

Abstract
Biological processes rely on transient interactions that govern assembly of biomolecules into higher order, multi‐component systems. A synthetic platform for the dynamic assembly of multicomponent complexes would provide novel entries to study and modulate the assembly of artificial systems into higher order topologies. Here, we establish a hybrid DNA origami‐based approach as an assembly platform that enables dynamic templating of supramolecular architectures. It entails the site‐selective recruitment of supramolecular polymers to the platform with preservation of the intrinsic dynamics and reversibility of the assembly process. The composition of the supramolecular assembly on the platform can be tuned dynamically, allowing for monomer rearrangement and inclusion of molecular cargo. This work should aid the study of supramolecular structures in their native environment in real‐time and incites new strategies for controlled multicomponent self‐assembly of synthetic building blocks.
Keywords: multicomponent assembly, polymers, self-assembly, supramolecular synthesis
DNA origami platforms were used to assemble synthetic supramolecular polymers, retaining their dynamic nature with control over monomer composition and loading and dynamic exchange of molecular cargo. The DNA origami‐templated self‐assembly allows for novel strategies for controlled multicomponent self‐assembly and characterization of synthetic supramolecular systems.
Cellular processes are governed by complex molecular networks, whose structure and ability to process biological information relies on transient non‐covalent interactions. [1] In many processes, supramolecular assemblies operate in response to extra‐ and intracellular stimuli and dynamically regulate cell morphology, [2] intracellular transport of molecular cargo, and proximity‐induced activation of enzymes in cell fate regulation including differentiation and cell death. [3] Signaling pathways adopt open‐ended supramolecular organizing centers that recruit effector proteins, enabling complex regulatory functions such as signal amplification, ultrasensitivity, and feedback control. [4] Synthetic supramolecular polymers are inspired by these natural architectures [5] and their modular design and synthetic accessibility enables the bottom‐up construction of biomimetic systems with adaptive and responsive behavior.[ 6 , 7 ] Supramolecular assemblies have concomitantly been used for the dynamic recruitment of proteins, [8] as scaffolds for cell receptor targeting,[ 9 , 10 ] and as a modular platform for cellular uptake.[ 11 , 12 ] Although the assembly behavior of one‐dimensional synthetic supramolecular systems has been thoroughly investigated,[ 5 , 13 , 14 ] real‐time characterization of the dynamics of individual supramolecular assemblies, the controlled positioning of supramolecular building blocks along an assembly, and the generation of higher ordered, well‐defined architectures consisting of multiple different types of building blocks remain outstanding challenges.
The combination of synthetic supramolecular systems with the predictable assembly of DNA elements allows for the generation of hybrid molecular systems,[ 15 , 16 , 17 ] which have included programmable supramolecular polymers [18] and a dynamic enzyme activation platform. [19] Rational design of DNA nanostructures, and in particular DNA origami‐based structures, enables controlled nanoscale organization of a large number of molecular species,[ 20 , 21 ] including metal nanoparticles with tunable optical properties, [22] seeded growth of gold nanostructures, [23] and shape‐controlled synthesis of covalent polymers. [24] The combination of DNA nanotechnology and covalent polymers has resulted in a new class of hybrid materials [25] with exciting properties through templated routing. [26] Manipulation of the nanoscale patterning of individual DNA‐conjugated synthetic polymers on a DNA origami platform offers exciting possibilities for polymer nanosynthesis. [27] Templated one‐dimensional aggregation of protein‐based building blocks on DNA origami has opened up a new avenue to analyze nucleated fibril growth, [28] but similar advances in synthetic supramolecular polymers have not been reported.
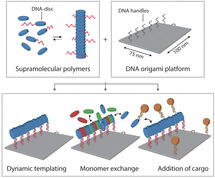
Here, we employ DNA origami as an organizing platform for the templated assembly of multicomponent, one dimensional supramolecular assemblies. Addition of a small fraction of DNA‐functionalized monomers directs the supramolecular assembly process to the surface of the DNA nanostructure while retaining the dynamic character of the system (Figure 1 a). We show recruitment of non‐DNA‐functionalized building blocks with various functionalities into the DNA origami‐templated assemblies. We propose that our hybrid DNA origami‐based approach can provide a controlled nanoscale environment for the supramolecular synthesis of multicomponent assemblies, opening up new possibilities for systematic non‐covalent synthesis and investigations into the mechanistic principles behind self‐assembly, which will be the cornerstones of follow‐up studies.
Figure 1.
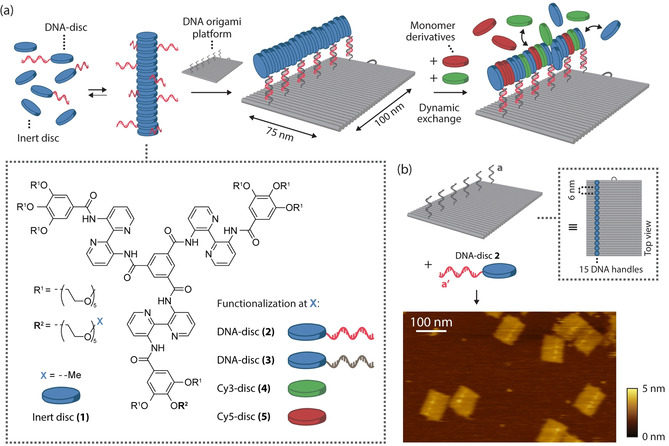
General concept for the assembly of one‐dimensional supramolecular assemblies on a DNA origami platform. a) DNA‐functionalized and non‐functionalized monomeric building blocks co‐assemble into one‐dimensional supramolecular polymers in aqueous solution. A two‐dimensional 75×100‐nm2 DNA origami platform is programmed to include single‐stranded DNA handles at specific locations, enabling hybridization to the DNA‐functionalized monomers within these aggregates. The dynamic nature of the multicomponent assembly allows for the templated exchange of various monomer derivatives. An overview of the utilized mono‐functionalized monomers, including the molecular structure of the monomer's core, is provided in the dashed inset (for reactions schemes and DNA sequences, see Figure S1). b) The rectangular DNA origami structure was designed to incorporate 15 copies of DNA‐disc 2 through DNA hybridization of ODN (a′) and the single‐stranded handles (a) protruding from the surface, following the long side of the nanostructures as indicated by the blue circles in the top view. (For clarity only 6 handles are shown here; for details on the DNA origami design, see Figure S3). AFM analysis revealed successful incorporation of DNA‐disc 2 onto DNA nanostructures as indicated by spots of high intensity at the programmed positions.
The supramolecular system consists of bipyridine‐based C3‐symmetrical amphiphilic discotic molecules that self‐assemble in water to form intrinsically fluorescent columnar aggregates, presumably via an isodesmic mechanism. [18] A library of differently functionalized monomers (Figure S1) includes fully glycol‐decorated monomer 1 (termed inert “disc”, due to the disc‐like molecular structure, Figure 1 a) and 1 a, with a single amine‐functionalization on one of the nine ethylene glycol tails as versatile starting point for the synthesis of other derivatives. Reaction with dibenzocyclooctyne‐N‐hydroxysuccinimidyl ester (DBCO‐NHS) afforded disc 1 b, allowing straightforward conjugation to azide‐functionalized oligonucleotides (ODNs, Supplementary Table 1) using strain‐promoted alkyne‐azide cycloaddition, affording DNA‐discs 2 and 3 (Figure 1 a, Figure S2). To enable spectroscopic investigation, also dye‐labelled monomers were synthesized by coupling of disc 1 a to the NHS esters of the sulfonated cyanine dyes (Cy3 and Cy5), yielding Cy3‐disc 4 and Cy5‐disc 5.
To construct a nanoscale platform for the recruitment of supramolecular assemblies, we employed the two‐dimensional 75×100‐nm2 DNA origami rectangle [29] and positioned 15 handle strands along the 100‐nm side of the DNA origami rectangle with 6‐nm spacing for hybridization with complementary DNA‐functionalized monomers (Figure 1 b). Given an intermolecular distance of <0.4 nm between monomers in a supramolecular aggregate, a distance of 6 nm between adjacent handle sites should leave enough room for the incorporation of other, non‐DNA‐functionalized, monomers. Incorporation of DNA‐functionalized disc 2 was characterized by atomic force microscopy (AFM), which revealed spots of high intensity at the programmed positions indicating successful hybridization (Figure 1 b). We attribute the observed patchy morphology to clustering of tethered monomers by intermolecular stacking (for polymeric mixture incorporation, see Figure S5a).
Structural evidence for the supramolecular recruitment of functionalized monomers to the DNA origami‐templated assembly platform was obtained by use of transmission electron microscopy (TEM) in combination with gold nanoparticles (AuNPs) as high‐contrast markers of the nanoscale supramolecular‐DNA hybrid architecture (Figure 2 a). DNA origami platforms were designed for site‐specific incorporation of a single AuNP to two corners of the nanostructure (Figure 2 b). Subsequently, multicomponent supramolecular assemblies were prepared by co‐assembly of non‐DNA‐functionalized inert disc 1, DNA‐disc 2 for incorporation onto the DNA origami platform and DNA‐disc 3 for hybridization to AuNPs (Figure 2 a). After incubation with DNA‐functionalized AuNPs (for images without AuNPs, see Figure S5c), analysis by TEM revealed that on average 4–6 AuNPs were aligned along the supramolecular assembly on the DNA origami platform (Figure 2 c). Given a concentration of 5 % DNA‐disc 3 and assuming an intermolecular distance of 0.3–0.5 nm between monomers, statistically a single DNA origami‐templated supramolecular polymer could contain 10–17 binding sites for AuNPs. We assume that steric interactions between the 10‐nm AuNPs in the polymers limit the actual number of incorporated nanoparticles. This structural characterization thus shows that supramolecular polymers can indeed be templated onto the DNA origami platform and that functionality can be introduced by co‐assembly of monomeric derivatives with molecular cargo.
Figure 2.
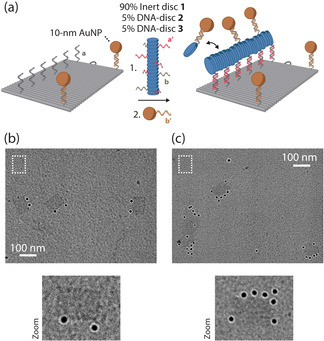
TEM analysis confirms supramolecular recruitment of gold nanoparticles (AuNPs) to DNA nanostructures. a) Schematic overview of both direct and supramolecular site‐selective recruitment of AuNPs to DNA nanostructures. DNA origami structures were designed to include 15 handles for DNA‐functionalized monomer assembly and two binding sites for AuNPs with three handles each (for design, see Figure S3). First, incubation of 4 nM DNA origami with 8 nM of DNA‐functionalized AuNPs and subsequent purification by gel extraction was performed to directly incorporate two AuNPs as visual markers. Then, a 0.3 μM pre‐annealed supramolecular polymer mixture consisting of 90 % inert disc 1, 5 % DNA‐disc 2, and 5 % DNA‐disc 3 was added to 0.5 nM purified DNA origami. Finally, 1 nM DNA‐functionalized AuNPs was added (See Figure S4). All incubation steps were performed for 1 h at 18 °C. TEM images of DNA origami nanostructures before functionalization (see Figure S5b), after direct incorporation of AuNPs at programmed positions (b), and after supramolecular recruitment of AuNPs (c). The expected size of a 75×100‐nm2 nanostructure is indicated in each image as a reference (dashed white rectangles). Scale bars, 100 nm.
Next, we utilized agarose gel electrophoresis to analyze the assembly of supramolecular polymers onto DNA origami nanostructures in solution. To this end, we assembled multicomponent polymers containing inert disc 1, 10 % Cy3‐disc 4 for visualization and 5 % DNA‐disc 2 on DNA origami nanostructures with complementary handles (Figure S6a). Analysis revealed a gel band with increasing Cy3 fluorescence from Cy3‐disc 4 co‐migrating with the DNA origami structure upon increasing concentration of monomers (Figure S6b; compare top gel stained for DNA with bottom gel displaying Cy3 fluorescence). An additional slow‐migrating band appeared at high monomer concentration (≥2.5 μM) containing both DNA origami and Cy3‐disc 4. We attribute this species to two DNA origami platforms bridged by a supramolecular assembly binding both DNA nanostructures simultaneously via an excess of monomers. Indeed, the ratio between both species observed on gel is concentration‐dependent and could be varied by changing the monomer concentration (Figure S6).
Non‐covalent interactions in supramolecular assemblies enable dynamic rearrangement and tuning of assembly composition. [13] To investigate the exchange of monomeric components in the aggregates assembled on the DNA origami platform, we performed a two‐step assembly protocol with monomers capable of fluorescence resonance energy transfer (FRET). First, supramolecular assemblies containing DNA‐disc 2 were assembled on the DNA origami platform, as described in Figure S6. Then, a solution containing a supramolecular polymer formed by dye‐labelled, non‐DNA‐functionalized, Cy3‐disc 4 was added to the mixture (Figure 3). Gel electrophoresis demonstrated Cy3 fluorescence at positions corresponding to the DNA origami nanostructures, illustrating that monomeric components can also be exchanged from the supramolecular polymer in solution into the supramolecular assembly on the DNA nanostructure (Figure S7). Importantly, both addition of Cy5‐disc 5 to DNA origami‐templated assemblies containing Cy3‐disc 4, and vice versa of Cy3‐disc 4 to assemblies containing Cy5‐disc 5 resulted in FRET (Figure 3 b,c) between both components on the DNA origami in the agarose gel. This indicates the mutual incorporation and close proximity of these two fluorescent monomers on the assembly platform, and further testifies to the dynamic intermixing of the supramolecular components on the DNA origami platform.
Figure 3.
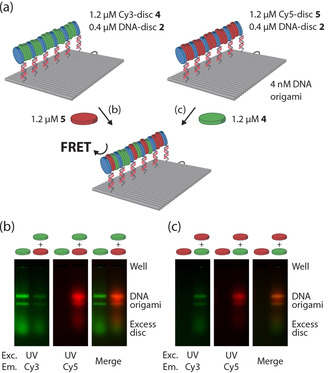
DNA‐guided recruitment and dynamic mixing of multicomponent supramolecular assemblies onto DNA nanostructures. a) Schematic overview of the assembly. DNA origami (4 nM) with 15 single‐stranded handles was incubated with a 1.6 μM mixture of 25 % DNA‐disc 2, and 75 % Cy3‐disc 4 (left) or 75 % Cy5‐disc 5 (right) building blocks for 1 h at 18 °C. Subsequently, 1.2 μM Cy5‐disc 5 (b) or Cy3‐disc 4 (c) was added and incubated for 1 h at 18 °C. The assembled complexes were subjected to gel electrophoresis in parallel on 1.5 % agarose gels without staining, to independently visualize the co‐migration of the supramolecular assemblies formed on the DNA origami and to observe FRET between the dye‐labeled monomers co‐assembled on the DNA origami platform. Images were obtained by illumination with UV light source (exc.) and emission through either a Cy3 (left, green) or Cy5 (middle, red) filter (em).
The kinetics of the supramolecular building block exchange on the DNA assembly platform was studied in more detail using FRET studies on engineered DNA origami platforms with specifically positioned Cy3 fluorophore for energy transfer to Cy5‐disc 5. To this end, 15 Cy3 fluorophores were incorporated at 6 nm from the monomeric recruitment sites (Figure 4 a). As reference a control system was included where the fluorophores are located at 39 nm from the polymer, too far away for efficient FRET to occur (Figure 4 b). Addition of a solution of supramolecular Cy5‐disc 5 assemblies to the supramolecular polymers assembled on DNA origami produced a time‐dependent increase in FRET, but only when the Cy3 fluorophores were placed in close proximity to the supramolecular assembly (Figure 4; compare red curves in a and b). The FRET signal approaches equilibrium after 60 min, which corresponds to reported mixing kinetics between similar supramolecular assemblies, purely assembled in solution.[ 8 , 18 ] No increase in FRET was observed when DNA‐disc 2, anchoring the supramolecular polymer to the DNA, was omitted from multicomponent mixture, confirming the absence of nonspecific interactions with the DNA nanostructure (gray curves). Collectively, these results demonstrate that the monomers incorporate site‐selectively out of the supramolecular polymer in solution into the supramolecular assembly templated on the DNA origami nanostructure and that multicomponent assemblies can be co‐assembled by stepwise addition of components to the platform.
Figure 4.
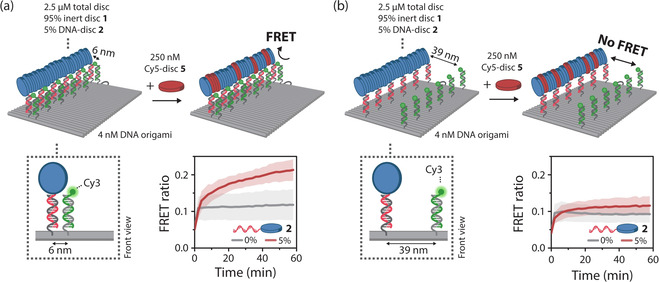
Supramolecular recruitment of fluorescent monomers occurs site‐selectively. To demonstrate site‐selective incorporation of monomers onto the DNA platform using origami‐to‐monomer FRET, the DNA origami structures were adapted to include 15 Cy3 fluorophores positioned at either 6 nm (a) or 39 nm (b) from the 15 handles for DNA‐monomer incorporation. To pre‐assemble supramolecular polymers, 4 nM of each DNA origami variant was incubated with a 2.5 μM pre‐annealed supramolecular polymer mixture of inert disc 1 containing 5 % DNA‐disc 2 for 1 h at 18 °C. After incubation, 250 nM Cy5‐disc 5 was added directly to the reaction mixtures. Time‐resolved FRET measurements in the absence (gray curves) or presence (red curves) of 5 % DNA‐disc 2 revealed an increase in FRET when the Cy3 fluorophores are positioned at 6 nm from the DNA‐monomer incorporation sites, and no increase at 39‐nm spacing. Data is represented as mean ± s.d. of three independent experiments.
In this work, we have presented a nanoscale DNA origami‐based platform for the dynamic templated assembly of synthetic supramolecular architectures. Our approach entailed the site‐specific immobilization of a small fraction of the supramolecular building blocks by their functionalization with an oligonucleotide complementary to the origami platform. Subsequently, a dynamic and reversible assembly of additional, cargo‐loaded supramolecular building blocks could be achieved on the platform, guided by DNA hybridization. We anticipate such platforms that retain the dynamic nature of the assembled system harbor a large potential in dynamic templating of supramolecular polymers and other reversible assemblies and for the generation of higher order topologies, while simultaneously facilitating real‐time characterization of supramolecular assembly events. In addition, templated synthesis of supramolecular materials with molecular‐scale accuracy could enable rational non‐covalent synthesis, providing orthogonal handles for hierarchical self‐assembly, controlled supramolecular block copolymerization,[ 30 , 31 ] directional nucleation‐elongation, [32] and nano‐actuated polymers. [27]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The research was supported by funding from HTSM through STW grant 12859‐FluNanoPart and the Netherlands Organization for Scientific Research (NWO) via Gravity program 024.001.035 and VICI grant 016.150.366. We thank Lorenzo Albertazzi for fruitful discussions. The ICMS Animation Studio contributed the cartoons of DNA strands and the DNA origami structure.
J. Schill, B. J. H. M. Rosier, B. Gumí Audenis, E. Magdalena Estirado, T. F. A. de Greef, L. Brunsveld, Angew. Chem. Int. Ed. 2021, 60, 7612.
Contributor Information
Prof. Dr. Tom F. A. de Greef, Email: t.f.a.d.greef@tue.nl.
Prof. Dr. Luc Brunsveld, Email: l.brunsveld@tue.nl.
References
- 1. Kholodenko B. N., Hancock J. F., Kolch W., Nat. Rev. Mol. Cell Biol. 2010, 11, 414–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pollard T. D., Cooper J. A., Science 2009, 326, 1208–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roostalu J., Surrey T., Nat. Rev. Mol. Cell Biol. 2017, 18, 702–710. [DOI] [PubMed] [Google Scholar]
- 4. Wu H., Fuxreiter M., Cell 2016, 165, 1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Greef T. F. A., Smulders M. M. J., Wolffs M., Schenning A. P. H. J., Sijbesma R. P., Meijer E. W., Chem. Rev. 2009, 109, 5687–5754. [DOI] [PubMed] [Google Scholar]
- 6. Aida T., Meijer E. W., Stupp S. I., Science 2012, 335, 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Uhlenheuer D. A., Petkau K., Brunsveld L., Chem. Soc. Rev. 2010, 39, 2817–2826. [DOI] [PubMed] [Google Scholar]
- 8. Petkau-Milroy K., Uhlenheuer D. A., Spiering A. J. H., Vekemans J. A. J. M., Brunsveld L., Chem. Sci. 2013, 4, 2886–2891. [Google Scholar]
- 9. Lee S. S., Fyrner T., Chen F., Álvarez Z., Sleep E., Chun D. S., Weiner J. A., Cook R. W., Freshman R. D., Schallmo M. S., et al., Nat. Nanotechnol. 2017, 12, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Müller M. K., Brunsveld L., Angew. Chem. Int. Ed. 2009, 48, 2921–2924; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 2965–2968. [Google Scholar]
- 11. van Dun S., Schill J., Milroy L. G., Brunsveld L., Chem. Eur. J. 2018, 24, 16445–16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Webber M. J., Langer R., Chem. Soc. Rev. 2017, 46, 6600–6620. [DOI] [PubMed] [Google Scholar]
- 13. Albertazzi L., Van Der Zwaag D., Leenders C. M. A., Fitzner R., Van Der Hofstad R. W., Meijer E. W., Science 2014, 344, 491–495. [DOI] [PubMed] [Google Scholar]
- 14. Ten Eikelder H. M. M., Adelizzi B., Palmans A. R. A., Markvoort A. J., J. Phys. Chem. B 2019, 123, 6627–6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wijnands S. P. W., Meijer E. W., Merkx M., Bioconjugate Chem. 2019, 30, 1905–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McLaughlin C. K., Hamblin G. D., Sleiman H. F., Chem. Soc. Rev. 2011, 40, 5647–5656. [DOI] [PubMed] [Google Scholar]
- 17. Vybornyi M., Vyborna Y., Häner R., Chem. Soc. Rev. 2019, 48, 4347–4360. [DOI] [PubMed] [Google Scholar]
- 18. Alemán García M. Á., Magdalena Estirado E., Milroy L. G., Brunsveld L., Angew. Chem. Int. Ed. 2018, 57, 4976–4980; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5070–5074. [Google Scholar]
- 19. Wijnands S. P. W., Engelen W., Lafleur R. P. M., Meijer E. W., Merkx M., Nat. Commun. 2018, 9, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seeman N. C., Sleiman H. F., Nat. Rev. Mater. 2018, 3, 17068. [Google Scholar]
- 21. Wang P., Meyer T. A., Pan V., Dutta P. K., Ke Y., Chem 2017, 2, 359–382. [Google Scholar]
- 22. Kuzyk A., Yang Y., Duan X., Stoll S., Govorov A. O., Sugiyama H., Endo M., Liu N., Nat. Commun. 2016, 7, 10591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Helmi S., Ziegler C., Kauert D. J., Seidel R., Nano Lett. 2014, 14, 6693–6698. [DOI] [PubMed] [Google Scholar]
- 24. Tokura Y., Jiang Y., Welle A., Stenzel M. H., Krzemien K. M., Michaelis J., Berger R., Barner-Kowollik C., Wu Y., Weil T., Angew. Chem. Int. Ed. 2016, 55, 5692–5697; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5786–5791. [Google Scholar]
- 25. Hannewald N., Winterwerber P., Zechel S., Ng D. Y. W., Hager M. D., Weil T., Schubert U. S., Angew. Chem. Int. Ed. 2021, 10.1002/anie.202005907; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 10.1002/ange.202005907. [DOI] [Google Scholar]
- 26. Knudsen J. B., Liu L., Kodal A. L. B., Madsen M., Li Q., Song J., Woehrstein J. B., Wickham S. F. J., Strauss M. T., Schueder F., et al., Nat. Nanotechnol. 2015, 10, 892–898. [DOI] [PubMed] [Google Scholar]
- 27. Krissanaprasit A., Madsen M., Knudsen J. B., Gudnason D., Surareungchai W., Birkedal V., Gothelf K. V., ACS Nano 2016, 10, 2243–2250. [DOI] [PubMed] [Google Scholar]
- 28. Udomprasert A., Bongiovanni M. N., Sha R., Sherman W. B., Wang T., Arora P. S., Canary J. W., Gras S. L., Seeman N. C., Nat. Nanotechnol. 2014, 9, 537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rothemund P. W. K., Nature 2006, 440, 297–302. [DOI] [PubMed] [Google Scholar]
- 30. Wagner W., Wehner M., Stepanenko V., Würthner F., J. Am. Chem. Soc. 2019, 141, 12044–12054. [DOI] [PubMed] [Google Scholar]
- 31. van der Zwaag D., de Greef T. F. A., Meijer E. W., Angew. Chem. Int. Ed. 2015, 54, 8334–8336; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8452–8454. [Google Scholar]
- 32. Bieling P., Laan L., Schek H., Munteanu E. L., Sandblad L., Dogterom M., Brunner D., Surrey T., Nature 2007, 450, 1100–1105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary