

Abstract
Cholestatic liver disease denotes any situation associated with impaired bile flow concomitant with a noxious bile acid accumulation in the liver and/or systemic circulation. Cholestatic liver disease can be subdivided into different types according to its clinical phenotype, such as biliary atresia, drug‐induced cholestasis, gallstone liver disease, intrahepatic cholestasis of pregnancy, primary biliary cholangitis and primary sclerosing cholangitis. Considerable effort has been devoted to elucidating underlying mechanisms of cholestatic liver injuries and explore novel therapeutic and diagnostic strategies using animal models. Animal models employed according to their appropriate applicability domain herein play a crucial role. This review provides an overview of currently available in vivo animal models, fit‐for‐purpose in modelling different types of cholestatic liver diseases. Moreover, a practical guide and workflow is provided which can be used for translational research purposes, including all advantages and disadvantages of currently available in vivo animal models.
Keywords: drug‐induced cholestasis, in vivo modelling, intrahepatic cholestasis of pregnancy, primary biliary cholangitis, primary sclerosing cholangitis
Abbreviations
- AE2
anion exchange protein 2
- AMA
antimitochondrial antibody
- ANIT
α‐naphtylisothiocyanate
- ARE(‐Del)
(deletion in) adenylate‐uridylate‐rich element
- ATP8B1
ATPase phospholipid transporting 8B1
- BDL
bile duct ligation
- BRIC
benign recurrent intrahepatic cholestasis
- BSEP
bile salt export pump
- CD
cluster of differentiation
- CFTR
cystic fibrosis transmembrane conductance regulator
- DDC
3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine
- DIC
drug‐induced cholestasis
- DSS
dextran sodium sulphate
- (dn)TGF‐β(RII) mice
(dominant negative form of) transforming growth factor‐β (receptor restricted to T cells)
- Foxp3
Forkhead box protein 3
- IBD
inflammatory bowel diseases
- ICP
intrahepatic cholestasis of pregnancy
- idd
insulin‐dependent diabetes
- IL‐2(Rα)
interleukin 2 (receptor α)
- MDR
multidrug resistance protein
- MRP
multidrug resistance‐associated protein
- NOD(c3c4)
non‐obese diabetic (with B6/B10 region on chromosomes 3 and 4)
- PBC
primary biliary cholangitis
- PDH‐E2
E2 subunit of the pyruvate dehydrogenase
- PFIC
progressive familiar intrahepatic cholestasis
- PSC
primary sclerosing cholangitis
- TNBS
2,4,6‐trinitrobenzenesulfonice acid
- Treg cells
regulatory T cells
Key points.
There are still significant gaps in the mechanistic understanding of different types of cholestasis, including biliary atresia, drug‐induced cholestasis, gallstone liver disease, intrahepatic cholestasis of pregnancy, primary biliary cirrhosis and primary sclerosing cholangitis.
A better understanding of currently available animal models is urged with their specific properties, since inadequate models may inherently determine a biased research outcome.
This review provides a practical guide for researchers in the field of translational hepatology by providing a schematic overview of available animal models for cholestasis, including their individual and general advantages and disadvantages relevant for a specific applicability domain.
1. INTRODUCTION
Cholestatic liver diseases arise from any situation associated with impaired bile flow, as a result of disturbed hepatobiliary production or bile excretion, accompanied by noxious bile acid accumulation in hepatocytes or systemic circulation. 1 The word cholestasis is derived from Greek (ie ‘chole’ and ‘stasis’) and literally means ‘bile halting’. 2 A schematic overview of the pathogenesis of cholestasis is provided in Figure 1. Different types of cholestasis can be divided depending on specific underlying mechanisms and aetiology, such as biliary atresia, benign recurrent intrahepatic cholestasis (BRIC), intrahepatic drug‐induced cholestasis (DIC), gallstone disease, iatrogenic cholestatic liver diseases, intrahepatic cholestasis of pregnancy (ICP), primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC) and progressive familiar intrahepatic cholestasis (PFIC). 3 , 4 , 5 , 6 , 7 The multifaceted liver disease biliary atresia can have devastating consequences including progression into end‐stage cirrhosis. 8 The diagnosis of biliary atresia is based on excluding various causes of neonatal cholestasis, consequently by the time of diagnosis the extrahepatic bile ducts are often already completely obstructed. 8 , 9 The incidence of neonatal cholestasis has been estimated to be 1 in 2500 new‐born children, from which about 34%‐43% have biliary atresia. 9 , 10 , 11 PFIC and BRIC are two different forms of familial intrahepatic cholestasis with autosomal recessive inheritance. 7 PFIC can be subdivided into three different types, from which type 1 is cause by a mutation in the ATPase phospholipid transporting 8B1 (ATP8B1) gene, type 2 by a mutation in the gene encoding the bile salt export pump (BSEP) and type 3 by a mutation in the gene encoding multidrug resistance protein 3 (MDR3). 6 PFIC is considered a rare disease typically presenting with cholestasis during infancy or childhood. 12 BRIC was also found associated with mutations in ATP8B1 and BSEP, defined as BRIC type 1 and type 2 respectively. BRIC represents a less severe cholestatic phenotype characterized by intermittent recurrent cholestatic episodes, with extensive pruritus. 7 Iatrogenic cholestatic liver diseases constitute a group of liver diseases resulting from an invasive surgery, including biliary complications evoked by a living donor liver transplantation. Adult recipients from living donor liver transplantation were reported to develop biliary complications in 15%‐40% of the cases, including biliary stricture (anastomotic or non‐anastomotic), bile leakage, bile duct obstruction, sphincter of Oddi dysfunction, etc Importantly, biliary complications remain the major cause of morbidity and mortality after living donor liver transplantation. 5 , 13 Additionally, biliary complications may also occur after invasive abdominal (ie gastrectomy and oesophagectomy), orthopaedic or cardiovascular surgeries and carry significant incidence of morbidity and mortality. 14 , 15 , 16 Gallstone liver disease is reported as major medical problem, especially in Western countries where it has a prevalence of about 15% in adults. Moreover, in Europe, gallstone liver disease is one of the common causes of hospitalization resulting from a gastrointestinal disease. 17 , 18 , 19 , 20 Approximately 20% of the patients will develop the typical symptoms, such as jaundice and biliary pain, or other types of complications in a time span of 15 years. 19 , 21 , 22 PBC and PSC have a prevalence ranging from 2 to 40 per 100,000 inhabitants and 0 to 16 per 100,000 inhabitants. 3 , 23 Ursodeoxycholic acid is the golden standard for safely and effectively treating PBC patients. Nevertheless, still about 15% of the PBC patients show a progressive disease that requires liver transplantation or in some cases results in death, despite ursodeoxycholic acid treatment. 24 , 25 For PSC, effective treatment is, thus far, still non‐existing, presumably due to considerable gaps in the mechanistic understanding of the disease, which hampers the identification of potential targets. 26 More atypical causes of cholestasis, albeit not less important, include DIC and ICP. 3 ICP is of high clinical relevance considering their worldwide incidence ranging from 0.2% up to 25% and risk on preterm birth, respiratory distress syndrome and still birth. 27 , 28 , 29 , 30 DIC is in 73% of the cases evoked by single‐prescription medication, mainly cardiovascular, anti‐inflammatory, antidiabetic and anti‐infectious drugs. 31 DIC can cause a tremendous financial loss for pharmaceutical companies, mostly due to early drug retraction from clinical trials. This can partly be explained by the fact that current preclinical animal models as well as preclinical in vitro models only predict 50%‐60% of all drug‐induced liver injuries, including DIC. 31 , 32 , 33 , 34 A better understanding of currently available animal models with their specific properties is, therefore, highly demanded, since an inadequate model will inherently determine a biased research outcome.
FIGURE 1.
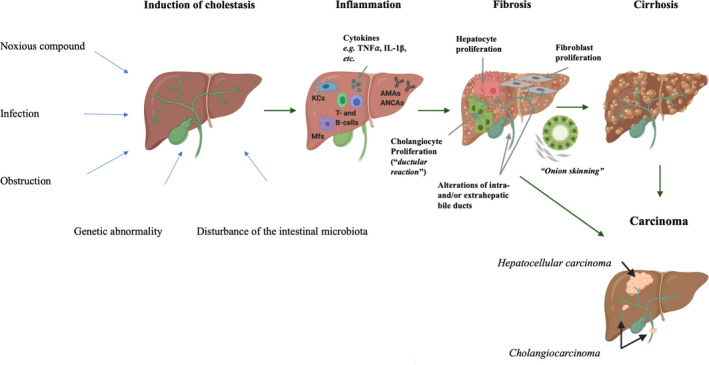
Schematic overview of the pathogenesis of cholestatic liver injury. Cholestasis can be induced by a number of factors including, noxious compounds, infection, obstruction of bile flow, disturbance in the intestinal microbiota or genetic abnormalities. 26 , 244 These initiating factors evoke an inflammatory injury on hepatocytes and/or cholangiocytes, which may result in activation of hepatocytes, fibroblasts and cholangiocytes. 48 , 244 These mature cells, may, in its turn, start to proliferate and cause fibrosis in the liver and/or bile duct. 244 In primary sclerosing cholangitis, fibrosis is typically accompanied by the ‘onion skinning’ around the bile ducts. 186 Moreover, fibrosis might in some cases progress into cirrhosis or even carcinoma (hepatocellular carcinoma or cholangiocarcinoma). 245 This figure was created with Biorender Software. IL‐1β, interleukin 1β; PSC, primary sclerosing cholangitis; TNF⍺, tumour necrosis factor ⍺
This review provides a state‐of‐the‐art overview of currently available surgery‐induced, genetically modified, chemical‐induced, viral‐induced and combination rodent models for different types of cholestasis (ie biliary atresia, DIC, gallstone liver disease, ICP, PBC and PSC) together with their characteristics, individual assets, drawbacks and applicability domain, which serves as a tool for researchers in the field of translational hepatology (Tables 1 and 2).
TABLE 1.
Reported applicability domain(s), advantages and disadvantages per in vivo rodent model
| Cholestatic liver injury | Type of experimental model | Reported rodent species/strains | Applicability domain(s) | Advantages | Disadvantages | References | |
|---|---|---|---|---|---|---|---|
| Biliary atresia | Surgery‐induced rodent models | Bile duct ligation |
Male and female Sprague‐Dawley rat pups, male Wistar rat pups (21‐30 days) |
Acute and chronic mechanistic studies. Therapeutic studies. |
Relatively easy surgical procedure. Progression into fibrosis. |
Rat pups have smaller size of biliary structures. Increased mortality. |
78, 221 |
|
Bile duct injection technique |
Male C57BL/6 mice |
Acute and chronic mechanistic studies. Therapeutic studies. |
Minor or no involvement of other tissues. Novel route of administration for testing therapeutic strategies directly in the bile ducts. |
Risk of peritonitis. Leakage of bile or the injective solvent into the abdominal cavity. Difficult procedure due to the small anatomic proportions which complicate access to the biliary tree. Model needs to be further characterized. |
90, 91 | ||
| Viral‐induced rodent models | Intraperitoneal inoculation of mice with rhesus rotavirus | Balb/c mouse pups (1‐2days) |
Acute mechanistic studies. Therapeutic studies. |
Affects both intra‐ as extrahepatic bile ducts. |
No (or delayed) progression into fibrosis. Bad reproducibility. Injection‐related injury. Cannibalization of pups. Variable time of infection and virus dosage. Survival rate is 10% at 3 weeks. |
222, 223 | |
| Drug‐induced cholestasis | Genetically modified mice models | Bsep‐/‐mouse model | Male C57BL/6 and FVB/N mice |
Acute mechanistic studies (related to BSEP malfunctioning). Therapeutic studies. Specific studies focused on the role BSEP in bile acid homeostasis and alternative bile acid transport systems. |
BSEP malfunctioning is a well‐known triggering factor of drug‐induced cholestasis. |
Time‐consuming (model of 60‐180 days). Focus on 1 transporter. Not applicable for drugs that simultaneously interfere with multiple hepatobiliary transporters. No development of severe cholestasis. |
48, 49, 97, 203 |
| Chemical‐induced rodent models | Chlorpromazine‐induced cholestasis | Male albino and Wistar rats |
(Acute) and chronic mechanistic studies. Therapeutic studies. |
Short model (5‐7 days). Well‐known hepatotoxicant. |
Acute effects are limited. Interindividual differences in the susceptibility. |
139, 143, 144 | |
| Cyclosporin A‐induced cholestasis |
Male Wistar rats and Sprague‐Dawley rats Male C57BL/6 mice |
Acute and chronic mechanistic studies. Therapeutic studies. |
Short model (11‐25 days). Well‐known hepatotoxicant. |
Development of steatosis. Immunosuppressive effect of cyclosporin A. | 145, 146, 148, 149, 151 | ||
|
Combination rodent models |
Bsep‐/‐mouse model with cholic acid feeding | Male FVB/N mice |
Acute mechanistic studies (related to BSEP malfunctioning). Therapeutic studies. Specific studies focused on the role BSEP in bile acid homeostasis and alternative bile acid transport systems. |
BSEP malfunctioning is a well‐known triggering factor of drug‐induced cholestasis. Development of severe cholestasis. More resemblance with cholestasis than single hit Bsep ‐/‐ mice model. |
Time‐consuming (model of 66‐189 days). Male predominance. High mortality rate. |
97, 203 | |
| Gallstone liver disease | Chemical‐induced rodent models | Lithogenic diet |
Male albino NMRI‐mice Male C57BL/6 mice |
Acute and chronic mechanistic studies. Therapeutic studies. Specific studies focused on the genetic susceptibility. |
Easy model. |
Only cholesterol gallstones. Long model (8 weeks). High variability across different strains and gender. |
155, 224, 225 |
| Intrahepatic cholestasis of pregnancy | Chemical‐induced rodent models | Ethinylestradiol and estradiol‐17β‐D‐glucuronide‐induced cholestasis |
Female Sprague‐Dawley and Wistar rats |
Acute mechanistic studies. Therapeutic studies. |
Short model (5 days). Mechanisms of hepatotoxicity are well known. |
Strict legislation in purchasing hormones. | 165, 226, 227 |
| Primary biliary cholangiopathy (PBC) | Surgery‐induced rodent models |
Bile duct injection technique |
Female C57BL/6 mice |
Acute and chronic mechanistic studies. Therapeutic studies. |
Minor or no involvement of other tissues. Novel route of administration for testing therapeutic strategies directly in the bile ducts. |
Risk of peritonitis. Leakage of bile or the injective solvent into the abdominal cavity. Difficult procedure due to the small anatomic proportions which complicate access to the biliary tree. Model needs to be further characterized. |
91 |
| Genetically modified mice models | Ae2 a,b ‐/‐ mouse model | Male FVB/N mice |
Acute and chronic mechanistic studies. Specific studies focused on the role of AE2 in PBC. |
Selective damage of bile ducts. |
Time‐consuming (model of 180‐450 days). Impaired gastric acid secretion, male sterility and osteoporosis. Interindividual differences in the severity of cholangitis. Relatively late onset. Only 30%‐80% AMA. No female predominance. No granulomas. Difficulty in breeding. |
105, 113, 114 | |
| ARE Del‐/‐ mouse model | Female C57BL/6 mice |
Acute and chronic mechanistic studies. Therapeutic studies. Specific studies focused on the role of interferon γ in PBC. Specific studies focused on the female predominance in PBC. |
Female predominance. |
Time‐consuming (model of 70 days). Lupus‐like autoimmune features. |
114, 118, 119 | ||
| dnTGF‐βRII mouse model | Female and male C57BL/6 mice |
Acute mechanistic studies. Specific studies focused on the role of (regulatory) T cells and TGF‐β pathways in PBC. |
100% AMA. |
Time‐consuming (model of 154‐196 days). Development of intestinal inflammation. No granulomas. No progression into chronic cholestasis. No female predominance. |
105, 112, 113 | ||
| IL‐2Rα ‐/‐ mouse model | Female and male C57BL/6 mice |
Acute mechanistic studies. Specific studies focused on the role of (regulatory) T cells in PBC. |
