

Abstract
We report the population pharmacokinetic (PK) and exposure‐response analyses of a novel subcutaneous formulation of daratumumab (DARA) using data from 3 DARA subcutaneous monotherapy studies (PAVO Part 2, MMY1008, COLUMBA) and 1 combination therapy study (PLEIADES). Results were based on 5159 PK samples from 742 patients (DARA 1800 mg subcutaneously, n = 487 [monotherapy, n = 288; combination therapy, n = 199]; DARA 16 mg/kg intravenously, n = 255 [all monotherapy, in COLUMBA]; age, 33‐92 years; weight, 28.6‐147.6 kg). Subcutaneous and intravenous DARA monotherapies were administered once every week for cycles 1‐2, once every 2 weeks for cycles 3‐6, and once every 4 weeks thereafter (1 cycle is 28 days). The subcutaneous DARA combination therapy was administered with the adaptation of corresponding standard‐of‐care regimens. PK samples were collected between cycle 1 and cycle 12. Among monotherapy studies, throughout the treatment period, subcutaneous DARA provided similar/slightly higher trough concentrations (Ctrough) versus intravenous DARA, with lower maximum concentrations and smaller peak‐to‐trough fluctuations. The PK profile was consistent between subcutaneous DARA monotherapy and combination therapies. The exposure‐response relationship between daratumumab PK and efficacy or safety end points was similar for subcutaneous and intravenous DARA. Although the ≤65‐kg subgroup reported a higher incidence of neutropenia, no relationship was found between the incidence of neutropenia and exposure, which was attributed, in part, to the preexisting imbalance in neutropenia between subcutaneous DARA (45.5%) and intravenous DARA (19%) in patients ≤50 kg. A flat relationship was observed between body weight and any grade and at least grade 3 infections. The results support the DARA 1800‐mg subcutaneous flat dose as an alternative to the approved intravenous DARA 16 mg/kg.
Keywords: daratumumab, subcutaneous, pharmacokinetics, multiple myeloma, biologics
The incidence of multiple myeloma in the United States and United Kingdom is 7.0 and 9.0 per 100 000 men and women per year, respectively, and the 5‐year survival rate of multiple myeloma is 54% and 52%, respectively. 1 , 2 Daratumumab (DARA) is a human IgGκ monoclonal antibody targeting CD38 with a direct on‐tumor 3 , 4 , 5 , 6 and immunomodulatory 7 , 8 , 9 mechanism of action. The results from pivotal clinical trials 10 , 11 , 12 , 13 , 14 , 15 , 16 in multiple myeloma led to the approval of intravenous DARA 16 mg/kg as monotherapy and in combination with standard‐of‐care therapies for newly diagnosed multiple myeloma and relapsed or refractory multiple myeloma. 17 , 18
Intravenous DARA has a median infusion duration of 7 hours for the first infusion and of 3 to 4 hours for the second and subsequent infusions. 17 To reduce patient and health care provider burden and improve safety for DARA, a subcutaneous formulation of DARA coformulated with recombinant human hyaluronidase PH20 (rHuPH20; ENHANZE drug delivery technology; Halozyme, Inc., San Diego, California) given as an 1800‐mg flat dose was developed. 19 , 20 Dose range for the subcutaneous formulation was evaluated in patients with relapsed or refractory multiple myeloma in the phase 1b PAVO study, which demonstrated that the safety, efficacy, and pharmacokinetics (PK) of subcutaneous DARA monotherapy administered over 3 to 5 minutes was noninferior to the corresponding historical data on intravenous DARA. 19 , 20 The formulation was also evaluated in Japanese patients with relapsed or refractory multiple myeloma in the phase 1 MMY1008 study.
Two recent studies evaluated subcutaneous DARA monotherapy (COLUMBA) and subcutaneous DARA in combination with standard‐of‐care therapies (PLEIADES) in patients with multiple myeloma. 21 , 22 , 23 In the phase 3 COLUMBA study, subcutaneous DARA was noninferior to intravenous DARA based on the predefined noninferiority criteria of the coprimary end points, overall response rate (ORR) and DARA maximum trough concentration (Ctrough; day 1 of cycle 3, which strongly correlates with ORR), 24 with a comparable safety profile, lower rates of infusion‐related reactions (IRRs), and a shorter duration of administration in patients with heavily pretreated relapsed or refractory multiple myeloma. 21 , 22 One patient (<1%) was positive for anti‐DARA antibodies with intravenous DARA, and no patients were positive with subcutaneous DARA; other clinical trials of intravenous DARA also reported low frequency of anti‐DARA antibodies (monotherapy, 0%; combination therapy, <1%). 17 In the phase 2 PLEIADES study, subcutaneous DARA in combination with bortezomib, lenalidomide, and dexamethasone (D‐VRd) and bortezomib, melphalan, and prednisone (D‐VMP) in patients with newly diagnosed multiple myeloma and subcutaneous DARA in combination with lenalidomide and dexamethasone (D‐Rd) in patients with relapsed or refractory multiple myeloma demonstrated similar activity and safety to the corresponding historical data for intravenous DARA combination therapies, with lower rates of IRRs and a shorter duration of administration. 23 Based on the results from the COLUMBA and PLEIADES studies, a Biologics License Application was submitted for subcutaneous DARA in the United States; subcutaneous DARA was approved by the United States Food and Drug Administration in May 2020 followed by approvals in the European Union, Canada, South Korea, and Brazil. 25
A previous population PK (popPK) model showed that the PK of DARA after intravenous administration could be described by a 2‐compartment popPK model with parallel linear and nonlinear Michaelis‐Menten elimination pathways. 24 The maximum peak concentration (Cmax) after the first dose increased dose‐proportionally, and the volume of distribution was consistent with initial distribution into the plasma compartment. After multiple doses, Cmax increased in a greater than dose‐proportional manner, consistent with target‐mediated drug disposition. 26 The covariates found to have a statistically significant but clinically not relevant effect on daratumumab PK include body weight, albumin, type of myeloma, and sex. 27 Findings from a popPK analysis of intravenous DARA combination therapies exhibited a similar PK profile. The exposure‐response analysis suggested that daratumumab efficacy was significantly correlated with daratumumab exposure, and maximal preinfusion trough concentration had the strongest correlation with efficacy end points among the investigated exposure metrics. 24
We present results from the popPK analysis for subcutaneous DARA results based on data from 3 monotherapy studies (PAVO Part 2, MMY1008, and COLUMBA) and 1 combination therapy study (PLEIADES) in patients with multiple myeloma. The key objectives of this analysis were to characterize the popPK of subcutaneous DARA and to assess the relationship of DARA exposure with efficacy and safety.
Methods
Patients and Study Designs
As part of the original studies, all patients provided written informed consent, and the trials were approved by all relevant review bodies. All trials were conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice guidelines.
The popPK and exposure‐response analyses included data (clinical cutoff date: January 8, 2019) from 4 clinical studies: 3 monotherapy studies — PAVO (MMY1004; ClinicalTrials.gov Identifier: NCT02519452) Part 2, MMY1008 (NCT03242889), COLUMBA (MMY3012; NCT03277105) — and 1 combination study, PLEIADES (MMY2040; NCT03412565). In all studies, subcutaneous DARA was administered by injection over 3 to 5 minutes at alternating left/right abdominal sites. Additional details on the study designs of each of these studies are presented in Table 1 and Supplementary Methods.
Table 1.
Summary of Study Designs
| Clinical Trial | Phase | Patient Population | Treatment Arm(s) | Serum Sample Collection Schedule | Study Sites |
|---|---|---|---|---|---|
| PAVO Part 2 | 1b | Relapsed or refractory multiple myeloma | DARA SC 1800 mg: once weekly during cycles 1 and 2, every 2 weeks for cycles 3 through 6, and every 4 weeks thereafter until disease progression or unacceptable toxicity; each cycle was 28 days. | Serum samples were collected postinfusion during the first and last weekly dose on day 1 of cycle 1 and day 22 of cycle 2 and on days without a dose on days 2, 3, and 4 of cycle 1 and days 23 and 25 of cycle 2. | Denmark (1 site), France (2 sites), Spain (3 sites), Sweden (1 site), the Netherlands (1 site), and the United States (3 sites). |
| MMY1008 | 1 | Japanese patients with relapsed or refractory multiple myeloma | DARA SC 1800 mg: once weekly during cycles 1 and 2, every 2 weeks for cycles 3 through 6, and every 4 weeks thereafter until disease progression or unacceptable toxicity; each cycle was 28 days. | Serum samples were collected postinfusion during the first and last weekly doses on day 1 of cycle 1 and day 22 of cycle 2 and on days without a dose on days 2, 3, and 4 of cycle 1 and days 23 and 25 of cycle 2. | Japan (4 sites). |
| COLUMBA | 3 | Relapsed or refractory multiple myeloma | DARA SC 1800 mg or DARA IV 16 mg/kg: once weekly during cycles 1 and 2, every 2 weeks for cycles 3 through 6, and every 4 weeks thereafter until disease progression or unacceptable toxicity; each cycle was 28 days. |
Serum samples were collected preinfusion on days 1 and 15 of cycle 1; day 1 of cycles 2, 3, 5, 7, and 12; and 4 and 8 weeks after the last DARA dose for both the DARA IV and DARA SC arms. Serum samples were also collected postinfusion on day 1 of cycles 1 and 3 for the DARA IV arm and postinfusion on day 4 of cycles 1 and 4 for the DARA SC arm. |
Australia (8 sites), Brazil (11 sites), Canada (7 sites), the Czech Republic (7 sites), France (7 sites), Greece (1 site), Israel (7 sites), Italy (8 sites), Japan (16 sites), Poland (9 sites), Russia (13 sites), Spain (12 sites), South Korea (8 sites), Sweden (6 sites), Taiwan (6 sites), Ukraine (10 sites), the United States (2 sites), and the United Kingdom (9 sites). |
| PLEIADES | 2 | Newly diagnosed multiple myeloma or relapsed or refractory multiple myeloma |
D‐VRd: DARA SC 1800 mg every week for cycles 1 through 3 and every 3 weeks thereafter; each cycle was 3 weeks. D‐VMP: DARA SC 1800 mg every week for cycle 1, every 3 weeks for cycles 2 through 9, and every 4 weeks thereafter; each cycle was 6 weeks for cycles 1 through 9. D‐Rd: DARA SC 1800 mg every week for cycles 1 and 2, every 2 weeks for cycles 3 through 6, and every 4 weeks thereafter; each cycle was 4 weeks. |
D‐VRd: serum samples were collected preinfusion on day 1 of cycles 1, 3, and 4 and postinfusion on day 4 of cycles 1 and 4. D‐VMP: serum samples were collected preinfusion on day 1 of cycles 1, 2, 3, 6, and 9 and postinfusion on day 4 of cycles 1 and 2. D‐Rd: serum samples were collected preinfusion on day 1 of cycles 1, 3, 6, 9, and 12 and postinfusion on day 4 of cycles 1 and 3. All arms also had serum samples collected 4 and 8 weeks after the last DARA dose. |
Brazil (3 sites), the Czech Republic (4 sites), the United States (8 sites), France (5 sites), Israel (5 sites), Spain (9 sites), the United Kingdom (6 sites), and Japan (3 sites). |
DARA, daratumumab; SC, subcutaneous; IV, intravenous; D‐VRd, DARA SC plus bortezomib/lenalidomide/dexamethasone; D‐VMP, DARA SC plus bortezomib/melphalan/prednisone; D‐Rd, DARA SC plus lenalidomide/dexamethasone.
Bioanalytical Methods
An enzyme‐linked immunosorbent assay (ELISA; lower limit of quantitation, 0.2 μg/mL; BioAnalytical Research Corporation Global Central Laboratory, Ghent, Belgium; Janssen Research & Development, LLC, Spring House, Pennsylvania) was used to determine serum DARA concentrations. The ELISA method was validated according to the European Medicines Agency, the US Food and Drug Administration, bioanalytical method validation guidance, and industry white papers (EMEA/CHMP/EWP 2011, Guidance for Industry 2018). 28 , 29
Antidrug Antibody Assessment
A validated antidrug antibody (ADA) electrochemiluminescence drug tolerant immunoassay (on the Meso Scale Discovery platform, Rockville, Maryland) was used to determine the presence or absence of anti‐DARA antibodies. Serum samples were first analyzed with a screening assay. Samples testing positive for ADAs in the screening assay were further evaluated in a confirmatory assay. For the samples that tested positive for ADA in the confirmatory assay, ADA titer was determined as the reciprocal of the highest dilution of the sample that yielded a positive ADA test result.
PopPK Analyses
Analyses were conducted on the combined PK‐evaluable data set from studies in which subcutaneous DARA was administered to a total of 487 patients (PAVO Part 2, n = 25; MMY1008, n = 6; COLUMBA, n = 257; PLEIADES, n = 199), and intravenous DARA was administered to a total of 255 patients.
The subcutaneous DARA data were described based on the previously developed final structural and covariate model from intravenous DARA, with an additional absorption phase, as no major differences in systemic PK characteristics and covariates were expected between intravenous and subcutaneous administration. 30 The base popPK model for intravenous DARA included a 2‐compartment structure with parallel linear and Michaelis‐Menten nonlinear elimination pathways. The linear clearance represents the nonspecific clearance for immunoglobulin G (IgG), and the Michaelis‐Menten nonlinear elimination represents the saturable target‐mediated clearance. Because of the treatment effect of DARA, the total target (CD38) number may decrease over time, which leads to time‐dependent clearance of DARA. The base popPK model for subcutaneous DARA was similar to intravenous DARA but with an additional depot compartment to account for the subcutaneous absorption process. The absorption of the subcutaneous DARA formulation was modeled with a first‐order absorption process. The subcutaneous DARA model was further evaluated by the goodness‐of‐fit (GOF) plots and visual predictive checks. The final model had no systematic bias in the relevant GOF plots and provided a good description of the observed data. Compared with the base popPK model, the estimated interindividual variability (IIV) for clearance decreased from 85% to 59%, whereas the IIV for volume of distribution in the central compartment decreased from 46% to 37%. The developed subcutaneous DARA monotherapy popPK model was used to estimate individual PK parameters for combination therapies (D‐VMP, D‐Rd, and D‐VRd) in the PLEIADES study through a maximum a posteriori probability Bayesian approach. 31
Serum concentration‐time data were used for nonlinear mixed‐effects modeling (NONMEM; version 7.2; ICON plc, Dublin, Ireland). The first‐order conditional estimation method was used for continuous dependent variables. The software R (version 3.4.1 or higher) was used for any data management, postprocessing of the NONMEM run, simulations, and all other analyses. The package Xpose (version 4.6.1) was used for model evaluation.
The intrinsic factors explored as covariates in the pooled population PK analysis were body weight, age, sex, race, baseline creatinine clearance, baseline albumin, alanine aminotransferase, alkaline phosphatase, and hepatic dysfunction categories using the National Cancer Institute criteria (based on aspartate aminotransferase and total bilirubin). Type of myeloma at baseline (IgG vs non‐IgG) was also investigated, as production of IgG in patients with multiple myeloma may affect the clearance of DARA. Exposure to DARA was compared between subgroups for baseline disease status (ie, number of prior lines of therapy, refractory status, and Eastern Cooperative Oncology Group [ECOG] performance status at baseline). The cutoff to define covariates significant to DARA PK was ±20% of the geometric mean of predicted Ctrough (predose on day 1 of cycle 3). Further clinical subgroup analyses were performed to identify any differences in efficacy and/or safety between these subpopulations.
Exposure‐Response Analyses
The exposure‐response analysis for subcutaneous DARA monotherapy included data from PAVO Part 2, MMY1008, and COLUMBA; the exposure‐response analysis for subcutaneous DARA in combination therapies was based on data from PLEIADES. Responders were defined as patients who achieved partial response or better based on International Myeloma Working Group response criteria. 32 The exposure‐efficacy analysis for ORR was evaluated graphically and compared between subcutaneous and intravenous DARA. The exposure‐response analysis for safety was explored for selected adverse events (AEs), including overall serious AEs (SAEs), overall grade ≥3 treatment‐emergent AEs (TEAEs), neutropenia, and IRRs. Peak DARA concentrations after the first dose and overall peak concentrations (except for IRRs) were investigated for their potential relationship with AEs by exposure quartiles and evaluated graphically. Logistic regression models were used to graphically evaluate the relationship between body weight and the incidence of the AEs and compare the body weight–AE relationship for subcutaneous daratumumab and intravenous daratumumab using the formula logit(Pr) = intercept + slope × body weight.
Results
Patient Demographics and Disease Characteristics
The popPK data set comprised 5159 measurable PK samples from 742 patients, with 487 patients in the subcutaneous DARA group (monotherapy, n = 288; combination therapy, n = 199) and 255 patients in the intravenous DARA monotherapy group. Data from 3 patients with no measurable DARA concentrations were excluded from the popPK analysis. Patient baseline demographics and disease covariates are summarized in Table 2.
Table 2.
Descriptive Statistics of Baseline Covariates
| PAVO Part 2 | MMY1008 | COLUMBA | PLEIADES | Total | |
|---|---|---|---|---|---|
| N = 25 | N = 6 | N = 512 | N = 199 | N = 742 | |
| Age (years), median (range) | 68 (51‐85) | 73 (42‐81) | 67 (33‐92) | 69 (33‐86) | 67 (33‐92) |
| Sex, n (%) | |||||
| Male | 14 (56) | 2 (33) | 281 (55) | 124 (62) | 421 (57) |
| Race, n (%) | |||||
| White | 19 (76) | 0 | 386 (75) | 121 (61) | 526 (71) |
| Black | 2 (8) | 0 | 14 (3) | 8 (4) | 24 (3) |
| Asian | 0 | 6 (100) | 68 (13) | 5 (3) | 79 (11) |
| Other a | 4 (16) | 0 | 44 (9) | 65 (33) | 113 (15) |
| Body mass index (kg/m2), median (range) | 26.3 (20.3‐43.6) | 23.4 (21.2‐25.9) | 26.8 (12.3‐47.5) | 26.3 (16.8‐48.0) | 26.6 (12.3‐48.0) |
| Baseline ECOG PS score, n (%) | |||||
| 0 | 11 (44) | 5 (83) | 149 (29) | 101 (51) | 266 (36) |
| 1 | 13 (52) | 1 (17) | 277 (54) | 93 (47) | 384 (52) |
| 2 | 1 (4) | 0 | 85 (17) | 5 (3) | 91 (12) |
| 3 | 0 | 0 | 1 (<1) | 0 | 1 (<1) |
| Lines of prior therapy, n (%) | |||||
| ≤4 lines | 20 (80) | 3 (50) | 351 (69) | 198 (99) | 572 (77) |
| >4 lines | 5 (20) | 3 (50) | 161 (31) | 1 (1) | 170 (23) |
| Type of myeloma at baseline b | |||||
| Non‐IgG | 11 (44) | 3 (50) | 211 (41) | 89 (45) | 314 (42) |
| IgG | 13 (52) | 3 (50) | 301 (59) | 110 (55) | 427 (58) |
| Missing | 1 (4) | 0 | 0 | 0 | 1 (<1) |
| Refractory status | |||||
| None | 5 (20) | 0 | 67 (13) | 184 (92) | 256 (35) |
| PI only | 4 (16) | 0 | 48 (9) | 13 (7) | 65 (9) |
| IMiD only | 2 (8) | 0 | 146 (29) | 1 (1) | 149 (20) |
| Both PI and IMiD | 14 (56) | 0 | 251 (49) | 1 (1) | 266 (36) |
| Missing | 0 | 6 (100) | 0 | 0 | 6 (1) |
| ISS stage | |||||
| I | 13 (52) | 4 (67) | 173 (34) | 79 (40) | 269 (36) |
| II | 5 (20) | 2 (33) | 188 (37) | 72 (36) | 267 (36) |
| III | 6 (24) | 0 | 150 (29) | 47 (24) | 203 (27) |
| Missing | 1 (4) | 0 | 1 (<1) | 1 (1) | 3 (<1) |
| eGFR (mL/min/1.73 m2), median (range) | 68.3 (26.2‐110.3) | 70.4 (62.2‐110.6) | 67.8 (21.1‐187.8) | 76.2 (19.8‐158.4) | 69.7 (19.8‐187.8) |
| Hepatic function | |||||
| Normal | 24 (96) | 6 (100) | 450 (88) | 175 (88) | 655 (88) |
| Mild dysfunction | 1 (4) | 0 | 58 (11) | 23 (12) | 82 (11) |
| Moderate dysfunction | 0 | 0 | 4 (1) | 1 (1) | 5 (1) |
| Albumin (g/L), median (range) | 41 (26‐47) | 40 (34‐44) | 39.5 (19‐53) | 38 (22‐51) | 39 (19‐53) |
ECOG PS, Eastern Cooperative Oncology Group performance status; PI, proteasome inhibitor; IMiD, immunomodulatory drug; ISS, International Staging System; eGFR, estimated glomerular filtration rate.
Includes “Hispanic or Latino,” “native Hawaiian or other Pacific Islander,” and “other” categories.
Based on immunofixation.
Observed PK of Subcutaneous DARA
Subcutaneous DARA (1800 mg) monotherapy and combination therapies achieved similar concentrations over time.
Model Parameters
The parameter estimates of the final model resulting from covariate analysis are provided in Table S1. The estimated bioavailability for subcutaneous DARA was approximately 70%. The estimated linear clearance was 0.005 L/h, and the volume of distribution of the central compartment (5.25 L) approached the plasma volume; both volumes were related to body weight, as expected for monoclonal antibodies. The model‐derived geometric mean (coefficient of variation %) half‐life associated with linear elimination was 20.4 days (22.4%) based on post hoc PK estimates for monotherapy and 23 to 27 days for combination therapies. Steady state was achieved approximately 5 months after the first dose of the once‐every‐4‐week dosing schedule.
PopPK of Subcutaneous DARA Monotherapy
The simulated typical PK profiles for subcutaneous and intravenous DARA monotherapy are presented in Figure 1A. The administration of subcutaneous DARA resulted in smaller peak‐to‐trough fluctuation compared with intravenous DARA. The mean peak‐to‐trough ratio on day 1 of cycle 3 was 1.2 versus 1.7 for subcutaneous DARA versus intravenous DARA, respectively. Similar to the PK profile of intravenous DARA, the concentrations following weekly subcutaneous DARA administration continued to increase until the dosing frequency was reduced to a once‐every‐2‐week dose schedule. Subsequently, concentration decreased when the dosing frequency was decreased to once every 4 weeks. Given the long half‐life and changing dosing frequency, steady state is reached approximately 5 months into the once‐every‐4‐week dosing schedule. Subcutaneous DARA resulted in similar or slightly higher Ctrough throughout the dose schedule after cycle 1 (Figure 1B), with lower Cmax compared with intravenous DARA (Figure 1A). The geometric mean ratio for DARA subcutaneous/intravenous Ctrough was consistently slightly >1 throughout the treatment period. The simulated target saturation over time with subcutaneous and intravenous DARA is shown in Figure S1. The target saturation with subcutaneous DARA was similar to that with intravenous DARA and was consistently above 96% over time. The dynamics of the simulated clearance profile after subcutaneous and intravenous DARA are shown in Figure S2. Total clearance decreased over time and approached the nonspecific linear clearance after approximately 8 weeks. The clearance profiles with subcutaneous DARA are similar to those with intravenous DARA; the only difference seen was on the first administration because of the slower absorption of DARA into systemic circulation.
Figure 1.
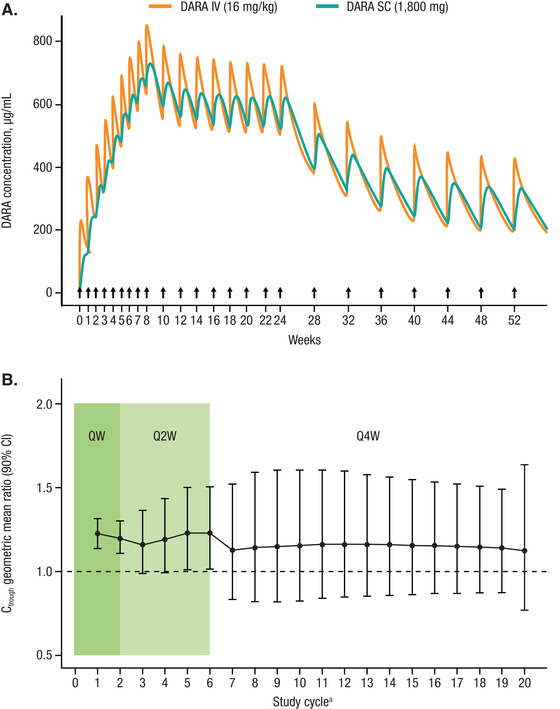
(A) Typical PK profile of subcutaneous DARA 1800 mg or intravenous DARA 16 mg/kg per the approved dose and schedulea for intravenous monotherapy. (B) Subcutaneous and intravenous DARA Ctrough geometric mean ratios over time. PK, pharmacokinetics; DARA, daratumumab; Ctrough, predicted trough concentration; CI, confidence interval. Note: Black arrows represent dose events. Dotted line represents ratio of 1. Point and bar represent geometric mean ratio and CI. aApproved dose schedule consisted of weekly administration for 8 weeks (8 doses), once every 2 weeks for 16 weeks (8 doses), and once every 4 weeks thereafter (eg, 8 doses).
PopPK of Subcutaneous DARA Combination Therapies
Subcutaneous DARA dosing regimens were different for monotherapy and combination therapy beyond week 6. Therefore, simulated Ctrough comparisons following 6 weekly doses, which is a point common to both monotherapy and combination therapies and closest to day 1 of cycle 3, were chosen for assessment of the impact of backbone therapies on subcutaneous DARA PK without the confounding influence of dose regimens across different combination therapies. The simulated Ctrough of subcutaneous DARA for combination therapies was similar to that of monotherapy following 6 weekly doses (Figure S3).
Exposure‐Response for Efficacy
ORR with intravenous DARA monotherapy has been shown to correlate with increasing maximum Ctrough. 24 Subcutaneous DARA monotherapy produced higher maximum Ctrough in both responders and nonresponders, and slightly higher ORRs compared with the approved intravenous DARA (Figure 2). The relationship between maximum Ctrough and ORR was similar for both subcutaneous and intravenous DARA monotherapy. For combination therapy, a high ORR was observed consistently across the studied concentration range, indicating that maximum efficacy in terms of ORR had been attained with subcutaneous DARA. Cross‐study comparisons of the subcutaneous DARA plus lenalidomide and dexamethasone (Rd) arm with POLLUX (intravenous DARA plus Rd) and for the subcutaneous DARA plus bortezomib, melphalan, and prednisone (VMP) arm with ALCYONE (intravenous DARA plus VMP) indicated a similar exposure‐response relationship for efficacy between subcutaneous and intravenous DARA for both D‐Rd and D‐VMP combinations (Figure S4).
Figure 2.
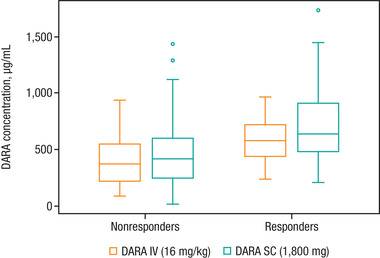
Box plot of DARA maximum Ctrough for nonresponders and responders after subcutaneous DARA 1800 mg or intravenous DARA 16 mg/kg monotherapy. DARA, daratumumab; Ctrough, predicted trough concentration. Note: The horizontal line in the box represents the median value of daratumumab concentration. The length of the box denotes the interquartile range (IQR), and whiskers represent the range (within 1.5 × IQR from the median). Outlier values are shown as circles.
Exposure‐Response for Safety
In the exposure‐safety analysis, no relationship was apparent between exposure and safety end points (SAEs, grade ≥3 TEAEs, neutropenia, and IRRs) using the peak concentrations after the first dose and overall peak concentrations for monotherapy and combination therapies. Although subcutaneous DARA showed a higher cycle 3 day 1 Ctrough in the lower‐body‐weight subgroups than did intravenous DARA, the probability of SAEs and grade ≥3 TEAEs was comparable for subcutaneous and intravenous DARA monotherapy (Figure 3).
Figure 3.
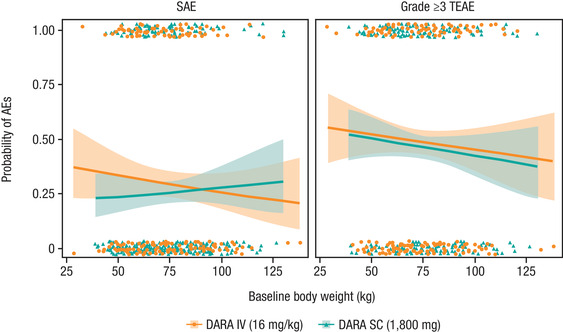
Rate of SAEs and grade ≥3 TEAEs in relation to baseline body weight after subcutaneous DARA 1800 mg or intravenous DARA 16 mg/kg monotherapy. SAE, serious adverse event; TEAE, treatment‐emergent adverse event; DARA, daratumumab; AE, adverse event. Note: The lines represent the predicted mean curves, and the shaded regions are the 95% confidence intervals. Dots represent the observed rate of SAEs and TEAEs.
Effect of Covariates
An exploratory covariate analysis was performed to assess the impact of intrinsic factors on DARA PK based on different routes of administration (intravenous vs subcutaneous). None of the investigated intrinsic factors (ie, age, sex, race, region, renal impairment, hepatic impairment, and ECOG performance status) had clinically relevant effects on exposure (predicted maximum Ctrough) to DARA monotherapy or combination therapy regimens. The covariate effects on exposure were generally smaller (<25% difference among subgroups) in combination therapy compared with monotherapy. Although patients with IgG myeloma or with lower baseline albumin concentrations appeared to have lower exposure, clinical analyses demonstrated that the lower DARA concentrations in patients with IgG myeloma and with lower baseline albumin values (subcutaneous DARA arm) had no clinically relevant effect on efficacy in terms of ORR. Patients from COLUMBA with abnormal albumin in the intravenous DARA arm appeared to have a higher incidence of SAEs despite achieving lower concentrations of DARA. Thus, increasing the DARA dose in patients with abnormal albumin is unlikely to improve the overall risk‐benefit profile. Therefore, no dose adjustment is recommended based on any of these factors.
Influence of Body Weight on PK, Efficacy, and Safety
Within each body weight subgroup, there was considerable overlap in the observed cycle 3 day 1 Ctrough (defined as maximum Ctrough) for both treatment groups (Figure 4A,B). Further simulation was conducted to compare the effect of body weight on exposure. Among monotherapy studies, the mean simulated maximum Ctrough in the low‐body‐weight subgroup (≤65 kg) was approximately 67% higher in the subcutaneous DARA group (721 μg/mL) than in the intravenous DARA group (432 μg/mL). The mean maximum Ctrough in the high‐body‐weight subgroup (>85 kg) was approximately 14% lower in the subcutaneous DARA group (472 μg/mL) than in the intravenous DARA group (546 μg/mL). The mean maximum Ctrough in the body weight subgroup >65 to 85 kg was comparable between subcutaneous and intravenous DARA. The simulated Cmax (cycle 3 day 1) for the low‐body‐weight subgroup (≤65 kg) for subcutaneous DARA monotherapy was comparable to the Cmax in the higher‐body‐weight subgroup (>85 kg) for the intravenous DARA group, but, in general, the Cmax values for the overall population were lower for subcutaneous DARA. Similar trend and comparable DARA concentrations across body weight groups were found for subcutaneous DARA combination therapies compared with monotherapy.
Figure 4.
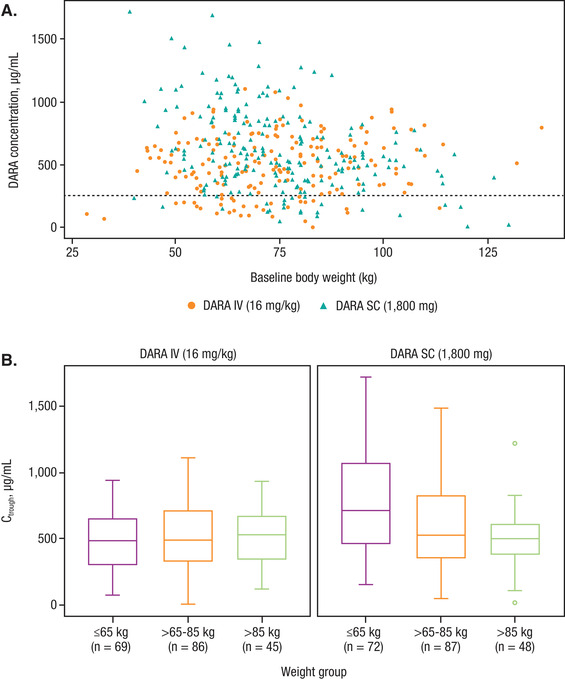
(A) Observed DARA on day 1 of cycle 3 Ctrough (after 8 weekly doses) across the range of body weights. (B) Box plots of observed DARA on day 1 of cycle 3 Ctrough (after 8 weekly doses) by body weight subgroups in subcutaneous or intravenous DARA monotherapy regimens. DARA, daratumumab; Ctrough, predicted trough concentration. Note: Black dashed line represents the concentrations of DARA at which the 99% target saturation is achieved.
Clinical analysis demonstrated consistent efficacy of the drug in all body weight subgroups. Specifically, in the high‐body‐weight subgroup (>85 kg), the ORR with subcutaneous DARA (43.9%; 95% confidence interval [CI], 31.7%‐56.7%) was similar to the ORR with intravenous DARA (32.8%; 95% CI, 21.3%‐46.0%).
In the pivotal monotherapy study (COLUMBA), the incidence of grade 3/4 neutropenia was higher with subcutaneous DARA (13.1%) compared with intravenous DARA (7.8%), primarily driven by the higher incidence (subcutaneous DARA, 20.4%; intravenous DARA, 8.7%) in the lower‐body‐weight subgroup. However, among the pooled monotherapy data, the exposure‐safety analysis using the exposure metrics of Cmax after the first dose (Figure 5A) and overall Cmax (Figure 5B) demonstrated no upward trend in the incidence of neutropenia with increased DARA exposure. In the highest exposure quartile (quartile 4) for first‐dose Cmax (Figure 5A), the mean probability of any grade and grade ≥3 neutropenia in the subcutaneous DARA group was similar to the intravenous DARA group. Importantly, the probability for any grade and grade ≥3 neutropenia for patients in the highest exposure quartile was similar or lower compared with the lower exposure quartiles (quartiles 1, 2, and 3) for both treatment groups. Additional analyses using area under the curve during the first week or maximum Ctrough also did not show any relationship with neutropenia (data on file). Notably, in COLUMBA, the incidence of preexisting grade 2 neutropenia in the subcutaneous DARA ≤50 kg group was higher with subcutaneous DARA (45.5%) versus intravenous DARA (19%). The imbalance in preexisting neutropenia may partly explain the higher neutropenia rate observed at lower‐body‐weight with subcutaneous DARA. In relation to IRRs, in quartile 4 for first‐dose Cmax (Figure 6), the mean probability of any grade and grade ≥3 IRRs was lower in the subcutaneous DARA group versus the intravenous DARA group.
Figure 5.
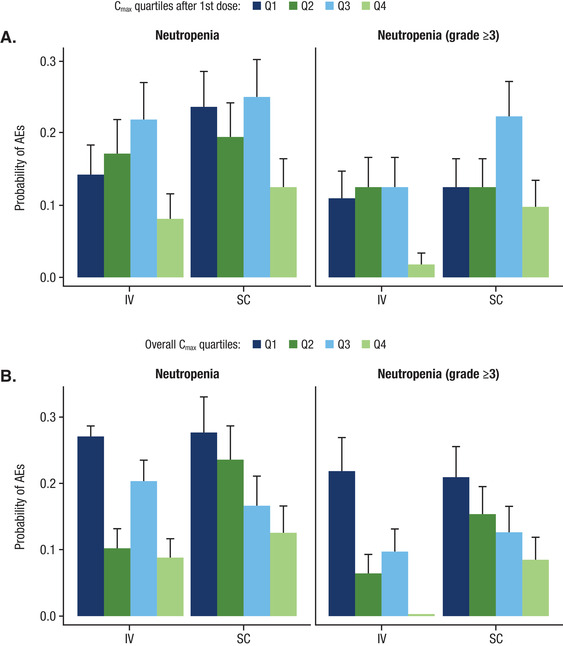
Rate of any grade and grade ≥3 neutropenia in relation to (A) Cmax after the first dose and (B) overall Cmax after subcutaneous DARA 1800 mg or intravenous DARA 16 mg/kg monotherapy. Cmax, maximum peak concentration; DARA, daratumumab; Q, quartile; AE, adverse event.
Figure 6.
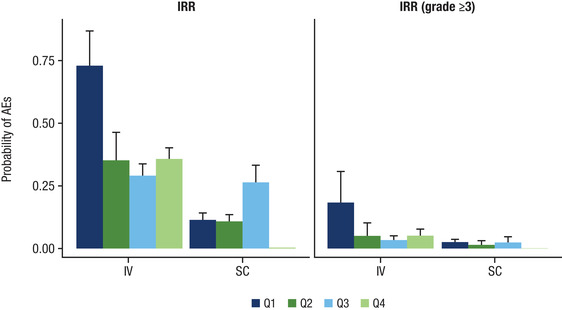
Rate of IRRs in relation to DARA Cmax (by quartiles) after the first dose of subcutaneous or intravenous DARA monotherapy. IRR, infusion‐related reaction; DARA, daratumumab; Cmax, maximum peak concentration; AE, adverse event; Q, quartile. Note: The quartiles for Cmax after the first dose were: Q1, 8.68‐124 μg/mL; Q2, 124‐194 μg/mL; Q3, 194‐254 μg/mL; and Q4, 254‐807 μg/mL.
Impact of ADAs on Exposure
None of the patients who received subcutaneous DARA were positive for ADA. One patient who received intravenous DARA was positive for anti‐DARA antibody 8 weeks after the last intravenous DARA dose. The ADA‐positive sample was not associated with an IRR, and the patient did not report any clinically relevant TEAEs.
A total of 18 of 420 patients (4.3%) were positive for anti‐rHuPH20 antibodies at baseline prior to administration of subcutaneous DARA. The incidence of treatment‐emergent, nonneutralizing anti‐rHuPH20 antibodies was 6.4% (27 of 420 patients): 6.9% (16 of 233 patients) with subcutaneous DARA monotherapy and 5.9% (11 of 187 patients) with subcutaneous DARA combination therapy. Subcutaneous DARA exposure was comparable between antibody‐negative patients and those with anti‐rHuPH20 antibodies. No relationship was apparent between the presence of anti‐rHuPH20 antibodies and safety.
Discussion
In the last few years, based on results from pivotal trials and subsequent regulatory approvals, DARA has changed the treatment paradigm and has expanded treatment options for patients with multiple myeloma. The results from prior popPK and exposure‐response analyses of intravenous DARA provided support for the recommended intravenous 16‐mg/kg dose in monotherapy and combination therapy for the treatment of multiple myeloma. 27 PK profiles were similar for intravenous DARA monotherapy and combination therapy, and covariates had no clinically important effects on intravenous DARA exposure; maximum clinical benefit on progression‐free survival was achieved by approximately 75% of patients with an acceptable safety profile. 27 Overall, the observed concentration‐time data of DARA after subcutaneous administration were well described by a 2‐compartment popPK model with a first‐order absorption and parallel linear and nonlinear Michaelis‐Menten eliminations.
The model parameter estimates of subcutaneous DARA were consistent with previously published literature. The estimated bioavailability for subcutaneous DARA was consistent with other subcutaneous monoclonal antibodies coadministered with rHuPH20, 33 , 34 and the estimated linear clearance was similar to the clearance of nonspecific endogenous IgG. 35 Similar to intravenous DARA, steady state was achieved approximately 5 months after first dose of the once‐every‐4‐week dosing schedule. 27
Among monotherapy studies, the subcutaneous DARA formulation consistently produced lower peak‐to‐trough fluctuations, similar or slightly higher Ctrough over time, and lower Cmax compared with the approved intravenous DARA formulation. Target saturation with the subcutaneous DARA dose was similar to that with intravenous DARA (>96% over time); a target saturation >99% at the end of weekly intravenous DARA dosing may be required to induce clinical effect. 24 These results suggest sufficient concentrations are attained by subcutaneous DARA. None of the investigated covariates (ie, age, sex, race, region, renal impairment, hepatic impairment, baseline albumin, ECOG performance status, and type of myeloma) had clinically relevant effects. Similar to previous intravenous DARA studies, the covariate effects on exposure were generally smaller in combination therapy compared with monotherapy, 27 and the lower DARA concentrations in patients with IgG myeloma and with lower baseline albumin values had no clinically relevant effect on efficacy in terms of ORR. Furthermore, as observed in previous intravenous DARA studies, 36 patients from COLUMBA with abnormal albumin in the intravenous DARA arm appeared to have a higher incidence of SAEs despite achieving lower concentrations of DARA. Therefore, no dose adjustment is recommended for subcutaneous DARA based on these factors. The simulated Ctrough values following 6 weekly doses of subcutaneous DARA combination therapies (D‐VMP, D‐Rd, and D‐VRd) were similar to monotherapy. This is consistent with findings from a popPK analysis of intravenous DARA, in which the PK of intravenous DARA in combination with other treatment therapies were consistent with the PK of intravenous DARA administered as monotherapy. 27
Subcutaneous DARA given as monotherapy or in combination therapy retained a similar exposure‐response relationship for both efficacy and safety end points as intravenous DARA. The probability of ORR was comparable for subcutaneous and intravenous DARA. In relation to safety, there was a flat relationship between exposure and the safety end points of SAEs, grade ≥3 TEAEs, neutropenia, and IRRs. In the head‐to‐head comparative study COLUMBA, treatment‐emergent neutropenia occurred early, was not sustained or did not deteriorate with continued treatment, and was manageable. 21 In addition, treatment discontinuation because of AEs was comparable between subcutaneous and intravenous DARA. SAEs, including infections, for the overall COLUMBA population and within the lower‐body‐weight subgroup were lower with subcutaneous DARA compared with intravenous DARA.
As previously seen in the popPK model for body weight‐based dosing of intravenous DARA, for subcutaneous DARA, body weight had a significant effect on linear clearance and central volume of distribution, but not on target‐mediated nonlinear clearance. 30 As expected, flat dosing with subcutaneous DARA resulted in higher concentrations for the lower‐body‐weight subgroup (≤65 kg), whereas body weight dosing with intravenous DARA resulted in higher concentrations for the higher‐body‐weight subgroup (>85 kg); however, the range of concentrations with subcutaneous DARA were within the range previously observed with intravenous DARA. Although the variability of the intravenous DARA maximum Ctrough was lower than expected, the spread of maximum Ctrough across body weight groups with subcutaneous DARA monotherapy was similar to previously observed data of intravenous DARA 16 mg/kg (36‐1764 μg/mL). 37 These exposure trends observed with subcutaneous DARA administered as a flat dose are consistent with previously reported data for subcutaneous administration of monoclonal antibodies. 38 , 39 , 40 In COLUMBA, subcutaneous DARA demonstrated similar efficacy in terms of ORR across the range of body weight subgroups, as achieved by intravenous DARA. 37 Given the wide therapeutic window of DARA and similar target saturation, no dose adjustment is recommended based on body weight.
The incidence of treatment‐emergent anti‐DARA and anti‐rHuPH20 antibodies for subcutaneous DARA was low and consistent with the literature 17 , 41 and did not appear to impact efficacy or safety. This is consistent with the findings in COLUMBA. 21
Conclusion
Subcutaneous DARA, both as a monotherapy and in combination therapies, demonstrated a profile comparable and favorable to intravenous DARA. These data support the use of subcutaneous DARA 1800 mg for the treatment of multiple myeloma.
Conflicts of Interest
S.Z.U. reports grants and personal fees from Amgen, Celgene, Sanofi, Seattle Genetics, Janssen, Takeda, SkylineDX, and Merck; personal fees from AbbVie and MundiPharma; and grants from Bristol Myers Squibb and Pharmacyclics. M.‐V.M. reports personal fees from Janssen, Celgene, Amgen, Takeda, AbbVie, GlaxoSmithKline, Sanofi, Oncopeptides, Roche, Genentech, Adaptive, Pfizer, and Regeneron. A.C. reports grants and personal fees from Janssen, Celgene, Novartis, Amgen, Seattle Genetics, and Millenium/Takeda; personal fees from Oncopeptides, Antengene, GlaxoSmithKline, Secura Bio, Bristol Myers Squibb, Karyopharm, and Sanofi; and grants from Pharmacyclics. J.S.‐M. reports consulting and serving on advisory boards for Amgen, Bristol Myers Squibb, Celgene, Janssen, Merck, Novartis, Takeda, Sanofi, Roche, AbbVie, GlaxoSmithKline, and Karyopharm. M.K. reports grants and personal fees from Bristol Myers Squibb/Celgene and Janssen and personal fees from Amgen, AbbVie, GlaxoSmithKline, and Takeda. M.L., C.H., M.Q., H.Z., Y.‐N.S., and D.A.P. are employees of Janssen Pharmaceuticals and own equity in Johnson & Johnson. R.C. is an employee of Janssen Pharmaceuticals. K.S., H.N., and C.T. have no conflicts of interest to disclose.
Funding
The clinical studies and the analyses presented here were supported by research funding from Janssen Research & Development, LLC. All authors had full access to all the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Author Contributions
M.L., M.‐V.M., H.N., R.C., C.H., H.Z., Y.‐N.S., and D.A.P. contributed to the study design and conduct, data analysis and interpretation, and drafting and revising of the article. S.Z.U. contributed to the data analysis and interpretation and drafting and revising of the article. A.C., C.T., and M.K. contributed to the study conduct, data analysis and interpretation, and drafting and revising of the article. J.S.‐M. and M.Q. contributed to the study design, data analysis and interpretation, and drafting and revising of the article. K.S. contributed to study conduct and drafting and revising of the article. All authors approved the final version before submission.
Data Availability
The data‐sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
Supporting information
Supporting information
Supporting information
Supporting information
Acknowledgments
The authors thank the patients who participated in the PAVO Part 2, MMY1008, COLUMBA, and PLEIADES studies and their families, as well as the study coinvestigators, research nurses, and coordinators at each of the clinical sites. Medical writing and editorial support were provided by Grace Wang, PharmD, of MedErgy and funded by Janssen Global Services, LLC.
Honghui Zhou is a fellow of the American College of Clinical Pharmacology.
References
- 1. National Cancer Institute . SEER Cancer Stat Facts: Myeloma. https://seer.cancer.gov/statfacts/html/mulmy.html. Accessed August 17, 2020.
- 2. Cancer Research UK . Myeloma statistics. https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/myeloma. Accessed August 17, 2020.
- 3. de Weers M, Tai YT, van der Veer MS, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186(3):1840‐1848. [DOI] [PubMed] [Google Scholar]
- 4. Lammerts van Bueren J, Jakobs D, Kaldenhoven N, et al. Direct in vitro comparison of daratumumab with surrogate analogs of CD38 antibodies MOR03087, SAR650984 and Ab79. Blood. 2014;124(21):3474. [Google Scholar]
- 5. Overdijk MB, Verploegen S, Bogels M, et al. Antibody‐mediated phagocytosis contributes to the anti‐tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs. 2015;7(2):311‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Overdijk MB, Jansen JH, Nederend M, et al. The therapeutic CD38 monoclonal antibody daratumumab induces programmed cell death via Fcgamma receptor‐mediated cross‐linking. J Immunol. 2016;197(3):807‐813. [DOI] [PubMed] [Google Scholar]
- 7. Krejcik J, Casneuf T, Nijhof IS, et al. Daratumumab depletes CD38+ immune‐regulatory cells, promotes T‐cell expansion, and skews T‐cell repertoire in multiple myeloma. Blood. 2016;128(3):384‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chiu C, Casneuf T, Axel A, et al. Daratumumab in combination with lenalidomide plus dexamethasone induces clonality increase and T‐cell expansion: results from a phase 3 randomized study (POLLUX). Blood. 2016;128(22). Abstract 4531. [Google Scholar]
- 9. Adams HC, III , Stevenaert F, Krejcik J, et al. High‐parameter mass cytometry evaluation of relapsed/refractory multiple myeloma patients treated with daratumumab demonstrates immune modulation as a novel mechanism of action. Cytometry A. 2019;95(3):279‐289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373(13):1207‐1219. [DOI] [PubMed] [Google Scholar]
- 11. Lonial S, Weiss BM, Usmani S, et al. Daratumumab monotherapy in patients with treatment‐refractory multiple myeloma (SIRIUS): an open‐label, randomised, phase 2 trial. Lancet. 2016;387(10027):1551‐1560. [DOI] [PubMed] [Google Scholar]
- 12. Palumbo A, Chanan‐Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754‐766. [DOI] [PubMed] [Google Scholar]
- 13. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319‐1331. [DOI] [PubMed] [Google Scholar]
- 14. Mateos MV, Dimopoulos MA, Cavo M, et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med. 2018;378(6):518‐528. [DOI] [PubMed] [Google Scholar]
- 15. Facon T, Kumar S, Plesner T, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019;380(22):2104‐2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moreau P, Attal M, Hulin C, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem‐cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open‐label, phase 3 study. Lancet. 2019;394(10192):29‐38. [DOI] [PubMed] [Google Scholar]
- 17. DARZALEX (daratumumab) injection, for intravenous use [package insert]. Horsham, PA: Janssen Biotech, Inc.; 2020. [Google Scholar]
- 18. European Medicines Agency . DARZALEX 20 mg/mL concentrate for solution for infusion [summary of product characteristics]. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004077/WC500207296.pdf. Accessed August 17, 2020.
- 19. Usmani SZ, Nahi H, Mateos MV, et al. Subcutaneous delivery of daratumumab in relapsed or refractory multiple myeloma. Blood. 2019;134(8):668‐677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. San‐Miguel J, Usmani SZ, Mateos MV, et al. Subcutaneous daratumumab in patients with relapsed or refractory multiple myeloma: Part 2 of the open‐label, multicenter, dose‐escalation phase 1b study (PAVO). Haematologica. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mateos MV, Nahi H, Legiec W, et al. Subcutaneous versus intravenous daratumumab in patients with relapsed or refractory multiple myeloma (COLUMBA): a multicentre, open‐label, non‐inferiority, randomised, phase 3 trial. Lancet Haematol. 2020;7(5):e370‐e380. [DOI] [PubMed] [Google Scholar]
- 22. Usmani SZ, Mateos M‐V, Nahi H, et al. Randomized, open‐label, non‐inferiority, phase 3 study of subcutaneous (SC) versus intravenous (IV) daratumumab (DARA) administration in patients with relapsed or refractory multiple myeloma: Columba update. Blood. 2019;134(suppl_1):1865‐1865. [Google Scholar]
- 23. Chari A, Rodriguez‐Otero P, McCarthy H, et al. Subcutaneous daratumumab plus standard treatment regimens in patients with multiple myeloma across lines of therapy (PLEIADES): an open‐label Phase II study. Br J Haematol. 2020. [DOI] [PubMed] [Google Scholar]
- 24. Xu XS, Yan X, Puchalski T, et al. Clinical implications of complex pharmacokinetics for daratumumab dose regimen in patients with relapsed/refractory multiple myeloma. Clin Pharmacol Ther. 2017;101(6):721‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. DARZALEX FASPRO™ (daratumumab and hyaluronidase‐fihj) [package insert]. Horsham, PA: Janssen Biotech, Inc.; 2020. [Google Scholar]
- 26. Clemens PL, Yan X, Lokhorst HM, et al. Pharmacokinetics of daratumumab following intravenous infusion in relapsed or refractory multiple myeloma after prior proteasome inhibitor and immunomodulatory drug treatment. Clin Pharmacokinet. 2017;56(8):915‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu XS, Dimopoulos MA, Sonneveld P, et al. Pharmacokinetics and exposure‐response analyses of daratumumab in combination therapy regimens for patients with multiple myeloma. Adv Ther. 2018;35(11):1859‐1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. European Medicines Agency . Guideline of bioanalytical method validation. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf. Accessed August 17, 2020.
- 29. U.S. Department of Health and Human Services . US Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM). Bioanalytical Method Validation: Guidance for Industry. https://www.fda.gov/media/70858/download. Accessed August 17, 2020.
- 30. Yan X, Clemens PL, Puchalski T, et al. Target‐mediated drug disposition of daratumumab following intravenous infusion in relapsed or refractory multiple myeloma after prior proteasome inhibitors and immunomodulatory drugs: a population pharmacokinetic analysis. Blood. 2015;126(23):4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beal SS , Boeckmann L , Bauer RJ , Sheiner LB . NONMEM User's Guides (1989‐2009). Ellicott City. MD: Icon Development Solutions; 2009. [Google Scholar]
- 32. Rajkumar SV, Harousseau JL, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood. 2011;117(18):4691‐4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gibiansky L, Giraudon M, Rayner CR, et al. Population pharmacokinetic analysis of oseltamivir and oseltamivir carboxylate following intravenous and oral administration to patients with and without renal impairment. J Pharmacokinet Pharmacodyn. 2015;42(3):225‐236. [DOI] [PubMed] [Google Scholar]
- 34. Quartino AL, Hillenbach C, Li J, et al. Population pharmacokinetic and exposure‐response analysis for trastuzumab administered using a subcutaneous “manual syringe” injection or intravenously in women with HER2‐positive early breast cancer. Cancer Chemother Pharmacol. 2016;77(1):77‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol. 2017;6(9):576‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yan X, Clemens PL, Puchalski T, et al. Influence of disease and patient characteristics on daratumumab exposure and clinical outcomes in relapsed or refractory multiple myeloma. Clin Pharmacokinet. 2018;57(4):529‐538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mateos MV, Usmani SZ, Grosicki S, et al. Non‐inferiority, Phase 3 Study of Subcutaneous (SC) Versus Intravenous (IV) Daratumumab (DARA) Administration in Patients (Pts) With Relapsed or Refractory Multiple Myeloma (RRMM): Body Weight Subgroup Analysis of COLUMBA. Poster presented at: The 61st American Society of Hematology (ASH) Annual Meeting & Exposition; December 7–10, 2019; Orlando, Florida. Abstract 1906. [Google Scholar]
- 38. Wang DD, Zhang S, Zhao H, Men AY, Parivar K. Fixed dosing versus body size‐based dosing of monoclonal antibodies in adult clinical trials. J Clin Pharmacol. 2009;49(9):1012‐1024. [DOI] [PubMed] [Google Scholar]
- 39. Bai S, Jorga K, Xin Y, et al. A guide to rational dosing of monoclonal antibodies. Clin Pharmacokinet. 2012;51(2):119‐135. [DOI] [PubMed] [Google Scholar]
- 40. Hendrikx JMA, Haanen JBAG, Voest EE, Schellens JHM, Huitema ADR, Beijnen JH. Fixed dosing of monoclonal antibodies in oncology. Oncologist. 2017;22(10):1212‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rosengren S, Dychter SS, Printz MA, et al. Clinical immunogenicity of rHuPH20, a hyaluronidase enabling subcutaneous drug administration. AAPS J. 2015;17(5):1144‐1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information
Data Availability Statement
The data‐sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.