

Abstract
Background and Aims
Nonalcoholic steatohepatitis (NASH) is considered as a pivotal stage in nonalcoholic fatty liver disease (NAFLD) progression, given that it paves the way for severe liver injuries such as fibrosis and cirrhosis. The etiology of human NASH is multifactorial, and identifying reliable molecular players and/or biomarkers has proven difficult. Together with the inappropriate consideration of risk factors revealed by epidemiological studies (altered glucose homeostasis, obesity, ethnicity, sex, etc.), the limited availability of representative NASH cohorts with associated liver biopsies, the gold standard for NASH diagnosis, probably explains the poor overlap between published “omics”‐defined NASH signatures.
Approach and Results
Here, we have explored transcriptomic profiles of livers starting from a 910‐obese‐patient cohort, which was further stratified based on stringent histological characterization, to define “NoNASH” and “NASH” patients. Sex was identified as the main factor for data heterogeneity in this cohort. Using powerful bootstrapping and random forest (RF) approaches, we identified reliably differentially expressed genes participating in distinct biological processes in NASH as a function of sex. RF‐calculated gene signatures identified NASH patients in independent cohorts with high accuracy.
Conclusions
This large‐scale analysis of transcriptomic profiles from human livers emphasized the sexually dimorphic nature of NASH and its link with fibrosis, calling for the integration of sex as a major determinant of liver responses to NASH progression and responses to drugs.
Abbreviations
- ALAT
alanine transaminase
- ANXA2P2
Annexin A2 pseudogene 2
- ASAT
aspartate aminotransferase
- AUC
area under the curve
- BMI
body mass index
- BP
Biological Processes
- CCND1
cyclin D1
- CYP2C19
cytochrome P450 family 2 subfamily C member 19
- DE
differentially expressed
- DEGs
differentially expressed genes
- DU
Duke University cohort
- FAT1
FAT tumor suppressor homolog 1
- FC
fold change
- FDR
false discovery rate
- GEO
Gene Expression Omnibus
- GO
Gene Ontology
- HbA1c
glycated hemoglobin
- HDL‐C
high‐density lipoprotein‐cholesterol
- HL
healthy liver
- HOMA‐IR
homeostatic model assessment of insulin resistance
- HUL
Hopital Universitaire de Lille cohort
- NAFL
nonalcoholic fatty liver
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NCBI
National Center for Biotechnology Information
- PCA
principal component analysis
- RF
random forest
- RRM2B
ribonucleotide reductase regulatory TP53‐inducible subunit M2B
- SVM
support vector machine
- TYMS
thymidylate synthetase
- UBD
ubiquitin D
- UKD
Universitätsklinikum Dresden cohort
- UZA
Universitair Ziekenhuis Antwerpen cohort
Nonalcoholic fatty liver disease (NAFLD) is a growing health burden initially developing in Western countries and spreading to areas in which lifestyle and diet changes increase the prevalence of obesity and insulin resistance.( 1 ) NAFLD is now the most common chronic liver condition, with a worldwide prevalence of ≈25% of the total population.( 2 ) NAFLD encompasses a spectrum of liver histological manifestations, from relatively benign hepatic steatosis (nonalcoholic fatty liver; NAFL) to more‐severe liver injuries leading to nonalcoholic steatohepatitis (NASH). Lobular inflammation and ballooning degeneration of hepatocytes are histological characteristics of NAFL progression toward NASH.( 3 ) NASH is strongly associated with fibrosis,( 4 , 5 , 6 ) which is itself, even at early stages, predictive of increased overall and liver‐related mortality.( 7 , 8 , 9 )
A number of genome‐wide–scale transcriptomic analyses described hepatic gene expression pattern alterations in NAFL and NASH patients versus “healthy obese” or lean individuals,( 10 , 11 , 12 , 13 , 14 , 15 , 16 ) prompting meta‐analysis to define NASH and/or fibrosis core molecular signatures.( 12 , 13 , 17 ) Although identifying unique or confirming established players in NASH progression, these studies did not allow the definition of a predictive core gene signature, given that little overlap between each meta‐analysis was observed. Multiple confounding factors and technical biases may account for this inconsistency, such as differences in genetic origin, unappreciated environmental factors, and cohort stratification criteria. In addition, stratification did not always take into account major risk factors for NASH revealed by epidemiological studies, such as metabolic status.( 18 , 19 ) Most important, many human pathological manifestations are sex dependent,( 20 , 21 ) and NAFLD‐induced liver injuries are mostly reported as more severe in men.( 22 ) However, despite obvious sex dimorphic traits in metabolic regulations,( 23 ) sex was not considered as a factor neither in the design nor upon interpretation of the above‐mentioned studies.
Generating a global overview of biological processes involved in human disease requires genome‐wide analysis of large cohorts containing hundreds of patients to insure the robustness of results.( 24 ) Transcriptomic signatures are generally defined through ”top‐down” approaches,( 25 ) starting from a comparative analysis to identify differentially expressed (DE) genes (DEGs) between healthy and pathological conditions to further select, within this DEG set, several genes supposedly constituting the ”disease” signature. However, the imbalance between the population size (a few hundred) and the number of quantified RNA transcripts (several thousand) generates unstable results, which displays a high sensitivity to the genetic, biological, and biometric characteristics of the studied cohort.( 26 ) Despite being long established, this variability remained ignored in previous studies, in which single differential analysis were performed on nonsegmented cohorts. Furthermore, machine learning methods, such as logistic regressions, are traditionally used to guarantee an unbiased selection of signature genes among DEGs.( 27 ) Although they may lead to comprehensive models, these methods fail to integrate nonlinear gene interactions reflecting biological complexity.( 28 ) Nonlinear approaches, such as random forests (RFs) or support vector machine (SVM), can model such complex interactions, but remain rarely used in such studies because of methodological complexity.( 29 , 30 , 31 ) Furthermore, variability in computational and statistical methods applied to DEG selection also contributes, especially in a small‐cohort‐size context, to observed discrepancies between identified signatures.
The main objective of this study was to define, using unbiased and robust bioinformatic approaches, NASH molecular signatures through a transcriptomic study starting from a large cohort of morbidly obese patients (n = 910). In addition to highlighting optimal bioinformatic approaches for biological signature identification, this study identified sex as the main parameter affecting NASH signature definition and associated altered biological processes.
Materials and Methods
Cohorts and Data Sets
The Hopital Universitaire de Lille (HUL) cohort (ABOS; ClinicialTrials.gov: NCT 01129297) was recruited among obese patients visiting the Obesity Surgery Department at the Centre Hospitalier Universitaire de Lille (Lille, France). All patients fulfilled criteria for, and were willing to undergo, weight‐loss surgery. More details on the constitution and characterization of this prospective cohort are in the Supporting Information section. Liver needle biopsies were obtained at the time of surgery from 910 patients undergoing bariatric surgery.( 32 ) Anthropometric, histological, and metabolic characteristics are indicated in Table 1. RNA extraction, purification, labeling, and hybridization procedures for microarray analysis have been reported.( 32 ) Transcriptome analysis was performed using Affymetrix Human Transcriptome Array (HTA) 2.0, and .CEL files were normalized in a single run of the apt‐probeset‐summarize command (gc‐scale‐rma analysis using meta probesets and full quantile normalization, APT program 2.10.0; ThermoFisher Scientific, Waltham, MA). Expression data are available at the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO; GSE130991).
TABLE 1.
Characteristics of NASH/NoNASH 620 Patients From the HUL Cohort
| Characteristics | HL | NAFL | NASH |
|---|---|---|---|
| n = 118 | n = 431 | n = 71 | |
| Biometric parameters | |||
| Women (n; %) | 110; 85 | 300; 72 | 44; 60 |
| Age (mean ± SD) | 35.6 ± 11.0 | 35.6 ± 11.0 | 35.6 ± 11.0 |
| BMI (mean ± SD) | 46.2 ± 7.0 | 46.2 ± 7.0 | 46.2 ± 7.0 |
| Body mass (kg) (mean ± SD) | 128.4 ± 23.0 | 128.4 ± 23.0 | 128.4 ± 23.0 |
| Liver histology | |||
| Steatosis grade (n; %) 0 | 118; 100 | 0; 0 | 0; 0 |
| 1 | 0; 0 | 310; 72 | 18; 25 |
| 2 | 0; 0 | 86; 20 | 28; 40 |
| 3 | 0; 0 | 35; 8 | 25; 35 |
| Lobular inflammation (n; %) 0 | 118; 100 | 431; 100 | 0; 0 |
| 1 | 0; 0 | 0; 0 | 50; 70 |
| 2 | 0; 0 | 0; 0 | 21; 30 |
| Ballooning (n; %) 0 | 118; 100 | 431; 100 | 0; 0 |
| 1 | 0; 0 | 0; 0 | 50; 70 |
| 2 | 0; 0 | 0; 0 | 21; 30 |
| Fibrosis (Kleiner) (n; %) 0 | 107; 87 | 321; 74 | 9; 12 |
| 1a | 4; 2 | 21; 3 | 9; 12 |
| 1b | 2; 1 | 17; 1 | 9; 12 |
| 1c | 5; 4 | 44; 9 | 4; 5 |
| 2 | 0; 0 | 13; 3 | 12; 16 |
| 3q | 0; 0 | 10; 2 | 13; 18 |
| 3s | 0; 0 | 4; 1 | 13; 18 |
| 4 | 0; 0 | 0; 0 | 5; 7 |
| Liver functions | |||
| ASAT (IU/L) (median; IQR) | 21; 9 | 23; 9 | 38; 23 |
| ALAT (IU/L) (median; IQR) | 20; 11 | 27; 16 | 47; 31 |
| GGT (IU/L) (median; IQR) | 25; 21 | 30; 27 | 57; 44 |
| Metabolic parameters | |||
| Diabetes (n; %) | 20; 16 | 145; 35 | 63; 86 |
| Treated diabetes (n; %) | 16; 12 | 121; 29 | 56; 77 |
| Fasting blood glucose (mM) (mean ± SD) | 5.4 ± 0.9 | 5.4 ± 0.9 | 5.4 ± 0.9 |
| Fasting insulin (IU/mL) (median; IQR) | 12.2; 8.3 | 13.7; 11.3 | 23.5; 25.8 |
| HbA1c (%) (median; IQR) | 5.5; 0.5 | 5.9; 0.9 | 7.8; 3.6 |
| HOMA‐IR (median; IQR) | 2.9; 2.2 | 3.5; 3.2 | 9.3; 10.9 |
| Total cholesterol (mmol/L) (mean ± SD) | 4.9 ± 1.0 | 4.9 ± 1.0 | 4.9 ± 1.0 |
| LDL‐C (mmol/L) (mean ± SD) | 3.1 ± 0.9 | 3.1 ± 0.9 | 3.1 ± 0.9 |
| HDL‐C (mmol/L) (mean ± SD) | 1.2 ± 0.2 | 1.2 ± 0.2 | 1.2 ± 0.2 |
| Triglycerides (mmol/L) (mean ± SD) | 1.3 ± 0.5 | 1.3 ± 0.5 | 1.3 ± 0.5 |
| Others | |||
| Diastolic blood pressure (mm Hg) (mean ± SD) | 72.6 ± 14.0 | 77.1 ± 14.0 | 76.6 ± 13.0 |
| Systolic blood pressure (mm Hg) (mean ± SD) | 130.3 ± 15.0 | 137.1 ± 19.0 | 139.2 ± 19.0 |
Abbreviations: GGT, gamma‐glutamyl transferase; IQR, interquartile range.
The Universitair Ziekenhuis Antwerpen (UZA) cohort was recruited among overweight patients visiting the Obesity Clinic at UZA suspected to have NAFLD based on imaging and blood biochemistry assays (see a previous work( 33 ) for details). Liver biopsies were from 178 obese patients, of which 79 further underwent gastric bypass surgery. Anthropometric, histological, and metabolic characteristics and RNA extraction, purification, labeling, and hybridization procedures have been reported.( 12 ) Transcriptome analysis was performed using Affymetrix Human Gene (HuGene) 2.0ST, and .CEL files were normalized as above. Expression data are available at NCBI GEO (GSE83452).
Ahrens et al. described the Universitätsklinikum Dresden (UKD) cohort, which includes lean (control, n = 18) and morbidly obese patients (body mass index [BMI] >42 kg/m2, n = 45) either classified as healthy (no steatosis, n = 18), NAFL (steatosis only, n = 12), or NASH (steatosis and inflammation, n = 15) with low fibrosis stage (F ≤ 1, mild).( 11 ) Transcriptome analysis was performed by the researchers using Affymetrix HuGene 1.1ST arrays, and .CEL files were normalized as above. Gene expression data are available at NCBI GEO (GSE48452).
Moylan et al. described the Duke University (DU) cohort composed of 72 overweight or obese patients (29 kg/m2 < BMI < 46 kg/m2) who were stratified according to the fibrosis stage (F ≤ 1, moderate, n = 40 and F ≥ 3, severe, n = 32) as recommended in a published work.( 34 ) The NASH phenotype (inflammation and ballooning) was also more pronounced in the severe branch of the cohort.( 10 ) Transcriptome analysis was performed by the researchers using Affymetrix HG U133 Plus 2.0 arrays, and .CEL files were normalized as above. Gene expression data are available at NCBI GEO (GSE49541).
Cohort Stratification, Data Analysis, and Bioinformatic Procedures
Detailed information can be found in the Supporting Information.
Data Availability Statement
The transcriptomic data sets generated during and/or analyzed during the current study are available in the NCBI GEO repository. Clinical data that support the findings of this study are available from the corresponding author upon reasonable request.
Results
Definition of the Learning Cohort
The prospective HUL cohort includes morbidly obese patients presenting all grades of liver steatosis, lobular inflammation, and ballooning. Validated liver transcriptomic profiles were obtained from 910 biopsies, which were classified on the basis of histological parameters (steatosis, hepatocyte ballooning, and lobular inflammation; Fig. 1) to yield a fully characterized 620‐patient cohort with healthy (HL; n = 118 [19%]), steatotic [NAFL; n = 431 [70%]), or NASH livers [NASH; n = 71 [11%]; Table 1). “Borderline” samples to which an unambiguous classification could not be attributed were excluded (n = 199; Fig. 1 and Supporting Fig. S1). Of note, the HL and NAFL categories were mostly associated with no or moderate fibrosis (F0‐F2, 98%; Table 1), whereas NASH patients exhibited an important proportion of severe fibrosis (F3‐F4, 43%; Table 1). With the aim of eliminating selection bias and confounding factors when assessing the effect of NASH on gene expression profiles as a function of sex, we defined a so‐called learning cohort from the 620‐patient cohort as follows. First, biopsies were selected according to stringent quality and biological criteria (Fig. 1). These criteria were: (1) defining a minimal length >10 mm and a number of portal areas per biopsy >8, leading to a 420‐biopsy subset (characteristics of this subcohort are detailed in Supporting Table S1); (2) excluding from the analysis patients with normal hepatic insulin sensitivity given that they are virtually absent from the NASH category (Fig. 2) by using a homeostatic model assessment of insulin resistance (HOMA‐IR) index >2.4, which is, after exclusion of patients taking “rapid insulin,” an arbitrary threshold in accordance with common practices( 35 , 36 ); and (3) defining a “NoNASH” group including healthy (HL) and steatotic (NAFL) livers. To eliminate or reduce any referral or unidentified bias as well as confounding factors, patients were then propensity matched within each subgroup (NoNASH or NASH) based on sex, BMI, HOMA‐IR, and fibrosis grade. This defined the HUL learning cohort composed of 124 matched male or female NoNASH patients and of 46 male or female matched NASH patients (Supporting Table S2), allowing to investigate the importance of the sex factor in balanced groups (Table 2). NASH patients displayed higher fibrosis scores and HOMA‐IR than NoNASH patients (F3‐F4 = 29% vs. 5% and mean HOMA‐IR = 27.7 vs. 6.8, respectively; Supporting Table S2).
FIG. 1.
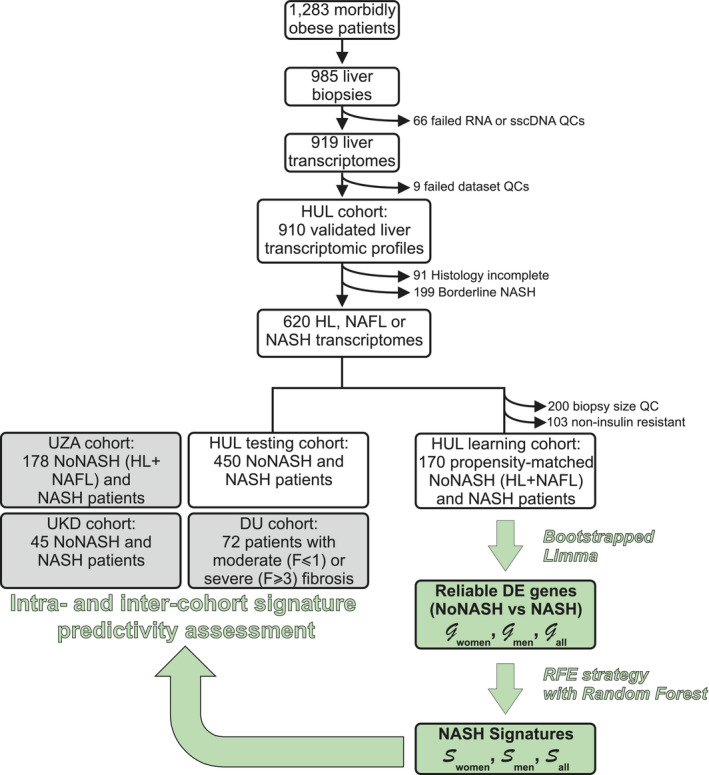
HUL cohort analysis. The main steps of the HUL cohort transcriptomic analysis, stratification, and bioinformatic analysis are indicated, as well as the steps during which definition and validation of proposed sex‐specific NASH signatures were undertaken. Details can be found in the Materials and Methods and Results sections. Abbreviations: QC, quality control; sscDNA, single‐stranded complementary DNA.
FIG. 2.
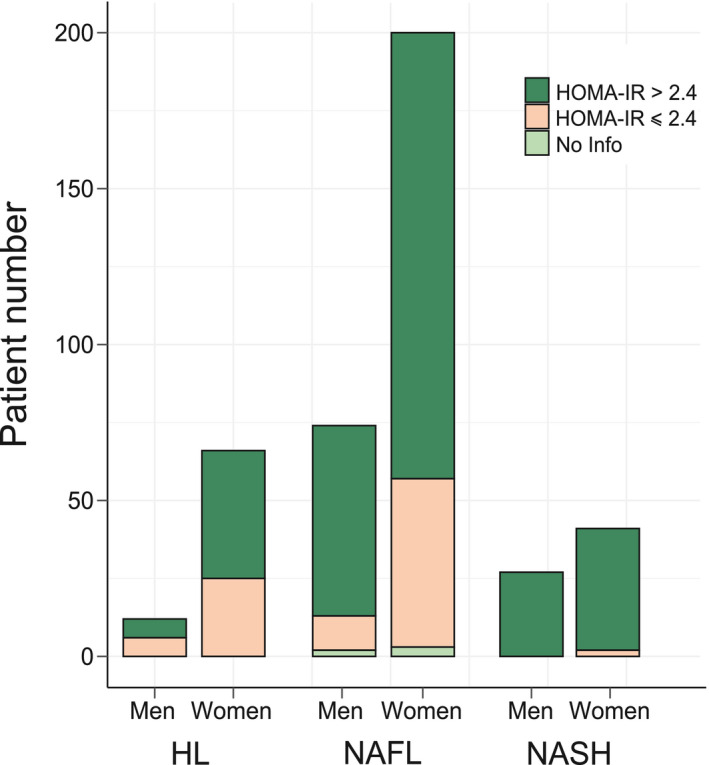
Insulin sensitivity and β‐cell function in the HUL cohort. The proportion in the HUL cohort of insulin‐resistant (HOMA‐IR index >2.4, in red) and non‐insulin‐resistant (HOMA‐IR index ≤ 2.4, in blue) patients among HL (n = 78), NAFL (n = 274), and NASH (n = 68) groups is displayed as a function of sex.
TABLE 2.
Main Biometric and Biochemical Parameters of the Learning Cohort
| Men | Women | |||
|---|---|---|---|---|
| NoNASH | NASH | NoNASH | NASH | |
| Population size | 62 | 23 | 62 | 23 |
| Age | 43.7 ± 12.0 | 48.5 ± 9.0 | 43.2 ± 12.0 | 47.3 ± 10.0 |
| BMI | 47.9 ± 7.4 | 46.5 ± 6.7 | 48.8 ± 7.0 | 46.8 ± 5.6 |
| HOMA‐IR | 7.79 ± 11.00 | 28.7 ± 56.0 | 5.69 ± 7.80 | 26.6 ± 69.0 |
| HbA1c | 6.46 ± 1.60 | 8.35 ± 1.90*** | 6.03 ± 0.80 | 7.92 ± 1.90*** |
| Steatosis grade | 1.23 ± 0.70 | 2.17 ± 0.80*** | 1.16 ± 0.80 | 2.04 ± 0.70*** |
| Lob. inflammation | 0 | 1.17 ± 0.40*** | 0 | 1.43 ± 0.50*** |
| Ballooning | 0 | 1.26 ± 0.40*** | 0 | 1.13 ± 0.30*** |
| NAS score | 1.23 ± 0.70 | 4.61 ± 0.90*** | 1.16 ± 0.80 | 4.61 ± 0.80*** |
| Fibrosis score | 0.52 ± 0.90 | 1.52 ± 1.10*** | 0.29 ± 0.60 | 1.61 ± 1.10*** |
| ASAT (IU/L) | 29.7 ± 11.0 | 42.5 ± 17.0*** | 22.2 ± 7.4.0$$$ | 40.7 ± 21.0*** |
| ALAT (IU/L) | 42.5 ± 23.0 | 57.8 ± 27.0** | 24.7 ± 9.7.0$$$ | 48.6 ± 25.0*** |
Mean ± SD of clinical parameters for each patient category are indicated. NoNASH vs. NASH comparison: **P < 0.05; ***P < 0.01. Women vs. men comparison: $$$ P < 0.01.
Abbreviation: NAS, NAFLD Activity Score.
Differential Gene Expression Analysis
The source of variation in gene expression was investigated using multivariate analysis of variance (ANOVA) on normalized log2‐transformed gene expression signals. Computation of F‐ratio (variation explained by the test variable/unexplained variation) for each factor considered in the differential model clearly confirmed sex as the factor explaining the highest expression variance to the data set (Fig. 3). Accordingly, DEGs were first identified, as usually performed in signature discovery studies, by a single Limma run comparing either NoNASH to NASH gene profiles irrespective of sex, or considering only female or male patients (thereafter referred to as “All,” “Women,” or “Men,” respectively). A variable number of genes was found significantly DE when comparing NASH to NoNASH patients in the men (3,083), women (297), and all patients (3,466) strata (false discovery rate [FDR], <10%).
FIG. 3.
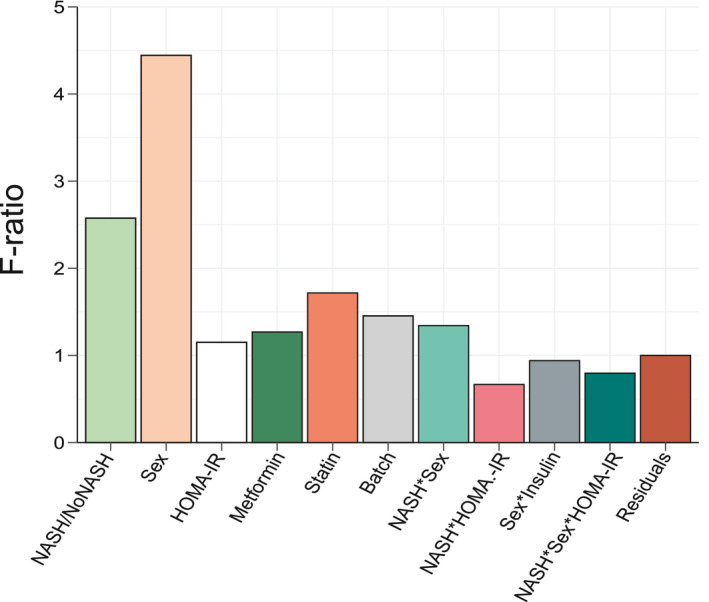
ANOVA. F‐ratio values of factors included in the Limma model were calculated. High F‐ratio values indicate a strong linear relationship between a given factor and gene expression values. Interaction term between factors A and B are indicated as an A*B annotation. Factors were selected on the basis of published reports.
The robustness of DEGs identification was assessed by a bootstrap procedure based on a random subsampling rate = 0.9 (100 iterations) of the learning cohort subgroups, followed by Limma differential analysis. This procedure generated three groups (G) of DEGs reliably detected in more than 75 bootstrap runs (FDR < 10%, , , and ) and revealed important qualitative and quantitative discrepancies with DEGs detected by a unique Limma run (Fig. 4A,B and Supporting Fig. S2). As an example, CHIL3L1 was detected as DE in the single Limma run for both men and women contrasts, with high fold changes (FCs; FCmen = 3.30; FCwomen = 2.14). However, the bootstrap procedure reliably detected CHIL3L1 as overexpressed only in male NASH patients ( ), given that it was found significantly DE in all 100 Limma runs. In contrast, it was dismissed from the women contrast ( ) because it was found significantly DE in only 32 of 100 runs despite a high mean FC (σFCw = 2.14). This bootstrap analysis thus attributed 1,325 (vs. 3,083 in the single Limma run), 55 (vs. 297), and 1,868 (vs. 3,466) DEGs to , , and contrasts, respectively (Fig. 5A and Supporting Fig. S3). Variance analysis did not reveal significant differences in global gene expression between men and women, suggesting that the difference in DEG number was not linked to difference in gene expression heterogeneity (Supporting Fig. S4). Thus, a strong influence of patient/sample heterogeneity on the differential analysis process was observed.
FIG. 4.
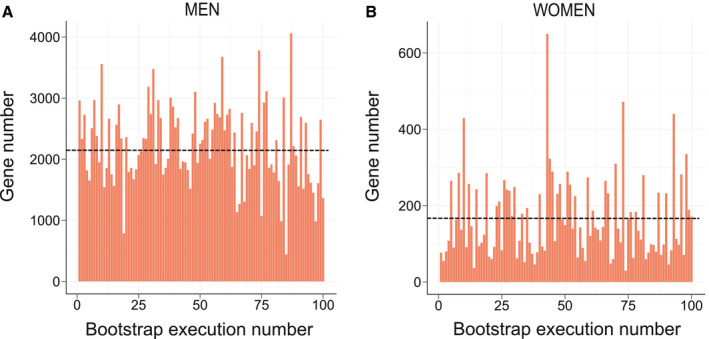
Instability of the Limma‐based determination of DE genes. The number of DEGs between NoNASH and NASH patients (FDR < 10%) for (A) men and (B) women was assessed after 100 subsamplings (rate = 0.9) of the learning cohort followed Limma analysis. Mean DEG number is represented by a black dotted line.
FIG. 5.
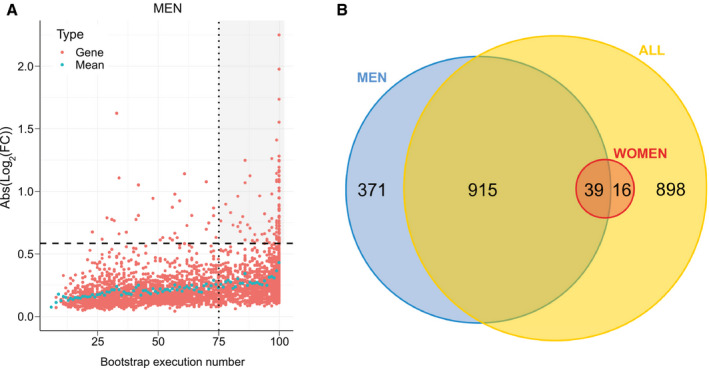
Identification of reliable DE genes. (A) The absolute log2FC of DEGs was computed for the men learning cohort ( , n = 85). Each significantly DEG (FDR < 10%) is represented by a red dot. Gene reliability is established by the number of bootstrap runs for which the gene remains significantly DE (75%). Blue dots represent the mean absolute log2FC for a given bootstrap run count. Dashed line, FC = 1.5; dotted line, occurrence = 75. The gray‐shaded area includes reliable DEGs (FC > 1.5) with occurrences ≥75. (B) Number of reliably identified DEGs between NoNASH and NASH groups (men [blue], women [red], and all patients [yellow]).
The incomplete overlap between and suggested a contribution of a sex‐specific factor to differential analysis results, given that only 39 common transcripts were identified (71% of and 3% of ; Fig. 5B). When raising the FC threshold of reliable DEGs up to 1.5 (Supporting Table S3), this overlap increased in proportion (13 overlapping transcripts; 87% of and 15% of ), whereas 74 and two genes remained men and women specific, respectively (Supporting Fig. S5). Men‐specific genes with highest absolute log2FC values were solute carrier family 22 member 10 (σFCm = 0:42), chitinase‐3‐like protein 1 (CHI3L1; σFCm = 3.36), and Hermansky‐Pudlak syndrome 5 protein (σFCm = 2.24), and the two women‐specific genes were HYDIN1 (σFCw = 0.52) and HYDIN2 (σFCw = 0.39).
Although suggesting a clear dichotomy between male and female patients, this latter analysis did not provide an overview of altered liver functions in NASH. A Gene Ontology (GO) term enrichment was thus performed on reliable , , and using the Biological Processes (BP) database (Table 3). Interestingly, term enrichment within identified cell‐cell contact, immune cell migration, inflammatory response, and extracellular matrix remodeling as the most prominent processes, in agreement with the literature.( 37 ) However, GO BP term enrichment of DEGs in pointed to cell‐cycle regulation processes, whereas revealed a pattern more related to metabolic and inflammation processes (Table 3). This dichotomy was also observed when restricting gene lists to genes with absolute log2FC > log2(1.2) to reduce technical noise, which additionally revealed a specific enrichment of cholesterol‐related genes in the men gene set (Supporting Table S4). Because of the low number of dysregulated genes passing this FC threshold (n = 41), the women subcohort did not allow a statistically significant enrichment in any GO BP term, but visual inspection of the gene list did not highlight any gene involved in cholesterol metabolism.
TABLE 3.
Biological Term Enrichment
|
Gene Set GO Terms |
(1,325 Genes) | (55 Genes) | (1,868 Genes) | |||
|---|---|---|---|---|---|---|
| Rank | P Value | Rank | P Value | Rank | P Value | |
| Cell‐cell adhesion | 1 | 2.9 × 10−9*** | ∅ | ∅ | 2 | 5.7 × 10−11*** |
| ATP hydrolysis‐coupled proton transport | 2 | 4.3 × 10−7*** | ∅ | ∅ | ∅ | ∅ |
| ER to Golgi vesicle‐mediated transport | 3 | 5.9 × 10−7*** | ∅ | ∅ | 130 | 1.0 × 10−2 |
| Regulation of cellular amino acid metabolic process | 4 | 6.0 × 10−7*** | ∅ | ∅ | ∅ | ∅ |
| Transferrin transport | 5 | 1.3 × 10−6** | ∅ | ∅ | ∅ | ∅ |
| Negative regulation of apoptotic process | 6 | 2.2 × 10−6** | ∅ | ∅ | 33 | 1.7 × 10−4* |
| Tumor necrosis factor–mediated signaling pathway | 7 | 2.4 × 10−6** | ∅ | ∅ | 38 | 3.5 × 10−4* |
| Regulation of macroautophagy | 8 | 3.2 × 10−6** | ∅ | ∅ | ∅ | ∅ |
| T‐cell‐receptor signaling pathway | 9 | 4.1 × 10−6** | ∅ | ∅ | 31 | 1.5 × 10−4* |
| NIK/NF‐kappa‐B signaling | 10 | 4.3 × 10−6** | ∅ | ∅ | 102 | 5.9 × 10−3 |
| G1/S transition of mitotic cell cycle | ∅ | ∅ | 1 | 1.6 × 10−4* | 17 | 1.1 × 10−5** |
| Response to organonitrogen compound | ∅ | ∅ | 2 | 6.3 × 10−4 | ∅ | ∅ |
| Cell adhesion | 50 | 1.1 × 10−3* | 3 | 1.3 × 10−3 | 5 | 4.2 × 10−8*** |
| Triglyceride catabolic process | ∅ | ∅ | 4 | 2.0 × 10−3 | ∅ | ∅ |
| Negative regulation of G1/S transition of mitotic cell cycle | ∅ | ∅ | 5 | 2.0 × 10−3 | 172 | 2.2 × 10−2 |
| Liver regeneration | 111 | 1.0 × 10−2 | 6 | 2.7 × 10−3 | 81 | 3.5 × 10−3 |
| Response to drug | 93 | 6.1 × 10−3 | 7 | 8.8 × 10−3 | 110 | 6.9 × 10−3 |
| Cellular response to hydrogen peroxide | 174 | 3.3 × 10−2 | 8 | 1.0 × 10−2 | 210 | 3.1 × 10−2 |
| Intestinal epithelial cell maturation | 161 | 2.4 × 10−2 | 9 | 1.1 × 10−2 | 241 | 4.4 × 10−2 |
| Aggresome assembly | ∅ | ∅ | 10 | 1.3 × 10−2 | ∅ | ∅ |
| Movement of cell or subcellular component | 23 | 4.0 × 10−5** | ∅ | ∅ | 1 | 3.7 × 10−11*** |
| Cell‐cell adhesion | 1 | 2.9 × 10−9*** | ∅ | ∅ | 2 | 5.7 × 10−11*** |
| Leukocyte migration | 30 | 1.4 × 10−4* | ∅ | ∅ | 3 | 1.0 × 10−10*** |
|
Fc‐gamma receptor signal (pathway in phagocytosis) |
36 | 2.4 × 10−4* | ∅ | ∅ | 4 | 5.6 × 10−9*** |
| Cell adhesion | 50 | 1.1 × 10−3* | 3 | 1.3 × 10−3 | 5 | 4.2 × 10−8*** |
| Actin cytoskeleton organization | 17 | 1.3 × 10−5** | ∅ | ∅ | 6 | 1.3 × 10−7*** |
| Leukocyte cell‐cell adhesion | 29 | 1.4 × 10−4* | ∅ | ∅ | 7 | 5.2 × 10−7*** |
| Regulation of cell shape | 86 | 5.0 × 10−3 | ∅ | ∅ | 8 | 8.1 × 10−7*** |
| Inflammatory response | 169 | 2.8 × 10−2 | ∅ | ∅ | 6 | 8.5 × 10−7*** |
| Extracellular matrix organization | 55 | 1.2 × 10−3* | ∅ | ∅ | 10 | 8.7 × 10−7*** |
Top 10 enriched GO terms for reliable DEGs in NASH vs. NoNASH men, women, and all patients subcohorts. P values and Benjamini‐Hochberg FDR were computed using DAVID and the Biological Process Direct GO terms database; enrichments were ranked following P values. Enrichments with corresponding FDR < 10%, 1%, and 0.1% are tagged with *, **, and ***, respectively.
Abbreviations: NIK, nuclear factor kappa‐B‐inducing kinase; NF‐kappa‐B, nuclear factor kappa‐B; DAVID, The Database for Annotation, Visualization and Integrated Discovery.
RF‐Based Identification of Signatures
A recursive feature elimination (RFE) strategy coupled to RF models was used to select an optimal gene subset from , , and (Figs. 1 and 6A and Supporting Fig. S6) to predict NoNASH and NASH patients. By progressively eliminating genes with lowest classification power, a minimal gene set yielding a maximized area under the curve (AUC) was defined. Signatures corresponding to these optimal subsets contained 20, 15, and 108 genes extracted from , , and and are thereafter referred to as , , and , respectively (Supporting Table S5). The overlap between these three signatures indicated that men‐ and women‐specific signatures shared only one gene (thymidylate synthetase [TYMS]; Fig. 6C). The larger signature obtained when considering all patients ( ) largely overlapped with and with , albeit to a lesser extent, highlighting the need of both sex‐specific signature genes in the RF model to efficiently classify an heterogeneous population.
FIG. 6.
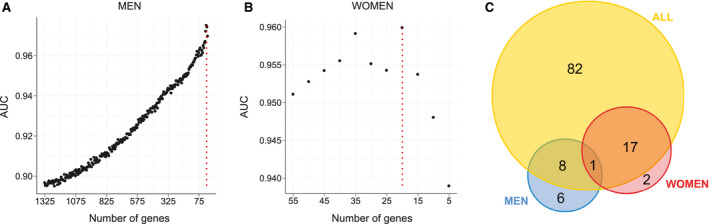
RF models. (A,B) Classification power (AUC) of RF models. RFs were trained with a progressively reduced number of genes to identify an optimal subset of genes corresponding to the proposed signature, for men and women, established by the second step of RFE strategy. Maximal AUC is indicated by a vertical dotted red line. (C) Number of genes composing men (blue), women (red), and all patients’ (yellow) RF‐based signatures.
Classification Power of Signatures
A principal component analysis (PCA) was first used to validate , , and as tools to separate NASH from NoNASH patients. Separations between NoNASH and NASH patients using gene expression values from reference signatures were sharper than when considering all DEG expression values (Fig. 7 and Supporting Fig. S7), suggesting that these signatures are highly efficient in discriminating NoNASH versus NASH patients.
FIG. 7.
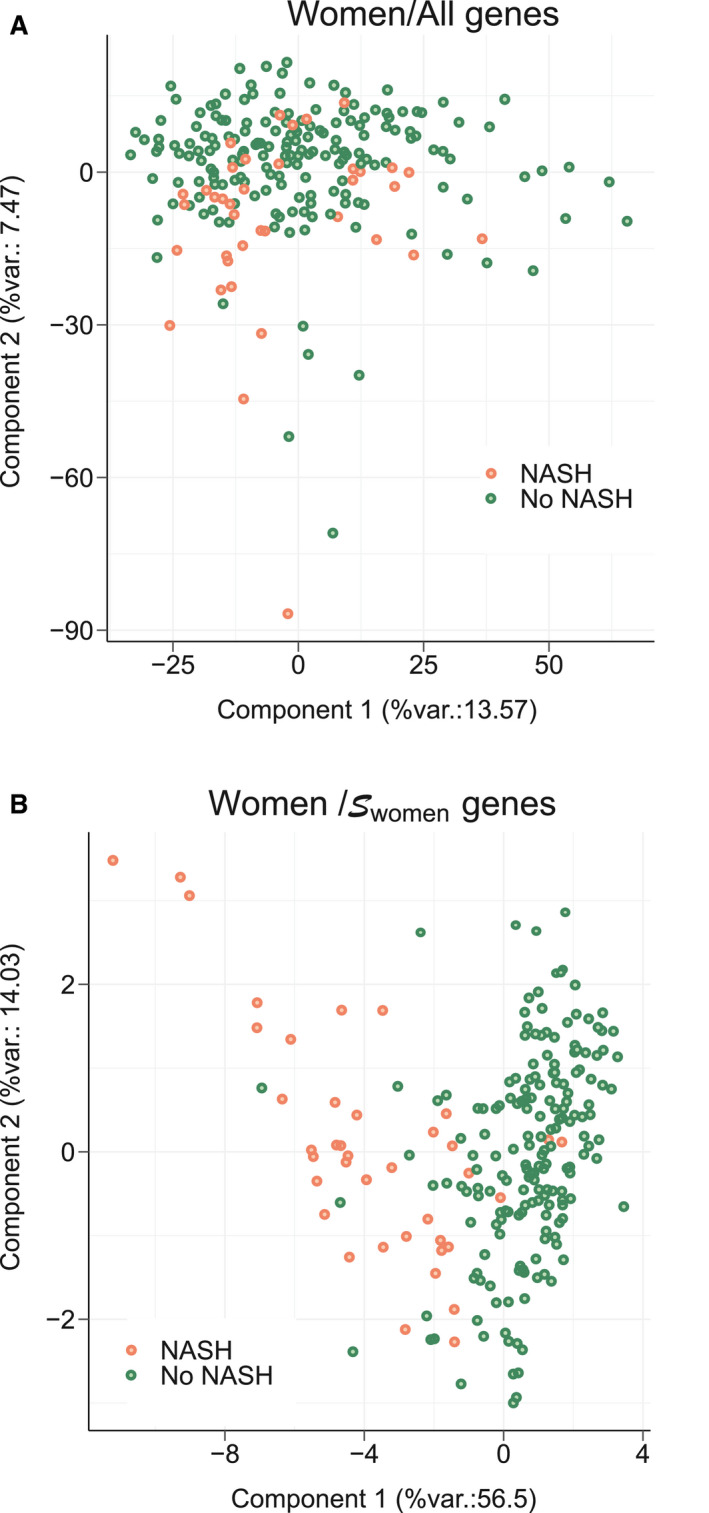
PCA. A PCA was run using gene expression values from women patients included in learning cohort based on: (A) all genes expression values or (B) genes. The percentage of the global data variance explained by each component is indicated by X and Y axis labels (%var.). Each dot represents a patient (NoNASH [blue] or NASH [yellow]).
The classification power of these three signatures was then evaluated through 200 cross‐validation runs using the learning cohort. The distribution of AUCs determined from these runs (Fig. 8A,B and Supporting Fig. S8) showed that the highest AUC values were achieved by RF models learnt from , , and to predict women (AUC RF‐ = 0.957), men (AUC RF‐ = 0.970), and all patients (AUC RF‐ = 0.952), respectively. In comparison, mean AUC reached when using randomly selected signature models built from , , and (AUC Rdm‐Gx) were strictly lower than those determined using reference signatures, however with AUCs > 0.8. Among guided random signatures, those built from reached higher AUCs to predict NASH when classifying all patients and, more especially, the women‐only cohort, with performances close to the reference signature (Fig. 8A; AUC Rdm‐ = 0.939). It is worth noting here that these guided random signatures are composed of 20 genes randomly selected among 55 reliable DEGs from , thereby inducing a frequent overlap between reference and guided random signatures. Thus, the high AUCs achieved by these random signatures did not suggest the uniqueness of a predictive NASH signature in RF models, but are rather indicative of a set of similar signatures built from a limited list (here n = 55) of predictive genes. Unguided random signatures built from the full list of genes (Rdm‐All) reached poor AUCs, ~0.62.
FIG. 8.
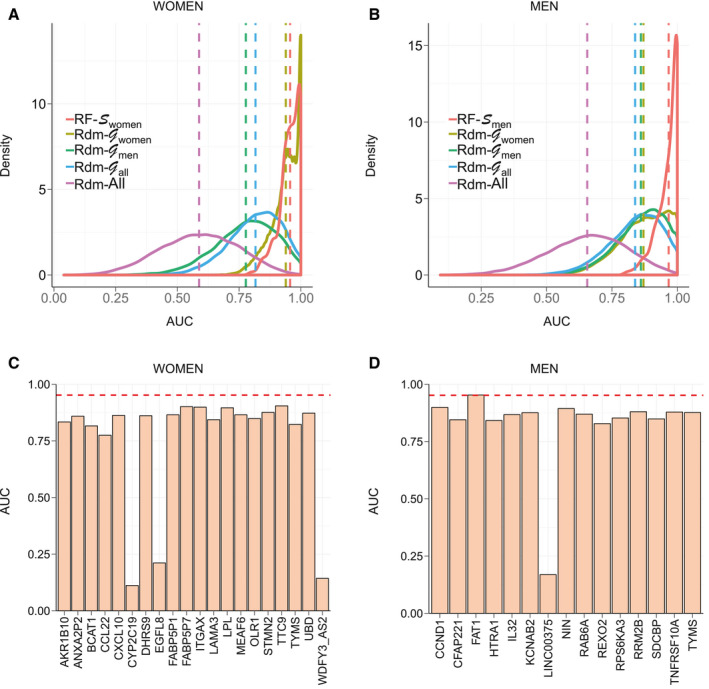
AUC values of signatures and single gene predictors. (A,B) AUC distribution of RF models to predict women (left) and men (right) of the learning cohort in a cross‐validation scheme. RF models learnt using, respectively, and (red) were compared in each plot to RF models learnt using random signatures built from (khaki), (green), (blue), and the full list of available genes (purple). Distribution means are represented as vertical dashed lines. (C,D) AUC of single gene predictors to predict NASH status of women (left) and men (right) patients of the learning cohort for each gene composing corresponding signatures ( and ). Mean AUCs reached by RF models learnt from corresponding signature in a cross‐validation scheme are represented through a red horizontal dashed line. Abbreviations: BCAT1, branched chain amino acid transaminase 1; CCL22, C‐C motif chemokine ligand 22; CFAP221, cilia‐ and flagella‐associated protein 221; CXCL10, C‐X‐C motif chemokine ligand 10; DHRS9, dehydrogenase/reductase SDR family member 9; EGFL8, EGF‐like domain multiple 8; HTRA1, HtrA serine peptidase 1; IL32, interleukin‐32; KCNAB2, potassium voltage‐gated channel subfamily A regulatory beta subunit 2; LAMA3, laminin subunit alpha 3; LINC00375, long intergenic non‐protein coding RNA 375; LPL, lipoprotein lipase; OLR1, oxidized low‐density lipoprotein receptor 1; RAB6A, RAB6A, member RAS oncogene family; REXO2, RNA exonuclease 2; RPS6KA3, ribosomal protein S6 kinase, 90 kDa, polypeptide 3; TNFRSF10A, TNF receptor superfamily member 10a; TTC9, tetratricopeptide repeat domain 9; WDFY3_AS2, WDFY3 antisense RNA 2.
Single Gene Predictors
Reference signatures were also compared to single gene predictors to classify NASH versus NoNASH patients in the learning cohort. The classification power of each gene composing , , and to classify women, men, and all patients, respectively, were evaluated (Fig. 8C,D and Supporting Fig. S9). All genes from and reached absolute AUC >0.77 and 0.82, respectively, especially for FAT1 with an AUC close to the model (AUC = 0.970; AUCFAT1 = 0.953). For genes, AUCs were low and fluctuated from 0.62 to 0.86, suggesting a higher complexity of the corresponding predictive model.
We also tested ad‐hoc signatures including a number of genes equal to reference ones and displaying the highest FC in , , and of the learning cohort (Supporting Fig. S10). For some genes, despite high FC, corresponding individual prediction for NASH remained poor (3‐hydroxy‐3‐methylglutaryl‐CoA synthase 1 in the men group, FCmen = 2.4; AUCmen = 0.65). Of note, RF‐based models using these ad‐hoc signatures most often reached better prediction than individual gene predictors. Taken together, these analyses demonstrate that RF‐calculated signals have a predictive power superior to random and “single gene” predictors.
Independent Cohort Validation
Reference signatures were used to classify NASH and NoNASH patients from other cohorts (Table 4). When confronted to the HUL “testing” cohort (excluding the 170‐patient learning cohort, n = 450), RF models learnt from , , and reached similar AUCs when classifying men as NASH or NoNASH patients (AUCmen = 0.87/0.87/0.93), women (AUCwomen = 0.86/0.84/0.87), and all patients (AUCall = 0.88/0.84/0.88). The similarly high AUC values reached when classifying the HUL cohort by these three signatures demonstrated the capacity of RF to learn efficient classification rules from various gene signatures. A validation using independent cohorts was, however, required to more precisely assess the ability of such signatures to identify NASH patients. Classification predictions ran on the UKD cohort yielded improved AUC values in the women and all patients subcohorts when compared to HUL AUC, whereas HUL‐generated signatures were slightly less accurate at classifying UKD men as NASH or NoNASH patients (AUCmen = 0.79/0.75/0.79). HUL‐generated signatures performed only fairly to classify the UZA cohort (56 NoNASH; 122 NASH), with the best AUC being reached when using to predict women, men, and all patients (AUCwomen = 0.73; AUCmen = 0.76; AUCall = 0.75), whereas yielded an AUC ~0.65. Given that these values remained significantly lower than AUCs obtained with other cohorts, we assessed signature performances after restricting the NASH category to highly fibrotic patients (F ≥ 3; n = 60). RF model performances on this UZAhigh fib subcohort increased the classification prediction power to the same extent for men, women, and all patients. and models reached similar performances, with AUCs in the 0.78‐0.84 range, and remained higher than models learnt from . These values remained, however, below those reached with other tested cohorts, suggesting a peculiar—yet undetermined—biological feature of the UZA cohort or a center effect.
TABLE 4.
AUC of RF Models
| Prediction of: | Women | Men | All | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| by: |
|
|
|
|
|
|
|
|
|
|||||||||
| HUL | 0.86 | 0.84 | 0.87 | 0.87 | 0.87 | 0.93 | 0.88 | 0.84 | 0.88 | |||||||||
| UZA | 0.73 | 0.62 | 0.70 | 0.76 | 0.67 | 0.76 | 0.75 | 0.63 | 0.71 | |||||||||
| UZAHigh Fib. | 0.82 | 0.69 | 0.78 | 0.83 | 0.71 | 0.84 | 0.82 | 0.69 | 0.79 | |||||||||
| UKD | 0.90 | 0.87 | 0.93 | 0.83 | 0.83 | 0.83 | 0.89 | 0.88 | 0.91 | |||||||||
| DU | ∅ | ∅ | ∅ | ∅ | ∅ | ∅ | 0.80 | 0.89 | 0.87 |
The ability of signatures to classify women, men, and all patients of the HUL, UZA, UZAHigh Fib., and UKD cohorts and all patients of the DU cohort as NASH or NoNASH was evaluated. For each classified population, RF models were learnt from , , and .
The DU cohort has been stratified according to extreme histological phenotypes, distinguishing a “mild NAFLD” group with a low fibrosis grade (F ≤ 1) and a “severe NAFLD” group with more‐pronounced liver damages associated with NASH (inflammation and ballooning) and strong fibrosis (F ≥ 3),( 10 ) thus ressembling the UZAhigh fib subcohort. We assessed the predictive power of signatures for all patients because sex was not discriminated in this cohort. RF models learnt from , , and showed similar or better AUCs for the DU cohort (AUCall = 0.80/0.89/0.87) when compared to HUL patient classification. This shows that signature performances extend beyond NASH prediction and may identify patients with severe, clinically relevant fibrotic lesions.
Signature Correlation Networks
To quantify gene relationships within each signature, gene expression correlation networks were built by computing Pearson correlations between each transcript pair in the learning cohort. The three resulting gene coexpression networks shared a similar structure, displaying a core of “central” highly correlated genes and more loosely correlated “peripheral” genes (Supporting Fig. S11). The signature contains two core gene sets, the first one, including karyopherin subunit alpha 2, Annexin A2 pseudogene 2 (ANXA2P2), Annexin A2, MYST/Esa1‐associated factor 6 (MEAF6), integrin, alpha X (complement component 3 receptor 4 subunit) (ITGAX), and TNF receptor superfamily member 12a, and the second one, containing damage‐specific DNA binding protein 2, mouse double minute 2 homolog, zinc finger matrin‐type 3 (ZMAT3), TYMS, ribosomal protein S27 like, and ribonucleotide reductase regulatory TP53‐inducible subunit M2B (RRM2B). The most highly correlated genes in were ubiquitin D (UBD), stathmin 2 (STMN2), ANXA2P2, fatty acid binding protein 5 pseudogene 1 (FABP5P1), and fatty acid binding protein 5 pseudogene 7 (FABP5P7), whereas ninein (NIN), syndecan binding protein (SDCBP), cyclin D1 (CCND1), RRM2B, and FAT tumor suppressor homolog 1 (FAT1) were most correlated in the network computed from . All correlations were positive except for cytochrome P450 family 2 subfamily C member 19 (CYP2C19) in the network and SFP1 and CYP2C19 in . Thus, this correlation study again emphasized the male‐female dichotomy in human NASH.
Correlation With Clinical Parameters
To assess whether RF‐built signatures, in addition to identifying NASH patients, also segregate patients according to biochemical or biometric parameters, Spearman’s correlation coefficients were computed between RF‐based classification predictions learnt from , , and and clinical parameters for the 620 NoNASH/NASH patient cohort (Table 1) to increase statistical power (549 NoNASH/71 NASH). Highest correlation was observed for HOMA‐IR and glycated hemoglobin (HbA1c) as expected because of higher values for both parameters in NASH patients compared to NoNASH patients of the learning cohort (Table 5). In contrast, no clear correlations were observed between BMI or high‐density lipoprotein‐cholesterol (HDL‐C) and any signature prediction. Other clinical parameters were correlated in a more sex‐specific way, with age, low‐density lipoprotein‐cholesterol (LDL‐C), HbA1c, and HOMA‐IR levels being more markedly correlated with model prediction, whereas liver enzyme levels (alanine aminotransferase [ALAT], aspartate aminotransferase [ASAT]) were more strongly correlated with . Taken together, this correlation analysis suggests that despite being based on distinct gene sets, RF‐built signatures identify patients with altered liver enzyme levels and altered glucose homeostasis.
TABLE 5.
Correlation Analysis
| Signature |
|
|
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Clinical parameter | Correlation | P Value | Correlation | P Value | Correlation | P Value | |||
| Age | 0.275 | 3.3 × 10−4*** | 0.153 | 1.1 × 10−3** | 0.21 | 1.3 × 10−7*** | |||
| BMI | 0.002 | 9.7 × 10−1 | 0.110 | 1.9 × 10−2 | 0.065 | 1.0 × 10−1 | |||
| LDL‐C | −0.205 | 8.6 × 10−3 | −0.069 | 1.4 × 10−1 | −0.098 | 1.5 × 10−2 | |||
| HDL‐C | −0.084 | 2.8 × 10−1 | −0.078 | 9.7 × 10−2 | −0.077 | 5.7 × 10−2 | |||
| Triglycerides | 0.243 | 1.6 × 10−3** | 0.205 | 1.1 × 10−5*** | 0.272 | 5.7 × 10−12*** | |||
| HbA1c | 0.471 | 1.9 × 10−10*** | 0.279 | 1.5 × 10−9*** | 0.353 | 1.4 × 10−19*** | |||
| HOMA‐IR | 0.423 | 1.8 × 10−8*** | 0.307 | 3.2 × 10−11*** | 0.294 | 1.3 × 10−13*** | |||
| ASAT | 0.253 | 1.0 × 10−3** | 0.312 | 1.2 × 10−11*** | 0.322 | 2.5 × 10−16*** | |||
| ALAT | 0.170 | 2.9 × 10−2 | 0.352 | 1.1 × 10−14*** | 0.306 | 7.2 × 10−15*** | |||
Correlation between prediction of RF models learnt from reference signatures on the learning cohort and clinical parameters of the HUL cohort. Spearman correlation coefficient and corresponding P value were computed in R (R Foundation for Statistical Computing, Vienna, Austria). Bonferroni’s correction was applied to deal with a multiple‐comparisons situation; P values with corresponding FWER <1% and 0.1% are tagged with ** and ***, respectively.
Abbreviation: FWER, family‐wise error rate.
Discussion
Several studies have already reported liver transcriptomic signatures of NASH, but their reliability and stability can be questioned because of limited cohort sizes and the lack of the evaluation of signature robustness. In this study, 170 propensity‐score–matched liver biopsies were selected with rigorous biological and statistical criteria, from which we determined DEGs using a robust bioinformatic protocol. Several methodological pitfalls have been addressed in our analysis. The use of a bootstrap method to increase the robustness of DEG identification by Limma, a commonly differential analysis approach, has previously evidenced a high sensitivity of the differential analysis to cohort composition.( 26 ) In our study, such a bootstrap analysis led to the exclusion of 50%‐82% of transcripts initially identified as DEGs by a single Limma run. This instability was noted for the three contrasts ( , , and ). Importantly, exclusion was not restricted to poorly expressed or weakly modulated genes. Thus, to avoid misinterpretation, a stability analysis using a bootstrap approach should be systematically performed in cohort studies.
Numerous studies reported the sexual dimorphic nature of metabolic regulations.( 38 , 39 ) In the liver, they have been mostly ascribed to the growth hormone/Janus kinase 2/signal transducer and activator of transcription 5 pathway.( 40 ) NASH has also a strong sex‐specific component, with men generally displaying a more‐severe phenotype than nonmenopausal women.( 41 , 42 ) On the basis of RF models, we identified sex‐specific NASH signatures whose predictive power was evaluated against independent cohorts. We further compared the robustness of such signatures to that of single gene predictors and random signatures. We found a larger number of reliable DEGs in men than in women, whose median age is 45 ± 11 years (SD). A distinct menopausal status among women aged ~40‐50 years could explain such a difference by increasing biological noise to the differential analysis and impairing DEG detection. The low overlap between and and associated signatures hints at sex specificity as well. Indeed, GO BP enrichment of reliable DEGs in men or women did not reveal recurrent biological themes, with the exception of ”cellular adhesion,” a rather broad terminology unable to pin down specific biological pathways. The 39 commonly dysregulated genes are not associated with a specific biological process, leaving open the question of a potentially (dis)similar natural history of male or female NASH. Interestingly, we observed that GO BP term enrichment of DEGs in pointed to cell‐cycle regulation processes, a feature which may be related to the higher propensity of female hepatocytes to proliferate.( 43 )
Three signatures were identified using RF models, with and encompassing a similar number of transcripts. was larger because of a higher sample number used for training, thereby enabling more sophisticated classification rules to be used with this more heterogeneous population. A careful evaluation of bioinformatically defined signatures is required given that algorithms that are used may use FCs that do not seem significant from a biological point of view. Thus, although several signatures can reach similarly high classification performances in RF, the identification of a unique signature surpassing all others in various conditions or cohorts remains unlikely. Additionally, such algorithms limit information redundancy when selecting signature genes, thus hindering the detection of significantly enriched GO terms in signature gene lists.
We compared the predictive performances of our signatures to those of randomly generated signatures or single gene predictors. RF‐based signatures were consistently more efficient at classifying NASH versus NoNASH patients from independent cohorts. A single gene predictor could perform better than a signature for a given data set, but not as efficiently on other cohorts. For example, FAT1, a gene regulating cell‐cell contact, which was highly predictive of NASH in male patients from the HUL cohort, turned out to be inefficient in the UKD cohort (Supporting Fig. S12). Thus, signatures are required to extrapolate classification performances to other cohorts by reducing prediction variability of single gene predictors. Among the tested three signatures elaborated from , , and , was the more robust with a limited size (n = 20). Reason(s) for this better performance are yet unclear.
The prognostic performance of signatures was improved in the UZA cohort when stratifying patients according to the fibrosis grade. In line with this, predictivity of signatures on the fibrosis‐stratified DU cohort was in the highest range, suggesting that our analysis integrates features of the fibrotic response, which is clinically relevant when considering long‐term outcomes.( 7 , 8 ) Interestingly, some genes constituting the NASH signatures were also identified when defining a cross‐species transcriptomic signature of fibrosis.( 12 ) Indeed, 12 of 34 (35%) genes constituting this fibrotic signature( 12 ) were identified as strongly up‐regulated (absolute log2FC, >1.5) in the bootstrapped Limma analysis (Supporting Table S3), and six of them are common to both the cross‐species fibrotic signature and NASH signatures reported here (Supporting Table S5). These genes are UBD/FAT10, CCDN1/cyclinD1, FAT1, secreted phosphoprotein 1/osteopontin, ZMAT3, and fatty acid binding protein 4/adipocyte P2.
Machine learning approaches like RF extract information and outperform linear approaches. They identified signatures in the HUL cohort that reached area under the receiver operating characteristic curves in the 0.62‐0.93 range when diagnosing independent cohorts, therefore being comparable to, or better than, other NASH signatures based on lipidomic( 44 , 45 ) or combining multiple proteomic, biometric, and genomic characteristics.( 46 ) Importantly, our study clearly points to sex as an often‐neglected,( 22 ) but nevertheless important, factor in liver and NASH biology.( 42 ) Indeed, liver physiopathological responses to various challenges are sex dependent in rodents.( 47 , 48 , 49 ) Although the translatability of these findings to our human‐based analysis is not straightforward, all these data converge toward a definition of human NASH as a sexually dimorphic disease. This is a potentially important and relevant finding in terms of biomarker research (given that, e.g., YKL‐40/CHI3L1 is included in a biomarker panel in development), as well as for risk stratification and pharmacological therapy. Liver pathophysiology displays sex‐linked disparities, suggesting that liver‐targeted drugs may exhibit distinct mechanisms of action in men and women. In this respect, we note that the recent randomized phase II clinical trial evaluating the effect of cenicriviroc (a dual C‐C chemokine receptor 2‐5 antagonist) in the treatment of NASH with fibrosis resulted in a positive effect in men, but not in women, on the improvement of fibrosis after 1 year.( 50 ) Although a limitation of our work is the comparison of a rather small NASH population to a larger non‐NASH cohort, our data and others call for a careful design of preclinical and clinical studies integrating sex as a major determinant of liver responses.
Author Contributions
C.G. and B.D. performed experiments and collected data; J.V., J.D.C., J.E., and P.L. analyzed data; J.V. and P.L. wrote the manuscript; D.T. provided expertise in statistical analysis; L.V.G., S.F., V.R., and F.P. constituted and managed UZA and HUL cohorts and collected biological data and samples; J.V., B.S., and P.L. coordinated the study; J.V., J.D.C., B.S., S.F., J.E., and P.L. revised the manuscript.
Supporting information
Supplementary Material
Supported by grants from Agence Nationale pour la Recherche (ANR‐16‐RHUS‐0006‐PreciNASH and ANR‐10‐LBEX‐46), the European Union (FP6 Hepadip FP6‐018734 and FP7 Resolve, FP7‐305707), Fondation de France (Grant 2014 00047965), and Fondation pour la Recherche Médicale (Equipe labellisée, DEQ20150331724). B.S. is a recipient of an Advanced ERC Grant (694717).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018;15:11‐20. [DOI] [PubMed] [Google Scholar]
- 2. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease—meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73‐84. [DOI] [PubMed] [Google Scholar]
- 3. Friedman SL, Neuschwander‐Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Argo CK, Northup PG, Al‐Osaimi AM, Caldwell SH. Systematic review of risk factors for fibrosis progression in non‐alcoholic steatohepatitis. J Hepatol 2009;51:371‐379. [DOI] [PubMed] [Google Scholar]
- 5. Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta‐analysis of paired‐biopsy studies. Clin Gastroenterol Hepatol 2015;13:643‐654.e1‐e9; quiz, e39‐e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kleiner DE, Brunt EM, Wilson LA, Behling C, Guy C, Contos M, et al. Association of histologic disease activity with progression of nonalcoholic fatty liver disease. JAMA Netw Open 2019;2:e1912565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Angulo P, Kleiner DE, Dam‐Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long‐term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149:389‐397.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: systematic review and meta‐analysis. Hepatology 2017;65:1557‐1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schuppan D, Surabattula R, Wang XY. Determinants of fibrosis progression and regression in NASH. J Hepatol 2018;68:238‐250. [DOI] [PubMed] [Google Scholar]
- 10. Moylan CA, Pang H, Dellinger A, Suzuki A, Garrett ME, Guy CD, et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014;59:471‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ahrens M, Ammerpohl O, von Schonfels W, Kolarova J, Bens S, Itzel T, et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease‐specific and remodeling signatures after bariatric surgery. Cell Metab 2013;18:296‐302. [DOI] [PubMed] [Google Scholar]
- 12. Lefebvre P, Lalloyer F, Bauge E, Pawlak M, Gheeraert C, Dehondt H, et al. Interspecies NASH disease activity whole‐genome profiling identifies a fibrogenic role of PPARalpha‐regulated dermatopontin. JCI Insight 2017;2:92264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Teufel A, Itzel T, Erhart W, Brosch M, Wang XY, Kim YO, et al. Comparison of gene expression patterns between mouse models of nonalcoholic fatty liver disease and liver tissues from patients. Gastroenterology 2016;151:513‐525.e0. [DOI] [PubMed] [Google Scholar]
- 14. Hyotylainen T, Jerby L, Petaja EM, Mattila I, Jantti S, Auvinen P, et al. Genome‐scale study reveals reduced metabolic adaptability in patients with non‐alcoholic fatty liver disease. Nat Commun 2016;7:8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suppli MP, Rigbolt KTG, Veidal SS, Heeboll S, Eriksen PL, Demant M, et al. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal‐weight individuals. Am J Physiol Gastrointest Liver Physiol 2019;316:G462‐G472. [DOI] [PubMed] [Google Scholar]
- 16. Haas JT, Vonghia L, Mogilenko DA, Verrijken A, Molendi‐Coste O, Fleury S, et al. Transcriptional network analysis implicates altered hepatic immune function in NASH development and resolution. Nat Metab 2019;1:604‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ryaboshapkina M, Hammar M. Human hepatic gene expression signature of non‐alcoholic fatty liver disease progression, a meta‐analysis. Sci Rep 2017;7:12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Subichin M, Clanton J, Makuszewski M, Bohon A, Zografakis JG, Dan A. Liver disease in the morbidly obese: a review of 1000 consecutive patients undergoing weight loss surgery. Surg Obes Relat Dis 2015;11:137‐141. [DOI] [PubMed] [Google Scholar]
- 19. Souto KP, Meinhardt NG, Ramos MJ, Ulbrich‐Kulkzynski JM, Stein AT, Damin DC. Nonalcoholic fatty liver disease in patients with different baseline glucose status undergoing bariatric surgery: analysis of intraoperative liver biopsies and literature review. Surg Obes Relat Dis 2018;14:66‐73. [DOI] [PubMed] [Google Scholar]
- 20. Gershoni M, Pietrokovski S. The landscape of sex‐differential transcriptome and its consequent selection in human adults. BMC Biol 2017;15:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Labonte B, Engmann O, Purushothaman I, Menard C, Wang J, Tan C, et al. Sex‐specific transcriptional signatures in human depression. Nat Med 2017;23:1102‐1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lonardo A, Nascimbeni F, Ballestri S, Fairweather D, Win S, Than TA, et al. Sex differences in NAFLD: state of the art and identification of research gaps. Hepatology 2019;70:1457‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tramunt B, Smati S, Grandgeorge N, Lenfant F, Arnal JF, Montagner A, et al. Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia 2020;63:453‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ein‐Dor L, Zuk O, Domany E. Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer. Proc Natl Acad Sci U S A 2006;103:5923‐5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chibon F. Cancer gene expression signatures—the rise and fall? Eur J Cancer 2013;49:2000‐2009. [DOI] [PubMed] [Google Scholar]
- 26. Michiels S, Koscielny S, Hill C. Prediction of cancer outcome with microarrays: a multiple random validation strategy. Lancet 2005;365:488‐492. [DOI] [PubMed] [Google Scholar]
- 27. Liao JG, Chin KV. Logistic regression for disease classification using microarray data: model selection in a large p and small n case. Bioinformatics 2007;23:1945‐1951. [DOI] [PubMed] [Google Scholar]
- 28. Higgins JP. Nonlinear systems in medicine. Yale J Biol Med 2002;75:247‐260. [PMC free article] [PubMed] [Google Scholar]
- 29. Diaz‐Uriarte R, Alvarez de Andres S. Gene selection and classification of microarray data using random forest. BMC Bioinform 2006;7:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boulesteix AL, Janitza S, Kruppa J, König IR. Overview of random forest methodology and practical guidance with emphasis on computational biology and bioinformatics. Wiley Interdiscip Rev Data Min Knowl Discov 2012;2:493‐507. [Google Scholar]
- 31. Guyon I, Weston J, Barnhill S, Vapnik V. Gene selection for cancer classification using support vector machines. Mach Learn 2002;46:389‐422. [Google Scholar]
- 32. Margerie D, Lefebvre P, Raverdy V, Schwahn U, Ruetten H, Larsen P, et al. Hepatic transcriptomic signatures of statin treatment are associated with impaired glucose homeostasis in severely obese patients. BMC Med Genomics 2019;12:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Verrijken A, Francque S, Mertens I, Prawitt J, Caron S, Hubens G, et al. Prothrombotic factors in histologically proven nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2014;59:121‐129. [DOI] [PubMed] [Google Scholar]
- 34. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 35. Wallace TM, Levy JC, Matthews DR. Use and abuse of HOMA modeling. Diabetes Care 2004;27:1487‐1495. [DOI] [PubMed] [Google Scholar]
- 36. Tang Q, Li X, Song P, Xu L. Optimal cut‐off values for the homeostasis model assessment of insulin resistance (HOMA‐IR) and pre‐diabetes screening: developments in research and prospects for the future. Drug Discov Ther 2015;9:380‐385. [DOI] [PubMed] [Google Scholar]
- 37. Diehl AM, Day C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N Engl J Med 2017;377:2063‐2072. [DOI] [PubMed] [Google Scholar]
- 38. Morselli E, Frank AP, Santos RS, Fatima LA, Palmer BF, Clegg DJ. Sex and gender: critical variables in pre‐clinical and clinical medical research. Cell Metab 2016;24:203‐209. [DOI] [PubMed] [Google Scholar]
- 39. Mauvais‐Jarvis F. Sex differences in metabolic homeostasis, diabetes, and obesity. Biol Sex Differ 2015;6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lichanska AM, Waters MJ. How growth hormone controls growth, obesity and sexual dimorphism. Trends Genet 2008;24:41‐47. [DOI] [PubMed] [Google Scholar]
- 41. Hashimoto E, Tokushige K. Prevalence, gender, ethnic variations, and prognosis of NASH. J Gastroenterol 2011;46(Suppl. 1):63‐69. [DOI] [PubMed] [Google Scholar]
- 42. Ballestri S, Nascimbeni F, Baldelli E, Marrazzo A, Romagnoli D, Lonardo A. NAFLD as a sexual dimorphic disease: role of gender and reproductive status in the development and progression of nonalcoholic fatty liver disease and inherent cardiovascular risk. Adv Ther 2017;34:1291‐1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen F, Jimenez RJ, Sharma K, Luu HY, Hsu BY, Ravindranathan A, et al. Broad distribution of hepatocyte proliferation in liver homeostasis and regeneration. Cell Stem Cell 2019;26:27‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mayo R, Crespo J, Martínez‐Arranz I, Banales JM, Arias M, Mincholé I, et al. Metabolomic‐based noninvasive serum test to diagnose nonalcoholic steatohepatitis: results from discovery and validation cohorts. Hepatol Commun 2018;2:807‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barr J, Caballeria J, Martinez‐Arranz I, Dominguez‐Diez A, Alonso C, Muntane J, et al. Obesity‐dependent metabolic signatures associated with nonalcoholic fatty liver disease progression. J Proteome Res 2012;11:2521‐2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wood GC, Chu X, Argyropoulos G, Benotti P, Rolston D, Mirshahi T, et al. A multi‐component classifier for nonalcoholic fatty liver disease (NAFLD) based on genomic, proteomic, and phenomic data domains. Sci Rep 2017;7:43238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jacobs SAH, Gart E, Vreeken D, Franx BAA, Wekking L, Verweij VGM, et al. Sex‐specific differences in fat storage, development of non‐alcoholic fatty liver disease and brain structure in Juvenile HFD‐induced obese Ldlr‐/‐.Leiden mice. Nutrients 2019;11:1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ande SR, Nguyen KH, Gregoire Nyomba BL, Mishra S. Prohibitin‐induced, obesity‐associated insulin resistance and accompanying low‐grade inflammation causes NASH and HCC. Sci Rep 2016;6:23608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kurt Z, Barrere‐Cain R, LaGuardia J, Mehrabian M, Pan C, Hui ST, et al. Tissue‐specific pathways and networks underlying sexual dimorphism in non‐alcoholic fatty liver disease. Biol Sex Differ 2018;9:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo‐controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018;67:1754‐1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The transcriptomic data sets generated during and/or analyzed during the current study are available in the NCBI GEO repository. Clinical data that support the findings of this study are available from the corresponding author upon reasonable request.