100% AMA. |
Time‐consuming (model of 56‐154 days). Development of Inflammatory bowel diseases, haemolytic anaemia and lymphoproliferative autoimmune disorder. High mortality rate. Short life span. No granulomas and eosinophilic infiltration. No progression into chronic cholestasis. No female predominance. |
105, 111, 113 | ||
| MRL/lpr mouse model | Female and male C57BL/6 mice |
Acute mechanistic studies. Specific studies focused on the role of T cells in PBS. |
First spontaneous model developed for PBC. |
Time‐consuming (model of 140 days). No progression to cirrhosis. No significant increase in bilirubin and hepatobiliary enzymes. No female predominance. Only 50% of mice develop a PBC‐like disease. |
98, 99 | ||
| NOD c3c4 mouse model | Female and male NOD c3c4 mice |
Acute and chronic mechanistic studies. Specific studies focused on the role of B and T cells in PBC. Specific studies focused on the peculiar switch from diabetes to a PBC‐like disease. |
Only existing model that could unravel the genetic switch from diabetes to a PBC‐like disease. |
Time‐consuming (model of 67 days). Biliary dilatation and cystic lesions. Only 50%‐60% AMA. Granulomas are rare. No progression to chronic cholestasis. No female predominance. |
105, 114, 116, 117 | ||
| Scurfy mice model | Male C57BL/6 mice |
Acute mechanistic studies. Specific studies focused on the role of regulatory T cells in PBC. |
Short model (21‐28days). 100% AMA. |
Lupus‐like autoimmune features. High mortality rate. No female predominance. |
102, 104, 105 | ||
| Chemical‐induced rodent models | 2‐octynoic acid‐and 2‐nonynoic acid‐induced cholestasis | Female C57BL/6 mice | Chronic mechanistic studies. |
Short model (28 days). 100% AMA. Easy model. |
Peritonitis. No female predominance. Less pronounced portal inflammation. |
105, 114, 170 | |
| Primary sclerosing cholangitis (PSC) | Surgery‐induced rodent models | Bile duct ligation |
Male C57BL/6 and 129/Sv mice Male Sprague‐Dawley rats |
Chronic mechanistic studies. |
Short model (14‐56 days). Relatively easy surgical procedure. |
High variability across different strains, species and gender. Technical pitfalls. No development of inflammatory bowel diseases. |
|
| Bile duct injection technique | Female C57BL/6 mice |
Acute and chronic mechanistic studies. Therapeutic studies. |
Minor or no involvement of other tissues. Novel route of administration for testing therapeutic strategies directly in the bile ducts. |
Risk of peritonitis. Leakage of bile or the injective solvent into the abdominal cavity. Difficult procedure due to the small anatomic proportions which complicate access to the biliary tree. Model needs to be further characterized. |
91 | ||
| Genetically modified mice models | Cftr ‐/‐ mouse model |
Male C57BL/6 mice |
Chronic mechanistic studies. Therapeutic studies. Specific studies focused on the long‐term pro‐tumorigenic aspects of PSC. |
Only existing model that could unravel the link between cystic fibrosis liver disease and PSC. |
Time‐consuming (model of 30‐728 days). High variability across strains, gender and age. No development of inflammatory bowel diseases. Intestinal obstruction. No progression into hepatic fibrosis. Contrasting results from different studies. |
81, 82, 127, 129, 200, 210 | |
| fch/fch mouse model | Male BALB/c mice |
Acute and chronic mechanistic studies. Specific studies focused on the long‐term pro‐tumorigenic aspects of PSC and the coincidence of cholestasis in erythropoietic protoporphyria. |
Only existing model that could unravel the link of cholestasis in erythropoietic protoporphyria. |
Time‐consuming (model of 84‐496 days). Model needs to be further characterized. |
81, 94, 135, 136 | ||
| Mdr2 ‐/‐ mouse model |
Male BALB/c, and Female FVB/N mice. |
Chronic mechanistic studies. Test novel therapeutic strategies. Biomarker discovery. |
Short model (28 days). Male predominance (only in the BALB/c strain). |
No development of inflammatory bowel diseases. | 123, 124, 201 | ||
| Chemical‐induced rodent models | α‐naphtylisothiocyanate‐induced cholestasis |
Male Sprague‐Dawley rats. Female and male C57BL/6 mice |
Acute and chronic mechanistic studies. Therapeutic studies. Diagnostic studies (biomarkers). |
Single dose suffices. Well‐known hepatotoxicant. Very short model (2 days). Easy model. |
No large bile duct injury. No development of inflammatory bowel diseases. |
79, 171, 172, 174 | |
| 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine‐induced cholestasis |
Male 129 DV mice, C57BL/6, FVB/N mice and Swiss albino mice. |
Chronic mechanistic studies. Therapeutic studies. |
Short model (7‐56 days). Available in 4 mice strains. Easy model. |
No development of inflammatory bowel diseases. | 82, 175, 176 | ||
| Lithocholic acid‐induced cholestasis |
Male 129 DV mice, C57BL/6, FVB/N mice and Swiss albino mice. Female and male Sprague‐Dawley rats. |
Acute mechanistic studies. Therapeutic studies. Specific studies focused on the role of bile acids in PSC. |
Very short model (4 days). Available in 4 mice strains and rats. Easy model. |
High mortality rate. Short lifespan. No development of inflammatory bowel diseases. |
81, 184, 185 | ||
| 2,4,6‐trinitrobenzenesulfonice acid‐induced cholestasis (intraportal administration) | Female Lewis rats and male Sprague‐Dawley rats. |
Acute and chronic mechanistic studies. |
Short model (7‐56 days). |
Mild phenotype. No development of inflammatory bowel diseases. AMA not directed towards PDH‐E2. Biliary tree remains intact upon administration in the portal vein. |
205, 207 | ||
|
Combinational rodent models |
Cftr ‐/‐ mice with dextran sodium sulphate feeding |
Male C57BL/6 mice |
Chronic mechanistic studies. Specific studies focused on the role of the coincidence of an inflammatory bowel disease in PSC. |
Better representation of the coincidence of an inflammatory bowel disease in PSC. Better resemblance of PSC cholestasis than the single hit Cftr ‐/‐ mice model. |
Time‐consuming (model of 54 days). No progression to hepatic fibrosis. |
81, 127, 129 | |
| Cftr ‐/‐ or Cftr ± mice with dextran sodium sulphate feeding and 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine feeding |
Male C57BL/6 mice |
Chronic mechanistic studies. Therapeutic studies. Specific studies focused on the role of the gut‐liver axis in PSC. Specific studies focused on the role of CFTR dysfunction in developing cholestatic liver diseases. |
Better representation of the coincidence of an inflammatory bowel disease in PSC. Progression into hepatic fibrosis. Better resemblance of PSC cholestasis than the double hit Cftr ‐/‐ mice model with dextran sodium sulphate feeding. |
Time‐consuming (model of 70 days). |
129, 200 | ||
| Mdr2 ‐/‐ mice with dextran sodium sulphate feeding | Male C57BL/6 mice |
Acute and chronic mechanistic studies. Specific studies focused on the role of the gut‐liver axis in PSC. |
Better representation of the coincidence of an inflammatory bowel disease in PSC |
Time‐consuming (model of 49‐56 days). |
201 | ||
| Bile duct ligated rats with 2,4,6‐trinitrobenzenesulfonic acid administration | Female and male Lewis rats | Acute and chronic mechanistic studies. | Better resemblance of PSC cholestasis than the single hit 2,4,6‐trinitrobenzenesulfonic acid administration model and bile duct ligation model. |
Time‐consuming (model of 42‐378 days). High mortality rate. No development of inflammatory bowel diseases. |
202, 208, 209 | ||
TABLE 2.
Serum and histological characteristics including the relevant disease phenotypes per in vivo model
| Type of experimental model | Experimental in vivo model | Serum characteristics | Histological characteristics | Relevant human disease phenotype | References |
|---|---|---|---|---|---|
| Chemical‐induced models | 2‐octynoic acid‐ and 2‐nonynoic acid‐induced cholestasis |
AMA: IgM↑ and IgG↑ |
Mild portal inflammation. Granuloma formation. Mild necrosis in hepatic parenchyma. |
Primary biliary cholangitis. |
168, 170 |
| 2,4,6‐trinitrobenzenesulfonice acid‐induced cholestasis (intraportal administration) |
ALP↑ AST↑ Bilirubin↑ SBA↑ |
Focal non‐parenchymal necrosis. Fibrous scarring of liver. Mild ductular reaction. |
Primary sclerosing cholangitis. |
207 | |
| 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine‐induced cholestasis |
ALP↑ AST↑ ALT↑ Bilirubin↑ SBA↑ |
Necrosis in liver lobules. Intraductal porphyrin plug. Portal inflammation. Ductular reaction. Damaged biliary epithelia. Periductular fibrosis (onion‐skin type). Increased wall thickness ductus hepatocholedocus. |
Primary sclerosing cholangitis. |
177, 228 | |
| α‐naphtylisothiocyanate‐induced cholestasis |
ALP↑ AST↑ ALT↑ Bilirubin↑ SBA↑ ɣGT↑ |
Inflammation. Multifocal periportal necrosis. Fatty metamorphosis. Sinusoid congestion. Vacuole degeneration. |
Primary sclerosing cholangitis. |
174, 229, 230 | |
| Chlorpromazine‐induced cholestasis |
Albumin↓ ALT↑ AST↑ Bilirubin↑ SBA↑ |
Expansion hepatic sinus. Hepatocellular necrosis. Ductular reaction. |
Drug‐induced cholestasis. | 143 | |
| Cyclosporin A‐induced cholestasis |
ALP↓ Bilirubin↑/= SBA↑ |
Vacuolization of liver cells Submembranous vesicle formation. | Drug‐induced cholestasis. | 145, 149, 151 | |
| Ethinylestradiol and estradiol‐17β‐D‐glucuronide‐induced cholestasis |
ALP↑ AST↑ ALT↑ Bilirubin↑ SBA↑ |
Inflammation. Widening of intercellular spaces. Oedema. Nuclear pyknosis of cells. Rearranged hepatocytes. |
Drug‐induced cholestasis. Intrahepatic cholestasis of pregnancy. |
165, 231 | |
| Lithocholic acid‐induced cholestasis |
ALP↑ AST↑ ALT↑ Bilirubin↑ SBA↑ |
Inflammation. Hepatocellular necrosis. Bile infarcts. Crystals obstructing interlobular bile ducts. Larger bile ducts with periductal oedema. Percholangitis. Periductal fibrosis. Ulceration of bile duct epithelium. |
Primary sclerosing cholangitis. Role of BA accumulation in cholestasis. |
185, 232, 233 | |
| Lithogenic diet | ND |
Liver Vacuolar degeneration. Neutrophil infiltration into acini and portal area of liver. Gallbladder Inflammation. Thickened muscular walls. Altered mucosal papillary architecture. Reactive epithelial changes. |
Gallstone liver disease. | 225, 234 | |
| Genetically modified mice models | Ae2 a,b ‐/‐ mouse model |
ALP↑ ALT↑ AMA: IgM↑ and IgG↑ |
Mild to intense portal inflammation. (Slight portal fibrosis) |
Primary biliary cholangitis. | 113 |
| ARE Del‐/‐ mouse model |
AST↑ ALT↑ SBA↑ AMA: IgM↑ and IgG↑ |
Portal and lobular inflammation. Small bile duct damage. Granuloma formation. Mild fibrosis. |
Primary biliary cholangitis. | 113, 119 | |
| Bsep‐/‐mouse model | No significant changes of albumin, ALP, AST, ALT, bilirubin and ɣGT |
Only ultrastructural changes: Dilatation of canalicular lumens. Loss of microvilli. Increased peroxisomes, lysosomes and lipid droplets in hepatocytes. |
Benign recurrent intrahepatic cholestasis type 2 Drug‐induced cholestasis. Type 2 progressive familial intrahepatic cholestasis. |
203, 235 | |
| Cftr ‐/‐ mouse model | No significant increase in ALP |
Ductular cell proliferation. Portal inflammation. (Periportal and bridging fibrosis.) Hepatic steatosis. (Cirrhosis). Cave: contrasting studies described Cftr ‐/‐ mice to only exhibit an intestinal phenotype with mild or non‐existing pathologies in other organs. 81 , 130 , 131 , 132 |
Cystic fibrosis liver disease. Primary sclerosing cholangitis. |
129 | |
| dnTGF‐βRII mouse model |
AMA: IgM no increase Other cholestatic parameters ND |
Portal inflammation. Bile ductular destruction. Inflammation in the parenchyma of the liver. |
Primary biliary cholangitis. | 106, 112 | |
| fch/fch mouse model |
Bilirubin↑ SBA↑ |
Ductular reaction. Periportal or septal fibrosis. Protophyrin deposits in small bile ductules. |
Erythropoietic protoporphyria (link with primary sclerosing cholangitis). | 135, 236 | |
| IL‐2Rα ‐/‐ mouse model |
AMA: IgA↑ and IgG↑ Other cholestatic parameters ND |
Liver Portal inflammation. Mild interface hepatitis. Bile ductular destruction. Colon Colon inflammation. |
Primary biliary cholangitis. | 111, 237 | |
| Mdr2 ‐/‐ mouse model |
AST↑ ALT↑ ALP↑ Bilirubin↑ SBA↑ |
Focal necrosis and eosinophilic bodies. Portal inflammation. Ductular reaction. Slight fibrosis. Dilatation of large bile ducts. |
Intrahepatic cholestasis of pregnancy. Primary sclerosing cholangitis. Progressive familial intrahepatic cholestasis type 3. |
76, 121, 122, 238 | |
| MRL/lpr mouse model |
AMA: IgA↑ and IgG↑ No significant increase in the other cholestatic parameters. |
Portal inflammation. Granuloma formation. Bile ductular destruction. Focal necrosis and/or acidophilic bodies. |
Primary biliary cholangitis | 98, 99 | |
| NOD c3c4 mouse model |
AST↑ ALT↑ AMA: IgG↑ (females) and IgM↑ |
Peribiliary lymphocytic infiltration. Bile ductular destruction. Intrahepatic biliary cyst formation. (Fibrosis and granuloma formation). Dilated bile ducts. |
Primary biliary cholangitis | 117, 239 | |
| Scurfy mice model | AMA: IgA↑, IgG↑ and IgM↑ |
(Peri)portal and perisinusoidal inflammation. Bile ductular destruction. Interface hepatitis. Focal necrosis. |
Primary biliary cholangitis. Systemic lupus erythematosus. |
102, 104 | |
| Surgical‐induced model | Bile duct ligation |
Albumin ↓ ALP ↑/↓ AST ↑ ALT ↑/= Bilirubin ↑ SBA ↑ ɣGT ↑ |
Liver fibrosis (F1 → F4). Portal hypertension. Biliary infarcts. Dilatation of bile canaliculi. Portal tract enlargement. Portal inflammation. Ductular reaction. |
Biliary atresia. (Extrahepatic cholestasis) Gallstone liver disease. Hepatic encephalopathy. Iatrogenic cholestatic liver diseases Obstructive cholestasis. Primary sclerosing cholangitis. Secondary biliary cirrhosis. |
|
| Bile duct injection technique |
ALT ↑ at day 1 (normalization after 7 days) No significant changes in AST, ALP and bilirubin. |
Necrosis Fibrosis Portal inflammation Cave: these histological features occur after 1 week postoperation and resolve after 2‐6 weeks. |
Biliary atresia. Biliary cirrhosis. Cholangiocarcinoma. Primary biliary cholangitis. Primary sclerosing cholangitis. |
90, 91 | |
| Viral‐induced rodent models | Intraperitoneal inoculation of mice with rhesus rotavirus | Bilirubin ↑ |
Obstruction of extrahepatic bile ducts by inflammatory cells. Portal inflammation. Ductular reaction. Focal stenosis of the common bile duct. (Distal cystic dilatation) |
Biliary atresia | 196, 223, 242 |
|
Combinational rodent models |
Bsep‐/‐mouse model and cholic acid feeding |
ALP ↑ AST ↑ Bilirubin ↑ SBA ↑ 5’‐nucleotidase ↑ |
Mild ductular reaction. Dilated bile ducts with infiltration of inflammatory cells. Necrosis in liver parenchyma. |
Drug‐induced cholestasis. Type 2 progressive familial intrahepatic cholestasis. |
97 |
| Cftr ‐/‐ mice with dextran sodium sulphate feeding | ALT↑ |
Liver Portal inflammation. Biliary epithelial damage. Ductular reaction. Colon Mononuclear cell infiltrates. Loss of crypts. Mucosal ulcerations in the colonic resection specimens. |
Primary sclerosing cholangitis. | 127, 210 | |
| Cftr ‐/‐ or Cftr ± mice with dextran sodium sulphate feeding and 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine feeding |
ALP↑ ALT↑ AST↑ |
Periportal and sinusoidal inflammation. Ductular reaction. Periductular fibrosis. ‘Onion skin’ fibrosis. Bridging fibrosis. Porphyrin bile plugs. |
Primary sclerosing cholangitis. | 200 | |
| Mdr2 ‐/‐ mice with dextran sodium sulphate feeding |
ALT↑ |
Liver Ductular reaction. Bridging fibrosis. Colon Colonic inflammation. Colonic shortening. |
Primary sclerosing cholangitis. | 201, 243 |
Abbreviations: ALP, alkaline phosphatase, ALT, alanine aminotransferase, AMA, antimitochondrial antibody, AST, aspartate aminotransferase; Ig, immunoglobulin; ND, not determined; (S)BA, (total serum) bile acids; ɣGT, gamma glutamyltransferase.
2. PATHOPHYSIOLOGY OF CHOLESTATIC LIVER DISEASES
2.1. Gallstone liver disease
Gallstones are hardened deposits of digestive fluid bile, which develop within the gallbladder. They develop when there is an imbalance in the chemical constituents of bile, which results in precipitation of the components. 20 Three main pathways have been identified in the formation of gallstones, cholesterol supersaturation, excess of bilirubin and disturbance in the functionality of the gallbladder. In a healthy situation, bile is able to dissolve the amount of cholesterol excreted through the liver, but in case of excessive cholesterol biosynthesis or excretion cholesterol may precipitate as crystals and evolve to stones (ie cholesterol supersaturation). 20 , 21 , 35 The yellow pigment bilirubin is released upon degradation of red blood cells. In some haemolytic conditions (eg hereditary spherocytosis and sickle cell disease), red blood cell disruption is abnormally high resulting in an overload of bilirubin, which sometimes lead to gallstone formation, referred to as pigment stones. 20 , 21 , 36 , 37 Finally, gallbladder hypomotility or even impaired contractility can perturb the effective clearance of bile in the gallbladder. The latter may result in a highly concentrated bile, which may, in turn, lead to gallstone formation. 21 , 38 Symptoms and complications of these gallstones can occur when the stones obstruct bile ducts and/or cystic ducts. A temporary obstruction of the cystic duct characterized as cholelithiasis is believed to be short‐lived and results in biliary pain. A more persistent obstruction occurs when a large stone gets permanently lodged in the neck of the gall bladder (cystic duct) often leading to acute cholecystitis. Other types of complications can also develop depending on the location of gallstone obstruction. As such, obstruction of the common bile duct causes choledocholithiasis and jaundice while obstructing the ampulla in the distal portion of the bile duct may result in gallstone pancreatitis. 21 , 39
2.2. Biliary atresia
Biliary atresia is a severe inflammatory and fibrosing cholangiopathy of infancy with an obstruction of the extrahepatic bile ducts, which can rapidly progress into end‐stage cirrhosis. Intrahepatic bile ducts are not hindered yet, are observed to be hyperplastic and embedded in portal tracts with varying levels of inflammation and fibrosis, and lobules with features of cholestasis and increased multinucleated hepatocytes. 8 , 40 Clinically, the disease is typified by pathological jaundice with direct or conjugated hyperbilirubinaemia, hepatosplenomegaly, acholic stool and the onset of symptoms in the first months of life. 8 , 41 Precise clinical phenotyping might differ between the perinatal and embryonic form, and cyst‐associated or cytomegalovirus‐associated variants of the disease. Perinatal biliary atresia also referred to as non‐syndromic biliary atresia represents the biggest subgroup with approximately 80% of the biliary atresia patients, characterized by a jaundice‐free period after birth. 8 , 42 The embryonic form occurs in about 10% of the affected infants and show an earlier onset of jaundice (at birth) together with non‐hepatic congenital malformation, such as splenic abnormalities. 8 , 43 , 44 Cystic biliary atresia is defined by a cystic malformation near the obstructed common bile duct, occurring in about 8% of the patients. Cytomegalovirus‐associated biliary atresia shows a poor bile drainage and carries the highest risk of death. The incidence of this form is highly variable based on geography as well as the detection methodology used (eg lymphocyte activation to cytomegalovirus proteins with or without other biochemical markers). 8 , 45 Several factors have been described in the pathogenesis, including abnormal foetal or prenatal circulation, genetic factors, embryogenesis defects, an abnormal inflammatory response, autoimmunity, viral infection and environmental toxins. 8
2.3. Drug‐induced cholestasis
Different types of DIC can be distinguished depending on (i) the location of the insult, intrahepatic DIC versus extrahepatic DIC, (ii) the reversibility of the insult, acute DIC versus chronic DIC and (iii) the clinical phenotype of the insult, cholestasis without hepatitis, with hepatitis or with bile duct injury. 46 Clinical symptoms may include jaundice or, when associated with parenchymal liver injury, non‐specific symptoms, such as anorexia, nausea, fatigue and malaise. 46 Moreover, chronic DIC may also be accompanied by xanthomas, pruritus and melanoderma. 46 , 47 Increasing efforts have been made to identify a set of mechanisms involved in all these different types of DIC. In this regard, inhibition of the BSEP was earlier denoted as a key molecular initiating event. Inhibiting the transporter involved in bile acid export may result in an accumulation of bile acids in the hepatocytes. In turn, bile acid accumulation was observed to trigger two types of cellular responses, namely a deteriorative response and an adaptive response. The deteriorative response is characterized by inflammation, oxidative stress, opening of the mitochondrial membrane permeability pore and cell death. The adaptive response is the curious phenomenon of cholestasis, as this type of cellular response strives to counteract cholestasis by regulating nuclear receptors involved in bile acid homeostasis. 48 , 49
2.4. Intrahepatic cholestasis of pregnancy
ICP is a cholestatic liver disease that refers to the sporadic occurrence of recurrent jaundice, pruritus, increased bile acid levels and/or elevated alanine/aspartate aminotransferase during the last trimester of pregnancy, sometimes resulting in foetal risks, such as preterm birth, respiratory distress syndrome or stillbirth. 27 , 28 , 29 There are still a lot of uncertainties in the aetiology of ICP. Genetic, environmental and hormonal factors are all believed to contribute to the pathogenesis of ICP. Accordingly, mutations in the MDR3 have been estimated to account for up to 15% of all ICP cases. 29 , 50 , 51 Additionally, oestrogen is proven to play an important role in ICP. Oestrogen reaches its maximum concentration in the last trimester of pregnancy, in which ICP occurs most frequently. Twin and triplet pregnancies with increased oestrogen levels appear to be more susceptible for ICP compared to single gestations. Finally, women with a family or personal history of ICP were seen to develop ICP when treated with high doses of oestrogen oral contraceptive. 52 , 53 Environmental factors that have been identified include an increased incidence of ICP associated with increased plasma levels of selenium, which varies over different seasons. 54 ICP has a mainly symptomatic treatment, since the pathophysiology is still unresolved. Ursodeoxycholic acid is being supported as first‐line therapy in ICP with beneficial effects on the mother, foetus and new‐borns. 55 , 56
2.5. Primary biliary cholangitis
PBC is a slowly progressive cholestatic liver disease with an important autoimmune aspect. This autoimmune disease is characterized by immune‐mediated destruction of the intrahepatic bile ducts and portal inflammation. In a more advanced stage of the disease, loss of bile ducts may lead to diminished bile secretion, accumulating noxious substances within the liver, fibrosis, cirrhosis and ultimately liver failure. Accordingly, four different histological stages can be distinguished during PBC: (i) portal inflammation with or without ‘florid’ bile duct lesions (ie intense inflammatory infiltration and necrosis around bile ducts), (ii) enhanced periportal lesions accompanied by interface hepatitis, (iii) abnormalities in the hepatic architecture and several fibrous septa and finally (iv) cirrhosis. Typical for PBC is the seropositivity for antimitochondrial antibodies (AMAs) towards the E2 subunits of 2‐oxo‐acid dehydrogenase complexes, such as pyruvate dehydrogenase (PDH‐E2). 57 AMAs are present in 90%‐95% of the patients and can often be detected years before clinical symptoms, including fatigue, pruritus, jaundice, xanthomas, osteoporosis and dyslipidaemia. 57 , 58 , 59 The pathogenesis of PBC still remains to be further elucidates; however, an important genetic aspect has already been identified. The latter is related to an increased susceptibility in first‐degree relatives 60 but a full characterization of the exact genetic influences is still missing. 61 , 62 PBC primarily affects woman, with a ratio 10 to 1 versus men, presumably related to a higher incidence of X‐chromosome monosomy in lymphoid cells. 57 , 63 Environmental factors play an additional important role in PBC. Several causatives have been proposed to initiate a strong autoimmune response due to molecular mimicry and cross reactivity with human pyruvate dehydrogenase complex autoepitopes, including bacteria, viruses and environmental chemicals. 57 , 64
2.6. Primary sclerosing cholangitis
PSC is a long‐term, chronic liver disease featured by inflammation and fibrosis of intra‐ and/or extrahepatic bile ducts, leading to end‐stage liver disease and reduced life expectancy. PSC primarily affects young, middle‐aged men. Most patients complain of right upper quadrant abdominal pain, pruritis, fatigue and jaundice. In addition, hepatomegaly and splenomegaly are regularly observed in PSC patients. 10%‐20% of PSC patients spontaneously develop cholangiocarcinoma, making this a very dangerous liver disease. 65 Interestingly, PSC is also closely associated with inflammatory bowel diseases (IBD), with up to 70% of the PSC patients having underlying IBD, particularly ulcerative colitis. PSC‐IBD represents a phenotypically different disease with a higher risk of hepatobiliary and colorectal malignancy compared to non‐IBD‐PSC. Unfortunately, the link between PSC and IBD is incompletely understood. 66 , 67 , 68 , 69 Both hereditary and environmental factors were shown to contribute to PSC development. It has been hypothesized that genetically predisposed pathways may initiate a persistent injury on cholangiocytes lining the bile ducts after being exposed to an environmental source. 26 , 65 With respect to the genetic susceptibility, a large cohort genome‐wide association study showed a strong correlation with specific human leukocyte antigens (HLA) class I, II and III regions. 26 , 70 Recently, novel insights were also gathered with regard to the role of peribiliary glands and the biliary tree stem cell compartment in the pathogenesis of PSC and related carcinogenesis. 71 , 72 , 73 There are three subtypes established for PSC being (i) classic PSC, affecting small and large bile ducts; (ii) small‐duct PSC, affecting only small bile ducts and (iii) PSC associated with autoimmune hepatitis, affecting small and large bile ducts. 65
3. SURGERY‐INDUCED RODENT MODELS
3.1. Obstructive cholestasis
The most frequently used surgery‐induced animal model of cholestasis relies on bile duct ligation (BDL), where a ligature is placed or surgical ligation is performed on the common bile duct. This surgical procedure creates an extrahepatic obstruction of the biliary system, which results in cholestasis and inflammation. 74 , 75 The BDL model induces acute extrahepatic obstructive biliary lesions, reflecting the clinical setting of gallstone liver disease and biliary atresia. 76 , 77 , 78 However, while BDL is stricto sensu an extrahepatic obstruction, the model has also been extensively used as platform for investigating the subsequent pathophysiological processes in hepatic morphology and function resulting from obstructive cholestasis. The BDL model has therefore also been widely used to study PSC. 74 , 75 , 79 , 80 , 81 The model was initially developed for rats, since they lack the gallbladder 82 , 83 and later successfully adapted to mice. 84 , 85 BDL in mice causes jaundice, release of transaminases, elevated bilirubin serum levels accompanied by development of bile duct proliferation with leukocyte infiltration, which eventually results in liver fibrosis. 86 In contrast to rats and humans, mice develop a biliary type of hepatocytic necrosis (bile infarcts) instead of apoptosis. 86 , 87 Additionally, unlike rats, the gallbladder of mice can drastically dilate after BDL and subsequently perforate resulting in bilioperitoneum, which may burgeon into death. Two strategies are followed to avoid these events, namely removal of the gall bladder via a cholecystectomy and placement of a surgical clip on the cystic duct. 88
In the last decade, modifications were also described of total BDL, including partial BDL and selective BDL. 85 , 87 , 89 Selective BDL is defined as ligating the left hepatic bile duct before it enters the common bile duct, while in partial BDL, a single ligation around the common bile duct is performed with leaving a defined lumen that allows a limited degree of bile flow. 87 The advantage of selective BDL is that the cholestatic injury is solely located in the left hepatic lobe, wherein bile infarcts develops as well as infiltrating neutrophils. 85 , 87 Partial BDL exhibits a less extensive tissue injury compared to selective BDL, believed to better mimic obstructive cholestasis in humans compared to total BDL. It should however be noted that cholestasis in this model seemed to spontaneously resolve after already 5 days, as a result partial BDL is particularly eligible for modelling acute cholestasis. 85
3.2. Cholangiopathies
Recently, the bile duct injection technique has been reported in which bile ducts are directly accessed without causing harm to the hepatobiliary tissue. 90 , 91 , 92 Studies have been reported injecting different types of solutions in the bile ducts varying from a harmless phosphate buffered saline solution to toxic dimethyl sulfoxide solutions, plasmid solutions and exogenous compounds including oxazolone. 90 , 91 , 92 The model has great potential to mimic several cholangiopathies, such as biliary atresia, PBC and PSC, depending on the causative agent that is being injected. The biggest advantage of this surgical model is the minimal injury to neighbouring tissue of the biliary tree. However, an important obstacle is the small anatomic proportion of mice (gall bladder has a diameter of 1‐2 mm), which complicate access to the biliary tree. Other drawbacks are the risk of peritonitis and possible leakage of bile or the injected solvent into the abdominal cavity. Noteworthy, a clear sex difference has been observed in this model. Female mice are described with a higher postoperative morbidity and mortality compared to male mice. 91
4. GENETICALLY MODIFIED MICE MODELS
Researchers have entered a new era of modelling human diseases concomitant with the emergence of genetically manipulating the laboratory mouse (Mus musculus). The mouse is particularly relevant due to its high degree of conversation with humans regarding its anatomy, physiology and genetics. 93 Genetically modified models are sometimes referred to as ‘spontaneous’ models or described to ‘spontaneously develop’ the respective liver pathology. The word ‘spontaneous’ indicates the absence of surgical or toxic/infectious insults to induce cholestasis. 52 , 94 , 95
4.1. Drug‐induced cholestasis
Malfunctioning of the BSEP, via drug inhibition, internalization, mutations or altered expression, drastically affects the bile acid homeostasis and inhibition of this transporter has been denoted as the key triggering factor of DIC. 48 , 49 Consequently, Bsep (Abcb11)‐/‐ mice may serve as an essential tool elaborating the consecutive effects of Bsep perturbance while identifying possible alternative bile acid transport systems. A shortcoming of this model is the development of mild non‐progressive cholestasis, presumably due to the more hydrophilic properties of endogenous bile acids in mice compared to humans. 96 The latter can be tackled by administering additional cholic acids to Bsep‐/‐ mice. 97 Finally, Bsep‐/‐ mice can additionally serve as a model for studying different genetic forms of cholestasis characterized by Bsep mutations, such as progressive familial cholestasis 2 and benign recurrent intrahepatic cholestasis 2. However, this goes beyond the scope of this review paper and thus will not further be discussed. 82 , 96
4.2. Primary biliary cholangitis
The first model created to mimic PBC consists of MRL/lpr mice bearing the lymphoproliferative (lpr) gene. 98 , 99 This gene results in the spontaneous development of severe autoimmune diseases, including lymphadenopathy, hypergammaglobulinaemia, glomerulonephritis, arthritis and Sjögren's syndrome. 98 The latter is frequently associated with PBC. 100 In accordance, MRL/lpr mice present PBC‐like histological features and infiltration of inflammatory cells, such as cluster differentiation 4 (CD4) + T cells and AMAs. The latter, however, only occurs in about 50% of the mice. In addition, MRL/lpr mice exhibit several other clinical features, incompatible with human PBC, including divergent bilirubin serum levels and hepatobiliary enzymes. 98 , 99 Ergo, development of additional models to study PBC urged, among them are mouse strains based on deficiencies in regulatory T (Treg) cells, including scurfy mice. 101 Scurfy mice contain a missense mutation in the transcription factor Forkhead box protein 3 (Foxp3) gene, essential in development, maintenance and function of Treg cells, resulting in complete abolition of CD4+ Foxp3+ Treg cells. 102 , 103 , 104 This experimental model was constructed on the assumption that mice genetically deficient in regulatory mechanisms that provide immune homeostasis, including naturally occurring Treg cells, would gain increased susceptibility for developing autoimmune diseases, such as PBC. 102 Indeed, scurfy mice manifest a similar serological, immunological and histopathological profile, albeit on the background of a multi‐system autoimmunity. 101 , 104 , 105 A similar disease phenotype was achieved in transgenic mice that express a dominant negative form of transforming growth factor‐β receptor restricted to T cells (dnTGF‐βRII mice), and in knockout mice with homozygous interleukin 2 receptor α (IL‐2Rα) deficiency, both containing a prominent dysfunction in Treg cell function. 101 Dysfunctional TGF‐β signalling results in reduced tolerance towards autoantigenic proteins present in the liver and leads to a PBC‐like liver disease, including the characteristic AMAs directed to mitochondrial autoantigens, such as PDH‐E2. 106 , 107 , 108 The discovery of PBC‐like disease in a child with homozygous IL‐2Rα deficiency 109 gave rise to the construction of the IL‐2Rα ‐/‐ mice. 110 Accordingly, both human PBC and IL‐2Rα ‐/‐ mice exhibit portal lymphoid infiltrates and increased numbers of cytokines. 111 dnTGF‐βRII and IL‐2Rα ‐/‐ mice are especially interesting to study acute mechanisms and address the role of Treg cells in PBC. 111 , 112 However, both models exhibit a coinciding IBD, absence of granulomas and lack of progression into chronic cholestasis. 105 , 111 , 113 , 114
An additional model was later developed to reduce the number of Treg cells, by introducing a mutation that disables the anion exchange protein 2 (AE2) gene. 101 AE2 encodes the Cl‐/HCO3 ‐ anion exchanger 2, which regulates the intracellular pH and transepithelial acid‐base transport, including secretin‐stimulated biliary bicarbonate excretion and proton gastric secretion. Thus, disturbed AE2 activity may have an influence on the bile acid equilibrium, but also gastric acid equilibrium. Regulation of pH appears an important aspect in modulating lymphocyte function. Indeed, mature Ae2a,b‐/‐ mice show immunological and hepatobiliary characteristics similar to PBC, but equally also PBC‐unlike alterations, including impaired gastric acid secretion and only 30%‐80% AMA. 113 A different strategy was used based on altered T‐cell functionality, called non‐obese diabetic (NOD)c3c4.mice. NOD mice develop an immune‐mediated destruction of pancreatic β cells, which underlies human type I diabetes and were created in the hope of identifying specific loci linked to the occurrence of type I diabetes. 115 This led to the discovery of complete abolishment of diabetes development when congenic segments from chromosomes 3 and 4 (NOD.c3 and NOD.c4) were used to replace the identified specific loci for insulin‐dependent diabetes (idd) in NOD mice. Rather than diabetes, these NOD.c3c4 mice develop an autoimmune biliary disease similar to PBC with a comparable immunologic and serologic profile (ie AMAs towards PDH‐E2). 95 , 116 Nevertheless, NODc3c4.mice also exhibit some PBC‐unspecific features, including biliary dilation and cystic lesions and absence of chronic destructive cholangitis. 114 , 117
No murine model was able to fully recapitulate the female preponderance present in human PBC, until the a new ‘designer’ mouse consisting of an adenylate‐uridylate‐rich element (ARE) deletion in the interferon γ gene that results in chronic expression of interferon γ. 118 ARE‐Del‐/‐ mice represent a phenotype very comparable to human PBC, particularly on histopathological, immunological and serological levels. Above all, this mouse model is the pioneer in exhibiting a female predominance in the matter of developing PBC, making this characteristic its biggest asset. Moreover, this specific advantage may finally open the research field in terms of elucidating the crucial mechanisms behind increased susceptibility of women in developing PBC. 114 , 119 , 120
4.3. Primary sclerosing cholangitis
One of the first genetically modified models to study hepatobiliary diseases related to PSC was described in 1994, the Mdr2 ‐/‐ mice. 121 The Mdr2 gene, orthologue of the human MDR3 (ABCB4) gene, encodes a transporter that secretes phospholipids into bile across the canalicular membrane. 122 As such, homozygous disruption of Mdr2 leads to absence of phospholipids (ie phosphatidylcholine) in bile and, thus interferes with the formation of biliary micelles resulting in liver injury. 122 This injury particularly translates into cholelithiasis and sclerosing cholangitis. The model was initially used to investigate the role of PFIC3 but also showed several histological lesions resembling human features of PSC, including the rapid progression into hepatic fibrosis. 52 , 122 , 123 , 124 The main drawback of the Mdr2 ‐/‐ mouse model for PSC is the absence of coexisting IBD and the spontaneous development of hepatocellular carcinoma in in vivo models. 94 , 123
A second model was created based on the observation that PSC and cystic fibrosis liver disease share several features being cholestasis, chronic inflammation and portal tract injury. 125 , 126 , 127 Moreover, abnormalities in the liver cystic fibrosis transmembrane conductance regulator (CFTR) gene product were earlier denoted to play a key role in the development of cholestasis in cystic fibrosis patients and, vice versa, mutations in the CFTR gene were also reported in PSC patients. 127 , 128 Hence, it was hypothesized that Cftr ‐/‐ mice, harbouring an exon 10 deletion in the Cftr gene, might also develop a similar cholestatic liver injury. Cftr ‐/‐ mice have been reported to, indeed, develop a progressive liver disease with focal cholangitis, inspissated bile and bile duct proliferation. 129 However, contrasting studies described Cftr ‐/‐ mice to only exhibit an intestinal phenotype with mild or non‐existing pathologies in other organs. 81 , 130 , 131 , 132 Moreover, this intestinal phenotype in Cftr ‐/‐ mice resembles more a distal intestinal obstruction syndrome and meconium ileus rather than an IBD phenotype typical for PSC. 69 , 129 , 133 Cftr ‐/‐ mice, as such, are therefore rarely preferred as experimental model for PSC.
Erythropoietic protoporphyria is an inherited disorder typified by accumulating protoporphyrins as a result of reduced ferrochelatase activity. Severe malfunctioning in this ferrochelatase activity predisposes to the development of severe liver diseases with a progressive character, which frequently require liver transplantation. 134 Mice with a homogeneous mutation in genes encoding the ferrochelatase enzyme (fch/fch) display an increasing number of protoporphyrin deposits in the lumen of small and large bile ducts, resulting in an incomplete obstruction. The toxic effect of the accumulating protoporhyrin additionally causes damage and hepatocyte death. 81 , 135 fch/fch mice present an extreme cholestatic phenotype with increased serum liver enzymes, increased bile salt levels and conjugated hyperbilirubinaemia, yet bile formation remains unchanged. 136 Not much is known about this model. A more in‐depth study is needed to characterize the longitudinal changes of fch/fch mice as well as underlying pathogenetic mechanisms.
5. CHEMICAL‐INDUCED RODENT MODELS
Chemical‐induced cholestasis is a well‐recognized problem attributed to compounds, such as drugs, pesticides, food additives, cosmetic ingredients or industrial compounds. 137 , 138 Different types of cholestatic liver injuries have been identified depending on the causative agent, varying from (hepato‐)canalicular, hepatocellular to ductular cholestasis. 139
5.1. Drug‐induced cholestasis
Chlorpromazine is an antipsychotic drug, which belongs to the largest class of first‐generation phenothiazines. 140 Although chlorpromazine is one of the three listed medicines in the World Health Organization's Essential Drug List for treating psychotic disorders, the drug contains many adverse effects, among which cholestasis. 141 Chlorpromazine is believed to induce cholestasis via direct BSEP inhibition in accessory to repressing BSEP and MDR3 gene expression. 142 Chlorpromazine has been commonly used to induce DIC in rodents while investigating its underlying mechanisms and the protective effect of chemicals against cholestasis. 143 , 144
Cyclosporin A is used to prevent graft rejection after organ transplantation. The two most serious side effects observed during cyclosporin A treatment are nephrotoxicity and hepatotoxicity, from which the latter is delineated as cholestasis with low to moderate hyperbilirubinaemia. 145 Cholestasis is presumably triggered by a number of factors, including direct inhibition and internalization of BSEP, disruption of the cytoskeleton and altered permeability of the canalicular membrane. 145 , 146 , 147 , 148 Similar to chlorpromazine, cyclosporin‐A‐treated rodents have been extensively employed as experimental model for DIC. 145 , 148 , 149 , 150 , 151
5.2. Gallstone liver disease
In 1964, Tepperman et al discovered that feeding mice a cholesterol‐cholic acid containing diet results in the formation of cholesterol gall stones in mice. 152 This lithogenic diet contains high fat, high cholesterol and 0.5% cholic acid. The combination of cholic acid with cholesterol is essential, cholesterol alone cannot induce gallstones. 152 Fujihera et al revealed the significant genetic contribution to gallstone formation in inbred mice. 153 Resultantly, numerous studies were performed on different mouse strains striving to identify the candidate genes behind the increased susceptibility towards gallstone development, among which Lith1 and Lith2 genes are believed to be major players. 154 , 155 , 156 , 157 Additional research are focussed on the impact of nuclear receptors, intestinal microbiota, gender and comorbidities, such as non‐alcoholic fatty liver disease, diabetes and obesity in the occurrence of gallstones. 157
5.3. Intrahepatic cholestasis of pregnancy
The first case of unexplained pruritus and jaundice in the last trimester of pregnancy was notified in 1883. 158 Still, the disease remained unnoticed until mid‐1950s. 159 Researchers started to highlight its importance and spread some of the drastic consequences the disease withholds, including premature births, fetal distress or even stillbirths. 160 As a result, research related to this subject was increased and resulted in the discovery of oestrogens, like estradiol‐17β‐D‐glucuronide, as inducers of reversible cholestasis in animals. 27 , 161 Estradiol‐17β‐D‐glucuronide inhibits BSEP and MDR1 transporters, initiates an internalization of transporters BSEP and multidrug resistance‐associated protein 2 (MRP2) and increases the permeability of tight junctions. 146 , 161 , 162 Administration of the synthetic ethinylestradiol or estradiol‐17β‐D‐glucuronide to rodents can be used to study the underlying mechanisms of ICP as well as DIC, since oestrogens are often used in oral contraceptives and hormone substitution therapies (ie menopausal hormone therapy). 163 , 164 , 165
5.4. Primary biliary cholangitis
2‐octynoic acid and 2‐nonynoic acid are chemicals regularly used as ingredients in food additives and perfumed cosmetics, such as toilet waters, soaps, detergents, lipstick, perfumes and facial creams, thanks to their violet scent. 137 , 166 2‐octynoic and 2‐nonynoic acid are mimics of lipoic acid and replace the lipoyl group of the immunodominant E2 domain of PDH (ie PDH‐E2), thereby generating a high specific and reactive antimitochondrial response. 166 This was also demonstrated in patients suffering from PBC, who presented an increased reactivity towards PDH‐E2 coupled with 2‐octynoic acid. 167 Accordingly, a xenobiotic‐induced model was designed by means of administering 2‐octynoic acid coupled with bovine serum album to mice. 168 , 169 , 170 These mice manifest auto‐immune cholangitis, concurrent with the typical AMAs of PBC. 170
5.5. Primary sclerosing cholangitis
α‐naphtylisothiocyanate (ANIT) is a chemical used for preparation of cationic aromatic urethane. 137 The compound is also well known among researchers for adequately inducing cholestasis in rats and mice, 79 , 171 by specifically targeting bile duct epithelial cells followed by hepatocellular necrosis. 137 , 172 Chronic and recurrent exposure to ANIT via the diet resembles an experimental setting of chronic cholangitis, bile duct hyperplasia and peribiliary fibrosis. 172 , 173 , 174 Moreover, mechanisms underlying peribiliary fibrosis in chronic ANIT models show similarities with other chronic cholestasis models, such as long‐term BDL and Mdr2 ‐/‐ mice. 172
3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine (DDC) is a second compound found to induce cholestasis. DDC is a porphyrinogenic agent and strong initiator of δ‐aminolevulinate synthetase. 137 , 175 Administration of DDC induces an increasing secretion of hepatotoxic protoporphyrins, concomitant with formation of protoporhyrin plugs. The latter induces an obstruction in the small bile ducts, which initiates cholestasis. 176 Moreover, DDC‐induced cholestasis is typified by sclerosing cholangitis and pronounced biliary fibrosis accompanied by ductular proliferation. 135 , 176 , 177 DDC feeding to rodents can therefore be applied to investigate (xenobiotic‐induced) chronic cholangiopathies. 82 , 177 Furthermore, lithocholic acid, a noxious endogenous bile acid, also induces cholestasis when present in abnormally high concentrations. 178 , 179 Accordingly, the effect of this endogenous bile acid has been widely studied in rodents to understand the potential role of the bile acid in the pathogenesis of cholestasis. 180 , 181 , 182 , 183 , 184 It has been postulated that cholestasis in lithocholic‐acid‐administered mice results from partial obstruction of the bile ducts, due to crystal formation (presumably lithocholic acid precipitates), along with bile infarcts, destructive cholangitis and periductal fibrosis within days. 185 The lithocholic acid rodent model is claimed a valid short‐term model in terms of investigating early changes in sclerosing cholangitis, thanks to the rapid induced changes after lithocholic acid administration. 186
6. VIRAL‐INDUCED MODELS
6.1. Biliary atresia
Although the aetiology of biliary atresia remains unclear, viral agents were proposed as one of the root causes of neonatal obstructive cholangiopathies, including biliary atresia. 187 Moreover, several studies discovered the presence of pathologic viruses inside the liver of biliary atresia patients namely, rotavirus, 188 , 189 reovirus, 190 , 191 cytomegalovirus 192 , 193 and human papillomavirus. 194 Together with these findings, a first murine model was developed based on the inoculation of the rhesus rotavirus to new‐born mice, which rendered a biliary atresia‐like obstruction of extrahepatic bile ducts. 195 Later on, this model has been frequently used to study disease mechanisms of biliary atresia. 196 , 197 , 198 Unfortunately, this model contains several practical limitations, including timing and dosing of the virus, which harms the reproducibility in addition to injection‐related injury to abdominal organs, inter‐strain differences and a low survival rate. 199 Bile duct proliferation and lymphocyte infiltration inside the portal triads are apparent in the model, resembling features of human biliary atresia, albeit without liver fibrosis. 199
7. COMBINATION MODELS
To achieve a better reflection of the in vivo situation, well‐acknowledged rodent models can be combined and yield an improved rodent model of cholestasis. These combination models regularly consist of genetically modified rodent models or a surgery‐induced model merged with a chemical‐induced rodent model. 97 , 127 , 200 , 201 , 202
7.1. Drug‐induced cholestasis
BSEP disturbance is well known as one of the key players in DIC, yet Bsep ‐/‐ mice only show mild non‐progressive cholestasis and lack important histopathological features of cholestasis. Adding cholic acids to the diet of Bsep ‐/‐ mice can be used to overcome this drawback by (i) altering the hydrophobicity of the bile acid pool, (ii) saturating bile acid hydroxylation and (iii) resulting in a more pronounced intrahepatic accumulation of hydrophobic bile acids. Accordingly, male Bsep ‐/‐ mice supplemented with cholic acids manifested severe progressive cholestasis associated with periportal fibrosis, inflammation and cell death. 97 , 203
7.2. Primary sclerosing cholangitis
Next to liver injury, about 70%‐86% of the PSC patients present an IBD, most frequently ulcerative colitis. 26 To investigate concomitant IBD presentation, additional toxics are used, including water‐soluble dextran sodium sulphate (DSS). 127 , 200 , 201 DSS is negatively charged, water‐soluble and contains a highly variable molecular weight between 5 and 1400 kilodalton. This sulphated polysaccharide can be administered via drinking water, after which it can damage the monolayer of epithelial cells lining the large intestine, hereby disrupting the upper barrier. As a result, intestinal contents, including bacteria and their products, can disseminate into underlying tissue and cause an inflammatory response resulting in an IBD phenotype. 204 2,4,6‐trinitrobenzenesulfonice acid (TNBS) is a chemical that can act as a hapten upon coupling with lysine moieties in proteins and provoke a cell‐mediated immune response. 205 Initially, the compound was used for inducing inflammatory colitis in rats by means of intracolonic instillation of TNBS. 206 The successful use of TNBS for evoking an inflammatory disease of the colon raised the question what the inflammatory effect of TNBS would be on the liver, when locally administered. Consequently, new models were created that inject TNBS in the portal vein and in the ductus choledochus (ie intracholedochal). TNBS administration in the portal vein yields an active liver injury in rats with PSC‐characteristic antineutrophil cytoplasmic antibodies, albeit without the involvement of the biliary tree 207 while intracholedochal injection does involve the biliary tree and induces a rather mild chronic cholangitis. 205 Orth and colleagues made a first attempt in combining models by administering TNBS to BDL rats. 202 The latter resulted in a PSC‐like appearance featured with irregularities in the intra‐ and extrahepatic bile ducts, inflammation and the development of antineutrophil cytoplasmic antibodies. Although this model clearly shows an improvement compared to the single hit model, coinciding IBDs are still missing. 202 , 208 , 209
To integrate the IBD component, previously defined in vivo models can also be conjugated with DSS feeding. To date, three such combinational models have been described in Cftr ‐/‐ mice or Mdr2 ‐/‐ mice. 127 , 176 , 200 , 201 , 204 , 210 , 211 Administering DSS to Mdr2‐/‐ mice results in an exacerbated sclerosing cholangitis along with increased ductular reaction and bridging fibrosis. 201 Similarly, DSS can be administered to Cftr ‐/‐ mice, producing a significant increase in bile duct injury compared to the phenotype observed in Cftr ‐/‐ mice. Yet, fibrosis remains absent and a clear association with CFTR dysfunction cannot be identified, since homozygote, heterozygote and wild‐type mice manifest similar phenotypes. 127 A follow‐up study was performed and indicates the need of three separate factors to elicit bile duct injury accompanied by progressive periductal fibrosis in Cftr ‐/‐ mice. Firstly, CFTR functionality should be harmed (ie Cftr ‐/‐ mice). Secondly, mice should be exposed to profibrogenic activators (ie DDC feeding). Thirdly, intestinal permeability needs to be disturbed (ie DSS feeding). Altogether, this one‐of‐a‐kind model may serve as an ideal tool to elaborate the full pathogenic mechanism by which CFTR dysfunction predisposes fibrotic liver diseases. 200
8. DRAWBACKS OF CURRENT IN VIVO RODENT MODELS
Liver diseases are a major cause of illness and mortality accounting for up to 2 million deaths per year. 212 , 213 Many researchers have focused on unravelling the aetiology and pathogenesis of liver diseases, including cholestatic liver disease. In vivo models have led to numerous breakthroughs in this field. However, they are also associated with inter‐ and intraspecies differences as well as model‐specific bottlenecks, which urges an additional consideration to facilitate proper selection and application of the described models.
8.1. Intraspecies differences
Three important aspects deserve specific attention, namely strain, gender and age. To exemplify, conflicting results were reported using Cftr ‐/‐ mice and DDC‐fed mice (partially) according to their genetic background. Cftr ‐/‐ mice present a progressive hepatobiliary disease in C57BL/6 strains while other strains only exhibit a strong intestinal phenotype, but lack any abnormality in the liver. 81 In addition, DDC feeding appears to specifically result in the highest degree of large duct disease when using Swiss albino mice. 177 Similarly, inadequate gender selection can interfere considerably in establishing a sensitive model of cholestasis. Indeed, male mice were considered more suitable for BDL than female counterparts because of lower mortality rates; however, the latter does not relate to Sv129 mice. 86 , 214 Gender differences may also enhance the modelling of PBC or PSC, considering their female or male preponderance, as is the case for the ARE Del‐/‐ mice model and Mdr2 ‐/‐ mice model respectively. 58 , 118 , 124 Likewise, the age of an animal should not be neglected, as the inflammatory response may alter in function of the rodents’ age. 215
8.2. Interspecies differences
The interspecies differences that should be encountered when modelling cholestasis include (i) a more hydrophilic bile acid composition in rodents compared to humans, (ii) differences in the substrate specificity of bile acid transporters, (iii) alterations in metabolic detoxification and (iv) hepatocellular excretion of bile acids and drugs. 216 , 217 Moreover, Cftr ‐/‐ knockouts and DDC feeding are not feasible in rats; hence, variation in the clinical phenotype between mice and rats may also occur. 129 , 177 , 218 , 219 Experimental in vivo models should preferably be benchmarked towards human samples. This strategy could also identify additional crucial interspecies differences, not yet established, and hence complement some missing gaps in the mechanistic understanding of the cholestatic pathology. It would be highly relevant to use human‐derived in vitro models and in silico modelling, based on structural and/or physical‐chemical parameters, in addition to in vivo models. These models lack the burden of a complex environment, allow high‐throughput screening and may further improve the accuracy in predicting cholestasis while keeping interspecies differences in mind. 49 , 220
8.3. Specific bottleneck within a model
In addition to inter‐and intraspecies differences, it is of the utmost importance to be adequately informed about specific aspects of a certain model relevant for the respective applicability domain(s) (Table 1). Additionally, a practical workflow is provided that guides researchers to make an informed decision in the most appropriate in vivo rodent model currently available while considering the specific bottlenecks (Figure 2).
FIGURE 2.

Practical workflow for selecting of the most applicable in vivo rodent model considering the applicability domain, advantages and disadvantages. This figure was created with Lucidchart software. The selection of experimental models was based on the information available in literature, as such the most appropriate well‐described models were integrated. AE2, anion exchange protein 2; AMA, antimitochondrial antibody; ARE‐Del, (deletion in) adenylate‐uridylate‐rich element; BSEP, bile salt export pump; CFTR, cystic fibrosis transmembrane conductance regulator; DDC, 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine; DSS, dextran sodium sulphate; dnTGF‐βRII mice, dominant negative form of transforming growth factor‐β receptor restricted to T cells; IL‐2Rα, interleukin 2 receptor α; NODc3c4, non‐obese diabetic with B6/B10 region on chromosomes 3 and 4; PBC, primary biliary cholangitis; PDH‐E2, E2 subunit of the pyruvate dehydrogenase; PSC, primary sclerosing cholangitis; TNBS, 2,4,6‐trinitrobenzenesulfonice acid
9. CONCLUSION
Considerable fundamental clues have been unveiled with the generation of experimental animal models related to the pathophysiological mechanisms of biliary and cholestatic diseases. Although extensive information has been gathered in the last couple of decades with regard to the mechanistic understanding of cholestatic liver diseases, still considerable gaps are left behind due to inadequate animal models exemplified by the difficulty of inducing the female preponderance of PBC and the coincidence of PSC and IBD. Although breakthroughs are made in the design of new models, which try to tackle these setbacks, still no experimental animal is able to perfectly mimic the real cholestasis phenotype seen in humans. For this reason, we should acknowledge the specific limitation of each model and design novel models by means of combining multiple in vivo rodent models, hereby circumventing limitations while increasing the assets. A number of these combination models have already been described in the last decade but are still limited. Further exploration of these combination models will undoubtedly pave the way for improved resemblance of human cholestasis in vivo and provide the possibility of more accurate mechanistic and therapeutic studies.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
Gijbels E, Pieters A, De Muynck K, Vinken M, Devisscher L. Rodent models of cholestatic liver disease: A practical guide for translational research. Liver Int. 2021;41:656–682. 10.1111/liv.14800
Mathieu Vinken and Lindsey Devisscher share equal seniorship.
Funding information
This work was supported by grants of the Research Foundation Flanders, Belgium and the Scientific Fund Willy Gepts, Belgium and the Center for Alternatives to Animal Testing at Johns Hopkins University, USA.
Handling Editor: Emma Andersson
REFERENCES
Author names in bold designate shared co‐first authorship.
- 1. European Association for the Study of the Liver (EASL) . Clinical Practice Guidelines: Management of cholestatic liver diseases. J Hepatology. 2009;51:237‐267. [DOI] [PubMed] [Google Scholar]
- 2. Noor F. A shift in paradigm towards human biology‐based systems for cholestatic‐liver diseases. J Physiol. 2015;593:5043‐5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nguyen KD, Sundaram V, Ayoub WS. Atypical causes of cholestasis. World J Gastroenterol. 2014;20:9418‐9426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sundaram V, Björnsson ES. Drug‐induced cholestasis. Hepatol Commun. 2017;1:726‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kochhar G, Parungao JM, Hanouneh IA, et al. Biliary complications following liver transplantation. World J Gastroenterol. 2013;19:2841‐2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Amer S, Hajira A. A comprehensive review of progressive familial intrahepatic cholestasis (PFIC): genetic disorders of hepatocanalicular transporters. Gastroenterology Res. 2014;7:39‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Strubbe B, Geerts A, Van Vlierberghe H, et al. Progressive familial intrahepatic cholestasis and benign recurrent intrahepatic cholestasis: a review. Acta Gastroenterol Belg. 2012;75:405‐410. [PubMed] [Google Scholar]
- 8. Asai A, Miethke A, Bezerra JA. Pathogenesis of biliary atresia: defining biology to understand clinical phenotypes. Nat Rev Gastroenterol Hepatol. 2015;12:342‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Govindarajan KK. Biliary atresia: where do we stand now? World journal of hepatology. 2016;8:1593‐1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Poddar U, Thapa BR, Das A, et al. Neonatal cholestasis: differentiation of biliary atresia from neonatal hepatitis in a developing country. Acta Paediatr. 2009;98:1260‐1264. [DOI] [PubMed] [Google Scholar]
- 11. Shah I, Bhatnagar S, Dhabe H. Clinical and biochemical factors associated with biliary atresia. Trop Gastroenterol. 2012;33:214‐217. [DOI] [PubMed] [Google Scholar]
- 12. Davit‐Spraul A, Gonzales E, Baussan C, et al. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009;4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shin M, Joh J‐W. Advances in endoscopic management of biliary complications after living donor liver transplantation: comprehensive review of the literature. World J Gastroenterol. 2016;22:6173‐6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mastoraki A, Kriaras I, Sfirakis P, et al. Biliary complications after cardiovascular procedures. Crit Care. 2006;10:P269. [Google Scholar]
- 15. Oida T, Kano H, Mimatsu K, et al. Cholecystitis or cholestasis after total gastrectomy and esophagectomy. Hepatogastroenterology. 2012;59:1455‐1457. [DOI] [PubMed] [Google Scholar]
- 16. Peer A, Hendel D, Halperin N. Acute cholecystitis and cholestasis as a complication in hip surgery. Injury. 1982;14:159‐161. [DOI] [PubMed] [Google Scholar]
- 17. Farthing M, Roberts SE, Samuel DG, et al. Survey of digestive health across Europe: Final report. Part 1: The burden of gastrointestinal diseases and the organisation and delivery of gastroenterology services across Europe. United European Gastroenterol J. 2014;2:539‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Everhart JE, Ruhl CE. Burden of digestive diseases in the United States Part III: Liver, biliary tract, and pancreas. Gastroenterology. 2009;136:1134‐1144. [DOI] [PubMed] [Google Scholar]
- 19. Di Ciaula A, Portincasa P. Recent advances in understanding and managing cholesterol gallstones. F1000Res. 2018;7:1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Njeze GE. Gallstones. Niger J Surg. 2013;19:49‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tanaja J, Lopez RA, Meer JM. Cholelithiasis. In: StatPearls, ed. Treasure Island, FL: StatPearls Publishing, 2020. [PubMed] [Google Scholar]
- 22. Chen X, Yan X‐R, Zhang L‐P. Ursodeoxycholic acid after common bile duct stones removal for prevention of recurrence: a systematic review and meta‐analysis of randomized controlled trials. Medicine. 2018;97:e13086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boonstra K, Beuers U, Ponsioen CY. Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: a systematic review. J Hepatology. 2012;56:1181‐1188. [DOI] [PubMed] [Google Scholar]
- 24. Lee YM, Kaplan MM. The natural history of PBC: has it changed? Semin Liver Dis. 2005;25:321‐326. [DOI] [PubMed] [Google Scholar]
- 25. Combes B, Carithers RL Jr, Maddrey WC, et al. A randomized, double‐blind, placebo‐controlled trial of ursodeoxycholic acid in primary biliary cirrhosis. Hepatology. 1995;22:759‐766. [PubMed] [Google Scholar]
- 26. Lazaridis KN, LaRusso NF. Primary sclerosing cholangitis. N Engl J Med. 2016;375:1161‐1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Forker EL. The effect of estrogen on bile formation in the rat. J Clin Invest. 1969;48:654‐663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wood AM, Livingston EG, Hughes BL, et al. Intrahepatic cholestasis of pregnancy: a review of diagnosis and management. Obstet Gynecol Surv. 2018;73:103‐109. [DOI] [PubMed] [Google Scholar]
- 29. Pusl T, Beuers U. Intrahepatic cholestasis of pregnancy. Orphanet J Rare Dis. 2007;2:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Floreani A, Gervasi MT. New insights on intrahepatic cholestasis of pregnancy. Clin Liver Dis. 2016;20:177‐189. [DOI] [PubMed] [Google Scholar]
- 31. Bhamidimarri KR, Schiff E. Drug‐induced cholestasis. Clin Liver Dis. 2013;17(4):519‐531. [DOI] [PubMed] [Google Scholar]
- 32. Olson H, Betton G, Robinson D, et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul Toxicol Pharmacol. 2000;32:56‐67. [DOI] [PubMed] [Google Scholar]
- 33. Ozer J, Ratner M, Shaw M, et al. The current state of serum biomarkers of hepatotoxicity. Toxicology. 2008;245:194‐205. [DOI] [PubMed] [Google Scholar]
- 34. Xu JJ, Henstock PV, Dunn MC, et al. Cellular imaging predictions of clinical drug‐induced liver injury. Toxicol Sci. 2008;105:97‐105. [DOI] [PubMed] [Google Scholar]
- 35. Johnston DE, Kaplan MM. Pathogenesis and treatment of gallstones. N Engl J Med. 1993;328:412‐421. [DOI] [PubMed] [Google Scholar]
- 36. Trotman BW, Bernstein SE, Bove KE, et al. Studies on the pathogenesis of pigment gallstones in hemolytic anemia: description and characteristics of a mouse model. J Clin Invest. 1980;65:1301‐1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stewart L, Oesterle AL, Erdan I, et al. Pathogenesis of pigment gallstones in Western societies: the central role of bacteria. J Gastrointest Surg. 2002;6(6):891‐904. [DOI] [PubMed] [Google Scholar]
- 38. Everson GT. Gallbladder function in gallstone disease. Gastroenterol Clin North Am. 1991;20:85‐110. [PubMed] [Google Scholar]
- 39. Hazem ZM. Acute biliary pancreatitis: diagnosis and treatment. Saudi J Gastroenterol. 2009;15:147‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Russo P, Magee JC, Boitnott J, et al. Design and validation of the biliary atresia research consortium histologic assessment system for cholestasis in infancy. Clin Gastroenterol Hepatol. 2011;9(357–362):e352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chardot C. Biliary atresia. Orphanet journal of rare diseases. 2006;1:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Harpavat S, Finegold MJ, Karpen SJ. Patients with biliary atresia have elevated direct/conjugated bilirubin levels shortly after birth. Pediatrics. 2011;128:e1428‐e1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Davenport M, Savage M, Mowat AP, et al. Biliary atresia splenic malformation syndrome: an etiologic and prognostic subgroup. Surgery. 1993;113:662‐668. [PubMed] [Google Scholar]
- 44. Schwarz KB, Haber BH, Rosenthal P, et al. Extrahepatic anomalies in infants with biliary atresia: results of a large prospective North American multicenter study. Hepatology. 2013;58:1724‐1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brindley SM, Lanham AM, Karrer FM, et al. Cytomegalovirus‐specific T‐cell reactivity in biliary atresia at the time of diagnosis is associated with deficits in regulatory T cells. Hepatology. 2012;55:1130‐1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Padda MS, Sanchez M, Akhtar AJ, et al. Drug‐induced cholestasis. Hepatology. 2011;53:1377‐1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Walker CO, Combes B. Biliary cirrhosis induced by chlorpromazine. Gastroenterology. 1966;51:631‐640. [PubMed] [Google Scholar]
- 48. Vinken M, Landesmann B, Goumenou M, et al. Development of an adverse outcome pathway from drug‐mediated bile salt export pump inhibition to cholestatic liver injury. Toxicol Sci. 2013;136:97‐106. [DOI] [PubMed] [Google Scholar]
- 49. Gijbels E, Vilas‐Boas V, Deferm N, et al. Mechanisms and in vitro models of drug‐induced cholestasis. Arch Toxicol. 2019;93:1169‐1186. [DOI] [PubMed] [Google Scholar]
- 50. Jacquemin E, Cresteil D, Manouvrier S, et al. Heterozygous non‐sense mutation of the MDR3 gene in familial intrahepatic cholestasis of pregnancy. Lancet. 1999;353:210‐211. [DOI] [PubMed] [Google Scholar]
- 51. Pauli‐Magnus C, Lang T, Meier Y, et al. Sequence analysis of bile salt export pump (ABCB11) and multidrug resistance p‐glycoprotein 3 (ABCB4, MDR3) in patients with intrahepatic cholestasis of pregnancy. Pharmacogenetics. 2004;14:91‐102. [DOI] [PubMed] [Google Scholar]
- 52. Lammert F, Wang DQ, Hillebrandt S, et al. Spontaneous cholecysto‐ and hepatolithiasis in Mdr2‐/‐ mice: a model for low phospholipid‐associated cholelithiasis. Hepatology. 2004;39:117‐128. [DOI] [PubMed] [Google Scholar]
- 53. Germain AM, Carvajal JA, Glasinovic JC, et al. Intrahepatic cholestasis of pregnancy: an intriguing pregnancy‐specific disorder. J Soc Gynecol Investig. 2002;9:10‐14. [DOI] [PubMed] [Google Scholar]
- 54. Reyes H, Báez ME, González MC, et al. Selenium, zinc and copper plasma levels in intrahepatic cholestasis of pregnancy, in normal pregnancies and in healthy individuals, in Chile. J Hepatol. 2000;32:542‐549. [DOI] [PubMed] [Google Scholar]
- 55. Bacq Y, le Besco M, Lecuyer AI, et al. Ursodeoxycholic acid therapy in intrahepatic cholestasis of pregnancy: results in real‐world conditions and factors predictive of response to treatment. Dig Liver Dis. 2017;49:63‐69. [DOI] [PubMed] [Google Scholar]
- 56. Joutsiniemi T, Timonen S, Linden M, et al. Intrahepatic cholestasis of pregnancy: observational study of the treatment with low‐dose ursodeoxycholic acid. BMC Gastroenterol. 2015;15:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261‐1273. [DOI] [PubMed] [Google Scholar]
- 58. Marchioni Beery RM, Vaziri H, Forouhar F. Primary biliary cirrhosis and primary sclerosing cholangitis: a review featuring a women's health perspective. J Clin Transl Hepatol. 2014;2:266‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Purohit T, Cappell MS. Primary biliary cirrhosis: pathophysiology, clinical presentation and therapy. World journal of hepatology. 2015;7:926‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bittencourt PL, Farias AQ, Abrantes‐Lemos CP, et al. Prevalence of immune disturbances and chronic liver disease in family members of patients with primary biliary cirrhosis. J Gastroenterol Hepatol. 2004;19:873‐878. [DOI] [PubMed] [Google Scholar]
- 61. Jones DE, Donaldson PT. Genetic factors in the pathogenesis of primary biliary cirrhosis. Clin Liver Dis. 2003;7:841‐864. [DOI] [PubMed] [Google Scholar]
- 62. Springer JE, Cole DE, Rubin LA, et al. Vitamin D‐receptor genotypes as independent genetic predictors of decreased bone mineral density in primary biliary cirrhosis. Gastroenterology. 2000;118:145‐151. [DOI] [PubMed] [Google Scholar]
- 63. Invernizzi P, Selmi C, Ranftler C, et al. Antinuclear antibodies in primary biliary cirrhosis. Semin Liver Dis. 2005;25:298‐310. [DOI] [PubMed] [Google Scholar]
- 64. Kouroumalis E, Notas G. Pathogenesis of primary biliary cirrhosis: a unifying model. World J Gastroenterol. 2006;12:2320‐2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rawla P, Samant H. Primary sclerosing cholangitis. In: StatPearls, ed. Treasure Island, FL: StatPearls Publishing, 2020. [PubMed] [Google Scholar]
- 66. Silveira MG, Lindor KD. Primary sclerosing cholangitis. Can J Gastroenterol. 2008;22:689‐698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Talwalkar JA, Lindor KD. Primary sclerosing cholangitis. Inflamm Bowel Dis. 2005;11:62‐72. [DOI] [PubMed] [Google Scholar]
- 68. Wiesner RH, LaRusso NF. Clinicopathologic features of the syndrome of primary sclerosing cholangitis. Gastroenterology. 1980;79:200‐206. [PubMed] [Google Scholar]
- 69. Mertz A, Nguyen NA, Katsanos KH, et al. Primary sclerosing cholangitis and inflammatory bowel disease comorbidity: an update of the evidence. Ann Gastroenterol. 2019;32:124‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Karlsen TH, Franke A, Melum E, et al. Genome‐wide association analysis in primary sclerosing cholangitis. Gastroenterology. 2010;138:1102‐1111. [DOI] [PubMed] [Google Scholar]
- 71. Carpino G, Nevi L, Overi D, et al. Peribiliary gland niche participates in biliary tree regeneration in mouse and in human primary sclerosing cholangitis. Hepatology. 2020;71:972‐989. [DOI] [PubMed] [Google Scholar]
- 72. Carpino G, Cardinale V, Renzi A, et al. Activation of biliary tree stem cells within peribiliary glands in primary sclerosing cholangitis. J Hepatol. 2015;63:1220‐1228. [DOI] [PubMed] [Google Scholar]
- 73. Carpino G, Cardinale V, Folseraas T, et al. Neoplastic transformation of the peribiliary stem cell niche in cholangiocarcinoma arisen in primary sclerosing cholangitis. Hepatology. 2019;69:622‐638. [DOI] [PubMed] [Google Scholar]
- 74. Tag CG, Sauer‐Lehnen S, Weiskirchen S, et al. Bile duct ligation in mice: induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J Vis Exp. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Van Campenhout S, Van Vlierberghe H, Devisscher L. Common bile duct ligation as model for secondary biliary cirrhosis. In: Vinken M, ed. Methods Mol Biol, vol. 1981. New York: Humana Press; 2019:237‐247. [DOI] [PubMed] [Google Scholar]
- 76. Mariotti V, Strazzabosco M, Fabris L, et al. Animal models of biliary injury and altered bile acid metabolism. Biochim Biophys Acta Mol Basis Dis. 2018;1864(4):1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Geerts AM, Vanheule E, Praet M, et al. Comparison of three research models of portal hypertension in mice: macroscopic, histological and portal pressure evaluation. Int J Exp Pathol. 2008;89:251‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Garrido M, Escobar C, Zamora C, et al. Bile duct ligature in young rats: a revisited animal model for biliary atresia. Eur J Histochem. 2017;61:2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Plaa GL, Priestly BG. Intrahepatic cholestasis induced by drugs and chemicals. Pharmacol Rev. 1976;28:207‐273. [PubMed] [Google Scholar]
- 80. Kountouras J, Billing BH, Scheuer PJ. Prolonged bile duct obstruction: a new experimental model for cirrhosis in the rat. Br J Exp Pathol. 1984;65:305‐311. [PMC free article] [PubMed] [Google Scholar]
- 81. Pollheimer MJ, Trauner M, Fickert P. Will we ever model PSC? ‐ "it's hard to be a PSC model!". Clin Res Hepatol Gastroenterol. 2011;35:792‐804. [DOI] [PubMed] [Google Scholar]
- 82. Mariotti V, Strazzabosco M, Fabris L, et al. Animal models of biliary injury and altered bile acid metabolism. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1254‐1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rodriguez‐Garay EA, Aguero RM, Pisani G, et al. Rat model of mild stenosis of the common bile duct. Res Exp Med. 1996;196:105‐116. [DOI] [PubMed] [Google Scholar]
- 84. Miyoshi H, Rust C, Roberts PJ, et al. Hepatocyte apoptosis after bile duct ligation in the mouse involves Fas. Gastroenterology. 1999;117:669‐677. [DOI] [PubMed] [Google Scholar]
- 85. Heinrich S, Georgiev P, Weber A, et al. Partial bile duct ligation in mice: a novel model of acute cholestasis. Surgery. 2011;149:445‐451. [DOI] [PubMed] [Google Scholar]
- 86. Georgiev P, Jochum W, Heinrich S, et al. Characterization of time‐related changes after experimental bile duct ligation. Br J Surg. 2008;95:646‐656. [DOI] [PubMed] [Google Scholar]
- 87. Fickert P, Zollner G, Fuchsbichler A, et al. Ursodeoxycholic acid aggravates bile infarcts in bile duct‐ligated and Mdr2 knockout mice via disruption of cholangioles. Gastroenterology. 2002;123:1238‐1251. [DOI] [PubMed] [Google Scholar]
- 88. Starkel P, Leclercq IA. Animal models for the study of hepatic fibrosis. Best Pract Res Clin Gastroenterol. 2011;25:319‐333. [DOI] [PubMed] [Google Scholar]
- 89. Yokota S, Ono Y, Nakao T, et al. Partial bile duct ligation in the mouse: A controlled model of localized obstructive cholestasis. JoVE. 2018:56930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yamada D, Rizvi S, Razumilava N, et al. IL‐33 facilitates oncogene‐induced cholangiocarcinoma in mice by an interleukin‐6‐sensitive mechanism. Hepatology. 2015;61:1627‐1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Berntsen NL, Fosby B, Valestrand L, et al. Establishment of a surgical bile duct injection technique giving direct access to the bile ducts for studies of the murine biliary tree. Am J Physiol Gastrointest Liver Physiol. 2018;314:G349‐G359. [DOI] [PubMed] [Google Scholar]
- 92. Berntsen NL, Fosby B, Tan C, et al. Natural killer T cells mediate inflammation in the bile ducts. Mucosal Immunol. 2018;11:1582‐1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gurumurthy CB, Lloyd KCK. Generating mouse models for biomedical research: technological advances. Dis Model Mech. 2019;12(1):dmm029462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Fickert P, Pollheimer MJ, Beuers U, et al. Characterization of animal models for primary sclerosing cholangitis (PSC). J Hepatology. 2014;60:1290‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Koarada S, Wu Y, Fertig N, et al. Genetic control of autoimmunity: protection from diabetes, but spontaneous autoimmune biliary disease in a nonobese diabetic congenic strain. J Immunol. 2004;173:2315‐2323. [DOI] [PubMed] [Google Scholar]
- 96. Gooijert KE, Havinga R, Wolters H, et al. The mechanism of increased biliary lipid secretion in mice with genetic inactivation of bile salt export pump. Am J Physiol Gastrointest Liver Physiol. 2015;308:G450‐G457. [DOI] [PubMed] [Google Scholar]
- 97. Wang R, Lam P, Liu L, et al. Severe cholestasis induced by cholic acid feeding in knockout mice of sister of P‐glycoprotein. Hepatology. 2003;38:1489‐1499. [DOI] [PubMed] [Google Scholar]
- 98. Tsuneyama K, Nose M, Nisihara M, et al. Spontaneous occurrence of chronic non‐suppurative destructive cholangitis and antimitochondrial autoantibodies in MRL/lpr mice: possible animal model for primary biliary cirrhosis. Pathology Int. 2001;51:418‐424. [DOI] [PubMed] [Google Scholar]
- 99. Ohba K, Omagari K, Murase K, et al. A possible mouse model for spontaneous cholangitis: serological and histological characteristics of MRL/lpr mice. Pathology. 2002;34:250‐256. [DOI] [PubMed] [Google Scholar]
- 100. Ito M, Kojima T, Miyata M, et al. Primary biliary cirrhosis (PBC)‐CREST (calcinosis, Raynaud's phenomenon, esophageal dysfunction, sclerodactyly and telangiectasia) overlap syndrome complicated by Sjögren's syndrome and arthritis. Intern Med. 1995;34:451‐454. [DOI] [PubMed] [Google Scholar]
- 101. Webb GJ, Siminovitch KA, Hirschfield GM. The immunogenetics of primary biliary cirrhosis: a comprehensive review. J Autoimmun. 2015;64:42‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhang W, Sharma R, Ju S‐T, et al. Deficiency in regulatory T cells results in development of antimitochondrial antibodies and autoimmune cholangitis. Hepatology. 2009;49:545‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Brunkow ME, Jeffery EW, Hjerrild KA, et al. Disruption of a new forkhead/winged‐helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68‐73. [DOI] [PubMed] [Google Scholar]
- 104. Hadaschik EN, Wei X, Leiss H, et al. Regulatory T cell‐deficient scurfy mice develop systemic autoimmune features resembling lupus‐like disease. Arthritis Res Ther. 2015;17:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Tsuneyama K, Moritoki Y, Kikuchi K, et al. Pathological features of new animal models for primary biliary cirrhosis. Int J Hepatol. 2012;2012:403954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Oertelt S, Lian ZX, Cheng CM, et al. Anti‐mitochondrial antibodies and primary biliary cirrhosis in TGF‐ receptor II dominant‐negative mice. J Immunol. 2006;177:1655‐1660. [DOI] [PubMed] [Google Scholar]
- 107. Gressner AM, Weiskirchen R, Breitkopf K, et al. Roles of TGF‐beta in hepatic fibrosis. Front Biosci. 2002;7:d793‐807. [DOI] [PubMed] [Google Scholar]
- 108. Moritoki Y, Zhang W, Tsuneyama K, et al. B cells suppress the inflammatory response in a mouse model of primary biliary cirrhosis. Gastroenterology. 2009;136:1037‐1047. [DOI] [PubMed] [Google Scholar]
- 109. Sharfe N, Dadi HK, Shahar M, et al. Human immune disorder arising from mutation of the alpha chain of the interleukin‐2 receptor. Proc Natl Acad Sci USA. 1997;94:3168‐3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Aoki CA, Roifman CM, Lian Z‐X, et al. IL‐2 receptor alpha deficiency and features of primary biliary cirrhosis. J Autoimmun. 2006;27:50‐53. [DOI] [PubMed] [Google Scholar]
- 111. Wakabayashi K, Lian Z‐X, Moritoki Y, et al. IL‐2 receptor α−/− mice and the development of primary biliary cirrhosis. Hepatology. 2006;44:1240‐1249. [DOI] [PubMed] [Google Scholar]
- 112. Chuang Y‐H, Lian Z‐X, Yang G‐X, et al. Natural killer T cells exacerbate liver injury in a transforming growth factor β receptor II dominant‐negative mouse model of primary biliary cirrhosis. Hepatology. 2008;47:571‐580. [DOI] [PubMed] [Google Scholar]
- 113. Salas JT, Banales JM, Sarvide S, et al. Ae2a, b‐deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis. Gastroenterology. 2008;134:1482‐1493. [DOI] [PubMed] [Google Scholar]
- 114. Tanaka A, Leung PSC, Young HA, et al. Therapeutic and immunological interventions in primary biliary cholangitis: from mouse models to humans. Arch Med Sci. 2018;14:930‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bach JF, Mathis D. The NOD mouse. Res Immunol. 1997;148:285‐286. [DOI] [PubMed] [Google Scholar]
- 116. Mason AL. An autoimmune biliary disease mouse model for primary biliary cirrhosis: something for everyone. Hepatology. 2006;44:1047‐1050. [DOI] [PubMed] [Google Scholar]
- 117. Irie J, Wu Y, Wicker LS, et al. NOD.c3c4 congenic mice develop autoimmune biliary disease that serologically and pathogenetically models human primary biliary cirrhosis. J Exp Med. 2006;203:1209‐1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Vergani D, Mieli‐Vergani G. Mouse model of primary biliary cholangitis with a striking female predominance: a new powerful research tool. Hepatology. 2016;64:1024‐1027. [DOI] [PubMed] [Google Scholar]
- 119. Bae HR, Leung PSC, Tsuneyama K, et al. Chronic expression of interferon‐gamma leads to murine autoimmune cholangitis with a female predominance. Hepatology. 2016;64:1189‐1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hodge DL, Berthet C, Coppola V, et al. IFN‐gamma AU‐rich element removal promotes chronic IFN‐gamma expression and autoimmunity in mice. J Autoimmun. 2014;53:33‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Mauad TH, van Nieuwkerk CM, Dingemans KP, et al. Mice with homozygous disruption of the mdr2 P‐glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am J Pathol. 1994;145:1237‐1245. [PMC free article] [PubMed] [Google Scholar]
- 122. Smit JJ, Schinkel AH, Oude Elferink RP, et al. Homozygous disruption of the murine mdr2 P‐glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451‐462. [DOI] [PubMed] [Google Scholar]
- 123. Fickert P, Fuchsbichler A, Wagner M, et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2004;127:261‐274. [DOI] [PubMed] [Google Scholar]
- 124. Ikenaga N, Liu SB, Sverdlov DY, et al. A new Mdr2(‐/‐) mouse model of sclerosing cholangitis with rapid fibrosis progression, early‐onset portal hypertension, and liver cancer. Am J Pathol. 2015;185:325‐334. [DOI] [PubMed] [Google Scholar]
- 125. O'Brien S, Keogan M, Casey M, et al. Biliary complications of cystic fibrosis. Gut. 1992;33:387‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Colombo C, Battezzati PM, Strazzabosco M, et al. Liver and biliary problems in cystic fibrosis. Semin Liver Dis. 1998;18:227‐235. [DOI] [PubMed] [Google Scholar]
- 127. Blanco PG, Zaman MM, Junaidi O, et al. Induction of colitis in cftr−/− mice results in bile duct injury. Am J Physiol Gastrointest Liver Physiol. 2004;287:G491‐G496. [DOI] [PubMed] [Google Scholar]
- 128. Sheth S, Shea JC, Bishop MD, et al. Increased prevalence of CFTR mutations and variants and decreased chloride secretion in primary sclerosing cholangitis. Hum Genet. 2003;113:286‐292. [DOI] [PubMed] [Google Scholar]
- 129. Durie PR, Kent G, Phillips MJ, et al. Characteristic multiorgan pathology of cystic fibrosis in a long‐living cystic fibrosis transmembrane regulator knockout murine model. Am J Pathol. 2004;164:1481‐1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Snouwaert JN, Brigman KK, Latour AM, et al. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257:1083‐1088. [DOI] [PubMed] [Google Scholar]
- 131. Dorin JR, Dickinson P, Alton EWFW, et al. Cystic fibrosis in the mouse by targeted insertional mutagenesis. Nature. 1992;359:211‐215. [DOI] [PubMed] [Google Scholar]
- 132. Colledge WH, Abella BS, Southern KW, et al. Generation and characterization of a delta F508 cystic fibrosis mouse model. Nat Genet. 1995;10:445‐452. [DOI] [PubMed] [Google Scholar]
- 133. Bazett M, Honeyman L, Stefanov AN, et al. Cystic fibrosis mouse model‐dependent intestinal structure and gut microbiome. Mamm Genome. 2015;26:222‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Bloomer J, Bruzzone C, Zhu L, et al. Molecular defects in ferrochelatase in patients with protoporphyria requiring liver transplantation. J Clin Invest. 1998;102:107‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Libbrecht L, Meerman L, Kuipers F, et al. Liver pathology and hepatocarcinogenesis in a long‐term mouse model of erythropoietic protoporphyria. J Pathol. 2003;199:191‐200. [DOI] [PubMed] [Google Scholar]
- 136. Meerman L, Koopen NR, Bloks V, et al. Biliary fibrosis associated with altered bile composition in a mouse model of erythropoietic protoporphyria. Gastroenterology. 1999;117:696‐705. [DOI] [PubMed] [Google Scholar]
- 137. Vilas‐Boas V, Gijbels E, Cooreman A, et al. Industrial, biocide, and cosmetic chemical inducers of cholestasis. Chem Res Toxicol. 2019;32:1327‐1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Vilas‐Boas V, Gijbels E, Jonckheer J, et al. Cholestatic liver injury induced by food additives, dietary supplements and parenteral nutrition. Environ Int. 2020;136:105422. [DOI] [PubMed] [Google Scholar]
- 139. Zimmerman HJ, Lewis JH. Drug‐induced cholestasis. Med Toxicol. 1987;2:112‐160. [DOI] [PubMed] [Google Scholar]
- 140. Ros E, Small DM, Carey MC. Effects of chlorpromazine hydrochloride on bile salt synthesis, bile formation and biliary lipid secretion in the Rhesus monkey: a model for chlorpromazine‐induced cholestasis. Eur J Clin Invest. 1979;9:29‐41. [DOI] [PubMed] [Google Scholar]
- 141. de Haan S, Liu X. Chlorpromazine dose for people with schizophrenia. Schizophr Bull. 2009;35:491‐492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Anthérieu S, Azzi PB‐E, Dumont J, et al. Oxidative stress plays a major role in chlorpromazine‐induced cholestasis in human HepaRG cells. Hepatology. 2013;57:1518‐1529. [DOI] [PubMed] [Google Scholar]
- 143. Yang Q, Yang F, Tang X, et al. Chlorpromazine‐induced perturbations of bile acids and free fatty acids in cholestatic liver injury prevented by the Chinese herbal compound Yin‐Chen‐Hao‐Tang. BMC Complement Altern Med. 2015;15:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. El‐Sisi A, Hegazy S, El‐Khateeb E. Effects of three different fibrates on intrahepatic cholestasis experimentally induced in rats. PPAR Res. 2013;2013:781348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Stone BG, Udani M, Sanghvi A, et al. Cyclosporin A‐induced cholestasis. Gastroenterology. 1987;93:344‐351. [PubMed] [Google Scholar]
- 146. Stieger B, Fattinger K, Madon J, et al. Drug‐ and estrogen‐induced cholestasis through inhibition of the hepatocellular bile salt export pump (Bsep) of rat liver. Gastroenterology. 2000;118:422‐430. [DOI] [PubMed] [Google Scholar]
- 147. Roman ID, Fernandez‐Moreno MD, Fueyo JA, et al. Cyclosporin A induced internalization of the bile salt export pump in isolated rat hepatocyte couplets. Toxicol Sci. 2003;71:276‐281. [DOI] [PubMed] [Google Scholar]
- 148. Yasumiba S, Tazuma S, Ochi H, et al. Cyclosporin A reduces canalicular membrane fluidity and regulates transporter function in rats. Biochem J. 2001;354:591‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Kienhuis AS, Vitins AP, Pennings JLA, et al. Cyclosporine A treated in vitro models induce cholestasis response through comparison of phenotype‐directed gene expression analysis of in vivo Cyclosporine A‐induced cholestasis. Toxicol Lett. 2013;221:225‐236. [DOI] [PubMed] [Google Scholar]
- 150. Pan Y, Cao M, You D, et al. Research progress on the animal models of drug‐induced liver injury: current status and further perspectives. Biomed Res Int. 2019;2019:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Fernández E, Muñoz ME, Román ID, et al. Cyclosporin A‐induced cholestasis in the rat. Drug Invest. 1992;4:54‐63. [Google Scholar]
- 152. Tepperman J, Caldwell FT, Tepperman HM. Induction of gallstones in mice by feeding a cholesterol‐cholic acid containing diet. Am J Physiol. 1964;206:628‐634. [DOI] [PubMed] [Google Scholar]
- 153. Fujihira E, Kaneta S, Ohshima T. Strain difference in mouse cholelithiasis and the effect of taurine on the gallstone formation in C57BL/C mice. Biochem Med. 1978;19:211‐217. [DOI] [PubMed] [Google Scholar]
- 154. Paigen B, Schork NJ, Svenson KL, et al. Quantitative trait loci mapping for cholesterol gallstones in AKR/J and C57L/J strains of mice. Physiol Genomics. 2000;4:59‐65. [DOI] [PubMed] [Google Scholar]
- 155. van Erpecum KJ, Wang DQ, Lammert F, et al. Phenotypic characterization of Lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice: soluble pronucleating proteins in gallbladder and hepatic biles. J Hepatol. 2001;35:444‐451. [DOI] [PubMed] [Google Scholar]
- 156. Wang DQ, Lammert F, Paigen B, et al. Phenotypic characterization of lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice. Pathophysiology Of biliary lipid secretion. J Lipid Res. 1999;40:2066‐2079. [PubMed] [Google Scholar]
- 157. Wang TY, Portincasa P, Liu M, et al. Mouse models of gallstone disease. Curr Opin Gastroenterol. 2018;34:59‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Ahlfeld JF. Bericht und arbeiten aus der geburtshilflich‐gynäkologischen klinik zu Giessen 1881–1882: FW Grunow. 1883.
- 159. Kreek MJ. Female sex steroids and cholestasis. Semin Liver Dis. 1987;7:8‐23. [DOI] [PubMed] [Google Scholar]
- 160. Lammert F, Marschall H‐U, Glantz A, et al. Intrahepatic cholestasis of pregnancy: molecular pathogenesis, diagnosis and management. J Hepatology. 2000;33:1012‐1021. [DOI] [PubMed] [Google Scholar]
- 161. Crocenzi FA, Mottino AD, Cao J, et al. Estradiol‐17beta‐D‐glucuronide induces endocytic internalization of Bsep in rats. Am J Physiol Gastrointest Liver Physiol. 2003;285:G449‐459. [DOI] [PubMed] [Google Scholar]
- 162. Mottino AD, Hoffman T, Crocenzi FA, et al. Disruption of function and localization of tight junctional structures and Mrp2 in sustained estradiol‐17beta‐D‐glucuronide‐induced cholestasis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G391‐402. [DOI] [PubMed] [Google Scholar]
- 163. Elias E, Iqbal S, Knutton S, et al. Increased tight junction permeability: a possible mechanism of oestrogen cholestasis. Eur J Clin Invest. 1983;13:383‐390. [DOI] [PubMed] [Google Scholar]
- 164. Mottino AD, Cao J, Veggi LM, et al. Altered localization and activity of canalicular Mrp2 in estradiol‐17β‐D‐glucuronide–induced cholestasis. Hepatology. 2002;35:1409‐1419. [DOI] [PubMed] [Google Scholar]
- 165. Muchova L, Vanova K, Suk J, et al. Protective effect of heme oxygenase induction in ethinylestradiol‐induced cholestasis. J Cell Mol Med. 2015;19:924‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Rieger R, Leung PSC, Jeddeloh MR, et al. Identification of 2‐nonynoic acid, a cosmetic component, as a potential trigger of primary biliary cirrhosis. J Autoimmun. 2006;27:7‐16. [DOI] [PubMed] [Google Scholar]
- 167. Amano K, Leung PSC, Rieger R, et al. Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: Identification of antibodies against a common environmental, cosmetic, and food Additive, 2‐octynoic Acid. J Immunol. 2005;174:5874. [DOI] [PubMed] [Google Scholar]
- 168. Wu S‐J, Yang Y‐H, Tsuneyama K, et al. Innate immunity and primary biliary cirrhosis: activated invariant natural killer T cells exacerbate murine autoimmune cholangitis and fibrosis. Hepatology. 2011;53:915‐925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Chang CH, Chen YC, Yu YH, et al. Innate immunity drives xenobiotic‐induced murine autoimmune cholangitis. Clin Exp Immunol. 2014;177:373‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Wakabayashi K, Lian Z‐X, Leung PSC, et al. Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease. Hepatology. 2008;48:531‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Becker BA, Plaa GL. The nature of α‐naphthylisothiocyanate‐induced cholestasis. Toxicol Appl Pharmacol. 1965;7:680‐685. [DOI] [PubMed] [Google Scholar]
- 172. Joshi N, Ray JL, Kopec AK, et al. Dose‐dependent effects of alpha‐naphthylisothiocyanate disconnect biliary fibrosis from hepatocellular necrosis. J Biochem Mol Toxicol. 2017;31:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Vierling JM. Animal models for primary sclerosing cholangitis. Best Pract Res Clin Gastroenterol. 2001;15:591‐610. [DOI] [PubMed] [Google Scholar]
- 174. Yu H, Li Y, Xu Z, et al. Identification of potential biomarkers in cholestasis and the therapeutic effect of melatonin by metabolomics, multivariate data and pathway analyses. Int J Mol Med. 2018;42:2515‐2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Gayathri AK, Padmanaban G. Biochemical effects of 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine in mouse liver. Biochem Pharmacol. 1974;23:2713‐2725. [DOI] [PubMed] [Google Scholar]
- 176. Fickert P, Pollheimer MJ, Christoph HÖ, et al. Chapter 15: Animal models of cholestasis. Animal models for the study of human disease. 2013:331‐349.
- 177. Fickert P, Stöger U, Fuchsbichler A, et al. A new xenobiotic‐induced mouse model of sclerosing cholangitis and biliary fibrosis. Am J Pathol. 2007;171:525‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Matsubara T, Tanaka N, Patterson AD, et al. Lithocholic acid disrupts phospholipid and sphingolipid homeostasis leading to cholestasis in mice. Hepatology. 2011;53:1282‐1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Festi D, Labate AMM, Roda A, et al. Diagnostic effectiveness of serum bile acids in liver diseases as evaluated by multivariate statistical methods. Hepatology. 1983;3:707‐713. [DOI] [PubMed] [Google Scholar]
- 180. Hofmann AF. Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity. Drug Metab Rev. 2004;36:703‐722. [DOI] [PubMed] [Google Scholar]
- 181. Palmer RH, Ruban Z. Production of bile duct hyperplasia and gallstones by lithocholic acid. J Clin Invest. 1966;45:1255‐1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Miyai K, Richardson AL, Mayr W, et al. Subcellular pathology of rat liver in cholestasis and choleresis induced by bile salts. 1. Effects of lithocholic, 3beta‐hydroxy‐5‐cholenoic, cholic, and dehydrocholic acids. Lab Invest. 1977;36:249‐258. [PubMed] [Google Scholar]
- 183. Hofmann AF. The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med. 1999;159:2647‐2658. [DOI] [PubMed] [Google Scholar]
- 184. Villalon L, Tuchweber B, Yousef IM. Low protein diets potentiate lithocholic acid‐induced cholestasis in rats. J Nutr. 1992;122:1587‐1596. [DOI] [PubMed] [Google Scholar]
- 185. Fickert P, Fuchsbichler A, Marschall H‐U, et al. Lithocholic acid feeding induces segmental bile duct obstruction and destructive cholangitis in mice. Am J Pathol. 2006;168:410‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Pollheimer MJ, Halilbasic E, Fickert P, et al. Pathogenesis of primary sclerosing cholangitis. Best Pract Res Clin Gastroenterol. 2011;25:727‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Landing BH. Considerations of the pathogenesis of neonatal hepatitis, biliary atresia and choledochal cyst–the concept of infantile obstructive cholangiopathy. Prog Pediatr Surg. 1974;6:113‐139. [PubMed] [Google Scholar]
- 188. Qiao H, Zhaori G, Jiang Z, et al. Detection of group C rotavirus antigen in bile duct and liver tissues of an infant with extrahepatic biliary atresia. Chin Med J (Engl). 1999;112:93‐95. [PubMed] [Google Scholar]
- 189. Riepenhoff‐Talty M, Gouvea V, Evans MJ, et al. Detection of group C rotavirus in infants with extrahepatic biliary atresia. J Infect Dis. 1996;174:8‐15. [DOI] [PubMed] [Google Scholar]
- 190. Glaser JH, Balistreri WF, Morecki R. Role of reovirus type 3 in persistent infantile cholestasis. J Pediatr. 1984;105:912‐915. [DOI] [PubMed] [Google Scholar]
- 191. Morecki R, Glaser JH, Cho S, et al. Biliary atresia and reovirus type 3 infection. N Engl J Med. 1982;307:481‐484. [DOI] [PubMed] [Google Scholar]
- 192. Domiati‐Saad R, Dawson DB, Margraf LR, et al. Cytomegalovirus and human herpesvirus 6, but not human papillomavirus, are present in neonatal giant cell hepatitis and extrahepatic biliary atresia. Pediatr Dev Pathol. 2000;3:367‐373. [DOI] [PubMed] [Google Scholar]
- 193. Fischler B, Ehrnst A, Forsgren M, et al. The viral association of neonatal cholestasis in Sweden: a possible link between cytomegalovirus infection and extrahepatic biliary atresia. J Pediatr Gastroenterol Nutr. 1998;27:57‐64. [DOI] [PubMed] [Google Scholar]
- 194. Drut R, Drut RM, Gómez MA, et al. Presence of human papillomavirus in extrahepatic biliary atresia. J Pediatr Gastroenterol Nutr. 1998;27:530‐535. [DOI] [PubMed] [Google Scholar]
- 195. Riepenhoff‐Talty M, Schaekel K, Clark HF, et al. Group A rotaviruses produce extrahepatic biliary obstruction in orally inoculated newborn mice. Pediatr Res. 1993;33:394‐399. [DOI] [PubMed] [Google Scholar]
- 196. Mohanty SK, Donnelly B, Temple H, et al. A rotavirus‐induced mouse model to study biliary atresia and neonatal cholestasis. Methods Mol Biol. 2019;1981:259‐271. [DOI] [PubMed] [Google Scholar]
- 197. Mohanty SK, Ivantes CAP, Mourya R, et al. Macrophages are targeted by rotavirus in experimental biliary atresia and induce neutrophil chemotaxis by Mip2/Cxcl2. Pediatr Res. 2010;67:345‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Qiao H, DeVincentes A, Alashari M, et al. Pathogenesis of rotavirus‐induced bile duct obstruction (a model for human biliary atresia) in normal Balb/C and CB17 scidMice with severe combined immunodeficiency (SCID). Pediatr Res. 1999;45:116. [Google Scholar]
- 199. Petersen C. Biliary atresia: the animal models. Semin Pediatr Surg. 2012;21:185‐191. [DOI] [PubMed] [Google Scholar]
- 200. Martin CR, Zaman MM, Ketwaroo GA, et al. CFTR dysfunction predisposes to fibrotic liver disease in a murine model. Am J Physiol Gastrointest Liver Physiol. 2012;303:G474‐G481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Peng Z‐W, Rothweiler S, Wei G, et al. The ectonucleotidase ENTPD1/CD39 limits biliary injury and fibrosis in mouse models of sclerosing cholangitis. Hepatology Commun. 2017;1:957‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Orth T, Neurath M, Schirmacher P, et al. A novel rat model of chronic fibrosing cholangitis induced by local administration of a hapten reagent into the dilated bile duct is associated with increased TNF‐α production and autoantibodies. J Hepatol. 2000;33:862‐872. [DOI] [PubMed] [Google Scholar]
- 203. Wang R, Salem M, Yousef IM, et al. Targeted inactivation of sister of P‐glycoprotein gene (spgp) in mice results in nonprogressive but persistent intrahepatic cholestasis. Proc Natl Acad Sci USA. 2001;98:2011‐2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Chassaing B, Aitken JD, Malleshappa M, et al. Dextran sulfate sodium (DSS)‐induced colitis in mice. Curr Protoc Immunol. 2014;104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Mourelle M, Salas A, Vilaseca J, et al. Induction of chronic cholangitis in the rat by trinitrobenzenesulfonic acid. J Hepatol. 1995;22:219‐225. [DOI] [PubMed] [Google Scholar]
- 206. Morris GP, Beck PL, Herridge MS, et al. Hapten‐induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology. 1989;96:795‐803. [PubMed] [Google Scholar]
- 207. Orth T, Neurath M, Schirmacher P, et al. Anti‐neutrophil cytoplasmic antibodies in a rat model of trinitrobenzenesulphonic acid‐induced liver injury. Eur J Clin Invest. 1999;29:929‐939. [DOI] [PubMed] [Google Scholar]
- 208. Mariotti V, Cadamuro M, Spirli C, et al. Animal models of cholestasis: An update on inflammatory cholangiopathies. Biochim Biophys Acta Mol Basis Dis. 2019;1865:954‐964. [DOI] [PubMed] [Google Scholar]
- 209. Goetz M, Lehr HA, Neurath MF, et al. Long‐term evaluation of a rat model of chronic cholangitis resembling human primary sclerosing cholangitis. Scand J Immunol. 2003;58:533‐540. [DOI] [PubMed] [Google Scholar]
- 210. Fiorotto R, Scirpo R, Trauner M, et al. Loss of CFTR affects biliary epithelium innate immunity and causes TLR4‐NF‐κB‐mediated inflammatory response in mice. Gastroenterology. 2011;141:1498‐1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Tebbi A, Levillayer F, Jouvion G, et al. Deficiency of multidrug resistance 2 contributes to cell transformation through oxidative stress. Carcinogenesis. 2016;37:39‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Asrani SK, Devarbhavi H, Eaton J, et al. Burden of liver diseases in the world. J Hepatol. 2019;70:151‐171. [DOI] [PubMed] [Google Scholar]
- 213. Wang F‐S, Fan J‐G, Zhang Z, et al. The global burden of liver disease: the major impact of China. Hepatology. 2014;60:2099‐2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Costas‐Rodríguez M, Van Campenhout S, Hastuti A, et al. Body distribution of stable copper isotopes during the progression of cholestatic liver disease induced by common bile duct ligation in mice. Metallomics. 2019;11:1093‐1103. [DOI] [PubMed] [Google Scholar]
- 215. Martínez AK, Maroni L, Marzioni M, et al. Mouse models of liver fibrosis mimic human liver fibrosis of different etiologies. Curr Pathobiol Rep. 2014;2:143‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Thakare R, Alamoudi JA, Gautam N, et al. Species differences in bile acids I. Plasma and urine bile acid composition. J Appl Toxicol. 2018;38:1323‐1335. [DOI] [PubMed] [Google Scholar]
- 217. Martignoni M, Groothuis GM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP‐mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol. 2006;2:875‐894. [DOI] [PubMed] [Google Scholar]
- 218. Dreano E, Bacchetta M, Simonin J, et al. Characterization of two rat models of cystic fibrosis—KO and F508del CFTR—Generated by Crispr‐Cas9. Animal Model Exp Med. 2019;2:297‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Jelnes P, Santoni‐Rugiu E, Rasmussen M, et al. Remarkable heterogeneity displayed by oval cells in rat and mouse models of stem cell–mediated liver regeneration. Hepatology. 2007;45:1462‐1470. [DOI] [PubMed] [Google Scholar]
- 220. He S, Ye T, Wang R, et al. An in silico model for predicting drug‐induced hepatotoxicity. Int J Mol Sci. 2019;20:1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Sokal EM, Baudoux MC, Collette E, et al. Branched chain amino acids improve body composition and nitrogen balance in a rat model of extra hepatic biliary atresia. Pediatr Res. 1996;40:66‐71. [DOI] [PubMed] [Google Scholar]
- 222. Chan RYY, Tan CEL, Czech‐Schmidt G, et al. Computerized three‐dimensional study of a rotavirus model of biliary atresia: comparison with human biliary atresia. Pediatr Surg Int. 2005;21:615‐620. [DOI] [PubMed] [Google Scholar]
- 223. Tucker RM, Hendrickson RJ, Mukaida N, et al. Progressive biliary destruction is independent of a functional tumor necrosis factor‐alpha pathway in a rhesus rotavirus‐induced murine model of biliary atresia. Viral Immunol. 2007;20:34‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Reihnér E, Ståhlberg D. Lithogenic diet and gallstone formation in mice: integrated response of activities of regulatory enzymes in hepatic cholesterol metabolism. Br J Nutr. 1996;76:765‐772. [DOI] [PubMed] [Google Scholar]
- 225. Li Y, Li M, Wu S, et al. Combination of curcumin and piperine prevents formation of gallstones in C57BL6 mice fed on lithogenic diet: whether NPC1L1/SREBP2 participates in this process? Lipids Health Dis. 2015;14:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Meyers M, Slikker W, Pascoe G, et al. Characterization of cholestasis induced by estradiol‐17 beta‐D‐glucuronide in the rat. J Pharmacol Exp Ther. 1980;214:87‐93. [PubMed] [Google Scholar]
- 227. Sano N, Takikawa H, Yamanaka M. Estradiol‐17β‐glucuronide‐induced cholestasis: Effects of ursodeoxycholate‐3‐O‐glucuronide and 3,7‐disulfate. J Hepatology. 1993;17:241‐246. [DOI] [PubMed] [Google Scholar]
- 228. Pose E, Sancho‐Bru P, Coll M. 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine diet: a rodent model in cholestasis research. Methods Mol Biol. 2019;1981:249‐257. [DOI] [PubMed] [Google Scholar]
- 229. Wang T, Zhou ZX, Sun LX, et al. Resveratrol effectively attenuates α‐naphthyl‐isothiocyanate‐induced acute cholestasis and liver injury through choleretic and anti‐inflammatory mechanisms. Acta Pharmacol Sin. 2014;35:1527‐1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Yao X, Li Y, Cheng X, et al. ER stress contributes to alpha‐naphthyl isothiocyanate‐induced liver injury with cholestasis in mice. Pathol Res Pract. 2016;212:560‐567. [DOI] [PubMed] [Google Scholar]
- 231. Yang J, Xiang D, Xiang D, et al. Baicalin protects against 17α‐ethinylestradiol‐induced cholestasis via the sirtuin 1/Hepatic nuclear receptor‐1α/Farnesoid X receptor pathway. Front Pharmacol. 2019;10:1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Woolbright BL, Li F, Xie Y, et al. Lithocholic acid feeding results in direct hepato‐toxicity independent of neutrophil function in mice. Toxicol Lett. 2014;228:56‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Alkhedaide AQ, Ismail TA, Alotaibi SH, et al. Preventive effect of artemisinin extract against cholestasis induced via lithocholic acid exposure. Biosci Rep. 2018;38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Shahid RA, Wang DQH, Fee BE, et al. Endogenous elevation of plasma cholecystokinin does not prevent gallstones. Eur J Clin Invest. 2015;45:237‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Sohn MJ, Woo MH, Seong M‐W, et al. Benign recurrent intrahepatic cholestasis type 2 in siblings with novel ABCB11 mutations. Pediatr Gastroenterol Hepatol Nutr. 2019;22:201‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Meerman L, Koopen NR, Bloks V, et al. Biliary fibrosis associated with altered bile composition in a mouse model of erythropoietic protoporphyria. Gastroenterology. 1999;117:696‐705. [DOI] [PubMed] [Google Scholar]
- 237. Hsu W, Zhang W, Tsuneyama K, et al. Differential mechanisms in the pathogenesis of autoimmune cholangitis versus inflammatory bowel disease in interleukin‐2Ralpha(‐/‐) mice. Hepatology (Baltimore, MD). 2009;49:133‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. van Nieuwerk CM, Groen AK, Ottenhoff R, et al. The role of bile salt composition in liver pathology of mdr2 (‐/‐) mice: differences between males and females. J Hepatol. 1997;26:138‐145. [DOI] [PubMed] [Google Scholar]
- 239. Koarada S, Wu Y, Fertig N, et al. Genetic control of autoimmunity: protection from diabetes, but spontaneous autoimmune biliary disease in a nonobese diabetic congenic strain. J Immunol. 2004;173:2315‐2323. [DOI] [PubMed] [Google Scholar]
- 240. Cho I, Koo B‐N, Kam EH, et al. Bile duct ligation of C57BL/6 mice as a model of hepatic encephalopathy. Anesth Pain Med. 2020;15:19‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Zhang Y, Hong J‐Y, Rockwell CE, et al. Effect of bile duct ligation on bile acid composition in mouse serum and liver. Liver Int. 2012;32:58‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Shivakumar P, Campbell KM, Sabla GE, et al. Obstruction of extrahepatic bile ducts by lymphocytes is regulated by IFN‐gamma in experimental biliary atresia. J Clin Investig. 2004;114:322‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Gao RY, Shearn CT, Orlicky DJ, et al. Bile acids modulate colonic MAdCAM‐1 expression in a murine model of combined cholestasis and colitis. Mucosal Immunol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Hirschfield GM, Heathcote EJ, Gershwin ME. Pathogenesis of cholestatic liver disease and therapeutic approaches. Gastroenterology. 2010;139:1481‐1496. [DOI] [PubMed] [Google Scholar]
- 245. Massarweh NN, El‐Serag HB. Epidemiology of hepatocellular carcinoma and intrahepatic cholangiocarcinoma. Cancer Control. 2017;24:1073274817729245. [DOI] [PMC free article] [PubMed] [Google Scholar]