

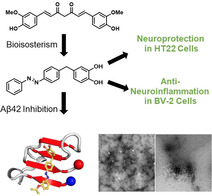
Abstract
Many (poly‐)phenolic natural products, for example, curcumin and taxifolin, have been studied for their activity against specific hallmarks of neurodegeneration, such as amyloid‐β 42 (Aβ42) aggregation and neuroinflammation. Due to their drawbacks, arising from poor pharmacokinetics, rapid metabolism, and even instability in aqueous medium, the biological activity of azobenzene compounds carrying a pharmacophoric catechol group, which have been designed as bioisoteres of curcumin has been examined. Molecular simulations reveal the ability of these compounds to form a hydrophobic cluster with Aβ42, which adopts different folds, affecting the propensity to populate fibril‐like conformations. Furthermore, the curcumin bioisosteres exceeded the parent compound in activity against Aβ42 aggregation inhibition, glutamate‐induced intracellular oxidative stress in HT22 cells, and neuroinflammation in microglial BV‐2 cells. The most active compound prevented apoptosis of HT22 cells at a concentration of 2.5 μm (83 % cell survival), whereas curcumin only showed very low protection at 10 μm (21 % cell survival).
Keywords: amyloid beta, bioisosterism, natural products, neuroprotectivity, replica-exchange molecular dynamics
Improving on nature: Synthetic bioisosteres of the natural product curcumin are designed, synthesized, and computationally evaluated by molecular dynamics and replica‐exchange molecular dynamics simulations for their interaction with amyloid‐β 42 aggregation. Biological evaluation shows that such compounds greatly exceed the anti‐neuroinflammatory and neuroprotective properties of their parent compounds.
Introduction
Alzheimer's disease (AD) is the most common form of dementia and causes progressive deterioration in cognitive behavior. [1] One of the main pathogenic hallmarks of AD is the deposition of senile plaques, which consist of aggregates of amyloid‐β (Aβ) peptides, generally containing 40 (Aβ40) or 42 (Aβ42) residues. [2] These plaques are linked to neurotoxicity, oxidative stress, and neurodegeneration.[ 3 , 4 ] Neuroinflammation also contributes to neurodegeneration and accelerates the progression of AD. [5] Because the aggregation of Aβ peptides is believed to be the initial event of AD, the identification of potential inhibitors of amyloid aggregation has attracted much interest. [6] Among these compounds, curcumin (Figure 1) is a diarylheptanoid natural product that has shown positive effects on counteracting oxidative stress and inflammation, as well as preventing Aβ aggregation. [7] Structure–activity relationship (SAR) studies have shown that methylation of the free hydroxy groups of curcumin leads to a loss of activity. [8] Nevertheless, the therapeutic potential of curcumin is limited by poor pharmacokinetics, high rate of metabolism, and low stability in an aqueous environment. [9] Additionally, curcumin is considered as a pan‐assay interference compound (PAIN), [9] which can possibly interfere with the assay readout or bind nonspecifically to proteins, leading to false positive results. [10]
Figure 1.
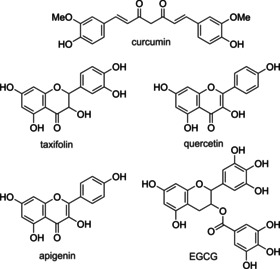
Chemical structures of curcumin, taxifolin, quercetin, apigenin, and epigallocatechin gallate (EGCG).
Other (poly‐)phenolic compounds, such as apigenin, quercetin, and taxifolin (Figure 1), have also shown positive effects in counteracting the causative events of neurodegeneration.[ 7c , 11 ] In particular, flavonoids, a class of polyphenolic natural products, are promising compounds against amyloid aggregation, neuroinflammation, and oxidative stress. [12] Recent studies have shown that chemical hybrids of taxifolin exhibit pronounced neuroprotectivity in vitro and in vivo. [13] Furthermore, through the development of chemical probes for proteomic studies, it was shown that these compounds seemed to specifically address mitochondrial targets. [14] Several studies revealed the importance of the catechol unit of flavonoids for their activity against age‐related disease processes, especially in the context of AD. [15] However, flavonoids suffer from similar drawbacks to those of curcumin, such as poor pharmacokinetics and low metabolic stability. [16]
A clear understanding of the precise mechanism of action of Aβ42 aggregation inhibitors is challenging due to the complexity of the conformational space of Aβ42 monomers, the occurrence of distinct oligomeric species in early aggregates, and the timescale of different events implicated in the formation of Aβ42 fibrils. Furthermore, whether a given compound can exert inhibitory activity acting at different stages of Aβ42 aggregation is also unclear. [17] The antiaggregating activity of small organic compounds has been related to specific chemical features, such as the hydroxylation profile, the presence of carboxyl moieties that may form salts bridges with Aβ42, and the molecular planarity conferred by aromatic rings. [18] Thus, it has been proposed that curcumin could intercalate in Aβ42 assemblies and destabilize preformed fibrils; [19] this effect is improved upon through 1) expansion of the aromatic rings; 2) integration of large conjugated structures; 3) the presence of aromatic rings connected by nitrogen‐containing bridge; and 4) hydroxyl groups on aromatic, conjugated rings. [19] Apart from that, (poly‐)phenolic compounds bearing a catechol unit, such as taxifolin, were observed to undergo autoxidation, and thus, produce a site‐specific covalent inhibition of Aβ42 aggregation by acting on K16 and/or K28 residues of preformed amyloid fibrils, according to an aza‐Michael addition mechanism. [20] EGCG (Figure 1), another catechol type (poly‐)phenolic compound, naturally occurring in green tea, was observed to produce covalent adducts through a Schiff base mechanism. [21] Interestingly, the reduced form of EGCG was proposed to act at early aggregation stages by redirecting toxic Aβ oligomers towards off‐pathway nontoxic oligomers. [22]
A common strategy to improve the pharmacological profile of bioactive molecules is bioisosterism. This applies changes in the molecular structure of a lead compound to improve their physicochemical properties, while preserving the relevant pharmacophoric features of the lead structure. [23] If combined with photopharmacology, which represents an emerging strategy that enables the photochemical control of biologically active molecules and biosensors, [24] bioisoteric compounds might offer a fine‐tuning of the antiaggregation activity. In particular, our strategy in this study has been to characterize the pharmacological profile of azobenzene bioisosteres of curcumin suitably modified to incorporate the pharmacophoric catechol moiety of flavonoids. These compounds were conceived by hybridizing relevant structural elements present in curcumin and taxifolin by following the rationale summarized in Figure 2. This led to a new class of compounds that successfully incorporated the previously cited Aβ‐related pharmacological properties (bioactive catechol ring, aromaticity, and planarity).
Figure 2.
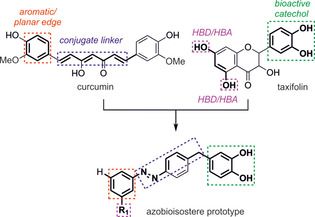
Rationalization for the azobioisostere prototype from the Aβ42 inhibitors curcumin and taxifolin. HBD=hydrogen‐bond donor; HBA=hydrogen‐bond acceptor.
Herein, we report the design and synthesis of azobenzene‐containing bioisosteric analogues of curcumin and investigate their antiaggregation ability against Aβ42 in vitro and in a bacterial model. [25] Furthermore, we evaluate their neuroprotective properties against intracellular oxidative stress in murine hippocampal HT22 cells and their anti‐neuroinflammatory potential in microglial BV‐2 cells.
Results and Discussion
Synthesis of the target compounds
The synthesis of the target compounds started with the Friedel–Crafts acylation of 4‐nitrobenzoyl chloride (1) with dimethoxybenzene (2) to yield the corresponding acetophenone 3. Ether cleavage of the methoxy groups was achieved in a mixture of concentrated hydrobromic acid and acetic acid, followed by hydrogenation of the keto group with H2 on Pd/C, to obtain compound 5 a as the first building block. Partial oxidation of anilines with oxone yielded the respective nitroso derivatives, which were combined with 5 a in a Baeyer–Mills reaction to form the desired target compounds 8 a–f (Scheme 1). Compound 8 g, with both catechol hydroxyl groups methylated, was also synthesized to explore the role of the catechol moiety by comparison (see below) with the activity of target compounds 8 a–f. It was synthesized in analogy to compounds 8 a–f, but without cleavage of the methoxy groups of compound 3.
Scheme 1.
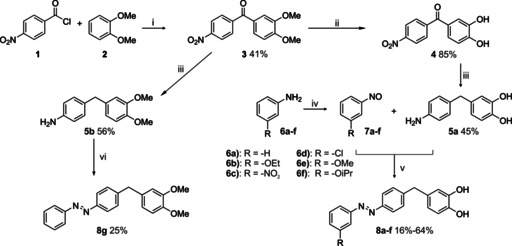
Synthesis of target compounds 8 a–f and comparison compound 8 g. Reagents and conditions: i) FeCl3, 60 °C, 16 h; ii) 48 % HBr, AcOH, reflux, 3.5 h; iii) H2, Pd/C, MeOH, 10 bar, RT, 16 h; iv) oxone, CH2Cl2/H2O, RT, 3.5 h; v) AcOH, RT, 16 h, vi) nitrosobenzene, AcOH, RT, 16 h.
In vitro inhibition of Aβ42 and tau aggregation
Because the aggregation of amyloidogenic proteins, such as Aβ42, and deposits of hyperphosphorylated tau protein in neurofibrillary tangles are associated with neurodegenerative diseases, such as AD,[ 26 , 27 ] compounds with antiaggregation properties may be a viable option for modifying the disease. To evaluate the antiaggregation activity of the target compounds, a rapid in vitro screening method in bacterial cells was applied.[ 7c , 25 ] This method is based on Escherichia coli overexpressing the respective protein (Aβ42, human tau), which forms inclusion bodies (IBs). IBs are consequently stained by thioflavin‐S (Th‐S) to assess the amount of aggregated protein.
The evaluation of the antiamyloid aggregation activity of the novel compounds displayed good potencies, with an aggregation inhibition between 65 and 80 % tested at a concentration of 10 μm (Table 1). In general, similar antiaggregation activity was found against Aβ42 and tau. Compounds 8 a and 8 f display an average (Aβ42 and tau) inhibition of 75.8 and 75.7 %, respectively, against these proteins. Compound 8 c, and to a lesser extent 8 b, showed, however, a higher inhibitory potency against aggregation of Aβ42. Interestingly, compound 8 g, which has a protected catechol moiety, displays practically no activity (<10 %) in the bacterial system.
Table 1.
In vitro antiamyloid activity of taxifolin, curcumin, and 8 a–g. E. coli overexpresses the respective protein, which forms IBs and can be quantified by Th‐S staining. Compounds were tested at 10 μm.
|
Compound |
Aβ42 |
tau |
||
|---|---|---|---|---|
|
|
inhibition [%] |
SEM[a] |
inhibition [%] |
SEM[a] |
|
control |
0.0 |
2.0 |
0.0 |
2.1 |
|
taxifolin |
4.9 |
4.0 |
1.1 |
4.4 |
|
curcumin |
37.8 |
2.7 |
35.2 |
3.2 |
|
8 a |
80.4 |
2.1 |
71.0 |
2.1 |
|
8 b |
78.2 |
3.4 |
65.1 |
2.4 |
|
8 c |
81.3 |
1.6 |
58.0 |
3.9 |
|
8 d |
63.1 |
4.2 |
66.6 |
2.9 |
|
8 e |
67.5 |
2.9 |
73.6 |
3.6 |
|
8 f |
73.3 |
4.3 |
78.3 |
4.1 |
|
8 g |
9.6 |
3.9 |
5.7 |
3.8 |
[a] SEM=standard error of the mean.
Because the new compounds were conceived as bioisosteric mimics of curcumin and taxifolin (Figure 1), we also investigated the antiamyloid effect of these natural products against Aβ42 and tau. As shown in Table 1, a similar antiaggregation activity was observed for Aβ42 and tau, whereas taxifolin displayed practically no activity (<5 %) and curcumin was found to have a moderate inhibitory effect (38 %). Remarkably, the activity of the target compounds greatly exceeds the potency of curcumin and taxifolin, revealing the suitability of the bioisosteric design.
In vitro inhibition of Aβ42 detected by TEM
The detection of amyloid fibrils by fluorescent dyes can be biased by compounds with absorptive and fluorescent properties such as the molecules investigated in this study. [28] Hence, the inhibitory effect of these compounds was further examined by resorting to TEM, which provided a dye‐independent approach to assess the antiaggregating effect of the compounds. The results clearly confirmed the inhibitory effect on fibril formation of Aβ42 at 10 μm for curcumin and compounds 8 a–f (cf. Figure 3 and Figure S5 in the Supporting Information).
Figure 3.
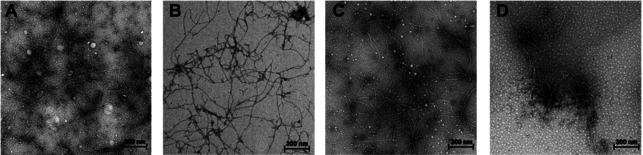
TEM analysis of the inhibitory effect on Aβ42. The Aβ monomer (100 μm) was incubated at 37 °C in phosphate‐buffered saline (PBS) for 24 h with or without 10 μm of the respective compound. A) Control; B) curcumin; C) 8 g; D) 8 f. Scale bar: 300 nm.
Interaction of the target compounds with Aβ42
In light of these results, molecular simulations that combine docking, classical molecular dynamics (MD) and replica‐exchange molecular dynamics (REMD) were carried out to investigate the potential mechanism of action responsible for Aβ42 aggregation inhibition. In particular, our aim was to examine the ability of the catechol‐containing target compounds to interfere with both early (oligomerization) and late (fibrillation) stages of Aβ42 aggregation.
Formation of covalent adducts with Aβ42 fibrils
To examine the ability of the target compounds to covalently interfere with Aβ42 aggregation, compounds 8 a–f were docked in their oxidized (o‐quinone) form into the 10 conformational states for the solid‐state NMR spectroscopy model of Aβ42 (PDB ID: 5KK3) [29] by using Glide. [30] The three top‐scoring docking solutions for compound 8 c were further investigated by means of 100 ns MD simulations with Amber18. [31] The choice of 8 c was motivated by the presence of the nitro group, which would help to stabilize the binding mode through electrostatic interactions with the protonated amino group of K16 residues along the binding groove. This would enhance the residence time around the reactive site and facilitate the proper arrangement for covalent adduct formation, as described for the aza‐Michael addition observed for taxifolin. [32] However, none of the three simulated poses for compound 8 c were able to maintain a proper orientation around the reactive site delimited by K16 and D22 in the binding groove (cf. distances d1 and d2 in Figure S1 in the Supporting Information). These results led us to exclude the possibility of a covalent Aβ42 inhibition mechanism for this class of compounds.
Interaction of the target compounds with Aβ42 monomer
As an alternative mechanism, we explored the ability of compound 8 f, one of the most potent Aβ42 inhibitors found in this study, on the early stage of Aβ42 aggregation by means of REMD simulations. Following previous studies, [22] the Aβ42 monomer (Aβ42mon), which crystallized in a nonpolar environment (PDB ID: 1IYT), [33] was selected to model the interaction with compound 8 f. A total of 30 μs of MD trajectory was collected and the first five replicas, corresponding to the Aβ42mon–8 f system at 315, 316.7, 318.4, 320.1, and 321.8 K, were analyzed (see the Experimental Section for a more complete discussion of the computational protocol).
Secondary‐structure analysis for the first five T‐replicas of Aβ42mon–8 f is reported in Figure 4. The results highlight the large conformational flexibility of Aβ42mon, which can adopt a variety of conformations that mediate the interaction with compound 8 f. In general, turn/coil are the most populated states, followed by β‐sheet and α‐helix arrangements. A high α‐helix content, especially for residues 15–18 and 24–36 of Aβ42mon, is observed at the beginning of all simulated replicas, although the α‐helical content is lost during the first 50–100 ns of REMD simulation. A transient α‐helix to β‐sheet conversion of the central (18–24) and C‐terminal residues is observed for the first three replicas. Interestingly, a different profile is observed for the fourth replica, in which a stable conformer characterized by an α‐helical motif for residues 14–24 and β‐sheet fold for residues at the N and C termini is found. This conformation seems to be the most populated one, as noted by the 2D root‐mean‐square (RMS) analysis (see Figure S2 D in the Supporting Information). Finally, a higher degree of conformational flexibility is observed for the last replica, in which the lack of well‐defined secondary structures is generally observed.
Figure 4.
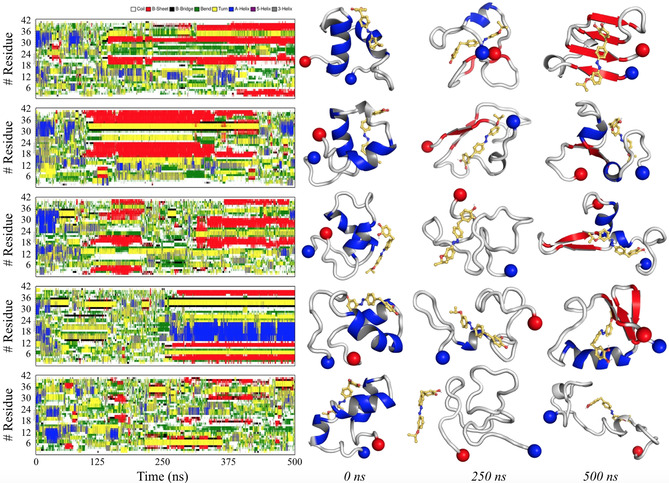
Left: Secondary‐structure analysis for the first five T‐replicas of Aβ42mon–8 f, according to the DSSP algorithm. Right: Representative geometries for Aβ42mon–8 f at 0, 250, and 500 ns of simulation. The N‐ and C‐terminal edges of Aβ42mon are reported as blue and red spheres, respectively.
Along the trajectories sampled in REMD simulations, compound 8 f exhibits a tendency to interact with the middle and C‐terminal regions of the Aβ42mon sequence (cf. Figure 3), thus affecting its conformational assembly. Interestingly, our data are in agreement with those observed by Zhang et al., [22a] who performed REMD simulations to study the conformational behavior of the Aβ42–EGCG complex. The present results, however, emphasize the role of hydrophobic interactions formed between 8 f and the apolar residues of Aβ42mon. Generally, the main interactions are formed with residues 12–20 and 32–38 of Aβ42mon (Aβ42mon Ca–8 f distances <1.0 nm), although there are differences between the different replicas (see Figure S3 in the Supporting Information).
Collectively, these data are in line with a putative Aβ42 antiaggregation mechanism, in which the presence of compound 8 f redirects the conformational landscape of Aβ42 oligomers toward less structured/off‐pathway oligomers. No evidence of stable and well‐formed β‐sheet configuration emerged from our simulations. In fact, the N terminus of Aβ42mon is mainly unstructured, a transient α‐helix propensity is often observed for residues 15–24, and a partial β‐sheet‐turn‐β‐sheet propensity is observed for the central and C‐terminal region. Compound 8 f is suggested to have a significant impact on this assembly because this compound would stably form contacts with the C‐terminal region of the Aβ42mon, intercalating the β‐sheet‐turn‐β‐sheet region.
Neurotoxicity and neuroprotection in HT22 cells
The effect of the target compounds on neuroprotection and neurotoxicity was examined by using the sensitivity of the murine hippocampal neuronal cell line HT22 to glutamate. High extracellular concentrations of glutamate lead to oxidative glutamate toxicity, so‐called oxytosis, a form of programmed cell death. [34] The inhibition of the cystine/glutamate antiporter causes glutathione (GSH) depletion, followed by accumulation of reactive oxygen species (ROS), calcium influx, and finally cell death by oxidative stress. [35] Similar features are observed in the brain during aging and are accelerated in AD. [36] Compounds 8 a–f showed very strong protection against intracellular oxidative stress at concentrations between 2.5 and 7.5 μm (Figure 5). The target compounds even exceeded the flavonol quercetin, which served as a positive control and prevented cell death at 25 μm. Curcumin and 8 g did not show distinct neuroprotection. At only 10 μm, weak protection with 21 % cell survival was observed. These data show the importance of the free catechol and are in good agreement with results reported by Maher et al., who showed that chemical alternation of the catechol structure of flavonoids of the plant Eriodictyon californicum (also known as yerba santa) led to a drastic reduction in activity in different phenotypic screening assays, including the oxytosis assay. [15a]
Figure 5.
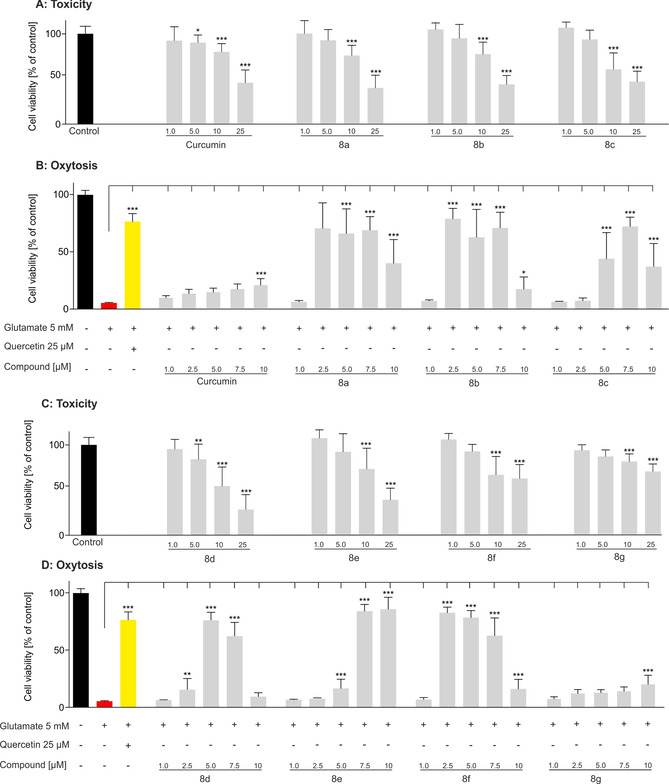
Neuroprotection and neurotoxicity were determined in HT22 cells. 5 mm glutamate (red) induced cell death, 25 μm quercetin (yellow) served as a positive control for cell survival: A) neurotoxicity of curcumin and 8 a–c; B) neuroprotection of curcumin and 8 a–c; C) neurotoxicity of 8 d–g; D) neuroprotection of 8 d–g. Data are presented as means±SEM of three independent experiments and results refer to untreated control cells (black). Statistical analysis was performed by using one‐way analysis of variance (ANOVA) followed by Dunnett's multiple comparison post‐test by using GraphPad Prism 5, with reference to cells treated with 5 mm glutamate. Level of significance: ***p<0.001, **p<0.01, *p<0.05.
DPPH radical scavenging assay
To evaluate (and exclude) unspecific protection against oxidative stress by radical scavenging, the direct antioxidant capacity was tested in a cell‐free system. The widely applied DPPH radical scavenging assay uses the stable radical 2,2‐diphenyl‐1‐picrylhydrazyl, which is decolorized upon reduction. [37] The known antioxidant ascorbic acid (vitamin C) served as a positive control with an IC50 value of 8.4 μm. The parent compound curcumin had an IC50 value of 10.5 μm. The target compounds were active over a similar range, from 5 to 10 μm (Table 2). Compound 8 g did not show any activity because there was no functionality to react with the free DPPH radical. Compound 8 a shows strong cellular protection against intracellular oxidative stress, but a weak radical scavenging activity compared with the other target compounds. This suggests that the neuroprotection in HT22 cells is based on a target‐specific mode of action. The pronounced differences in the activity of curcumin in the oxytosis assay (10 μm, 21 % cell survival) in comparison with the EC50 value of 10.5 μm in the DPPH assay strongly indicate a specific intracellular protective mechanism, rather than an unspecific protection due to radical scavenging.
Table 2.
Free radical scavenging capacity determined by the DPPH assay.
|
Compound |
EC50 [μm] |
SEM |
|---|---|---|
|
ascorbic acid |
8.4 |
0.5 |
|
curcumin |
10.5 |
0.2 |
|
8 a |
9.1 |
0.3 |
|
8 b |
7.7 |
0.4 |
|
8 c |
5.4 |
0.5 |
|
8 d |
5.6 |
0.1 |
|
8 e |
9.6 |
0.4 |
|
8 f |
5.4 |
0.1 |
|
8 g |
not active |
The lack of activity of dimethoxy compound 8 g in the DPPH radical scavenging assay shows the necessity of a catechol (or monohydroxyl) unit for reaction with free radicals.
Anti‐inflammatory effect on BV‐2 cells
Apart from amyloid plaques, neuroinflammation represents a key hallmark of AD. [38] Microglia cells act as a major immune defense in the central nervous system. [39] Activation by, for example, bacterial endotoxins to their proinflammatory phenotype results in the production of NO and several other inflammation‐promoting factors, such as cytokines, free radicals, and excitatory neurotransmitters. [38] Although active microglia cells are important for brain repair processes and response to immune challenge, chronic activation, such as that in AD, leads to neurodegeneration caused by inflammation and oxidative stress.[ 39b , 40 ] Therefore, inhibition of the proinflammatory microglia state is important in the context of AD. [41]
Mouse microglial BV‐2 cells were used to evaluate a possible anti‐inflammatory effect. Cells were treated with bacterial lipopolysaccharide (LPS) to induce inflammation and the production of NO was quantified in the Griess assay. All compounds reduced the NO production dose, with the strongest anti‐inflammatory effect at 10 μm (Figure 6). Similar to the results for neuroprotectivity on HT22 cells, the target compounds exceeded the activity of curcumin. Compound 8 c was the most active compound, with a decrease of inflammation down to 17 % relative to the LPS control. The other compounds tested reduced NO production in a similar manner to 31–42 % (compound 8 f is shown as a representative example; for more detailed information, see the Supporting Information). It is not surprising that compound 8 f was not the most active compound, as in the oxytosis assay or Aβ fibrilization inhibition, since the respective modes of action may well differ from each other. Nevertheless, protection of the catechol with methoxy groups (compound 8 g) led to a dramatic loss of activity, similar to in the other assays applied in this study. No effect on the production of NO was observed at 2.5 and 5 μm. At 10 μm, compound 8 g reduced the amount of NO to 65 % in comparison with the control.
Figure 6.
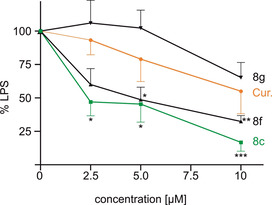
Effect of compounds 8 c, 8 f, 8 g, and curcumin on the production of NO as an inflammation marker. BV‐2 cells were treated with 50 ng mL−1 LPS alone or with the respective compound. NO was determined by the Griess assay in the supernatant. Data are presented as means±SEM of three independent experiments and results refer to LPS‐treated cells. Statistical analysis was performed by using one‐way ANOVA followed by Dunnett's multiple comparison post‐test by using GraphPad Prism 5. Level of significance: ***p<0.001, **p<0.01, *p<0.05.
Conclusion
In this study, a series of azobioisosteres of curcumin, suitably modified to incorporate the pharmacophoric catechol moiety of flavonoids, have been synthesized and their pharmacological profile against amyloid aggregation, intracellular oxidative stress, and neuroinflammation has been characterized. The synthetic bioisosteric compounds have shown higher aggregation inhibition of Aβ42 relative to the parent compound, curcumin. This could be observed in a bacterial in vitro assay with Th‐S staining, as well as dye‐independently in TEM experiments. Additionally, the compounds showed strong activity in AD‐related cell assays. In particular, there was higher protection against glutamate‐induced intracellular oxidative stress in murine hippocampal HT22 cells than that of curcumin. Moreover, the compounds revealed pronounced anti‐inflammatory properties in microglial BV‐2 cells. The observed effects seemed to underlie a specific mechanism, as the activity of the compounds in the DPPH radical scavenging assay did not show substantial differences.
Until now, drugs targeting Aβ have failed in clinical trials. The reasons for this failure include fluorescence interference during the commonly used thioflavin T assay, poor reproducibility of Aβ experiments in general, and the overall complexity of amyloid aggregation processes in AD with still unknown aspects of toxicity of amyloid species. [42] Therefore, the experimental design of this work includes a dye‐based readout for Aβ aggregation that applies to a highly replicable assay, and a dye‐independent setup, that is, electron microscopy. In addition to their antiaggregation properties, the compounds presented herein also act against oxidative stress and neuroinflammation.
It must be taken into account that azobenzenes are commonly suspected to cause long‐term toxicity due to instability towards bacterial azoreductases, which cleave the azobenzene in anilines, but there are several examples of food colorants and drugs, which support the safe use of molecules containing azobenzene moieties. [43]
Finally, because azobenzene compounds can undergo cis–trans photoisomerization upon irradiation with light of an appropriate wavelength, this paves the way to the photoinduced control of the antiaggregating activity of azobenzene bioisosteres. Currently, the use of the target compounds in a photopharmacological approach is under investigation.
Experimental Section
Computational methods
Docking and MD simulations of complexes with Aβ42 fibrils: The solid‐state NMR structure of the Aβ42 fibril (PDB ID 5KK3), [29] corresponding to a double S‐shaped Aβ42 fibrillar assembly, was used to model the interaction of compounds 8 a–f with Aβ42 fibrils, following the same protocol as that adopted in previous studies. [32] Briefly, 20 docking runs, one for each of the two monomers of the 10 NMR models deposited for 5KK3, were performed with Glide. [30] Protonation states for the protein were set at pH 7.4. According to previous pK a studies, [32] one K16 located in the middle of the fibril assembly was simulated in its neutral form. For each of the six tested compounds, the oxidized o‐quinone form was generated and used during docking. A total of 2000 poses were generated, and thus, analyzed to identify suitable candidates for further MD studies.
Among the best‐ranked poses obtained for compound 8 c, three docked arrangements were selected and investigated by means of MD simulations. The choice of this compound as a suitable candidate for MD simulation was dictated by the presence of a nitro group, which would be able to stabilize the protein–ligand complex by interacting with K16 residues along the Aβ fibrils. MD simulations were run with Amber18. [31] The Amber ff14SB‐ILDN force field [44] was used for the protein and modified parameters (see above) from the general Amber force field (GAFF) [45] were used to parameterize the ligand. Partial charges were derived at the B3LYP/6‐31G(d) level, after preliminary geometry optimization, by using the restrained electrostatic potential [46] fitting procedure. Torsional parameters for the C‐C‐N‐N dihedral angle, which defined the conformation of the benzene ring relative to the diazo group, were refined by using 4‐({4‐[(1E)‐2‐(3‐hydroxyphenyl)diazen‐1‐yl]phenyl}methyl)cyclohexa‐3,5‐diene‐1,2‐dione (see Table S1 in the Supporting Information) as a reference model in quantum mechanical (QM) calculations. To this end, the MM torsional potential energy of C‐C‐N‐N torsion were fitted to the QM‐derived potential energy profile obtained from a relaxed scan performed at the M062X/6‐31G(d) level in the gas phase with Gaussian 09. [47] The python package pyevolve [48] was used to fit the two profiles (see the Supporting Information)
The three 8 c–Aβ42 fibrils were solvated with TIP3P [49] water molecules by using a truncated octahedron box with a layer of 20 Å and neutralized by adding Na+ ions. [50] The systems were energy minimized in a three‐stage protocol, which involved hydrogen atoms, then water molecules, and finally the whole system, with a maximum number of 20 000 minimization cycles for the last stage. Then, the systems were gradually heated from 0 to 300 K in six steps; the first was performed at a constant volume and the rest at constant pressure. To avoid artifactual alterations in the ligand pose due to thermal equilibration, harmonic restraints with a force constant of 10 kcal mol−1 Å−2 were applied during equilibration to selected ligand–protein contacts. A Cartesian restraint of 2 kcal mol−1 Å−2 was also applied to the backbone atoms of the first and last Aβ42 monomers to preserve the structural integrity of the fibrils. These restraints were gradually eliminated during the first 50 ns of the MD simulation. The SHAKE algorithm [51] was applied to constrain bonds involving hydrogen atoms. Periodic boundary conditions were used during the MD simulations and a cutoff of 10 Å for the nonbonded interactions and the particle mesh Ewald (PME) method [52] was used for the treatment of electrostatic interactions beyond the cutoff. Langevin dynamics with a collision frequency of 1.0 ps−1 were applied for temperature regulation during heating. Finally, 100 ns of the MD production in the NVT ensemble (300 K) were run by using the weak‐coupling algorithm [53] (with a time constant of 10.0 ps) for each of the three complexes. The analysis was performed for the set of snapshots saved every 2 fs along the trajectories.
REMD simulation of the interaction between 8 f and the Aβ42 monomer: The solution NMR spectroscopy structure of the Aβ42 peptide (PDB ID: 1IYT) [33] was used to model the interaction between Aβ42 monomer and compound 8 f, which was simulated in its reduced (catechol) form. The ligand parameters were adopted from the GAFF force field, although both atomic charges (RESP charges derived from B3LYP/6‐31G(d) calculations) and torsional (C‐C‐N‐N) parameters were adjusted, and the ff14SB‐ILDN force field was used for the protein. Standard protonation states at pH 7.4 were adopted for ionizable residues. The system was embedded in a truncated octahedron box of TIP3P water molecules and counterions (Na+, Cl−) were added at a salt concentration of 0.15 m. The final system (18,386 atoms) contained an Aβ42 monomer, one molecule of 8 f randomly placed around the monomer, 5891 water molecules, 20 Na+ ions, and 17 Cl− ions.
REMD simulations were carried out on 60 T‐replicas ranging from 315 to 430 K with Gromacs2018. [54] The systems were energy minimized by applying 50 000 steps of the steepest descent algorithm followed by 5000 steps of conjugate gradient algorithm. 1 ns of MD simulation in the NVT ensemble by using the velocity‐rescaling thermostat (0.1 ps time coupling constant) [55] was run to heat the system to the final temperatures for production. Positional restraints with a force constant of 1000 kJ mol−1 mn−2 were applied to the Aβ42 backbone atoms to avoid unnatural distortions during heating. Finally, 5 ns of MD simulation in the NPT ensemble by using the Parrinello–Rahman barostat [56] with a 0.5 ps time constant for coupling were run to properly equilibrate density.
500 ns of REMD simulation in the NPT ensemble by using the Parrinello–Rahman barostat [56] under periodic boundary conditions were run for each T‐replica, leading to a total of 30 μs of sampled MD trajectory. The LINCS method [57] was applied to constraint bonds involving hydrogen atoms. A cutoff of 1.2 nm was used to treat short‐range nonbonded interactions, whereas the PME method was applied to manage long‐range electrostatic interactions. [52] A time step of 2 fs was applied to collect trajectories during the simulation. Exchanges between T‐replicas were attempted every 100 MD steps, leading to an acceptance ratio of about 45 %.
Demuxed trajectories for the first five replicas were considered for final analysis. Here, time‐dependent evolution of the secondary structure of Aβ42 was calculated by using the DSSP algorithm. [58] Finally, mdmap and rms commands implemented in Gromacs2018 were applied to generate the contact map and 2D‐RMS plots, respectively.
General
All reagents were used without further purification and bought from common commercial suppliers. TLC was performed on silica gel 60 (alumina foils with fluorescent indicator 254 nm). UV light (254 and 366 nm) was used for detection. For column chromatography, silica gel 60 (particle size 0.040–0.063 mm) was used. NMR spectra were recorded with a Bruker AV‐400 spectrometer (Bruker, Karlsruhe, Germany) in CDCl3 or [D6]DMSO, and chemical shifts are expressed in ppm relative to CDCl3 (δ=7.26 ppm for 1H and δ=77.16 ppm for 13C) or [D6]DMSO (δ=2.50 ppm for 1H and δ=39.52 ppm for 13C). Spectral data reported refer to the thermodynamically more stable trans isomer. The purity of the synthesized products was determined by means of HPLC (Shimadzu Products), containing a DGU‐20A3R degassing unit, a LC20AB liquid chromatograph, and an SPD‐20A UV/Vis detector. UV detection was measured at 254 nm. Mass spectra were obtained by using a LCMS 2020 instrument (Shimadzu Products). As a stationary phase, a Synergi 4U fusion‐RP (150 mm×4.6 mm) column was used and, as a mobile phase, a gradient of methanol/water with 0.1 % formic acid was used . Parameters: A=water, B=methanol, V(B)/[V(A)+V(B)]=from 5 to 90 % over 10 min, V(B)/[V(A)+V(B)]=90 % for 5 min, V(B)/[V(A)+V(B)]=from 90 to 5 % over 3 min. The method was performed at a flow rate of 1.0 mL min−1. Compounds were only used for biological evaluation if the purity was ≥95 %. Melting points were determined by using an OptiMelt automated melting point system (Scientific Instruments GmbH, Gilching, Germany).
(3,4‐Dimethoxyphenyl)(4‐nitrophenyl)methanone (3)
4‐Nitrobenzoyl chloride (500 mg, 2.69 mmol) was added to a suspension of FeCl3 (436 mg, 2.69 mmol) in veratrole (3 mL) and the reaction mixture was heated to 60 °C for 16 h. Water and methanol were added until the precipitant was dissolved, and the suspension was extracted with dichloromethane. The combined organic layers were washed with water and dried over Na2SO4. The solvent was removed under reduced pressure and the crude product was filtered through silica gel by using dichloromethane as the eluent. The solvent was removed under reduced pressure and the residue was kept in the fridge to crystallize the product. The precipitant was washed with ethanol and the product was obtained as a yellow solid (316 mg, 41 %). M.p. 148 °C; 1H NMR (400 MHz, CDCl3): δ=8.32 (d, 3 J=8.7 Hz, 2 H; Ph), 7.88 (d, 3 J=8.7 Hz, 2 H; Ph), 7.50 (d, 4 J=2.0 Hz, 1 H; Ph), 7.30 (dd, 3 J=8.4 Hz, 4 J=2.0 Hz, 1 H), 6.90 (d, 3 J=8.4 Hz, 1 H; Ph), 3.97 (s, 3 H; OCH 3), 3.95 ppm (s, 3 H; OCH 3); 13C NMR (100 MHz, CDCl3): δ=193.6 (Cq), 154.0 (+, Ph‐C), 149.6 (+, Ph‐C), 149.6 (+, Ph‐C), 143.9 (+, Ph‐C), 130.4 (+, 2×Ph‐C), 129.1 (+, Ph‐C), 125.9 (+, Ph‐C), 123.5 (+, 2×Ph‐C), 111.8 (+, Ph‐C), 110.0 (+, Ph‐C), 56.3 (+, ‐OCH3), 56.2 ppm (+, ‐OCH3); ESI‐MS: m/z: 288.28 [M+H]+.
(3,4‐Dihydroxyphenyl)(4‐nitrophenyl)methanone (4)
Compound 3 (1.00 g, 3.48 mmol) was suspended in acetic acid (10 mL), 48 % HBr (20.0 mL) was added, and the reaction mixture was heated to reflux for 3.5 h. After cooling, the precipitant was filtered, washed with water, and dried under vacuum. The product was obtained as a yellowish green solid (733 mg, 85 %). M.p. 165 °C; 1H NMR (400 MHz, [D6]DMSO): δ=10.14 (s, 1 H; OH), 9.56 (s, 1 H; OH), 8.34 (d, 3 J=8.7 Hz, 2 H), 7.86 (d, 3 J=8.8 Hz, 2 H; Ph), 7.26 (d, 4 J=2.2 Hz, 1 H; Ph), 7.11 (dd, 3 J=8.3, 4 J=2.1 Hz, 1 H; Ph), 6.87 ppm (d, 3 J=8.3 Hz, 1 H; Ph); 13C NMR (100 MHz, [D6]DMSO): 192.9 (Cq), 151.5 (Cq, Ph‐C), 148.8 (Cq, Ph‐C), 145.5 (Cq, Ph‐C), 144.1 (Cq, Ph‐C), 130.0 (+, 2×Ph‐C), 127.3 (Cq, Ph‐C), 124.0 (+, Ph‐C), 123.4 (+, 2×Ph‐C), 116.6 (+, Ph‐C), 115.2 ppm (+, Ph‐C); ESI‐MS: m/z: 259.90 [M+H]+.
4‐(4‐Aminobenzyl)benzene‐1,2‐diol (5 a)
Hydrogenation of 4 (200 mg, 0.772 mmol) was performed at room temperature for 16 h under a hydrogen atmosphere (10 bar) in methanol (10 mL) by using 20 wt % Pd/C. The reaction mixture was filtered through Celite and the solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel by using a mixture of dichloromethane/methanol/triethylamine (40:1:0.1) as the eluent. The product was obtained as a light brown solid (76 mg, 45 %). M.p. 205 °C; 1H NMR (400 MHz, [D6]DMSO): δ=8.67 (s, 1 H; OH), 8.55 (s, 1 H; OH), 6.81 (d, 3 J=8.02 Hz, 2 H; Ph), 6.60 (d, 3 J=7.9 Hz, 1 H; Ph), 6.51 (d, 4 J=2.1 Hz, 1 H), 6.47 (d, 3 J=8.3 Hz, 2 H; Ph), 6.41 (dd, 3 J=8.02, 4 J=2.10 Hz, 1 H; Ph), 4.82 (s, 2 H; NH 2), 3.55 ppm (s, 2 H; CH 2); 13C NMR (100 MHz, [D6]DMSO) δ=146.4 (Cq, Ph‐C), 144.9 (Cq, Ph‐C), 143.1 (Cq, Ph‐C), 133.2 (Cq, Ph‐C), 129.0 (+, 2×Ph‐C), 128.9 (Cq, Ph‐C), 119.1 (+, Ph‐C), 115.9 (+, Ph‐C), 115.3 (+, Ph‐C), 113.9 (+, 2×Ph‐C), 39.8 ppm (−, CH2); ESI‐MS: m/z: 216.00 [M+H]+.
4‐(3,4‐Dimethoxybenzyl)aniline (5 b)
Hydrogenation of 3 (400 mg, 1.57 mmol) was performed at room temperature for 16 h under a hydrogen atmosphere (10 bar) in methanol (30 mL) by using 20 wt % Pd/C. The reaction mixture was filtered through Celite and the solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel by using a mixture of ethyl acetate/cyclohexane (1:1) as the eluent. The product was obtained as a colorless oil (225 mg, 56 %). 1H NMR (400 MHz, CDCl3): δ=7.12 (d, 3 J=8.46, 2 H), 6.93 (d, 4 J=1.9 Hz, 1 H), 6.86 (dd, 3 J=8.2, 4 J=2.0 Hz, 1 H), 6.80 (d, 3 J=8.2 Hz, 1 H), 6.62 (d, 3 J=8.46, 2 H), 3.86 (s, 3 H; ‐OCH 3), 3.82 (s, 3 H, ‐OCH 3), 3.49–2.92 ppm (s, 2 H); 13C NMR (100 MHz, CDCl3): δ=148.9 (Cq, Ph‐C), 148.2 (Cq, Ph‐C), 145.8 (Cq, Ph‐C), 137.07 (Cq, Ph‐C), 134.32 (Cq, Ph‐C), 127.8 (+, 2×Ph‐C), 118.7 (+, Ph‐C), 115.1 (+, 2×Ph‐C), 110.9 (+, Ph‐C), 109.7 (+, Ph‐C), 75.6 (−, CH2), 55.9 (+, ‐OCH3), 55.8 ppm (+, ‐OCH3); ESI‐MS: m/z: 242.95 [M+H]+.
(E)‐4‐[4‐(Phenyldiazenyl)benzyl]benzene‐1,2‐diol (8 a)
Compound 5 a (48 mg, 0.22 mmol) and nitrosobenzene (28.7 mg, 0.27 mmol) were stirred in acetic acid (3 mL) at room temperature for 16 h. Ethyl acetate (50 mL) was added, and the organic layer was washed with a 1 m aqueous solution of NaOH (50 mL) and water (50 mL). The solvent was removed under reduced pressure and the crude product was purified by column chromatography on silica gel by using a mixture of ethyl acetate and cyclohexane (1:2→1:1→2:1) as the eluent. The product was obtained as an orange solid (17 mg, 25 %). M.p. 121 °C; 1H NMR (400 MHz, CDCl3): δ=7.92–7.87 (m, 2 H; Ph), 7.86–7.80 (m, 2 H; Ph), 7.55–7.43 (m, 3 H; Ph), 7.34–7.28 (m, 2 H; Ph), 6.80 (d, 3 J=8.00 Hz, 1 H; Ph), 6.70 (d, 4 J=2.1 Hz, 1 H; Ph), 6.65 (dd, 3 J=8.1, 4 J=2.1 Hz, 1 H), 5.20 (s, 2 H; OH), 3.93 ppm (s, 2 H; CH 2); 13C NMR (100 MHz, CDCl3): δ=152.8 (Cq, Ph‐C), 151.2 (Cq, Ph‐C), 145.0 (Cq, Ph‐C), 144.0 (Cq, Ph‐C), 142.4 (Cq, Ph‐C), 140.7 (Cq, Ph‐C), 133.3 (+, Ph‐C), 130.9 (+, Ph‐C), 129.6 (+, Ph‐C), 129.1 (+, Ph‐C), 128.7 (+, Ph‐C), 127.8 (+, Ph‐C), 127.1 (+, Ph‐C), 123.1 (+, Ph‐C), 122.8 (+, Ph‐C), 121.2 (+, Ph‐C), 116.1 (+, Ph‐C), 115.5 (+, Ph‐C), 41.23 ppm (−, CH2); ESI‐MS: m/z: 304.95 [M+H]+; HPLC purity: 98 % (retention time: cis=9.18 min, trans=10.47 min).
General procedure for the partial oxidation of aromatic amines
Amine 6 b–f (1 equiv) was dissolved in dichloromethane (0.3 m) and an aqueous solution of oxone (1 equiv, 0.17 m; commercially available mixture of 2 KHSO5 ⋅KHSO4 ⋅K2SO4) was added. The mixture was stirred for 3.5 h at room temperature. The aqueous layer was extracted with dichloromethane (50 mL); the combined organic layers were washed with a 5 % solution of HCl (50 mL), water (25 mL), and brine (25 mL); and dried over Na2SO4. The solvent was removed under reduced pressure and the crude product was directly used for the next reaction.
General procedure for the Mills reaction
The crude product of partial oxidation and 5 a were dissolved in acetic acid and stirred at room temperature for 16 h. After that time, the solvent was removed under reduced pressure; ethyl acetate was added; and the organic layer was washed with a saturated aqueous solution of NaHCO3, water, and brine. The solvent was removed under reduced pressure and the crude product was purified by column chromatography on silica gel by using a mixture of ethyl acetate and cyclohexane or cyclohexane and dichloromethane as the eluent.
(E)‐4‐{4‐[(3‐Ethoxyphenyl)diazenyl]benzyl}benzene‐1,2‐diol (8 b)
The product was purified by column chromatography on silica gel by using a mixture of ethyl acetate and cyclohexane (1:4) as the eluent. The product was obtained as an orange solid (24 mg, 21 %). M.p. 126 °C; 1H NMR (400 MHz, CDCl3): δ=7.83 (d, 3 J=8.4 Hz, 2 H; Ph), 7.51 (d, 3 J=7.8, 1 H; Ph), 7.42 (d, 3 J=7.95 Hz, 2 H; Ph), 7.30 (d, 3 J=8.4 Hz, 2 H; Ph), 7.02 (ddd, 3 J=8.2, 4 J=2.6, 4 J=1.0 Hz, 1 H; Ph), 6.80 (d, 3 J=8.1 Hz, 1 H; Ph), 6.70 (d, 4 J=2.0 Hz, 1 H; Ph), 6.66 (dd, 3 J=8.0, 4 J=2.1 Hz, 1 H; Ph), 5.15 (s, 2 H; OH), 4.13 (q, 3 J=7.0 Hz, 2 H; OCH 2CH3), 3.94 (s, 2 H; CH 2), 1.45 ppm (t, 3 J=7.0 Hz, 3 H; OCH2CH3); 13C NMR (100 MHz, CDCl3): 159.8 (Cq, Ph‐C), 154.0 (Cq, Ph‐C), 151.2 (Cq, Ph‐C), 144.8 (Cq, Ph‐C), 143.7 (Cq, Ph‐C), 142.1 (Cq, Ph‐C), 133.7 (Cq, Ph‐C), 129.8 (+, Ph‐C), 129.7 (+, 2×Ph‐C), 123.1 (+, 2×Ph‐C), 121.5 (+, Ph‐C), 118.2 (+, Ph‐C), 117.0 (+, Ph‐C), 116.2 (+, Ph‐C), 115.5 (+, Ph‐C), 106.5 (+, Ph‐C), 63.8 (−, OCH2CH3), 41.2 (−, CH2), 14.9 (+, OCH2 CH3); ESI‐MS: m/z: 349.00 [M+H]+; HPLC purity: 96 % (retention time: cis=9.63 min, trans=10.83 min).
(E)‐4‐{4‐[(3‐Nitrophenyl)diazenyl]benzyl}benzene‐1,2‐diol (8 c)
The product was purified by column chromatography on silica gel by using a mixture of ethyl acetate and cyclohexane (1:4→1:3) as the eluent. The product was obtained as an orange solid (59 mg, 52 %). M.p. 125 °C; 1H NMR (400 MHz, CDCl3): δ=8.72 (t, 4 J=2.1 Hz, 1 H; Ph), 8.31 (ddd, 3 J=8.2, 4 J=2.3, 4 J=1.0 Hz, 1 H; Ph), 8.23 (dt, 3 J=7.9, 4 J=1.0 Hz, 1 H; Ph), 7.89 (d, 3 J=8.32, 2 H; Ph), 7.69 (t, 3 J=8.0 Hz, 1 H; Ph), 7.35 (d, 3 J=8.1 Hz, 2 H; Ph), 6.81 (d, 3 J=8.0 Hz, 1 H; Ph), 6.72 (3d, J=2.0 Hz, 1 H; Ph), 6.67 (dd, 3 J=8.1, 4 J=2.0 Hz, 1 H), 5.22 (s, 2 H; OH), 3.96 ppm (s, 2 H; CH 2); 13C NMR (100 MHz, CDCl3): δ=153.2 (Cq, Ph‐C), 150.8 (Cq, Ph‐C), 146.2 (Cq, Ph‐C), 143.8 (Cq, Ph‐C), 142.1 (Cq, Ph‐C), 133.5 (Cq, Ph‐C), 130.0 (+, Ph‐C), 129.8 (+, 2×Ph‐C), 129.3 (+, Ph‐C), 124.8 (+, Ph‐C), 123.6 (+, 2×Ph‐C), 121.6 (+, Ph‐C), 117.1 (+, Ph‐C), 116.2 (+, Ph‐C), 115.6 (+, Ph‐C), 41.3 ppm (−, CH2); ESI‐MS: m/z: 350.10 [M+H]+; HPLC purity: 97 % (retention time: cis=11.60 min, trans=13.52 min).
(E)‐4‐{4‐[(3‐Chlorophenyl)diazenyl]benzyl}benzene‐1,2‐diol (8 d)
The crude product was purified by column chromatography on silica gel by using a mixture of ethyl acetate and cyclohexane (1:3) as the eluent. The product was obtained as an orange powder (150 mg, 64 %). M.p. 149 °C; 1H NMR (400 MHz, CDCl3): δ=7.90–7.79 (m, 4 H; Ph), 7.44 (d, 3 J=6.9 Hz, 2 H; Ph), 7.32 (d, 3 J=8.1 Hz, 2 H; Ph), 6.81 (d, 3 J=8.0 Hz, 1 H; Ph), 6.71 (d, 4 J=2.0 Hz, 1 H), 6.67 (dd, 3 J=8.1, 4 J=2.1 Hz, 1 H; Ph), 5.07 (s, 1 H; OH), 4.99 (s, 1 H; OH), 3.95 ppm (s, 2 H; CH2); 13C NMR (100 MHz, CDCl3): 148.8 (Cq, Ph‐C), 145.2 (Cq, Ph‐C), 141.8 (Cq, Ph‐C), 130.2 (+, Ph‐C), 129.7 (+, 2×Ph‐C), 123.3 (+, Ph‐C), 121.6 (+, 2×Ph‐C), 117.1 (+, Ph‐C), 115.6, 41.2 ppm (−, CH2); ESI‐MS: m/z: 335.05 [M+H]+; HPLC purity: 97 % (retention time: cis=9.65 min, trans=10.95 min).
(E)‐4‐{4‐[(3‐Methoxyphenyl)diazenyl]benzyl}benzene‐1,2‐diol (8 e)
The product was purified by column chromatography on silica gel by using a mixture of ethyl acetate and cyclohexane (1:4) as the eluent. The product was obtained as an orange solid (17 mg, 25 %). M.p. 122 °C; 1H NMR (400 MHz, CDCl3): δ=7.84 (d, 3 J=8.4 Hz, 2 H; Ph), 7.47–7.39 (m, 3 H; Ph), 7.31 (d, 3 J=7.95 Hz, 2 H; Ph), 7.05–7.01 (m, 1 H; Ph), 6.80 (d, 3 J=8.1, 1 H; Ph), 6.71 (d, 4 J=2.1 Hz, 1 H; Ph), 6.66 (dd, 3 J=8.10 Hz, 4 J=2.0 Hz, 1 H), 5.38 (s, 2 H; OH), 3.94 (s, 2 H; CH2), 3.90 (s, 3 H; OCH3); 13C NMR (100 MHz, CDCl3): 159.7 (Cq, Ph‐C), 154.2 (Cq, Ph‐C), 151.01 (Cq, Ph‐C), 144.89 (Cq, Ph‐C), 143.55 (Cq, Ph‐C), 142.1 (Cq, Ph‐C), 133.6 (Cq, Ph‐C), 129.9 (+, Ph‐C), 129.7 (+, 2×Ph‐C), 123.2 (+, 2×Ph‐C), 121.6 (+, Ph‐C), 117.7 (+, Ph‐C), 117.1 (+, Ph‐C), 116.2 (+, Ph‐C), 115.6 (+, Ph‐C), 105.8 (+, Ph‐C), 55.6 (−, OCH3), 41.2 ppm (−, CH2); ESI‐MS: m/z: 335.00 [M+H]+; HPLC purity: 96 % (retention time: cis=9.31 min, trans=10.57 min).
(E)‐4‐{4‐[(3‐Isopropoxyphenyl)diazenyl]benzyl}benzene‐1,2‐diol (8 f)
The product was purified by column chromatography on silica gel by using a mixture of ethyl acetate and cyclohexane (1:4) as the eluent. The product was obtained as an orange solid (37 mg, 16 %). M.p. 127 °C; 1H NMR (400 MHz, CDCl3): δ=7.82 (d, 3 J=8.1 Hz, 2 H; Ph), 7.49 (d, 3 J=7.8, 1 H; Ph), 7.44–7.36 (m, 2 H; Ph), 7.30 (d, 3 J=8.1 Hz, 2 H), 7.00 (m, 1 H; Ph), 6.80 (d, J=8.0 Hz, 1 H; Ph), 6.69 (d, 4 J=1.9 Hz, 1 H; Ph), 6.65 (dd, 3 J=8.1, 4 J=2.0 Hz, 1 H; Ph), 5.23 (s, 2 H; OH), 4.66 (p, 3 J=6.1 Hz, 1 H; OiPr‐H), 3.93 (s, 2 H; CH 2), 1.39 (s, 3 H; OiPr‐CH 3), 1.37 ppm (s, 3 H; OiPr‐CH 3); 13C NMR (100 MHz, CDCl3): 158.7 (Cq, Ph‐C), 154.1 (Cq, Ph‐C), 151.2 (Cq, Ph‐C), 144.8 (Cq, Ph‐C), 143.7 (Cq, Ph‐C), 142.1 (Cq, Ph‐C), 133.7 (Cq, Ph‐C), 129.9 (+, Ph‐C), 129.6 (+, 2×Ph‐C), 123.1 (+, 2×Ph‐C), 121.6 (+, Ph‐C), 119.3 (+, Ph‐C), 116.6 (+, Ph‐C), 115.6 (+, Ph‐C), 108.2 (+, Ph‐C), 70.3 (+, OiPr‐CH), 41.2 (−, CH2), 22.1 (+, 2×OiPr‐CH3); ESI‐MS: m/z: 363.15 [M+H]+; HPLC purity: 96 % (retention time: cis=9.74 min, trans=10.84 min).
(E)‐1‐[4‐(3,4‐Dimethoxybenzyl)phenyl]‐2‐phenyldiazene (8 g)
The product was purified by column chromatography on silica gel by using a mixture of ethyl acetate and cyclohexane (1:2→1:1→2:1) as the eluent. The product was obtained as an orange oil (17 mg, 25 %). 1H NMR (400 MHz, CDCl3): δ=7.93–7.88 (m, 4 H; Ph), 7.60–7.43 (m, 5 H; Ph), 6.95–6.81 (m, 3 H; Ph), 3.88–3.83, 3.87 (m, 2 H; CH 2), (s, 3 H; ‐OMe), 3.85 ppm (s, 3 H; ‐OMe); 13C NMR (100 MHz, CDCl3): 152.8 (Cq, Ph‐C), 152.2 (Cq, Ph‐C), 149.2 (Cq, Ph‐C), 149.1 (Cq, Ph‐C), 143.2 (Cq, Ph‐C), 132.4 (Cq, Ph‐C), 131.2 (+, Ph‐C), 129.2 (+, 2×Ph‐C), 127.6 (+, 2×Ph‐C), 123.1 (+, 2×Ph‐C), 123.01 (+, 2×Ph‐C), 120.2 (+, Ph‐C), 111.1 (+, Ph‐C), 110.8 (+, Ph‐C), 76.5 (−, CH2), 56.0 (+, OMe); ESI‐MS: m/z: 331.95 [M+H]+; HPLC purity: 97 % (retention time: cis=7.66 min, trans=11.28 min).
Aβ42 aggregation inhibition—TEM analysis
The procedure was adapted from a method reported by Murakami et al. [59] Aβ42, purchased from Amatek, was dissolved in a 0.1 % solution of NH4OH at a concentration of 1 mg mL−1. Each compound was dissolved in ethanol at a concentration of 1 mg mL−1. Samples were diluted to a final concentration of 100 μm Aβ42, and the desired concentration of compound (10 μm/50 μm) in PBS (50 mm sodium phosphate and 100 mm NaCl, pH 7.4). After 24 h incubation at 37 °C, solutions were applied on 200 mesh, fixed with 1.25 % glutaraldehyde solution, and negatively stained with 1 % uranyl acetate. The aggregates were observed with a JEOL JEM‐2100 transmission electron microscope. Images on the transmission electron microscope were acquired with a TemCamF416 camera (Tietz Video and Imaging Processing Systems, Gauting, Germany).
Inhibition assay in E. coli cells overexpressing Aβ42 and tau
Cloning and overexpression of Aβ42 peptide: E. coli competent cells BL21 (DE3) were transformed with the pET28a vector (Novagen, Inc., Madison, WI, USA) carrying the DNA sequence of Aβ42. Because of the addition of the initiation codon ATG in front of both genes, the overexpressed peptide contained an additional methionine residue at its N terminus. For overnight culture preparation, M9 minimal medium (10 mL) containing 50 μg mL−1 kanamycin was inoculated with a colony of BL21 (DE3) bearing the plasmid to be expressed at 37 °C. For expression of the Aβ42 peptide, the required volume of overnight culture to obtain 1:500 dilution was added to fresh M9 minimal medium containing 50 μg mL−1 kanamycin and 250 μm Th‐S. The bacterial culture was grown at 37 °C and 250 rpm. Once the cell density reached OD600=0.6, an aliquot of culture (980 μL) was transferred into 1.5 mL Eppendorf tubes with solutions (10 μL) of each compound to be tested in DMSO and isopropyl 1‐thio‐β‐d‐galactopyranoside (IPTG; 10 μL) at 100 mm. The final concentration of drug was fixed at 10 μm. The samples were grown overnight at 37 °C and 1400 rpm by using a Thermomixer (Eppendorf, Hamburg, Germany). As a negative control (maximal amyloid presence), the same amount of DMSO without drug was added to the sample. In parallel, noninduced samples (in the absence of IPTG) were also prepared and used as positive controls (nonamyloid presence). In addition, these samples were used to assess the potential intrinsic toxicity of the compounds and to confirm the correct bacterial growth.
Cloning and overexpression of tau protein: E. coli BL21 (DE3) competent cells were transformed with pTARA containing the RNA polymerase gen of T7 phage (T7RP) under the control of the promoter PBAD. E. coli BL21 (DE3) with pTARA competent cells were transformed with pRKT42 vector encoding four repeats of tau protein in two inserts. For overnight culture preparation, M9 medium (10 mL) containing 0.5 % glucose, 50 μg mL−1 ampicillin, and 12.5 μg mL−1 chloramphenicol were inoculated with a colony of BL21 (DE3) bearing the plasmids to be expressed at 37 °C. For the expression of tau protein, the required volume of overnight culture to obtain 1:500 dilution was added to fresh M9 minimal medium containing 0.5 % glucose, 50 μg mL−1 ampicillin, 12.5 μg mL−1 chloramphenicol, and 250 μm Th‐S. The bacterial culture was grown at 37 °C and 250 rpm. Once the cell density reached OD600=0.6, an aliquot of culture (980 μL) was transferred into 1.5 mL Eppendorf tubes with solutions (10 μL) of each compound to be tested in DMSO and arabinose (10 μL, 25 %). The final concentration of compound was fixed at 10 μm. The samples were grown overnight at 37 °C and 1400 rpm by using a Thermomixer (Eppendorf, Hamburg, Germany). As a negative control (maximal presence of tau), the same amount of DMSO without drug was added to the sample. In parallel, noninduced samples (in the absence of arabinose) were also prepared and used as positive controls (absence of tau). In addition, these samples were used to assess the potential intrinsic toxicity of the compounds and to confirm the correct bacterial growth.
Th‐S steady‐state fluorescence: Th‐S (T1892) and other chemical reagents were purchased from Sigma (St. Louis, MO). Th‐S stock solution (2500 mm) was prepared in double‐distilled water purified through a Milli‐Q system (Millipore, USA). Th‐S fluorescence and absorbance were tracked by using a DTX 800 plate reader multimode detector equipped with multimode analysis software (Beckman‐Coulter, USA). Filters of 430/35 and 485/20 nm were used for the excitation and emission wavelengths, respectively. 535/25 nm filters were also used for absorbance determination. To normalize the Th‐S fluorescence as a function of the bacterial concentration, OD600 was obtained by using a Shimadzu UV‐2401 PC UV/Vis spectrophotometer (Shimadzu, Japan). Notably, fluorescence normalization was carried out by considering the Th‐S fluorescence of the bacterial cells expressing the peptide or protein in the absence of drug as 100 % and the Th‐S fluorescence of the bacterial cells nonexpressing the peptide or protein as 0 %.
Cell culture general procedures
HT22 cells were grown in Dulbecco's modified Eagle medium (DMEM; Sigma Aldrich, Munich Germany) supplemented with 10 % (v/v) fetal calf serum (FCS) and 1 % (v/v) penicillin–streptomycin. BV‐2 cells were grown in low‐glucose DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10 % FCS and 1 % (v/v) penicillin–streptomycin. Cells were subcultured every 2 days and incubated at 37 °C with 5 % CO2 in a humidified incubator. Compounds were dissolved in DMSO (Sigma Aldrich, Munich, Germany) as stock solutions and diluted further into culture medium. To determine cell viability, a colorimetric 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl tetrazolium bromide (MTT; Sigma Aldrich, Munich, Germany) assay was used. MTT solution (4 mg mL−1 in PBS) was diluted 1:10 with medium and added to the wells after removal of the old medium. Cells were incubated for 3 h and then lysis buffer (10 % sodium dodecyl sulfide) was applied. The next day, the absorbance at 560 nm was determined with a multiwell plate photometer (Tecan, SpectraMax 250).
Neurotoxicity and neuroprotection (oxytosis)
For the toxicity and oxytosis assay, 5×103 cells per well were seeded into sterile 96‐well plates and incubated overnight. For the neurotoxicity assay, medium was removed and 1, 5, 10, 25, or 50 μm of the compound diluted with medium from a 0.1 m stock solution was added to the wells. DMSO (0.05 %) in DMEM served as a control. Cells were incubated for 24 h if neurotoxicity was determined by using a colorimetric MTT assay.
For the oxytosis assay, 5 mm glutamate (monosodium l‐glutamate, Sigma Aldrich, Munich, Germany) together with 1, 2.5, 5, 7.5, or 10 μm of the respective compounds were added to the cells and incubated for 24 h. As a positive control, a mixture of 25 μm quercetin (Sigma Aldrich, Munich, Germany) and 5 mm glutamate was used. After 24 h incubation, cell viability was determined by using a colorimetric MTT assay, as described above. Results are presented as percentage of untreated control cells. Data are expressed as means±SEM of three independent experiments. Analysis was accomplished by using GraphPad Prism 5 software by applying one‐way ANOVA followed by Dunnett's multiple comparison post‐test. Levels of significance: *p<0.05; **p<0.01; ***p<0.001.
DPPH radical scavenging assay
Stock solutions of standard antioxidant, ascorbic acid, and compounds were prepared in DMSO (3 mm). DPPH solution was freshly prepared in methanol daily and stored in the dark. A dilution row of compound in methanol ranging over nine dilutions (1–500 μm) was prepared in a 96‐well plate by using a multichannel pipette. The blank was measured at 517 nm. To 100 μL compound dilution, a solution of DPPH (33.3 μL, 200 μm) in ethanol was added by using a multichannel pipette. The 96‐well plate was incubated at room temperature in the dark for 30 min. After incubation, the absorbance was measured at 517 nm. Methanol (100 μL) and DPPH (33.3 μL, 200 μm) served as the negative control. The percentage of DPPH radical scavenging activity (SCV) was calculated by using Equation 1:
| (1) |
Concentration‐dependent SCV curves were calculated by using a nonlinear fit and EC50 values were then determined graphically by using GraphPad Prism 5 software.
Microglial activity
For the anti‐neuroinflammation assay, 1×106 cells per well were seeded in a sterile six‐well plate. After overnight incubation, the medium was exchanged for fresh medium. The cells were pretreated with the respective compounds at the indicated concentrations for 30 min after 50 ng mL−1 bacterial LPS was added. After 24 h incubation, the medium was collected, spun briefly to remove floating cells, and the supernatant (100 μL) was assayed for nitrite by using the Griess Reagent (100 μL) in a 96‐well plate. After incubation for 10 min at room temperature, the absorbance at 550 nm was read on a microplate reader. Results were normalized to cell number as assessed by the MTT assay, as described above. Analysis was accomplished by using GraphPad Prism 5 software by applying one‐way ANOVA followed by Dunnett's multiple comparison post‐test. Levels of significance: *p<0.05; **p<0.01; ***p<0.001.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the Graduate School of Life Science (GSLS) Würzburg and the COST action CA15135 (Multitarget Paradigm for Innovative Ligand Identification in the Drug Discovery Process MuTaLig) to J.H. is gratefully acknowledged. We are also grateful for financial support by the Spanish Ministerio de Economía y Competitividad (MDM2017‐0767; AEI/FEDER UE) and the Generalitat de Catalunya (2017SGR1746; 2019LLAV00016). The Barcelona Supercomputer Center is acknowledged for providing computational resources (BCV‐2021‐1‐0022). We thank Dr. Pamela Maher (Cellular Neurobiology Laboratory, The Salk Institute for Biological Studies, La Jolla, USA) for providing HT22 and BV‐2 cells. We thank Claudia Gehrig and Daniela Bunsen (Biocenter/Theodor‐Boveri‐Institute) for technical assistance. Open access funding enabled and organized by Projekt DEAL.
J. Hofmann, T. Ginex, A. Espargaró, M. Scheiner, S. Gunesch, M. Aragó, C. Stigloher, R. Sabaté, F. J. Luque, M. Decker, Chem. Eur. J. 2021, 27, 6015.
Contributor Information
Prof. Dr. F. Javier Luque, Email: fjluque@ub.edu.
Prof. Dr. Michael Decker, Email: michael.decker@uni-wuerzburg.de.
References
- 1.E. Babusikova, A. Evinova, J. Jurecekova, M. Jesenak, D. Dobrota in Advanced Understanding of Neurodegenerative Diseases, IntechOpen, 2011.
- 2.
- 2a. Glenner G. G., Wong C. W., Biochem. Biophys. Res. Commun. 1984, 120, 885–890; [DOI] [PubMed] [Google Scholar]
- 2b. Masters C. L., Simms G., Weinman N. A., Multhaup G., McDonald B. L., Beyreuther K., Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haass C., Selkoe D. J., Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [DOI] [PubMed] [Google Scholar]
- 4. Sayre L. M., Perry G., Smith M. A., Chem. Res. Toxicol. 2008, 21, 172–188. [DOI] [PubMed] [Google Scholar]
- 5. Prior M., Chiruta C., Currais A., Goldberg J., Ramsey J., Dargusch R., Maher P. A., Schubert D., ACS Chem. Neurosci. 2014, 5, 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Giorgetti S., Greco C., Tortora P., Aprile F. A., Int. J. Mol. Sci. 2018, 19, 2677; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Panza F., Lozupone M., Logroscino G., Imbimbo B. P., Nat. Rev. Neurosci. 2019, 15, 73–88. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Fang L., Gou S., Liu X., Cao F., Cheng L., Bioorg. Med. Chem. Lett. 2014, 24, 40–43; [DOI] [PubMed] [Google Scholar]
- 7b. Venigalla M., Sonego S., Gyengesi E., Sharman M. J., Münch G., Neurochem. Int. 2016, 95, 63–74; [DOI] [PubMed] [Google Scholar]
- 7c. Espargaró A., Ginex T., Vadell M. d. M., Busquets M. A., Estelrich J., Muñoz-Torrero D., Luque F. J., Sabate R., J. Nat. Prod. 2017, 80, 278–289. [DOI] [PubMed] [Google Scholar]
- 8. Reinke A. A., Gestwicki J. E., Chem. Biol. Drug Des. 2007, 70, 206–215. [DOI] [PubMed] [Google Scholar]
- 9. Nelson K. M., Dahlin J. L., Bisson J., Graham J., Pauli G. F., Walters M. A., J. Med. Chem. 2017, 60, 1620–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baell J. B., Holloway G. A., J. Med. Chem. 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Li F., Gong Q., Dong H., Shi J., Curr. Pharm. Des. 2012, 18, 27–33; [DOI] [PubMed] [Google Scholar]
- 11b. Gunesch S., Schramm S., Decker M., Future Med. Chem. 2017, 9, 711–713; [DOI] [PubMed] [Google Scholar]
- 11c. Vrba J., Gažák R., Kuzma M., Papoušková B., Vacek J., Weiszenstein M., Křen V., Ulrichová J., J. Med. Chem. 2013, 56, 856–866; [DOI] [PubMed] [Google Scholar]
- 11d. Yang F., Lim G. P., Begum A. N., Ubeda O. J., Simmons M. R., Ambegaokar S. S., Chen P. P., Kayed R., Glabe C. G., Frautschy S. A., J. Biol. Chem. 2005, 280, 5892–5901; [DOI] [PubMed] [Google Scholar]
- 11e. Henríquez G., Gomez A., Guerrero E., Narayan M., ACS Chem. Neurosci. 2020, 11, 2915–2934. [DOI] [PubMed] [Google Scholar]
- 12. Pohl F., Kong Thoo Lin P., Molecules 2018, 23, 3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gunesch S., Kiermeier C., Hoffmann M., Fischer W., Pinto A. F. M., Maurice T., Maher P., Decker M., Redox Biol. 2019, 101378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gunesch S., Soriano-Castell D., Lamer S., Schlosser A., Maher P., Decker M., ACS Chem. Neurosci. 2020, 11, 3823–3837. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Fischer W., Currais A., Liang Z., Pinto A., Maher P., Redox Biol. 2019, 21, 101089; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Taguchi N., Yuriguchi M., Ando T., Kitai R., Aoki H., Kunisada T., Biol. Pharm. Bull. 2019, 42, 1446–1449; [DOI] [PubMed] [Google Scholar]
- 15c. Sato M., Murakami K., Uno M., Ikubo H., Nakagawa Y., Katayama S., Akagi K.-i., Irie K., Biosci. Biotech. Biochem. 2013, 77, 1100–1103. [DOI] [PubMed] [Google Scholar]
- 16. Singh M., Arseneault M., Sanderson T., Murthy V., Ramassamy C., J. Agric. Food Chem. 2008, 56, 4855–4873. [DOI] [PubMed] [Google Scholar]
- 17. Michaels T. C. T., Šarić A., Meisl G., Heller G. T., Curk S., Arosio P., Linse S., Dobson C. M., Vendruscolo M., Knowles T. P. J., Proc. Natl. Acad. Sci. USA 2020, 117, 24251–24257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murakami K., Irie K., Molecules 2019, 24, 2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chainoglou E., Hadjipavlou-Litina D., Int. J. Mol. Sci. 2020, 21, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sato M., Murakami K., Uno M., Nakagawa Y., Katayama S., Akagi K.-i., Masuda Y., Takegoshi K., Irie K., J. Biol. Chem. 2013, 288, 23212–23224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Palhano F. L., Lee J., Grimster N. P., Kelly J. W., J. Am. Chem. Soc. 2013, 135, 7503–7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Zhang T., Zhang J., Derreumaux P., Mu Y., J. Phys. Chem. B 2013, 117, 3993–4002; [DOI] [PubMed] [Google Scholar]
- 22b. Minh Hung H., Nguyen M. T., Tran P.-T., Truong V. K., Chapman J., Quynh Anh L. H., Derreumaux P., Vu V. V., Ngo S. T., J. Chem. Inf. Model. 2020, 60, 1399–1408. [DOI] [PubMed] [Google Scholar]
- 23. Lima L. M., Barreiro E. J., Curr. Med. Chem. 2005, 12, 23–49. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Fuchter M. J., J. Med. Chem. 2020, 63, 11436–11447; [DOI] [PubMed] [Google Scholar]
- 24b. Velema W. A., Szymanski W., Feringa B. L., J. Am. Chem. Soc. 2014, 136, 2178–2191; [DOI] [PubMed] [Google Scholar]
- 24c. Rodríguez-Soacha D. A., Decker M., Adv. Ther. 2018, 1, 1800037; [Google Scholar]
- 24d. Agnetta L., Decker M. in Design of Hybrid Molecules for Drug Development (Ed.: Decker M), Elsevier, Amsterdam, 2017, pp. 279–311. [Google Scholar]
- 25. Espargaró A., Medina A., Di Pietro O., Muñoz-Torrero D., Sabate R., Sci. Rep. 2016, 6, 23349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chiti F., Dobson C. M., Annu. Rev. Biochem. 2017, 86, 27–68. [DOI] [PubMed] [Google Scholar]
- 27. Invernizzi G., Papaleo E., Sabate R., Ventura S., Int. J. Biochem. Cell Biol. 2012, 44, 1541–1554. [DOI] [PubMed] [Google Scholar]
- 28. Hudson S. A., Ecroyd H., Kee T. W., Carver J. A., FEBS J. 2009, 276, 5960–5972. [DOI] [PubMed] [Google Scholar]
- 29. Colvin M. T., Silvers R., Ni Q. Z., Can T. V., Sergeyev I., Rosay M., Donovan K. J., Michael B., Wall J., Linse S., Griffin R. G., J. Am. Chem. Soc. 2016, 138, 9663–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.S. Release, Schrödinger, LLC, New York, NY 2018.
- 31.Amber18, T. Darden, R. Duke, D. Ghoreishi, M. Gilson, H. Gohlke, A. Goetz, D. Greene, R. Harris, N. Homeyer, S. Izadi, University of California, San Francisco 2018.
- 32. Ginex T., Trius M., Luque F. J., Chem. Eur. J. 2018, 24, 5813–5824. [DOI] [PubMed] [Google Scholar]
- 33. Crescenzi O., Tomaselli S., Guerrini R., Salvadori S., D'Ursi A. M., Temussi P. A., Picone D., Eur. J. Biochem. 2002, 269, 5642–5648. [DOI] [PubMed] [Google Scholar]
- 34. Tan S., Schubert D., Maher P., Curr. Top. Med. Chem. 2001, 1, 497–506. [DOI] [PubMed] [Google Scholar]
- 35. Currais A., Maher P., Redox. Signal. 2013, 19, 813–822. [DOI] [PubMed] [Google Scholar]
- 36. Sonnen J. A., Breitner J. C., Lovell M. A., Markesbery W. R., Quinn J. F., Montine T. J., Free Radical Biol. Med. 2008, 45, 219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blois M. S., Nature 1958, 181, 1199–1200. [Google Scholar]
- 38. Wyss-Coray T., Rogers J., Cold Spring Harbor Perspect. Med. 2012, 2, a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.
- 39a. Kreutzberg G. W., Trends Neurosci. 1996, 19, 312–318; [DOI] [PubMed] [Google Scholar]
- 39b. Minghetti L., Levi G., Prog. Neurobiol. 1998, 54, 99–125. [DOI] [PubMed] [Google Scholar]
- 40.
- 40a. González-Scarano F., Baltuch G., Annu. Rev. Neurosci. 1999, 22, 219–240; [DOI] [PubMed] [Google Scholar]
- 40b. Lenz K. M., Nelson L. H., Front. Immunol. 2018, 9, . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Scheiner M., Dolles D., Gunesch S., Hoffmann M., Nabissi M., Marinelli O., Naldi M., Bartolini M., Petralla S., Poeta E., Monti B., Falkeis C., Vieth M., Hübner H., Gmeiner P., Maitra R., Maurice T., Decker M., J. Med. Chem. 2019, 62, 9078–9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.
- 42a. Doig A. J., Derreumaux P., Curr. Opin. Struct. Biol. 2015, 30, 50–56; [DOI] [PubMed] [Google Scholar]
- 42b. Abbott A., Dolgin E., Nature 2016, 540, 15–16; [DOI] [PubMed] [Google Scholar]
- 42c. Doig A. J., del Castillo-Frias M. P., Berthoumieu O., Tarus B., Nasica-Labouze J., Sterpone F., Nguyen P. H., Hooper N. M., Faller P., Derreumaux P., ACS Chem. Neurosci. 2017, 8, 1435–1437. [DOI] [PubMed] [Google Scholar]
- 43. Broichhagen J., Frank J. A., Trauner D., Acc. Chem. Res. 2015, 48, 1947–1960. [DOI] [PubMed] [Google Scholar]
- 44. Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J. L., Dror R. O., Shaw D. E., Proteins 2010, 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., Case D. A., J. Comput. Chem. 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- 46. Wang J., Cieplak P., Kollman P. A., J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar]
- 47.Gaussian 09, Revision D1, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian Inc., Wallingford CT, 2016.
- 48. Perone C. S., SIGEVOlution 2009, 4, 12–20. [Google Scholar]
- 49. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L., J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- 50. Joung I. S., Cheatham T. E., J. Phys. Chem. B 2008, 112, 9020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ryckaert J.-P., Ciccotti G., Berendsen H. J., J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- 52. Darden T., York D., Pedersen L., J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- 53. Berendsen H. J. C., Postma J. P. M., v. Gunsteren W. F., DiNola A., Haak J. R., J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar]
- 54.M. Abraham, D. Van Der Spoel, E. Lindahl, B. Hess, GROMACS user manual version 2014, 5, 1–298.
- 55. Bussi G., Donadio D., Parrinello M., J. Chem. Phys. 2007, 126, 014101. [DOI] [PubMed] [Google Scholar]
- 56. Parrinello M., Rahman A., J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar]
- 57. Hess B., Bekker H., Berendsen H. J. C., Fraaije J. G. E. M., J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar]
- 58. Kabsch W., Sander C., Biopolymers 1983, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- 59. Murakami K., Irie K., Morimoto A., Ohigashi H., Shindo M., Nagao M., Shimizu T., Shirasawa T., J. Biol. Chem. 2003, 278, 46179–46187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary