

Abstract
Background
The presence of lupus anticoagulant (LA) is an independent risk factor for thrombosis. This laboratory phenomenon is detected as a phospholipid‐dependent prolongation of the clotting time and is caused by autoantibodies against beta2‐glycoprotein I (β2GPI) or prothrombin. How these autoantibodies cause LA is unclear.
Objective
To elucidate how anti‐β2GPI and anti‐prothrombin antibodies cause the LA phenomenon.
Methods
The effects of monoclonal anti‐β2GPI and anti‐prothrombin antibodies on coagulation were analyzed in plasma and with purified coagulation factors.
Results
Detection of LA caused by anti‐β2GPI or anti‐prothrombin antibodies required the presence of the procofactor factor V (FV) in plasma. LA effect disappeared when FV was replaced by activated FV (FVa), both in a model system and in patient plasma, although differences between anti‐β2GPI and anti‐prothrombin antibodies were observed. Further exploration of the effects of the antibodies on coagulation showed that the anti‐β2GPI antibody attenuated FV activation by activated faxtor X (FXa), whereas the anti‐prothrombin antibody did not. Binding studies showed that β2GPI‐‐antibody complexes directly interacted with FV with high affinity. Anti‐prothrombin complexes caused the LA phenomenon through competition for phospholipid binding sites with coagulation factors as reduced FXa binding to lipospheres was observed with flow cytometry in the presence of these antibodies.
Conclusion
Anti‐β2GPI and anti‐prothrombin antibodies cause LA through different mechanisms of action: While anti‐β2GPI antibodies interfere with FV activation by FXa through a direct interaction with FV, anti‐prothrombin antibodies compete with FXa for phospholipid binding sites. These data provide leads for understanding the paradoxical association between thrombosis and a prolonged clotting time in the antiphospholipid syndrome.
Keywords: antiphospholipid antibodies, beta2‐glycoprotein I, factor V, lupus anticoagulant, prothrombin
Essentials.
How antibodies against β2GPI and prothrombin cause lupus anticoagulant is unclear.
Effects of anti‐β2GPI and prothrombin antibodies on coagulation were compared.
Anti‐β2GPI antibodies bind to FV, interfering with FXa‐mediated FV activation.
Anti‐prothrombin antibodies compete for binding sites on phospholipids.
1. INTRODUCTION
Antiphospholipid syndrome (APS) is a rare autoimmune disease that is defined by recurrent arterial and venous thrombosis or pregnancy‐related complications in combination with the laboratory detection of antiphospholipid (aPL) antibodies. 1 , 2 , 3 aPL are a heterogeneous but overlapping group of autoantibodies, which include anti‐beta2‐glycoprotein I (β2GPI) and anti‐cardiolipin antibodies, and antibodies that prolong the plasma clotting time in vitro in a phospholipid‐dependent manner, a phenomenon known as lupus anticoagulant (LA). 4 The persistent presence of one or more of these aPL in patients with thrombosis or pregnancy morbidity is sufficient for the diagnosis of APS. 1 , 2 , 3
Of all aPL, only the presence of LA is an independent risk factor for thrombosis and adverse pregnancy outcomes. 5 , 6 Although the laboratory tests for LA do not allow identification of the responsible antibodies, LA has been attributed to autoantibodies against the phospholipid‐binding plasma proteins β2GPI or prothrombin. 7 , 8 , 9 Antibodies causing LA induce an in vitro prolongation of the clotting time, which is corrected after the addition of an excess of phospholipids. 2 The clear association of LA with thrombosis represents a paradox, as prolonged clotting times are usually indicative of a bleeding tendency. In order to solve this paradox, we need to understand how these autoantibodies cause LA.
Up to now, the underlying mechanism of LA caused by aPL has not been elucidated. The general assumption is that aPL induce LA through competition with coagulation factors for binding sites on anionic phospholipids. Addition of an excess of phospholipids would dilute the aPL, thereby lowering the likelihood that they will compete with clotting factors. 10 , 11 However, this explanation is too simple for the LA phenomenon. Although competition might play a role for anti‐prothrombin antibodies, this is less likely for anti‐β2GPI antibodies, because the affinity of β2GPI for anionic phospholipids at physiological calcium ion concentrations is much lower than the affinity of the vitamin K‐dependent coagulation factors. While antibody binding enhances the avidity of β2GPI substantially, this is unlikely to cause competition with the high affinity binding of the vitamin K‐dependent coagulation factors. 11 , 12
To obtain a better understanding of how anti‐β2GPI and anti‐prothrombin antibodies cause LA, we need to understand the minimal requirements to detect LA. LA is detected with phospholipid‐sensitive coagulation assays, including the dilute Russell's viper venom time (dRVVT). This test is based on the activation of coagulation factor X (FX) by the venom of the Russell's viper, thereby bypassing the intrinsic and extrinsic coagulation pathways. 1 , 2 This shows that antibodies causing LA have an effect on the common pathway of coagulation. Coagulation reactions initiated with Russell's viper venom involve three phospholipid‐dependent reactions: The activation of coagulation factor V (FV) by activated FX (FXa), the assembly of the prothrombinase complex, and the activation of prothrombin by the prothrombinase complex. 13 Here, we investigated the mode of action of anti‐β2GPI and anti‐prothrombin antibodies.
2. MATERIALS AND METHODS
2.1. Materials
FV‐depleted plasma, affinity purified sheep anti‐human FV, and factor VIII (FVIII) polyclonal antibodies were from Affinity Biologicals. Phospholipids (Coagulation reagent I) were from Avanti polar lipids. Silica microspheres of 1.5 µm nominal diameter were from Bangs Laboratories. Alexa Fluor 546 streptavidin was from Invitrogen. Horseradish peroxidase (HRP)‐conjugated rabbit anti‐goat IgG and goat anti‐rabbit IgG were from DAKO. Rabbit anti‐FX polyclonal antibody was from Abcam. Human prothrombin and α‐thrombin were from Enzyme Research Laboratories. Human FV, FVa, FX, and FXa; biotinylated EGR‐chloromethylketone; and monoclonal anti‐FV antibody 5146 against FV heavy chain were from Haematologic Technologies. Recombinant human FVIII was obtained as pharmaceutical formulation from Advate, Baxalta. QUANTA Lite® aPS/PT IgG/IgM was from Inova Diagnostics. IBL Cardiolipin IgG/IgM ELISA kit was from IBL International GmbH. IMTEC β2GPI antibodies IgG/IgM ELISA kit was from Clindia Benelux BV. Chromogenic substrate S2238 was from Instrumentation Laboratories. Pefabloc TH was from Pentapharm. dRVVT reagents (screen and confirm) were from Stago. Protifar was from Nutricia). Immobilon‐FL Polyvinylidene fluoride membrane was from Merck Biochemicals. nuPAGE Tris‐Acetate 3–8% gel was from Thermo Fisher Scientific. Factor V activator enzyme (RVV‐V) was from ImmBiomed. Human β2GPI was purified from fresh citrated plasma as described previously; 9 purity was checked with SDS‐PAGE and Coomassie blue staining. All β2GPI‐preparations yielded a single band of 50 kDa and were >99% pure. Pooled normal plasma (PNP) was prepared from at least 250 healthy hospital workers. Monoclonal anti‐β2GPI antibody 3B7 and anti‐prothrombin antibody 3B1 were produced in‐house and purified from hybridoma culture medium with protein G and protein A Sepharose, respectively. Monoclonal anti‐β2GPI antibody 27G7 and monoclonal anti‐prothrombin antibody 28F4 were prepared as previously described. 14 Prothrombin active site mutant (FIIi S525A) was produced in human embryonic kidney 293 cells in the presence of Vitamin K1 and purified from serum‐free medium with a calcium‐dependent monoclonal antibody against prothrombin.
2.2. LA‐positive patient plasma samples
Residual anonymous plasma samples from APS screening diagnostics at the University Medical Center (UMC) Utrecht were used. These samples were derived from thrombotic APS patients and had been tested for LA using dRVTT and activated partial thromboplastin time (APTT). Samples were mixed with PNP to correct for coagulation factor deficiencies. Anti‐β2GPI antibodies were determined with the IMTEC β2GPI antibodies IgG/IgM ELISA kit and anti‐cardiolipin antibodies with the IBL Cardiolipin IgG/IgM ELISA kit. Samples were deemed positive for anti‐β2GPI antibodies when they were positive for anti‐β2GPI antibodies, anti‐cardiolipin antibodies, or both. Anti‐prothrombin antibodies were detected using a homemade ELISA as described before, 15 following the guidance from the Scientific Standardization Committee on Lupus Anticoagulant/Antiphospholipid Antibodies of the ISTH. 16 Anti‐phosphatidylserine‐prothrombin complex (aPS/PT) IgG and IgM were measured with a commercial kit according to the instructions of the manufacturer.
2.3. Coagulation assays
All coagulation assays were performed on an MC10‐plus coagulometer at 37°C. LA activity was measured with dRVTT screen and confirm reagents. PNP was incubated with different concentrations of monoclonal anti‐β2GPI antibodies (3B7 or 27G7) or anti‐prothrombin antibodies (28F4 or 3B1) for 2 minutes at 37°C prior to the addition of dRVVT screen and confirm reagents. Normalized LA (nLA) ratios were expressed as: (screen clotting time with antibody/screen clotting time without antibody)/(confirm clotting time with antibody/confirm clotting time without antibody). A nLA‐ratio >1.2 was considered clinically relevant.
2.4. Preparation of phospholipid vesicles
Phospholipid vesicles were prepared from coagulation reagent I, a mixture of 1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine (DOPE), 1,2‐dioleoyl‐sn‐glycero‐3‐phospho‐L‐serine (DOPS), and 1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC) with a 3:2:5 molar lipid ratio. Dry lipid films were made by evaporation and were hydrated with HEPES‐buffered saline (HBS): 10 mM HEPES, 150 mM NaCl, pH 7.4. Phospholipid suspensions were incubated in a sonication water bath to yield unilamellar vesicles. Phospholipid concentration was determined with a phosphate quantitation assay as described. 17 Phospholipids were stored at −80 °C. Prior to each experiment, phospholipids were incubated at 37°C in a sonication water bath to ensure homogeneity and presence of unilamellar vesicles.
2.5. FXa‐initiated coagulation tests
FV‐depleted plasma was incubated with 2.5 nM FV or FVa supplemented with or without 100 µg/mL anti‐β2GPI antibody (3B7) or anti‐prothrombin antibody (28F4). Coagulation was initiated with 250 pM FXa in HBS, 0.1% bovine serum albumin (BSA), 8.3 mM CaCl2 containing either 4 or 500 µM phospholipids. In some experiments, coagulation was initiated with preformed prothrombinase complex: 250 pM FXa and 2.5 nM FVa in HBS, 0.1% BSA, 8.3 mM CaCl2 containing either 4 or 500 µM phospholipids was incubated for 10 minutes at 37 °C and added to FV‐depleted plasma with or without antibodies.
2.6. FV activation with RVV‐V
LA‐positive patient plasma mixed 1:1 with PNP, and PNP supplemented with either 100 µg/mL anti‐β2GPI (3B7) or anti‐prothrombin (28F4) antibody, were incubated with the FV‐activating snake venom enzyme RVV‐V (5 units/mL plasma) for 3 minutes at 37°C. Coagulation was initiated using dRVVT screen and confirm reagents.
2.7. Factor Xa‐mediated factor V activation
The effect of FXa‐mediated FV activation in the presence of antibody‐β2GPI or antibody‐prothrombin complexes was performed as described. 18 Briefly, 0.5 nM FV was activated with 0.1 nM FXa in coagulation buffer (10 mM HEPES, 140 mM NaCl, 5 mM CaCl2, 0.1% BSA, pH 7.5) and either 4 or 500 µM phospholipids with or without 50 µg/mL β2GPI or prothrombin active site mutant (prothrombin‐S525A; FIIi), and 12.5–100 µg/mL 3B7 or 100 µg/mL 28F4 for 10 minutes at 37 °C. Subsequently, 200 µL (to β2GPI samples) or 400 µL (to prothrombin samples) coagulation buffer was added containing 0.5 nM FXa, 0.5 µM prothrombin, 10 µM phospholipids, 1 µM Pefablock TH, and incubated for 3 minutes at 37 °C. β2GPI, prothrombin, 3B7, or 28F4 were supplemented to FVa samples generated in the absence of these proteins to correct for confounding effects of these antibodies on prothrombinase reaction rates. Prothrombinase reactions were stopped by transferring 50 µL samples to 150 µL stop‐buffer (10 mM HEPES, 175 mM NaCl, 20 mM EDTA, 0.1% BSA, pH 7.7). Thrombin formation was measured by the addition of 100 µL of this mixture to 100 µL 1 mM chromogenic substrate S2238. Thrombin concentrations were deduced from a thrombin calibration curve with known concentrations. FVa concentration was calculated assuming a turnover number for prothrombinase of 6000 mole thrombin per minute per mole FXa‐FVa. 18
2.8. Western blot of FV
For visualization of FV activation with western blot, 10 nM FV was activated with 0.1 nM FXa in 10 mM HEPES, 16.6 mM CaCl2, 175 mM NaCl, 0.1% BSA, pH 7.7, containing 4 µM phospholipids and 50 µg/ml 3B7 with or without 1 µM β2GPI at 37°C. At the indicated time points, samples were taken, mixed with reducing Laemmli sample buffer, and put on ice. Samples were heated at 95°C for 5 minutes, subjected to SDS‐PAGE on a nuPAGE Tris‐Acetate 3–8% gel, blotted overnight at 4°C onto Immobilon‐FL Polyvinylidene fluoride membrane, blocked with Odyssey blocking buffer, and incubated with mouse anti‐FV heavy chain antibody (4 µg/mL) in Odyssey blocking buffer. After extensive washing, blots were incubated with IRDye680‐conjugated donkey anti‐mouse antibodies in Odyssey blocking buffer and rinsed thoroughly. Bands were visualized on an Odyssey imager.
2.9. Binding of antibody‐β2GPI and antibody‐prothrombin complexes to coagulation factors V, Va, VIII, X, and Xa
NUNC maxisorp microtiter plates were coated with 10 µg/mL anti‐β2GPI antibody (3B7) or anti‐prothrombin antibody (28F4) in 50 mM carbonate buffer, pH 9.6, overnight at 4°C. Plates were blocked with 1% milk powder (Protifar) in HBS and incubated with 10 µg/mL β2GPI or prothrombin in blocking buffer. After washing with 0.1% Tween in HBS, plates were incubated with several dilutions of PNP. Plates were washed and incubated with either sheep anti‐FV or sheep anti‐FVIII antibodies (1:5000 in blocking buffer), followed by washing and incubation with HRP‐conjugated rabbit anti‐goat antibodies (1:5000 in blocking buffer). After a final washing step, 100 µL TMB was added to each well and colorimetric reactions were stopped by addition of 50 µL 0.2 M H2SO4. Plates were read at 450 nm in a Spectramax M2e device (Molecular Devices). Binding of anti‐β2GPI complexes to purified coagulation factors was also studied in which PNP was substituted with FV, FVa, FX, FXa, and FVIII diluted in HBS with 0.1% Protifar and 8 mM CaCl2. FX and FXa were detected with rabbit anti‐factor X/Xa antibodies, followed by incubation with HRP‐conjugated goat anti‐rabbit IgG (1:2000).
2.10. Flow cytometry binding studies on phospholipids
Competition of antibody‐β2GPI or antibody‐prothrombin complexes and FXa on phospholipid binding sites was studied on lipospheres as described. 19 , 20 In short, silica microspheres of 1.5 µm nominal diameter (5 × 109 microspheres/mL) were cleaned and incubated with 2 mM sonicated PS:PC:PE (5:3:2) vesicles for 5 minutes in a sonification water bath, followed by a 30‐minute incubation without sonification. Formed lipospheres were washed five times by centrifuging 3 minutes at 80xg and resuspending them in TBS/BSA/PLV buffer (0.15 M NaCl, 50 mM Tris‐HCl, 0.1% fatty acid free bovine albumin and 10 µM sonicated PC vesicles, pH 7.4). Washed lipospheres were incubated with either 2 µM β2GPI or prothrombin and different concentrations of 3B7 or 28F4 for 10 minutes at 37oC in TBS/BSA/PLV buffer supplemented with biotinylated‐FXa 21 and either 5 mM CaCl2 or 20 mM EDTA. Next, beads were incubated with 5 µg/mL Alexa Fluor 546 streptavidin for 10 minutes in dark. FXa binding to lipospheres was assessed with flow cytometry using a BD FACS Canto II.
2.11. Statistical analyses
Results are reported as mean and standard deviation and all data analyses were performed with GraphPad Prism 8.3 software. Differences between conditions were analyzed with Student’s t‐tests, or Mann‐Whitney tests for non‐Gaussian distributed data. P‐values <0.05 were considered statistically significant.
3. RESULTS
3.1. The detection of the lupus anticoagulant phenomenon depends on the presence of factor V
To identify the phospholipid‐dependent reactions in the common pathway in which aPL interfere, clotting was initiated with FXa or the prothrombinase complex in FV‐depleted plasma supplemented with either FV or activated FV (FVa) in the presence or absence of aPL. A mouse monoclonal antibody (mAb) against β2GPI (3B7) and a mAb against prothrombin (28F4) were used as models for human aPL. In the presence of FV, both the anti‐β2GPI (Figure 1A) and the anti‐prothrombin (Figure 1B) mAb caused an increase in FXa‐initiated clotting time at limiting phospholipid concentrations (4 µM), resulting in an increased clotting time ratio (calculated between clotting times with and without mAbs). The addition of an excess of phospholipids (500 µM) normalized the FXa‐initiated clotting times, resulting in an nLA ratio of 2.14 ± 0.74 for 3B7 and 2.19 ± 0.57 for 28F4, which is indicative of the LA phenomenon. However, the anti‐β2GPI mAb did not prolong FXa‐initiated clotting times in the presence of FVa (nLA ratio of 1.10 ± 0.15), or when coagulation was initiated with preformed prothrombinase complex (nLA ratio 1.01 ± 0.04). Contrary to the anti‐β2GPI mAb, the anti‐prothrombin mAb still caused a prolonged clotting time in FV‐depleted plasma supplemented with FVa or when coagulation was initiated with preformed prothrombinase complex, but this prolongation persisted in the presence of an excess of phospholipids. As a consequence, the LA phenomenon was not detected under these conditions (nLA ratio of 1.15 ± 0.15 and 1.12 ± 0.08, respectively). Combined, these data suggest that anti‐β2GPI interfere with FV activation, but that anti‐prothrombin antibodies interfere with either FV activation, assembly of the prothrombinase complex, prothrombin conversion, or a combination of these processes.
Figure 1.
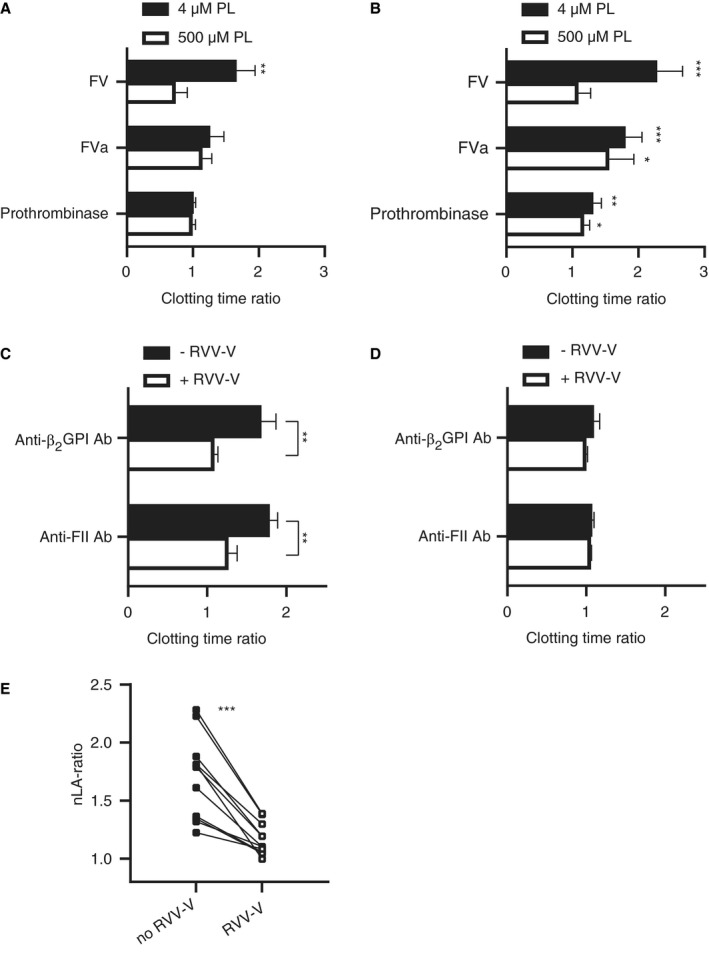
Lupus anticoagulant induced by anti‐beta2‐glycoprotein I (β2GPI) and anti‐prothrombin antibodies requires factor V (FV). A, B, FV‐depleted plasma was reconstituted with FV, activated FV (FVa), or preformed prothrombinase complex in the presence or absence of monoclonal anti‐β2GPI (3B7) (A) or anti‐prothrombin (anti‐FII; 28F4; B) antibodies. Clotting was initiated with activated factor X (FXa) and CaCl2 at 4 µM or 500 µM phospholipids. Clotting time ratios at low (4 µM) and high (500 µM) phospholipids were calculated between clotting times with and without monoclonal antibodies (mAbs; n = 4–5). Significant differences were determined between clotting times with and without mAbs using an unpaired Student’s t‐test. C, D, Pooled normal plasma was supplemented with the FV‐activating snake venom enzyme RVV‐V in the presence or absence of mAbs. Clotting times were obtained using dilute Russell's viper venom time (dRVVT) screen (C) and confirm (D) reagents and ratios were calculated between clotting times with and without mAbs (n = 3). Significant differences were determined between clotting time ratios with and without RVV‐V using an unpaired Student’s t‐test. E, Similar experiments were performed with mixed lupus anticoagulant (LA)‐positive patients plasma (n = 11) positive for anti‐β2GPI (n = 9) or anti‐prothrombin antibodies (n = 11). Results are shown as individual LA ratios of patients’ plasma incubated with or without RVV‐V and were calculated as described in the Method section. Connected lines represent paired data. Significant differences were determined using a paired sample t‐test. *P‐value<0.05, **P‐value<0.01, ***P‐value<0.005
To confirm that detection of the LA phenomenon requires the presence of the procofactor FV in plasma, clotting tests were performed with dRVTT screen (low phospholipid concentrations) and confirm (high phospholipid concentrations) reagents in PNP, before and after treatment with the FV‐activating snake venom enzyme RVV‐V. As expected, addition of the anti‐β2GPI or anti‐prothrombin mAb to PNP caused a prolonged clotting time measured with screen reagent (Figure 1C), but not when clotting time was measured with confirm reagent (Figure 1D; nLA ratio of 1.54 ± 0.23 for 3B7 and 1.67 ± 0.09 for 28F4). Activation of FV with RVV‐V attenuated the effects of the anti‐β2GPI and the anti‐prothrombin antibodies on the screen clotting time (Figure 1C), supporting the observations in FV‐depleted plasma (nLA ratio of 1.09 ± 0.03 for 3B7 and 1.20 ± 0.11 for 28F4). Next, we assessed whether FV activation interferes with LA detection in plasma from APS patients (n = 11). Hereto, dRVVT clotting times were determined in plasma from nine patients with LA that had IgM or IgG anti‐β2GPI and IgM or IgG anti‐prothrombin antibodies (of which five patients were positive for anti‐cardiolipin, anti‐β2GPI, and anti‐prothrombin antibodies), and two patients with LA that only had IgM or IgG anti‐prothrombin antibodies (Figure 1E). Activation of FV with RVV‐V strongly reduced the strength of the LA phenomenon in patients with anti‐β2GPI or anti‐prothrombin antibodies. No differences in nLA ratio were observed between the two plasma samples of patients that only had anti‐prothrombin antibodies and plasma samples of patients that had both anti‐β2GPI or anti‐prothrombin antibodies.
3.2. Anti‐β2GPI antibodies but not anti‐prothrombin antibodies, attenuate FXa‐dependent FV activation
To determine whether anti‐β2GPI and anti‐prothrombin antibodies interfere with FXa‐mediated FV activation, we activated purified FV with FXa in the presence or absence of the model anti‐β2GPI or anti‐prothrombin mAbs and their antigens. β2GPI or 3B7 alone had no effect on FV activation at 4 μM phospholipids, but 3B7 inhibited FV activation in a dose‐dependent manner in the presence of β2GPI (Figure 2A). Consistent with data obtained in clotting experiments, 3B7‐β2GPI complexes had no effect on FXa‐mediated FV activation in the presence of 500 μM phospholipids (Figure 2B). In contrast to 3B7‐β2GPI complexes, 28F4‐prothrombin complexes had no effect on FXa‐dependent FV activation at limiting phospholipid concentrations (Figure 2C). Analysis of FV cleavage products over time indicated a delay in formation of the 105 kDa band of the FVa‐heavy chain in the presence of both anti‐β2GPI antibody and β2GPI, confirming the inhibitory effect of these immune complexes on FV activation (Figure 3A‐C). These results suggest that anti‐β2GPI antibodies cause the LA phenomenon through interference with FV activation, while anti‐prothrombin antibodies affect the common pathway through a different mechanism.
Figure 2.
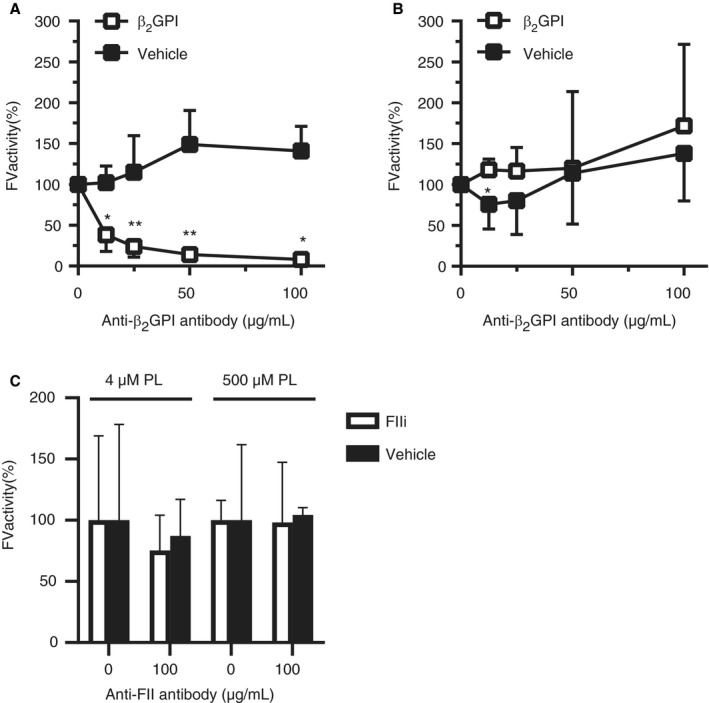
Antibody‐beta2‐glycoprotein I (β2GPI) but not antibody‐prothrombin complexes attenuate activated factor X (FXa)‐dependent factor V (FV) activation. A, B, FV was activated with FXa at either 4 (A) or 500 (B) µM phospholipids, with or without β2GPI and a monoclonal anti‐β2GPI antibody (3B7). C, FV was activated with FXa at either 4 or 500 µM phospholipids, with or without prothrombin active site mutant (FIIi) or a monoclonal anti‐prothrombin antibody (28F4). Data are expressed as relative FV activity compared to the conditions without antibodies (n = 3–5). Significant differences were determined between samples with and without β2GPI or FIIi using a Mann‐Whitney test. *P‐value<0.05, **P‐value<0.01
Figure 3.
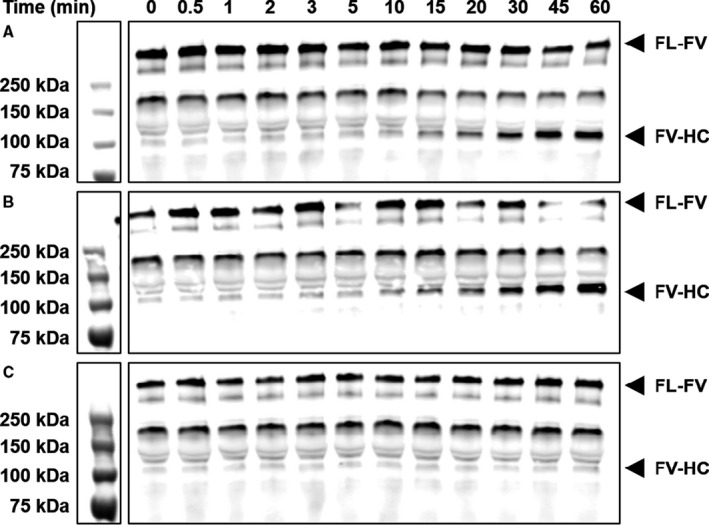
Antibody‐beta2‐glycoprotein I (β2GPI) complexes delay activated factor X (FXa)‐dependent factor V (FV) activation over time. 10 nM FV was activated with 0.1 nM FXa in the presence of phospholipids and CaCl2 and anti‐β2GPI antibody (3B7; A), β2GPI (B), or both (C) at 37°C. Samples were taken at the indicated time points and subjected to SDS‐PAGE and western blot
3.3. Antibody‐β2GPI complexes bind to FV with high affinity
The observation that anti‐β2GPI antibodies interfere with FXa‐dependent FV activation suggests a direct interaction between antibody‐β2GPI complexes and the individual components of the prothrombinase complex: FV(a) or FXa. Therefore, the interaction between antibody‐β2GPI complexes and FV, FVa, FX, and FXa was investigated with solid phase binding assays. Binding experiments with immobilized 3B7‐β2GPI complexes and FV and FVa showed that both proteins interacted with 3B7‐β2GPI complexes with high affinity: The KD was 17 ± 3 nM for FV and 46 ± 10 nM for FVa (Figure 4A). No binding was observed on immobilized 3B7 in the absence of β2GPI, or between 3B7‐β2GPI complexes and FX or FXa (data not shown). Interestingly, FVIII, which is homologous to FV, also bound with high affinity (KD was 2.7 ± 0.3 nM) to 3B7‐β2GPI complexes. However, as plasma concentrations of FVIII are estimated to range from 0.3 to 1 nM, this interaction is not likely to be meaningful under physiological conditions. This was supported by binding experiments in plasma: Whereas plasma FV (20–30 nM) interacts with immobilized 3B7‐β2GPI complexes, FVIII does not (Figure 4B). In addition, no interaction was found between 28F4‐prothrombin complexes and FV (data not shown).
Figure 4.
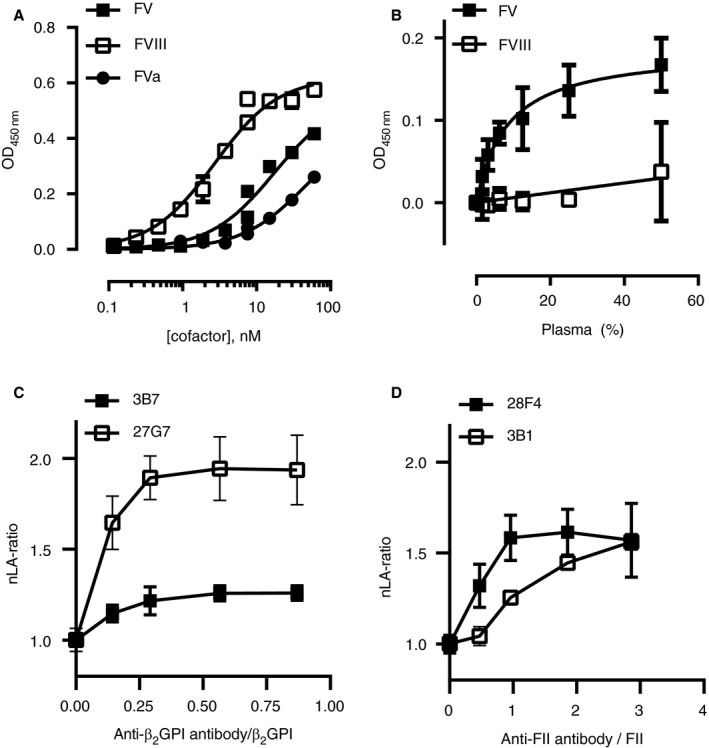
Antibody‐beta2‐glycoprotein I (β2GPI) complexes bind to factor V (FV) with high affinity and have an indirect effect on lupus anticoagulant. 3B7 was immobilized on a microtiter plate and saturated with β2GPI. A, Plates were incubated with purified FV, factor VIII (FVIII), or activated FV (FVa) at the indicated concentrations and binding was assessed with polyclonal antibodies (n = 2). B, Plates were incubated with several dilutions of pooled normal plasma and binding of FV or FVIII was assessed (n = 3). Data were analyzed with non‐linear regression assuming a 1:1 interaction. C, D, Normalized lupus anticoagulant (nLA) ratios were obtained with dilute Russell's viper venom time (dRVVT) screen and confirm reagents in pooled normal plasma supplemented with increasing concentrations of monoclonal anti‐β2GPI (3B7 or 27G7; C) or anti‐prothrombin (28F4 or 3B1; D) antibodies (25, 50, 100, and 150 µg/mL; n = 3). Antibody concentrations were expressed relative to the concentrations of β2GPI or prothrombin in plasma
As FV has a much lower plasma concentration than β2GPI (3–4 μM), we investigated whether the stoichiometry of the antibody‐antigen complexes supports indirect inhibition of coagulation through inhibition of FV activation (Figure 4C,D). While the effect of two different anti‐β2GPI mAbs (3B7 and 27G7) on the LA‐ratio reached a plateau phase long before β2GPI was saturated, two anti‐prothrombin mAbs (28F4 and 3B1) had a maximal effect on the LA ratio at saturating prothrombin concentrations, or higher. These data further support a role for FV in LA induced by anti‐β2GPI antibodies.
3.4. Antibody‐prothrombin complexes compete with coagulation factors on phospholipid binding sites
Competition for binding sites on phospholipids could explain the effects of anti‐prothrombin antibodies on clotting. Moreover, the direct interaction between β2GPI‐antibody complexes and FV cannot rule out contribution of competition for phospholipid binding sites to the LA phenomenon induced by anti‐β2GPI antibodies. To investigate if competition for phospholipid binding sites contributes to the LA phenomenon induced by aPL, we labeled FXa in its active site with biotinylated EGR‐chloromethylketone and assessed its binding to silica microspheres coated with PS:PC:PE membranes (lipospheres) with flow cytometry (Figure 5). Binding of FXa to lipospheres was Ca2+‐ and phospholipid‐dependent, as no binding to lipospheres was observed in the presence of EDTA, or when the silica beads were coated with PC only (data not shown). Antibody‐prothrombin complexes caused a strong dose‐dependent reduction of FXa binding to lipospheres, with a 66 ± 17% reduction in FXa binding at 100 μg/mL mAb. In contrast, the anti‐β2GPI mAb caused a 31 ± 19% reduction of FXa binding at the same concentration. As the anti‐β2GPI antibody almost completely attenuated FXa‐mediated FV activation at this concentration, these data suggest that competition for phospholipid binding sites plays a minor role in the attenuation of FV activation by anti‐β2GPI antibodies.
Figure 5.
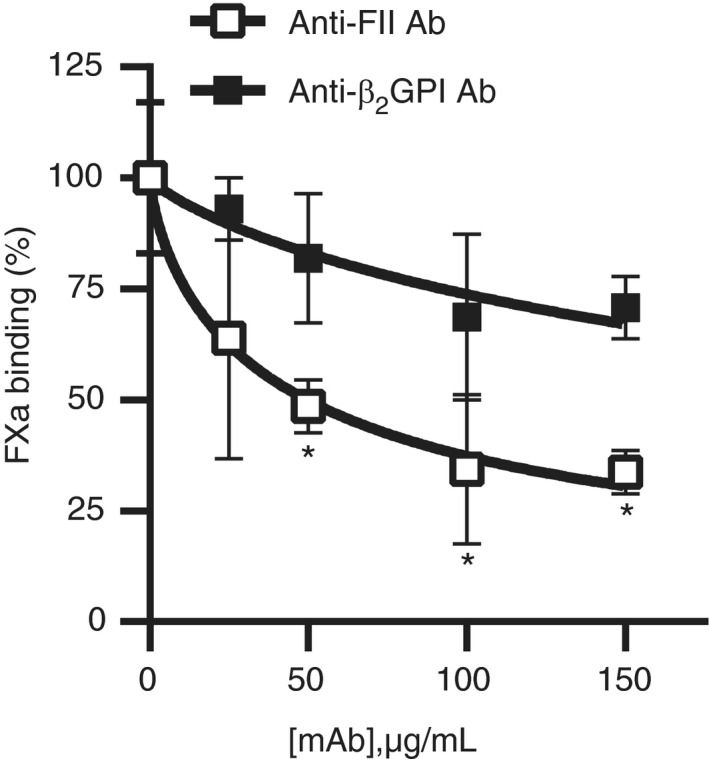
Antibody‐prothrombin complexes compete with coagulation factors on phospholipid binding sites. Biotinylated activated factor X (FXa) was incubated with lipospheres and competition with antibody‐beta2‐glycoprotein I (β2GPI) or antibody‐prothrombin complexes was assessed with flow cytometry. Significant differences were determined between baseline FXa binding at 0 µg/mL and other concentrations of monoclonal antibodies using an unpaired Student’s t‐test (n = 3). *P‐value<0.05
4. DISCUSSION
Here we provide evidence that anti‐β2GPI and anti‐prothrombin antibodies cause the LA phenomenon through different mechanisms of action: While antibody‐β2GPI complexes bind to FV and interfere with FXa‐dependent FV activation, anti‐prothrombin antibodies prolong clotting times through competition with coagulation factors on phospholipid binding sites.
Several studies showed that antibodies against β2GPI and prothrombin enhance the binding of these proteins to negatively charged phospholipids. 10 , 11 , 22 , 23 , 24 , 25 , 26 Consequently, it has always been assumed that aPL induce LA through competition with coagulation factors for binding sites on anionic phospholipids. However, only anti‐prothrombin antibodies have been shown to actually compete with coagulation factors for phospholipid binding sites. 27 Data presented here indicate that competition with coagulation factors can explain LA caused by anti‐prothrombin antibodies, but not LA caused by anti‐β2GPI antibodies.
Our data indicate that anti‐β2GPI antibodies inhibit the initiation phase of the coagulation reaction. During the initiation of coagulation, the procofactor FV is activated through limited proteolysis by FXa, leading to assembly of the prothrombinase complex and subsequent thrombin generation. 28 The activation of FV by FXa is much less efficient than the activation of FV by thrombin, but minute amounts of FVa are sufficient for substantial thrombin formation. By binding to FV, antibody‐β2GPI complexes interfere with the activation of FV by FXa. Although our binding experiments show that antibody‐β2GPI complexes bind to both purified coagulation FV and FVa, binding to FVa is much weaker than binding to FV, which explains why anti‐β2GPI antibodies do not prolong the clotting time when the prothrombinase complex has already been formed. The inhibitory effect of anti‐β2GPI antibodies on the clotting time is only apparent at limiting phospholipid concentrations, not when the phospholipid concentration is high. It has been shown that β2GPI cannot bind to phospholipids and the lipoprotein receptor ApoER2 simultaneously. 29 We hypothesize that a similar mechanism applies here. When phospholipids are present in excess, β2GPI distributes over the entire phospholipid surface, lowering the likelihood that β2GPI binds FV.
How β2GPI interacts with FV is currently unclear. Our data show that antibody‐β2GPI complexes bind to both FV and FVa, suggesting that the binding site for β2GPI resides on either the light or the heavy chain of FV. However, as the affinity for FV differs from the affinity for FVa, we cannot completely rule out that the B‐domain of FV contributes to the binding site for β2GPI. Surprisingly, we found that antibody‐β2GPI complexes also bind to the FV‐homologue FVIII, although our data suggest the strength of this interaction is not relevant under physiological conditions. This suggests that the epitope on FV to which β2GPI binds is conserved in both proteins. Further experiments are required to identify the exact binding epitope.
FV activation strongly reduced the inhibitory effect of anti‐prothrombin antibody on clotting at limiting phospholipid concentrations, while the effect of the anti‐β2GPI antibodies was completely abrogated. The observation that most plasma samples from LA‐positive patients showed incomplete correction of LA after FV activation with RVV‐V, suggests that anti‐prothrombin antibodies contribute substantially to the LA phenomenon. This is in line with a recent report showing that the LA phenomenon in plasma from LA‐positive patients with antibodies against cardiolipin, β2GPI, and prothrombin is large attributable to the anti‐prothrombin antibodies. 30 Whether anti‐prothrombin antibodies are also responsible for the prothrombotic phenotype associated with LA remains to be determined. In our study, we did not investigate how antibodies directed against other phospholipid‐binding proteins cause LA. One can assume that these antibodies cause LA through similar mechanisms as described here. However, it remains to be determined whether these antibodies contribute to thrombosis observed in APS patients.
The data presented here were obtained with model mAbs against β2GPI (3B7) and prothrombin (28F4). As the majority of the experiments are performed with only one mAb against β2GPI and one mAb against prothrombin, it remains to be determined to what extent these results can be applied to other mAbs directed against β2GPI or prothrombin and whether the polyclonal heterogeneous antibody population found in patients with APS induce LA in a similar manner. In support of the applicability of our data to patients with a β2GPI‐dependent LA, the mAb we used binds to the first domain of β2GPI, similar to pathogenic aPL antibodies. 31 , 32 , 33 Moreover, the mechanism described here does not seem to be limited to mAbs, as similar results were obtained in plasma from 11 patients positive for LA and anti‐β2GPI or anti‐prothrombin antibodies treated with RVV‐V.
Despite the clear inhibitory effects of anti‐β2GPI and anti‐prothrombin antibodies on coagulation in vitro, patients with APS suffer from thrombosis. The central role of FV in the LA phenomenon caused by anti‐β2GPI antibodies might explain this increased risk of thrombosis as FV has a dual role in coagulation. It has procoagulant properties as the cofactor for FXa in the prothrombinase complex during thrombin formation, and anticoagulant properties as the binding site for tissue factor pathway inhibitor (TFPI)‐α during prothrombinase inhibition 34 , 35 and, together with protein S, as the cofactor for activated protein C (APC) during FVIIIa inactivation. 36 One can hypothesize that the binding of antibody‐β2GPI complexes to FV impairs its function in its anticoagulant role. However, this does not explain thrombosis caused by anti‐prothrombin antibodies. The increased affinity of prothrombin for phospholipids in the presence of its antibody might result in an accumulation of prothrombin at the site of vascular injury and thereby enhances platelet activation and a pro‐coagulant endothelial phenotype. As shown in an in vitro flow model, anti‐prothrombin antibodies concentrate prothrombin on the phospholipid surface resulting in increased thrombin production. 25 , 26 In addition, high levels of prothrombin cause interference with the anticoagulant protein C pathway by inhibiting protein S function. 37 It is conceivable that antibody‐mediated accumulation of prothrombin on the phospholipid surface interferes with protein S function in a similar manner. In addition, antibody‐prothrombin complexes can cause acquired protein C resistance through interference with the protein C anticoagulant pathway by inhibiting APC or protein S function. How the interactions described here contribute to thrombosis in APS patients remains to be determined.
CONFLICTS OF INTEREST
There are no conflicts of interest reported by any of the authors.
AUTHOR CONTRIBUTIONS
T. Noordermeer, J.E. Molhoek, S.A.E. Sebastian, S. Drost‐Verhoef, and A.C.W. van Wesel performed research and analyzed data. T. Noordermeer, J.E. Molhoek, R.E.G. Schutgens, P.G. de Groot, J.C.M. Meijers, and R.T. Urbanus designed the study, interpreted data, and wrote the manuscript. All authors read and approved the manuscript.
ACKNOWLEDGMENTS
This study was supported by the Netherlands Thrombosis Foundation (Grant number 2018‐03).
Manuscript handled by: Patricia Liaw
Final decision: Patricia Liaw, 29 December 2020
REFERENCES
- 1. Keeling D, Mackie I, Moore GW, Greer IA, Greaves M. Guidelines on the investigation and management of antiphospholipid syndrome. Br J Haematol. 2012;157(1):47‐58. [DOI] [PubMed] [Google Scholar]
- 2. Limper M, de Leeuw K, Lely AT, et al. Diagnosing and treating antiphospholipid syndrome: A consensus paper. Neth J Med. 2019;77(3):98‐108. [PubMed] [Google Scholar]
- 3. Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4(2):295‐306. [DOI] [PubMed] [Google Scholar]
- 4. Pengo V, Banzato A, Denas G, et al. Correct laboratory approach to APS diagnosis and monitoring. Autoimmun Rev. 2013;12(8):832‐834. [DOI] [PubMed] [Google Scholar]
- 5. Lockshin MD, Kim M, Laskin CA, et al. Lupus anticoagulant, but not anticardiolipin antibody, predicts adverse pregnancy outcome in patients with antiphospholipid antibodies. Arthritis Rheum. 2012;64(7):2311‐2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ruffatti A, Del Ross T, Ciprian M, et al. Risk factors for a first thrombotic event in antiphospholipid antibody carriers: A prospective multicentre follow‐up study. Ann Rheum Dis. 2011;70(6):1083‐1086. [DOI] [PubMed] [Google Scholar]
- 7. Roubey RAS, Pratt CW, Buyon JP, Winfield JB. Lupus anticoagulant activity of autoimmune antiphospholipid antibodies is dependent upon β2‐glycoprotein I. J Clin Invest. 1992;90(3):1100‐1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bevers EM, Galli M, Barbui T, Comfurius P, Zwaal RFA. Lupus anticoagulant IgG’s (LA) are not directed to phospholipids only, but to a complex of lipid‐bound human prothrombin. Thromb Haemost. 1991;66(6):629‐632. [PubMed] [Google Scholar]
- 9. Oosting JD, Derksen RHWM, Entjes HTI, Bouma BN, De Groot PG. Lupus anticoagulant activity is frequently dependent on the presence of β2‐glycoprotein I. Thromb Haemost. 1992;67(5):499‐502. [PubMed] [Google Scholar]
- 10. Arnout J, Wittevrongel C, Vanrusselt M, Hoylaerts M, Vermylen J. Beta‐2‐glycoprotein I dependent lupus anticoagulants form stable bivalent antibody Beta‐2‐glycoprotein I complexes on phospholipid surfaces. Thromb Haemost. 1998;79(1):79‐86. [PubMed] [Google Scholar]
- 11. Willems GM, Janssen MP, Pelsers MMAL, et al. Role of divalency in the high‐affinity binding of anticardiolipin antibody‐β2‐glycoprotein I complexes to lipid membranes. Biochemistry. 1996;35(43):13833‐13842. [DOI] [PubMed] [Google Scholar]
- 12. Bevers EM, Janssen MP, Comfurius P, et al. Quantitative determination of the binding of β2‐ glycoprotein I and prothrombin to phosphatidylserine‐exposing blood platelets. Biochem J. 2005;386(2):271‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Qureshi SH, Yang L, Manithody C, Rezaie AR. Membrane‐dependent Interaction of Factor Xa and Prothrombin with Factor Va in the Prothrombinase Complex. Biochemistry. 2009;48(22):5034‐5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arnout J, Meijer P, Vermylen J. Lupus anticoagulant testing in Europe. an analysis of results from the first European Concerted Action on Thrombophilia (ECAT) survey using plasmas spiked with monoclonal antibodies against human b2‐glycoprotein 1. Thromb Haemost. 1999;81(34):929. [PubMed] [Google Scholar]
- 15. Atsumi T, Ieko M, Bertolaccini ML, et al. Association of autoantibodies against the phosphatidylserine‐prothrombin complex with manifestations of the antiphospholipid syndrome and with the presence of lupus anticoagulant. Arthritis Rheum. 2000;43(9):1982‐1993. [DOI] [PubMed] [Google Scholar]
- 16. Devreese KMJ, Pierangeli SS, de Laat B, Tripodi A, Atsumi T, Ortel TL. Testing for Antiphospholipid antibodies with Solid Phase Assays: Guidance from the SSC of the ISTH. J Thromb Haemost. 2014;12(5):792‐795. [DOI] [PubMed] [Google Scholar]
- 17. Rouser G, Fkeischer S, Yamamoto A. Two dimensional thin layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids. 1970;5(5):494‐496. [DOI] [PubMed] [Google Scholar]
- 18. Segers K, Dahlbäck B, Bock PE, Tans G, Rosing J, Nicolaes GAF. The Role of Thrombin Exosites I and II in the Activation of Human Coagulation Factor V. J Biol Chem. 2007;282(47):33915‐33924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shi J, Gilbert GE. Lactadherin inhibits enzyme complexes of blood coagulation by competing for phospholipid‐binding sites. Blood. 2003;101(7):2628‐2636. [DOI] [PubMed] [Google Scholar]
- 20. Gilbert GE. Use of Microsphere‐Supported Phospholipid Membranes for Analysis of Protein‐Lipid Interactions. Curr Protoc Cytom. 2005;34:1. [DOI] [PubMed] [Google Scholar]
- 21. Srivasatava KR, Majumder R, Kane WH, Quinn‐allen MA. Phosphatidylserine and FVa Regulate FXa Structure. Biochem J. 2014;459(1):229‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roubey RAS, Eisenberg RA, Harper MF, Winfield JB. “Anticardiolipin” autoantibodies recognize β2‐glycoprotein I in the absence of phospholipid: Importance of Ag density and bivalent binding. J Immunol. 1995;154(2):954‐960. [PubMed] [Google Scholar]
- 23. Takeya H, Mori T, Gabazza EC, et al. Anti‐β2‐glycoprotein I (β2GPI) monoclonal antibodies with lupus anticoagulant‐like activity enhance the β2GPI binding to phospholipids. J Clin Invest. 1997;99(9):2260‐2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Willems GM, Janssen MP, Comfurius P, Galli M, Zwaal RFA, Bevers EM. Kinetics of prothrombin‐mediated binding of lupus anticoagulant antibodies to phosphatidylserine‐containing phospholipid membranes: An ellipsometric study. Biochemistry. 2002;41(48):14357‐14363. [DOI] [PubMed] [Google Scholar]
- 25. Field SL, Hogg PJ, Daly EB, et al. Lupus anticoagulants form immune complexes with prothrombin and phospholipid that can augment thrombin production in flow. Blood. 1999;94(10):3421‐3431. [PubMed] [Google Scholar]
- 26. Field SL, Chesterman CN, Dai Y‐P, Hogg PJ. Lupus Antibody Bivalency Is Required to Enhance Prothrombin Binding to Phospholipid. J Immunol. 2001;166(10):6118‐6125. [DOI] [PubMed] [Google Scholar]
- 27. Simmelink MJA, Horbach DA, Derksen RHWM, et al. Complexes of anti‐prothrombin antibodies and prothrombin cause lupus anticoagulant activity by competing with the binding of clotting factors for catalytic phospholipid surfaces. Br J Haematol. 2001;113(3):621‐629. [DOI] [PubMed] [Google Scholar]
- 28. Schuijt TJ, Bakhtiari K, Daffre S, et al. Factor Xa Activation of Factor V is of Paramount Importance in Initiating the Coagulation System: Lessons from a Tick Salivary Protein. Circulation. 2013;128:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee CJ, De Biasio A, Beglova N. Mode of Interaction between β2GPI and Lipoprotein Receptors Suggests Mutually Exclusive Binding of β2GPI to the Receptors and Anionic Phospholipids. Structure. 2010;18(3):366‐376. [DOI] [PubMed] [Google Scholar]
- 30. Cattini MG, Bison E, Pontara E, Cheng C, Denas G, Pengo V. Tetra positive thrombotic Antiphospholipid syndrome: major contribution of anti phosphatidyl‐serine/prothrombin antibodies to Lupus anticoagulant activity. J Thromb Haemost. 2020;18:1‐9. [DOI] [PubMed] [Google Scholar]
- 31. Dienava‐Verdoold I, Boon‐Spijker MG, De Groot PG, et al. Patient‐derived monoclonal antibodies directed towards beta2 glycoprotein‐1 display lupus anticoagulant activity. J Thromb Haemost. 2011;9(4):738‐747. [DOI] [PubMed] [Google Scholar]
- 32. De Laat B, Derksen RHWM, Urbanus RT, De Groot PG. IgG antibodies that recognize epitope Gly40‐Arg43 in domain I of β2‐glycoprotein I cause LAC, and their presence correlates strongly with thrombosis. Blood. 2005;105(4):1540‐1545. [DOI] [PubMed] [Google Scholar]
- 33. Iverson GM, Victoria EJ, Marquis DM. Anti‐β2 glycoprotein I (β2GPI) autoantibodies recognize an epitope on the first domain of β2GPI. Proc Natl Acad Sci USA. 1998;95(26):15542‐15546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wood JP, Bunce MW, Maroney SA, Tracy PB, Camire RM, Mast AE. Tissue factor pathway inhibitor‐alpha inhibits prothrombinase during the initiation of blood coagulation. Proc Natl Acad Sci USA. 2013;110(44):17838‐17843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Al Dieri R, Bloemen S, Kelchtermans H, Wagenvoord R, Hemker HC. A new regulatory function of activated factor V: Inhibition of the activation by tissue factor/factor VII(a) of factor X. J Thromb Haemost. 2013;11(3):503‐511. [DOI] [PubMed] [Google Scholar]
- 36. Thorelli E, Kaufman RJ, Dahlbäck B. Cleavage of factor V at Arg 506 by activated protein C and the expression of anticoagulant activity of factor V. Blood. 1999;93(8):2552‐2558. [PubMed] [Google Scholar]
- 37. Brugge JM, Tans G, Rosing J, Castoldi E. Protein S levels modulate the activated protein C resistance phenotype induced by elevated prothrombin levels. Thromb Haemost. 2006;95(2):236‐242. [DOI] [PubMed] [Google Scholar]