

Abstract
Amine transaminases (ATAs) are used to synthesize enantiomerically pure amines, which are building blocks for pharmaceuticals and agrochemicals. R‐selective ATAs belong to the fold type IV PLP‐dependent enzymes, and different sequence‐, structure‐ and substrate scope‐based features have been identified in the past decade. However, our knowledge is still restricted due to the limited number of characterized (R)‐ATAs, with additional bias towards fungal origin. We aimed to expand the toolbox of (R)‐ATAs and contribute to the understanding of this enzyme subfamily. We identified and characterized four new (R)‐ATAs. The ATA from Exophiala sideris contains a motif characteristic for d‐ATAs, which was previously believed to be a disqualifying factor for (R)‐ATA activity. The crystal structure of the ATA from Shinella is the first from a Gram‐negative bacterium. The ATAs from Pseudonocardia acaciae and Tetrasphaera japonica are the first characterized (R)‐ATAs with a shortened/missing N‐terminal helix. The active‐site charges vary significantly between the new and known ATAs, correlating with their diverging substrate scope.
Keywords: amine transaminases, chiral amines, fold type IV, PLP-dependent enzymes, transferases
Diversifying: The toolbox of R‐selective amine transaminases (ATAs) has been expanded by an ATA containing a motif characteristic for d‐ATAs, two ATAs with a shortened/missing N‐terminal helix and the first ATA with known structure from a Gram‐negative bacterium. Their active site charges vary significantly compared to known ATAs correlating with their diverging substrate scope.

Introduction
Chiral amines have an everlasting place as important building blocks in pharmaceuticals, fine chemicals and agrochemicals.[ 1 , 2 , 3 ] In some applications, such as active pharmaceutical ingredients, it is essential that they are enantiopure. Chiral amines can be synthesized with a range of different enzymes.[ 3 , 4 , 5 ] Transaminases (TAs) are currently the most important example. [6] TAs belong to the protein superfamily of pyridoxal 5’‐phosphate (PLP)‐dependent enzymes and reversibly transfer an amino group from a suitable donor to a carbonyl acceptor using PLP as co‐factor. According to the substrate they convert, TAs can be grouped into α‐TAs and ω‐TAs. Whereas α‐TAs only accept α‐amino and α‐keto acids, ω‐TAs have a broader substrate scope and also transfer amino groups to non‐α positions. [7] A subgroup of ω‐TAs are amine transaminases (ATAs). They accept substrates that entirely lack a carboxylate group.[ 3 , 7 ] The toolbox of ATAs provides solutions and a promising potential for the synthesis of different amine enantiomers. For S enantiomers, there is a growing number of ATAs available, most of them belonging to fold type I of the PLP dependent enzymes. [7] Fold type IV ATAs, which are used for the R enantiomers, are less investigated and therefore not as well understood as their S‐selective counterpart, which can hamper protein engineering efforts. Thus, it is important to broaden the knowledge on this enzyme class by identifying and characterizing new members, thereby also supporting protein engineering efforts to expand the toolbox of technically useful enzymes.
R‐selective ATAs are not the only members of fold type IV PLP dependent enzymes. 4‐Amino‐4‐deoxychorismate lyases (ADCLs), d‐amino acid aminotransferases (d‐ATAs) and l‐branched chain aminotransferases (BCATs) have a similar overall structure and therefore belong to the same family. Apart from their function, the different subfamilies can be distinguished by key motifs assigned by Höhne et al. [8] These motifs have driven the progress on the identification and characterization of new R‐selective ATAs and a handful of structures provided valuable knowledge about this fold type IV class.[ 9 , 10 , 11 , 12 , 13 ] Fold type IV ATAs (Figure 1) are usually homodimers. [14] The single monomers can be divided into different domains: the small domain with an α/β‐structure and the large domain with a pseudo‐barrel structure. The small domain can include a characteristic N‐terminal helix that is only found in (R)‐ATAs and is lacking in BCATs and d‐ATAs. [10] The small and the large domain are separated by an inter‐domain loop.[ 10 , 11 ]
Figure 1.
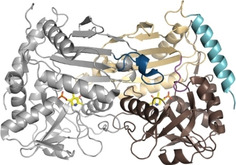
Structure of the R‐selective ATA from Aspergillus terreus (AT‐ωTA, PDB ID: 4CE5; [9] single monomer: gray, active‐site loop: dark blue, small domain: beige, large domain: brown, interdomain loop: purple, N‐terminal helix: turquoise, PLP: yellow).
The two active sites of (R)‐ATAs are located at the dimer interface and are formed by amino acids of both monomers. Each active site can accommodate ketones as well as amines and consists of the large (O‐pocket) and the small binding pocket (P‐pocket) and a PLP binding site. The large pocket harbors the bulky substituent of the ketone substrate or the α‐amine of the co‐substrate. The small binding pocket accommodates the smaller substituent, which is usually a methyl‐group.[ 8 , 9 , 10 , 11 , 12 , 15 , 16 , 17 ] While the small binding pocket is solely part of one monomer, the large binding pocket constitutes of amino acids from both monomers. The second monomer provides a flexible loop that reaches into the large binding pocket of the first monomer and is suggested to interact with the substrate carboxylate by forming a salt bridge with a guanidinium moiety, usually an arginine. This arginine is believed to be involved in the dual substrate recognition of (R)‐ATAs.[ 9 , 17 , 18 ] The co‐factor PLP is located in the PLP binding site and gets activated by the active site lysine. [9]
The structural characteristics of (R)‐ATAs are derived from a limited number of expressed and characterized (R)‐ATAs with a strong bias for (R)‐ATAs of fungal origin. We attempted to identify and characterize new (R)‐ATAs from diverse origins (fungal, Gram‐positive and Gram‐negative bacteria) with novel features to get more insight into fold type IV (R)‐ATAs and find new (R)‐ATAs with extended substrate scope and/or stability.
Results and Discussion
Multiple sequence alignment
In a first selection round, published (R)‐ATA sequences[ 9 , 10 , 11 , 16 , 17 , 19 ] were used to search for related uncharacterized sequences in the NCBI database (pBLAST). [20] The resulting sequences were combined in a multiple sequence alignment (MSA) and irrelevant sequences were removed using the genome mining approach described by Höhne et al. [8] The remaining sequences (495) were screened for potential interesting features. Here, we especially paid attention to putative (R)‐ATAs with underrepresented amino acids at the motif positions. We further had a look at the carboxylate trap motif which is characteristic for d‐ATAs and whose presence is believed to be a disqualifying factor for (R)‐ATA activity. [8] At last, we considered the length of the N‐terminal helix and active site loop.
The genome mining approach (Figure S1 in the Supporting Information, left workflow) by Höhne et al. [8] comprises a couple of steps leading to putative (R)‐ATAs (numbering according to Höhne et al. [8] ). First, an alignment of fold‐type IV PLP‐dependent enzymes is built, followed by the removal of unfunctional proteins which can be identified by long gaps or the missing catalytically important lysine. Then, putative BCATs are excluded by removing sequences with the 95‐YxR motif. Putative d‐ATAs are identified by the 97‐YxQ motif and ADCLs by K or R at position 97 and an RGY at position 107. Furthermore, the remaining sequences should not contain R, K, D, or E at position 95 and if Y is located at this position, R or K are not permitted at position 97. Furthermore, R or K are also not allowed at position 40. The last step is the removal of sequences containing RxH at position 107. All sequences left are putative (R)‐ATAs.
Often, the whole process can be shortened and simplified by looking for sequences containing the following two motifs (Figure S1, right workflow): 1) position 31 to 40: [HR]‐x(4)‐Y‐x‐[VT]‐x‐[HSAPT] and 2) position 95 to 97: [YF]‐V‐[EAQN]. These motifs contain the amino acids that are found most often in putative (R)‐ATAs. However, putative (R)‐ATAs with amino acids that are underrepresented at those positions would be missed. In our MSA (for sequence logo, see Figure S2), approximately 2 % of the sequences have K at position 31 (third most frequent after H and R), 3 % Q at position 36 (second most common amino acid after Y). Interestingly, all the sequences with Q36 are from Gram‐negative bacteria. Furthermore, at position 38 A is found in 1 % of the cases and 8 % of the sequences contain G at position 40. The first motif should therefore be extended to [HRK]‐x(4)‐[YQ]‐x‐[VTA]‐x‐[HSAPTG]. The extension of the second motif is not as straight forward since many amino acids are underrepresented. 1 % of the sequences in the MSA contains L, C, G, Q, or W at position 95. Similarly, 6 % of the sequences have A, C, I, or L at position 96 of which 3 % are I and 2.5 % are C. At position 97, 11 % of the sequences contain G, D, F, L, M, S, T, W, or C with 1 % D, 2 % M, 3 % S and 3 % W. To include most of the possible amino acids at motif 2, it could be extended to [YF]‐[VIC]‐[EAQSNWMD].
One of Höhne's criteria for excluding (R)‐ATA function is the RxH motif at position 107. Instead, RxH is an important characteristic of d‐ATAs. Together with Y36, these amino acids form the carboxylate trap and coordinate the α‐carboxyl group of a substrate in the O‐pocket thereby determining the pro‐d‐position of keto acids.[ 8 , 13 , 21 ] In (R)‐ATAs, the O‐pocket is responsible for the dual substrate recognition. It accommodates both, the α‐carboxylate of keto acids and the hydrophobic moiety of (R)‐amines. Here, only the arginine on the flexible active site loop is assumed to coordinate the carboxylate.
We identified 12 sequences in the MSA (2.4 %) that contain the (R)‐ATA motifs but also the RxH motif (Figure S3). Thus, the question arose if they still code for putative (R)‐ATAs. In this context, a recent publication from the Bornscheuer group became interesting. [21] They enabled (R)‐ATA activity in a d‐ATA from Bacillus subtilis. Initial (R)‐ATA activity was achieved in several variants with single amino acid exchanges. Subsequent variants with multiple exchanges showed improved (R)‐ATA activity while d‐ATA activity decreased. Interestingly, most of these variants still had the RxH motif intact indicating that the existence of this motif is not a disqualifying factor for (R)‐ATA activity. However, it seems that (R)‐ATA activity can be improved significantly if the motif is disrupted by exchanging the histidine residue. The exchange of H to L in their final variant resulted in doubled (R)‐ATA activity.
Another feature we looked at was the N‐terminal helix which can be found in (R)‐ATAs and is lacking in BCATs and d‐ATAs. Thomsen et al. postulated that this unique N‐terminal helix (first 20 amino acids in Aspergillus fumigatus) significantly affects protein stability and is crucial for the soluble expression of fungal (R)‐ATAs. A deletion of this helix led to the nonfunctional and insoluble expression of the (R)‐ATA from A. fumigatus. [10] However, bacterial (R)‐ATAs with low activity lacking the N‐terminal helix were functionally expressed when Höhne et al. [8] established the characteristic sequence motifs. Nevertheless, no (R)‐ATA lacking the N‐terminal helix has been properly characterized. In our MSA, it became clear that the length of the N terminus of fungal (R)‐ATAs was rather conserved which supports Thomsen's hypothesis. The only exception was the putative (R)‐ATA from the fungus Sphaerulina musiva. It lacks a substantial part of the N terminus including the helix and 18 additional amino acids (Figure S4). Bacterial (R)‐ATAs have an N‐terminal helix with varying length. In the MSA, the N terminus of some bacterial sequences have the same length as the fungal ones; others are longer or shorter, or completely missing.
The last feature is the length of the active site loop. Guan et al. compared the three crystal structures of the wild‐type (R)‐ATA from Arthrobacter sp. KNK168, its variant G136F, and the further evolved variant ATA‐117‐Rd11. [17] Based on this comparison and differences in activity, they proposed that the active site loop has a strong influence on the substrate scope and is therefore a promising target for protein engineering. [17] With regard to that, Iglesias et al. [22] identified insertions in the loop of five fungal (R)‐ATAs and chose the (R)‐ATA from Capronia semiimersa for expression and characterization. However, the correlation between insertion and substrate scope remained unclear. As opposed to Iglesias’ MSA, ours was not restricted to putative fungal (R)‐ATAs (Figure S5). In general, the loop length is rather conserved and usually comprises 16 or 17 amino acids. A couple of putative (R)‐ATAs, such as the (R)‐ATA from C. semiimersa, [22] contain two additional amino acids. We further found two sequences with three additional amino acids and the longest insertion comprised four additional amino acids. This putative (R)‐ATA was again from the fungus S. musiva.
Selection and expression of putative (R)‐ATAs
As pointed out before, we wanted to characterize (R)‐ATAs with potential novel features and from Gram‐positive and Gram‐negative bacteria in addition to the better characterized fungal enzymes to reduce the strong bias for fungal (R)‐ATAs. The final choice consisted of eight (R)‐ATAs, six bacterial and two fungal with 76–38 % sequence identity compared to AT‐ωTA and between themselves (Table S1). In the evolutionary tree except for Shinella the bacterial sequences are distant from the fungal sequences (Figure S6).
The fungal (R)‐ATAs originate from Exophiala sideris (ES‐ωTA) and S. musiva (SM‐ωTA). ES‐ωTA comprises the RxH motif. It was further interesting due to the habitat of its organism. E. sideris was isolated from environments polluted with toxic alkyl benzenes and arsenic. [23] These toxic compounds might have initiated evolution and enabled the acceptance of new substrates. SM‐ωTA was chosen for multiple reasons: the characteristic N‐terminal helix is missing, the second motif comprises under‐represented amino acids (Table 1), and the active site loop contains an insertion of four amino acids but lacks the arginine residue that was proposed to be important for dual substrate recognition.[ 9 , 17 , 18 ]
Table 1.
Conserved amino acids of fold type IV PLP‐dependent enzymes according to Höhne et al. [8] and corresponding amino acids of the selected new (R)‐ATAs (numbering of amino acid residues according to Höhne et al. [8] ). Shaded amino acids mark underrepresented amino acids of the motif, insertions in the region of the flexible loop and the RxH motif in ES‐ωTA. The arginine (red R) is supposedly the conserved arginine in the flexible loop that is proposed to play a role in dual substrate recognition.
|
Protein |
Sequence motif 1 |
Sequence motif 2 |
|||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
31 |
36 |
38 |
40 |
95 ff |
105 ff |
|
||||||||||||||
|
conserved amino acids |
ADCLs |
[FY] |
F |
T |
X |
zxK |
R |
G |
Y |
|
|
|
|
|
|
|
|
|
|
|
|
|
d‐ATAs |
F |
Y |
V |
K(R/X) |
zYzQ |
R |
x |
H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BCATs |
Y |
F |
G |
R(K) |
YzR |
z |
G |
z |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(R)‐ATAs |
[HR] |
Y |
[VT] |
[STAHP] |
[FY]V[EANQ] |
not conserved (active site loop) |
|||||||||||||||
|
amino acids found in novel putative (R)‐ATAs |
ES‐ωTA |
H |
Y |
V |
S |
FVE |
V |
R |
G |
– |
– |
– |
– |
H |
T |
P |
G |
E |
T |
F |
|
|
FF‐ωTA |
H |
Y |
V |
H |
YVE |
P |
G |
S |
– |
– |
– |
– |
– |
R |
D |
L |
R |
T |
C |
|
|
|
MeV‐ωTA |
R |
Y |
V |
G [a] |
YVA |
A |
G |
A |
P |
– |
– |
– |
– |
Y |
H |
P |
A |
H |
A |
|
|
|
PA‐ωTA |
R |
Y |
V |
A |
YVE |
H |
G |
S |
– |
– |
– |
– |
– |
R |
D |
P |
R |
T |
F |
|
|
|
RA‐ωTA |
R |
Y |
V |
H |
YVE |
Y |
G |
S |
– |
– |
– |
– |
– |
R |
D |
P |
R |
E |
C |
|
|
|
SH‐ωTA |
H |
Y |
V |
A |
YVM[ a] |
V |
R |
Q |
– |
– |
– |
– |
– |
Y |
A |
P |
E |
E |
C |
|
|
|
SM‐ωTA |
R |
Y |
V |
S |
YVC [a] |
V |
S |
N |
N |
P |
A |
I |
G |
K |
K |
A |
E |
E |
L |
|
|
|
TJ‐ωTA |
R |
Y |
V |
A |
YVE |
A |
G |
Q |
– |
– |
– |
– |
– |
R |
D |
P |
R |
L |
L |
||
[a] Indicated bold amino acids are underrepresented amino acids of the (R)‐ATA motifs published by Höhne et al. [8]
The (R)‐ATAs from Gram‐positive bacteria originate from Pseudonocardia acaciae (PA‐ωTA), Rubrobacter aplysinae (RA‐ωTA) and Tetrasphaera japonica (TJ‐ωTA). The sequences of these ATAs exhibit low sequence identity to known (R)‐ATA structures (45–52 %). Furthermore, PA‐ωTA lacks the N‐terminal helix while TJ‐ωTA has a shortened one.
The remaining three (R)‐ATAs are from Gram‐negative bacteria. They originate from Fodinicurvata fenggangensis (FF‐ωTA), Methylobacterium variabile (MeV‐ωTA) and Shinella (SH‐ωTA). FF‐ωTA was chosen due to the lack of the N‐terminal helix and the habitat of its organism. F. fenggangensis was isolated from a salt mine and tolerates up to 20 % sodium chloride. [24] MeV‐ωTA has an underrepresented amino acid in the first sequence motif and is lacking the arginine in the active site loop (Table 1). SH‐ωTA comprises an underrepresented amino acid in the second sequence motif (Table 1).
ES‐ωTA, PA‐ωTA, TJ‐ωTA and SH‐ωTA were well expressed. SM‐ωTA, RA‐ωTA, FF‐ωTA and MeV‐ωTA did not give any soluble expression (Figure S7, Table S2). Spectrophotometric activity assays [25] with the cleared cell lysates confirmed these results (Table S2).
For the four functionally expressed putative (R)‐ATAs, we aimed for solving their crystal structure, thus making purification necessary. For this purpose, C‐ and N‐terminal His‐tags were added to ES‐ωTA, PA‐ωTA, TJ‐ωTA and SH‐ωTA. The purification buffers were chosen based on a differential scanning fluorimetry screen to ensure proper protein folding. Whereas TJ‐ωTA was functionally expressed in both His‐tagged versions, ES‐ωTA was only soluble with the C‐terminal tag and PA‐ωTA and SH‐ωTA only accepted the N‐terminal tag. These five soluble His‐tagged (R)‐ATA versions were used for purification and crystallization trials.
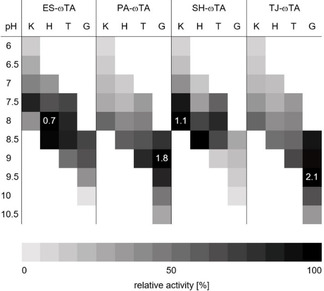
Influence of buffer, pH and temperature on the activity
The soluble and active (R)‐ATAs were characterized with respect to their pH‐optimum, buffer preference, temperature optimum and stability. R‐selective ATAs are usually most active in a pH range of 7 to 9. ES‐ωTA was most active (0.7 U mg−1) in HEPES buffer at pH 8 but showed comparable activity in potassium phosphate (KPi) buffer at pH 7.5. Similarly, the highest activity (1.1 U mg−1) of SH‐ωTA was obtained in KPi at pH 8. The two (R)‐ATAs from Gram‐positive bacteria, PA‐ωTA (1.8 U mg−1) and TJ‐ωTA (2.1 U mg−1), both prefer a higher pH with an optimum at 9 and 9.5, respectively. They also remained active at higher pH (Table 2).
Table 2.
Overview of the relative activities in different buffers with a concentration of 50 mM and at different pH. The activities were determined according to a spectrophotometric assay [25] (K: potassium phosphate buffer, H: HEPES buffer, T: Tris HCl buffer, G: Glycine NaOH buffer; white areas: activity not determined; numbers in the fields give the highest activity in U mg−1 total lysate protein; for absolute values, see Table S3).
|
|
The new putative (R)‐ATAs also had their individual temperature optima (Table S4). The standard reaction temperature chosen for most assays and biotransformations employing (R)‐ATAs is 30 °C and is usually a compromise between their highest temperature‐dependent activity and their limited stability at higher temperatures. For ES‐ωTA, an increase of the standard temperature by 2.5 °C rendered the (R)‐ATA double as active. For reactions with this (R)‐ATA, adjusting the temperature was crucial, especially since the stability of ES‐ωTA was rather low. Incubation at 40 °C led to 50 % inactivation after only six minutes (Table S2, Figure S8). In contrast to ES‐ωTA, PA‐ωTA worked over a broad temperature range. Its optimum was also at 32.5 °C but changes of the temperature had a minor effect on the activity. PA‐ωTA even retained 50 % of its activity at 15 °C. PA‐ωTA's activity dropped at 40 °C and incubation at this temperature revealed moderate stability. SH‐ωTA and TJ‐ωTA were more stable at 40 °C and do not reach their thermal inactivation half‐life within an hour of incubation (Figure S8). Additionally, both (R)‐ATAs reached their highest activity at elevated temperatures: SH‐ωTA was most active at 35 °C, TJ‐ωTA at 37.5 °C.
PA‐ωTA, SH‐ωTA and TJ‐ωTA were also incubated at 45 °C (Figure S8). Surprisingly, SH‐ωTA lost its activity very fast. In contrast, PA‐ωTA and TJ‐ωTA seemed to tolerate higher temperatures. These two (R)‐ATAs lack the N‐terminal helix which might be a reason for this tolerance. Their structure should be more compact and less flexible than the ones from (R)‐ATAs containing the N‐terminal helix. The fact that other PLP fold type IV transaminases without the helix show higher thermotolerance than published (R)‐ATAs containing the helix[ 26 , 27 , 28 , 29 ] supports this hypothesis. However, simple deletion of the helix of an (R)‐ATA originally containing it supposedly leads to a non‐functional enzyme. [10]
Substrate scope
For the synthesis of amines, different strategies were developed to shift the equilibrium of the transamination reaction.[ 1 , 30 , 31 , 32 , 33 ] The use of the amine donor isopropylamine (IPA) leads to the volatile co‐product acetone. Acetone can be evaporated, thus leading to the desired equilibrium shift. In an alternative approach, in which alanine is used as amine donor, a lactate dehydrogenase (LDH) and a glucose dehydrogenase (GDH) are used to convert the co‐product pyruvate and therefore remove it from the reaction. We tested both systems (Table 3 and 4) since IPA is preferred in industrial applications but is often incompatible with wild‐type (R)‐ATAs. [34] In general, the substrate scope which was determined using the LDH/GDH system (Table 3), was in the same range as the one from already published (R)‐ATAs. [35] This finding confirmed that all tested enzymes are indeed (R)‐ATAs.
Table 3.
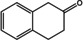
Substrate scope of the novel (R)‐ATAs with d‐alanine as amino donor and addition of LDH and GDH to drive the equilibrium to the products side.
|
Substrate |
Analytical yield [%] |
||||
|---|---|---|---|---|---|
|
|
ES‐ωTA |
PA‐ωTA |
SH‐ωTA |
TJ‐ωTA |
|
|
1 a |
|
5 |
42 |
51 |
20 |
|
1 b |
|
25 |
95 |
80 |
62 |
|
1 c [a] |
|
72 |
58 |
72 |
59 |
|
1 d |
|
10 |
– |
– |
– |
|
1 e [a] |
|
– |
– |
– |
– |
|
1 f |
|
18 |
45 |
95 |
13 |
|
1 g |
|
15 |
– |
– |
<5 |
|
1 h |
|
15 |
– |
– |
– |
|
1 i [a] |
|
– |
<5 |
– |
– |
|
1 j [a] |
|
– |
– |
– |
– |
|
1 k [a] |
|
10 |
81 |
13 |
28 |
|
1 l |
|
– |
– |
17 |
– |
|
ee [%][b] |
≥99 (R) |
≥99 (R) |
≥99 (R) |
≥99 (R) |
|
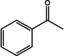
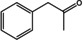
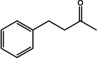
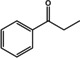
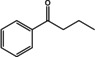
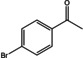
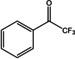
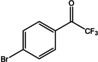
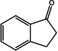
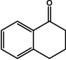
[a] ee not determined; [b] Measured by chiral HPLC; the ee is the same for all substrates; [c] Reaction conditions (not optimized): 20 mM ketone dissolved in DMSO (5 % v/v), 100 mM potassium phosphate buffer, pH 7.5, 1 mM PLP, 0.1 mM NAD+, cell free Escherichia coli lysate containing 4–8 U (R)‐ATA (according to the spectrophotometric assay), 130 mM d‐alanine, 60 mM glucose, 30 U GDH, and 90 U LDH, shaken for 24 h at 250 rpm and 30 °C. Yields were measured by HPLC.
Table 4.
Substrate scope of the new (R)‐ATAs with IPA as amino donor.[a]
|
Substrate |
Analytical yield [%] |
|||
|---|---|---|---|---|
|
|
ES‐ωTA |
PA‐ωTA |
SH‐ωTA |
TJ‐ωTA |
|
1 a |
4 |
23 |
2 |
2 |
|
1 b |
5 |
92 |
85 |
79 |
|
1 d |
7 |
– |
– |
– |
|
1 f |
16 |
32 |
20 |
– |
|
1 g |
– |
– |
– |
– |
|
1 h |
15 |
– |
– |
– |
|
ee [%][b] |
≥99 (R) |
≥99 (R) |
≥99 (R) |
≥99 (R) |
[a] Reaction conditions (not optimized): 20 mM ketone dissolved in DMSO (5 % v/v), 100 mM potassium phosphate buffer, pH 7.5, 1 mM PLP, 1 M isopropylamine and cell free E. coli lysate containing 8 U ATA (according to the spectrophotometric assay), shaken for 24 h at 250 rpm and 30 °C. Yields were measured by HPLC; [b] Measured by chiral HPLC; the ee is the same for all substrates.
ES‐ωTA showed low activity in the initial photometric assay and was also not very active on conventional substrates such as 1 a or 1 b. Interestingly, the analytical yield increased when a larger moiety was accommodated in the small binding pocket. Usually, substituents bigger than a methyl group prevent or significantly reduce the yield. For ES‐ωTA, the opposite was the case: the yield was low for the methyl substituent (1 a) and an exchange to a ‐CF3 group (1 g) or an ethyl group (1 d) improved the yield from 5 to 15 or 10 %, respectively. However, a propyl group was not accepted.
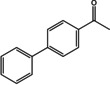
Similarly to the recently published (R)‐ATA from Exophiala xenobiotica (EX‐ωTA), SH‐ωTA was one of the rare (R)‐ATAs that converted the biaryl ketone 1 l. However, the yield of the corresponding amine was much higher when EX‐ωTA was used, reaching a value of 83 %. [12]
Special attention needs to be drawn to PA‐ωTA and TJ‐ωTA. To our knowledge, they represent the first characterized R‐selective ATAs that lack the N‐terminal helix and convert ketones to the corresponding amines. Our group recently discovered potential (R)‐ATAs also lacking the helix, but amination of ketones could not be shown and significant differences in the active site suggested that these enzymes belong to a new subgroup of the fold type IV family. [36] However, PA‐ωTA and TJ‐ωTA can clearly be classified as (R)‐ATAs according to the sequence motif and their activity.
We also tested the novel (R)‐ATAs for acceptance of the amine donor IPA (Table 4). In comparison to the LDH/GDH system, the analytical yields increased or decreased depending on the substrate. For instance, 1 b in combination with IPA reached yields in the same range as with the LDH/GDH system. Again, ES‐ωTA was an exception with lower yields for 1 b but similar ones for almost all other amine acceptors. Yields of 1 a and 1 f with SH‐ωTA and TJ‐ωTA decreased tremendously when IPA was used. PA‐ωTA worked moderately with IPA and gave the highest analytical yields compared to the other new (R)‐ATAs. The ability to use IPA is especially interesting for PA‐ωTA (and theoretically also for TJ‐ωTA) as it can easily be used at high pH. The LDH/GDH system becomes incompatible at high pH due to low pH optima of the LDH and GDH. [37]
Structural characterization
Of the four purified (R)‐ATAs with a His‐tag, only ES‐ωTA, PA‐ωTA and SH‐ωTA crystallized. Although TJ‐ωTA could be expressed and purified with N‐ and C‐terminal His‐Tag, both versions did not result in the formation of crystals. The diffraction data for PA‐ωTA were of poor quality, thus preventing structure determination. Optimization of the crystallization conditions did not improve crystal quality. For ES‐ωTA and SH‐ωTA, the obtained crystal structures were used for analysis and structures were determined to 3.1 and 2.1 Å respectively (for detailed information, see Table S5). The asymmetric unit of ES‐ωTA comprises six chains, the asymmetric unit of SH‐ωTA has four chains.
The overall structures of ES‐ωTA and SH‐ωTA show all the common characteristics found in deposited (R)‐ATA structures. The single chains or monomers of both (R)‐ATAs form homodimers with the two active sites at the dimer interface. The flexible loop pointing into the active site pocket contains the flipping arginine. Generally, this loop can take an open‐loop or closed‐loop conformation. All chains of the two structures have a closed loop. Every active site accommodates a PLP. Whereas the PLP in the active sites of ES‐ωTA is covalently bound to the active site lysine (internal aldimine, Figure S9), the PLP in the active sites of SH‐ωTA shows different conformations. In chain A and D, the PLP is partly bound to K182 and partly in its unbound form (Figure S10A). Chain B and C have a PLG (glycine bound to PLP) with alternative conformation in their active site (Figure S10B).
In a recent publication, [12] we postulated that the charge of the active site (Figure 2) plays a role in the acceptance of certain substrates. Published (R)‐ATAs are usually negatively charged at the entrance to the active site (e. g., 4CE5, [9] 4CHI, [10] 4CMD [11] ). However, EX‐ωTA has a reduced active site charge, thus allowing the conversion of bulky hydrophobic and uncharged biaryl ketones. [12] The active site charge of ES‐ωTA is in between the ones of EX‐ωTA and other (R)‐ATAs with a known crystal structure. In contrast to this, the whole active site of SH‐ωTA is positively charged. This makes SH‐ωTA not only the first (R)‐ATA from a Gram‐negative bacterium with an existing crystal structure, but also the first one with a positive active site charge. With this positive active site charge, SH‐ωTA does not provide an ideal environment for the conversion of the hydrophobic biaryl ketone 1 l but it is better than the repelling negative charge found in most (R)‐ATA crystal structures. Accordingly, SH‐ωTA converted 17 % of the ketone 1 l. Recently, we postulated that a second factor contributes to the conversion of biaryl ketones: the presence of sulfur‐π (S‐π) interactions between the sulfur of a methionine in the active site and the π aromatic system of the biaryl ketone. [12] S‐π interactions have energy minima at a distance of 3.6 Å when the sulfur is above the aromatic ring, [38] and at a distance of about 5 Å in an in‐plane configuration.[ 38 , 39 ] In SH‐ωTA, M119 might play a role in positioning the biaryl substrate. However, the interactions are quite weak (Figure 3) according to the results of a VINA docking performed with YASARA. The docking results show that M119 is located above the ring next to the keto group at a distance of 7.1 Å. Since the optimal distance is 3.6 Å, this interaction is probably neglectable. Additionally, M119 is in plane with the second ring and interacts with it over a distance of 7.8 Å. This might indicate a weak S‐π interaction, but it has to be added that there are discrepancies between dockings and actual substrate positioning in the protein crystal structure. In addition to the possible S‐π interactions of M119, H57 also helps to position the biaryl ketone via π‐stacking at a distance of 4.7 Å.
Figure 2.
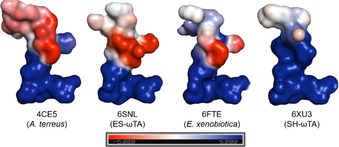
Active‐site charge of published and new (R)‐ATAs. Most published (R)‐ATA structures[ 9 , 10 , 11 ] (represented by 4CE5) have a negatively charged entrance to the active site and a positively charged area for PLP binding. The recently published structure 6FTE showed reduced active‐site charge. Whereas ES‐ωTA has a similar active site charge as (R)‐ATAs with published structures, SH‐ωTA has a positive overall charge.
Figure 3.
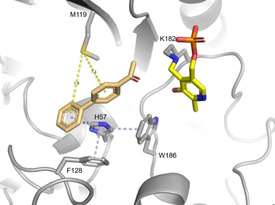
Docking results (VINA performed with YASARA) of the biaryl ketone 1 l into the active site of SH‐ωTA. Possible interactions allowing the conversion of 1 l are potential S‐π interactions between M119 and 1 l (yellow) and π‐stacking interactions between H57 and 1 l (light blue).
Looking at the distance of the keto group of 1 l relative to the amine of the internal aldimine, the position of the biaryl ketone in the active site of SH‐ωTA is also not optimal for an efficient reaction. The keto group points away from the amine, which probably also contributes to the low conversion.
According to the MSA, ES‐ωTA contains the sequence motif RxH that is common for d‐ATAs. A look at the structure (Figure 4) reveals that even though the amino acids forming the carboxylate trap are present, the coordination of the trap is disrupted in ES‐ωTA. Both, the arginine and the histidine of the motif are located on the flexible active site loop. In d‐ATAs, this loop completely points into the active site and positions the residues R and H close to the keto substrate and the carboxylate trap partner tyrosine. Contrary to this, the ES‐ωTA loop in its closed state is moved to the side so that only the arginine remains pointing into the active site further allowing the dual substrate recognition. The histidine faces away preventing the formation of the carboxylate trap as found in d‐ATAs.
Figure 4.
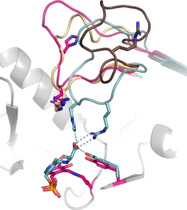
Loop conformation of ES‐ωTA (pink) compared to the open‐ (brown; chain B of 4UUG) and closed‐loop conformations (beige; chain A of 4UUG) of the (R)‐ATA from A. fumigatus. ES‐ωTA contains part of the d‐ATA motif RxH. In d‐ATAs (turquoise, PDB ID 3DAA), this R and H (together with Y) coordinate the carboxylate of the d‐keto acid. In ES‐ωTA, the flexible loop in its closed state is moved to the side so that the histidine faces away from the active site and prevents the interactions found in d‐ATAs.
Conclusion
The expansion of knowledge of the (R)‐ATA fold IV type of PLP‐dependent enzymes still remains a challenge. Addressing this issue, in this study, we broadened the available toolbox of (R)‐ATAs and decreased the bias for fungal (R)‐ATAs. SH‐ωTA is the first reported (R)‐ATA with a positive active site charge and from a Gram‐negative bacterium for which the crystal structure was solved. PA‐ωTA and TJ‐ωTA are both from Gram‐positive bacteria and lack the N‐terminal helix or have a shortened one, therefore demonstrating that the helix is not needed for soluble expression and is also not a means to distinguish (R)‐ATAs from other members of the fold type IV family. Furthermore, these two (R)‐ATAs have a high pH tolerance with an optimum of pH 9 and 9.5, respectively. This feature is also of organic‐synthetic interest as it should enable the access to new applications such as combined transamination reactions with chemical reactions requiring an elevated pH. Finally, it was confirmed that ES‐ωTA shows (R)‐ATA activity despite comprising the RxH motif normally occurring in d‐ATAs.
With our contribution to expand the knowledge about (R)‐ATAs, we only scratched the surface. Looking back at our selection criteria, very interesting candidates could not be investigated due to non‐functional protein expression. For instance, SM‐ωTA would have combined features such as a missing N‐terminal helix, a longer active site loop and a missing arginine residue on the same loop. Thus, a subsequent study could focus on functional expression of difficult to express (R)‐ATAs in different expression hosts, such as Pichia pastoris or other bacterial expression systems like Bacillus or Pseudomonas species. Further investigations remain to be carried out to fully understand (R)‐ATAs and obtain a toolbox providing a broad diversity of applications for the synthesis of chiral amines.
Experimental Section
Data mining and identification of amine transaminases: Published fold IV amine transaminase (ATA) sequences[ 9 , 10 , 11 , 15 , 16 , 17 , 19 ] were used to search for related sequences in the NCBI database (pBLAST). [20] The first 1000 hits of each search were downloaded and redundant sequences were removed with CD‐HIT Suite [40] using a sequence identity cut‐off of 0.95. All sequences were combined in one file and again, redundant sequences were removed with a cut‐off of 0.9. The obtained protein sequences were aligned by MUSCLE with Mega 7.0.26, [41] and hits comprising the motifs characteristic for BCATs, d‐ATAs or ADCLs [8] (Table 1) were excluded. Chosen proteins:
PLP‐dependent enzyme from F. fenggangensis (WP_081816415)
PLP‐dependent enzyme from M. variabile (WP_048444767)
PLP‐dependent enzyme from P. acaciae (WP_028922477)
PLP‐dependent enzyme from R. aplysinae (WP_047865756)
PLP‐dependent enzyme from Shinella sp. (WP_050745125)
PLP‐dependent enzyme from S. musiva (XP_016761339)
PLP‐dependent enzyme from T. japonica (WP_048556467)
Cloning: All genes were ordered codon‐optimized for expression in E. coli from Geneart/ThermoFisher Scientific. The genes were cloned into pMS470Δ8 [42] by restriction digestion (NdeI/HindIII) and ligation (T4 ligase). For purification purposes, His‐tags were added to ES‐ωTA, PA‐ωTA, SH‐ωTA and TJ‐ωTA. The genes were amplified by PCR (Table S6) and subcloned by restriction digest (NcoI/XhoI) and ligation (T4 ligase) into pET28a(+) from Novagen/Merck or pEHISTEV. [43] Subcloning into pET28a(+) adds a C‐terminal His‐tag whereas subcloning into pEHISTEV adds an N‐terminal His‐tag which can be cleaved off with the TEV protease.
Protein expression: E. coli Top10F’ cells (Invitrogen/ThermoFisher Scientific) were transformed with the pMS470Δ8 vectors encoding the (R)‐ATAs and E. coli BL21‐Gold(DE3) (Stratagene) were transformed with the pET‐based vectors. The cells were grown at 37 °C in terrific broth medium supplemented with ampicillin (Amp: 100 μg mL−1) or kanamycin (Kan: 40 μg mL−1). Expression was induced with 0.1 mM isopropyl β‐d‐1‐thiogalactopyranoside (IPTG) at an OD600 between 0.6 and 0.8 and carried out at 25 °C overnight. The cells were harvested at 4000 g for 15 min and resuspended in buffer (pMS470Δ8 constructs: 50 mM potassium phosphate (KPi) buffer, pH 7.5, 0.1 mM PLP; or pET‐based constructs in buffer A (Table 5) containing 10 mM imidazole). The cells were disrupted by sonication (Branson Sonifier S‐250, 6 min, 80 % duty cycle, 70 % output) and centrifuged at 50 000 g for 1 h. The cleared lysates were filtered through 0.45 μm syringe filters. The protein concentration of the lysate was determined by Bradford protein assay (ThermoFisher Scientific). The expression and solubility of the proteins was analyzed by SDS‐PAGE (NuPAGE Bis‐Tris PreCast Gels/ThermoFisher Scientific).
Table 5.
Overview of the vectors used for expression with His‐tags, buffers used for purification and extinction coefficients of the purified (R)‐ATAs. The decision for pET28(+) or pEHISTEV was based on soluble expression (TJ‐ωTA was soluble with both vectors). The buffers were chosen based on a differential scanning fluorimetry screen, [45] with which the folding state of a protein can be determined in different buffers (results not shown).
|
|
ES‐ωTA |
PA‐ωTA |
SH‐ωTA |
TJ‐ωTA |
|---|---|---|---|---|
|
Vector |
pET28(+) |
pEHISTEV |
pEHISTEV |
pET28(+)/pEHISTEV |
|
Buffer A |
50 mM KPi, pH 7.5, 500 mM NaCl, 0.1 mM PLP |
50 mM MES, pH 6, 200 mM NaCl, 0.1 mM PLP |
50 mM MES, pH 6, 50 mM NaCl, 0.1 mM PLP |
50 mM MOPS, pH 7, 50 mM NaCl, 0.1 mM PLP |
|
Buffer B |
50 mM Tris‐HCl, pH 8, 50 mM NaCl, 10 % v/v glycerol, 0.1 mM PLP |
50 mM MES, pH 6, 50 mM NaCl, 0.1 mM PLP |
50 mM MES, pH 6, 50 mM NaCl, 0.1 mM PLP |
50 mM MOPS, pH 7, 50 mM NaCl, 0.1 mM PLP |
|
Cleavage His‐tag |
no |
yes |
yes |
no/yes |
|
extinction coefficient [M−1cm−1] |
1.28 |
1.18 |
1.41 |
1.23 |
Protein purification: For the purification of functionally expressed His‐tagged (R)‐ATAs (Table 5), the filtered cell‐free lysate was incubated with Ni Sepharose 6 Fast Flow resin (GE Healthcare) for 20 min. The Ni Sepharose resin was then filled into empty PD‐10 columns (GE Healthcare). After removal of impurities with buffer A containing 30 mM imidazole, the target protein was eluted with 300 mM imidazole in buffer A. Fractions were analyzed by SDS‐PAGE, pooled, concentrated (Vivaspin 20 Centrifugal Filter Units; 10 000 Da molecular weight cutoff; Sartorius, Göttingen, Germany) and desalted into buffer B (for composition, see Table 5) on PD‐10 desalting columns (GE Healthcare).
The His‐tags of proteins with an N‐terminal tag were cleaved off by an in‐house produced TEV protease. 1 mg TEV protease was used to cleave 10 mg (R)‐ATA in 50 mM MOPS buffer, pH 7, supplemented with 200 mM NaCl, 1 mM DTT and 0.1 mM PLP. The whole mixture was slowly shaken at 4 °C overnight. Precipitated protein was removed by centrifugation for 10 min at 4 °C, 2800 g and filtration with 0.45 μm syringe filters. The resulting solution was again loaded on a Ni Sepharose column and the (R)‐ATA lacking the His‐tag was collected in the flow‐through. The flow‐through was further purified with PD‐10 desalting columns and the buffer changed to buffer B.
All purified proteins were concentrated (Vivaspin 20 Centrifugal Filter Units; 10 000 Da molecular weight cutoff; Sartorius, Göttingen, Germany) to a volume of less than 5 mL and subjected to a final size exclusion purification step with an ÄKTA pure protein purification system (GE healthcare) and a Sepharose 16/60 200 pg column. The size exclusion chromatography was carried out with 20 mM buffer B and peak fractions were collected, pooled and concentrated to a protein concentration of approximately 10 mg mL−1.
The concentration of the purified proteins was determined with a Nanodrop spectrophotometer (model 2000c, Peqlab, Erlangen, Germany), using the extinction coefficients at 280 nm given in Table 5. The coefficients were calculated on the basis of the amino acid sequence using ProtParam. [44] Purified proteins were aliquoted, frozen in liquid nitrogen and stored at −80 °C until further use.
Crystallization and structure determination: Screening for crystallization conditions was performed with a mosquito robot (TTP Labtech) using the protein crystallization screens JCSG+ and PACT premier (Molecular Dimensions) by the sitting drop vapor‐diffusion method in MRC 3‐well plates.
The stock solution of ES‐ωTA contained 20 mM Tris ⋅ HCl buffer, pH 8, 50 mM NaCl, 10 % v/v glycerol, 0.1 mM PLP and had a protein concentration of 10.7 mg mL−1. SH‐ωTA had a protein concentration of 8.9 mg mL−1 in 20 mM MES buffer, pH 6, supplemented with 50 mM NaCl and 0.1 mM PLP. For the screenings, drops of 400 nL were pipetted with a 1 : 1 ratio of protein and screening solution. The crystallization plates were incubated in the dark at room temperature. For both (R)‐ATAs, crystals appeared in several crystallization conditions. The crystal from ES‐ωTA was mounted from condition H8 from the JCSG+ screen (0.2 M NaCl, 0.1 M Bis ⋅ Tris, pH 5.5, 25 % w/v PEG 3350) and transferred to a cryoprotectant solution of the same composition supplemented with 20 % v/v glycerol and finally flash‐frozen in liquid nitrogen. A crystal of SH‐ωTA was mounted from condition E5 of the PACT screen (0.2 M sodium nitrate, 20 % w/v PEG 3350). The crystal was subjected to soaking for 2 h at room temperature. The soaking solution contained 0.2 M sodium nitrate, 20 % w/v PEG 3350, 20 % v/v glycerol and 0.1 mM gabaculine. This crystal was also flash frozen and stored in liquid nitrogen. Data collection was performed on the synchrotron beamlines i04 (Diamond Light Source, Didcot, UK) and MX‐14‐1 (BESSY, Berlin, Germany). The data sets were processed and scaled using the XDS program package. [46] Molecular replacement (search template 4CE5 [9] ), model building and refinement were carried out with Phenix (version 1.14‐3260), [47] whereas final model building was performed in Coot. [48] Data collection and processing statistics are summarized in Table S5. The final structures were checked using MolProbity.[ 49 , 50 ] The coordinates have been deposited in the Protein Data Bank with accession codes 6SNL (ES‐ωTA) and 6XU3 (SH‐ωTA).
As crystallization trials remained unsuccessful for PA‐ωTA and TJ‐ωTA, further conditions were tested with the Index (Hampton Research) and Morpheus (Molecular Dimensions) protein crystallization screens at 17 °C. TJ‐ωTA did not crystallize in any condition. PA‐ωTA crystallized in several conditions of the Morpheus screen and in condition 22 of the Index screen. However, PA‐ωTA crystals diffracted with poor quality and prevented us from proceeding with structure determination.
Substrate docking and visualization of active site electrostatics: Docking studies were performed with YASARA (version 18.4.24). To prepare the structure 6XU3 for docking, any waters, and other ligands were removed. The cofactor PLP was kept in the structure. After adding any missing hydrogens, the YASARA standard protocol for energy minimization was applied to the dimeric SH‐ωTA structure. Structures of the substrates were drawn with ChemDraw (version 17.0.0.206(121)), and the energy was minimized with YASARA. A simulation cell was created around one active site of the dimeric structure of SH‐ωTA and VINA docking was performed with the standard mcr_dock with 999 runs. The docking result was chosen according to the best energy but also manual visualization and realistic positioning of the substrate.
For the visualization of the active site charge, PDB structures were loaded into PyMOL (version 2.1.1). The active site charge was calculated with the APBS electrostatics plugin for PyMOL and visualized accordingly.
Spectrophotometric activity assay: A standard photometric assay [25] was adjusted to determine the activity of the new (R)‐ATAs. In the assay, (R)‐ATAs convert phenylethylamine to acetophenone, which can be detected spectrophotometrically at 300 nm. A rising product concentration leads to an increasing absorbance and can be monitored. Standard reactions were set up with a volume of 1 mL containing 50 mM KPi buffer pH 7.5, 0.1 mM PLP, 5 mM phenylethylamine, and 5 mM pyruvic acid. 50 μg to 250 μg of total protein were added to the reaction. The increase of acetophenone was measured for several minutes at 300 nm at 30 °C. The Beer‐Lambert law was used to calculate the volumetric activity [U mL−1] and the specific activity [U mg−1 total lysate protein]. For that, the molar extinction coefficient of acetophenone ϵ 300=0.28 cm2 μmol−1 was used. As indicated in the results section, the (R)‐ATA activities were also tested in other buffers. The buffer concentration was always 50 mM. Concentrations of other components were not changed.
(R)‐ATA stability at 40 or 45 °C: The stability of the (R)‐ATAs was studied in KPi buffer, pH 7.5 and at 40 or 45 °C. The protein lysate (protein concentration 10 mg mL−1) was incubated at 40 or 45 °C and samples were taken at different time points. To remove denatured protein, these samples were centrifuged for 3 min at maximum speed. The activity of the supernatant was measured with the spectrophotometric assay at 30 °C.
Biotransformations
Enzymes. LDH (lactate dehydrogenase) from rabbit muscle (Sigma‐Aldrich, Type II, ammonium sulfate suspension, 800–1200 U mg−1 protein). GDH (glucose dehydrogenase) from Bacillus megaterium was expressed in E. coli. The activity was determined as 1884 U mL−1 according to the assay described in literature. [51]
Reagents. d‐(+)‐Glucose was purchased from VWR. d‐Alanine, PLP (Pyridoxal 5’‐phosphate hydrate) and NAD+ were purchased from Sigma‐Aldrich.
General methods. HPLC analyses to determine the degree of conversion were carried out in an Agilent RR1200 HPLC system, using a reversed phase column (Zorbax Eclipse XDBC18, RR, 18 μm, 4.6×50 mm, Agilent) or with a Hewlett Packard 1100 liquid chromatographic system, using a reversed‐phase column (Prevail 150×4.6 mm, particle size 3 μm, Nr 75). HPLC analyses to determine ee values were performed on a Hewlett‐Packard 1100 LC liquid chromatograph. Details of HPLC methods and analyses can be found in the Supporting Information.
General procedure for the screening of ketones 1 a–l using alanine as amino donor. The ketone (20 mM) was first dissolved in DMSO (32 μL, 5 % v/v) in a 2 mL reaction tube (Eppendorf). Then (R)‐ATA lysate (1 a, b, d, f–h, l: 8 U; 1 c, e, i–k: 8 mg lysate protein per reaction (corresponds to 4–8 U)), 100 mM KPi, pH 7.5, 1 mM PLP, 0.1 mM NAD+, d‐glucose (57 mM), d‐alanine (130 mM), LDH (90 U for ketones 1 a, b, d, f–i, l; 40 U for 1 c, e, j, k), and GDH (30 U) were added. The reaction was shaken at 30 °C and 250 rpm for 24 h. To determine the non‐isolated yield of the formed amine, 10 μL of the mixture were diluted with 90 μL of DMSO and analyzed by achiral reversed‐phase HPLC with previous centrifuging and filtering of the sample. To determine the enantiomeric excess for reactions employing the ketones 1 a, b, f–h, the reaction was quenched by addition of aqueous 10 N NaOH (400 μL). The mixture was then extracted with ethyl acetate (2×500 μL), and the organic layers were separated by centrifugation (90 s, 13 000 rpm, Eppendorf Microcentrifuge 5418), combined and dried over anhydrous Na2SO4. The enantiomeric excess of amines was measured by chiral HPLC.
General procedure for the screening of ketones 1 a, b, d, f–h using IPA as amino donor. In a 2 mL reaction tube, ketone (20 mM) was first dissolved in DMSO (32 μL, 5 % v/v) and then (R)‐ATA lysate (8 U), 100 mM KPi buffer, pH 7.5, 1 M isopropylamine and 1 mM PLP were added. The reaction was shaken at 30 °C and 250 rpm for 24 h. To determine the non‐isolated yield of the formed amine, 10 μL of the mixture were diluted with 90 μL of DMSO and analyzed by achiral reverse phase HPLC with previous centrifuging and filtering of the sample. To determine the enantiomeric excess the reaction was quenched by addition of aqueous 10 N NaOH (400 μL). The mixture was then extracted with ethyl acetate (2×500 μL), and the organic layers were separated by centrifugation (90 s, 13 000 rpm), combined, and dried over anhydrous Na2SO4. The enantiomeric excess of amines was measured by chiral HPLC.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The Biocascades project is supported by the European Commission under the Horizon 2020 program through the Marie Skłodowska‐Curie actions: ITN‐EID under the Environment Cluster (grant no. 634200). We further thank Math Boesten for assistance with HPLC analysis and Tea Pavkov‐Keller for additional crystallization trials of PA‐ωTA.
A. Telzerow, J. Paris, M. Håkansson, J. González-Sabín, N. Ríos-Lombardía, H. Gröger, F. Morís, M. Schürmann, H. Schwab, K. Steiner, ChemBioChem 2021, 22, 1232.
Contributor Information
Prof. Dr. Helmut Schwab, Email: helmut.schwab@tugraz.at.
Dr. Kerstin Steiner, kerstin.steiner11@googlemail.com.
References
- 1. Ferrandi E. E., Monti D., World J. Microbiol. Biotechnol. 2018, 34, 1–10. [DOI] [PubMed] [Google Scholar]
- 2. Kelly S. A., Pohle S., Wharry S., Mix S., Allen C. C. R., Moody T. S., Gilmore B. F., Chem. Rev. 2018, 118, 349–367. [DOI] [PubMed] [Google Scholar]
- 3. Gomm A., O'Reilly E., Curr. Opin. Chem. Biol. 2018, 43, 106–112. [DOI] [PubMed] [Google Scholar]
- 4.N. J. Turner, M. D. Truppo in Chiral Amine Synthesis, Wiley-VCH, Weinheim, Germany, 2010, pp. 431–459.
- 5. Aleku G. A., France S. P., Man H., Mangas-Sanchez J., Montgomery S. L., Sharma M., Leipold F., Hussain S., Grogan G., Turner N. J., Nat. Chem. 2017, 9, 961–969. [DOI] [PubMed] [Google Scholar]
- 6. Adams J. P., Brown M. J. B., Diaz-Rodriguez A., Lloyd R. C., Roiban G., Adv. Synth. Catal. 2019, 361, 2421–2432. [Google Scholar]
- 7. Steffen-Munsberg F., Vickers C., Kohls H., Land H., Mallin H., Nobili A., Skalden L., van den Bergh T., Joosten H. J., Berglund P., Höhne M., Bornscheuer U. T., Biotechnol. Adv. 2015, 33, 566–604. [DOI] [PubMed] [Google Scholar]
- 8. Höhne M., Schätzle S., Jochens H., Robins K., Bornscheuer U. T., Nat. Chem. Biol. 2010, 6, 807–813. [DOI] [PubMed] [Google Scholar]
- 9. Łyskowski A., Gruber C., Steinkellner G., Schürmann M., Schwab H., Gruber K., Steiner K., PLoS One 2014, 9, e87350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thomsen M., Skalden L., Palm G. J., Höhne M., Bornscheuer U. T., Hinrichs W., Acta Crystallogr. Sect. D 2014, 70, 1086–1093. [DOI] [PubMed] [Google Scholar]
- 11. Sayer C., Martinez-Torres R. J., Richter N., Isupov M. N., Hailes H. C., Littlechild J. A., Ward J. M., FEBS J. 2014, 281, 2240–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Telzerow A., Paris J., Håkansson M., González-Sabín J., Ríos-Lombardía N., Schürmann M., Gröger H., Morís F., Kourist R., Schwab H., Steiner K., ACS Catal. 2019, 9, 1140–1148. [Google Scholar]
- 13. Bezsudnova E. Y., Popov V. O., Boyko K. M., Appl. Microbiol. Biotechnol. 2020, 104, 2343–2357. [DOI] [PubMed] [Google Scholar]
- 14. Liang J., Han Q., Tan Y., Ding H., Li J., Front. Mol. Biosci. 2019, 6, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schätzle S., Steffen-Munsberg F., Thontowi A., Höhne M., Robins K., Bornscheuer U. T., Adv. Synth. Catal. 2011, 353, 2439–2445. [Google Scholar]
- 16. Jiang J., Chen X., Zhang D., Wu Q., Zhu D., Appl. Microbiol. Biotechnol. 2015, 99, 2613–2621. [DOI] [PubMed] [Google Scholar]
- 17. Guan L.-J., Ohtsuka J., Okai M., Miyakawa T., Mase T., Zhi Y., Hou F., Ito N., Iwasaki A., Yasohara Y., Tanokura M., Sci. Rep. 2015, 5, 10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Skalden L., Thomsen M., Höhne M., Bornscheuer U. T., Hinrichs W., FEBS J. 2015, 282, 407–415. [DOI] [PubMed] [Google Scholar]
- 19. Savile C. K., Janey J. M., Mundorff E. C., Moore J. C., Tam S., Jarvis W. R., Colbeck J. C., Krebber A., Fleitz F. J., Brands J., Devine P. N., Huisman G. W., Hughes G. J., Science 2010, 329, 305–309. [DOI] [PubMed] [Google Scholar]
- 20. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J., J. Mol. Biol. 1990, 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 21. Voss M., Xiang C., Esque J., Nobili A., Menke M. J., André I., Höhne M., Bornscheuer U. T., ACS Chem. Biol. 2020, 15, 416–424. [DOI] [PubMed] [Google Scholar]
- 22. Iglesias C., Panizza P., Rodriguez Giordano S., Appl. Microbiol. Biotechnol. 2017, 101, 5677–5687. [DOI] [PubMed] [Google Scholar]
- 23. Seyedmousavi S., Badali H., Chlebicki A., Zhao J., Prenafeta-boldú F. X., De Hoog G. S., Fungal Genet. Biol. 2011, 115, 1030–1037. [DOI] [PubMed] [Google Scholar]
- 24. Wang Y.-X., Liu J.-H., Zhang X.-X., Chen Y.-G., Wang Z.-G., Chen Y., Li Q.-Y., Peng Q., Cui X.-L., Int. J. Syst. Evol. Microbiol. 2009, 59, 2575–2581. [DOI] [PubMed] [Google Scholar]
- 25. Schätzle S., Höhne M., Redestad E., Robins K., Bornscheuer U. T., Anal. Chem. 2009, 81, 8244–8248. [DOI] [PubMed] [Google Scholar]
- 26. Bezsudnova E. Y., Dibrova D. V., Nikolaeva A. Y., Rakitina T. V., Popov V. O., J. Biotechnol. 2018, 271, 26–28. [DOI] [PubMed] [Google Scholar]
- 27. Bezsudnova E. Y., Boyko K. M., Nikolaeva A. Y., Zeifman Y. S., Rakitina T. V., Suplatov D. A., Popov V. O., Biochimie 2019, 158, 130–138. [DOI] [PubMed] [Google Scholar]
- 28. Zeifman Y. S., Boyko K. M., Nikolaeva A. Y., Timofeev V. I., Rakitina T. V., Popov V. O., Bezsudnova E. Y., Biochim. Biophys. Acta Proteins Proteomics 2019, 1867, 575–585. [DOI] [PubMed] [Google Scholar]
- 29. Kelly S. A., Magill D. J., Megaw J., Skvortsov T., Allers T., McGrath J. W., Allen C. C. R., Moody T. S., Gilmore B. F., Appl. Microbiol. Biotechnol. 2019, 103, 5727–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guo F., Berglund P., Green Chem. 2017, 19, 333–360. [Google Scholar]
- 31. Simon R. C., Richter N., Busto E., Kroutil W., ACS Catal. 2014, 4, 129–143. [Google Scholar]
- 32. Brundiek H., Höhne M. in Applied Biocatalysis: From Fundamental Science to Industrial Applications (Eds.: Hilterhaus L., Liese A., Kettling U., Antranikian G.), 2016, Wiley-VCH, Weinheim, Germany, pp. 199–218. [Google Scholar]
- 33. Patil M. D., Grogan G., Bommarius A., Yun H., Catalysts 2018, 8, 254. [Google Scholar]
- 34. Kelefiotis-Stratidakis P., Tyrikos-Ergas T., Pavlidis I. V., Org. Biomol. Chem. 2019, 17, 1634–1642. [DOI] [PubMed] [Google Scholar]
- 35. Calvelage S., Dörr M., Höhne M., Bornscheuer U., Adv. Synth. Catal. 2017, 359, 4235–4243. [Google Scholar]
- 36. Pavkov-Keller T., Strohmeier G. A., Diepold M., Peeters W., Smeets N., Schürmann M., Gruber K., Schwab H., Steiner K., Sci. Rep. 2016, 6, 38183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nagao T., Mitamura T., Wang X. H., Negoro S., J. Bacteriol. 1992, 174, 5013–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ringer A. L., Senenko A., Sherrill C. D., Protein Sci. 2007, 16, 2216–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zauhar R. J., Colbert C. L., Morgan R. S., Welsh W. J., Biopolymers 2000, 53, 233–248. [DOI] [PubMed] [Google Scholar]
- 40. Huang Y., Niu B., Gao Y., Fu L., Li W., Bioinformatics 2010, 26, 680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kumar S., Stecher G., Tamura K., Mol. Biol. Evol. 2016, 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Balzer D., Ziegelin G., Pansegrau W., Kruft V., Lanka E., Nucleic Acids Res. 1992, 20, 1851–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huanting L., Naismith J. H., Protein Expression Purif. 2009, 63, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., Bairoch A., Proteomics Protoc. Handb. 2005, 571–607. [Google Scholar]
- 45. Seabrook S. A., Newman J., ACS Comb. Sci. 2013, 15, 387–392. [DOI] [PubMed] [Google Scholar]
- 46. Kabsch W., Acta Crystallogr. Sect. D 2010, 66, 125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., Acta Crystallogr. Sect. D 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Emsley P., Lohkamp B., Scott W. G., Cowtan K., Acta Crystallogr. Sect. D 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen V. B., Arendall W. B., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., Acta Crystallogr. Sect. D 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Williams C. J., Headd J. J., Moriarty N. W., Prisant M. G., Videau L. L., Deis L. N., Verma V., Keedy D. A., Hintze B. J., Chen V. B., Jain S., Lewis S. M., W. B. Arendall III , Snoeyink J., Adams P. D., Lovell S. C., Richardson J. S., Richardson D. C., Protein Sci. 2017, 27, 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Smith L. D., Budgen N., Bungard S. J., Danson M. J., Hough D. W., Biochem. J. 1989, 261, 973–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary