

Abstract
An open‐cage fullerene incorporating phosphorous ylid and carbonyl group moieties on the rim of the orifice can be filled with gases (H2, He, Ne) in the solid state, and the cage opening then contracted in situ by raising the temperature to complete an intramolecular Wittig reaction, trapping the atom or molecule inside. Known transformations complete conversion of the product fullerene to C60 containing the endohedral species. As well as providing an improved synthesis of large quantities of 4He@C60, H2@C60, and D2@C60, the method allows the efficient incorporation of expensive gases such as HD and 3He, to prepare HD@C60 and 3He@C60. The method also enables the first synthesis of Ne@C60 by molecular surgery, and its characterization by crystallography and 13C NMR spectroscopy.
Keywords: endohedral fullerene, NMR spectroscopy, phosphorus ylid, synthetic methods, X-ray diffraction
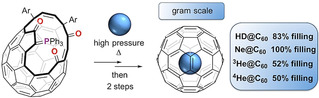
Solid‐state filling of a stable phosphorus ylid open‐cage fullerene is followed by heating, inducing an in situ intramolecular Wittig reaction to reduce the size of the cage opening and trapping a single gas atom or molecule inside. Expensive gases can be compressed to high pressure, leading to high incorporation of the endohedral species. Cage closure steps allow preparation of gram‐scale endohedral fullerenes including 3He@C60, HD@C60, and Ne@C60.
Introduction
Compounds in which atoms or small molecules (A) are trapped in the cavity of cage fullerenes such as C60 are known as endofullerenes and denoted A@C60. They are of interest for study of their material properties and the properties of the isolated endohedral species.[ 1a , 1b , 1c ]
The noble gas endofullerenes of C60 have been the object of sustained theoretical studies[ 2a , 2b , 2c , 2d , 2e , 2f , 2g , 2h , 2i , 2j , 2k ] of their geometry, reactivity and electronic structure, and subject to a recent review. [3] Encapsulation of a noble gas in C60 was first detected by mass spectrometry in the formation of 4He@C60 from collision of accelerated C60 +. with helium gas, [4] and 4He@C60 was also observed at the part‐per‐million level in C60 formed by the carbon arc discharge method using He as the buffer gas. [5] High temperature and pressure exposure of C60 to the noble gases allows direct incorporation, and isolation of samples containing approximately 0.1 % of the endohedral atom He, Ne, Ar, or Kr, or 0.03 % of Xe. [6] Incorporation is improved by addition of KCN, to levels of 1 % for He and circa 0.3 % for Ar, Kr, or Xe, albeit at the cost of lower recovery.[ 7a , 7b , 7c ] High‐energy helium bombardment of C60 under explosive conditions has also been used for direct encapsulation of the noble gas. [8] Removal of empty C60 using many cycles of preparative HPLC has been reported for the heavier noble gas endofullerenes (Ar@C60, 1.3 mg, 98 % filled;[ 7c , 9 ] Kr@C60, 0.14 mg, 90 % filled; [10] Xe@C60 0.32 mg, 50 % filled [7b] ), but direct encapsulation methods are not practical for synthesis of larger amounts of a noble gas endofullerene, and cannot be used to prepare molecular endofullerenes such as H2@C60.
Yet, many applications exist to benefit from the availability of larger‐scale synthetic methods. Noble gas endofullerenes encapsulating the 3He or 129Xe isotopes, with nuclear spin= , are potentially valuable as biosensors for detection by magnetic resonance, or as tools to monitor the course of fullerene reactions by NMR.[ 11a , 11b , 11c , 11d ] Determining the quantised rotational and translational energy levels of an endohedral species, using inelastic neutron scattering and IR/THz spectroscopy, provides a powerful test of current models of non‐bonding interactions.[ 12a , 12b , 12c ] Endofullerenes in which a trapped molecule exhibits nuclear spin isomerism are of importance for the study of spin isomer interconversion, allotrope enrichment, and have potential applications to chemical and clinical magnetic resonance. [13] Here, nuclear spin conversion in H2@C60[ 14a , 14b , 14c , 14d ] and H2O@C60[ 15a , 15b ] has been studied to date.
To address these needs, a great deal of progress has been made in the synthesis of endofullerenes by multi‐step routes (termed “molecular surgery”) in which a hole is chemically opened in the fullerene, an atom or small molecule enters the cavity, and the opening is then repaired to restore the original carbon cage with the atom or small molecule entrapped. Following the first insertion of He and H2 into an open fullerene by Rubin et al., [16] cage closure was pioneered by Komatsu and Murata who reported syntheses of H2@C60 and 4He@C60 from open fullerene 1, by insertion of H2 directly into 1 (Figure 1), and of 4He into the sulfoxide derivative, under high pressure (4He at 1230 atm, H2 at 800 atm) followed by a series of chemical reactions to re‐form the C60 cage.[ 17a , 17b , 17c ] The method allows larger‐scale synthesis of the noble gas endofullerene than is possible using direct encapsulation, 38 mg of 4He@C60 with 30 % 4He filling was obtained. Similarly, the molecular endofullerene H2@C60 was prepared with 118 mg mass recovery and >90 % H2 filling. Murata and co‐workers also developed the synthesis and orifice‐suture of open‐cage fullerene 2 in their preparation of H2O@C60, [18] and we have since reported an optimised procedure to obtain H2O@C60, and syntheses of H2@C60 and HF@C60 that also rely upon encapsulation of the endohedral molecule by 2.[ 19a , 19b ] Recently, we used an open‐cage fullerene with a larger opening to prepare Ar@C60 and CH4@C60.[ 20a , 20b ]
Figure 1.

Open‐cage fullerenes used in the reported syntheses of 4He@C60, H2@C60, H2O@C60, and HF@C60.
We now report a phosphorus ylid derivative of 2 that can be filled in the solid state and the orifice closed in situ by raising the temperature, enabling efficient synthesis of H2@C60 and 4He@C60, and their expensive isotopologues HD@C60, D2@C60, and 3He@C60, as well as the first “molecular surgery” synthesis of Ne@C60.
Results and Discussion
Our reported synthesis of H2@C60 involved trapping of H2 inside open‐cage fullerene 2. [19b] In this key step, 2 was formed in situ upon heating its hydrate, bis(hemiketal) 3, in solution with 3 Å molecular sieves under a high‐pressure atmosphere of H2, to give H2@2 with 60 % H2 incorporation. Then, heating H2@2 with Ph3P induced the first stage of ring closure, giving H2@5, but also had to be conducted under a high pressure of H2 to avoid loss of the endohedral molecule (Scheme 1). We later showed that HF@3 undergoes dehydration and slow reaction with Ph3P at room temperature, to form a stable phosphorus ylid that is an intermediate in the ring closure leading to 5 but, unfortunately, only undergoes the necessary intramolecular Wittig reaction upon heating—leading to complete loss of HF. The structure of the phosphorus ylid, HF@4, was suggested by close agreement of a calculated 13C NMR spectrum of 4 with the experimental spectrum of the regioisomer shown in Scheme 1, [19a] and we have now confirmed the structure of 4 by X‐ray crystallography (Figure 2). The crystal structure reveals that the C=O bond (1.223 Å) of the carbonyl group adjacent to the phosphorus ylid moiety is longer than those of the remote carbonyl groups (1.209 Å and 1.202 Å), suggesting that the ylid has substantial enolate character which may explain its good stability. [21]
Scheme 1.

Phosphorus ylid 4 is a precursor in reported syntheses of endofullerenes, including H2@C60, and may be prepared from known (bis)hemiketal 3 under mild conditions. Stable, and isolable, 4 is a suitable intermediate for encapsulation and in situ entrapment of an endohedral species A.
Figure 2.

Crystal structure of phosphorus ylid 4. Thermal ellipsoids are shown at the 50 % probability level and hydrogen atoms are omitted for clarity. Deposition Number 1953259 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures. Structure details are reported in Section S3.1 of the Supporting Information.
If entry of H2 or another species into 4 occurred at a temperature lower than that required for the intramolecular Wittig reaction, it would be possible to fill the ylid 4 then induce the Wittig reaction that traps the endohedral species simply by raising the temperature. This would avoid the need to vent gas, add Ph3P reagent, and re‐pressurise as in our synthesis of H2@C60. Comparison of the activation enthalpies for entry of some small atoms and molecules (He, Ne and H2) through the 16‐membered openings of 2 and 4 was made by density functional theory calculations and, in each case, the barrier to entry into 4 was only around 10 kJ mol−1 higher than that for entry into 2 (Table 1).
Table 1.
Binding and activation energies for entry and exit of He, Ne, and H2 into open fullerenes 2 and 4.[a]
|
|
ΔH (binding) [kJ mol−1] |
ΔH ≠ (entry) [kJ mol−1] |
ΔH ≠ (exit) [kJ mol−1] |
|---|---|---|---|
|
H2+2 a⇌H2@2 a |
−22.3 |
50.6 |
72.9 |
|
He+2 a⇌He@2 a |
−10.4 |
31.5 |
41.9 |
|
Ne+2 a⇌Ne@2 a |
−20.1 |
52.8 |
72.9 |
|
H2+4 a⇌H2@4 a |
−21.0 |
60.6 |
81.6 |
|
He+4 a⇌He@4 a |
−10.3 |
39.0 |
49.4 |
|
Ne+4 a⇌Ne@4 a |
−19.2 |
64.6 |
83.8 |
[a] Energies were calculated with density functional theory using the M06‐2X functional and cc‐pVTZ basis set at M06‐2X/cc‐pVDZ geometries. Model structures 2 a and 4 a, in which the 6‐tert‐butylpyridyl groups are replaced by methyl substituents, were used to represent 2 and 4. For full details and references see the Supporting Information.
So, under the same conditions of our solution‐phase H2 filling of 2 (120 atm of H2, 120 °C in 1,2‐dichlorobenzene) [19b] we obtained comparable 62 % H2 encapsulation by 4, implying that closure of 4 does not occur before equilibration of H2 between the cavity and outside. Since the Wittig closure reaction is now unimolecular, a solvent should not be necessary and, indeed, heating solid 4 gave 5 in excellent yield. The closure was significantly slower than in solution, for example, heating 4 for 3 h at 120 °C gave 55 % conversion to 5 in 1,2‐dichlorobenzene‐d 4 solution, but only 4 % conversion in the solid state.
At 160 °C closure of solid 4 was complete in less than 1 h, and heating the solid phosphorus ylid 4 under approximately 500 atm H2 at 160 °C gave H2@5 in 88 % yield with 80 % encapsulation, demonstrating that endohedral incorporation of the gas is faster than ring closure in the solid state.
Open fullerenes have been filled in the solid state before,[ 17b , 17c , 22a , 22b , 22c ] but this is the first example where contraction of the cage opening can be carried out in situ, and there are huge practical advantages. The pressure reactor can be small in volume, allowing higher pressures to be used safely, and the volume of gas needed to achieve the high pressure is much smaller—essential if the gas is expensive and/or rare (such as for encapsulation of 3He, see below). To apply this method of solid‐state filling and in situ closure for large‐scale synthesis, stainless steel pressure reactors with volumes between 1.2–5.0 mL and with pressure ratings of 2400–4000 atm were constructed as part of a bespoke apparatus for compression of gas using a manual intensifier (see Supporting Information). Results reporting the preparation of A@5, where A=Ne and isotopologues of H2 and He, are summarised in Table 2, and described in detail below.
Table 2.
Solid‐state filling of 4 and in situ ring contraction.[a]
|
Entry |
Gas A |
Pressure [atm] |
A@5 filling factor [%][b] |
Yield of isolated A@5 [%][c] |
|---|---|---|---|---|
|
1 |
HD |
520 |
83 |
75 |
|
2 |
D2 |
423 |
73 |
72 |
|
3 |
H2 |
1806 |
95 |
79 |
|
4 |
4He |
2374 |
50 |
84 |
|
5 |
3He |
2315 |
52 |
79 |
|
6 |
Ne |
1742 |
63 |
82 |
[a] All reactions were performed under the stated pressure of gas A, at temperatures in the range 140–186 °C and for 0.75–14 h. For full details see the Supporting Information. [b] Filling factors were calculated by comparison of integrals in the 1H NMR spectrum (A=H2) or by comparison of peak intensities for the filled and empty species in the 13C NMR spectrum (A=Ne, He, HD, or D2). [c] Yield of isolated product, following purification by column chromatography.
H2@C60, HD@C60, and D2@C60
HD@C60 is an interesting isotopologue of H2@C60 as it lacks the nuclear symmetry, and thus selection rules in the rotational energy levels. HD@C60 has been prepared before, and its IR and inelastic neutron scattering (INS) spectra acquired, but as a mixture with H2@C60 and D2@C60.[ 23a , 23b ] Detailed predictions of the variable temperature INS spectra of HD@C60 have been made, [24] but to test them experimentally requires a large sample of pure material. Although the use of solid‐state filling helps overcome the problem of the high cost of the gas, the low pressure (10 atm) at which HD is available limits the pressure our intensifier could generate to 80–85 atm. With an initial pressure of 82 atm HD gas at room temperature, heating with 4 at 160 °C for 2 h gave HD@5 with only 44 % incorporation. The problem was alleviated by cooling the pressure reactor in liquid nitrogen before charging to 119 atm HD. Upon warming to room temperature, a pressure of 420 atm was achieved and at 140 °C, 520 atm. HD@5 was obtained in 75 % yield and with an 83 % filling factor of HD (Table 2, entry 1). Of concern was the known disproportionation of HD to H2+D2 in contact with iron. [25] Under the described conditions, disproportionation was limited to <1 %, but when higher temperatures and pressures were used for the filling/closure it became significant (after overnight exposure of 4 to 800 atm of HD at 180 °C, 35 % disproportion, as measured inside the cage, had occurred).
Similarly, cryogenic charging of the pressure reactor enabled heating of phosphorus ylid 4 under 423 atm of D2 gas, or circa 1800 atm of H2. Respectively, D2@5 was obtained with 73 % filling and H2@5 with 95 % filling, both in good yield (Table 2, entries 2 and 3).
Closure of the cage opening of each compound, HD@5, D2@5, and H2@5, was carried out according to the two‐step procedure previously reported (Scheme 2). [19b] From a single high‐pressure filling experiment we were easily able to prepare 100–150 mg of HD@C60 (83 % filling), D2@C60 (73 % filling), or H2@C60 (approx. 95 % filling), and obtain these endofullerenes on gram scale over several batches.
Scheme 2.

Cage closure of A@5 to prepare A@C60, where A=Ne, 3He, 4He H2, D2, or HD. After reduction to A@6, the second step involves sequential [4+2], retro[4+2], and [2+2+2] cycloaddition. [19b]
4He@C60 and 3He@C60
Helium has much less favourable enthalpy of binding into open fullerene 4 than H2 (Table 1) so a higher pressure is required for good incorporation. Our pressure intensifier has a limit of 1000 atm, but the cryogenic method described above enabled pressures well above 2000 atm to be generated in the reactor at a temperature of 180 °C. Filling and ring contraction of 4 under almost 2400 atm of 4He lead to 50 % helium incorporation in 4He@5 (Table 2, entry 4) while, for comparison, 600 atm gave a filling factor of only 21 %. Synthesis of 4He@C60 with 30 % 4He encapsulation has been reported by Komatsu and Murata, [17b] although the method we describe herein is more easily scalable for preparation of the endofullerene in larger (gram) quantities. The rarity and expense of 3He has previously been an obstacle to the preparation of 3He@C60—only 10 L at STP was available to us. So, a compressor was designed and built to minimise the dead volumes in our existing apparatus, and enabled 3He compression into a 2.4 mL capacity pressure reactor using the cryogenic charging method. This gave a pressure in excess of 2300 atm under an elevated temperature of >170 °C for the intramolecular Wittig reaction, and 3He@5 was obtained with 52 % 3He filling (Table 2, entry 5). Importantly, we were able to recover >99.8 % of the 3He gas back into the apparatus and source cylinder after each experiment. Around 40 % of the unrecovered gas had been encapsulated by the fullerene. Once again, conversion of He@5 to He@C60 was conducted as described in Scheme 2, and >1 g material with circa 50 % filling was prepared for each isotope.
Ne@C60
The DFT calculations given in Table 1 suggested that incorporation of Ne into 2 or 4 should be possible. The synthesis of Ne@C60 was first achieved by filling solid fullerene 2 (under Ne gas at 380 atm, 150 °C, 17 h), and isolation of the product as its hydrate, Ne@3. The filling factor of Ne@3 was estimated to be 15–20 % from the 1H NMR spectrum. Although an attempt to convert Ne@3 to Ne@5 was made using the established method of heating with Ph3P at 120 °C, complete loss of neon occurred. Changing the conditions to PhP(2‐furyl)2 at 60 °C led to isolation of Ne@5 without loss of endohedral neon, and Ne@C60 was subsequently obtained with 16 % filling following the two‐step procedure of Scheme 2. Next, we examined the solid‐state filling and in situ ring contraction of ylid 4 for preparation of Ne@5 with higher incorporation of neon. From exposure of 4 to 600 atm Ne at 160 °C for 2 h, Ne@5 was recovered with approximately 40 % filling estimated from the high resolution ESI+ mass spectrum by comparison of peak intensities for the filled and empty species. Furthermore, using the cryogenic method described earlier a pressure of >1700 atm could be attained, and a filling of 63 % in Ne@5 was achieved (Table 2, entry 6). As expected, Ne@C60 with the same filling factor of 63 % was ultimately obtained, in 43 % yield from Ne@5.
Ne@C60 has been previously prepared only using the direct insertion method, by heating C60 at high temperature and pressure with the gas, and resulting in unreported (small) quantities of material with 0.1–0.3 % Ne incorporation.[ 6 , 7a ] With >0.4 g in hand, we carried out enrichment of Ne@C60 (63 % filled) to a sample with >99.5 % incorporation of neon, by recycling preparative HPLC, and obtained a crystal structure of the nickel(II) octaethylporphyrin/benzene solvate [26] in which the C60 cage is indistinguishable from that of empty C60 (Figure 3). [27] The structure shows the neon atom at the centre of the cage.
Figure 3.
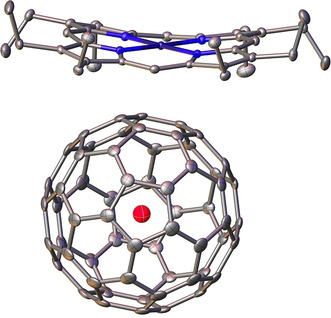
Crystal structure for the nickel(II) octaethylporphyrin/benzene solvate of Ne@C60. Thermal ellipsoids are shown at the 50 % probability level. Hydrogen atoms and benzene molecules are omitted for clarity. Deposition Number 1953465 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures. Structure details are reported in Section S3.2 of the Supporting Information.
In the noble gas@C60 series studied by 13C NMR spectroscopy to date, deshielding of the cage resonance with respect to empty C60 increases with the van der Waals radius of the entrapped atom: He@C60, Δδ=+0.02 ppm; [17b] Ar@C60, Δδ=+0.18 ppm; [20a] Kr@C60, Δδ=+0.39 ppm; [10] and Xe@C60, Δδ=+0.96 ppm. [7b] Shown in Figure 4 a, the 13C NMR resonance of Ne@C60 was measured with a chemical shift of δ C=142.83 ppm in 1,2‐dichlorobenzene‐d 4 at 298 K, deshielded by Δδ=+0.024 ppm relative to empty C60. The absence of a visible peak for empty C60 attests to the high purity of the Ne@C60 sample, and displacement of the 13C peak of empty C60 by 23.6 ppb in the shielding (negative δ) direction, relative to the 13C peak of Ne@C60, was measured from a mixed sample of Ne@C60 and C60. The slight deshielding of the cage 13C resonance in Ne@C60 is within 1.2 ppb of that measured for He@C60 (Δδ=+0.025 ppm relative to C60), and is much less pronounced than in Ar@C60 and the heavier noble gas endofullerenes.
Figure 4.

a) 13C NMR spectrum of Ne@C60 (>99.5 % neon filling), 31–32 mm solution in degassed 1,2‐dichlorobenzene‐d 4 at a field of 176 MHz and 298 K, acquired with 152 transients. There is no visible peak from unfilled C60. b) Expanded view of the base of the Ne@C60 resonance, acquired at 298 K with 680 transients, to show side peaks arising from minor isotopomers with adjacent 13C nuclei that share either a hexagon–pentagon (HP) or hexagon–hexagon (HH) edge.
Figure 4 b shows two side peaks with an intensity ratio of 2:1 that are assigned to minor isotopomers of Ne@C60 that each contain a pair of neighbouring 13C nuclei separated by one bond. [28] Two peaks are observed since there are two types of carbon−carbon bond in C60, either a hexagon–pentagon (HP) or shorter hexagon–hexagon (HH) shared edge, present in a 2:1 ratio, respectively. The shifts of the side peaks relative to the main Ne@C60 resonance correspond to one‐bond isotope shifts of 1ΔHP=12.53±0.01 ppb for the inner peak, and 1ΔHH=19.94±0.03 ppb for the outer peak. Side peaks of this kind were also observed for the helium endofullerenes, 3He@C60 and 4He@C60. [29]
Large‐scale synthesis of phosphorus ylid 4
The methods described above allow the filling of large amounts of open‐cage fullerene 4 with the various endohedral species described. Synthesis of bis(hemiketal) 3, the precursor to 4, is well described by Murata[ 18 , 30 ] but optimisation of the early cage‐opening steps was necessary to more efficiently supply material for our scaled‐up filling procedure (Scheme 3). After opening the C60 cage by reaction with 3,6‐bis(6‐(tert‐butyl)pyridin‐2‐yl)pyridazine, [18] the existing method for expanding the orifice by photo‐oxygenation with singlet oxygen required irradiation of 6 in a mixture of CS2 (flashpoint circa 30 °C, auto‐ignition temperature 100 °C) and 1‐chloronaphthalene, using a 500 W Xenon lamp as O2 is passed through the mixture over 23 h. Although recent improvements in both the reaction yield and safety have resulted from use of an LED light source and replacement of CS2 with CCl4, a reaction time of 48 h is required (from 3 g C60). [30]
Scheme 3.

Optimised cage opening of C60.
We found that use of 2 molar equiv of C60 (relative to the pyridazine reagent) in the first (cycloaddition) step gave a cleaner reaction—presumably by suppressing the formation of multiple addition products. Simple dilution of the product reaction mixture with toluene and direct transfer to the photoreaction vessel enabled the excess remaining C60 to serve as an efficient photosensitizer for the formation of 1O2, and the reaction time of photo‐oxygenation was correspondingly reduced. Indeed, use of a 400 W high‐pressure sodium lamp in a water‐cooled immersion well led to completion of the photochemical reaction in just 1 h (on a scale using 10 g C60). Excess C60 was readily recovered during purification of 5 by column chromatography and the overall yield was 70 %, based on the portion of C60 that was consumed in the reaction (see Supporting Information). Conversion of 5 to bis(hemiketal) 3 is carried out by oxidative cleavage using 4‐methylmorpholine 4‐oxide following the published procedure, [18] and 3 is converted to phosphorus ylid 4 upon treatment with 16 molar equiv Ph3P at 60 °C. [19a] For this final step, an excellent and reproducible yield (85–90 %) of 4 is achieved at multi‐gram scale. Overall, the improved methods allow a 10 g batch of ylid 4 to be prepared in a few days.
Conclusion
Optimised synthesis and solid‐state filling of a phosphorus ylid 4 with H2, He, or Ne, with in situ contraction of the cage opening via an intramolecular Wittig reaction, traps the gas inside. Large amounts of material can be processed in a small pressure reactor, allowing the use of high‐pressure conditions and expensive gases. Further transformations gave up to 1 g each of endofullerenes 3He@C60, 4He@C60, Ne@C60, H2@C60, D2@C60, and HD@C60, which will enable experimental studies of these interesting species, for example using NMR, IR, and inelastic neutron‐scattering spectroscopy. These studies are underway.
The new availability of a large quantity of 3He@C60 offers the potential to extend its use in magnetic resonance imaging, where 3He gas is already used to visualise air passages in the lung by attachment to other species—although the development of methods for hyperpolarisation of the 3He nucleus will be needed to achieve this practical application. The first synthesis of pure HD@C60 will permit the rotational and translational energy levels to be determined by INS, and compared with the established predictions as a test of these theoretical methods.
Synthesis of pure Ne@C60 completes the series of noble gas endofullerenes to be prepared in macroscopic quantities (up to Xe@C60). The absolute chemical shift, and relative endohedral shift, of the cage 13C NMR resonance of Ne@C60 indicates very similar interactions between the noble gas atom and the fullerene cage to that in He@C60.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (EP/M001962/1, EP/P009980/1), including core capability (EP/K039466); and the European Research Council (786707‐FunMagResBeacons). We acknowledge the use of the IRIDIS High Performance Computing Facility and associated support services at the University of Southampton. The authors thank Prof. G. John Langley and Julie Herniman for the APPI mass spectrometry data reported in the Supporting Information.
G. Hoffman, M. C. Walkey, J. Gräsvik, G. R. Bacanu, S. Alom, S. Bloodworth, M. E. Light, M. H. Levitt, R. J. Whitby, Angew. Chem. Int. Ed. 2021, 60, 8960.
References
- 1.
- 1a. Lawler R. G., Nanostruct. Sci. Technol. 2017, 25, 229–263; [Google Scholar]
- 1b. Levitt M. H., Horsewill A. J., Philos. Trans. R. Soc. London Ser. A 2013, 371, 20130124; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Popov A. A., Yang S. F., Dunsch L., Chem. Rev. 2013, 113, 5989–6113. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Albert V. V., Sabin J. R., Harris F. E., Int. J. Quantum Chem. 2007, 107, 3061–3066; [Google Scholar]
- 2b. Bühl M., Patchkovskii S., Thiel W., Chem. Phys. Lett. 1997, 275, 14–18; [Google Scholar]
- 2c. Chaudhuri S. K., Chaudhuri R. K., Mukherjee P. K., Chattopadhyay S. A., J. Chem. Phys. 2017, 147, 034111; [DOI] [PubMed] [Google Scholar]
- 2d. Cimpoesu F., Ito S., Shimotani H., Takagi H., Dragoe N., Phys. Chem. Chem. Phys. 2011, 13, 9609–9615; [DOI] [PubMed] [Google Scholar]
- 2e. Darzynkiewicz R. B., Scuseria G. E., J. Phys. Chem. A 1997, 101, 7141–7144; [Google Scholar]
- 2f. Hesselmann A., Korona T., Phys. Chem. Chem. Phys. 2011, 13, 732–743; [DOI] [PubMed] [Google Scholar]
- 2g. Jiménez-Vázquez H. A., Cross R. J., J. Chem. Phys. 1996, 104, 5589–5593; [Google Scholar]
- 2h. Koner A., Kumar C., Sathyamurthy N., Mol. Phys. 2018, 116, 2728–2735; [Google Scholar]
- 2i. Osuna S., Swart M., Sola M., Chem. Eur. J. 2009, 15, 13111–13123; [DOI] [PubMed] [Google Scholar]
- 2j. Son M. S., Sung Y. K., Chem. Phys. Lett. 1995, 245, 113–118; [Google Scholar]
- 2k. Sure R., Tonner R., Schwerdtfeger P. A., J. Comput. Chem. 2015, 36, 88–96. [DOI] [PubMed] [Google Scholar]
- 3. Jalife S., Arcudia J., Pan S., Merino G., Chem. Sci. 2020, 11, 6642–6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weiske T., Wong T., Kratschmer W., Terlouw J. K., Schwarz H., Angew. Chem. Int. Ed. Engl. 1992, 31, 183–185; [Google Scholar]; Angew. Chem. 1992, 104, 242–244. [Google Scholar]
- 5. Saunders M., Jiménez-Vázquez H. A., Cross R. J., Poreda R. J., Science 1993, 259, 1428–1430. [DOI] [PubMed] [Google Scholar]
- 6. Saunders M., Jiménez-Vázquez H. A., Cross R. J., Mroczkowski S., Gross M. L., Giblin D. E., Poreda R. J., J. Am. Chem. Soc. 1994, 116, 2193–2194. [Google Scholar]
- 7.
- 7a. Cross R. J., Khong A., Saunders M., J. Org. Chem. 2003, 68, 8281–8283; [DOI] [PubMed] [Google Scholar]
- 7b. Syamala M. S., Cross R. J., Saunders M., J. Am. Chem. Soc. 2002, 124, 6216–6219; [DOI] [PubMed] [Google Scholar]
- 7c. Takeda A., Yokoyama Y., Ito S., Miyazaki T., Shimotani H., Yakigaya K., Kakiuchi T., Sawa H., Takagi H., Kitazawa K., Dragoe N., Chem. Commun. 2006, 912–914. [DOI] [PubMed] [Google Scholar]
- 8. Peng R. F., Chu S. J., Huang Y. M., Yu H. J., Wang T. S., Jin B., Fu Y. B., Wang C. R., J. Mater. Chem. 2009, 19, 3602–3605. [Google Scholar]
- 9. DiCamillo B. A., Hettich R. L., Guiochon G., Compton R. N., Saunders M., Jiménez-Vázquez H. A., Khong A., Cross R. J., J. Phys. Chem. 1996, 100, 9197–9201. [Google Scholar]
- 10. Yamamoto K., Saunders M., Khong A., Cross R. J., Grayson M., Gross M. L., Benedetto A. F., Weisman R. B., J. Am. Chem. Soc. 1999, 121, 1591–1596. [Google Scholar]
- 11.
- 11a. Birkett P. R., Buhl M., Khong A., Saunders M., Taylor R., J. Chem. Soc. Perkin Trans. 2 1999, 2037–2039; [Google Scholar]
- 11b. Kupka T., eMagRes 2016, 5, 959–965; [Google Scholar]
- 11c. Saunders M., Cross R. J., Jiménez-Vázquez H. A., Shimshi R., Khong A., Science 1996, 271, 1693–1697; [Google Scholar]
- 11d. Wang G. W., Saunders M., Cross R. J., J. Am. Chem. Soc. 2001, 123, 256–259. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Bačić Z., J. Chem. Phys. 2018, 149, 100901; [DOI] [PubMed] [Google Scholar]
- 12b. Levitt M. H., Philos. Trans. R. Soc. London Ser. A 2013, 371, 20120429; [DOI] [PubMed] [Google Scholar]
- 12c. Xu M. Z., Felker P. M., Bačić Z., Int. Rev. Phys. Chem. 2020, 39, 425–463. [Google Scholar]
- 13. Chen J. Y. C., Li Y. J., Frunzi M., Lei X. G., Murata Y., Lawler R. G., Turro N. J., Philos. Trans. R. Soc. London Ser. A 2013, 371, 20110628. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Chen J. Y. C., Marti A. A., Turro N. J., Komatsu K., Murata Y., Lawler R. G., J. Phys. Chem. B 2010, 114, 14689–14695; [DOI] [PubMed] [Google Scholar]
- 14b. Frunzi M., Jockusch S., Chen J. Y. C., Calderon R. M. K., Lei X. G., Murata Y., Komatsu K., Guldi D. M., Lawler R. G., Turro N. J., J. Am. Chem. Soc. 2011, 133, 14232–14235; [DOI] [PubMed] [Google Scholar]
- 14c. Li Y. J., Lei X., Jockusch S., Chen J. Y. C., Frunzi M., Johnson J. A., Lawler R. G., Murata Y., Murata M., Komatsu K., Turro N. J., J. Am. Chem. Soc. 2010, 132, 4042–4043; [DOI] [PubMed] [Google Scholar]
- 14d. Turro N. J., Marti A. A., Chen J. Y. C., Jockusch S., Lawler R. G., Ruzzi M., Sartori E., Chuang S. C., Komatsu K., Murata Y., J. Am. Chem. Soc. 2008, 130, 10506–10507. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Beduz C., Carravetta M., Chen J. Y. C., Concistrè M., Denning M., Frunzi M., Horsewill A. J., Johannessen O. G., Lawler R., Lei X. G., Levitt M. H., Li Y. J., Mamone S., Murata Y., Nagel U., Nishida T., Ollivier J., Rols S., Room T., Sarkar R., Turro N. J., Yang Y. F., Proc. Natl. Acad. Sci. USA 2012, 109, 12894–12898; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Mamone S., Concistrè M., Carignani E., Meier B., Krachmalnicoff A., Johannessen O. G., Lei X. G., Li Y. J., Denning M., Carravetta M., Goh K., Horsewill A. J., Whitby R. J., Levitt M. H., J. Chem. Phys. 2014, 140, 194306. [DOI] [PubMed] [Google Scholar]
- 16. Rubin Y., Jarrosson T., Wang G. W., Bartberger M. D., Houk K. N., Schick G., Saunders M., Cross R. J., Angew. Chem. Int. Ed. 2001, 40, 1543–1546; [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 1591–1594. [Google Scholar]
- 17.
- 17a. Komatsu K., Murata M., Murata Y., Science 2005, 307, 238–240; [DOI] [PubMed] [Google Scholar]
- 17b. Morinaka Y., Tanabe F., Murata M., Murata Y., Komatsu K., Chem. Commun. 2010, 46, 4532–4534; [DOI] [PubMed] [Google Scholar]
- 17c. Murata M., Murata Y., Komatsu K., J. Am. Chem. Soc. 2006, 128, 8024–8033. [DOI] [PubMed] [Google Scholar]
- 18. Kurotobi K., Murata Y., Science 2011, 333, 613–616. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Krachmalnicoff A., Bounds R., Mamone S., Alom S., Concistrè M., Meier B., Kouřil K., Light M. E., Johnson M. R., Rols S., Horsewill A. J., Shugai A., Nagel U., Rõõm T., Carravetta M., Levitt M. H., Whitby R. J., Nat. Chem. 2016, 8, 953–957; [DOI] [PubMed] [Google Scholar]
- 19b. Krachmalnicoff A., Levitt M. H., Whitby R. J., Chem. Commun. 2014, 50, 13037–13040. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Bloodworth S., Hoffman G., Walkey M. C., Bacanu G. R., Herniman J. M., Levitt M. H., Whitby R. J., Chem. Commun. 2020, 56, 10521–10524; [DOI] [PubMed] [Google Scholar]
- 20b. Bloodworth S., Sitinova G., Alom S., Vidal S., Bacanu G. R., Elliott S. J., Light M. E., Herniman J. M., Langley G. J., Levitt M. H., Whitby R. J., Angew. Chem. Int. Ed. 2019, 58, 5038–5043; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5092–5097. [Google Scholar]
- 21.Similar bond length observations have been made from the single-crystal X-ray structure of a related phosphorus ylid, prepared from another open fullerene with the 1,2-dicarbonyl moiety on the cage opening. See: Hashikawa Y., Okamoto S., Murata Y., Commun. Chem. 2020, 3, 90. [Google Scholar]
- 22.
- 22a. Futagoishi T., Aharen T., Kato T., Kato A., Ihara T., Tada T., Murata M., Wakamiya A., Kageyama H., Kanemitsu Y., Murata Y., Angew. Chem. Int. Ed. 2017, 56, 4261–4265; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4325–4329; [Google Scholar]
- 22b. Hasegawa S., Hashikawa Y., Kato T., Murata Y., Angew. Chem. Int. Ed. 2018, 57, 12804–12808; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12986–12990; [Google Scholar]
- 22c. Murata Y., Murata M., Komatsu K., J. Am. Chem. Soc. 2003, 125, 7152–7153. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Ge M., Nagel U., Huvonen D., Rõõm T., Mamone S., Levitt M. H., Carravetta M., Murata Y., Komatsu K., Lei X. G., Turro N. J., J. Chem. Phys. 2011, 135, 114511; [DOI] [PubMed] [Google Scholar]
- 23b. Horsewill A. J., Rols S., Johnson M. R., Murata Y., Murata M., Komatsu K., Carravetta M., Mamone S., Levitt M. H., Chen J. Y. C., Johnson J. A., Lei X., Turro N. J., Phys. Rev. B 2010, 82, 081410. [Google Scholar]
- 24. Xu M. Z., Ye S. F., Lawler R., Turro N. J., Bačić Z., Philos. Trans. R. Soc. London Ser. A 2013, 371, 20110630. [DOI] [PubMed] [Google Scholar]
- 25. Murata Y., Chuang S. C., Tanabe F., Murata M., Komatsu K., Philos. Trans. R. Soc. London Ser. A 2013, 371, 20110629. [DOI] [PubMed] [Google Scholar]
- 26. Olmstead M. M., Costa D. A., Maitra K., Noll B. C., Phillips S. L., Van Calcar P. M., Balch A. L., J. Am. Chem. Soc. 1999, 121, 7090–7097. [Google Scholar]
- 27. Lee H. M., Olmstead M. M., Suetsuna T., Shimotani H., Dragoe N., Cross R. J., Kitazawa K., Balch A. L., Chem. Commun. 2002, 1352–1353. [DOI] [PubMed] [Google Scholar]
- 28. Bacanu G. R., Hoffman G., Amponsah M., Concistrè M., Whitby R. J., Levitt M. H., Phys. Chem. Chem. Phys. 2020, 22, 11850–11860. [DOI] [PubMed] [Google Scholar]
- 29. Bacanu G. R., Rantaharju J., Hoffman G., Walkey M. C., Bloodworth S., Concistrè M., Whitby R. J., Levitt M. H., J. Am. Chem. Soc. 2020, 142, 16926–16929. [DOI] [PubMed] [Google Scholar]
- 30. Hashikawa Y., Murata M., Wakamiya A., Murata Y., J. Am. Chem. Soc. 2017, 139, 16350–16358. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary