

Abstract
Ash1l potentially contributes to neurodevelopmental diseases. Although specific Ash1l mutations are rare, they have led to informative studies in animal models that may bring therapeutic advances. Ash1l is highly expressed in the brain and correlates with the neuropathology of Tourette syndrome (TS), autism spectrum disorder, and intellectual disability during development, implicating shared epigenetic factors and overlapping neuropathological mechanisms. Functional convergence of Ash1l generated several significant signaling pathways: chromatin remodeling and transcriptional regulation, protein synthesis and cellular metabolism, and synapse development and function. Here, we systematically review the literature on Ash1l, including its discovery, expression, function, regulation, implication in the nervous system, signaling pathway, mutations, and putative involvement in TS and other neurodevelopmental traits. Such findings highlight Ash1l pleiotropy and the necessity of transcending a single gene to complicated mechanisms of network convergence underlying these diseases. With the progress in functional genomic analysis (highlighted in this review), and although the importance and necessity of Ash1l becomes increasingly apparent in the medical field, further research is required to discover the precise function and molecular regulatory mechanisms related to Ash1l. Thus, a new perspective is proposed for basic scientific research and clinical interventions for cross‐disorder diseases.
Keywords: Ash1l, gene pleiotropy, histone methylation, neurodevelopment, Tourette syndrome
1. INTRODUCTION
Tourette syndrome (TS) is a highly inheritable, non‐Mendelian neuropsychiatric disorder, which is characterized by the presence and continued existence of movement and vocal tics and caused by abnormal development in the early brain (Browne et al., 2015; Mataix‐Cols et al., 2015; Pauls et al., 2014). Clinically, tic usually onsets at 4–6 years of age, exhibiting a waxing and waning course, and typically improving in adolescence (Browne et al., 2015). Remarkably, TS patients are commonly associated with co‐occurring comorbidities and overlapping behavioral and emotional conditions, including obsessive–compulsive disorder, attention deficit/hyperactivity disorder (ADHD), other possible autism spectrum disorder (ASD) symptoms, and co‐existing mental illnesses (depression, anxiety, antagonistic defiant disease, conduct disorder, and/or personality disorder) (Freeman et al., 2000; Robertson, 2006, 2015; Robertson et al., 2015; Clarke et al., 2012; Tsetsos et al., 2016), highlighting the overlapping circuitry alterations and the existence of connectional co‐substrates in brain networks that are potentially linked in TS and other neuropsychiatric disorders (Cauda et al., 2015; Church et al., 2009; Cravedi et al., 2017; Cross‐Disorder Group of the Psychiatric Genomics Consortium, et al., 2013; Harrison & Weinberger, 2005; Hirschtritt et al., 2015; Huisman‐van Dijk et al., 2016; Cross‐Disorder Group of the Psychiatric Genomics Consortium et al., 2013; Lee et al., 2019; Mathews & Grados, 2011; Robertson et al., 2017; Robertson et al., 2015; van den Heuvel & Sporns, 2019; Vissers et al., 2012). Neurobiologically, convergent results strongly support brain connectivity abnormalities and dysfunction of cortico‐striato‐thalamo‐cortical (CSTC) networks and dopaminergic systems in individuals with TS (Church et al., 2009), which play a critical role in movement control and output and are consistent with imaging evidence.
Genetic factors contribute to the pathogenesis and phenotypic heterogeneity in TS patients. Evidence based on familial aggregation studies has demonstrated that the relative risk of TS in siblings of TS individuals is 10‐ to 100‐fold greater compared to that in the general population (Browne et al., 2015; Mataix‐Cols et al., 2015; Pauls et al., 2014), making TS one of the most heritable neuropsychiatric conditions (Pauls et al., 2014). In addition, twin studies have shown that monozygotic twins are five times more likely to have TS than dizygotic twins (Price et al., 1985). Although these lines of evidence suggest that TS is primarily of genetic origin, the genetic susceptibility factors have remained largely elusive.
TS is a complex, multifactorial, and highly polygenic disease that involves the coexistence of multiple common risk variants combined with rare, inherited, or de novo mutations (Robertson et al., 2017). In fact, candidate gene studies, genome‐wide association studies, and copy number variation studies have consistently screened and identified reoccurring mutual mutations overlapping across TS and other neuropsychiatric disorders. The identified genes (ASH1L, RIMS1, NRXN1, NLGN4X, SHANK3, LRRTMs, DISC1, SLC6A3, GRIN2b, SLITRK1, HDC, and CNTNAP2) (De Rubeis et al., 2014; Kim et al., 2009; Kosmicki et al., 2017; Peñagarikano et al., 2011; Pittenger, 2017; Abelson et al., 2005) encode proteins that converge onto certain conserved pathways, transcriptional control, and chromatin remodeling (ASH1L) (Taniguchi & Moore, 2014), synapse development and function (NRXN1, RIMS1, NLGN4X, LRRTMs, and SHANK3) (Ebrahimi‐Fakhari & Sahin, 2015; Kelleher & Bear, 2008; Südhof, 2008), molecules involved in neuronal development (DISC1 and CNTNAP2), or neurotransmitter receptors (SLC6A3, GRIN2b, and HDC), and thus, supporting the evidence of common biologic pathways underlying TS and other neuropsychiatric disorders (Karagiannidis et al., 2016; Georgitsi et al., 2016). However, results from genetic studies and reoccurrence trials in populations have been mixed so far, and no well‐defined TS‐associated risk gene with conclusive effect has been identified (Georgitsi et al., 2016; Paschou, 2013). Although there are no individual candidate genes in genetic studies that meet the statistical criteria for identified TS risk factors, these potentially susceptible genes may provide neurobiological clues to the development of this disease.
We first reported that mutations in Ash1l confer susceptibility to TS, underlying the significance of this gene in the molecular mechanism underlying TS (Liu et al., 2020). In addition to TS, Ash1l has been assumed to play a pathogenic role in other, more common neuropsychiatric illnesses. As a member of the Trithorax (TRX) group family, Ash1l encodes histone H3‐methyltransferases that counteract Polycomb group repression by specifically catalyzing H3K36me2 (Gregory et al., 2007; Schuettengruber et al., 2011), regulating transcription and chromatin remodeling. Evidence showed that Ash1l is active in the early brain development, and occupies the transcribed region of active genes, including Hox genes and other genes important for chromatin modification (An et al., 2011; Huang, Yu, et al., 2017; Miyazaki et al., 2013; Schmähling et al., 2018; Tanaka et al., 2007; Yuan et al., 2011). The pleiotropic loci are located within genes and are increasingly expressed throughout its lifetime (from the mid‐prenatal period) in the brain and plays a prominent role in the process of neural development. Hence, mutations and deregulation of human Ash1l are linked to various developmental diseases (Rogawski et al., 2016; Wagner & Carpenter, 2012).
In this review, we briefly describe recent work in Ash1l, including its discovery, expression, function, regulation, implication in nervous system, signaling pathways, and mutations in Ash1l potentially involved in TS and other neuropsychiatric traits (summarized in Table 1). Such findings highlight the pleiotropy of Ash1l and the necessity of transcending a single gene to complicated mechanisms of network convergence underlying these diseases. With the progress in functional genomic analysis—as highlighted in this review—although the importance and necessity of Ash1l becomes increasingly apparent in the medical field, more work needs to be done to discover the precise function and molecular regulatory mechanism related to Ash1l. Thus, a new perspective is proposed for basic scientific research and clinical interventions for cross‐disorder diseases.
TABLE 1.
We briefly described recent work in several mutations in Ash1l attributed putative involvement in TS and other neuropsychiatric traits
| Disease | Traits | Mutations |
|---|---|---|
| TS | Ash1l knockout mice line exhibits typical tic behavior and over‐grooming phenotypes, causing changes in neurotransmitters that parallel findings in TS patients | p.S74L, p.Y2077F, p.R1516C, p.R2630T, c.A4639G_p. K1547E (Liu et al., 2020) |
| Among the five variants, three transmitted variants (p.S74L, p.R1516C, and p.R2630T) were previously reported in ASD cases (Stessman et al., 2017) | ||
| ID/MCA | Almost all de novo mutations in Ash1l result in mental retardation, usually accompanied with developmental delays, facial deformities, and spinal deformities | (c.8868_8869delAAinsAAA, NM_018489.2) |
| (c.3704_3705delCTinsC, NM_018489.2) | ||
| (c.7764_7768dup,NM_018489.2) | ||
| (c.6427G‐T, NM_018489.2) (Krumm et al., 2015; Stessman et al., 2017) | ||
| p. (Lys808TyrfsTer40) (Shen et al., 2019) | ||
| p. (Ala724Ser) (de Ligt et al., 2012) | ||
| de novo mutation (c.8356G > C:p.(Ala2786Pro) (Okamoto et al., 2017) | ||
| ASD include | ||
| OCD and ADHD | A varied condition characterized by impaired social skills, communication problems, and repetitive behaviors | A de novo heterozygous missense variant, causing a val2080‐to‐ile substitution (p. Val2080Ile) (Wang et al., 2016) |
| Mounting genetic studies have reported Ash1l is associated with ASD (Cauda et al., 2015; De Rubeis et al., 2014; Krumm et al., 2015; Satterstrom et al., 2019) | ||
| SCZ | The study has not shown clinical traits in schizophrenia | De Rubeis et al. (2014) identified that Ash1l mutation was related to autism and schizophrenia, consistent with Franco's study (Cauda et al., 2015) |
We briefly described recent work in several mutations in Ash1l to which putative involvement in TS and other neuropsychiatric traits was attributed.
Abbreviations: ADHD, attention deficit/hyperactivity disorder; ASD, Autism spectrum disorders; ID, Intellectual disability; MCA, multiple congenital malformation; OCD, obsessive disorder; SCZ, Schizophrenia; TS, Tourette syndrome.
2. MUTATIONS IN Ash1l CONFER SUSCEPTIBILITY TO TOURETTE SYNDROME
The heterozygous mutations in Ash1l, previously identified by whole exome sequencing (WES), are strongly enriched for variants likely to increase nervous system disease risk (Faundes et al., 2018), including TS, ASD, ADHD, multiple congenital malformation (MCA)/intellectual disability (ID), and SCZ (Cravedi et al., 2017; Kern et al., 2015; Liu et al., 2020; Okamoto et al., 2017; Penzes et al., 2013; Reay & Cairns, 2020; Satterstrom et al., 2019; Toro et al., 2010; Wang et al., 2016). Ash1l is a methyltransferase that catalyzes H3K36me2 at specific locations on the histone tail and plays a critical role in maintaining active gene expression (Gregory et al., 2007). However, the definite role of Ash1l at cellular and molecular levels during brain neurodevelopment remains largely unknown.
We conducted WES of 100 TS‐affected trios to perform a de novo mutation analysis and RV‐TDT and identified candidate genes that increase the risk for TS (Liu et al., 2020). Overall, we found that 76 genes were likely associated with TS (p value ≤ .05), and further screened five promising candidate genes that are intolerant to variants and highly expressed in the brain. Five damaging missense variants in Ash1l had been identified: four of them were transmitted variants (p.S74L, p.Y2077F, p.R1516C, and p.R2630T) and one was a de novo variant (c.A4639G_p.K1547E). Three transmitted variants (p.S74L, p.R1516C, and p.R2630T) were also previously reported in ASD cases (Stessman et al., 2017). We performed follow‐up targeted sequencing of Ash1l in additional 524 unrelated TS samples and replicated the association (p value = .001). The point mutations (p.Y2077F and p.S2200G) in Ash1l affect its enzymatic activity. Furthermore, an Ash1l +/− mouse line has been created and exhibits typical tics and over‐grooming behaviors. A leading member of our research group, Professor Guan, performed sequencing of total RNA from Ash1l +/− mouse brain and re‐identified Ash1l which encodes a nuclear protein that is highly expressed in the central nervous system, especially in the striatum, primary motor cortex, primary somatosensory cortex, and medio‐dorsal nucleus of thalamus, matching the neuropathological brain regions in TS. Combining cHIP data of H3K36me2, we identified the enrichment of related genes in specific brain tissues, especially genes involved in chemical neurotransmission and synaptic transmission, neuron projection, and axon growth. Zhu et al. (2016) found that Ash1l binds to the promoter region of NRXN1 when screening for neural activity‐dependent epigenetic regulatory factors, thereby inhibiting the transcriptional activity of NRXN1 and causing synapse formation disorder, which causes mental illness. Huang, Yu, et al. (2017) conducted a rare gene copy number variation study in 2,434 European TS patients and 4,093 healthy controls and found that the deleted fragments of NRXN1 in TS patients were significantly longer than those in the control group, suggesting that NRXN1 and Ash1l participate in the development of TS.
Taken together, although there is convincing evidence that Ash1l plays a pathogenic role in TS, more research needs to be done on well‐defined patient cohorts and further uncover the functional consequences of these disruptions.
3. DISCOVERY, LOCATION, AND EXPRESSION OF Ash1l
Nakamura et al. (2000) first identified and cloned Ash1l expressed sequence tags (EST) sharing homology with Drosophila Ash1 in 2000. Ash1l, located on chromosome 1q21 (Gregory et al., 2006), produced a 10.5 kb transcript expressed in all examined tissues, and delineated an ORF between 320 and 9,280 bp, which encodes a protein of 2,962 residues. The Ash1l protein consists of four AT hooks, a SET domain, a PHD finger motif, and a homeodomain to the bromodomain, and resembles most closely to ALL‐1 (Yano et al., 1997). The sequence encoding the AT hook, upstream of the main homology region, is not conserved, whereas the SET and PHD finger domains show 66% and 77% similarity, respectively, with Drosophila proteins. However, no homology with the bromine domain was found in the Drosophila Ash1 product, and this domain determines the specificity of binding to specific chromatin or protein targets via variant amino acids (Winston & Allis, 1999). Reader proteins exhibit its site specificity owing to recognition of neighboring residues in the bromodomain (Kouzarides, 2007). The protein–protein interaction of Ash1l possibly results in fine tuning of Ash1 activity.
Ash1l is widely expressed in various tissues, with the majority of expression being found in the brain, heart, and kidney in the GTEx Consortium data (Melé et al., 2015), implicating its critical role in neurological disease. Temporally, the highest level of Ash1l expression was found in the brain from 9 to 17 weeks after conception, at year 1 postnatally, and from adolescence to adulthood (years 13 to 40 postnatally). Spatially, Ash1l exhibited the highest expression in prefrontal cortical (PFC) structures, including the dorsolateral, ventrolateral, and medial PFC (DFC, VFC, and MFC) (Cheon et al., 2020). Consistent with GTEx data, the transcriptome data of BrainSpan (http://www.brainspan.org) showed that brain regions with high expression levels of Ash1l (e.g., striatum, primary motor cortex, primary somatosensory cortex, and medio‐dorsal nucleus of thalamus) are related to TS neuropathology.
Ash1l localizes to the mitotic spindle in dividing cells and in the Golgi apparatus. Furthermore, Ash1l was detected in distinct cellular compartments: in small speckles uniformly distributed throughout the nucleoplasm and in cell‐cell tight junctions. The dual location of Ash1l implicates that it has two unrelated functions. Similar to its Drosophila homolog, its location in the nucleus is associated with polygene chromosomes (Rozovskaia et al., 1999; Tripoulas et al., 1996). In this way, Ash1l is involved in histone lysine methylation, recruiting downstream reader proteins involved in gene activation or repression. Immunostaining showed that Ash1l is located in cell‐cell tight junctions exactly matching that of Zo‐1 and cingulin (Keon et al., 1996) and might function as a transcription factor in most or all cells or be recruited for the assembly of epithelial and endothelial cells.
The possibility of Ash1l translocating from the cell junction to the nucleus is comparable to the adhesion‐mediated signaling pathway, such as the Notch receptor. The protein is an evolutionary conserved transmembrane receptor that regulates cell fate decisions performed through intercellular communication (Kopan & Turner, 1996). Upon ligand activation, the intracellular domain of the Notch protein may be cleaved and migrated to the nucleus to bind and activate DNA‐binding transcription factors (Kidd et al., 1998; Schroeter et al., 1998).
4. FUNCTION AND SIGNALING PATHWAY OF Ash1l
Interaction with members of the repressor Polycomb group and activator TRX can modify the nucleosome histones or reshape chromatin to maintain the heritable state of gene transcription “ON” or “OFF” (Brookes & Shi, 2014). Ash1l, an epigenetic regulator, encodes a TRX histone methyltransferase to antagonize Polycomb silencing. Dorighi and Tamkun (2013) showed that mutations in Ash1 significantly decrease H3K36 demethylation levels, suggesting that Ash1 specially catalyzes H3K36 demethylation. They further identified that the KIS protein promotes transcriptional elongation to facilitate the binding of Ash1 and TRX to active genes and counteracts the inhibition of the methylation of H3K27 by the Polycomb group protein. As histone methylation has site‐specific selectivity, depending on the methylation site and degree, which recruits downstream reader proteins to perform transcriptional activation or repression (Brookes & Shi, 2014), the lack of functional histone lysine‐specific methyltransferase disrupts histone methylation. Mechanistically, methylation of H3K36 inhibits the methylation of H3K27 to counteract the catalytic activity of PRC2 (Schmitges et al., 2011; Yu et al., 2016), and results in the methylation of H3K36 and H3K27 to be mutually exclusive along chromatin in many biological systems (De & Müller, 2019; Gaydos et al., 2012; Lu et al., 2016; Popovic et al., 2014; Yu et al., 2016). Gregory et al. (2007) suggested that Ash1l, occupying the transcribed region of active genes including Hox genes, is active in early brain development along with other genes important for chromatin modification. Conversely, Tanaka et al. (2011) indicated that H3K36 methyltransferase activity of Ash1l induces inhibition rather than activation of the Hox genes. They also showed that the functions of Ash1 and MLL1 are essential for the normal development of bone marrow monocytic lineage from murine hematopoietic stem cells in vivo.
The bromodomain in Ash1l protein bestows the binding specificity to particular chromatin or protein targets, resulting in more flexible regulation of Ash1l activity. Huang, Yang, et al. (2017) showed that Mrg15, recruited to the common transcription target by Ash1, reinforced the enzymatic activity in facilitating the proper deposition of H3K36me2, and thus, antagonizing Polycomb silencing and maintaining the transcriptional region at “ON” state of the target gene. The authors created a point mutation (Ash1‐R1288A) in Ash1l to attenuate interaction with Mrg15, and the knocked‐in flies display abnormal homeotic transformation phenotypes. These findings confirmed that Mrg15 allosterically activates Ash1 by inducing a conformational change and eliminates the obstruction of the catalytic center by the auto‐inhibitory loop of Ash1, which is controlled by the interaction partners among H3K36‐specific methyltransferases.
Despite many studies associating Ash1l to human neuropsychiatric diseases and other characteristics, few published reports described its function, neither on a cellular nor on a molecular level. Bagnell (2019) found that a co‐expression gene network of Ash1l is enriched in development of neuronal projections, protein ubiquitination, and neurotrophic signaling pathways. Cauda et al. (2015) found that the overlapped expression profile between Ash1l and other risk genes, such as SHANK3, DISK1, and NRXN1, is involved in chromatic regulation and neurodevelopment in early life. Mutations in Ash1l may change early brain developmental events, which is associated with neuropathological abnormalities such as migration, stratification, and proliferative defects. Consistently, Lalli et al. (2020) revealed the functional convergent modules of a set of 13 ASD‐associated genes via high‐throughput single‐cell sequencing, and found that the inhibition of ADNP, ASH1L, CHD2, and DYRK1A delayed neuronal differentiation through a shared transcriptional pathway involved in cell cycle control and neural progenitor cell proliferation. The authors confirmed that ADNP, CHD2, and Ash1l are also enriched in maturing neurons, implying the continued sharing of downstream molecular targets in the regulation of synaptic tissue, neuron projection, and axon growth.
Ash1l has multiple epigenetic regulation targets that affect the connectivity of neurons. Cheon et al. (2020) found that Ash1l regulates neuronal morphogenesis by modulating neurotrophin signaling, especially the BDNF‐TrkB pathway, to affect neuronal arborization and modulating growth cone size in human neurons. BDNF is a prototypic protein that regulates various developmental events from selection of neural progenitor cells to terminal dendritic differentiation and connectivity of neurons (Rojas Vega et al., 2006). Activity‐dependent synaptic regulation via BDNF and its receptors involves various stages of synaptic development and the transcription, translation, and transport of several forms of synaptic plasticity proteins (Wong et al., 2015). A mutation in Ash1l reduces the expression of TrkB in the brain, which renders neurons unable to respond to exogenous BDNF; hence, the neuronal morphogenetic phenotype is not able to be rescued following exposure to BDNF—reducing neurite outgrowth. Therefore, Ash1l regulates neuronal connections epigenetically by regulating the BDNF‐TrkB signaling pathway, which may contribute to the neurodevelopmental pathogenesis associated with ASD patients with Ash1l mutations. This also indicates that there is a balance between the TRX histone, Ash1l, and PRC2 activity related to neuronal morphogenesis.
We applied the string database to search for the interacting proteins with Ash1l and performed gene ontology (GO) and KEGG pathway enrichment analyses in R. In the function analysis results, we selected nerve‐related pathways and found that Ash1l and its interacting proteins are mainly related to synaptic plasticity. Furthermore, we used the more function of the string database to find more proteins interacting with Ash1l and applied the clusterProfiler to perform GSEA analysis of the obtained target genes in the network, showing that Ash1l and its interacting proteins participate in development‐related pathways (Figure 1).
FIGURE 1.
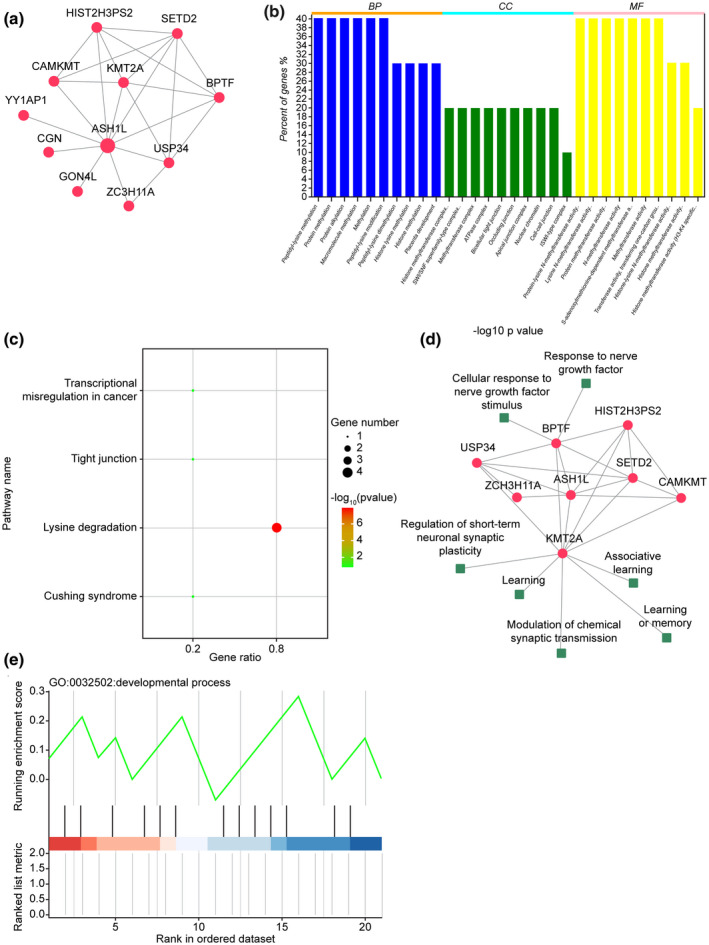
Bioinformatics analysis of the Ash1l gene networks. (a) The string database was used to search for Ash1l interaction proteins, visualizing the protein interaction network via cytoscape software. (b,c) GO and KEGG pathway enrichment analyses were conducted in R through enrichment analysis of gene sets, visualizing the protein interaction network via cytoscape software. (d) Nerve‐related pathways in functional analysis results are highlighted using cytoscape software for network visualization. Functional analysis showed that Ash1l and its interacting proteins are mainly related to synaptic plasticity. (e) The “more” function of the string database was used to search for more proteins that interact with Ash11l. ClusterProfiler was used to perform GSEA analysis of the target genes obtained in the network, indicating that Ash11l and its interacting proteins are involved in development
5. Ash1l‐KNOCKOUT MOUSE AS A PATHOPHYSIOLOGICAL MODEL OF TS
Ash1l is involved in activity‐dependent suppression of Nrxn1α expression, which plays an important role in synapse elimination (Zhu et al., 2016). Activity‐dependent spine maintenance or elimination is the most important factor for the remodeling of established neuronal circuits during postnatal and adolescence periods, thus, placing the early brain in a susceptible context (Sahin & Sur, 2015). Our research group identified mutations (p.S74L and p.Y2077F) in Ash1l of TS patients, resulting in defects in enzymatic activity and conferring susceptibility to TS (Liu et al., 2020). Furthermore, an Ash1l+/− mouse line has been created which exhibits tics and repetitive grooming behaviors that are rescued by haloperidol. Excessive activation in the dorsal striatum and elevated dopamine release events indicate hyperactivity in striatal neurons in Ash1l mutant mice. Mutations of Ash1l result in excessive spines in the neocortex and aberrant activities in the neocortex and subcortical nuclei, inducing defects in motor function and higher functions in the brain, which might cause overconnectivity in local microcircuitry and active abnormality. As spiny synapses are the basic units of excitatory neural circuits and are directly involved in the signaling of neurotransmitters, dendritic spinal dysfunction may play an important role in TS (Gao & Penzes, 2015; Glausier & Lewis, 2013; Penzes et al., 2011). We also demonstrated that the densities of Chat and SST interneurons are increased and activated PV interneurons are slightly decreased in the basal ganglia in Ash1l mutant mice. These results suggest that there is an excitatory and inhibitory imbalance (E/I imbalance) in the basal ganglia, as previously reported in TS patients (Kataoka et al., 2010; Lerner et al., 2012). Our findings demonstrate a critical role of Ash1l in the normal development of neuronal connectivity and established causality between mutations in Ash1l and the genesis of tic‐like behaviors in mice. Ash1l mutations in TS lead to loss of enzymatic activity, which affects the regulation of dopamine signaling transmission and synapse pruning, causing continued existence of synapses that should be eliminated, and thus, synaptic dysfunction, resulting in E/I imbalance.
Mounting evidence implies that dysregulation in E/I balance might be a shared pathophysiological mechanism (Gao & Penzes, 2015). Satterstrom et al. (2019) analyzed the single‐cell gene expression of risk genes in ASD and ADHD, including Ash1l, in cells from the cortex, and found these genes enriched in excitatory and inhibitory neuronal lines, consistent with multiple paths to an E/I imbalance underlying TS, ASD, and other neurodevelopmental disorders. When E/I balance is disrupted, the local microcircuits are altered, resulting in long‐distance and even whole‐brain effects.
Changes in E/I imbalance in cortical activity may explain similar behavioral and cognitive deficits in these diseases (Gao & Penzes, 2015). Genes derived from the whole‐genome association and genetic linkage studies are involved in initial formation or pruning of synaptic contacts in neural circuits (Penzes et al., 2013; Toro et al., 2010; Yin et al., 2012). In addition, structural or functional alterations in glutamatergic excitatory and GABAergic inhibitory circuits have been reported in postmortem examination of TS, ASD, and SCZ (Ebrahimi‐Fakhari & Sahin, 2015; Kataoka et al., 2010; Yin et al., 2012). Although the molecules leading to connectivity changes and their roles in mediating E/I balance between neural and cognitive/behavioral manifestations still require further study, two main pathways of protein synthesis, the PI3K/mTOR and Ras‐MAPK pathways, are related to synaptic dysfunction and E/I imbalance, and may be new research avenues for TS (Gao & Penzes, 2015; Yin et al., 2012). Therefore, understanding the molecular underpinnings of E/I imbalance may provide insights into the mechanisms of these diseases and discover potential targets for future drug discovery in TS.
6. Ash1l AND OTHER NEUROLOGICAL CONDITIONS
TS, ASD, ADHD, and SCZ, with overlapping behavioral phenotypes, are hereditary diseases with complex genetic etiologies (de Lacy & King, 2013; Harrison & Weinberger, 2005; Penzes et al., 2011; Rapoport et al., 2009), showing that multiple susceptible genes may work together to trigger disease onset along different nodes of a common pathway or network (Lakraj et al., 2014). Meta‐analysis studies identified 109 loci with pleiotropic effects, including Ash1l, associated with TS and co‐occurrence with psychiatric disorders (Brinkmeier et al., 2015; Lee et al., 2019). These conditions have the same neurobiological origin, originating from the common root of a unique neurodevelopmental tree (Cauda et al., 2015), and the shared etiology and susceptibility show considerable overlap in the common network of brain connectomes, neuroanatomically, in functional or structural aspects (Crossley et al., 2014; van den Heuvel & Sporns, 2019), indicating that an individual mutation and molecular mechanism is unlikely to underlie these diseases (Harrison & Weinberger, 2005; Toro et al., 2010). Convergent evidence shows that connectivity abnormalities, neuronal circuit problems, brain networks, and E/I imbalance are core parts of the pathology in TS, ASD, ADHD, and SCZ. In addition, disruption of a highly centralized hub node in the shared networks across disorders may play key roles.
In a neuropathological context, functional magnetic resonance imaging (fMRI) studies have unanimously reported abnormalities of long‐distance “underconnectivity” with short‐range “overconnectivity” in TS, ASD, ADHD, and SCZ patients (Cauda et al., 2015; Faundes et al., 2018; Kern et al., 2015; Lakraj et al., 2014; Lerner et al., 2012; Martino et al., 2018; Sahin & Sur, 2015; Vissers et al., 2012) that correlate with symptom severity (Cauda et al., 2015; Faundes et al., 2018; Kern et al., 2015; Lakraj et al., 2014; Lerner et al., 2012; Martino et al., 2018; Sahin & Sur, 2015; Vissers et al., 2012). The similarity in connectivity abnormalities in these disorders indicates a shared final pathway, providing further support for a common brain connectome and mis‐wired neural circuits in these disorders (Church et al., 2009; Kern et al., 2015; Vissers et al., 2012). As the majority of connectivity studies have only reported network architecture alterations in a single disorder, more is needed to be known about the underlying mechanism of multiple neuropsychiatric disorders that are potentially linked. Therefore, we might construct a common framework which links the changes in the connectome among these diseases, placing them into a cross‐diseases environment that might disclose the common biological mechanisms and explain the overlapped symptomatology and shared developmental and genetic mechanisms. We further postulate that Ash1l may be located in the epicenter of the shared networks of connectional co‐substrates in multiple neuropsychiatric disorders virtually linked (Cauda et al., 2015; Crossley et al., 2014; Lakraj et al., 2014; Taniguchi & Moore, 2014; van der Heuvel & Sporns, 2019).
At least seven Ash1l mutations have been identified in people with autism. The disease is characterized by impaired social skills, communication problems, and repetitive behaviors. Wang et al. (2016) screened a de novo heterozygous missense variant in Ash1l, a val2080‐to‐ile (V2080I) substitution, using single‐molecule molecular inversion probes (smMIPs) to sequence 189 candidate genes in a large cohort of over 1,045 Chinese ASD trios confirmed by Sanger sequencing (p = .019). The authors also noted that other Ash1l variants have been found in several autistic patients of European descent in other large studies of autism. de Rubeis et al. (2014) identified seven candidate genes including Ash1l, ML13, ETFB, NAA15, MYO9B, MIB1, and VIL in the data of WES from 3,871 ASD patients and 9,937 parents' controls, and found that the case group had more loss‐of‐function mutations than the control group. Krumm et al. (2015) reported that Ash1l is associated with autism and identified candidate genes RIMS1, CUL7, and LZTR1 in 2,377 autistic families, which is consistent with our RNA‐seq data (Liu et al., 2020). Cauda et al. (2015) found that the expression profile of Ash1l overlaps with other risk genes in ASD and SCZ, including SHANK3, DISK1, CYFIP1, SCN2A, NRXN1, and RELN, which are involved in epigenetic regulation and early brain development. Satterstrom et al. (2020) performed the largest exome sequencing study in 11,986 patients with ASD and identified 102 risk genes, including Ash1l, at a false discovery rate of 0.1 or less. Their data also showed that these genes are highly expressed in early brain development and play critical roles in gene regulation and neuronal communication. These Ash1l mutations related to ASD may change a single amino acid in a specific lysine methyltransferase, leading to the loss of genetic material in the Ash1l gene sequence, or to prematurely terminate signals that lead to the expression of abnormal enzyme (Cauda et al., 2015; Cheon et al., 2020; De Rubeis et al., 2014; Satterstrom et al., 2020). The normal variation of other genes, as well as undetermined environmental risk factors such as parental age and birth complications, can also affect an individual's risk of developing this complex disease.
In given reports, almost all de novo mutations in Ash1l induce mental retardation, usually accompanied with developmental delays, facial deformities, and spinal deformities (Faundes et al., 2018; Krumm et al., 2015; Stessman et al., 2017). Stessman et al. (2017) described three unrelated ID patients and reviewed two patients, previously reported (Krumm et al., 2015) from a large genetic study on autism, with a variety of cognitive neurodevelopmental disorders associated with heterozygous variants in Ash1l. The patients ranged in ages 6 to 17 years and clinical details were limited, but all had developmental delays with mild to moderate ID, 2 of 3 (67%) had delayed speech, 2 of 3 (67%) had ASD, and 2 of 3 (67%) had seizures. Additional variable features included sleep difficulties and dysmorphic facial features. de Ligt et al. (2012) found six missense mutations from re‐sequencing in 765 ID patients to verify new candidate genes and, coincidentally, identified a de novo heterozygous missense variant (p.Ala724Ser) in Ash1l in a male patient with severe ID, abnormal behavior, and facial deformities. Okamoto et al. (2017) reported a case of MCA/ID with obvious mental and growth disorders, microcephaly, and development retardation in white matter sheath and identified an Ash1l de novo mutation (c.8356G > C: p.Ala2786Pro), resulting in an ala2786‐to‐pro (A2786P) substitution in the highly conserved BAH domain. Shen et al. (2019) verified a frameshift mutation p. (Lys808TyrfsTer40) which leads to loss‐of‐function in Ash1l in a patient with MCA, fine motor developmental retardation, learning difficulties, ADHD, sleep apnea, and scoliosis. The MRI also showed a significant abnormality in the formation of white matter myelin, indicating the critical role of Ash1l in the development of the central nervous system. Brinkmeier et al. (2015) showed that Ash1l mutant mice show postnatal death and growth retardation, whereas surviving mice show marked growth deficiency, bone deformation, spinal abnormalities (thoracic abnormalities), and infertility. These studies further indicate that Ash1 mutations are related to neurogenetic abnormalities. Ash1l catalyzes the methylation of H3K36 and occupies the transcribed region of all active genes including Hox during its development. These abnormalities may be a result of insufficient Hox expression during the fetal period.
7. CONCLUDING REMARKS
Although TS, ADHD, ASD, and SCZ have complicated genetic etiologies, behavioral phenotypes and heterogeneity, a significant overlap in symptoms exists, proposing the idea that these different diseases may share common pathogenic mechanisms. The pathways involved in neural development networks provide a framework for understanding how different genetic disturbances of different diseases interact in a convergent way to disrupt neuronal structure, synaptic function, and neuronal circuit organization and behavior. Disease‐related changes rather than causing a wide‐spread damage, may not be specific, but may be subtle—affecting only a subset of synapses in a selective neuron group. As a result, different changes in shared cell substrates may be the basis for phenotypic variability and classified as different neurological diseases. Given the considerable evidence, we propose a reasonable hypothesis that these diseases may have a common network of pathogenesis, and Ash1l may be located at the epicenter hub of neural co‐networks that are the center of pathological damage. Mutations in Ash1l interacting with the local environment may result in transcriptional abnormalities, causing physiological, metabolic, and/or structural damage of neurons and synapses in specific networks. The disease process may initially involve a high degree of regional change, thereby exposing areas in the topological neighborhood to alter functional input or communication, resulting in a series of changes throughout the brain and subsequent neural circuit and E/I imbalance reorganization mechanisms. Owing to the high interconnectivity of the networks, the adjacent topological structure nodes quickly respond, causing cascading network reactions, thus, suggesting that these disorders are part of a continuum.
The elucidation of the shared function of newly discovered disease‐related genes is a key step in translating genetic discoveries into clinical applications, and discussing how the destruction of these molecules related to TS disrupts shared pathways and contributes to the urgency of these diseases. Therefore, further work is required to determine the specific cell types that play a key role in these circuits and common intercellular signaling pathways connecting different risk genes. Only when we are able to predispose the heterogeneity of neurodevelopmental disorders in a shared landscape can the shared molecular regulatory mechanisms of overlapped symptoms and common developmental and/or genetic mechanisms be unlocked.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ACKNOWLEDGMENTS
We thank all the persons for their participation. This work was supported by funds from the National Natural Science Foundation of China (Grant 81371499).
Zhang C, Xu L, Zheng X, Liu S, Che F. Role of Ash1l in Tourette syndrome and other neurodevelopmental disorders. Develop Neurobiol.2021;81:79–91. 10.1002/dneu.22795
Contributor Information
Shiguo Liu, Email: liushiguo2002@126.com.
Fengyuan Che, Email: che1971@126.com.
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- Abelson, J. F. , Kwan, K. Y. , O'Roak, B. J. , Baek, D. Y. , Stillman, A. A. , Morgan, T. M. , Mathews, C. A. , Pauls, D. L. , Rasin, M. R. , Gunel, M. , Davis, N. R. , Ercan‐Sencicek, A. G. , Guez, D. H. , Spertus, J. A. , Leckman, J. F. , Dure, L. S., 4th , Kurlan, R. , Singer, H. S. , Gilbert, D. L. , … State, M. W. . (2005). Sequence variants in SLITRK1 are associated with Tourette's syndrome. Science, 310(5746), 317–320. [DOI] [PubMed] [Google Scholar]
- An, S. , Yeo, K. J. , Jeon, Y. H. , & Song, J. J. (2011). Crystal structure of the human histone methyltransferase ASH1L catalytic domain and its implications for the regulatory mechanism. The Journal of Biological Chemistry, 286(10), 8369–8374. 10.1074/jbc.M110.203380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnell, A. (2019). The role of ASH1L during human neurodevelopment. Senior Theses, 273. https://scholarcommons.sc.edu/senior_theses/273
- Brinkmeier, M. L. , Geister, K. A. , Jones, M. , Waqas, M. , Maillard, I. , & Camper, S. A. (2015). The histone methyltransferase gene absent, small, or homeotic discs‐1 like is required for normal hox gene expression and fertility in mice1. Biology of Reproduction, 93(5), 121. 10.1095/biolreprod.115.131516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes, E. , & Shi, Y. (2014). Diverse epigenetic mechanisms of human disease. Annual Review of Genetics, 48, 237–268. 10.1146/annurev-genet-120213-092518 [DOI] [PubMed] [Google Scholar]
- Browne, H. A. , Hansen, S. N. , Buxbaum, J. D. , Gair, S. L. , Nissen, J. B. , Nikolajsen, K. H. , Schendel, D. E. , Reichenberg, A. , Parner, E. T. , & Grice, D. E. (2015). Familial clustering of tic disorders and obsessive‐compulsive disorder. JAMA Psychiatry, 72(4), 359–366. 10.1001/jamapsychiatry.2014.2656 [DOI] [PubMed] [Google Scholar]
- Cauda, F. , Costa, T. , Fava, L. , Palermo, S. , Bianco, F. , Duca, S. , … Keller, R. . (2015). Predictability of autism, schizophrenic and obsessive spectra diagnosis: Toward a damage network approach. bioRxiv, 014563. 10.1101/014563 [DOI] [Google Scholar]
- Cheon, S. H. , Culver, A. M. , Bagnell, A. M. , Ritchie, F. D. , Clytus, J. M. , McCord, M. , Papendorp, C. M. , Chukwurah, E. , Smith, A. J. , Cowen, M. H. , & Lizarraga, S. B. . (2020). Ash1l regulates the structural development of neuronal circuitry by modulating BDNF/TrkB signaling in human neurons. bioRxiv, 2020.02.18.954586. 10.1101/2020.02.18.954586 [DOI] [Google Scholar]
- Church, J. A. , Fair, D. A. , Dosenbach, N. U. , Cohen, A. L. , Miezin, F. M. , Petersen, S. E. , & Schlaggar, B. L. (2009). Control networks in paediatric Tourette syndrome show immature and anomalous patterns of functional connectivity. Brain: A Journal of Neurology, 132(Pt 1), 225–238. 10.1093/brain/awn223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, R. A. , Lee, S. , & Eapen, V. (2012). Pathogenetic model for Tourette syndrome delineates overlap with related neurodevelopmental disorders including Autism. Translational Psychiatry, 2(9), e158. 10.1038/tp.2012.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravedi, E. , Deniau, E. , Giannitelli, M. , Xavier, J. , Hartmann, A. , & Cohen, D. (2017). Tourette syndrome and other neurodevelopmental disorders: A comprehensive review. Child and Adolescent Psychiatry and Mental Health, 11, 59. 10.1186/s13034-017-0196-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross‐Disorder Group of the Psychiatric Genomics Consortium . (2013). Identification of risk loci with shared effects on five major psychiatric disorders: A genome‐wide analysis. Lancet, 381(9875), 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross‐Disorder Group of the Psychiatric Genomics Consortium , Lee, S. H. , Ripke, S. , Neale, B. M. , Faraone, S. V. , Purcell, S. M. , Perlis, R. H. , Mowry, B. J. , Thapar, A. , Goddard, M. E. , Witte, J. S. , Absher, D. , Agartz, I. , Akil, H. , Amin, F. , Andreassen, O. A. , Anjorin, A. , Anney, R. , Anttila, V. , … International Inflammatory Bowel Disease Genetics Consortium (IIBDGC) . (2013). Genetic relationship between five psychiatric disorders estimated from genome‐wide SNPs. Nature Genetics, 45(9), 984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley, N. A. , Mechelli, A. , Scott, J. , Carletti, F. , Fox, P. T. , McGuire, P. , & Bullmore, E. T. (2014). The hubs of the human connectome are generally implicated in the anatomy of brain disorders. Brain: A Journal of Neurology, 137(Pt 8), 2382–2395. 10.1093/brain/awu132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De, I. , & Müller, C. W. (2019). Unleashing the power of ASH1L methyltransferase. Structure, 27(5), 727–728. [DOI] [PubMed] [Google Scholar]
- de Lacy, N. , & King, B. H. (2013). Revisiting the relationship between autism and schizophrenia: Toward an integrated neurobiology. Annual Review of Clinical Psychology, 9, 555–587. 10.1146/annurev-clinpsy-050212-185627 [DOI] [PubMed] [Google Scholar]
- de Ligt, J. , Willemsen, M. H. , van Bon, B. W. , Kleefstra, T. , Yntema, H. G. , Kroes, T. , Vulto‐van Silfhout, A. T. , Koolen, D. A. , de Vries, P. , Gilissen, C. , del Rosario, M. , Hoischen, A. , Scheffer, H. , de Vries, B. B. , Brunner, H. G. , Veltman, J. A. , & Vissers, L. E. (2012). Diagnostic exome sequencing in persons with severe intellectual disability. The New England Journal of Medicine, 367(20), 1921–1929. 10.1056/NEJMoa1206524 [DOI] [PubMed] [Google Scholar]
- De Rubeis, S. , He, X. , Goldberg, A. P. , Poultney, C. S. , Samocha, K. , Cicek, A. E. , Kou, Y. , Liu, L. , Fromer, M. , Walker, S. , Singh, T. , Klei, L. , Kosmicki, J. , Shih‐Chen, F. , Aleksic, B. , Biscaldi, M. , Bolton, P. F. , Brownfeld, J. M. , Cai, J. , Campbell, N. G. , … Buxbaum, J. D. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature, 515(7526), 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorighi, K. M. , & Tamkun, J. W. (2013). The trithorax group proteins Kismet and ASH1 promote H3K36 dimethylation to counteract Polycomb group repression in Drosophila. Development, 140(20), 4182–4192. 10.1242/dev.095786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi‐Fakhari, D. , & Sahin, M. (2015). Autism and the synapse: Emerging mechanisms and mechanism‐based therapies. Current Opinion in Neurology, 28(2), 91–102. 10.1097/WCO.0000000000000186 [DOI] [PubMed] [Google Scholar]
- Faundes, V. , Newman, W. G. , Bernardini, L. , Canham, N. , Clayton‐Smith, J. , Dallapiccola, B. , Davies, S. J. , Demos, M. K. , Goldman, A. , Gill, H. , Horton, R. , Kerr, B. , Kumar, D. , Lehman, A. , McKee, S. , Morton, J. , Parker, M. J. , Rankin, J. , Robertson, L. , … . Hurles, M. E. (2018). Histone lysine methylases and demethylases in the landscape of human developmental disorders. American Journal of Human Genetics, 102(1), 175–187. 10.1016/j.ajhg.2017.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman, R. D. , Fast, D. K. , Burd, L. , Kerbeshian, J. , Robertson, M. M. , & Sandor, P. (2000). An international perspective on Tourette syndrome: Selected findings from 3,500 individuals in 22 countries. Developmental Medicine and Child Neurology, 42(7), 436–447. 10.1017/S0012162200000839 [DOI] [PubMed] [Google Scholar]
- Gao, R. , & Penzes, P. (2015). Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Current Molecular Medicine, 15(2), 146–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaydos, L. J. , Rechtsteiner, A. , Egelhofer, T. A. , Carroll, C. R. , & Strome, S. (2012). Antagonism between MES‐4 and Polycomb repressive complex 2 promotes appropriate gene expression in C. elegans germ cells. Cell Reports, 2(5), 1169–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgitsi, M. , Willsey, A. J. , Mathews, C. A. , State, M. , Scharf, J. M. , & Paschou, P. (2016). The genetic etiology of Tourette syndrome: Large‐scale collaborative efforts on the precipice of discovery. Frontiers in Neuroscience, 10, 351. 10.3389/fnins.2016.00351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glausier, J. R. , & Lewis, D. A. (2013). Dendritic spine pathology in schizophrenia. Neuroscience, 251, 90–107. 10.1016/j.neuroscience.2012.04.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory, G. D. , Vakoc, C. R. , Rozovskaia, T. , Zheng, X. , Patel, S. , Nakamura, T. , Canaani, E. , & Blobel, G. A. (2007). Mammalian ASH1L is a histone methyltransferase that occupies the transcribed region of active genes. Molecular and Cellular Biology, 27(24), 8466–8479. 10.1128/MCB.00993-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory, S. G. , Barlow, K. F. , McLay, K. E. , Kaul, R. , Swarbreck, D. , Dunham, A. , Scott, C. E. , Howe, K. L. , Woodfine, K. , Spencer, C. C. , Jones, M. C. , Gillson, C. , Searle, S. , Zhou, Y. , Kokocinski, F. , McDonald, L. , Evans, R. , Phillips, K. , Atkinson, A. , Cooper, R. , … Prigmore, E. (2006). The DNA sequence and biological annotation of human chromosome 1. Nature, 441(7091), 315–321. [DOI] [PubMed] [Google Scholar]
- Harrison, P. J. , & Weinberger, D. R. (2005). Schizophrenia genes, gene expression, and neuropathology: On the matter of their convergence. Molecular Psychiatry, 10(1), 40–45. 10.1038/sj.mp.4001558 [DOI] [PubMed] [Google Scholar]
- Hirschtritt, M. E. , Lee, P. C. , Pauls, D. L. , Dion, Y. , Grados, M. A. , Illmann, C. , King, R. A. , Sandor, P. , McMahon, W. M. , Lyon, G. J. , Cath, D. C. , Kurlan, R. , Robertson, M. M. , Osiecki, L. , Scharf, J. M. , Mathews, C. A. , & Tourette Syndrome Association International Consortium for Genetics . (2015). Lifetime prevalence, age of risk, and genetic relationships of comorbid psychiatric disorders in Tourette syndrome. JAMA Psychiatry, 72(4), 325–333. 10.1001/jamapsychiatry.2014.2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, A. Y. , Yu, D. , Davis, L. K. , Sul, J. H. , Tsetsos, F. , Ramensky, V. , Zelaya, I. , Ramos, E. M. , Osiecki, L. , Chen, J. A. , McGrath, L. M. , Illmann, C. , Sandor, P. , Barr, C. L. , Grados, M. , Singer, H. S. , Nöthen, M. M. , Hebebrand, J. , King, R. A. , … Smit, J. (2017). Rare copy number variants in NRXN1 and CNTN6 increase risk for Tourette syndrome. Neuron, 94(6), 1101–1111.e7. 10.1016/j.neuron.2017.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C. , Yang, F. , Zhang, Z. , Zhang, J. , Cai, G. , Li, L. , Zheng, Y. , Chen, S. , Xi, R. , & Zhu, B. (2017). Mrg15 stimulates Ash1 H3K36 methyltransferase activity and facilitates Ash1 Trithorax group protein function in Drosophila. Nature Communications, 8(1), 1649. 10.1038/s41467-017-01897-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisman‐van Dijk, H. M. , Schoot, R. V. D. , Rijkeboer, M. M. , Mathews, C. A. , & Cath, D. C. (2016). The relationship between tics, OC, ADHD and autism symptoms: A cross‐disorder symptom analysis in Gilles de la Tourette syndrome patients and family‐members. Psychiatry Research, 237, 138–146. 10.1016/j.psychres.2016.01.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannidis, I. , Tsetsos, F. , Padmanabhuni, S. S. , Alexander, J. , Georgitsi, M. , & Paschou, P. (2016). The genetics of gilles de la tourette syndrome: a common aetiological basis with comorbid disorders?. Current Behavioral Neuroence Reports, 3(3), 1–14. 10.1007/s40473-016-0088-z [DOI] [Google Scholar]
- Kataoka, Y. , Kalanithi, P. S. , Grantz, H. , Schwartz, M. L. , Saper, C. , Leckman, J. F. , & Vaccarino, F. M. (2010). Decreased number of parvalbumin and cholinergic interneurons in the striatum of individuals with Tourette syndrome. The Journal of Comparative Neurology, 518(3), 277–291. 10.1002/cne.22206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher, R. J., 3rd , & Bear, M. F. (2008). The autistic neuron: Troubled translation? Cell, 135(3), 401–406. 10.1016/j.cell.2008.10.017 [DOI] [PubMed] [Google Scholar]
- Keon, B. H. , Schäfer, S. , Kuhn, C. , Grund, C. , & Franke, W. W. (1996). Symplekin, a novel type of tight junction plaque protein. The Journal of Cell Biology, 134(4), 1003–1018. 10.1083/jcb.134.4.1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern, J. K. , Geier, D. A. , King, P. G. , Sykes, L. K. , Mehta, J. A. , & Geier, M. R. (2015). Shared Brain connectivity issues, symptoms, and comorbidities in autism spectrum disorder, attention deficit/hyperactivity disorder, and Tourette syndrome. Brain Connectivity, 5(6), 321–335. 10.1089/brain.2014.0324 [DOI] [PubMed] [Google Scholar]
- Kidd, S. , Lieber, T. , & Young, M. W. (1998). Ligand‐induced cleavage and regulation of nuclear entry of Notch in Drosophila melanogaster embryos. Genes & Development, 12(23), 3728–3740. 10.1101/gad.12.23.3728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. Y. , Duan, X. , Liu, C. Y. , Jang, M. H. , Guo, J. U. , Pow‐anpongkul, N. , Kang, E. , Song, H. , & Ming, G. L. (2009). DISC1 regulates new neuron development in the adult brain via modulation of AKT‐mTOR signaling through KIAA1212. Neuron, 63(6), 761–773. 10.1016/j.neuron.2009.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan, R. , & Turner, D. L. (1996). The Notch pathway: Democracy and aristocracy in the selection of cell fate. Current Opinion in Neurobiology, 6(5), 594–601. 10.1016/S0959-4388(96)80090-0 [DOI] [PubMed] [Google Scholar]
- Kosmicki, J. A. , Samocha, K. E. , Howrigan, D. P. , Sanders, S. J. , Slowikowski, K. , Lek, M. , Karczewski, K. J. , Cutler, D. J. , Devlin, B. , Roeder, K. , Buxbaum, J. D. , Neale, B. M. , MacArthur, D. G. , Wall, D. P. , Robinson, E. B. , & Daly, M. J. (2017). Refining the role of de novo protein‐truncating variants in neurodevelopmental disorders by using population reference samples. Nature Genetics, 49(4), 504–510. 10.1038/ng.3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides, T. (2007). SnapShot: Histone‐modifying enzymes. Cell, 128(4), 802. 10.1016/j.cell.2007.02.018 [DOI] [PubMed] [Google Scholar]
- Krumm, N. , Turner, T. N. , Baker, C. , Vives, L. , Mohajeri, K. , Witherspoon, K. , Raja, A. , Coe, B. P. , Stessman, H. A. , He, Z. X. , Leal, S. M. , Bernier, R. , & Eichler, E. E. (2015). Excess of rare, inherited truncating mutations in autism. Nature Genetics, 47(6), 582–588. 10.1038/ng.3303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakraj, A. D. , Jabbari, B. , & Machado, D. G. (2014). Chapter 25—Neuronal networks and therapeutics in neurodegenerative disorders. In Faingold C. L. & Blumenfeld H. (Eds.), Neuronal networks in brain function, CNS disorders, and therapeutics (pp. 335–348). Academic Press. [Google Scholar]
- Lalli, M. A. , Avey, D. , Dougherty, J. D. , Milbrandt, J. , & Mitra, R. D. (2020). High‐throughput single‐cell functional elucidation of neurodevelopmental disease‐associated genes reveals convergent mechanisms altering neuronal differentiation. Genome Research, 30(9), 1317–1331. 10.1101/gr.262295.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, P. H. , Anttila, V. , Won, H. , Feng, Y.‐C. , Rosenthal, J. , Zhu, Z. , Tucker‐Drob, E. M. , Nivard, M. G. , Grotzinger, A. D. , Posthuma, D. , Wang, M.‐J. , Yu, D. , Stahl, E. A. , Walters, R. K. , Anney, R. J. L. , Duncan, L. E. , Ge, T. , Adolfsson, R. , Banaschewski, T. , … Smoller, J. W. (2019). Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell, 179(7), 1469–1482. 10.1016/j.cell.2019.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner, A. , Bagic, A. , Simmons, J. M. , Mari, Z. , Bonne, O. , Xu, B. , Kazuba, D. , Herscovitch, P. , Carson, R. E. , Murphy, D. L. , Drevets, W. C. , & Hallett, M. (2012). Widespread abnormality of the γ‐aminobutyric acid‐ergic system in Tourette syndrome. Brain: A Journal of Neurology, 135(Pt 6), 1926–1936. 10.1093/brain/aws104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S. , Tian, M. , He, F. , Li, J. , Xie, H. , Liu, W. , Zhang, Y. , Zhang, R. U. , Yi, M. , Che, F. , Ma, X. U. , Zheng, Y. I. , Deng, H. , Wang, G. , Chen, L. , Sun, X. , Xu, Y. , Wang, J. , Zang, Y. , … Guan, J.‐S. (2020). Mutations in ASH1L confer susceptibility to Tourette syndrome. Molecular Psychiatry, 25(2), 476–490. 10.1038/s41380-019-0560-8 [DOI] [PubMed] [Google Scholar]
- Lu, C. , Jain, S. U. , Hoelper, D. , Bechet, D. , Molden, R. C. , Ran, L. , Murphy, D. , Venneti, S. , Hameed, M. , Pawel, B. R. , Wunder, J. S. , Dickson, B. C. , Lundgren, S. M. , Jani, K. S. , De Jay, N. , Papillon‐Cavanagh, S. , Andrulis, I. L. , Sawyer, S. L. , Grynspan, D. , … Lewis, P. W. (2016). Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science, 352(6287), 844–849. 10.1126/science.aac7272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino, D. , Ganos, C. , & Worbe, Y. (2018). Neuroimaging applications in Tourette's syndrome. International Review of Neurobiology, 143, 65–108. [DOI] [PubMed] [Google Scholar]
- Mataix‐Cols, D. , Isomura, K. , Pérez‐Vigil, A. , Chang, Z. , Rück, C. , Larsson, K. J. , Leckman, J. F. , Serlachius, E. , Larsson, H. , & Lichtenstein, P. (2015). Familial risks of Tourette syndrome and chronic tic disorders. A population‐based cohort study. JAMA Psychiatry, 72(8), 787–793. 10.1001/jamapsychiatry.2015.0627 [DOI] [PubMed] [Google Scholar]
- Mathews, C. A. , & Grados, M. A. (2011). Familiality of Tourette syndrome, obsessive‐compulsive disorder, and attention‐deficit/hyperactivity disorder: Heritability analysis in a large sib‐pair sample. Journal of the American Academy of Child and Adolescent Psychiatry, 50(1), 46–54. 10.1016/j.jaac.2010.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melé, M. , Ferreira, P. G. , Reverter, F. , DeLuca, D. S. , Monlong, J. , Sammeth, M. , Young, T. R. , Goldmann, J. M. , Pervouchine, D. D. , Sullivan, T. J. , Johnson, R. , Segrè, A. V. , Djebali, S. , Niarchou, A. , GTEx Consortium , Wright, F. A. , Lappalainen, T. , Calvo, M. , Getz, G. , … Guigó, R. . (2015). Human genomics. The human transcriptome across tissues and individuals. Science, 348(6235), 660–665. 10.1126/science.aaa0355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki, H. , Higashimoto, K. , Yada, Y. , Endo, T. A. , Sharif, J. , Komori, T. , Matsuda, M. , Koseki, Y. , Nakayama, M. , Soejima, H. , Handa, H. , Koseki, H. , Hirose, S. , & Nishioka, K. (2013). Ash1l methylates Lys36 of histone H3 independently of transcriptional elongation to counteract polycomb silencing. PLoS Genetics, 9(11), e1003897. 10.1371/journal.pgen.1003897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, T. , Blechman, J. , Tada, S. , Rozovskaia, T. , Itoyama, T. , Bullrich, F. , Mazo, A. , Croce, C. M. , Geiger, B. , & Canaani, E. (2000). huASH1 protein, a putative transcription factor encoded by a human homologue of the Drosophila ash1 gene, localizes to both nuclei and cell‐cell tight junctions. Proceedings of the National Academy of Sciences of the United States of America, 97(13), 7284–7289. 10.1073/pnas.97.13.7284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto, N. , Miya, F. , Tsunoda, T. , Kato, M. , Saitoh, S. , Yamasaki, M. , Kanemura, Y. , & Kosaki, K. (2017). Novel MCA/ID syndrome with ASH1L mutation. American Journal of Medical Genetics. Part A, 173(6), 1644–1648. [DOI] [PubMed] [Google Scholar]
- Paschou, P. (2013). The genetic basis of Gilles de la Tourette syndrome. Neuroscience and Biobehavioral Reviews, 37(6), 1026–1039. 10.1016/j.neubiorev.2013.01.016 [DOI] [PubMed] [Google Scholar]
- Pauls, D. L. , Fernandez, T. V. , Mathews, C. A. , State, M. W. , & Scharf, J. M. (2014). The inheritance of tourette disorder: A review. Journal of Obsessive‐Compulsive and Related Disorders, 3(4), 380–385. 10.1016/j.jocrd.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peñagarikano, O. , Abrahams, B. S. , Herman, E. I. , Winden, K. D. , Gdalyahu, A. , Dong, H. , Sonnenblick, L. I. , Gruver, R. , Almajano, J. , Bragin, A. , Golshani, P. , Trachtenberg, J. T. , Peles, E. , & Geschwind, D. H. (2011). Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism‐related deficits. Cell, 147(1), 235–246. 10.1016/j.cell.2011.08.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes, P. , Buonanno, A. , Passafaro, M. , Sala, C. , & Sweet, R. A. (2013). Developmental vulnerability of synapses and circuits associated with neuropsychiatric disorders. Journal of Neurochemistry, 126(2), 165–182. 10.1111/jnc.12261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes, P. , Cahill, M. E. , Jones, K. A. , VanLeeuwen, J. E. , & Woolfrey, K. M. (2011). Dendritic spine pathology in neuropsychiatric disorders. Nature Neuroscience, 14(3), 285–293. 10.1038/nn.2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger, C. (2017). Histidine decarboxylase knockout mice as a model of the pathophysiology of tourette syndrome and related conditions. Handbook of Experimental Pharmacology, 241, 189–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic, R. , Martinez‐Garcia, E. , Giannopoulou, E. G. , Zhang, Q. , Zhang, Q. , Ezponda, T. , Shah, M. Y. , Zheng, Y. , Will, C. M. , Small, E. C. , Hua, Y. , Bulic, M. , Jiang, Y. , Carrara, M. , Calogero, R. A. , Kath, W. L. , Kelleher, N. L. , Wang, J. P. , Elemento, O. , & Licht, J. D. (2014). Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genetics, 10(9), e1004566. 10.1371/journal.pgen.1004566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, R. A. , Kidd, K. K. , Cohen, D. J. , Pauls, D. L. , & Leckman, J. F. (1985). A twin study of Tourette syndrome. Archives of General Psychiatry, 42(8), 815–820. 10.1001/archpsyc.1985.01790310077011 [DOI] [PubMed] [Google Scholar]
- Rapoport, J. , Chavez, A. , Greenstein, D. , Addington, A. , & Gogtay, N. (2009). Autism spectrum disorders and childhood‐onset schizophrenia: Clinical and biological contributions to a relation revisited. Journal of the American Academy of Child and Adolescent Psychiatry, 48(1), 10–18. 10.1097/CHI.0b013e31818b1c63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reay, W. R. , & Cairns, M. J. (2020). Pairwise common variant meta‐analyses of schizophrenia with other psychiatric disorders reveals shared and distinct gene and gene‐set associations. Translational Psychiatry, 10(1), 134. 10.1038/s41398-020-0817-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, M. M. (2006). Mood disorders and Gilles de la Tourette's syndrome: An update on prevalence, etiology, comorbidity, clinical associations, and implications. Journal of Psychosomatic Research, 61(3), 349–358. 10.1016/j.jpsychores.2006.07.019 [DOI] [PubMed] [Google Scholar]
- Robertson, M. M. (2015). A personal 35 year perspective on Gilles de la Tourette syndrome: Prevalence, phenomenology, comorbidities, and coexistent psychopathologies. The Lancet. Psychiatry, 2(1), 68–87. 10.1016/S2215-0366(14)00132-1 [DOI] [PubMed] [Google Scholar]
- Robertson, M. M. , Cavanna, A. E. , & Eapen, V. (2015). Gilles de la Tourette syndrome and disruptive behavior disorders: Prevalence, associations, and explanation of the relationships. The Journal of Neuropsychiatry and Clinical Neurosciences, 27(1), 33–41. 10.1176/appi.neuropsych.13050112 [DOI] [PubMed] [Google Scholar]
- Robertson, M. M. , Eapen, V. , Singer, H. S. , Martino, D. , Scharf, J. M. , Paschou, P. , Roessner, V. , Woods, D. W. , Hariz, M. , Mathews, C. A. , Črnčec, R. , & Leckman, J. F. (2017). Gilles de la Tourette syndrome. Nature Reviews. Disease Primers, 3, 16097. 10.1038/nrdp.2016.97 [DOI] [PubMed] [Google Scholar]
- Rogawski, D. S. , Grembecka, J. , & Cierpicki, T. (2016). H3K36 methyltransferases as cancer drug targets: Rationale and perspectives for inhibitor development. Future Medicinal Chemistry, 8(13), 1589–1607. 10.4155/fmc-2016-0071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas Vega, S. , Strüder, H. K. , Vera Wahrmann, B. , Schmidt, A. , Bloch, W. , & Hollmann, W. (2006). Acute BDNF and cortisol response to low intensity exercise and following ramp incremental exercise to exhaustion in humans. Brain Research, 1121(1), 59–65. 10.1016/j.brainres.2006.08.105 [DOI] [PubMed] [Google Scholar]
- Rozovskaia, T. , Tillib, S. , Smith, S. , Sedkov, Y. , Rozenblatt‐Rosen, O. , Petruk, S. , Yano, T. , Nakamura, T. , Ben‐Simchon, L. , Gildea, J. , Croce, C. M. , Shearn, A. , Canaani, E. , & Mazo, A. (1999). Trithorax and ASH1 interact directly and associate with the trithorax group‐responsive bxd region of the Ultrabithorax promoter. Molecular and Cellular Biology, 19(9), 6441–6447. 10.1128/MCB.19.9.6441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin, M. , & Sur, M. (2015). Genes, circuits, and precision therapies for autism and related neurodevelopmental disorders. Science, 350(6263), aab3897. 10.1126/science.aab3897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterstrom, F. K. , Kosmicki, J. A. , Wang, J. , Breen, M. S. , De Rubeis, S. , An, J.‐Y. , Peng, M. , Collins, R. , Grove, J. , Klei, L. , Stevens, C. , Reichert, J. , Mulhern, M. S. , Artomov, M. , Gerges, S. , Sheppard, B. , Xu, X. , Bhaduri, A. , Norman, U. , … Walters, R. K. (2020). Large‐scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell, 180(3), 568–584.e23. 10.1016/j.cell.2019.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterstrom, F. K. , Walters, R. K. , Singh, T. , Wigdor, E. M. , Lescai, F. , Demontis, D. , Kosmicki, J. A. , Grove, J. , Stevens, C. , Bybjerg‐Grauholm, J. , Bækvad‐Hansen, M. , Palmer, D. S. , Maller, J. B. , iPSYCH‐Broad Consortium , Nordentoft, M. , Mors, O. , Robinson, E. B. , Hougaard, D. M. , Werge, T. M. , … Daly, M. J. . (2019). Autism spectrum disorder and attention deficit hyperactivity disorder have a similar burden of rare protein‐truncating variants. Nature Neuroscience, 22(12), 1961–1965. 10.1038/s41593-019-0527-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmähling, S. , Meiler, A. , Lee, Y. , Mohammed, A. , Finkl, K. , Tauscher, K. , Israel, L. , Wirth, M. , Philippou‐Massier, J. , Blum, H. , Habermann, B. , Imhof, A. , Song, J. J. , & Müller, J . (2018). Regulation and function of H3K36 di‐methylation by the trithorax‐group protein complex AMC. Development, 145(7), dev163808. 10.1242/dev.16380819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitges, F. W. , Prusty, A. B. , Faty, M. , Stützer, A. , Lingaraju, G. M. , Aiwazian, J. , Sack, R. , Hess, D. , Li, L. , Zhou, S. , Bunker, R. D. , Wirth, U. , Bouwmeester, T. , Bauer, A. , Ly‐Hartig, N. , Zhao, K. , Chan, H. , Gu, J. , Gut, H. , … Thomä, N. H. (2011). Histone methylation by PRC2 is inhibited by active chromatin marks. Molecular Cell, 42(3), 330–341. 10.1016/j.molcel.2011.03.025 [DOI] [PubMed] [Google Scholar]
- Schroeter, E. H. , Kisslinger, J. A. , & Kopan, R. (1998). Notch‐1 signalling requires ligand‐induced proteolytic release of intracellular domain. Nature, 393(6683), 382–386. [DOI] [PubMed] [Google Scholar]
- Schuettengruber, B. , Martinez, A. M. , Iovino, N. , & Cavalli, G. (2011). Trithorax group proteins: Switching genes on and keeping them active. Nature Reviews. Molecular Cell Biology, 12(12), 799–814. 10.1038/nrm3230 [DOI] [PubMed] [Google Scholar]
- Shen, W. , Krautscheid, P. , Rutz, A. M. , Bayrak‐Toydemir, P. , & Dugan, S. L. (2019). De novo loss‐of‐function variants of ASH1L are associated with an emergent neurodevelopmental disorder. European Journal of Medical Genetics, 62(1), 55–60. 10.1016/j.ejmg.2018.05.003 [DOI] [PubMed] [Google Scholar]
- Stessman, H. A. F. , Xiong, B. O. , Coe, B. P. , Wang, T. , Hoekzema, K. , Fenckova, M. , Kvarnung, M. , Gerdts, J. , Trinh, S. , Cosemans, N. , Vives, L. , Lin, J. , Turner, T. N. , Santen, G. , Ruivenkamp, C. , Kriek, M. , van Haeringen, A. , Aten, E. , Friend, K. , … Eichler, E. E. (2017). Targeted sequencing identifies 91 neurodevelopmental‐disorder risk genes with autism and developmental‐disability biases. Nature Genetics, 49(4), 515–526. 10.1038/ng.3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof, T. C. (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature, 455(7215), 903–911. 10.1038/nature07456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, Y. , Katagiri, Z. , Kawahashi, K. , Kioussis, D. , & Kitajima, S. (2007). Trithorax‐group protein ASH1 methylates histone H3 lysine 36. Gene, 397(1–2), 161–168. 10.1016/j.gene.2007.04.027 [DOI] [PubMed] [Google Scholar]
- Tanaka, Y. , Kawahashi, K. , Katagiri, Z. , Nakayama, Y. , Mahajan, M. , & Kioussis, D. (2011). Dual function of histone H3 lysine 36 methyltransferase ASH1 in regulation of Hox gene expression. PLoS ONE, 6(11), e28171. 10.1371/journal.pone.0028171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi, H. , & Moore, A. W. (2014). Chromatin regulators in neurodevelopment and disease: Analysis of fly neural circuits provides insights: Networks of chromatin regulators and transcription factors underlie Drosophila neurogenesis and cognitive defects in intellectual disability and neuropsychiatric disorder models. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology, 36(9), 872–883. 10.1002/bies.201400087 [DOI] [PubMed] [Google Scholar]
- Toro, R. , Konyukh, M. , Delorme, R. , Leblond, C. , Chaste, P. , Fauchereau, F. , Coleman, M. , Leboyer, M. , Gillberg, C. , & Bourgeron, T. (2010). Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends in Genetics, 26(8), 363–372. 10.1016/j.tig.2010.05.007 [DOI] [PubMed] [Google Scholar]
- Tripoulas, N. , LaJeunesse, D. , Gildea, J. , & Shearn, A. (1996). The Drosophila ash1 gene product, which is localized at specific sites on polytene chromosomes, contains a SET domain and a PHD finger. Genetics, 143(2), 913–928. https://pubmed.ncbi.nlm.nih.gov/8725238/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsetsos, F. , Padmanabhuni, S. S. , Alexander, J. , Karagiannidis, I. , Tsifintaris, M. , Topaloudi, A. , Mantzaris, D. , Georgitsi, M. , Drineas, P. , & Paschou, P. (2016). Meta‐analysis of Tourette syndrome and attention deficit hyperactivity disorder provides support for a shared genetic basis. Frontiers in Neuroscience, 10, 340. 10.3389/fnins.2016.00340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Heuvel, M. P. , & Sporns, O. (2019). A cross‐disorder connectome landscape of brain dysconnectivity. Nature Reviews. Neuroscience, 20(7), 435–446. 10.1038/s41583-019-0177-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers, M. E. , Cohen, M. X. , & Geurts, H. M. (2012). Brain connectivity and high functioning autism: A promising path of research that needs refined models, methodological convergence, and stronger behavioral links. Neuroscience and Biobehavioral Reviews, 36(1), 604–625. 10.1016/j.neubiorev.2011.09.003 [DOI] [PubMed] [Google Scholar]
- Wagner, E. J. , & Carpenter, P. B. (2012). Understanding the language of Lys36 methylation at histone H3. Nature Reviews. Molecular Cell Biology, 13(2), 115–126. 10.1038/nrm3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, T. , Guo, H. , Xiong, B. O. , Stessman, H. A. F. , Wu, H. , Coe, B. P. , Turner, T. N. , Liu, Y. , Zhao, W. , Hoekzema, K. , Vives, L. , Xia, L. U. , Tang, M. , Ou, J. , Chen, B. , Shen, Y. , Xun, G. , Long, M. , Lin, J. , … Eichler, E. E. (2016). De novo genic mutations among a Chinese autism spectrum disorder cohort. Nature Communications, 7, 13316. 10.1038/ncomms13316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston, F. , & Allis, C. D. (1999). The bromodomain: A chromatin‐targeting module? Nature Structural Biology, 6(7), 601–604. [DOI] [PubMed] [Google Scholar]
- Wong, Y. H. , Lee, C. M. , Xie, W. , Cui, B. , & Poo, M. M. (2015). Activity‐dependent BDNF release via endocytic pathways is regulated by synaptotagmin‐6 and complexin. Proceedings of the National Academy of Sciences of the United States of America, 112(32), E4475–E4484. 10.1073/pnas.1511830112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano, T. , Nakamura, T. , Blechman, J. , Sorio, C. , Dang, C. V. , Geiger, B. , & Canaani, E. (1997). Nuclear punctate distribution of ALL‐1 is conferred by distinct elements at the N terminus of the protein. Proceedings of the National Academy of Sciences of the United States of America, 94(14), 7286–7291. 10.1073/pnas.94.14.7286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, D. M. , Chen, Y. J. , Sathyamurthy, A. , Xiong, W. C. , & Mei, L. (2012). Synaptic dysfunction in schizophrenia. Advances in Experimental Medicine and Biology, 970, 493–516. [DOI] [PubMed] [Google Scholar]
- Yu, Y. , Chen, J. , Gao, Y. , Gao, J. , Liao, R. , Wang, Y. , Oyang, C. , Li, E. , Zeng, C. , Zhou, S. , Yang, P. , Jin, H. , & Yi, W. (2016). Quantitative profiling of combinational K27/K36 modifications on histone H3 variants in mouse organs. Journal of Proteome Research, 15(3), 1070–1079. 10.1021/acs.jproteome.5b01164 [DOI] [PubMed] [Google Scholar]
- Yuan, W. , Xu, M. , Huang, C. , Liu, N. , Chen, S. , & Zhu, B. (2011). H3K36 methylation antagonizes PRC2‐mediated H3K27 methylation. The Journal of Biological Chemistry, 286(10), 7983–7989. 10.1074/jbc.M110.194027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Τ. , Liang, C. , Li, D. , Tian, M. , Liu, S. , Gao, G. , & Guan, J. S. (2016). Histone methyltransferase Ash1L mediates activity‐dependent repression of neurexin‐1α. Scientific Reports, 6, 26597. 10.1038/srep26597 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.