

Abstract
An enantioselective sulfimidation of 3‐thiosubstituted 2‐quinolones and 2‐pyridones was achieved with a stoichiometric nitrene source (PhI=NNs) and a silver‐based catalyst system. Key to the success of the reaction is the use of a chiral phenanthroline ligand with a hydrogen bonding site. The enantioselectivity does not depend on the size of the two substituents at the sulfur atom but only on the binding properties of the heterocyclic lactams. A total of 21 chiral sulfimides were obtained in high yields (44–99 %) and with significant enantiomeric excess (70–99 % ee). The sulfimidation proceeds with high site‐selectivity and can also be employed for the kinetic resolution of chiral sulfoxides. Mechanistic evidence suggests the intermediacy of a heteroleptic silver complex, in which the silver atom is bound to one molecule of the chiral ligand and one molecule of an achiral 1,10‐phenanthroline. Support for the suggested reaction course was obtained by ESI mass spectrometry, DFT calculations, and a Hammett analysis.
Keywords: asymmetric catalysis, enantioselectivity, hydrogen bonds, ligand design, silver
A picky ligand: A chiral phenanthroline ligand allows for the site‐ and enantioselective imidation (Ns=para‐nitrosulfonyl) of a variety of sulfides. Silver acts in concert with an achiral 1,10‐phenanthroline (1,10‐phen) ligand as the catalytically active center.

Introduction
Sulfur compounds with two carbon–sulfur single bonds and an additional ylide‐type sulfur double bond to a nitrogen substituent are called sulfimides. Alternatively, the compound class has been referred to as sulfilimines or iminosulfuraes. Their chemistry has attracted significant attention and has been studied for decades. [1] A key structural feature of sulfimides is the fact that they display a significant barrier to pyramidal inversion at the sulfur atom. As a consequence, sulfimides exist at ambient temperature as pairs of separable enantiomers provided that the two carbon substituents are different. The stereospecific oxidation of sulfimides to sulfoximines is feasible and enables access to another class of chiral sulfur compounds with intriguing properties. [2] Given the large interest in the synthesis of enantiopure compounds, enantioselective approaches to sulfimides have been investigated and the most frequently chosen route to chiral sulfimides rests on a nitrene transfer reaction to thioethers (sulfides). [3] Sulfimidation reactions have been mainly performed with N‐carboxylated and N‐sulfonylated nitrene precursors. The resulting N‐sulfonylated sulfimides I and N‐alkoxycarbonylated sulfimides II represent typical products and enantioselective imidation reactions have been developed based on copper, [4] manganese, [5] ruthenium,[ 6 , 7 ] and iron [8] as catalytically active metal centers (Scheme 1). The selectivity rests on the differentiation between the two enantiotopic lone pairs at the sulfur atom of sulfides. The exposure of the electron pair in turn depends for most catalytic enantioselective processes on the size difference of the substituents R. The best enantiomeric excess (ee) is typically achieved if this size difference is significant with one group being small (RS) and one being large (RL). However, if the steric demand of the substituents is similar, a low asymmetric induction is observed, for example, for diarylsulfides.
Scheme 1.

Top: Products I and II of previously reported enantioselective sulfimidation reactions obtained by metal catalysis (catalytically active metal and ee given). Bottom: Attempted enantioselective sulfimidation of substrates III to sulfimides IV with N‐sulfonylated phenyliminoiodinanes and a chiral silver catalyst.
In the present study we have approached the topic of enantioselective sulfimidation by using a yet unexplored catalytically active metal (silver). Even more importantly, we combined the metal with a ligand that pre‐coordinates to lactam substrates III by hydrogen bonding and thus displays one of the two enantiotopic lone pairs to the reactive nitrogen center. As a result, products of type IV have become readily available in high optical purity. The scope and the mechanism of the reaction have been studied and the results of our experiments are presented in this research article.
Results and Discussion
Ligand synthesis and preliminary studies. The development of chiral ligands with a lactam hydrogen bonding site continues to be a research focus of our group. [9] The goal is to achieve not only an improved site‐selectivity in a given transition‐metal catalyzed reaction [10] but also to induce a significant enantioselectivity. In a recent contribution, [11] we prepared chiral phenanthroline ligands 1 which are based on the readily available octahydro‐1H‐4,7‐methanoisoindol‐1‐one skeleton. [12] Their synthesis can be achieved by Sonogashira cross‐coupling of known alkyne 2 [13] with the respective bromo‐substituted phenanthroline (Scheme 2). Ligand 1 a was successfully employed in an enantioselective Ag‐catalyzed amination of C−H bonds which was assumed to occur by an enantioselective nitrene insertion [14] at the hydrogen‐bonded substrate. Although silver catalysis has been shown to be a viable approach for the amination[ 15 , 16 ] and aziridination[ 15 , 17 ] of organic substrates it has not been extensively applied to sulfides. Bolm and co‐workers found that silver nitrate acts in combination with an achiral terpyridine ligand as a catalyst to mediate the imidation of sulfoxides to generate sulfoximines.[ 18 , 19 ] Two sulfimides were obtained in high yields (77 % and 83 %) albeit in racemic form.
Scheme 2.
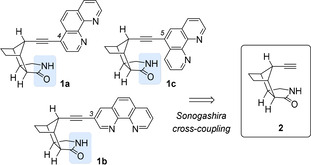
Structure of isomeric phenanthroline ligands 1 which are available from enantiopure alkyne 2 by Sonogashira cross‐coupling with the respective bromo‐substituted phenanthroline. The potential two‐point hydrogen bonding site is marked in blue.
Our initial experiments aimed to identify the ligand best suited for a defined prochiral sulfide. Although pre‐coordination by a given lactam to any of the three ligands 1 can be assumed, the trajectory of the nitrene transfer was expected to be different resulting in varying reactivity and enantioselectivity. We employed Ag(PF6) as the source of the catalytically active metal center (10 mol %) and added 1,10‐phenanthroline (10 mol %) as an achiral ligand. The chiral ligand was used in 12 mol % and N‐(p‐nitrophenylsulfonyl)iminophenyliodinane (PhI=NNs) was employed as the nitrene precursor and dichloromethane as the solvent. The first set of reactions (Scheme 3) was performed with 3‐phenylthio‐2‐quinolone (3 a). While all ligands 1 facilitated a complete conversion within 16 hours and yields varied only marginally (92–98 %), the enantioselectivity in the formation of sulfimide 4 a was most pronounced with catalysts 1 a (97 % ee) and 1 c (96 % ee). Although we had anticipated the reaction to be enantioselective, the high selectivity achieved in the reaction came as a very pleasant surprise. In the amination reactions, [11] enantioselectivities exceeding 90 % ee could only been achieved with cyclic substrates in which free rotations were completely restricted.
Scheme 3.

Evaluation of the regioisomeric chiral phenanthroline ligands 1 in the enantioselective Ag‐catalyzed sulfimidation of representative 3‐substituted 2‐quinolone 3 a and 2‐pyridone 5 a (phen=1,10‐phenanthroline).
With 3‐phenylthio‐2‐pyridone (5 a) a reactivity difference was notable for the three ligands and the yields of the respective sulfimide 6 a were only moderate when ligands 1 b and 1 c were used. The by far best result was recorded for ligand 1 a. Not only did the ligand provide the highest yield (70 %), but it also facilitated the formation of product 6 a with the highest enantioselectivity (92 % ee). Also, for the 2‐pyridones the degree of selectivity was remarkable given the flexibility of the substrate (see the Supporting Information for further details and other substrates).
Before applying the imidation by a nosyl‐protected nitrogen to a larger array of substrates two other iminophenyliodinanes were probed as reagents for a possible nitrene transfer. The related para‐toluenesulfonyl (tosyl, Ts) reagent gave product 7 in a much lower enantioselectivity of only 48 % ee (Scheme 4). The electronically different trichloroethoxysulfonyl (Tces)‐substituted reagent, however, displayed a similar degree of reactivity and selectivity as the nosyl‐based reagent. Product 8 was obtained in 94 % yield and with 90 % ee. The latter result bears some synthetic relevance since removal of the Tces group can be performed under conditions that are orthogonal to those employed for the Ns group. [20]
Scheme 4.

Enantioselective Ag‐catalyzed sulfimidation of compound 3 a with different N‐protected nitrene sources to give products 7 and 8.
Scope and site‐selectivity. The imidation of 3‐thiosubstituted 2‐quinolone 3 a suggested that various sulfides with a similar structure could be converted into the respective sulfimides irrespective of the nature of the thio substituent. Accordingly, 2‐quinolones 3 were prepared and subjected to a nitrene transfer reaction, both racemic [21] and enantioselective. Several aryl groups were introduced as group R in substrates 3 and the reaction turned out to be robust towards the electronic (products 4 b–4 f) and the steric situation (products 4 g–4 i). If the opposite enantiomer ent‐1 a of ligand 1 a was employed in the reactions, the ee remained identical but the opposite sulfimide enantiomer was produced. The reactions of 3 a in the presence of ent‐1 a for example delivered ent‐4 a on a scale of 0.4 mmol. For reasons of availability the absolute configuration of the sulfimides was representatively determined for dextrorotatory product ent‐4 a. Comparison of experimental and calculated vibrational circular dichroism (VCD) spectra allowed the assignment with high fidelity (see the SI for details).[ 22 , 23 ] The (R)‐configuration was assigned to compound ent‐4 a, its enantiomer 4 a is (S)‐configured. The assignment of the configuration for all other products which were obtained with 1 a and which were consistently levorotatory was based on analogy.
As mentioned in the introduction, the most remarkable feature of the present Ag‐catalyzed sulfimidation is the fact that it is applicable to diaryl sulfides irrespective of the relative size of the two substituents. As seen for the aryl‐substituted products 4 a–4 i the results fully met the expectations (Scheme 5). Interestingly, also a methyl group could be used as group R and 3‐methylthio‐2‐quinolone (3 j) gave sulfimide 4 j in 98 % yield and 96 % ee. However, if the size of the alkyl group increases, the enantioselectivity drops. With R=isopropyl (substrate 3 k) and R=cyclohexyl (substrate 3 l) the enantioselectivity remained moderate (70–73 % ee).
Scheme 5.

Enantioselective Ag‐catalyzed sulfimidation of various 3‐thiosubstituted 2‐quinolones 3 employing ligand 1 a as the source of chirality (60 μmol scale, substrate concentration c=10 mM). The enantiomeric excess (ee) was calculated from the ratio of enantiomers (4/ent‐4) as determined by chiral HPLC analysis.
In a second set of experiments, the scope of the sulfimidation was explored with 3‐thiosubstituted 2‐pyridones 5 (Scheme 6). The conditions remained unchanged as compared to 3 and a clean transfer of the nitrogen fragment from PhI=NNs to the sulfides was observed. If there was no additional substituent X in the pyridones, the enantioselectivities were in all cases higher than 83 % ee (products 6 a–6 g). There is a weak correlation between the selectivity and the reactivity. The most reactive substrate 5 e (vide supra) delivered most side products and the lowest ee. The high ee for ortho‐tolyl‐substituted product 6 g was unexpected given that this substituent had led in the quinolone case (product 4 h) to a decreased enantioselectivity. For substrate 5 h (X=methyl), the enantioselectivity of the sulfimidation was high but the yield suffered, for substrate 5 i (X=trifluoromethyl) the opposite scenario—high yield, diminished ee—applied.
Scheme 6.

Enantioselective Ag‐catalyzed sulfimidation of various 3‐thiosubstituted 2‐pyridones 5 employing ligand 1 a as the source of chirality (60 μmol scale, substrate concentration c=10 mM). The enantiomeric excess (ee) was calculated from the ratio of enantiomers (6/ent‐6) as determined by chiral HPLC analysis.
The observed enantiotopos differentiation in the Ag‐catalyzed sulfimidation were in agreement with the previous hypothesis that the heteroleptic complex 9 was the catalytically competent species (Figure 1). [11] Upon transfer of the protected nitrogen fragment from iminophenyliodinane to the metal center, pre‐coordination of a given substrate, for example, 3 a, would allow in the reactive complex 10 only to display the pro‐S electron pair towards the reactive nitrene center. The reaction would consequently lead to the (S)‐configured sulfimides as observed.
Figure 1.

Putative heteroleptic silver complex 9 formed from the silver source, one equivalent of ligand 1 a, and one equivalent of 1,10‐phenanthroline and enantioselective nitrene transfer within complex 10: Only the pro‐S electron pair at the sulfur atom of substrate 3 a is displayed towards the reactive center.
The constraints for a nitrene transfer as postulated in complex 10 stimulated further experiments which aimed to verify and to exploit the pre‐coordination by hydrogen bonds. A straightforward experiment along these lines involves the reaction of an N‐alkylated substrate that is not competent to form two hydrogen bonds to complex 9. The experiment was performed with 2‐quinolone 11, the N‐methylated derivative of substrate 3 a (Scheme 7). The sulfimidation led to a product which was completely racemic (product rac‐12).
Scheme 7.

Ag‐catalyzed sulfimidation of 2‐quinolone 11 under the conditions previously applied to substrates 3 and 5 (60 μmol scale, substrate concentration c=10 mM). Within the limits of error, product 12 was formed as the racemate (rac‐12).
An interesting consequence of the hydrogen bond‐mediated sulfimidation relates to the fact that the reaction should not only proceed with high enantioselectivity but also with exquisite site‐selectivity. [24] This issue was addressed with substrate 3 m that contains two potentially reactive sulfur atoms (Scheme 8). All reactions with this substrate were performed with a stoichiometric amount of the imidation reagent to avoid a two‐fold imidation. Achiral conditions which allowed for a relatively clean reaction included the use of 20 mol % Ag(PF6) and 60 mol % of 1,10‐phenanthroline. After 24 h the conversion was 60 % and sulfimidation had occurred with notable selectivity at the methylthio group in the periphery of the molecule. The site‐selectivity was determined as r.r. (regioisomeric ratio)=rac‐13/rac‐4 m=84/16 by 1H NMR spectroscopic analysis of the crude product. Product rac‐13 was isolated in 47 % yield. The outcome of the enantioselective reaction performed with ligand 1 a and the standard catalyst loading was strikingly different. Product 13 could not be detected, and the reaction exclusively occurred at the internal sulfur atom that is located at position C3 of the quinolone. The only product was sulfimide 4 m which was isolated in 53 % yield (58 % conversion) with 97 % ee.
Scheme 8.

Site‐selectivity in the sulfimidation of substrate 3 m: The hydrogen bonding ligand 1 a directs the reaction to a defined site (bottom) while an achiral ligand exerts the opposite site‐selectivity (top).
The fact that a bound substrate can only display a distinct lone pair to the catalytically active center (cf. Figure 1) should result for any substrate with a single reactive center in a kinetic resolution. Sulfoxides are like sulfimides chiral and carry a lone pair at the sulfur atom. If subjected to the conditions of the enantioselective sulfimidation, one sulfoxide enantiomer should be preferentially processed as its stereogenic sulfur atom is properly configured to match the chirality of the active site within the catalytically active silver complex. We probed this hypothesis with sulfoxide rac‐14 which was treated with the imidation reagent and the optimized catalyst mixture (Scheme 9). The resulting sulfoximine 15 was obtained in enantiomerically enriched form while the recovered sulfoxide also showed some optical purity. Depending on the equivalents n of the iminophenyliodinane the enantioselectivity could be optimized for the sulfoximine or the sulfoxide. Although we did not prove the absolute configuration in this case, it is highly likely that the (R)‐enantiomer of sulfoxide 14 is preferentially processed leaving the (S)‐enantiomer behind. Sulfoximine 15 should thus be (R)‐configured as shown.
Scheme 9.

Kinetic resolution of sulfoxide rac‐14 by the enantioselective Ag‐catalyzed sulfimidation in the presence of chiral ligand 1 a. Dependent on the equivalents n of the imidation reagent, product 15 or the recovered (recd.) substrate 14 can be isolated with high enantiomeric excess.
Mechanistic experiments and discussion. Despite the fact that the synthetic experiments delivered circumstantial evidence (see the SI for further details) for the postulated mechanism of the enantioselective sulfimidation we attempted to obtain further data to substantiate the proposed reaction pathway. A hint for the formation of the postulated heteroleptic complex 9 was obtained by high resolution electrospray ion mass spectrometric (ESI‐MS) analysis. Under conditions that mimicked the composition of the catalytically active mixture, a sample was subjected to ESI‐MS in the positive detection mode (detection of cationic species). It was clearly evident from the exact mass analysis (Figure 2) that the most intense peak at m/z=640.1254 (m/z calcd=640.1261) corresponds to the heteroleptic complex 9 with the formula C35H27AgN5O+ formed from one equivalent AgPF6, 1,10‐phenanthroline and ligand 1 a (see the SI for further details). Likewise, the other two signals at m/z=467.0411 (m/z calcd=467.0420) and m/z=813.2081 (m/z calcd=813.2102) could be matched with the formula C24H16AgN4 + and C46H38AgN6O2 +. The former mass correlates to the homoleptic silver complex 16 with two 1,10‐phenanthroline ligands while the latter mass can be assigned to the homoleptic complex 17 of silver and two chiral phenanthroline ligands 1 a.
Figure 2.

ESI mass spectrometric analysis of a mixture obtained by dissolving AgPF6 (3 μmol), 1,10‐phenanthroline (phen, 3 μmol), and ligand 1 a (3.6 μmol) in dichloromethane (1 mL). A 0.1 mL aliquot was diluted with 0.9 mL CH2Cl2 before subjecting the mixture to the analysis.
Diffusion Ordered Spectroscopy (DOSY) experiments [25] were performed with AgPF6 and 1,10‐phenanthroline in CD2Cl2 solution and corroborated the existence of homoleptic complex 16. Likewise, the homoleptic complex 17 could be identified when studying AgPF6 and ligand 1 a. Experiments with AgPF6, phen, and ligand 1 a, remained somewhat inconclusive, however. 1H‐NMR chemical shift data suggest its formation but the increase in the apparent volume as measured by the DOSY experiment was lower than expected (see the SI for further details).
In order to evaluate a possible transition state that would be accessible for heteroleptic complex 9 and substrate 3 a upon conversion of 9 to a nitrene complex, we turned to DFT calculations (M06L [26] ‐D3 [27] def2‐TZVP(D), [28] for further details see SI). Assuming triplet multiplicity, two transition states leading to product 4 a and its enantiomer (ent‐4 a) were identified. The former is approx. 26 kJ mol−1 more favorable in Gibbs free energy, in agreement with the experimental findings. The associated barrier of activation is 83 kJ mol−1, which is also in line with a reaction at room temperature.
The more favorable transition state is shown in Figure 3. It features N⋅⋅⋅S and Ag⋅⋅⋅N distances of 1.91 and 2.23 Å, respectively, and the substrate is also clearly bound via a two‐point hydrogen bonding motif (with O⋅⋅⋅H distances of 1.86 and 1.90 Å). Each phenanthroline ligand is coordinated to the Ag center via a shorter bond (2.22 and 2.25 Å, originating from the nitrogen atoms para to the alkynyl linker and trans to this one) and a somewhat longer bond (2.34 and 2.55 Å). The C3‐S single bond adapts an s‐trans orientation, that is, the phenyl group points away from the site of hydrogen bonding (cf. structure 10 in Figure 1). In addition, the sulfur center to be sulfimidated carries a noticeable positive charge of +0.98 according to an NBO [29] analysis. The less favorable transition state (leading to ent‐4 a) shares several structural similarities with the more favorable one, for example, with respect to the hydrogen bonding motif and the orientation of the phenanthroline ligands (see the SI for further information). However, the C3‐S single bond is s‐cis oriented and the phenyl group points towards the binding site. The transition state is overall somewhat later on the reaction coordinate, with a shorter N⋅⋅⋅S (1.89 Å) and a longer Ag⋅⋅⋅N (2.27 Å) distance. Apart from this, the two most striking differences are the already mentioned CO‐C3‐S‐CPh dihedral angle (−44° vs. −148°) and a narrower Ag‐N‐S angle (105° vs. 121°).
Figure 3.
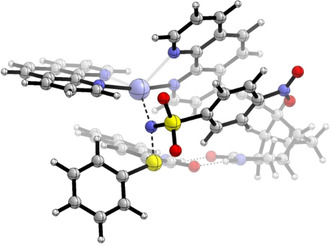
Favorable transition state for the conversion of substrate 3 a to product 4 a, according to DFT calculations. Graphics generated with CYLview20. [30]
The DFT calculation suggests that the sulfur atom carries a partial positive charge in the transition state. A Hammett analysis of the sulfimidation reaction was performed in order to interrogate the stereoelectronic situation at the sulfur atom and to potentially confirm the predicted positive charge increase (Figure 4). To this end, the six para‐substituted 3‐(phenylthio)quinolones 3 b–3 f were studied and the relative rates of the sulfimidation was determined in comparison with the parent compound 3 a. Depending on the substituent X we see a rate increase for electron donating substituent and a rate decrease for electron withdrawing groups. The decadic logarithm of the relative rates k X/k H reveals a good relation with the tabulated σ p values [31] of the substituents X. The slope of the linear function was determined as ρ=−0.52 (see the SI for more details) in agreement with a positive charge to be developed at the reactive center.[ 32 , 33 ]
Figure 4.

Hammett analysis performed for substrates 3 a–3 f. Conditions: Competing reactions were performed under typical catalytic conditions with 1 equiv of the respective para‐substituted 3‐(phenylthio)quinolone 3 and 1 equiv of 3 a at ambient temperature (2 equiv PhI=NNs). The reaction was stopped after two hours (<25 % conversion) and the relative ratio of the products 4 was determined by 1H NMR spectroscopy.
Conclusion
In summary, our study has revealed that the directing power of hydrogen bonding ligands is not only operative in defined metal complexes but also in an equilibrium scenario with reversible coordination of chiral and achiral ligands at a cationic metal center. The observed site‐ and enantioselectivity makes several heterocyclic sulfimides available which would be difficult to access in enantioselective form by known methods of asymmetric catalysis. The suggested reaction mechanism of the nitrene transfer which had been previously proposed to occur via a nitrene‐silver complex with distorted trigonal bipyramidal coordination [11] has been further corroborated and offers a useful model for the design of related phenanthroline ligands with which more remote positions of a given substrate can be aminated.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (grant Ba 1372/17‐2) is gratefully acknowledged. A.R.R. thanks the Alexander von Humboldt foundation for a research fellowship. J.G. thanks the Fonds der Chemischen Industrie (Kekulé fellowship). We thank M. Muhr (group of Prof. R. A. Fischer, TUM) for his help with the ESI‐MS measurements and Prof. R. M. Gschwind (Regensburg) for helpful discussions. O. Ackermann, F. Pecho, L. Koser, and J. Kudermann (all at TUM) are acknowledged for their help with HPLC and GLC analyses. Open access funding enabled and organized by Projekt DEAL.
R. R. Annapureddy, F. Burg, J. Gramüller, T. P. Golub, C. Merten, S. M. Huber, T. Bach, Angew. Chem. Int. Ed. 2021, 60, 7920.
Contributor Information
Dr. Rajasekar Reddy Annapureddy, http://www.oc1.ch.tum.de/home en/.
Prof. Dr. Thorsten Bach, Email: thorsten.bach@ch.tum.de.
References
- 1.For early reviews, see:
- 1a. Taylor P. C., Sulfur Rep. 1999, 21, 241–280; [Google Scholar]
- 1b. Koval I. V., Sulfur Rep. 1993, 14, 149–215; [Google Scholar]
- 1c. Furukawa N., Oae S., Ind. Eng. Chem. Prod. Res. Dev. 1981, 20, 260–270; [Google Scholar]
- 1d. Gilchrist T. L., Moody C. J., Chem. Rev. 1977, 77, 409–435. [Google Scholar]
- 2.Reviews:
- 2a. Mäder P., Kattner L., J. Med. Chem. 2020, 63, 14243–14275; [DOI] [PubMed] [Google Scholar]
- 2b. Matos P. M., Stockman R. A., Org. Biomol. Chem. 2020, 18, 6429–6442; [DOI] [PubMed] [Google Scholar]
- 2c. Wojaczyńska E., Wojaczyński J., Chem. Rev. 2020, 120, 4578–4611; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Wiezorek S., Lamers P., Bolm C., Chem. Soc. Rev. 2019, 48, 5408–5423; [DOI] [PubMed] [Google Scholar]
- 2e. Lücking U., Angew. Chem. Int. Ed. 2013, 52, 9399–9408; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9570–9580; [Google Scholar]
- 2f. Frings M., Bolm C., Blum A., Gnamm C., Eur. J. Med. Chem. 2017, 126, 225–245; [DOI] [PubMed] [Google Scholar]
- 2g. Reggelin M., Zur C., Synthesis 2000, 1–64. [Google Scholar]
- 3.Review: Bizet V., Hendriks C. M. M., Bolm C., Chem. Soc. Rev. 2015, 44, 3378–3390. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Takada H., Nishibayashi Y., Ohe K., Uemura S., Chem. Commun. 1996, 931–932; [Google Scholar]
- 4b. Takada H., Nishibayashi Y., Ohe K., Uemura S., Phosphorus Sulfur Silicon Relat. Elem. 1997, 120, 363–364; [Google Scholar]
- 4c. Takada H., Nishibayashi Y., Ohe K., Uemura S., Baird C. P., Sparey T. J., Taylor P. C., J. Org. Chem. 1997, 62, 6512–6518; [Google Scholar]
- 4d. Miyake Y., Takada H., Ohe K., Uemura S., J. Chem. Soc. Perkin Trans. 1 1998, 2373–2376. [Google Scholar]
- 5.
- 5a. Nishikori H., Ohta C., Oberlin E., Irie R., Katsuki T., Tetrahedron 1999, 55, 13937–13946; [Google Scholar]
- 5b. Nishikori H., Katsuki T., Appl. Catal. A 2000, 194, 475–477; [Google Scholar]
- 5c. Ohta C., Katsuki T., Tetrahedron Lett. 2001, 42, 3885–3888. [Google Scholar]
- 6.
- 6a. Murakami M., Uchida T., Katsuki T., Tetrahedron Lett. 2001, 42, 7071–7074; [Google Scholar]
- 6b. Murakami M., Katsuki T., Tetrahedron Lett. 2002, 43, 3947–3949; [Google Scholar]
- 6c. Murakami M., Uchida T., Saito B., Katsuki T., Chirality 2003, 15, 116–123; [DOI] [PubMed] [Google Scholar]
- 6d. Fujita H., Uchida T., Irie R., Katsuki T., Chem. Lett. 2007, 36, 1092–1093. [Google Scholar]
- 7.
- 7a. Tamura Y., Uchida T., Katsuki T., Tetrahedron Lett. 2003, 44, 3301–3303; [Google Scholar]
- 7b. Uchida T., Tamura Y., Ohba M., Katsuki T., Tetrahedron Lett. 2003, 44, 7965–7968; [Google Scholar]
- 7c. Yoshitake M., Hayashi H., Uchida T., Org. Lett. 2020, 22, 4021–4025. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Wang J., Frings M., Bolm C., Angew. Chem. Int. Ed. 2013, 52, 8661–8665; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 8823–8827; [Google Scholar]
- 8b. Farwell C. C., McIntosh J. A., Hyster T. K., Wang Z. J., Arnold F. H., J. Am. Chem. Soc. 2014, 136, 8766–8771; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Prier C. K., Hyster T. K., Farwell C. C., Huang A., Arnold F. H., Angew. Chem. Int. Ed. 2016, 55, 4711–4715; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4789–4793. [Google Scholar]
- 9.For a perspective, see: Burg F., Bach T., J. Org. Chem. 2019, 84, 8815–8836. [DOI] [PubMed] [Google Scholar]
- 10.Reviews:
- 10a. Proctor R. S. J., Colgan A. C., Phipps R. J., Nat. Chem. 2020, 12, 990–1004; [DOI] [PubMed] [Google Scholar]
- 10b. Fanourakis A., Docherty P. J., Chuentragool P., Phipps R. J., ACS Catal. 2020, 10, 10672–10714; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c. Kuninobu Y., Torigoe T., Org. Biomol. Chem. 2020, 18, 4126–4134; [DOI] [PubMed] [Google Scholar]
- 10d. Vidal D., Olivo G., Costas M., Chem. Eur. J. 2018, 24, 5042–5054; [DOI] [PubMed] [Google Scholar]
- 10e. Davis H. J., Phipps R. J., Chem. Sci. 2017, 8, 864–877; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10f. Dydio P., Reek J. N. H., Chem. Sci. 2014, 5, 2135–2145; [Google Scholar]
- 10g. Raynal M., Ballester P., Videl-Ferran A., Van Leeuwen P. W. N. M., Chem. Soc. Rev. 2014, 43, 1660–1733; [DOI] [PubMed] [Google Scholar]
- 10h. Carboni S., Gennari C., Pignatoro L., Piarulli U., Dalton Trans. 2011, 40, 4355–4373; [DOI] [PubMed] [Google Scholar]
- 10i. Das S., Brudvig G. W., Crabtree R. H., Chem. Commun. 2008, 413–424. [DOI] [PubMed] [Google Scholar]
- 11. Annapureddy R. R., Jandl C., Bach T., J. Am. Chem. Soc. 2020, 142, 7374–7378. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Lonergan D. G., Riego J., Deslongchamps G., Tetrahedron Lett. 1996, 37, 6109–6112; [Google Scholar]
- 12b. Lonergan D. G., Deslongchamps G., Tetrahedron 1998, 54, 14041–14052. [Google Scholar]
- 13.
- 13a. Fackler P., Berthold C., Voss F., Bach T., J. Am. Chem. Soc. 2010, 132, 15911–15913; [DOI] [PubMed] [Google Scholar]
- 13b. Burg F., Gicquel M., Breitenlechner S., Pöthig A., Bach T., Angew. Chem. Int. Ed. 2018, 57, 2953–2957; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3003–3007. [Google Scholar]
- 14.Reviews:
- 14a. Hayashi H., Uchida T., Eur. J. Org. Chem. 2020, 909–916; [Google Scholar]
- 14b. Park Y., Kim Y., Chang S., Chem. Rev. 2017, 117, 9247–9301; [DOI] [PubMed] [Google Scholar]
- 14c. Uchida T., Katsuki T., Chem. Rec. 2014, 14, 117–129; [DOI] [PubMed] [Google Scholar]
- 14d. Collet F., Lescot C., Liang C., Dauban P., Dalton Trans. 2010, 39, 10401–10413. [DOI] [PubMed] [Google Scholar]
- 15.For reviews on Ag-catalyzed nitrene transfer reactions, see:
- 15a. Dehghany M., Eshon J., Roberts J. M., Schomaker J. M. in Silver Catalysis in Organic Synthesis (Eds. Li C.-J., Bi Xi.), Wiley-VCH, Weinheim, 2019, pp. 439–532; [Google Scholar]
- 15b. Alderson J. M., Corbin J. R., Schomaker J. M., Acc. Chem. Res. 2017, 50, 2147–2158; [DOI] [PubMed] [Google Scholar]
- 15c. Zheng Q.-Z., Jiao N., Chem. Soc. Rev. 2016, 45, 4590–4627; [DOI] [PubMed] [Google Scholar]
- 15d. Li Z., He C., Eur. J. Org. Chem. 2006, 4313–4322. [Google Scholar]
- 16.For Ag-catalyzed C−H amination, see:
- 16a. Cui Y., He C., Angew. Chem. Int. Ed. 2004, 43, 4210–4212; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 4306–4308; [Google Scholar]
- 16b. Li Z., Capretto D. A., Rahaman R., He C., Angew. Chem. Int. Ed. 2007, 46, 5184–5186; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 5276–5278; [Google Scholar]
- 16c. Rigoli J. W., Weatherly C. D., Alderson J. M., Vo B. T., Schomaker J. M., J. Am. Chem. Soc. 2013, 135, 17238–17241; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16d. Ju M., Zerull E. E., Roberts J. M., Huang M., Guzei I. A., Schomaker J. M., J. Am. Chem. Soc. 2020, 142, 12930–12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.For Ag-catalyzed aziridination, see:
- 17a. Cui Y., He C., J. Am. Chem. Soc. 2003, 125, 16202–16203; [DOI] [PubMed] [Google Scholar]
- 17b. Llaveria J., Beltrán Á., Díaz-Requejo M. M., Matheu M. I., Castillón S., Pérez P. J., Angew. Chem. Int. Ed. 2010, 49, 7092–7095; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 7246–7249; [Google Scholar]
- 17c. Ju M., Weatherly C. D., Guzei I. A., Schomaker J. M., Angew. Chem. Int. Ed. 2017, 56, 9944–9948; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10076–10080; [Google Scholar]
- 17d. Elkoush T., Mak C. L., Paley D. W., Campbell M. G., ACS Catal. 2020, 10, 4820–4826. [Google Scholar]
- 18. Cho G. Y., Bolm C., Org. Lett. 2005, 7, 4983–4985. [DOI] [PubMed] [Google Scholar]
- 19. García Mancheño O., Bolm C., Chem. Eur. J. 2007, 13, 6674–6681. [DOI] [PubMed] [Google Scholar]
- 20. Lebel H., Piras H., Borduy M., ACS Catal. 2016, 6, 1109–1112. [Google Scholar]
- 21. Okamura H., Bolm C., Org. Lett. 2004, 6, 1305–1307. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Merten C., Golub T. P., Kreienborg N. M., J. Org. Chem. 2019, 84, 8797–8814; [DOI] [PubMed] [Google Scholar]
- 22b. Scholten K., Engelage E., Merten C., Phys. Chem. Chem. Phys. 2020, 22, 27979–27986. [DOI] [PubMed] [Google Scholar]
- 23.Gaussian 09, Rev E.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Wallingford CT, USA, 2013.
- 24.For a related study, see: Burg F., Breitenlechner S., Jandl C., Bach T., Chem. Sci. 2020, 11, 2121–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morris K. F., C. S. Johnson, Jr. , J. Am. Chem. Soc. 1992, 114, 3139–3141. [Google Scholar]
- 26. Zhao Y., Truhlar D. G., J. Chem. Phys. 2006, 125, 194101. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305; [DOI] [PubMed] [Google Scholar]
- 27b. Rappoport D., Furche F., J. Chem. Phys. 2010, 133, 134105. [DOI] [PubMed] [Google Scholar]
- 28. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 29. Reed A. E., Weinstock R. B., Weinhold F., J. Chem. Phys. 1985, 83, 735–746. [Google Scholar]
- 30.C. Y. Legault, CYLview20, Visualization and analysis software for computational chemistry, http://www.cylview.org (accessed December 12, 2020).
- 31. Hansch C., Leo A., Taft R. W., Chem. Rev. 1991, 91, 165–195. [Google Scholar]
- 32. van Leest N. P., van der Vlugt J. I., de Bruin B., Chem. Eur. J. 2021, 27, 371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stateman L. M., Wappes E. A., Nakafuku K. M., Edwards K. M., Nagib D. A., Chem. Sci. 2019, 10, 2693–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary