

Abstract
Apararenone is a long‐acting, nonsteroidal mineralocorticoid receptor antagonist (MRA). The safety, tolerability, and pharmacokinetic (PK) and pharmacodynamic (PD) profiles of single‐ and multiple‐dose apararenone were assessed in 3 phase 1 randomized, double‐blind studies in 223 healthy adults. Study 1 assessed the PK, safety/tolerability, and PD of single‐dose apararenone (3.75–640 mg) and multiple‐dose apararenone (10–40 mg/day on days 1–14, 320 mg loading dose on day 1 + 10 mg/day on days 2–14, or 40–320 mg loading dose on day 1 + 2.5–20 mg/day on days 2–14) in Caucasian and Black men and women. Study 2 assessed the PK and safety of single‐dose apararenone (5–320 mg) in healthy Japanese men. Study 3 assessed the PK, PD, and safety/tolerability of single‐dose apararenone (160 or 640 mg) or eplerenone (200 mg; only for 160 mg of apararenone), each after fludrocortisone challenge in Caucasian men. In studies 1 and 2, an approximately dose‐proportional increase was observed in PK parameters over the apararenone dose range of 3.75–40 mg; at higher doses, a less than dose‐proportional increase was observed. Food, sex, age, and race had no apparent effect on apararenone PK. A long half‐life was seen for apararenone and its principal metabolite; in addition, the exposure of the metabolite was lower than that of apararenone. Apararenone suppressed the decrease in urinary sodium and potassium ion ratio that occurs after loading with fludrocortisone. These studies support the mechanism of action of apararenone as an MRA, and further clinical development is warranted.
Keywords: apararenone, pharmacodynamics, pharmacokinetics, phase 1, safety
Diabetic nephropathy occurs in 20% to 30% of patients with type 1 or 2 diabetes mellitus. 1 Worldwide, diabetic nephropathy is the principal cause of chronic kidney disease requiring maintenance dialysis (ie, hemodialysis, peritoneal dialysis). 2 Incipient nephropathy progresses from microalbuminuria to overt nephropathy and gradual progression of renal impairment, 3 , 4 , 5 which ultimately leads to renal failure requiring renal replacement therapy. 6 , 7 Moreover, as the extent of albuminuria increases, the risks of cardiovascular events, renal events, and all‐cause mortality increase. 4 , 5 , 7 , 8
Several studies have reported that angiotensin‐converting enzyme inhibitors and angiotensin II‐receptor blockers restrict increases in proteinuria and the progression of renal impairment during early nephropathy. 9 , 10 , 11 However, other treatments such as mineralocorticoid receptor (MR) antagonists (MRAs) are under investigation for the management of diabetic nephropathy.
The principal endogenous mineralocorticoid, aldosterone, has been associated with hypertension and increased cardiovascular risk, obesity, and impaired insulin sensitivity and diabetes. 2 , 12 , 13 In various lifestyle‐related conditions (eg, hypertension, obstructive sleep apnea), MRs are activated in several body organs, such as the central nervous system, heart and vasculature, kidneys, pancreas, and skeletal and smooth muscle. 14 Further, MRs have been implicated in aging, inflammation, organ fibrosis, and oxidative stress, 15 and the traditional concept of MRAs as potassium ion (K+)‐sparing diuretics is evolving to the possibility of MRAs being therapeutic agents with wide‐ranging organ‐protective benefits in several clinical settings. 14 For instance, overactivation of MRs in the kidneys can cause renal damage, independently of blood pressure, 2 , 15 , 16 and clear evidence exists of clinical efficacy for the MRAs spironolactone and eplerenone in reducing renal damage and proteinuria in patients with diabetic nephropathy. 2 , 12
Disadvantages associated with therapeutic applications of existing steroidal MRAs include the poor specificity of spironolactone for MRs, which may therefore be associated with adverse events (AEs) related to sex hormone activity, such as breast tenderness, gynecomastia, and menstrual irregularity. 17 In addition, eplerenone has weaker affinity than spironolactone for androgen and progesterone receptors but is contraindicated in diabetic patients with proteinuria and moderate or severe renal dysfunction because of an increased risk of hyperkalemia. 2 , 18
In response, a new generation of nonsteroidal MRAs has recently been developed to obtain a selective receptor block, avoiding side effects like hyperkalemia. 19 The nonsteroidal MRA finerenone is now being assessed for the treatment of diabetic nephropathy and chronic heart failure, whereas esaxerenone is in development for diabetic nephropathy and, in some regions, is also marketed as an antihypertensive agent. 2 , 19 , 20 Apararenone (MT‐3995; Supplemental Figure 1) is a long‐acting nonsteroidal MRA with selective antagonist activity at MRs. Although apararenone had less inhibitory potential on MRs compared with spironolactone (half maximal inhibitory concentration [IC50], 0.28 vs 0.01 μmol/L), it had comparable inhibitory potential on estrogen receptors (both >100 μmol/L), and weaker inhibitory potential on androgen, progesterone, and glucocorticoid receptors (>100 vs 0.05–6.05 μmol/L). In adrenalectomized and uninephrectomized rat models, apararenone had favorable efficacy and safety profiles.
The purpose of the present studies was to define the safety, tolerability, and pharmacokinetic (PK) and pharmacodynamic (PD) profiles of apararenone. We evaluated the effects of food, sex, age, and race on apararenone PK, whereas the PD evaluation included confirmation of MR antagonism as the principal mechanism of apararenone action.
Methods
Study Ethics
This article describes the findings from 3 separate studies, all of which had their study protocols, participant information sheets, informed consent forms, and all other relevant study documentation approved by the appropriate independent ethics committee (Welwyn Clinical Pharmacology Ethics Committee, Welwyn Garden City, UK) or institutional review board (Kyushu Clinical Pharmacology Research Clinic, Fukuoka, Japan).
The studies were conducted in accordance with the principles of the Declaration of Helsinki (Seoul, Republic of Korea, October 2008), Guidelines of the International Conference on Harmonisation on Good Clinical Practice (Committee for Proprietary Medicinal Products/ICH/135/95), and other applicable regulatory requirements. All subjects provided written informed consent to participate in the study. Studies 1 and 3 were conducted at PAREXEL EPCU (Harrow, UK), and study 2 at Hakata Clinic (Fukuoka, Japan).
Participants
In all studies, subjects were healthy and free from clinically significant illness or disease, as determined by medical history, physical examination, and laboratory and other tests. In study 1, all subjects were male, except in study part 2.1, which was conducted in women; all subjects were aged 18–55 years, except in study part 2.2, which included men aged ≥65 years, and all subjects were Caucasian, except in study part 2.3, which was conducted in Blacks of African/Caribbean descent (see Table 1 and below for clarification of the study design). All study subjects had a body weight ≥50 kg (women) or ≥60 kg (men) and a body mass index (BMI) of 18–30 kg/m2.
Table 1.
Overview of the Apararenone Phase 1 Studies in This Analysis
| Study 1: Apararenone PK (single dose, effects of food, sex, age, and race; and multiple doses) and PD (multiple doses) | |
| Design | Randomized, double‐blind, placebo‐controlled |
| Subjects | Healthy male volunteers (except part 2.1, which was conducted in women); all were Caucasian (except part 2.3, which was conducted in Blacks of African/Caribbean descent) |
| Age | 18–55 years (except in part 2.2, which was conducted in subjects aged ≥65 years) |
| Body weight | Women ≥50 kg, men ≥60 kg |
| Data collection | PK, safety, tolerability, PD |
| Apararenone doses | Part 1 — single ascending doses of 3.75, 10, 20, 40, 80, 160, 320, and 640 mg (at each dose, n = 6 plus 2 matched placebo recipients; fasting state [part 1.1], then fed state [part 1.2]) |
| Part 2.1 — single dose of 20 mg (n = 6) or placebo (n = 2) in women aged 18–55 years | |
| Part 2.2 — single dose of 20 mg (n = 6) or placebo (n = 2) in men aged ≥65 years | |
| Part 2.3 — single dose of 20 mg (n = 6) or placebo (n = 2) in men of Black African/Caribbean origin | |
| Part 3.1 — daily doses of 10, 20, or 40 mg/day (days 1–14) in men; at each dose, n = 8 plus 2 matched placebo recipients | |
| Part 3.2 — loading dose of 320 mg (day 1), followed by 10 mg/day (days 2–14) in men; n = 8 plus 2 matched placebo recipients | |
| Part 3.3 — loading dose of 320, 80, or 40 mg (day 1), followed by 20, 5, or 2.5 mg/day, respectively (days 2–14) (males; at each dose, n = 8 plus 2 matched placebo recipients) | |
| Study 2: Single‐dose study in healthy Japanese men | |
| Design | Randomized, double‐blind, placebo‐controlled |
| Subjects | Healthy Japanese men |
| Age | 20–55 years |
| Body weight | ≥60 kg |
| Data collection | PK, safety |
| Apararenone doses | Single dose of 5, 10, 20, or 320 mg (at each dose, n = 6 plus 2 matched placebo recipients) |
| Study 3: Study of PD effects of single‐dose apararenone (after fludrocortisone challenge; eplerenone active control) | |
| Design | Randomized, double‐blind, placebo‐controlled |
| Subjects | Healthy Caucasian male volunteers |
| Age | 18–55 years |
| Body weight | ≥60 kg |
| Data collection | PK, safety, tolerability, PD |
| Apararenone doses | Part 1 — single dose of apararenone 160 mg (n = 9), placebo (n = 4), or eplerenone 200 mg (n = 9) |
| Part 2 — single dose of apararenone 640 mg (n = 9) or placebo (n = 2) | |
| Fludrocortisone challenge (in parts 1 and 2) — the first fludrocortisone dose (1.0 mg) was administered 9 hours predose (day −1), then fludrocortisone 0.5 mg was administered at the same time as the investigational product (day 1) and 16 hours postdose; and fludrocortisone 0.1 mg was administered 2, 4, 6, 8, 10, 12, 14, and 24 hours postdose | |
PD, pharmacodynamics; PK, pharmacokinetics.
Inclusion criteria for subjects in study 2 and study 3, who were Japanese men aged 20–55 years and Caucasian men aged 18–55 years, respectively, were the same as for this age and sex group in study 1.
All studies excluded subjects taking food supplements or prescribed or over‐the‐counter medicines, including salt substitutes, K+ supplements, K+‐sparing diuretics, high‐dose vitamins, and herbal and dietary supplements (eg, St John's wort) during the 7 days (or 14 days if the medicine was a potential enzyme inducer) or 5 half‐lives (whichever was longer) before first study drug administration. In addition, subjects consuming food or drink containing grapefruit, Seville oranges, or cranberry products (including marmalade and fruit juices), and/or food or drink containing licorice (confectionery products), from 7 to 14 days before baseline, or subjects using tobacco or nicotine‐containing products (snuff, chewing tobacco, cigars, pipes, or nicotine‐replacement products) within 6 months before the first study drug administration or a positive cotinine test at screening or baseline were excluded.
Study Design
The designs of all 3 studies are summarized in Table 1 and below.
Study 1 was a randomized, double‐blind, placebo‐controlled first‐in‐human study to determine the safety, tolerability, and PK of ascending and multiple doses of apararenone and the PD effects of apararenone after multiple doses. The study also investigated the effects of food, sex, age, and race on the PK of a single dose of apararenone. The study was divided into 3 parts. Blood samples for PK analysis of apararenone and the principal apararenone metabolite (termed 1118174; Supplemental Figure 1) were collected in part 1.1 (fasted state) and part 2 as follows: from predose to 336 hours postdose (3.75 mg/day), from predose to 672 hours postdose (10 mg/day), from predose to 1176 hours postdose (20 mg/day), and from predose to 2688 hours postdose (40–640 mg/day). In part 1.2 (fed state), blood samples for PK analysis were collected from predose until 1176 hours postdose.
In part 3, blood samples for PK analysis were collected on day 1 (from predose to 12 hours postdose); on days 3, 5, 7, 9, 11, and 13 (predose); on days 2, 4, 6, 8, 10, and 12 (predose and 4 hours postdose); and on day 14 (from predose to 2016 hours postdose). Urine samples for PK analysis of apararenone and 1118174 were also collected within 24 hours before administration to before the start of administration on day 1, 0–24 hours after administration on day 1, and 0–24, 24–48, and 48–72 hours after administration on day 14. In part 3, whole‐blood samples were collected to assess PD parameters. For the measurement of plasma aldosterone concentration (PAC), plasma renin concentration (PRC), and plasma renin activity (PRA), whole blood was collected at the following times: from −24 to −12 hours predose on day 1, predose on day 1, and from predose to 24 hours postdose on day 14.
Study 2 was a randomized, double‐blind, placebo‐controlled single‐oral‐dose study of apararenone 5, 10, 20, or 320 mg (Table 1). The collection of blood samples for PK analysis was similar to that in part 1 of study 1.
Study 3 was a randomized, double‐blind, placebo‐controlled study of apararenone to evaluate the PD effects of single oral doses after the administration of fludrocortisone, using eplerenone as a randomized, open‐label, active control. The study also investigated the safety and tolerability of single oral doses of apararenone and was divided into 2 parts (Table 1). Eplerenone was chosen as the active control because it has high MR selectivity and is safer than spironolactone with regard to the lower incidence of sex hormone‐related AEs. 21 , 22
In study 3, antialdosterone activity was determined by urine levels of Na+ and K+ after repeat administration of fludrocortisone, a valid method to confirm antialdosterone activity of potential drug candidates that reverse the urinary electrolyte effect induced after fludrocortisone challenge. 21 , 22 , 23 All subjects in both study parts also received a fludrocortisone challenge as follows: the first fludrocortisone dose (1.0 mg) was administered 9 hours predose (day −1), then fludrocortisone 0.5 mg was administered at the same time as the investigational product (day 1) and 16 hours postdose, and fludrocortisone 0.1 mg was administered 2, 4, 6, 8, 10, 12, 14, and 24 hours postdose.
In study 3, the blood samples for PK analysis of apararenone and eplerenone were collected from predose to 48 hours postdose. Urine samples for PD analysis were collected 24–9 hours predose, 9–0 hours predose, and at the following times postdose: 0–2, 2–4, 4–6, 6–8, 8–10, 10–12, 12–14, 14–16, 16–24, 24–32, 32–40, and 40–48 hours.
Bioanalytical Methods
Concentrations of apararenone, 1118174, and eplerenone in human plasma and urine samples were measured using validated bioanalytical methods for liquid chromatography‐tandem mass spectrometry, with solid‐phase extraction and relevant internal standards (Supplemental Table 1). The mobile phase was a mixture of 0.1% formic acid in acetonitrile and 0.1% formic acid in water for apararenone and 1118174 or a mixture of acetonitrile and 10 mM ammonium acetate for eplerenone. All instrument control, data collection, peak area integration, and storage were performed using Analyst (AB Sciex LLC., Framingham, Massachusetts). The mass spectrometer response (peak area ratio of analyte to internal standard) of each calibration standard was calculated by Watson LIMS (Thermo Fisher Scientific, Waltham, Massachusetts) and plotted against the nominal (prepared) concentration. A weighted (1/x2) least‐squares linear regression analysis was used to calculate an equation of the calibration line. Concentrations of apararenone, 1118174, and eplerenone in the samples were back‐calculated from the calibration lines to 3 significant figures.
During sample analysis, the back‐calculated concentrations of the apararenone, 1118174, and eplerenone calibration standards were within ±15% of the nominal value (±20% at the lower limit of quantification) for ≥75% of the calibration standards in each accepted analytical batch. The accuracy of the apararenone, 1118174, and eplerenone quality control (QC) samples was within ±15% of the nominal values for at least two‐thirds of the QC samples (≥50% at each concentration) in each accepted batch. Mean precision and accuracy for low‐, medium‐, and high‐quality control samples during analysis of all analytes indicated that the method performed reliably during the analysis of study samples.
PAC was measured by a solid‐phase 125I radioimmunoassay using the Siemens Coat‐A‐Count aldosterone assay (PITKAL‐10; Siemens, Los Angeles, California). PRC was determined using the LIAISON direct renin assay and LIAISON analyzer (DiaSorin Inc., Stillwater, Minnesota); this is a chemiluminescent immunoassay used for the quantitative determination of renin in human ethylenediaminetetraacwetic acid‐plasma specimens. The RENCTK radioimmunoassay kit (DiaSorin Inc., Stillwater, Minnesota) was used to determine PRA by quantitative determination of angiotensin I.
Safety and Tolerability Assessments
In all studies, the following parameters were assessed: physical examination; vital signs including supine blood pressure, pulse rate, and auricular body temperature; 12‐lead electrocardiogram parameters including PR, QRS, QT, and corrected QT intervals (QTc using Bazett's formula); clinical laboratory assessments including hematology, coagulation, biochemistry, thyroid function, and urinalysis; and AEs.
Pharmacokinetic, Pharmacodynamic, and Statistical Analyses
In all 3 parts of study 1, the principal PK end points for apararenone and its major metabolite (1118174) included peak plasma concentration (Cmax), time to peak plasma concentration (tmax), area under the plasma concentration‐time curve (AUC) from time zero to 24 hours postdose (AUC0–24), AUC from time zero to the last quantifiable concentration (AUC0–t), AUC from time zero extrapolated to infinity (AUC0–∞), elimination half‐life (t1/2), and terminal elimination rate constant (Kel). Apparent oral clearance (CL/F) was determined for apararenone only. The accumulation ratio was calculated as AUC0–24 on day 14/AUC0–24 on day 1. In part 3 of study 1, the principal PD parameters evaluated were AUC0–24 for PAC, PRA, and PRC.
In study 2 and study 3, PK parameters similar to those in part 1 of study 1 were evaluated for apararenone, 1118174, and eplerenone (study 3 only). In study 3, PD parameters assessed for apararenone and eplerenone were urinary sodium (Na+) and K+ excretion and the urinary log10 Na+/K+ ratio.
The plasma PK parameters and plasma PD parameters were determined by noncompartmental analysis from individual concentration‐time data for apararenone, 1118174, and eplerenone in plasma for all groups using validated software (WinNonLin Professional; Pharsight Corporation, Mountain View, California). All values below the limit of quantification were imputed with a value of zero.
All statistical analyses were performed using the latest available version of SAS (SAS Institute Inc., Cary, North Carolina). For the urine PD parameters in study 3, PD responses of apararenone and eplerenone were assessed by analysis of variance. The estimates of differences for placebo or eplerenone and 95% confidence intervals (CIs) were calculated.
Results
Baseline and Demographic Characteristics
Study 1 included 64 subjects of mean ± standard deviation (SD) age 30.9 ± 9.1 years and mean ± SD BMI of 24.7 ± 2.3 kg/m2. Study 2 included 32 subjects, and mean age, height, weight, and BMI were similar between the study groups; mean age was 24.2–26.5 years, and mean BMI was 21.3–23.0 kg/m2. Study 3 included 33 subjects, and mean age, height, weight, and BMI were similar between the study groups; mean age was 27.7–35.7 years, and mean BMI was 23.8–26.0 kg/m2 (Supplemental Table 2).
In study 1, 14 subjects withdrew, including 9 in part 1.1, 3 in part 1.2 (2 of these subjects were included in the withdrawals within part 1.1), and 4 in part 3 of the study. One subject in the placebo group withdrew from study 2, and there were no subject withdrawals in study 3.
Pharmacokinetics and Pharmacodynamics
In study 1, after single oral dosing of apararenone in Caucasian subjects in a fasted state, an approximately dose‐proportional increase was observed in Cmax and AUC0–∞ for apararenone over the dose range of 3.75–40 mg, whereas at higher doses, a less than dose‐proportional increase was observed (Supplemental Table 3 and Supplemental Figure 2). Median tmax was delayed with increasing dose. The mean t1/2 was similar for all doses. For 1118174, although the PK could not be adequately evaluated at lower doses, owing to the low concentration, similar trends to apararenone were observed for dose proportionality, tmax, and t1/2. Additionally, the metabolic ratios were similar among doses.
PK parameters for apararenone and 1118174 on day 1 and day 14 for the multiple‐dose assessments in study 1 are shown in Table 2, and the time course of mean plasma concentrations for apararenone and 1118174 are shown in Figure 1. After multiple oral dosing of apararenone between 10 and 40 mg, without a loading dose in the fed state, steady state was approached but generally not achieved, with the trough concentration still increasing on day 14 (336 hours). The accumulation ratio of AUC0–24 for apararenone was approximately 8‐fold. An approximately dose‐proportional increase was observed in Cmax and AUC0–24 on day 14. The mean t1/2 for apararenone and 1118174 ranged from 275 to 285 hours and from 1126 to 1250 hours, respectively; these values were comparable to the results observed after single‐dose administration.
Table 2.
Pharmacokinetic Parameters of Apararenone and 1118174 on Days 1 and 14 After Multiple Oral Administration of Apararenone (Study 1)
| Apararenone Dose (Day 1) | |||||||
|---|---|---|---|---|---|---|---|
| Pharmacokinetic Parameter | 10 mg (n = 8) | 20 mg (n = 8) | 40 mg (n = 8) | LD 40 mg/2.5 mg (n = 8) | LD 80 mg/5 mg (n = 8) | LD 320 mg/10 mg (n = 8) | LD 320 mg/20 mg (n = 8) |
| Apararenone | |||||||
| Cmax, ng/mL | 160.5 (21.1) | 310.8 (55.3) | 544.1 (88.9) | 562.3 (149.4) | 1077.3 (310.2) | 2006.3 (479.1) | 2255.0 (447.3) |
| tmax, h a | 4.0 (3.0–4.0) | 4.0 (4.0–10.0) | 4.0 (3.0–12.0) | 4.5 (4.0–23.9) | 5.0 (3.0–23.9) | 17.0 (4.0–23.9) | 10.0 (4.0–24.0) |
| AUC0–24, ng·h/mL | 2867 (522) | 5479 (932) | 10,022 (2718) | 10,535 (2370) | 21,055 (5946) | 40,056 (9472) | 42,599 (8733) |
| 1118174 | |||||||
| Cmax, ng/mL | 0.55 (1.02) | 2.93 (1.64) | 4.86 (4.67) | 5.59 (3.33) | 18.26 (11.92) | 29.38 (15.88) | 26.43 (10.95) |
| tmax, h a | 12.3 (0.7–23.9) b | 23.9 (23.9–23.9) c | 23.9 (23.9–24.0) d | 23.9 (23.9–24.0) c | 23.9 (23.9–23.9) | 23.9 (23.9–23.9) | 24.0 (23.9–24.0) |
| AUC0–24, ng·h/mL | 7 (9) b | 31 (21) c | 63 (53) d | 67 (41) c | 204 (130) | 335 (167) | 324 (127) |
| Apararenone Dose (Day 14) | |||||||
|---|---|---|---|---|---|---|---|
| 10 mg (n = 8) | 20 mg (n = 8) | 40 mg (n = 8) | LD 40 mg/2.5 mg (n = 7) | LD 80 mg/5 mg (n = 8) | LD 320 mg/10 mg (n = 8) | LD 320 mg/20 mg (n = 8) | |
| Apararenone | |||||||
| Cmax, ng/mL | 1156.3 (130.7) | 2083.8 (382.9) | 3653.8 (565.6) | 437.7 (70.9) | 799.6 (163.5) | 1703.8 (237.4) | 2677.5 (427.2) |
| tmax, h a | 7.5 (5.0–10.0) | 5.0 (4.0–10.0) | 5.0 (4.0–6.0) | 5.0 (0.0–10.0) | 4.5 (1.5–10.0) | 5.0 (0.0–10.0) | 4.5 (3.0–12.0) |
| AUC0–24, ng·h/mL | 23,956 (2452) | 43,838 (7683) | 77,034 (12,056) | 9494 (1909) | 17,027 (3425) | 36,429 (5065) | 57,948 (8757) |
| t1/2, h | 282 (74) | 285 (93) | 275 (99) | 269 (71) | 232 (63) | 282 (69) | 288 (64) |
| AR | 8.5 (0.8) | 8.1 (1.4) | 7.9 (1.2) | 0.92 (0.18) | 0.86 (0.24) | 0.94 (0.14) | 1.39 (0.21) |
| 1118174 | |||||||
| Cmax, ng/mL | 157.1 (53.6) | 262.9 (87.5) | 442.6 (148.3) | 76.0 (28.6) | 170.4 (62.7) | 318.8 (105.7) | 380.5 (103.5) |
| tmax, h a | 504.4 (72.0–1008.6) | 227.8 (119.1–1512.8) | 263.7 (190.7–1008.6) | 335.1 (10.0–599.8) | 192.1 (72.0–337.1) | 155.4 (10.0–193.0) | 263.7 (0.0–337.3) |
| AUC0–24, ng·h/mL | 2292 (1010) | 3660 (1346) | 5811 (3039) | 1178 (522) | 3198 (1544) | 5563 (2051) | 6194 (2575) |
| t1/2, h | 1250 (559) c | 1126 (393) d | 1147 (500) d | 1487 (595) e | 1049 (469) c | 1247 (405) | 1218 (454) |
| AR | 977 (959) b | 170 (84) c | 158 (81) d | 25.4 (12.3) d | 17.0 (2.8) | 17.9 (3.8) | 19.5 (3.7) |
AR, accumulation ratio; AUC0–24, area under the plasma concentration‐time curve from time 0 to 24 hours; Cmax, peak plasma concentration; LD, loading dose; tmax, time to Cmax.
Data shown are mean (standard deviation).
Median (range).
n = 2.
n = 7.
n = 6.
n = 5.
Figure 1.
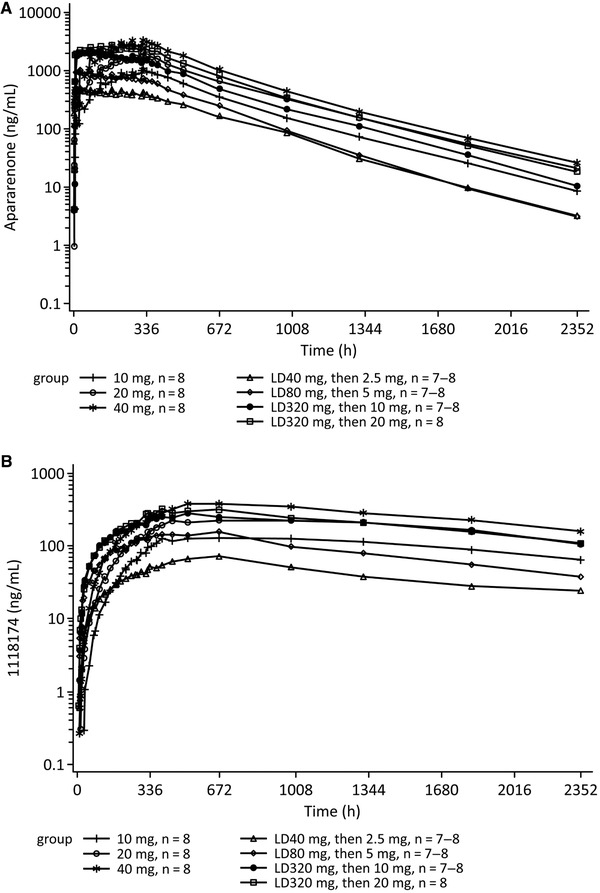
Time course (log‐linear scale) of mean plasma concentrations of apararenone (A) and 1118174 (B) after multiple oral administrations of apararenone (study 1). LD, loading dose.
When 40, 80, 320, and 320 mg of apararenone was administered for 1 day as a loading dose, followed by 2.5, 5, 10, and 20 mg of apararenone as a maintenance dose for 13 days, the mean trough concentration of apararenone was maintained at an almost constant level around 14 days at all the doses; however, the concentration of 1118174 continued to increase. An approximately dose‐proportional increase was observed in Cmax and AUC0–24 for apararenone on day 14, whereas values for 1118174 increased less than dose‐proportionally. The mean t1/2 values for apararenone and 1118174 were similar to t1/2 values in Caucasians (study 1). After 80 and 320 mg of apararenone was administered in a fed state, the Cmax and AUC0–24 for apararenone was higher than that observed after the administration of the same dose in a fasted state in part 1.1 of the study. The urinary excretion was minimal (up to 14% of the dose of apararenone and up to 0.5% of the dose of 1118174 on day 14).
For the PD assessment in study 1, the AUC0–24 of PAC, PRA, and PRC after multiple oral dosing increased in an approximately dose‐dependent manner, and the values were higher than baseline in all cohorts (Figure 2A–C).
Figure 2.
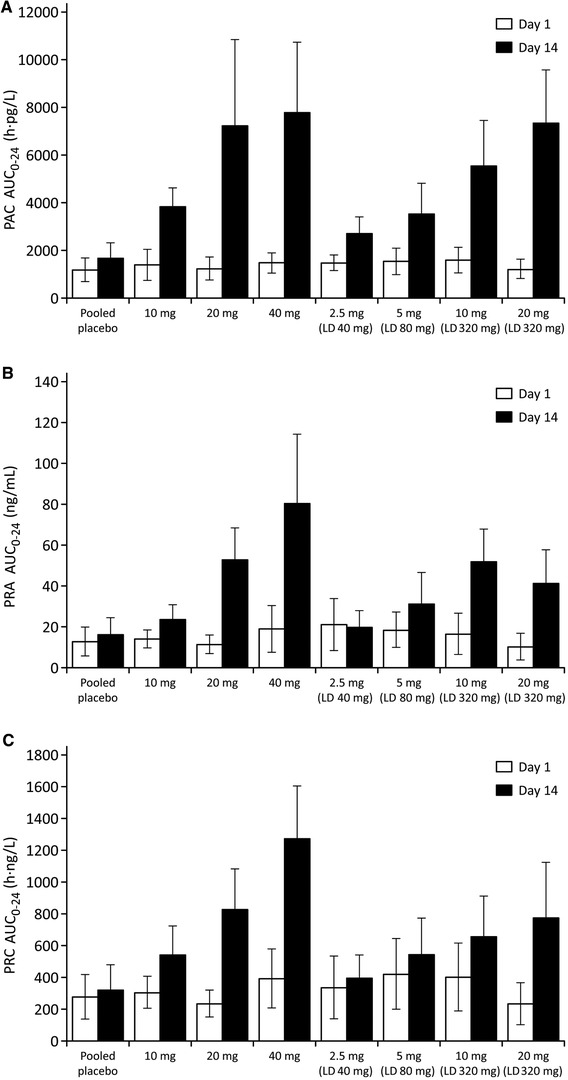
Arithmetic mean ± standard deviation area under the concentration‐time curve from 0 to 24 hours (AUC0–24) for (A) plasma aldosterone concentration (PAC), (B) plasma renin activity (PRA), and (C) plasma renin concentration (PRC) in study 1. LD, loading dose.
PK parameters for apararenone and 1118174 after single oral apararenone doses in Japanese subjects in the fasted state, as assessed in study 2, are shown in Supplemental Table 4. The results obtained in Japanese subjects in this study were similar to the results obtained in Caucasian subjects in study 1.
The effects of food, sex, age, and race (in Black and Japanese subjects) on apararenone PK after administration of a single oral dose of 20 mg were assessed in studies 1 and 2. Table 3 provides a summary of these effects. The effects of food, sex, age, and race on plasma apararenone concentrations are shown in Supplemental Figure 3. None of these factors appeared to affect the PK of apararenone after a single 20‐mg oral dose.
Table 3.
Effects of Food, Sex, Age, and Race on PK of Apararenone After Single Oral Administration of Apararenone 20 mg (Studies 1 and 2)
| Study 1 | ||||||
|---|---|---|---|---|---|---|
| Parameter | (Control) Caucasian Male; Young: Aged ≤55 Years; Fasted (n = 6) | Fed (n = 5) | Female (n = 6) | Elderly: Aged ≥65 Years; (n = 6) | Black (n = 6) | Study 2 Japanese (n = 6) |
| Cmax, ng/mL | 239.7 (48.9) | 234.2 (32.3) | 261.8 (45.4) | 257.8 (68.0) | 239.8 (28.8) | 293.5 (30.1) |
| tmax, h a | 13.0 (2.0–71.9) | 6.0 (5.0–10.0) | 3.0 (1.5–24.0) | 3.5 (3.0–48.0) | 7.0 (2.0–10.1) | 7.0 (1.5–24.0) |
| AUC0–24, ng·h/mL | 4726 (813) | 4417 (1056) | 4629 (1080) | 4362 (888) | 4937 (692) | 5789 (713) |
| AUC0–t, ng·h/mL | 84,205 (13,582) b | 86,253 (20,084) | 99,169 (14,950) | 94,475 (17,211) | 88,701 (22,738) | 90,097 (26,315) |
| AUC0–∞, ng·h/mL | 90,816 (16,671) b | 90,886 (22,059) | 100,702 (14,729) | 96,294 (17,607) | 90,468 (22,849) | 92,489 (25,539) |
| t1/2, h | 304 (79) b | 266 (62) | 304 (72) | 344 (52) | 270 (93) | 225 (53) |
AUC0–24, area under the concentration‐time curve from time zero until 24 hours postdose; AUC0–t, area under the concentration‐time curve from time zero until the last quantifiable concentration; AUC0–∞, AUC from time zero extrapolated to infinity; Cmax, peak plasma concentration; SD, standard deviation; t1/2, elimination half‐life; tmax, time to Cmax.
Data shown are mean (standard deviation).
Median (range).
n = 4
For study 3, the time courses of mean plasma concentrations of apararenone and eplerenone after single oral administration are shown in Figure 3 A. The time courses of the mean urinary Na+/K+ ratios are shown in Figure 3B. The eplerenone effects were observed immediately, with an increase in ratio between 0 and 8 hours, which decreased from 12 hours onward, whereas the effect of apararenone 640 mg increased mainly from 8 to 12 hours onward. This is consistent with the earlier Cmax of eplerenone and the long t1/2 of apararenone. From 0 to 24 hours postdose, significantly higher cumulative ratios were observed with eplerenone 200 mg and apararenone 640 mg compared with placebo, whereas an increase in ratio relative to placebo from 24 to 48 hours was only observed with apararenone 160 and 640 mg (Table 4). PK parameters for apararenone and eplerenone after single oral administration of 160 and 640 mg apararenone and 200 mg eplerenone (study 3) are shown in Supplemental Table 5.
Figure 3.
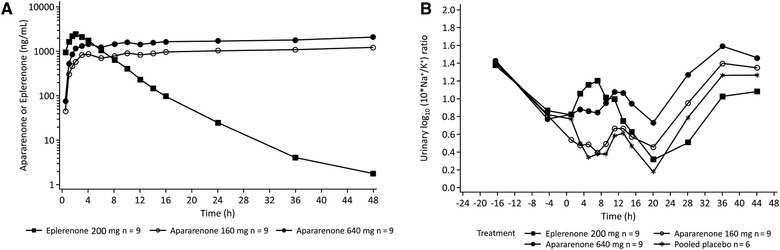
Time course (log‐linear scale) of mean plasma concentrations of apararenone and eplerenone after single oral administration of apararenone and eplerenone (A) and time course of mean urinary sodium/potassium (Na+/K+) ratio (B) in study 3.
Table 4.
Statistical Analysis of Urinary Sodium/Potassium Ratio (Study 3)
| Cumulative Time Interval | Statistics | Apararenone 160 mg (n = 9) | Apararenone 640 mg (n = 9) | Eplerenone 200 mg (n = 9) | Pooled Placebo (n = 9) |
|---|---|---|---|---|---|
| 0–24 hours postdose | LS means | 0.548 | 0.925 | 0.894 | 0.508 |
| Difference (95%CI) vs placebo | 0.041 (−0.136 to 0.218) | 0.418 (0.241–0.594) | 0.387 (0.210–0.564) | — | |
| P value vs placebo | .6410 | <.0001 | .0001 | — | |
| Difference (95%CI) vs eplerenone | −0.346 (−0.504 to −0.188) | 0.031 (−0.127 to 0.189) | — | — | |
| P value vs eplerenone | .0001 | 0.6932 | — | — | |
| 24–48 hours postdose | LS means | 1.232 | 1.441 | 0.911 | 1.117 |
| Difference (95%CI) vs placebo | 0.115 (−0.065 to 0.295) | 0.324 (0.144–0.504) | −0.206 (−0.386 to −0.025) | — | |
| P value vs placebo | .2016 | .0010 | .0269 | — | |
| Difference (95%CI) vs eplerenone | 0.321 (0.159–0.482) | 0.530 (0.368–0.691) | — | — | |
| P value vs eplerenone | .0003 | <.0001 | — |
CI, confidence interval; LS, least squares.
Safety
The treatment‐emergent AEs (TEAEs) occurring in study 1 are summarized in Supplemental Table 6. Across the entire study, dizziness was the most common TEAE in subjects receiving apararenone (n = 6 across doses and regimens). Other reported TEAEs with apararenone included headache (n = 3), thyroid function test abnormal (n = 3), abdominal pain (n = 1), urticaria (n = 1), nausea (n = 1), and hot flash (n = 1).
In study 2, 2 TEAEs were reported. These were moderate parotitis occurring in a subject in the placebo group and mild hepatic enzyme increase in a subject in the apararenone 5‐mg group.
In study 3, TEAEs were reported by 3.0% (1 of 33 subjects), 33.3% (3 of 9 subjects), 33.3% (3 of 9 subjects), 11.1% (1 of 9 subjects), and 16.7% (1 of 6 of subjects) in the fludrocortisone, apararenone 160‐mg, apararenone 640‐mg, eplerenone 200‐mg, and placebo groups, respectively. Headache was the most commonly occurring TEAE (fludrocortisone, 3.0% [1 of 33]; apararenone 160 mg, 33.3% [3 of 9]; apararenone 640 mg, 11.1% [1 of 9]). Single events of syncope (placebo, 16.7% [1 of 6]), back pain (apararenone 160 mg, 11.1% [1 of 9]), musculoskeletal chest pain (apararenone 160 mg, 11.1% [1 of 9]), toothache (apararenone 640 mg, 11.1% [1 of 9]), fatigue (eplerenone 200 mg, 11.1% [1 of 9]), nasopharyngitis (apararenone 640 mg, 11.1% [1 of 9]), and rhinorrhea (placebo, 16.7% [1 of 6]) were reported.
Discussion
This evaluation, which comprised data from 3 clinical studies of apararenone, characterized the safety, tolerability, PK, and PD profiles of apararenone, a long‐acting nonsteroidal MRA with selective antagonist activity at MRs in healthy volunteers. Safety data from the 3 studies indicated that apararenone was tolerated over the dose range administered.
As determined in study 1 and study 2, approximately dose‐proportional increases in Cmax and AUC were observed over the apararenone dose range of 3.75–40 mg, whereas at higher doses, a less than dose‐proportional increase was observed after single oral administration of apararenone in a fasted state. Considering that both the t1/2 of apararenone and the metabolic ratio were similar between doses, which means the elimination of apararenone was comparable at each dose, the nonlinear PK at higher doses was possibly because of reduced absorption rather than to metabolic saturation. Because an apararenone dose of approximately 10 mg was expected to be effective, based on the results of study 3 (as discussed below), and because the urinary albumin‐to‐creatinine ratio‐lowering effect of apararenone for 24 weeks in patients with stage 2 diabetic nephropathy was confirmed over the dose range 2.5–10 mg (data on file), apararenone PK in the area around the effective apararenone dose range appeared to be linear.
Multiple‐dose administration of apararenone was performed in the fed state over a period of 14 days in 7 dose groups, with or without a loading dose. Because of the long half‐life, steady state was generally not achieved for both apararenone and 1118174 within 14 days of multiple‐dose administration without a loading dose. In the dose groups with a loading dose, the plasma concentration of apararenone was maintained at an almost constant level for approximately 14 days; however, the plasma concentration of 1118174 was still increasing. PK parameters for apararenone and 1118174 after the first dose (day 1) up to 40 mg were comparable to those observed after single doses in part 1 of study 1 in the fasted state. For doses higher than 40 mg, exposure was higher in part 3 compared with an identical part 1 dose. This could possibly be attributed to a food effect at doses higher than 40 mg, which increased drug solubility in the gastric lumen in the fed state.
Study 1 showed 1118174 exposure amounted to approximately 50% of that of apararenone (metabolic ratio range, 0.421–0.652). The binding affinity of 1118174 for MR is approximately one‐fiftieth that of apararenone (inhibitory constant [ki], 0.106 μmol/L and 5.21 nmol/L, respectively), and the protein‐binding ratio of 1118174 was similar to that of apararenone (96.5%–97.1% and 94.2%–94.6%, respectively; data on file), suggesting that 1118174 does not contribute to apararenone efficacy.
The current evaluations indicated a long half‐life for both apararenone and 1118174. Apararenone circulates in enterohepatic circulation in rat models. After oral administration of [14C]‐apararenone to bile duct‐cannulated rats, excretion into bile and urine within 48 hours was 55.9%, and 8.4%, respectively. Moreover, cumulative excretion of radioactivity in bile, urine, and feces after intraduodenal injection of the bile, which was collected from other bile duct‐cannulated rats after oral administration of [14C]‐apararenone, was 33.3%, 7.4%, and 48.0% of the dose, respectively.
Apararenone is also excreted mainly by glucuronidation in humans. Specifically, the principal metabolite of apararenone in human plasma was determined to be 1118174 after administration of a single oral dose of radiolabeled apararenone in another clinical pharmacology study. Although minimal concentrations of apararenone glucuronide were detected in plasma, apararenone was predominantly excreted as apararenone glucuronide, suggesting that apararenone elimination in humans was mainly via metabolism by uridine 5'‐diphospho‐glucuronosyltransferase.
In addition, multiple administrations of activated charcoal resulted in reduced exposure and shortened elimination of apararenone and 1118174 in another (unpublished) clinical pharmacology study. Oral administration of activated charcoal reduced the half‐life of unchanged apararenone in plasma by about 4‐fold (56 and 240 hours during and after charcoal dosing, respectively) and that of 1118174 by about 9‐fold (96 and 865 hours during and after charcoal dosing, respectively). These findings suggested that, as supported by a rat model, absorbed apararenone was secreted into the gastrointestinal tract as unchanged apararenone or metabolites via enterohepatic circulation, and charcoal adsorbed to them in the intestine and enhanced the excretion.
Although partly based on findings from animal studies, we speculate that the long apparent half‐lives of apararenone and 1118174 in humans could be attributed to enterohepatic recycling. Importantly, the long half‐lives of apararenone and 1118174 do not appear to result in safety concerns within the parameters assessed in the current studies.
Considering the intrinsic and extrinsic factors affecting the PK profile of apararenone, no apparent differences were observed regarding the effects of food, sex, age, and race (Caucasian, Black, Japanese). Age is a risk factor for nephropathy and nonalcoholic steatohepatitis, and both diseases have a high prevalence in the elderly. 23 , 24 The lack of an age effect on apararenone PK suggests that apararenone dose adjustment is unlikely to be necessary in the elderly.
Although not assessed in the current evaluations, the impact of nephropathy (ie, renal function decline) on apararenone PK is expected to be small, owing to low urinary excretion of the drug. Therefore, strict apararenone dose adjustment is not expected to be necessary during clinical application.
From the results of study 1 and previous preclinical evaluations, a maintenance dose of 5–10 mg of apararenone was considered effective. Because apararenone has a long half‐life and accumulates with repeated dosing, it was considered that single doses of 160 and 640 mg were required to obtain the same plasma concentrations after repeated doses of 5 and 10 mg, respectively.
In study 3, a single dose of 640 mg showed the same effect (ie, Na+/K+ ratio) as eplerenone 200 mg. The eplerenone effect was observed immediately, with an increase in the Na+/K+ ratio between 0 and 8 hours, which decreased from 12 hours onward, whereas the effect of apararenone 640 mg increased mainly from 8 to 12 hours onward. This is consistent with the earlier Cmax of eplerenone and the long half‐life of apararenone. Of note, urine Na+/K+ ratio and PAC are an index of anti‐MR activity, 25 and it is thought that MR antagonism could be confirmed with the results of study 1 and study 3. These results suggest that apararenone 10 mg/day will ultimately be effective in the clinical setting, which is also supported by findings from phase 2 clinical studies (data on file).
Based on their mechanism of action, traditional MRAs present important side effects, such as hyperkalemia, that limit their use in clinical practice. It is suggested that nonsteroidal MRAs with higher MR selectivity decrease the risk of hyperkalemia. 20 , 26 The selectivity of apararenone for human MRs over 5 other human steroid receptors (ie, androgen, progesterone, glucocorticoid, and estrogen receptors alpha and beta) was superior to that for eplerenone and spironolactone in corresponding specific‐binding assays. 27 In addition, with the current findings for apararenone and eplerenone, plasma drug concentrations and urine Na+/K+ ratios were related, with serum K+ elevations appearing to depend on plasma drug concentrations. Therefore, apararenone, which has high selectivity for MRs and a small peak‐trough fluctuation, may show a low risk of hyperkalemia.
Limitations of the current evaluation include that the studies were conducted in healthy volunteers and were either single‐dose assessments or multiple‐dose assessments of short duration. However, it is expected that further clinical development of apararenone will yield additional insight into the drug's PK, PD, and safety and tolerability profiles, including in specific patient populations.
Conclusion
The findings of this evaluation of phase 1 studies in healthy volunteers, which characterized the PK of apararenone and its principal metabolite, 1118174, found no intrinsic and extrinsic effects of food, sex, age, and race on apararenone PK. In addition, apararenone suppressed the decrease in urinary Na+/K+ ratio that occurs after loading with fludrocortisone. These phase 1 studies support the mechanism of action of apararenone as an MRA, and further clinical development is warranted.
Conflicts of Interest
Both authors are full‐time employees of Mitsubishi Tanabe Pharma Corporation.
Data Sharing
Data sharing from these studies will be reviewed and considered on a case‐by‐case basis by the corresponding author.
Author Contributions
T.N. contributed to study design, conduct and/or collection, data analysis and interpretation, writing and/or review of the article, and final approval of the article for submission. A.K. contributed to the writing and/or review of the article and final approval of the article for submission.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Acknowledgments
Apararenone is being developed by Mitsubishi Tanabe Pharma Corporation. These studies were funded by Mitsubishi Tanabe Pharma Corporation. We thank David Murdoch (BSc, Hons.) and Tricia Newell (PhD) of Edanz Medical Writing for providing medical writing support, which was funded by Mitsubishi Tanabe Pharma Corporation.
References
- 1. American Diabetes Association . Nephropathy in diabetes. Diabetes Care. 2004;27(suppl 1):s79‐s83. [DOI] [PubMed] [Google Scholar]
- 2. Ito S, Shikata K, Nangaku M, Okuda Y, Sawanobori T. Efficacy and safety of esaxerenone (CS‐3150) for the treatment of type 2 diabetes with microalbuminuria: a randomized, double‐blind, placebo‐controlled, phase II trial. Clin J Am Soc Nephrol. 2019;14(8):1161‐1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sumida K, Molnar MZ, Potukuchi PK, et al. Changes in albuminuria and subsequent risk of incident kidney disease. Clin J Am Soc Nephrol. 2017;12(12):1941‐1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Toyama T, Furuichi K, Ninomiya T, et al. The impacts of albuminuria and low eGFR on the risk of cardiovascular death, all‐cause mortality, and renal events in diabetic patients: meta‐analysis. PLoS One. 2013;8(8):e71810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saito T, Mochizuki T, Uchida K, Tsuchiya K, Nitta K. Metabolic syndrome and risk of progression of chronic kidney disease: a single‐center cohort study in Japan. Heart Vessels. 2013;28(3):323‐329. [DOI] [PubMed] [Google Scholar]
- 6. Oshima M, Toyama T, Haneda M, et al. Estimated glomerular filtration rate decline and risk of end‐stage renal disease in type 2 diabetes. PLoS One. 2018;13(8):e0201535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wada T, Haneda M, Furuichi K, et al. Clinical impact of albuminuria and glomerular filtration rate on renal and cardiovascular events, and all‐cause mortality in Japanese patients with type 2 diabetes. Clin Exp Nephrol. 2014;18(4):613‐620. [DOI] [PubMed] [Google Scholar]
- 8. Zhang XL, Yuan MX, Wan G, et al. The effects of AER and eGFR on outcomes of CVD in patients with T2DM in an urban community over 8 years of multifactorial treatment: the Beijing Communities Diabetes Study 18. Ther Clin Risk Manag. 2018;14:1537‐1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Atkins RC, Briganti EM, Lewis JB, et al. Proteinuria reduction and progression to renal failure in patients with type 2 diabetes mellitus and overt nephropathy. Am J Kidney Dis. 2005;45(2):281‐287. [DOI] [PubMed] [Google Scholar]
- 10. Sano T, Kawamura T, Matsumae H, et al. Effects of long‐term enalapril treatment on persistent micro‐albuminuria in well‐controlled hypertensive and normotensive NIDDM patients. Diabetes Care. 1994;17(5):420‐424. [DOI] [PubMed] [Google Scholar]
- 11. Japanese Society of Nephrology . Evidence‐based clinical practice guideline for CKD 2013. Clin Exp Nephrol. 2014;18(3):346‐423. [DOI] [PubMed] [Google Scholar]
- 12. Hirsch JS, Drexler Y, Bomback AS. Aldosterone blockade in chronic kidney disease. Semin Nephrol. 2014;34(3):307‐322. [DOI] [PubMed] [Google Scholar]
- 13. Mavrakanas TA, Gariani K, Martin PY. Mineralocorticoid receptor blockade in addition to angiotensin converting enzyme inhibitor or angiotensin II receptor blocker treatment: an emerging paradigm in diabetic nephropathy: a systematic review. Eur J Intern Med. 2014;25(2):173‐176. [DOI] [PubMed] [Google Scholar]
- 14. Guichard JL, Clark D 3rd, Calhoun DA, Ahmed MI. Aldosterone receptor antagonists: current perspectives and therapies. Vasc Health Risk Manag. 2013;9:321‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jaisser F, Farman N. Emerging roles of the mineralocorticoid receptor in pathology: toward new paradigms in clinical pharmacology. Pharmacol Rev. 2016;68(1):49‐75. [DOI] [PubMed] [Google Scholar]
- 16. Schwenk MH, Hirsch JS, Bomback AS. Aldosterone blockade in CKD: emphasis on pharmacology. Adv Chronic Kidney Dis. 2015;22(2):123‐132. [DOI] [PubMed] [Google Scholar]
- 17. Kallistratos MS, Pittaras A, Theodoulidis I, Grassos C, Poulimenos LE, Manolis AJ. Adverse effects of mineralocorticoid receptor antagonist administration. Curr Pharm Des. 2018;24(46):5537‐5541. [DOI] [PubMed] [Google Scholar]
- 18. Vukadinovic D, Lavall D, Vukadinovic AN, Pitt B, Wagenpfeil S, Bohm M. True rate of mineralocorticoid receptor antagonists‐related hyperkalemia in placebo‐controlled trials: a meta‐analysis. Am Heart J. 2017;188:99‐108. [DOI] [PubMed] [Google Scholar]
- 19. Capelli I, Gasperoni L, Ruggeri M, et al. New mineralocorticoid receptor antagonists: update on their use in chronic kidney disease and heart failure. J Nephrol. 2020;33(1):37‐48. [DOI] [PubMed] [Google Scholar]
- 20. Katayama S, Yamada D, Nakayama M, et al. A randomized controlled study of finerenone versus placebo in Japanese patients with type 2 diabetes mellitus and diabetic nephropathy. J Diabetes Complications. 2017;31(4):758‐765. [DOI] [PubMed] [Google Scholar]
- 21. Colussi G, Catena C, Sechi LA. Spironolactone, eplerenone and the new aldosterone blockers in endocrine and primary hypertension. J Hypertens. 2013;31(1):3‐15. [DOI] [PubMed] [Google Scholar]
- 22. Sato A. Mineralocorticoid receptor antagonists: their use and differentiation in Japan. Hypertens Res. 2013;36(3):185‐190. [DOI] [PubMed] [Google Scholar]
- 23. Reutens AT, Atkins RC. Epidemiology of diabetic nephropathy. Contrib Nephrol. 2011;170:1‐7. [DOI] [PubMed] [Google Scholar]
- 24. Iqbal U, Perumpail BJ, Akhtar D, Kim D, Ahmed A. The epidemiology, risk profiling and diagnostic challenges of nonalcoholic fatty liver disease. Medicines (Basel). 2019;6(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eudy RJ, Sahasrabudhe V, Sweeney K, et al. The use of plasma aldosterone and urinary sodium to potassium ratio as translatable quantitative biomarkers of mineralocorticoid receptor antagonism. J Transl Med. 2011;9:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Barrera‐Chimal J, Girerd S, Jaisser F. Mineralocorticoid receptor antagonists and kidney diseases: pathophysiological basis. Kidney Int. 2019;96(2):302‐319. [DOI] [PubMed] [Google Scholar]
- 27. Sueta D, Yamamoto E, Tsujita K. Mineralocorticoid receptor blockers: novel selective nonsteroidal mineralocorticoid receptor antagonists. Curr Hypertens Rep. 2020;22(3):21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.