential adverse reactions to MTX, E6011 was found to generally be safe and well tolerated at any dose for 24 weeks. However, further accumulation of safety information is necessary through additional clinical studies.
Regarding laboratory data, an increased mean change in creatine kinase levels was observed in the 400/200‐mg group (18.4 IU/liter). This was due to included data on 1 patient in the 400/200‐mg group. Without this data point, the mean creatine kinase change from baseline in the 400/200‐mg group (9.0%) was consistent with that in other groups. Additionally, this patient’s high level of creatine kinase (240 IU/liter at 24 weeks) was subsequently improved to within normal range, despite continuation of the study drug.
Considering the mode of action of E6011, which primarily ameliorates local inflammation by regulating cell trafficking without direct suppression of the systemic inflammatory reaction (i.e., no change in CRP level), its safety profile may be preferable to that of other biologic agents. Anemic hemoglobin levels are generally expected to normalize with improvement in systemic inflammation, although E6011 did not confer an increase in hemoglobin level in this study. This may also suggest that E6011 exerts its biologic effect locally rather than systemically. In contrast, hemoglobin level increased slightly in responders to E6011, while it decreased in nonresponders (data not shown). However, such differences were not sufficient to affect the mean value in the cohort. It is therefore unlikely that E6011 reduced hemoglobin level in any of the patients. These results indicate that safety signals were similar among 100‐mg, 200‐mg, and 400‐mg doses of E6011. However, a longer and more detailed evaluation is required to fully establish the safety profile of E6011.
In conclusion, although the primary end point in this study was not met, our data suggest that E6011 may have modest efficacy for patients with active RA who had an inadequate response to MTX, especially if they showed a higher proportion of CD16+ monocytes at baseline. The effect of E6011 may primarily emerge at locally inflamed lesions rather than systemically, which may be due to its mode of action, and conventional measures for clinical evaluation may not be appropriate for evaluating this treatment. The proportion of CD16+ monocytes in peripheral blood at baseline may indicate which patients will respond well to E6011. Although further evidence is necessary, this may help determine who should be treated with E6011. Because only preliminary evidence was obtained in this study, and our work cannot be translated to real‐world clinical practice at present, further evaluation in future clinical trials is warranted to confirm the therapeutic benefit of E6011.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Mr. Tago had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Tanaka, Takeuchi, Yamanaka, Nanki, Umehara, Yasuda, Tago, Kitahara, Hojo, Kawano, Imai.
Acquisition of data
Kawakubo, Torii.
Analysis and interpretation of data
Tanaka, Takeuchi, Yamanaka, Nanki, Umehara, Yasuda, Tago, Kitahara, Kawakubo, Torii, Hojo, Kawano, Imai.
ROLE OF THE STUDY SPONSOR
This work was supported by funding from Eisai Company, Ltd. Eisai Company, Ltd. was involved in study design, data collection, analysis, and interpretation of data. The corresponding author had full access to all data and had responsibility for the decision to submit for publication. Publication of this article was not contingent upon approval by Eisai Company, Ltd.
Acknowledgments
The authors thank the study participants and all investigators. We thank Mark Abramovitz, PhD, and Richard Robins, PhD, from Edanz Group for editing a draft of the manuscript.
Clinicaltrials.gov identifier: NCT02960438.
Supported by Eisai Company, Ltd.
Dr. Tanaka has received consulting fees, speaking fees, and/or honoraria from Pfizer, Sanofi, Asahi‐kasei, GlaxoSmithKline, Mitsubishi‐Tanabe, Gilead, and Janssen (less than $10,000 each) and from Daiichi‐Sankyo, Eli Lilly, Novartis, YL Biologics, Bristol Myers Squibb, Eisai, Chugai, AbbVie, and Astellas (more than $10,000 each) and has received research grants from AbbVie, Mitsubishi‐Tanabe, Chugai, Asahi‐Kasei, Eisai, Takeda, and Daiichi‐Sankyo. Dr. Takeuchi has received consulting fees, speaking fees and/or honoraria from Daiichi‐Sankyo, Takeda, Ono Pharmaceutical, Gilead, Taiho Pharmaceutical, Taisho Pharmaceutical, A2 Healthcare Corporation, GlaxoSmithKline, Eli Lilly, Boehringer Ingelheim, Novartis, AYUMI, Pfizer, and Sanofi (less than $10,000 each) and from AbbVie, Asahi‐Kasei, Astellas, Eisai, JCR, Mitsubishi‐Tanabe, Chugai, Nippon Kayaku, UCB, and Bristol Myers Squibb (more than $10,000 each). Dr. Yamanaka has received consulting fees, speaking fees, and/or honoraria from AbbVie, Astellas, AYUMI, Bristol Myers Squibb, Chugai, Daiichi‐Sankyo, Eisai, Kaken, Mitsubishi‐Tanabe, Nippon Boehringer Ingelheim, Nippon Shinyaku, Novartis, Ono Pharmaceutical, Pfizer, Taisho Pharmaceutical, Takeda, Torii, and UCB (less than $10,000 each) and from Teijin Company and YL Biologics (more than $10,000 each). Dr. Nanki has received consulting fees, speaking fees, and/or honoraria from Daiichi Sankyo, Teijin, Takeda, Shionogi, UCB, Sanofi, Nippon Boehringer Ingelheim, Pfizer, Nihon Pharmaceutical, Yutoku Pharmaceutical, Janssen, Gilead, and Taiho Pharmaceutical (less than $10,000 each) and from Mitsubishi‐Tanabe, Chugai, Eisai, Asahi‐Kasei, Bristol Myers Squibb, Ono Pharmaceutical, AYUMI, Bayer Yakuhin, AbbVie, Eli Lilly, Novartis, Astellas, and Nippon Kayaku (more than $10,000 each). Dr. Umehara has received consulting fees from Eisai (less than $10,000). Drs. Yasuda, Kawano, and Imai own stock or stock options in KAN Research Institute, Inc. Mr. Tago, Dr. Kitahara, and Messers. Kawakubo, Torii, and Hojo own stock or stock options in Eisai.
References
- 1. Smolen JS, Landewé R, Bijlsma J, Burmester G, Chatzidionysiou K, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease‐modifying antirheumatic drugs: 2016 update. Ann Rheum Dis 2017;76:960–77. [DOI] [PubMed] [Google Scholar]
- 2. Smolen JS, Breedveld FC, Burmester GR, Bykerk V, Dougados M, Emery P, et al. Treating rheumatoid arthritis to target: 2014 update of the recommendations of an international task force. Ann Rheum Dis 2016;75:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tanaka Y. Intensive treatment and treatment holiday of TNF‐inhibitors in rheumatoid arthritis. Curr Opin Rheumatol 2012;24:319–26. [DOI] [PubMed] [Google Scholar]
- 4. Tanaka Y. Current concepts in the management of rheumatoid arthritis. Korean J Intern Med 2016;31:210–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Iannone F, Sinigaglia L, Favalli EG, Sarzi‐Puttini P, Atzeni F, Caporal R, et al. Drug survival of adalimumab in patients with rheumatoid arthritis over 10 years in the real‐world settings: high rate remission together with normal function ability. Clin Rheumatol 2016;35:2649–56. [DOI] [PubMed] [Google Scholar]
- 6. Iannone F, Ferraccioli G, Sinigaglia L, Favalli EG, Sarzi‐Puttini P, Atzeni F, et al. Real‐world experience of tocilizumab in rheumatoid arthritis: sub‐analysis of data from the Italian biologics’ register GISEA. Clin Rheumatol 2018;37:315–21. [DOI] [PubMed] [Google Scholar]
- 7. Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 1997;91:521–30. [DOI] [PubMed] [Google Scholar]
- 8. Yoshie O, Imai T, Nomiyama H. Chemokines in immunity. Adv Immunol 2001;78:57–110. [DOI] [PubMed] [Google Scholar]
- 9. Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, et al. The disintegrin‐like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1‐mediated cell‐cell adhesion. Blood 2003;102:1186–95. [DOI] [PubMed] [Google Scholar]
- 10. Sawai H, Park YW, Roberson J, Imai T, Goronzy JJ, Weyand CM. T cell costimulation by fractalkine‐expressing synoviocytes in rheumatoid arthritis. Arthritis Rheum 2005;52:1392–401. [DOI] [PubMed] [Google Scholar]
- 11. Nanki T, Imai T, Nagasaka K, Urasaki Y, Nonomura Y, Taniguchi K, et al. Migration of CX3CR1‐positive T cells producing type 1 cytokines and cytotoxic molecules into the synovium of patients with rheumatoid arthritis. Arthritis Rheum 2002;46:2878–83. [DOI] [PubMed] [Google Scholar]
- 12. Nishimura M, Umehara H, Nakayama T, Yoneda O, Hieshima K, Kakizaki M, et al. Dual functions of fractalkine/CX3C ligand 1 in trafficking of perforin+/granzyme B+ cytotoxic effector lymphocytes that are defined by CX3CR1 expression. J Immunol 2002;168:6173–80. [DOI] [PubMed] [Google Scholar]
- 13. Fong AM, Robinson LA, Steeber DA, Tedder TF, Yoshie O, Imai T, et al. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J Exp Med 1998;188:1413–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tsukamoto M, Seta N, Yoshimoto K, Suzuki K, Yamaoka K, Takeuchi T. CD14brightCD16+ intermediate monocytes are induced by interleukin‐10 and positively correlate with disease activity in rheumatoid arthritis. Arthritis Res Ther 2017;19:1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kawanaka N, Yamamura M, Aita T, Morita Y, Okamoto A, Kawashima M, et al. CD14+, CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheum 2002;46:2578–86. [DOI] [PubMed] [Google Scholar]
- 16. Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity 2010;33:375–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay‐Barrena G, et al. Nr4a1‐dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell 2013;153:362–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yano R, Yamamura M, Sunahori K, Takasugi K, Yamana J, Kawashima M, et al. Recruitment of CD16+ monocytes into synovial tissues is mediated by fractalkine and CX3CR1 in rheumatoid arthritis patients. Acta Med Okayama 2007;61:89–98. [DOI] [PubMed] [Google Scholar]
- 19. Tanaka Y, Takeuchi T, Umehara H, Nanki T, Yasuda N, Tago F, et al. Safety, pharmacokinetics, and efficacy of E6011, an anti‐fractalkine monoclonal antibody, in a first‐in‐patient phase 1/2 study on rheumatoid arthritis. Mod Rheumatol 2018;28:58–65. [DOI] [PubMed] [Google Scholar]
- 20. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315–24. [DOI] [PubMed] [Google Scholar]
- 21. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO III, et al. Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81. [DOI] [PubMed] [Google Scholar]
- 22. Fries JF, Spitz PW, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum 1980;23:137–45. [DOI] [PubMed] [Google Scholar]
- 23. Prevoo ML, van’t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty‐eight–joint counts: development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 1995;38:44–8. [DOI] [PubMed] [Google Scholar]
- 24. Aletaha D, Nell VP, Stamm T, Uffmann M, Pflugbeil S, Machold K, et al. Acute phase reactants add little to composite disease activity indices for rheumatoid arthritis: validation of a clinical activity score. Arthritis Res Ther 2005;7:R796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. White GE, McNeill E, Channon KM, Greaves DR. Fractalkine promotes human monocyte survival via a reduction in oxidative stress. Arterioscler Thromb Vasc Biol 2014;34:2554–62. [DOI] [PMC free article] [PubMed] [Google Scholar]