

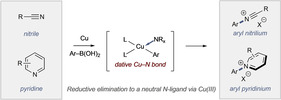
Abstract
Metal‐catalyzed C–N cross‐coupling generally forms C−N bonds by reductive elimination from metal complexes bearing covalent C‐ and N‐ligands. We have identified a Cu‐mediated C–N cross‐coupling that uses a dative N‐ligand in the bond‐forming event, which, in contrast to conventional methods, generates reactive cationic products. Mechanistic studies suggest the process operates via transmetalation of an aryl organoboron to a CuII complex bearing neutral N‐ligands, such as nitriles or N‐heterocycles. Subsequent generation of a putative CuIII complex enables the oxidative C–N coupling to take place, delivering nitrilium intermediates and pyridinium products. The reaction is general for a range of N(sp) and N(sp2) precursors and can be applied to drug synthesis and late‐stage N‐arylation, and the limitations in the methodology are mechanistically evidenced.
Keywords: arylation, boron, copper, cross-coupling, reaction mechanisms
Cu(OTf)2 promotes the oxidative arylation of nitriles and N‐heterocycles to generate ionic products. Mechanistic studies suggest the process operates via reductive elimination from a CuIII complex bearing neutral N‐ligands to generate nitrilium and pyridinium products. The reaction is general for a range of N(sp) and N(sp2) precursors and can be applied to drug synthesis and late‐stage N‐arylation.
Introduction
Transition metal‐mediated C‐N cross‐coupling is an essential synthetic method, used extensively throughout the chemical industry for the synthesis of pharmaceuticals, agrochemicals, natural products, and materials.[ 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 ] The development of new or improved processes for C−N bond construction remains a continual inspiration for metal‐based reaction development. Despite a broad diversity and subtlety in the mechanism of these methods, the basic premise of the reaction involves a series of individual mechanistic steps, e.g., oxidative addition, transmetalation, and/or deprotonation, to allow access to a key metal complex bearing formally anionic, covalently bound C‐ and N‐ligands (Scheme 1 a). This complex undergoes reductive elimination to deliver a neutral product, which is produced regardless of whether the catalysis itself is electroneutral (e.g., the Buchwald‐Hartwig or Ullmann‐Goldberg reactions) or oxidative (e.g., the Chan‐Lam reaction).[ 6 , 7 , 8 , 9 ]
Scheme 1.
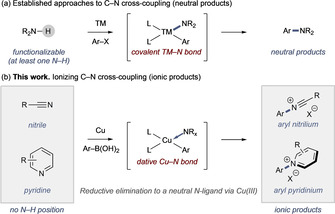
a) General approach to cross‐coupling. b) This work: cross‐coupling to unconventional substrates. Ar=aryl, TM=transition metal.
In these processes, the N‐ligand originates from a precursor amine or amine‐derived substrate bearing at least one functionalizable N−H, which undergoes deprotonation at some stage in the reaction mechanism to deliver the required anionic N‐ligand. This limits the scope of established processes to substrates with at least one N−H.
However, it should be noted that the direct N‐arylation of substrates without a functionalizable site is known. N‐arylation of nitriles and N‐heterocycles has been achieved with or without transition metal activators, for example, using diaryliodonium salts.[ 10 , 11 ] With Cu‐promoted processes,[ 10 , 11 ] these are mechanistically ambiguous, with no evidence for a metal‐centered reductive elimination. These processes have also been rationalized as direct arylation using the increased electrophilicity of the aryl transfer reagent via Lewis acid activation. [12] More specifically, while CuI has been shown to slightly accelerate aryl transfer with diaryliodoniums, these processes also proceed effectively without CuI,[ 13 , 14 ] consistent with observed general reactivity of this class of reagents [15] and related reactive aryl transfer reagents, such as aryldiazonium salts. [16]
Here, we report the discovery, mechanistic rationale, example scope, and limitations of a Cu‐mediated C‐N cross‐coupling method that promotes reductive elimination to neutral N‐ligands, such as nitriles and N‐heterocycles generating reactive cationic products (Scheme 1 b).
Results and Discussion
During investigations to rationalize the reactivity of CuII sources in standard Chan‐Lam reactions, the amide product 3 a was identified in good yield when the reaction of arylboronic acid 1 with aniline was attempted with Cu(OTf)2 in MeCN as solvent (Scheme 2 a). A similar observation was made by Sanford during studies of fluorodeboronation, which also used stoichiometric Cu(OTf)2 (4 equiv) in MeCN (Scheme 2 b). [17] We were intrigued by this observation since hydrolysis of MeCN to acetamide followed by Chan‐Lam‐type N‐arylation seemed unlikely—we have previously attempted Chan‐Lam arylations of amides using Cu(OTf)2 and found this to be problematic (vide infra). Consequently, we sought to understand the origin of this coupling process.
Scheme 2.
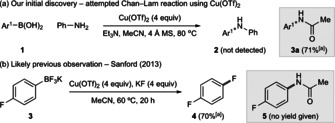
Observations of Cu‐mediated nitrile arylation. [a] Determined by HPLC. Ar1=p‐PhC6H4. Tf=trifluoromethylsulfonyl.
Control experiments indicated the possibility of an alternative pathway. The Chan‐Lam arylation of acetamide 6 using Cu(OTf)2 in PhMe does not provide the N‐aryl product. Instead, the products of aryl‐boronic acid oxidation and protodeboronation were observed and represented the almost complete mass balance (Scheme 3 a)—protodeboronation was also noted as an issue in Sanford's study [17] and is a common problem for Cu‐mediated reactions of organoborons.[ 18 , 19 ] Separate experiments (Table S1) indicated that the same conditions did not lead to nitrile hydrolysis. [20] The competition reaction of 1 with acetamide and D3CCN in the presence of Cu(OTf)2 led to the deuterated acetamide product 3 b exclusively, further supporting the absence of a Chan‐Lam pathway and indicating selectivity for nitrile (Scheme 3 b).
Scheme 3.
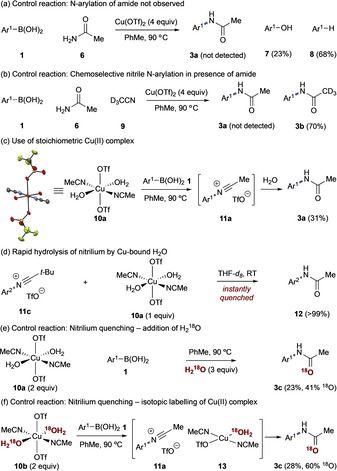
Control reactions. Ar1=p‐PhC6H4; Ar2=p‐(F3CO)C6H4.
To rationalize these initial observations, we considered a reaction pathway that proceeded via formation of a nitrilium intermediate formed by Cu‐mediated N‐arylation of the nitrile. N‐arylation of nitriles is known using highly reactive aryl transfer agents, such as iodonium and diazonium salts;[ 10 , 16 ] however, oxidative coupling of nitriles with arylboronic acids is unknown. Accordingly, we sought to establish if an oxidative coupling pathway was operational.
Treatment of Cu(OTf)2 with H2O in MeCN leads to a stable and isolable complex CuII(OTf)2(H2O)2(MeCN)2 (10 a, Scheme 3 c). [21] Heating this complex with 1 lead to the observed acetamide 3 a, which we propose proceeds through nitrilium 11 a, suggesting possible formation and involvement of 10 a in the reaction. [22]
Nitrilium ions are highly reactive electrophiles capable of a variety of bond forming processes with nucleophiles; [23] however, extensive experimentation to intercept the proposed nitrilium 11 a were unsuccessful and afforded a mixture of amide and returned starting material (Tables S2 and S3). We therefore attributed amide formation to hydrolysis of the nitrilium with H2O present in the reaction mixture, arising either from boroxine formation from 1 or Cu‐bound H2O in 10 a—H2O could not be excluded in preparation of stoichiometric Cu(OTf)2 nitrile complexes as noted above.
Independent preparation of stable nitrilium 11 c [24] and treatment with 10 a led to instantaneous hydrolysis, highlighting the lability of Cu‐bound H2O (Scheme 3 d). To probe the origin of H2O in the acetamide product, we undertook labelling experiments. Addition of H2 18O to the reaction of 10 a with 1 led to 41 % 18O incorporation in the product 3 c, consistent with the 16O:18O stoichiometry (Scheme 3 e). Preparation of 18O‐labelled complex 10 b was successful; however, the 18O incorporation could not be quantified due to lability of the dative ligands. Indeed, despite obtaining crystal structure data of 10 a and 10 b (identical), HRMS analyses were uniformly unsuccessful. Use of 10 b in the absence of additional H2O gave 3 c in comparable yield to the reaction of 10 a and with 60 % 18O incorporation (Scheme 3 f).
The inability to trap the nitrilium by any nucleophile other than H2O suggests that nitrilium quenching may be occurring from H2O in solution, a Cu aquo species (e.g., 10 a/10 b), or from a CuI complex liberated after reductive elimination (e.g., 13, Scheme 3 f).
To further substantiate this nitrilium proposal, HRMS analysis of reaction mixtures identified a series of mass ions that allowed the following mechanism to be proposed (Scheme 4). [25]
Scheme 4.
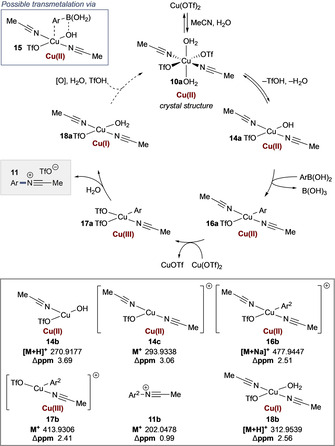
Proposed mechanism with observed HRMS mass ions.
We propose that Cu(OTf)2 forms 10 a (crystal structure obtained from reaction mixtures). Loss of TfOH and H2O gives 14 a–mass ions consistent with [14 a–MeCN] (14 b) and [14 a–H2O] (14 c) were found. Transmetalation then gives 16 a (16 b found), possibly via a pathway consistent to the Chan‐Lam amination (15). [26] Disproportionation of 16 a gives the key CuIII intermediate 17 a with [17 a–TfO−] (17 b) found,[ 26 , 27 , 28 ] allowing formation of the nitrilium product 11 (11 b found). Mass ions consistent with the proposed CuI aquo complex 18 a were detected (18 b), consistent with the quenching proposal outlined in Scheme 3 f. Stoichiometric Cu(OTf)2 was exclusively effective—other Cu sources failed to promote the reaction (Table S7). Extensive investigation failed to allow this process to operate with catalytic Cu(OTf)2—the addition of terminal oxidants led to issues of organoboron oxidation and rendering Cu turnover (Scheme 4, dotted line) irrelevant (Table S9). The same turnover issues in systems using Cu(OTf)2 and CuOTf have been observed in C‐F bond formation by Sanford [17] and Hartwig, [29] respectively, where 3–4 equivalents of Cu were necessary for reaction efficiency. This problem remains unresolved for many Cu(OTf)2‐based processes. [30]
The proposed nitrilium ions were observable by HRMS; however, the inability to intercept the proposed nitrilium with other nucleophiles was unsatisfactory. Specifically, this invites further scrutiny of the proposed key C−N bond forming event in Scheme 4—the potential for an on‐metal hydrolysis cannot be excluded. We therefore sought to demonstrate the C−N bond forming process using a system that would allow unambiguous identification C−N bond formation produced from reductive elimination to a neutral N‐ligand on CuIII.
Treatment of Cu(OTf)2 with DMAP allowed formation of CuII(OTf)2(DMAP)4 19 and its structure unambiguously confirmed by X‐ray (Scheme 5).
Scheme 5.
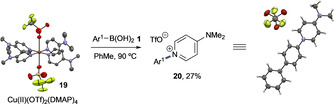
Cross‐coupling to N(sp2) via DMAP complex 19. Ar1=p‐PhC6H4, DMAP=4‐dimethylaminopyridine.
Complex 19 is similar to the nitrile complex 10 a; however, this can be prepared without aquo ligands. Under the same reaction conditions used in Scheme 3 c, 19 leads to a similar C−N bond formation giving N‐aryl pyridinium 20 and in similar yield to the nitrile process. 20 was characterized unambiguously by spectroscopy and X‐ray, providing strong support for C‐N cross‐coupling via CuIII. We propose this reaction to follow a similar course to that proposed in Scheme 4. Single electron pathways via oxidation of DMAP by CuII were proposed to be unlikely based on oxidation potentials and EPR analysis (vide infra).[ 33 , 34 , 35 ]
Despite evidence for the feasibility of reductive elimination from (aryl)CuIII complexes yielding N‐aryl ammonium products,[ 36 , 37 ] the equivalent N(sp3) cross‐coupling under the conditions reported here did not afford the desired C‐N(sp3) bond. We attribute this to competing amine oxidation by CuII; [38] this was substantiated by EPR studies, which showed quenching of CuII and, in the case of N‐methylpyrrolidine, a radical species could be observed (Scheme 6 a). Addition of tertiary amines to the optimized DMAP N‐arylation process had variable effects on the observed yield (Scheme 6 b). For example, PhNMe2 almost completely reduced CuII and lowered yield of 21 by approximately half; however, n‐butylaziridine reduced approx. 25 % of CuII yet had no impact on the yield of 21. Little reduction of CuII by TMEDA was observed by EPR and the arylation reaction was instead impaired by formation of a series of novel but unreactive bidentate complexes (Scheme S12). As expected, DMAP did not significantly reduce CuII.
Scheme 6.
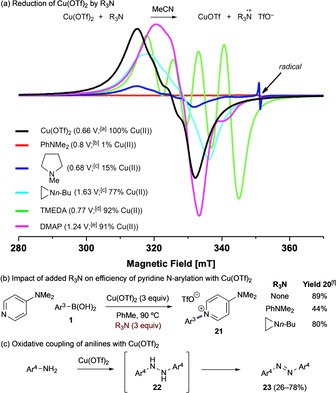
Limitations of the Cu‐mediated arylation with R3N. Ar3=4‐MeC6H4. Ar4=4‐FC6H4. [a] vs. Fc+/0. [39] [b] vs. SCE. [40] [c] vs. SCE. [41] [d] vs. SCE. [42] [e] vs. SCE. [43] [f] Determined by 1H NMR analysis. Fc=ferrocene, SCE=saturated calomel electrode.
Moreover, in the presence of unsubstituted anilines, an alternative oxidative coupling pathway becomes evident via formation of 1,2‐diarylhydrazines (22) and azobenzenes (23) (Scheme 6 c). This is clearly mechanistically related to previously reported Cu‐mediated N‐N coupling reactions.[ 44 , 45 ] Consistent with these previous reports, our EPR data suggests that these processes proceed via single electron oxidation of the aniline by CuII; however, importantly, the resulting aminium radical does not appear to be free in solution and attempts to intercept these species were universally unsuccessful (Table S6). In contrast to a previously proposed mechanism, [44] our data suggests formation of the N−N bond at the metal or within the solvent cage. This would deliver the symmetrical hydrazine product, consistent with previous observations.[ 44 , 45 ] As an adjunct to the main work described here, additional control experiments have shown facile oxidation of the hydrazine to the azobenzene by Cu(OTf)2 aligning with the experimental data observed across these separate studies (Scheme S14).[ 44 , 45 ]
Following optimization (Tables S7–S11), a general process was developed for the coupling of arylboronic acids with nitriles and N‐heterocycles—a selection of products is provided in Scheme 7 (for additional substrates see Scheme S15). [46] The process tolerates a variety of functional groups on both the nitrile and arylboronic acid, with standard structural and electronic variations examined in this example scope. The nitrilium process is an unusual amidation protocol (essentially an aryl Ritter reaction) providing a new approach to this ubiquitous motif; however, the heterocycle N‐arylation process allows access to products that cannot be made easily using any established method, providing novel opportunities for synthetic design. In general, the scope of the boronic acid was very good for arylboronic acids, with some lower yields observed using heteroaromatic species consistent with established limitations with these substrates. [47] Alkylboronic acids were tolerated only in the N‐heterocycle process (e.g., product 45); no desired products were observed in the equivalent nitrilium reactions. For the nitrilium process, the C‐N cross‐coupling could be achieved using the nitrile as solvent where practical (e.g., for MeCN, EtCN), otherwise PhMe was the preferred medium for both the nitrilium and N‐heterocycle processes. While generally effective, solubility issues can present with certain arylboronic acids in PhMe resulting in lower yields (e.g., 29–31). With regards the N‐heterocycle process, the reaction was broadly tolerant to the nature of the heterocycle, although higher yields were obtained with more electron‐rich compounds, which may be expected based on the oxidative coupling process. The issue of lower yields with substrates bearing ortho‐substitution was replicated (e.g., 27 and 40) and is again consistent with observations in Cu‐mediated oxidative coupling processes. [9] As discussed above for the nitrile process, stoichiometric Cu(OTf)2 was also needed for the heterocycle process, which perhaps offers some explanation for the lack of observable reinsertion into the N‐aryl pyridinium products. Additional demonstrations of utility are provided in Scheme 7 c‐g. The C‐N coupling process can be applied to the N‐arylation of non‐aryl N(sp2) including the common organic base DBU as well as the Lewis base organocatalyst (−)‐tetramisole to afford compounds 52 and 53, respectively (Schemes 7 c and d).
Scheme 7.
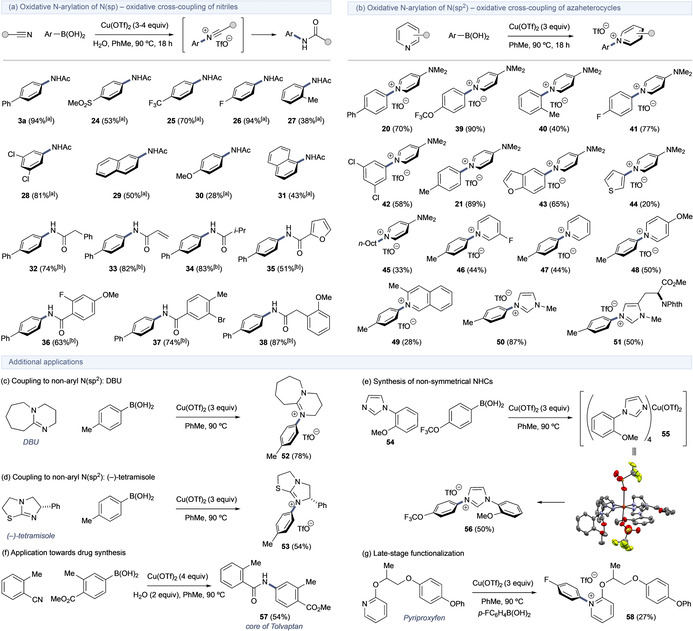
a) Cu‐mediated N‐arylation of N(sp) and N(sp2): representative examples. [a] Cu(OTf)2 (3 equiv), H2O (3 equiv), MeCN as solvent. [b] Cu(OTf)2 (4 equiv), H2O (2 equiv), PhMe as solvent. Ac=acetyl, Phth=phthalimide.
The ability to induce direct N‐arylation of N‐heterocycles allows a significantly shorter route to non‐symmetrical NHCs by N‐arylation of N‐aryl imidazoles such as 54, which proceeds via the expected complex 55 to deliver imidazolium salt 56 (Scheme 7 e; see also 50 and 51 in Scheme 7 b for alkyl/aryl imidazolium). [11] Lastly, the process can be used in synthesis, for example using the nitrilium process to access pharmaceutically relevant amides, such as the Tolvaptan intermediate 57 (Scheme 7 f) and for late‐stage functionalization, for example N‐arylation of the agrochemical Pyriproxyfen, giving product 58 (Scheme 7 g).
Conclusion
In summary, the data provided establishes a framework for oxidative C‐N cross‐coupling of arylboronic acids with neutral N‐ligands. Importantly, mechanistic data supports a CuIII‐based process and is distinct from Lewis acid‐assisted N‐arylations using reactive aryl transfer electrophiles (e.g., iodoniums). This expands the scope of oxidative coupling, allowing access to new products. The broader implications are that, assuming specific metal‐centered mechanistic events can be appropriately controlled, neutral N‐ligands may be effective partners for cross‐coupling more generally within transition metal catalysis, providing new opportunities for reaction design. [48]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Acknowledgements
N.L.B. thanks the EPSRC for postdoctoral funding (EP/R025754/1). C.X. and J.W.B.F. thank the Leverhulme Trust for postdoctoral funding (RPG‐2015‐308; RPG‐2018‐362). J.C.V. thanks GlaxoSmithKline for a PhD studentship. J.B. thanks AstraZeneca for a PhD studentship. S.B. thanks the University of St Andrews for a PhD studentship. We thank Tony Cook (GlaxoSmithKline) and Jim Tweedie (University of Glasgow) for assistance with HRMS analysis, Cameron L. Carpenter‐Warren for assistance with crystallography, and Malcolm J. Stirling for assistance with preliminary experiments.
N. L. Bell, C. Xu, J. W. B. Fyfe, J. C. Vantourout, J. Brals, S. Chabbra, B. E. Bode, D. B. Cordes, A. M. Z. Slawin, T. M. McGuire, A. J. B. Watson, Angew. Chem. Int. Ed. 2021, 60, 7935.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv.13399307.v1).
References
- 1. Catalyzed Carbon-Heteroatom Bond Formation (Ed.: Yudin A. K.), Wiley-VCH, Weinheim, 2011. [Google Scholar]
- 2. Bariwal J., der Eycken E., Chem. Soc. Rev. 2013, 42, 9283–9303. [DOI] [PubMed] [Google Scholar]
- 3.“Amination and Formation of sp2 C-N Bonds”: Topics in Organometallic Chemistry (Eds.: Taillefer M., Ma D.), Springer, Heidelberg, 2013. [Google Scholar]
- 4. Tasler S., Mies J., Lang M., Adv. Synth. Catal. 2007, 349, 2286–2300. [Google Scholar]
- 5. Magano J., Dunetz J. R., Chem. Rev. 2011, 111, 2177–2250. [DOI] [PubMed] [Google Scholar]
- 6. Ruiz-Castillo P., Buchwald S. L., Chem. Rev. 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sambiagio C., Marsden S. P., Blacker A. J., McGowan P. C., Chem. Soc. Rev. 2014, 43, 3525–3550. [DOI] [PubMed] [Google Scholar]
- 8.“Copper-Catalyzed Amination of Aryl and Alkenyl Electrophiles”: Shaughnessy K. H., Ciganek E., DeVasher R. B. in Organic Reactions (Eds.: Denmark S. E., Overman L. E.), Wiley, Hoboken, 2014. [Google Scholar]
- 9. West M. J., Fyfe J. W. B., Vantourout J. C., Watson A. J. B., Chem. Rev. 2019, 119, 12491–12523. [DOI] [PubMed] [Google Scholar]
- 10. Cao C. K., Sheng J., Chen C., Synthesis 2017, 49, 5081–5092. [Google Scholar]
- 11.For a related Cu/Fe-promoted process, see: Li S., Yang F., Lv T., Lan J., Gao G., You J., Chem. Commun. 2014, 50, 3941–3943. [DOI] [PubMed] [Google Scholar]
- 12. Hickman A. J., Sanford M. S., Nature 2012, 484, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Teskey C. J., Sohel S. M. A., Bunting D. L., Modha S. G., Greaney M. F., Angew. Chem. Int. Ed. 2017, 56, 5263–5266; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5347–5350. [Google Scholar]
- 14. Modha S. G., Popescu M. V., Greaney M. F., J. Org. Chem. 2017, 82, 11933–11938. [DOI] [PubMed] [Google Scholar]
- 15. Aradi K., Tóth B. L., Tolnai G. L., Novák Z., Synlett 2016, 27, 1456–1485. [Google Scholar]
- 16. Youn S. W., Yoo H. J., Lee E. M., Lee S. Y., Adv. Synth. Catal. 2018, 360, 278–283. [Google Scholar]
- 17. Ye Y., Schimler S. D., Hanley P. S., Sanford M. S., J. Am. Chem. Soc. 2013, 135, 16292–16295. [DOI] [PubMed] [Google Scholar]
- 18. Kuivila H. G., Reuwer J. F., Mangravite J. A., J. Am. Chem. Soc. 1964, 86, 2666–2670. [Google Scholar]
- 19. Cundy D. J., Forsyth S. A., Tetrahedron Lett. 1998, 39, 7979–7982. [Google Scholar]
- 20. Marcé P., Lynch J., Blacker A. J., Williams J. M. J., Chem. Commun. 2016, 52, 1436–1438. [DOI] [PubMed] [Google Scholar]
- 21. Irangu J., Ferguson M. J., Jordan R. B., Inorg. Chem. 2005, 44, 1619–1625. [DOI] [PubMed] [Google Scholar]
- 22. Rach S. F., Kühn F. E., Chem. Rev. 2009, 109, 2061–2080. [DOI] [PubMed] [Google Scholar]
- 23. van Dijk T., Slootweg J. C., Lammertsma K., Org. Biomol. Chem. 2017, 15, 10134–10144. [DOI] [PubMed] [Google Scholar]
- 24. van Dijk T., Burck S., Rong M. K., Rosenthal A. J., Nieger M., Slootweg J. C., Lammertsma K., Angew. Chem. Int. Ed. 2014, 53, 9068–9071; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9214–9217. [Google Scholar]
- 25.For a useful related mechanistic discussion, see: Tromp M., van Strijdonck G. P. F., van Berkel S. S., van den Hoogenband A., Feiters M. C., de Bruin B., Fiddy S. G., van der Eerden A. M. J., van Bokhoven J. A., van Leeuwen P. W. N. M., Koningsberger D. C., Organometallics 2010, 29, 3085–3097. [Google Scholar]
- 26. Vantourout J. C., Miras H. N., Isidro-Llobet A., Sproules S., Watson A. J. B., J. Am. Chem. Soc. 2017, 139, 4769–4779. [DOI] [PubMed] [Google Scholar]
- 27. King A. E., Brunold T. C., Stahl S. S., J. Am. Chem. Soc. 2009, 131, 5044–5045. [DOI] [PubMed] [Google Scholar]
- 28. King A. E., Ryland B. L., Brunold T. C., Stahl S. S., Organometallics 2012, 31, 7948–7957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fier P. S., Hartwig J. F., J. Am. Chem. Soc. 2012, 134, 10795–10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turnover is possible in Cu(OTf)2 systems using electron-rich ligands that are incompatible with the present system due to competing ligand arylation. For examples of systems where turnover has been achieved see refs [31] and [32].
- 31. Taylor N. J., Emer E., Preshlock S., Schedler M., Tredwell M., Verhoog S., Mercier J., Genicot C., Gouverneur V., J. Am. Chem. Soc. 2017, 139, 8267–8276. [DOI] [PubMed] [Google Scholar]
- 32. Preshlock S., Tredwell M., Gouverneur V., Chem. Rev. 2016, 116, 719–766. [DOI] [PubMed] [Google Scholar]
- 33. Rössler S. L., Jelier B. J., Tripet P. F., Shemet A., Jeschke G., Togni A., Carreira E. M., Angew. Chem. Int. Ed. 2019, 58, 526–531; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 536–541. [Google Scholar]
- 34. Casitas A., Ribas X., Chem. Sci. 2013, 4, 2301–2318. [Google Scholar]
- 35. DiMucci I. M., Lukens J. T., Chatterjee S., Carsch K. M., Titus C. J., Lee S. J., Nordlund D., Betley T. A., MacMillan S. N., Lancaster K. M., J. Am. Chem. Soc. 2019, 141, 18508–18520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ribas X., Jackson D. A., Donnadieu B., Mahía J., Parella T., Xifra R., Hedman B., Hodgson K. O., Llobet A., Stack T. D. P., Angew. Chem. Int. Ed. 2002, 41, 2991–2994; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 3117–3120. [Google Scholar]
- 37. Huffman L. M., Stahl S. S., J. Am. Chem. Soc. 2008, 130, 9196–9197. [DOI] [PubMed] [Google Scholar]
- 38. Weiss J. F., Tollin G., Yoke J. T., Inorg. Chem. 1964, 3, 1344–1348. [Google Scholar]
- 39. Inamo M., Kumagai H., Harada U., Itoh S., Iwatsuki S., Ishihara K., Takagi H. D., Dalton Trans. 2004, 1703–1707. [DOI] [PubMed] [Google Scholar]
- 40. Smith J. R. L., Masheder D., J. Chem. Soc. Perkin Trans. 2 1976, 47–51. [Google Scholar]
- 41. Macdonald T. L., Gutheim W. G., Martin R. B., Guengerich F. P., Biochemistry 1989, 28, 2071–2077. [DOI] [PubMed] [Google Scholar]
- 42. Masui M., Sayo H., Tsuda Y., J. Chem. Soc. B 1968, 973–976. [Google Scholar]
- 43. Lima F., Sharma U. K., Grunenberg L., Saha D., Johannsen S., Sedelmeier J., Van der Eycken E. V., Ley S. V., Angew. Chem. Int. Ed. 2017, 56, 15136–15140; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15332–15336. [Google Scholar]
- 44. Zhang C., Jiao N., Angew. Chem. Int. Ed. 2010, 49, 6174–6177; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 6310–6313. [Google Scholar]
- 45. Ryan M. C., Martinelli J. R., Stahl S. S., J. Am. Chem. Soc. 2018, 140, 9074–9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.For examples of the utility of pyridinium salts, see:
- 46a. Basch C. H., Liao J., Xu J., Piane J. J., Watson M. P., J. Am. Chem. Soc. 2017, 139, 5313–5316; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46b. Plunkett S., Basch C. H., Santana S. O., Watson M. P., J. Am. Chem. Soc. 2019, 141, 2257–2262; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46c. Moser D., Duan Y., Wang F., Ma Y., O'Neill M. J., Cornella J., Angew. Chem. Int. Ed. 2018, 57, 11035–11039; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11201–11205. [Google Scholar]
- 47. Cox P. A., Leach A. G., Campbell A. D., Lloyd-Jones G. C., J. Am. Chem. Soc. 2016, 138, 9145–9157. [DOI] [PubMed] [Google Scholar]
- 48.The research data underpinning this publication can be accessed at 10.17630/eee822ca-b387-4736-b499-b1bb9dc62bbf. Deposition Numbers 1971679 (for 10a/1 b), 1971680 (for 19), and 1971681 (for 55), 2042594 (for S1), and 2042593 (for S2) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary