

Summary
Pre‐mRNA splicing is an essential step for the regulation of gene expression. In order to specifically capture splicing variants in plants for genome‐wide association studies (GWAS), we developed a software tool to quantify and visualise Variations of Splicing in Population (VaSP).
VaSP can quantify splicing variants from short‐read RNA‐seq datasets and discover genotype‐specific splicing (GSS) events, which can be used to prioritise causal pre‐mRNA splicing events in GWAS. We applied our method to an RNA‐seq dataset with 328 samples from 82 genotypes from a rice diversity panel exposed to optimal and saline growing conditions.
In total, 764 significant GSS events were identified in salt stress conditions. GSS events were used as markers for a GWAS with the shoot Na+ accumulation, which identified six GSS events in five genes significantly associated with the shoot Na+ content. Two of these genes, OsNUC1 and OsRAD23 emerged as top candidate genes with splice variants that exhibited significant divergence between the variants for shoot growth under salt stress conditions.
VaSP is a versatile tool for alternative splicing analysis in plants and a powerful tool for prioritising candidate causal pre‐mRNA splicing and corresponding genomic variations in GWAS.
Keywords: alternative splicing (AS), genotype‐specific RNA splicing (GSS), prioritisation, rice (Oryza sativa), salt stress, splice site mutation, splicing quantitative trait locus (sQTL)
Introduction
The removal of introns and joining exons during the mRNA maturation process is an essential step in gene regulation for biological processes in most eukaryotes. Alternative splicing (AS) is an important co‐transcriptional and post‐transcriptional regulatory mechanism in plant response to abiotic stresses (Laloum et al., 2018; Zhu et al., 2018; Jabre et al., 2019; Zhu et al., 2020). There are five basic types of AS: alternative donor site (AltD), alternative acceptor (AltA) site, alternative position (AltP), exon skipping (ExonS) and intron retention (IntronR). ExonS is the most common type of AS in animals, while IntronR is the most prevalent form in plants (Gupta et al., 2004; Ner‐Gaon et al., 2004; Wang & Brendel, 2006; Kim et al., 2007; Braunschweig et al., 2014; Grau‐Bove et al., 2018; Chaudhary et al., 2019b). In plants, up to 60–70% of multi‐exon genes have AS, and up to a half of the AS events are IntronR (Wang & Brendel, 2006; Marquez et al., 2012; Chamala et al., 2015; Zhang et al., 2017). These differences suggest that the mechanism of splice site recognition may differ between plants (intron definition) and animals (exon definition) (Barbazuk et al., 2008). Although many studies on AS are based on expressed sequence tags (ESTs) and cDNAs in plants, only a few studies focus on AS variations at the plant population level (Chen et al., 2018; Khokhar et al., 2019). With advancements in next‐generation sequencing technology, it is feasible to obtain RNA‐seq datasets for AS analysis, but it is challenging to accurately determine and quantify AS due to short reads and the complexity of splicing when evaluating a large population. Our ability to obtain novel insights into the regulation and function of splicing is further hindered by the difficulty of estimating transcript abundance from short‐read RNA‐seq data.
At present, there are many tools for AS detection using short‐read RNA‐seq data (Hooper, 2014; Ding et al., 2017; Mehmood et al., 2019). Most of the tools estimate transcript isoform ratios or exon inclusion levels, which is statistically challenging and maybe inaccurate when applied to short‐read data (Vaquero‐Garcia et al., 2016; Li et al., 2018). A more accurate approach is to focus on reads that span exon–exon junctions (junction reads) in a local splicing region, such as implemented in tools like rMats (Shen et al., 2014), Majiq (Vaquero‐Garcia et al., 2016), and LeafCutter (Li et al., 2018). The software tool rMats was designed for the detection of differential AS between two groups, and Majiq proposes to estimate local splicing variation and identifies complex transcript variations. Both tools rely on existing annotations of genes and transcripts and were not designed for quantifying intron usage variation in a large natural population. LeafCutter is able to identify AS events in animal populations by using junction reads in a cluster region (overlapped introns), but it cannot detect AS events in a single intron region (Vaquero‐Garcia et al., 2018), such as IntronR, which is the most prevalent AS form in plants. Furthermore, the current tools for AS analysis are primarily designed for mammal genomic data (e.g. human or mouse), but plants and animals have different splicing mechanisms. Compared with human genes, plant genes are smaller with slightly longer exons and much shorter introns (Barbazuk et al., 2008; Keren et al., 2010), which may affect the output of these existing tools when applied to plant datasets. Therefore, an efficient and accurate method that will function for the features of pre‐mRNA splicing and to prioritise candidate causal pre‐mRNA splicing events for association studies in plants is needed for the comprehensive discovery of AS.
Rice (Oryza sativa) is a major staple food crop, and it is also a model plant for genomics research. Rice is one of the most salt‐sensitive major crops, and it is especially sensitive during the vegetative growth stage (Lutts et al., 1996; Munns & Tester, 2008). Salinity tolerance is a polygenic trait resulting from several underlying physiological and molecular mechanisms (Munns & Tester, 2008; Deinlein et al., 2014). Although many studies have investigated genome‐wide expression profiles in response to salt stress in rice (Kawasaki et al., 2001; Walia et al., 2005, 2007; Cotsaftis et al., 2011), only a few methods and datasets have been generated for exploration of natural variation using expression analysis (Du et al., 2019). A recent study showed that alternative splicing plays an important role in maintaining mineral nutrient homeostasis in rice, but the association of AS with salt stress or sodium homeostasis has not been examined (Dong et al., 2018). Thus, we reasoned that an analysis of AS variation in a diverse rice population would provide new insight into genetic variations associated with salt stress in rice.
To address the problem of a lack of tools that are adequate for the discovery of AS in plants, we developed an R package, VaSP (https://www.bioconductor.org/packages/VaSP), for quantification and visualisation of Variations of Splicing events in a Population. We applied VaSP on a rice population, using a large set of short‐read RNA‐seq samples. VaSP quantifies each intron splicing event using a novel statistical score, called the Single Splicing Strength (3S) score, based on the number of junction reads and gene‐level reads. We used VaSP to discover a set of genotype‐specific splicing (GSS) events and their associated splice site variations, which can prioritise candidate causal pre‐mRNA splicing events for the associate study. Genome‐wide association analysis between GSS variations, as markers, and shoot Na+ content, one of the key traits for salt tolerance, revealed potential genetic variations involved in salt stress response in rice, which demonstrates that the power of GSS for prioritising candidate causal pre‐mRNA splicing for stress response.
Materials and Methods
Populational genotype‐specific pre‐mRNA splicing discovery
The R package, VaSP (Fig. 1a), was developed to detect variations in splicing events, especially IntronR, in RNA‐seq data from a diverse set of genotypes. For each candidate junction in the sample, VaSP assigns a Single Splicing Strength (3S) score, which is defined as follows (Fig. 1b):
| (Eqn 1) |
where R is the count of junction reads for a single intron, and Ci is the average coverage of the i th isoform for the parent gene. The gene‐level read coverage, used for normalisation, is the sum of the average coverages for all transcript isoforms in the gene. In the R package, one can calculate 3S scores for all splicing junctions in the genome by the function of spliceGenome or in a particular gene by the function of spliceGene. VaSP package also provides the visualisation to AS events, and users can display differential splicing information by using the function of splicePlot (Fig. 1c,d).
Fig. 1.
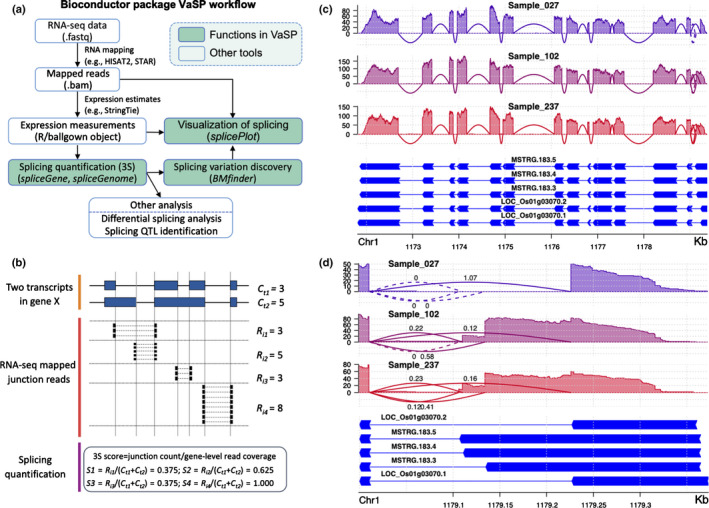
Overview of VaSP, an R package for quantification and visualisation of Variations of Splicing events in a Population. (a) The workflow and functions of VaSP. The input is an R data object ballgown produced by a standard RNA‐seq data analysis protocol (Pertea et al., 2016), including mapping with HISAT, assembling with StringTie and collecting expression information with Ballgown. VaSP calculates the Single Splicing Strength (3S) scores for all splicing junctions in the genome (spliceGenome) or in a particular gene (spliceGene), identifies genotype‐specific splicing (GSS) events (BMfinder, see the main text for details), and displays differential splicing information (splicePlot). The 3S scores can be also used for other analyses, such as differential splicing analysis or splicing QTL identification. (b) VaSP estimates 3S scores based on junction‐read counts normalised by gene‐level read coverage. In this example, VaSP calculates the splicing scores of four introns in a gene X with two transcript isoforms. Only the fourth intron is a full usage intron excised by both the two isoforms and the other three are alternative donor site (AltD) sites or intron retention (IntronR), respectively. (c) Visualisation of splicing information in gene MSTRG.183 (LOC_Os01g03070), whole gene without splicing scores. (d) Visualisation of differential splicing region of the gene MSTRG.183 with splicing scores displaying. In (c) and (d), the y‐axes are read depth and the arcs (lines between exons) indicate exon–exon junctions (introns). The dotted arcs indicate no junction reads spanning the intron (3S = 0) and solid arcs indicate 3S > 0. The transcripts labelled beginning with ‘LOC_Os’ indicate annotated transcripts by MSU7 and the ones beginning with ‘MSTRG’ are transcripts assembled by StringTie.
To identify a GSS event in a population, VaSP finds nonoverlapping bimodal distribution of 3S scores in the population and divides all samples into two groups. The bimodal distribution test implemented in the function of BMfinder was previously used in a single feature polymorphism (SFP) detection program (Wang et al., 2010). The splicing scores of an intron were divided into two clusters ‘around medoids’ with pam function in the R package cluster, a more robust version of K‐means (Reynolds et al., 2006), and the Z‐score test was used to make sure any member of a cluster did not belong to the other cluster at a probability of 99%. According to the Z‐score test, if the probability (P‐value) of a sample belonging to the other cluster was larger than 0.01, the sample was set to missing (did not belong to any cluster).
For GSS event discovery in the rice diversity panel 1 (RDP1) population (Famoso et al., 2011; Zhao et al., 2011; Eizenga et al., 2014), we only focused on reliable intron splicing events that were supported by at least five junction reads (Danan‐Gotthold et al., 2015) in at least 5% of all samples and in widely expressed genes: average per‐base read coverage ≥ 1 in more than 95% of all samples. Suitable cutoffs of minor allele frequency (MAF) and missing clustering rate (MISS) were used to control the stringency. For the analysis of RNA‐seq data from RDP1, the thresholds were set as MAF ≥ 0.05 and MISS ≤ 0.05. The GSS events under control and salt stress conditions were separately identified. The two replicates of all accessions under each condition were then merged, and any accession with the two replications clustered into different groups was set to missing. The splicing events with a missing rate > 0.05 were excluded and the splicing events located in assembled genes (MSTRG‐tag) spanning more than two annotated genes were also removed. These genes that included more than two annotated genes were largely due to assembly errors.
Genome‐wide analysis of sQTLs and eQTLs under salt stress
To identify genetic variations associated with GSS events, a genome‐wide association study (GWAS) was conducted using single nucleotide polymorphism (SNP) genotyping data and 3S scores of GSS events under salt stress conditions. The splicing scores of the two replicates for 82 accessions under salt stress conditions were averaged and used as a splicing trait (s‐trait). The SNP genotyping data were downloaded from imputed genotypes of RDP1 (http://www.ricediversity.org), and the genotypes of the 82 accessions used in this study were extracted with MAF ≥ 0.05. We applied the univariate linear mixed model with the score test (‐lmm 3) implemented in GEmma software for splicing quantitative trait locus (sQTL) analysis (Zhou & Stephens, 2012). The window size used for an sQTL was set to 500 kb and the threshold of significant P‐value was 1 × 10−5. For sQTL detection, we filtered significant SNPs from top to bottom according to the P‐values. For each GSS event, the most significant SNP with the smallest P‐value was picked as the first sQTL if there were at least three significant SNPs in the 500‐kb window of an sQTL region (each side 250‐kb) to avoid false‐positive outlier SNPs. We then conducted linkage disequilibrium (LD) analysis for the associated SNPs using R package snpStats (Clayton, 2020), and removed all other significant SNPs linked to the most significant SNP (lead SNP) (r2 ≥ 0.1) (Chen et al., 2018). Then, we stepped to the next independent significant SNP until there was no significant SNP left. An sQTL was defined as cis‐sQTL if it was located within 250 kb from both sides of the GSS event because, in rice, the LD decay is 100–350 kb (Zhang et al., 2019) and the recombination rate is about 230 kb/cM (Yu et al., 2011). The analysis of expression‐QTLs (eQTLs) was similar to the sQTLs except that the gene expression fragments per kilobase per million reads (FPKM) were log2 transformed, log2(FPKM + 1), and averaged for the two replicates, which were used as an expression trait (e‐trait).
Association analysis of GSS variations with salt tolerance‐related phenotypes under salt stress condition
To identify GSS events associated with shoot Na+ content, one of the key salt tolerance‐related phenotypes, a linear model was used to fit the phenotype with the genotype as follows:
| (Eqn 2) |
where y is the shoot Na+ content, the phenotype from our previous works (Campbell et al., 2017; Du et al., 2019), x is the clusters of each GSS event (genotype), PC1–PC4 are the first four principal components from clusters of all GSS events under salt stress conditions (control population structure), and ε is the error. The threshold of a significant P‐value was 0.01. The events due to mapping errors were manually removed from the result.
To test if a discovered GSS variation was associated with rice growth response to salinity, we leveraged 14‐d temporal RGB imaging data generated for the same population under 90 mM NaCl salt stress and control conditions (Campbell et al., 2015). These imaging data were used to derive a pixel count of the shoot tissue of the rice genotypes called the Projected Shoot Area (PSA), which shows a strong positive correlation with manual biomass‐related measurements (shoot area, fresh weight and dry weight). We used PSA to measure plant growth status, and the square root‐transformed ratio of the plant PSA under salt stress over that of plants under normal growth conditions to measure the salt‐induced growth response (Campbell et al., 2015). We extracted the data for the 82 accessions used in this study and did a pairwise comparison of growth responses between different GSS clusters.
Plant materials, growth conditions for RNA sequencing
The rice materials used in this study are a subset of the RDP1, which comprises 421 accessions collected from 85 countries (Famoso et al., 2011; Zhao et al., 2011; Eizenga et al., 2014). In total, 328 samples from 82 accessions of RDP1 with two conditions, control and salt stress conditions, and two replicates in each condition, were used for transcriptomics data analysis in this study (Du et al., 2019). The experiment was conducted in a walk‐in growth chamber as described previously by Campbell et al. with modifications (Campbell et al., 2017, 2020). Plant growth, salt stress treatment and RNA sequencing were conducted as described by Du et al. (2019).
RNA‐seq mapping and transcript assembly
All RNA‐seq data (BioProject: PRJNA385135), obtained from Illumina 101‐bp single‐end RNA sequencing, were examined using the software FastQC (Andrews, 2014). All raw short reads were screened and trimmed using Trimmomatic (Bolger et al., 2014) with single‐end‐mode. Trimmed short reads were mapped to the rice reference genome (Kawahara et al., 2013) using HISAT2 (Kim et al., 2015). The aligned reads were assembled to the reference annotation (MSU7) or novel transcripts with StringTie (Pertea et al., 2015). Assembled transcripts from all samples were merged with the transcripts from the reference annotation, and then the abundances of all the merged transcripts were re‐estimated for all samples, using StringTie (Pertea et al., 2015). The information about the coordinates and expression profiles of genes, transcripts, exons, and introns was stored in the object of ballgown using R Ballgown package (Frazee et al., 2015), which was used as the input of VaSP.
Results
VaSP can identify pre‐mRNA splicing events in a population
We developed VaSP (Fig. 1a) to detect variations in splicing events, especially IntronR, in RNA‐seq data from a diverse set of genotypes. After applying the standard RNA‐seq data analysis protocol (Pertea et al., 2016), VaSP obtained read mapping information, such as junction reads and read coverage, from the object of ballgown in R Ballgown package (Frazee et al., 2015). For each candidate junction in the sample, VaSP quantifies the splicing event by assigning a 3S score (Fig. 1b), which is the count of junction reads normalised by gene‐level read coverage. The 3S score histogram of all high‐confidence introns from our RNA‐seq data indicated that the majority of the values were around 1 or larger than 1, implying frequent usage of that intron, and some score 0, implying no usage of that intron (Fig. 2a). The 3S score is different from the traditional splicing qualification factor percent‐spliced‐in (PSI) in that a 3S score can be larger than one (100%), because the reads may be unevenly distributed along the gene and the number of junction reads could be larger than the average gene‐level coverage. For example, usually poly(A) RNA‐seq is biased toward the 3′ end of transcripts that results in more reads on the 3′ end than the 5′ end. The 3S scores calculated by VaSP reflect this characteristic in RNA‐seq samples (Fig. 2b). Therefore, in general, the larger the 3S score, the higher the splicing level. It is pertinent to point out that the comparison of 3S scores between two different intron splicing locations in a gene is not meaningful. Despite this limitation, 3S scores are useful for quantifying splicing among samples or in a population, and therefore can be used for differential AS analysis or splicing quantitative trait locus (sQTL) identification (Fig. 1a).
Fig. 2.
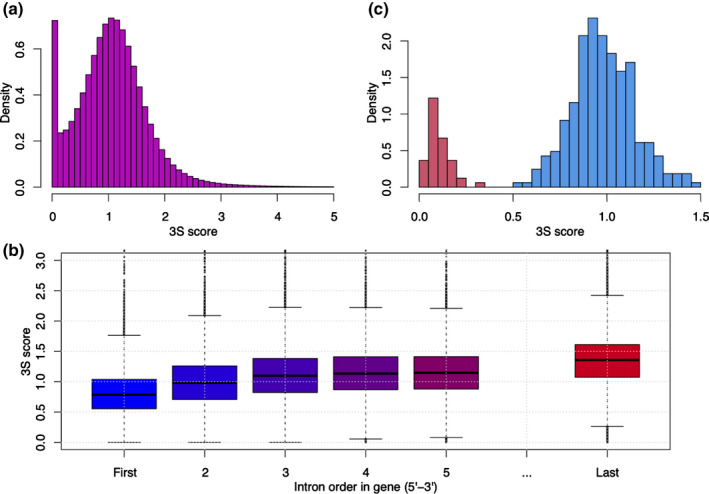
The distribution of Single Splicing Strength (3S) scores from RNA‐seq data in RDP1 population. (a) 3S score histogram of all high‐confidence introns. (b) The distribution of 3S scores of introns along with genes. For boxplots, the lower and the upper horizon lines are minimum and maximum 3S scores, respectively, and boxes have ranges from 25% to 75% quantiles and a centre line for 50% quantile (median). Dots indicate outliers. (c) An example of the nonoverlapping bimodal distribution of 3S scores for a splicing intron.
VaSP considers one intron splicing as a splicing event, and each intron is assigned a 3S score. Each intron can overlap with other introns from different transcript isoforms in the same gene. VaSP makes the identification of pre‐mRNA splicing straightforward after junction reads are counted and transcript isoforms are assembled. As the 3S score for each intron splicing is based on the normalisation of gene‐level average coverage, 3S scores for an intron in different samples are comparable. Moreover, as all reads are considered for normalisation, the effect caused by the variance of gene expression levels is reduced, compared with using a small portion of reads around junction sites. These advantages make VaSP suitable for population‐level analysis. Although VaSP does not directly distinguish different types of AS events like rMats (Shen et al., 2014), it can detect events from all basic AS types by a combination of individual VaSP‐detected introns. VaSP package also provides the visualisation to AS events, including novel ones, with publication‐quality multipanel figure output (Fig. 1c,d), to enhance AS annotation.
To prioritise high‐confidence causal pre‐mRNA splicing associated with a phenotype of interest, VaSP implemented an additional functional module to discover GSS events, which is based on the fact that the biallelic homozygous populations are frequently used in plants, especially inbred or natural populations. Using multiple samples or an inbred plant population, VaSP can identify large‐effect pre‐mRNA splicing variations without the need for genotypic information. VaSP evaluates the nonoverlapping bimodal distribution of 3S scores in the population and divides all samples into two groups by applying K‐means clustering and Z‐score testing (Fig. 2c). More specifically, for each candidate junction, VaSP conducts K‐means clustering to classify all samples (population) into two alleles based on 3S scores. Then, the Z‐score is calculated to test if the two groups of samples have a nonoverlapping bimodal distribution. A junction with significant nonoverlapping bimodal distribution 3S scores indicates that a given splicing event is a large‐effect pre‐mRNA splicing variation, and hence is called GSS. As all samples are clustered into two groups, corresponding to two homozygous genotypes, this method is particularly useful for biallelic homozygous populations in plants, that is either inbred or natural populations without heterozygous genotypes, which are frequently used for biological research. Moreover, the 3S scores calculated by VaSP can be used for differential splicing analysis between two sample groups (Dong et al., 2018) or the identification of sQTLs in large populations (Chen et al., 2018). Although the GSS event identification is limited to biallelic homozygous populations, the 3S scores can be used in different types of populations, including populations with heterozygous genotypes. Using GSS as markers can prioritise causal pre‐mRNA splicing in a GWAS.
Genome‐wide discovery of GSS variations in a rice diversity panel
We tested VaSP on an RNA‐seq data set of 328 RNA samples for 82 genotypes from the rice diversity panel 1 (RDP1) (Famoso et al., 2011; Zhao et al., 2011; Eizenga et al., 2014; McCouch et al., 2016; Wang et al., 2018). Shoot tissues were collected for RNA‐seq with two replicates for plants grown under control and salt stress conditions (Du et al., 2019). Using the criteria of being supported by at least five junction reads in at least 5% of all the 328 samples (Au et al., 2010; Edriss et al., 2013; Ding et al., 2017), VaSP discovered 108 180 significant intron splicing events, which were for any single intron splicing. Of these, 9396 (8.7%) were novel junctions, as they are not annotated by MSU7 or RAP‐DB/IRGSP1.0 (Kawahara et al., 2013) (Supporting Information Fig. S1a). To avoid false positives, we further limited this set to 94 856 intron splicing events (6781 are novel, 7.15%) in 13 787 widely expressed genes (their gene‐level average read coverage ≥ 1 in at least 95% of all 328 samples) (Emig et al., 2010; Law et al., 2018).
In total, 752 and 764 significant GSS events were identified by VaSP for control and salt stress conditions, respectively. Among these GSS variations, 633 events in 448 genes were common for both control and salt stress conditions and, when combined, there were in total, 883 events in 577 genes (Fig. S1b). These events included different types of AS, such as ExonS (65, 7.4%), IntronR (207, 23.4%), AltD (120, 13.6%), AltA (172, 19.5%), and AltP (88, 10.0%) (Fig. 3). In the distribution of genome‐wide 108 180 pre‐mRNA splicing events, the majority (84 876, 89.5%) was located in gene‐coding regions (Fig. S1c), while among the 883 GSS events, only 66.3% (585) were located in gene‐coding regions (Fig. S1d).
Fig. 3.
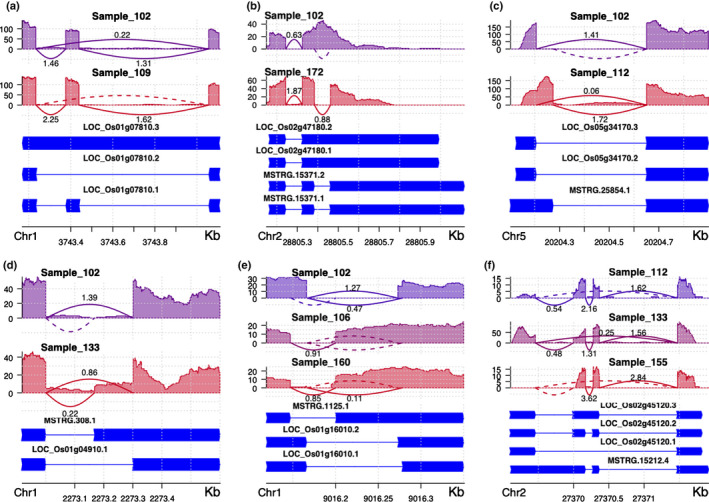
Examples of different alternative splicing types. (a) exon skipping (ExonS). (b) Intron retention (IntronR). (c) Alternative donor site (AltD). (d) Alternative acceptor site (AltA). (e) Alternative position (AltP). (f) Other types of complex splicing. The y‐axes are read depth and the arcs (lines between exons) indicate exon–exon junctions (i.e. introns). The dotted arcs indicate no junction reads spanning the introns and solid arcs indicate introns with junction reads.
For comparison using the same dataset, VaSP used the splicing scores calculated by LeafCutter (Li et al., 2018), Majiq (Vaquero‐Garcia et al., 2016), and a score based on transcript isoform ratio for clustering for identification of GSS events. Among all existing scores, the 3S scores calculated by VaSP identified the largest number of GSS events (Table 1). VaSP also identified more shared splicing events between the two conditions than the other methods. For quantifying pre‐mRNA splicing events, VaSP performed similarly to LeafCutter, but identified more IntronR events. Although the isoform quantification derived from the output of StringTie or Cufflinks for input into VaSP to calculate gene‐level read coverage may be inaccurate, using the gene‐level read coverage for normalisation is more reliable than using exon reads around splice sites, where reads could be ambiguously assigned (Vaquero‐Garcia et al., 2016). This makes the 3S scores calculated by VaSP more tolerant to incorrectly assembled transcript models, and hence, increases the ability to detect pre‐mRNA splicing variations in a population.
Table 1.
Numbers of genotype‐specific splicing (GSS) events and their corresponding genes identified in the RDP1 using Single Splicing Strength (3S) scores of VaSP, the scores of LeafCutter, the scores of Majiq and isoform ratio to perform clustering with the VaSP function.
| Method | VaSP | LeafCutter | Majiq | Isoform ratio | ||||
|---|---|---|---|---|---|---|---|---|
| Events | Genes | Events | Genes | Events | Genes | Events | Genes | |
| Control | 752 (85.2%) | 506 (87.7%) | 556 (80.2%) | 421 (83.7%) | 533 (74.4%) | 185 (80.0%) | 409 (74.9%) | 270 (76.3%) |
| Salt | 764 (86.5%) | 520 (90.1%) | 601 (86.7%) | 440 (87.5%) | 594 (83.0%) | 193 (83.5%) | 421 (77.1%) | 280 (79.0%) |
| Shared a | 633 (71.7%) | 449 (77.8%) | 464 (67.0%) | 358 (71.2%) | 411 (57.4%) | 147 (63.6%) | 284 (52.0%) | 196 (55.4%) |
| Total b | 883 (100%) | 577 (100%) | 693 (100%) | 503 (100%) | 716 (100%) | 231 (100%) | 546 (100%) | 354 (100%) |
Numbers in brackets are the percentages of GSS events under a certain condition in all discovered GSS events.
The common GSS events and genes discovered in both control and salt stress conditions.
The total number of nonredundant GSS events and genes for both control and salt stress conditions.
Most GSS events are cis‐regulated
GSS events reflect significant splicing variations between two genotypes. In order to find out how the large‐effect splicing variations are regulated, we selected all the GSS variations discovered by VaSP in RDP1 population under salt stress condition and conducted sQTL analysis for genomic variations within the population. For a given GSS event, the average 3S scores of the two replicates for each genotype were used as splicing traits (s‐traits). We used 3.34 million imputed SNPs for RDP1 as the genotypic data for the 82 accessions (Wang et al., 2018). As a result, in total, 1366 sQTLs were obtained for 764 GSS events with the cutoff of P‐value < 1 × 10−5, each with zero to four sQTLs (Fig. 4a). Among these, 551 (account for 72.1% of events) are cis‐sQTLs with the distances to affected GSS introns less than 250 kb, while the rest 815 are trans‐sQTLs (Fig. 4b). Furthermore, most of the cis‐sQTLs were located in or near the introns that they affect (Fig. 4c). Although there was also a large number of trans‐sQTLs identified, they were less significant with much smaller effects (Fig. 4d). As here we showed that GSS events are mainly cis‐regulated and genotype specific, one can easily determine the cis‐regulating SNPs for a GSS from the result of GWAS using GSS as markers.
Fig. 4.
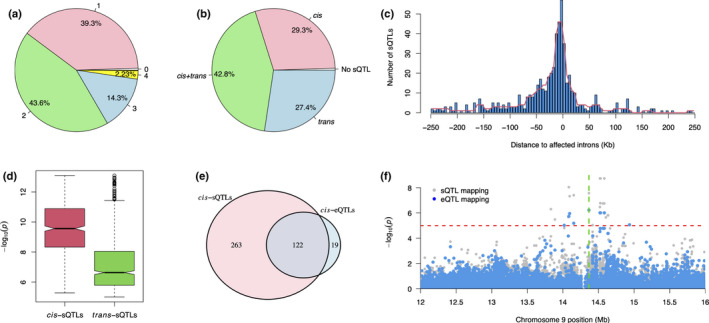
Splicing QTL (sQTL) analysis of genotype‐specific splicing (GSS) events under saline conditions. (a) The number of sQTLs identified for each GSS event. (b) Different types of sQTLs for each GSS event. (c) Distances of sQTLs to the introns they affect. (d) Significances of cis‐sQTLs and trans‐sQTLs. For boxplots, the lower and the upper horizon lines are minimum and maximum values of −log10(P), respectively, and the boxes have ranges from 25% to 75% quantiles and a centre line for 50% quantile (median). Dots indicate outliers. (e) Overlaps of genes with cis‐sQTLs and cis‐eQTLs. (f) An example of genes with overlapped cis‐sQTLs and cis‐eQTLs. The horizontal red dotted line indicates the threshold P‐value = 1×10−5 and the vertical green dotted line indicates the position of the target gene, OsRAD23 (LOC_Os09g24200).
An AS event can be reflected by the differential expression of transcripts or isoforms in a given gene (Wang et al., 2008). It has been shown that AS and gene‐level expression are under relatively independent genetic control (Chen et al., 2018). To check the relationship between AS and overall gene expression, we then performed eQTL analysis for the parent genes of GSS events under salt stress conditions. As a result, 141 (account for 27.1% of 520 GSS genes) cis‐eQTLs were obtained. We compared two sets of genes with cis‐sQTLs or cis‐eQTLs, and found that 122 (31.7%) out of 385 genes with cis‐sQTLs also have cis‐eQTLs (Fig. 4e). For example, there were both significant cis‐sQTLs and cis‐eQTLs in gene OsRAD23 (LOC_Os09g24200) (Fig. 4f).
GSS events naturally link to splice site mutations
sQTL analysis is only a primary step to find the regulation of AS. Due to the limitation of the population size, the large number of SNPs, and the large extent of LD decay in rice (Mather et al., 2007), the peak regions of sQTLs could be very large (Fig. 4f) and it is hard to find out the causal SNPs. As most of the GSS events are cis‐regulated, they are likely to be related to splicing cis‐regulatory elements, such as splice sites, exonic and intronic splicing regulators. Among variations in these splicing regulatory elements, a splice site mutation is the most consequential as it directly causes a GSS variation. To identify the causal SNP associated with each RNA intron splicing event from multiple linked SNPs in a sQTL peak region, we collected and screened all SNPs existing at the splice sites. By using all assembled transcripts as a reference, we identified 3825 SNPs located at splice donor or acceptor sites of 3335 genes from 3.34 million SNPs by SnpEff (Cingolani et al., 2012). Out of the 3335 genes, 2990 genes are annotated by MSU7, 1442 (48.23%) of which were annotated as transposable element (TE)‐related genes (Fig. S2). In the rice genome, only 30.35% (16 937/55 801) of all genes annotated by MSU7 are TE‐related genes. This result shows that splice site mutations tended to be enriched with TE‐related genes, which have much lower transcript abundance than non‐TE‐related genes (Jiao & Deng, 2007). Out of 3825 SNPs, only 240 (6.3%) were located at splice sites of 253 introns in 230 widely expressed genes (the gene‐level average read coverage ≥ 1 in at least 95% of all 328 samples) in the assayed rice seedlings. Most of the intron splicing site SNPs, 3585 (93.7%), were located in genes that were not widely expressed in all 82 accessions.
After studying SNPs existing at the splice sites to GSS events and non‐GSS events, one can find there were different effects on pre‐mRNA splicing regulation by these splice site variations. Based on their functions, we classified splice site mutations into three types: explicit‐effect, implicit‐effect and non‐effect, to help understand the regulation of pre‐mRNA splicing and to identify the most important and causal splice site mutations (Fig. S3). Explicit‐effect mutations produce a new splice site inside the original intron or IntronR where the mutant position can be identified in the new transcript. Implicit‐effect mutations produce a new splice site outside the original intron and the mutant splice sites do not appear on transcripts. Non‐effect mutations do not have significant effects on intron splice levels (Fig. S3). Among 240 splice site mutations located in widely expressed genes in the population, 21 (8.8%) were explicit‐effect mutations and 31 (12.9%) were implicit‐effect mutations, which are located in the cis‐sQTL peak regions and highly correlated with the lead SNPs (r2 ≥ 0.8). The rest of 188 (78.3% of 240 splice site mutations) were non‐effect mutations, as their effect on splicing variations was not detected by our algorithm (Table S1 for more details of explicit‐ and implicit‐effect SNPs). Most of these non‐effect mutations were located in introns with few junction reads supporting or low splice levels in the population. We examined the 22 non‐effect mutations located in high read coverage and high splicing level introns (in which the 3S score and junction count of at least 95% of the whole population are larger than 0.5 and 5, separately), and found that they were all located in splice donor sites, 19 of them were GT to GC mutations, which are just two types of canonical splice sites (GT–AG and GC–AG). Therefore, the effect of splice site mutations identified in this study could be consequential and these splice site mutations may play an essential role in regulating pre‐mRNA splicing.
Association analysis of GSS variations as markers with phenotypic outcomes to prioritise causal AS candidates
In our previous study, we reported the complex polygenic nature of salinity tolerance in rice using the same population (Campbell et al., 2017). There was only one large‐effect QTL for root sodium content identified by GWAS and the other salinity‐related phenotypes were regulated by a large number of loci with small effects. To identify AS associated with salt stress response, we employed the shoot Na+ content from RDP1 panel under salt stress conditions (Campbell et al., 2017). Although we can use 3S scores of all introns to perform an association study, there would be a high false‐positive rate and hence it is challenging to identify the causal SNPs linked to phenotypes. Therefore, we conducted an association study to associate GSS events, as markers, with phenotypes to prioritise the candidate causal AS and corresponding SNPs. This analysis identified six GSS events in five genes associated with shoot Na+ content (P < 0.01, Table 2). These GSS events included AltD, AltA, and IntronR, and most of them change the deduced protein sequence by changing the number of amino acid residues or producing a premature stop codon. To further confirm the function of these genes in salt stress response, we then asked whether these six GSS variations were associated with rice growth response to salinity. For this analysis, we leveraged temporal PSA from RGB imaging data generated for the same population for 14 d under salt stress and control conditions (Campbell et al., 2015). We found three out of six GSS events to be associated with significant differences in shoot growth response between different splicing clusters with P‐value < 0.01 (Table 2).
Table 2.
Information about genotype‐specific splicing (GSS) events in which the splicing variation was significantly associated with shoot Na+ content.
| Intron ID | Chr | Start | End | Strand | AS type | Effect | Growth response | MSU7 locus | MSU7 annotation |
|---|---|---|---|---|---|---|---|---|---|
| 91601 | 2 | 22 366 972 | 22 367 092 | ‐ | AltD | 2aa deletion | P = 0.59 | LOC_Os02g37030 | Protein binding protein, putative, expressed |
| 147728 | 4 | 31 546 251 | 31 546 895 | + | AltA | 2aa insertion | P = 0.0049* | LOC_Os04g52960 | Nucleolin, putative, expressed |
| 147745 | 4 | 31 546 251 | 31 546 901 | + | AltA | 2aa deletion | P = 0.0038* | LOC_Os04g52960 | Nucleolin, putative, expressed |
| 162765 | 5 | 19 785 085 | 19 785 537 | ‐ | AltA | 5′UTR | P = 0.76 | LOC_Os05g33630 | Inosine‐uridine preferring nucleoside hydrolase family protein, putative, expressed |
| 171645 | 6 | 1653 470 | 1653 637 | + | AltD | C‐terminal change | P = 0.29 | LOC_Os06g04040 | WD domain, G‐beta repeat domain containing protein, expressed |
| 232443 | 9 | 14 365 419 | 14 365 735 | + | IntronR | premature stop | P = 0.0043* | LOC_Os09g24200 | RAD23 DNA repair protein, putative, expressed |
Alternative splicing (AS) types include intron retention (IntronR), alternative donor site (AltD), and alternative acceptor site (AltA).
Indicates significant difference between two alleles (P < 0.01). aa, amino acid; 5′UTR, five‐prime untranslated region.
There were two GSS events, for the combination of Intron 147728 and Intron 147745 in the gene, OsNUC1 (LOC_Os04g52960), which has been identified as a salt‐responsive gene (Sripinyowanich et al., 2013; Udomchalothorn et al., 2017; Boonchai et al., 2018). Introns 147728 and 147745, located in between the third and fourth exons of OsNUC1, are a pair of AltA events with the same 5′ end and similar lengths (Table 2), but Intron 147728 is six nucleotides shorter than Intron 147745 at the 3′ end (Fig. 5a). There are two genotypes in the population: Genotype 1 (the reference genotype) produced only one transcript with Intron 147728 and had no transcript with Intron 147745 (Fig. 5, red samples); Genotype 2 produced two transcripts with the expression ratio of about 0.4 : 0.6 (Fig. 5, blue samples). These two transcripts differ in length, as the transcript with Intron 147728 is 6‐nt longer than transcript with Intron 147745, which results in a two amino acid (2 aa) deletion in the protein sequence. This 2 aa deletion is located in the amino terminal acidic serine‐rich (SR) region of OsNUC1 protein (Sripinyowanich et al., 2013). Although the mechanism of how the OsNUC1 protein with the 2 aa deletion directly alters the response of rice to salt stress is not known, Genotype 2 with two transcripts has a significantly better growth response than Genotype 1 which transcribes only the longer transcript (Fig. 5d). While the longer transcript with Intron 147728 was annotated by MSU7 (LOC_Os04g52960.1) and RAP‐DB/IRGSP1.0 (Os04t0620700‐01), the shorter transcript with Intron 147745 was not annotated. This suggests that the novel shorter transcript could enhance salt stress tolerance in rice. Our results provide a potential clue for a detailed functional analysis of OsNUC1 in salt response.
Fig. 5.
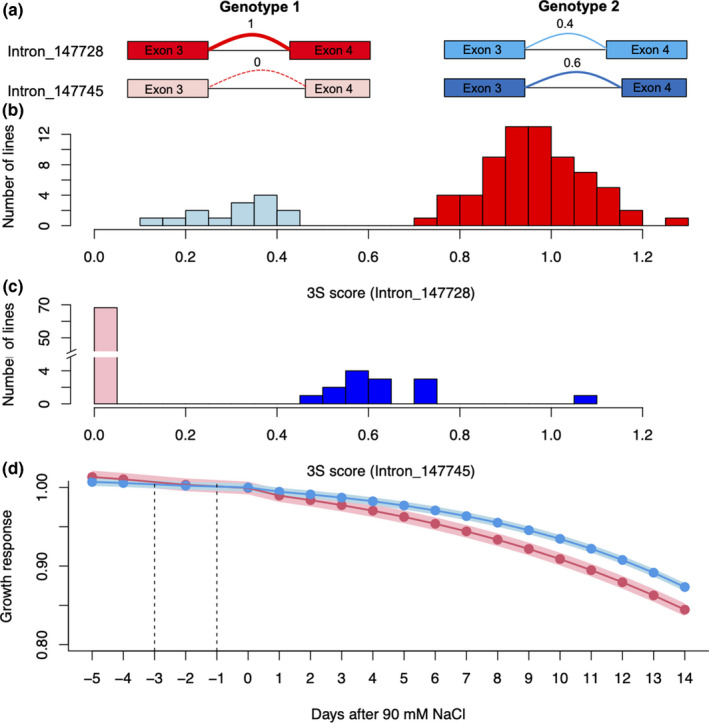
Genotype‐specific splicing (GSS) events in OsNUC1 comparison of salt‐induced growth response between different splice levels of GSS events. (a) Intron 147728 and Intron 147745 of OsNUC1 have two significantly different combinations of their Single Splicing Strength (3S) scores in the population, which forms two GSS events. In this manuscript, we used GSS events as markers, and the ‘Genotype 1’ and ‘Genotype 2’ in one GSS event indicates the two clusters of rice lines. The difference between ‘Genotype 1’ and ‘Genotype 2’ is the difference in splicing levels of two introns in OsNUC1. (b) The distribution of 3S scores for Intron 147728 and two clusters (two genotypes) were discovered. The cluster with light blue colour has lines of Genotype 2 with low 3S scores for Introns 147728, while lines in the red cluster (Genotype 1) have 3S scores close to 1, indicating transcripts having Intron 147728 only. (c) The distribution of 3S scores for Intron 147745 and the same two clusters were discovered. The cluster with pink colour has lines of Genotype 1 with almost zero 3S scores for Intron 147745, while the blue cluster (Genotype 2) has 3S scores close around 0.6. (d) The comparison of salt‐induced growth response between Genotypes 1 and 2. The red line indicates the growth response for Genotype 1 (only protein with a two amino acid insertion translated) and the blue line indicates the growth response for Genotype 2, and the shadow indicates the standard errors. The vertical dashed lines at −3 and −1 d indicate the time when 45 mM and 90 mM NaCl were treated, separately (Campbell et al., 2015).
Another significant GSS event, for Intron 232443, is in the gene OsRAD23 (LOC_Os09g24200), which encodes a RAD23 DNA repair protein. This GSS event was associated with alternative splicing (AS) caused by a GT to AT mutation, SNP ID: mlid0064466340, at the splice donor site of the sixth intron, resulting in an IntronR, represented by Sample 133 in Fig. 6a. As this IntronR was the largest effect AS found in the gene and the other AS in the 1st, 6th and 7th introns with small 3S scores had small effects on pre‐mRNA splicing, this IntronR is the main difference in the population (Fig. 6a). It should be noted that this evidently IntronR event was not identified using splicing scores of LeafCutter or Majiq. Interestingly, OsRAD23 also has a significant cis‐eQTLs (Fig. 4f). The overall expression of IntronR allele (A‐allele) was higher than the reference G‐allele. RAD23 proteins contain four domains: ubiquitin‐like (UBL), ubiquitin‐associated 1(UBA1), stress‐inducible‐1 (STI1), and UBA2 domains. The IntronR allele produces a premature stop codon and results in a truncated protein without the C‐terminal STI1 and UBA2 domains (Fig. 6b) (Farmer et al., 2010; Fu et al., 2010). Association analysis shows that the mutant allele (A‐allele) had higher shoot Na+ content under salt stress, indicating that the truncated protein increases salt sensitivity (Fig. 6c). Therefore, the IntronR allele (A‐allele) is more salt‐sensitive than the G‐allele, which is also supported by the growth response difference under salt stress (Fig. 6d).
Fig. 6.
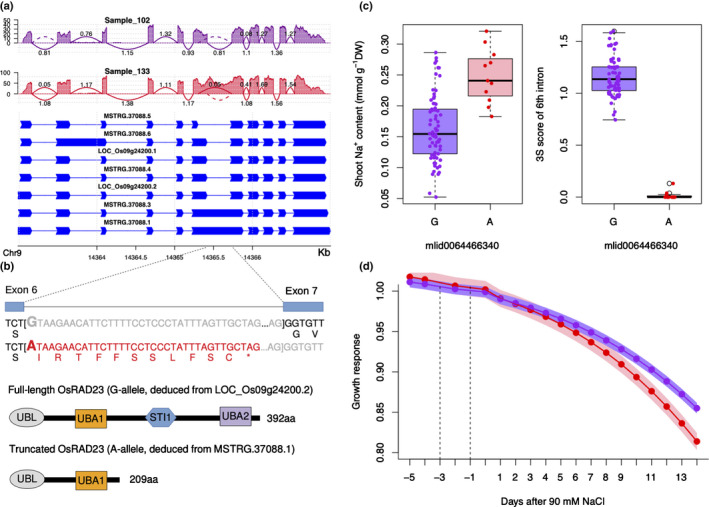
The genotype‐specific splicing (GSS) event in OsRAD23 and the association with shoot sodium content and salt‐induced growth response. (a) VaSP splice plot of OsRAD23 (LOC_Os09g24200) for transcript structures and splicing information in different genotypes, represented by Sample 102 (purple) and Sample 133 (red). The y‐axes are read depth and the arcs (lines between exons) indicate exon–exon junctions (introns). The dotted arcs indicate no junction reads spanning the introns and solid arcs indicate introns with junction reads. (b) Impact of intron retention (IntronR) on protein coding. The IntronR of the 6th intron causes a premature stop codon (TAG) and hence the corresponding transcripts (A‐allele) will be translated into a truncated protein without the C‐terminal region. (c) The association of splice site mutation with shoot Na+ content (left) and splicing levels of the 6th intron were indicated by Single Splicing Strength (3S) scores (right). For boxplots, the lower and the upper horizon lines are minimum and maximum values, respectively, and boxes have ranges from 25% to 75% quantiles and a centre line for 50% quantile (median). The points indicate values from individual accessions. (d) The comparison of salt‐induced growth response between the genotypes with IntronR (A‐allele) and without IntronR (G‐allele). The red line indicates the growth response for A‐allele and the purple line indicates the growth response for the G‐allele. The shadow indicates the standard errors. Two vertical dashed lines at −3 and −1 d indicate the time when 45 mM and 90 mM NaCl were treated, respectively (Campbell et al., 2015).
Discussion
Versatile applications of R package VaSP
We present a new software package, VaSP, with a novel 3S score feature to evaluate each intron splicing event. VaSP was able to identify novel splicing events independent of prior transcript annotations, especially IntronR, the most prevalent form in plants. Moreover, VaSP uses gene‐level read coverage of all isoforms for normalisation to avoid the challenges associated with estimating transcript isoform ratios or exon inclusion levels. The 3S scores are useful for quantifying splicing among samples or in a population, and therefore can be used for differential AS analysis or sQTL identification.
VaSP provides an optional further analysis in biallelic homozygous populations. The 3S score can be used for clustering to identify GSS variations without the need for genotypic information for multiple samples or in an inbred plant population, and two clusters of GSS events can differentiate two homozygous genotyping groups. This is particularly useful in plants, where inbred or natural populations, generally without heterozygous genotypes, are frequently used for biological research.
The VaSP package also provides visualisation of gene structure and AS events with publication‐quality multipanel figure output. The transcriptional isoforms, RNA‐seq read depths, and the splicing information can be shown in a single figure. One can use it to check the reliability of AS events at gene level for multiple samples.
AS for rice salt tolerance
Salinity is one of the major environmental factors limiting rice production globally, and there is a need for breeding salt tolerant rice with high yield potential. Like other types of abiotic stresses, however, salt tolerance is a polygenic trait (Munns & Tester, 2008; Deinlein et al., 2014). To identify salt‐responsive splicing variations, the GSS score feature of VaSP was applied to an association study and correlated with the shoot Na+ accumulation phenotype. Although 3S scores of all intron splicing events could be used for the study, we only focused on GSS events due to their large effects on splicing variations and the potential important biological functions. AS and salt‐related phenotypes are traits largely influenced by the environment. The direct association analysis of AS and salt‐related phenotypes would produce a large false‐positive rate, especially for a relatively small population. It is easier to find out the causal SNPs for large‐effect GSS events that are linked to phenotypes.
In our study, the efficiency of this analysis in RDP1 population was demonstrated by the identification of two salt‐responsive genes: OsNUC1 and OsRAD23 with splice variants that exhibit significant divergence between the variants for shoot growth under salt stress conditions. We identified six GSS events associated with rice shoot sodium content from our analysis of genotypes from the RDP1. Among these six GSS events, three GSS variations in two genes were associated with significant differences in salt‐induced growth response between two different isoforms. One of the genes, OsNUC1, has two AltA sites. OsNUC1 contains five domains: bipartite nuclear localisation signal (NLS), the SR region, two RNA recognition motifs (RRM), and a C‐terminal glycine‐ and arginine‐rich (GAR) domain (Sripinyowanich et al., 2013). Previous studies have reported that OsNUC1 is a salt‐responsive gene and its over‐expression enhances salt tolerance in transgenic Arabidopsis and rice (Sripinyowanich et al., 2013; Udomchalothorn et al., 2017; Boonchai et al., 2018). In our study, the transcript abundance of OsNUC1 was significantly downregulated under salt stress. We show that a novel 6‐nt shorter transcript (2 aa deletion in SR region of OsNUC1 protein) is associated with enhanced salt tolerance in the rice germplasm. Interestingly, a salt tolerant QTL, qSES4, was identified on rice chromosome 4 and OsNUC1 is one of candidate genes in the region (Pang et al., 2017).
An IntronR (GT to AT splice site mutation) in OsRAD23, which also has a cis‐eQTL, is associated with salt sensitivity. OsRAD23 is also responsive to salt stress (Nveawiah‐Yoho et al., 2013; Li et al., 2015; Lakra et al., 2019). RAD23 proteins play a role in stress response and development through their delivery of ubiquitinated substrates to the 26S proteasome (Farmer et al., 2010; Kang et al., 2017; Wang et al., 2017). Specifically, the UBA2 domain is important for ubiquitin binding and RAD23 stability in proteasomal degradation (Chen et al., 2001; Heessen et al., 2005; Jang et al., 2012). The UBL‐UBA protein OsDSK2a mediates seedling growth and salt responses in rice by combing with polyubiquitin chains, and interacts with the gibberellin (GA)‐deactivating enzyme EUI, resulting in its degradation through the ubiquitin‐proteasome system (Wang et al., 2020). The IntronR A‐allele we identified encodes a truncated RAD23 protein lacking the C‐terminal STI1 and UBA2 domains, which are essential for ubiquitin binding and RAD23 protein stability in proteasomal degradation (Chen et al., 2001; Heessen et al., 2005; Jang et al., 2012). Relative to the reference allele (the G‐allele) with a full‐length protein, genotypes with the A‐allele are more salt‐sensitive. Therefore, we hypothesise that replacing the A‐allele with the G‐allele could potentially improve the salt tolerance of currently A‐allele genotypes.
Splice site mutation, AS and evolution
AS increases the transcriptome and proteome diversity and, in plants, many alternative transcripts may not be translated into proteins or buffer against the stress‐responsive transcriptome (Nilsen & Graveley, 2010; Tress et al., 2017; Chaudhary et al., 2019a), therefore contributing to phenotypic diversity in eukaryotes. Although several studies have focused on the mechanism and the regulation of AS, (Lopez, 1998; Modrek & Lee, 2002; Black, 2003; Matlin et al., 2005; Irimia & Roy, 2014; Lee & Rio, 2015), even including the mechanism of chromatin remodeling in AS under stress (Yu et al., 2019), the evolution of AS is less explored (Keren et al., 2010). Although the basic ability of splicing introns is conserved throughout evolution, the splicing signals and their corresponding splicing factors evolved (Schwartz et al., 2008) and the prevalence of AS throughout evolution is important (Keren et al., 2010), for instance, in shaping the evolution of genomes (Gamazon & Stranger, 2014) and the evolution of novel phenotypes (Bush et al., 2017). There are four main splicing signals directing intron splicing: the 5′ donor and 3′ acceptor splice sites (5′ SS and 3′ SS), the branch site (BS), and the polypyrimidine tract (PPT) (Murray et al., 2008; Schwartz et al., 2008). Among them, 5′ SS and 3′ SS, especially the intronic terminal dinucleotides, are highly conserved and the most important splice sites (the three canonical splice site combinations are GT–AG, GC–AG and AT–AC, the first being by far the most common). Splice site mutations could affect splicing in multiple ways, including a change in splice junctions, and intron retention. These mutations can also affect splicing efficiency, reducing but not necessarily eliminating the normally spliced transcript. In the rice RDP1 population, 3825 SNPs were identified at splice donor or acceptor sites for 3335 genes, but only 230 of these genes were widely expressed (the gene‐level average read coverage ≥ 1 in at least 95% of all 328 samples) in this rice population. As about half of the genes with splice site mutations were TE related, which are an extensive source of mutations and genetic polymorphisms (Bourque et al., 2018) and usually have lower transcriptional activities (Jiao & Deng, 2007), the splice site mutations have low evolutionary pressure or selection pressure. Most of these TE‐related genes with splice site mutations are retrotransposons or Class 1 TEs (Fig. S2).
Author contributions
HY designed and performed research and developed algorithms; HY and QD analysed data; HY, CZ, BY and HW wrote the paper; MC conducted experiments and collected data; CZ, HW and BY acquired funding and supervised the project.
Supporting information
Fig. S1 Splicing events identified by VaSP in RDP1 population.
Fig. S2 Different types of TE (transposable element)‐related genes with splice site mutations.
Fig. S3 Classification of splice site mutations.
Table S1 Detailed information of explicit‐effect (ex) and implicit‐effect (im) of splice site mutations in our dataset.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
This work was completed utilising the Holland Computing Center of the University of Nebraska. We thank Qi Zhang for the discussion on the statistical methods used in the manuscript. This project was supported by the National Science Foundation (Award no. DBI‐1564621 and Award no. 1238125 to HW and CZ, OIA‐1736192 HW and CZ, Award no. OIA‐1557417 to CZ and BY, and Award no. MCB‐1818‐82 to CZ and BY) and the Nebraska Soybean Board (Award 20R‐09‐1/2 no. 1739 to CZ).
Contributor Information
Harkamal Walia, Email: hwalia2@unl.edu.
Chi Zhang, Email: czhang5@unl.edu.
Data availability
The R package, VaSP, is available on Bioconductor (https://bioconductor.org/packages/VaSP). The RNA‐seq data used in this study are available at NCBI GEO under the accession no. GSE98455.
References
- Andrews S. 2014. FastQC a quality control tool for high throughput sequence data . [WWW document] URL https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Au KF, Jiang H, Lin L, Xing Y, Wong WH. 2010. Detection of splice junctions from paired‐end RNA‐seq data by SpliceMap. Nucleic Acids Research 38: 4570–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbazuk WB, Fu Y, McGinnis KM. 2008. Genome‐wide analyses of alternative splicing in plants: opportunities and challenges. Genome Research 18: 1381–1392. [DOI] [PubMed] [Google Scholar]
- Black DL. 2003. Mechanisms of alternative pre‐messenger RNA splicing. Annual Review of Biochemistry 72: 291–336. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonchai C, Udomchalothorn T, Sripinyowanich S, Comai L, Buaboocha T, Chadchawan S. 2018. Rice overexpressing OsNUC1‐S reveals differential gene expression leading to yield loss reduction after salt stress at the booting stage. International Journal of Molecular Sciences 19: 3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque G, Burns KH, Gehring M, Gorbunova V, Seluanov A, Hammell M, Imbeault M, Izsvak Z, Levin HL, Macfarlan TS et al. 2018. Ten things you should know about transposable elements. Genome Biology 19: 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweig U, Barbosa‐Morais NL, Pan Q, Nachman EN, Alipanahi B, Gonatopoulos‐Pournatzis T, Frey B, Irimia M, Blencowe BJ. 2014. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Research 24: 1774–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush SJ, Chen L, Tovar‐Corona JM, Urrutia AO. 2017. Alternative splicing and the evolution of phenotypic novelty. Philosophical Transactions of the Royal Society B: Biological Sciences 372: 20150474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MT, Bandillo N, Al Shiblawi FRA, Sharma S, Liu K, Du Q, Schmitz AJ, Zhang C, Very AA, Lorenz AJ et al. 2017. Allelic variants of OsHKT1 ;1 underlie the divergence between indica and japonica subspecies of rice (Oryza sativa) for root sodium content. PLoS genetics 13: e1006823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MT, Du Q, Liu K, Sharma S, Zhang C, Walia H. 2020. Characterization of the transcriptional divergence between the subspecies of cultivated rice (Oryza sativa). BMC Genomics 21: 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MT, Knecht AC, Berger B, Brien CJ, Wang D, Walia H. 2015. Integrating image‐based phenomics and association analysis to dissect the genetic architecture of temporal salinity responses in rice. Plant Physiology 168: 1476–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamala S, Feng G, Chavarro C, Barbazuk WB. 2015. Genome‐wide identification of evolutionarily conserved alternative splicing events in flowering plants. Frontiers in Bioengineering and Biotechnology 3: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary S, Jabre I, Reddy ASN, Staiger D, Syed NH. 2019a. Perspective on alternative splicing and proteome complexity in plants. Trends in Plant Science 24: 496–506. [DOI] [PubMed] [Google Scholar]
- Chaudhary S, Khokhar W, Jabre I, Reddy ASN, Byrne LJ, Wilson CM, Syed NH. 2019b. Alternative splicing and protein diversity: plants versus animals. Frontiers in Plant Science 10: 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Shinde U, Ortolan TG, Madura K. 2001. Ubiquitin‐associated (UBA) domains in Rad23 bind ubiquitin and promote inhibition of multi‐ubiquitin chain assembly. EMBO Reports 2: 933–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Han Y, Liu H, Wang X, Sun J, Zhao B, Li W, Tian J, Liang Y, Yan J et al. 2018. Genome‐wide association analyses reveal the importance of alternative splicing in diversifying gene function and regulating phenotypic variation in maize. Plant Cell 30: 1404–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton D. 2020. snpStats: SnpMatrix and XSnpMatrix classes and methods . R package v.1.38.0. doi: 10.18129/B9.bioc.snpStats.
- Cotsaftis O, Plett D, Johnson AA, Walia H, Wilson C, Ismail AM, Close TJ, Tester M, Baumann U. 2011. Root‐specific transcript profiling of contrasting rice genotypes in response to salinity stress. Molecular Plant 4: 25–41. [DOI] [PubMed] [Google Scholar]
- Danan‐Gotthold M, Golan‐Gerstl R, Eisenberg E, Meir K, Karni R, Levanon EY. 2015. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Research 43: 5130–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deinlein U, Stephan AB, Horie T, Luo W, Xu G, Schroeder JI. 2014. Plant salt‐tolerance mechanisms. Trends in Plant Science 19: 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Rath E, Bai Y. 2017. Comparison of alternative splicing junction detection tools using RNA‐Seq data. Current Genomics 18: 268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, He F, Berkowitz O, Liu J, Cao P, Tang M, Shi H, Wang W, Li Q, Shen Z et al. 2018. Alternative splicing plays a critical role in maintaining mineral nutrient homeostasis in rice (Oryza sativa). Plant Cell 30: 2267–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Q, Campbell M, Yu H, Liu K, Walia H, Zhang Q, Zhang C. 2019. Network‐based feature selection reveals substructures of gene modules responding to salt stress in rice. Plant Direct 3: e00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edriss V, Guldbrandtsen B, Lund MS, Su G. 2013. Effect of marker‐data editing on the accuracy of genomic prediction. Journal of Animal Breeding and Genetics 130: 128–135. [DOI] [PubMed] [Google Scholar]
- Eizenga GC, Ali ML, Bryant RJ, Yeater KM, McClung AM, McCouch S. 2014. Registration of the rice diversity panel 1 for genomewide association studies. Journal of Plant Registrations 8: 109–116. [Google Scholar]
- Emig D, Salomonis N, Baumbach J, Lengauer T, Conklin BR, Albrecht M. 2010. AltAnalyze and DomainGraph: analyzing and visualizing exon expression data. Nucleic Acids Research 38: W755–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famoso AN, Zhao K, Clark RT, Tung CW, Wright MH, Bustamante C, Kochian LV, McCouch SR. 2011. Genetic architecture of aluminum tolerance in rice (Oryza sativa) determined through genome‐wide association analysis and QTL mapping. PLoS Genetics 7: e1002221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer LM, Book AJ, Lee KH, Lin YL, Fu H, Vierstra RD. 2010. The RAD23 family provides an essential connection between the 26S proteasome and ubiquitylated proteins in Arabidopsis . Plant Cell 22: 124–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazee AC, Pertea G, Jaffe AE, Langmead B, Salzberg SL, Leek JT. 2015. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nature Biotechnology 33: 243–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Lin YL, Fatimababy AS. 2010. Proteasomal recognition of ubiquitylated substrates. Trends in Plant Science 15: 375–386. [DOI] [PubMed] [Google Scholar]
- Gamazon ER, Stranger BE. 2014. Genomics of alternative splicing: evolution, development and pathophysiology. Human Genetics 133: 679–687. [DOI] [PubMed] [Google Scholar]
- Grau‐Bove X, Ruiz‐Trillo I, Irimia M. 2018. Origin of exon skipping‐rich transcriptomes in animals driven by evolution of gene architecture. Genome Biology 19: 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Zink D, Korn B, Vingron M, Haas SA. 2004. Genome wide identification and classification of alternative splicing based on EST data. Bioinformatics 20: 2579–2585. [DOI] [PubMed] [Google Scholar]
- Heessen S, Masucci MG, Dantuma NP. 2005. The UBA2 domain functions as an intrinsic stabilization signal that protects Rad23 from proteasomal degradation. Molecular Cell 18: 225–235. [DOI] [PubMed] [Google Scholar]
- Hooper JE. 2014. A survey of software for genome‐wide discovery of differential splicing in RNA‐Seq data. Human Genomics 8: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia M, Roy SW. 2014. Origin of spliceosomal introns and alternative splicing. Cold Spring Harbor Perspectives in Biology 6: a016071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabre I, Reddy ASN, Kalyna M, Chaudhary S, Khokhar W, Byrne LJ, Wilson CM, Syed NH. 2019. Does co‐transcriptional regulation of alternative splicing mediate plant stress responses? Nucleic Acids Research 47: 2716–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang IC, Niu QW, Deng S, Zhao P, Chua NH. 2012. Enhancing protein stability with retained biological function in transgenic plants. The Plant Journal 72: 345–354. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Deng XW. 2007. A genome‐wide transcriptional activity survey of rice transposable element‐related genes. Genome biology 8: R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M, Lee S, Abdelmageed H, Reichert A, Lee HK, Fokar M, Mysore KS, Allen RD. 2017. Arabidopsis stress associated protein 9 mediates biotic and abiotic stress responsive ABA signaling via the proteasome pathway. Plant, Cell, & Environment 40: 702–716. [DOI] [PubMed] [Google Scholar]
- Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, Schwartz DC, Tanaka T, Wu J, Zhou S et al. 2013. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki S, Borchert C, Deyholos M, Wang H, Brazille S, Kawai K, Galbraith D, Bohnert HJ. 2001. Gene expression profiles during the initial phase of salt stress in rice. Plant Cell 13: 889–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren H, Lev‐Maor G, Ast G. 2010. Alternative splicing and evolution: diversification, exon definition and function. Nature Reviews Genetics 11: 345–355. [DOI] [PubMed] [Google Scholar]
- Khokhar W, Hassan MA, Reddy ASN, Chaudhary S, Jabre I, Byrne LJ, Syed NH. 2019. Genome‐wide identification of splicing quantitative trait loci (sQTLs) in diverse ecotypes of arabidopsis thaliana . Frontiers in Plant Science 10: 1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12: 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Magen A, Ast G. 2007. Different levels of alternative splicing among eukaryotes. Nucleic Acids Research 35: 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakra N, Kaur C, Singla‐Pareek SL, Pareek A. 2019. Mapping the 'early salinity response' triggered proteome adaptation in contrasting rice genotypes using iTRAQ approach. Rice 12: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laloum T, Martin G, Duque P. 2018. Alternative splicing control of abiotic stress responses. Trends in Plant Science 23: 140–150. [DOI] [PubMed] [Google Scholar]
- Law CW, Alhamdoosh M, Su S, Dong X, Tian L, Smyth GK, Ritchie ME. 2018. RNA‐seq analysis is easy as 1‐2‐3 with limma, Glimma and edgeR. F1000Research 5: 1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Rio DC. 2015. Mechanisms and regulation of alternative pre‐mRNA splicing. Annual Review of Biochemistry 84: 291–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhao F, Fang W, Xie D, Hou J, Yang X, Zhao Y, Tang Z, Nie L, Lv S. 2015. Identification of early salt stress responsive proteins in seedling roots of upland cotton (Gossypium hirsutum L.) employing iTRAQ‐based proteomic technique. Frontiers Plant Science 6: 732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YI, Knowles DA, Humphrey J, Barbeira AN, Dickinson SP, Im HK, Pritchard JK. 2018. Annotation‐free quantification of RNA splicing using LeafCutter. Nature Genetics 50: 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez AJ. 1998. Alternative splicing of pre‐mRNA: developmental consequences and mechanisms of regulation. Annual Review of Genetics 32: 279–305. [DOI] [PubMed] [Google Scholar]
- Lutts S, Kinet JM, Bouharmont J. 1996. NaCl‐induced senescence in leaves of rice (Oryza sativaL.) cultivars differing in salinity resistance. Annals of Botany 78: 389–398. [Google Scholar]
- Marquez Y, Brown JW, Simpson C, Barta A, Kalyna M. 2012. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis . Genome Research 22: 1184–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mather KA, Caicedo AL, Polato NR, Olsen KM, McCouch S, Purugganan MD. 2007. The extent of linkage disequilibrium in rice (Oryza sativa L.). Genetics 177: 2223–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlin AJ, Clark F, Smith CW. 2005. Understanding alternative splicing: towards a cellular code. Nature Reviews Molecular Cell Biology 6: 386–398. [DOI] [PubMed] [Google Scholar]
- McCouch SR, Wright MH, Tung C‐W, Maron LG, McNally KL, Fitzgerald M, Singh N, DeClerck G, Agosto‐Perez F, Korniliev P et al. 2016. Open access resources for genome‐wide association mapping in rice. Nature Communications 7: 10532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehmood A, Laiho A, Venalainen MS, McGlinchey AJ, Wang N, Elo LL. 2019. Systematic evaluation of differential splicing tools for RNA‐seq studies. Briefings in Bioinformatics 21: 2052–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrek B, Lee C. 2002. A genomic view of alternative splicing. Nature Genetics 30: 13–19. [DOI] [PubMed] [Google Scholar]
- Munns R, Tester M. 2008. Mechanisms of salinity tolerance. Annual Review of Plant Biology 59: 651–681. [DOI] [PubMed] [Google Scholar]
- Murray JI, Voelker RB, Henscheid KL, Warf MB, Berglund JA. 2008. Identification of motifs that function in the splicing of non‐canonical introns. Genome Biology 9: R97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ner‐Gaon H, Halachmi R, Savaldi‐Goldstein S, Rubin E, Ophir R, Fluhr R. 2004. Intron retention is a major phenomenon in alternative splicing in Arabidopsis . The Plant Journal 39: 877–885. [DOI] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR. 2010. Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nveawiah‐Yoho P, Zhou J, Palmer M, Sauve R, Zhou S, Howe K, Fish T, Thannhauser T. 2013. Identification of proteins for salt tolerance using a comparative proteomics analysis of tomato accessions with contrasting salt tolerance. Journal of the American Society for Horticultural Science 138: 382–394. [Google Scholar]
- Pang Y, Chen K, Wang X, Wang W, Xu J, Ali J, Li Z. 2017. Simultaneous improvement and genetic dissection of salt tolerance of rice (Oryza sativa L.) by designed QTL pyramiding. Frontiers Plant Science 8: 1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. 2016. Transcript‐level expression analysis of RNA‐seq experiments with HISAT, StringTie and Ballgown. Nature Protocols 11: 1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. 2015. StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nature Biotechnology 33: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AP, Richards G, de la Iglesia B, Rayward‐Smith VJ. 2006. Clustering rules: a comparison of partitioning and hierarchical clustering algorithms. Journal of Mathematical Modelling and Algorithms 5: 475–504. [Google Scholar]
- Schwartz SH, Silva J, Burstein D, Pupko T, Eyras E, Ast G. 2008. Large‐scale comparative analysis of splicing signals and their corresponding splicing factors in eukaryotes. Genome Research 18: 88–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, Xing Y. 2014. rMats: robust and flexible detection of differential alternative splicing from replicate RNA‐Seq data. Proceedings of the National Academy of Sciences, USA 111: E5593–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sripinyowanich S, Chamnanmanoontham N, Udomchalothorn T, Maneeprasopsuk S, Santawee P, Buaboocha T, Qu LJ, Gu H, Chadchawan S. 2013. Overexpression of a partial fragment of the salt‐responsive gene OsNUC1 enhances salt adaptation in transgenic Arabidopsis thaliana and rice (Oryza sativa L.) during salt stress. Plant Science 213: 67–78. [DOI] [PubMed] [Google Scholar]
- Tress ML, Abascal F, Valencia A. 2017. Alternative splicing may not be the key to proteome complexity. Trends in Biochemal Science 42: 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udomchalothorn T, Plaimas K, Sripinyowanich S, Boonchai C, Kojonna T, Chutimanukul P, Comai L, Buaboocha T, Chadchawan S. 2017. OsNucleolin1‐L expression in arabidopsis enhances photosynthesis via transcriptome modification under salt stress sonditions. Plant Cell Physiology 58: 717–734. [DOI] [PubMed] [Google Scholar]
- Vaquero‐Garcia J, Barrera A, Gazzara MR, Gonzalez‐Vallinas J, Lahens NF, Hogenesch JB, Lynch KW, Barash Y. 2016. A new view of transcriptome complexity and regulation through the lens of local splicing variations. eLife 5: e11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquero‐Garcia J, Norton S, Barash Y. 2018. LeafCutter vs. MAJIQ and comparing software in the fast moving field of genomics. bioRxiv: 463927. [Google Scholar]
- Walia H, Wilson C, Condamine P, Liu X, Ismail AM, Zeng L, Wanamaker SI, Mandal J, Xu J, Cui X et al. 2005. Comparative transcriptional profiling of two contrasting rice genotypes under salinity stress during the vegetative growth stage. Plant Physiology 139: 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walia H, Wilson C, Zeng L, Ismail AM, Condamine P, Close TJ. 2007. Genome‐wide transcriptional analysis of salinity stressed japonica and indica rice genotypes during panicle initiation stage. Plant Molecular Biology 63: 609–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang BB, Brendel V. 2006. Genomewide comparative analysis of alternative splicing in plants. Proceedings of the National Academy of Sciences, USA 103: 7175–7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DR, Agosto‐Pérez FJ, Chebotarov D, Shi Y, Marchini J, Fitzgerald M, McNally KL, Alexandrov N, McCouch SR. 2018. An imputation platform to enhance integration of rice genetic resources. Nature Communications 9: 3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Qin H, Zhou S, Wei P, Zhang H, Zhou Y, Miao Y, Huang R. 2020. The ubiquitin‐binding protein OsDSK2a mediates seedling growth and salt responses by regulating gibberellin metabolism in rice. Plant Cell 32: 414–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Yu H, Xie W, Xing Y, Yu S, Xu C, Li X, Xiao J, Zhang Q. 2010. A global analysis of QTLs for expression variations in rice shoots at the early seedling stage. The Plant Journal 63: 1063–1074. [DOI] [PubMed] [Google Scholar]
- Wang N, Gong X‐q, Ma F‐w. 2017. Genome‐wide identification of the radiation sensitivity protein‐23 (RAD23) family members in apple (Malus × domestica Borkh.) and expression analysis of their stress responsiveness. Journal of Integrative Agriculture 16: 820–827. [Google Scholar]
- Yu H, Xie W, Wang J, Xing Y, Xu C, Li X, Xiao J, Zhang Q. 2011. Gains in QTL detection using an ultra‐high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS ONE 6: e17595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Meng X, Liu Y, Wang X, Wang TJ, Zhang A, Li N, Qi X, Liu B, Xu ZY. 2019. The chromatin remodeler ZmCHB101 impacts alternative splicing contexts in response to osmotic stress. Plant Cell Reports 38: 131–145. [DOI] [PubMed] [Google Scholar]
- Zhang P, Zhong K, Zhong Z, Tong H. 2019. Genome‐wide association study of important agronomic traits within a core collection of rice (Oryza sativa L.). BMC Plant Biology 19: 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Calixto CPG, Marquez Y, Venhuizen P, Tzioutziou NA, Guo W, Spensley M, Entizne JC, Lewandowska D, Ten Have S et al. 2017. A high quality Arabidopsis transcriptome for accurate transcript‐level analysis of alternative splicing. Nucleic Acids Research 45: 5061–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Tung CW, Eizenga GC, Wright MH, Ali ML, Price AH, Norton GJ, Islam MR, Reynolds A, Mezey J et al. 2011. Genome‐wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa . Nature Communications 2: 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Stephens M. 2012. Genome‐wide efficient mixed‐model analysis for association studies. Nature Genetics 44: 821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Mao F, Tian Y, Lin X, Gu L, Gu H, Qu LJ, Wu Y, Wu Z. 2020. The features and regulation of co‐transcriptional splicing in Arabidopsis . Molecular Plant 13: 278–294. [DOI] [PubMed] [Google Scholar]
- Zhu J, Liu M, Liu X, Dong Z. 2018. RNA polymerase II activity revealed by GRO‐seq and pNET‐seq in Arabidopsis . Nature Plants 4: 1112–1123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Splicing events identified by VaSP in RDP1 population.
Fig. S2 Different types of TE (transposable element)‐related genes with splice site mutations.
Fig. S3 Classification of splice site mutations.
Table S1 Detailed information of explicit‐effect (ex) and implicit‐effect (im) of splice site mutations in our dataset.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
The R package, VaSP, is available on Bioconductor (https://bioconductor.org/packages/VaSP). The RNA‐seq data used in this study are available at NCBI GEO under the accession no. GSE98455.