

Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is one of the most common types of muscular dystrophy affecting roughly 1 in 8,000 individuals. The complex underlying genetics and poor mechanistic understanding has caused a bottleneck in therapeutic development. Until the discovery of DUX4 and its causal role in FSHD, most trials were untargeted with limited results. Emerging approaches can learn from these early trials to increase their chance of success. Here, we explore the evolution of FSHD clinical trials from non-specific anabolic or anti-inflammatory/oxidant strategies, to cutting-edge molecular therapies targeting DUX4 and we discuss the importance of clinical outcome measures. With combined advances across multiple facets of FSHD research, the field is now poised to accelerate the process of therapeutic discovery and testing.
Keywords: Facioscapulohumeral muscular dystrophy, DUX4, Therapies, Clinical Trials, Anabolic, Clinical Outcome Measures
FACIOSCAPULOHUMERAL MUSCULAR DYSTROPHY
Facioscapulohumeral muscular dystrophy (FSHD) is an autosomal dominant myopathy affecting roughly 1 in 8000 individuals, making it one of the most common types of muscular dystrophy [1]. The disease is characterized by progressive, asymmetrical muscle atrophy that typically affects the face, upper limb and shoulder skeletal muscles and later, the lower limbs [2]. Symptoms can emerge anywhere from childhood to adulthood, but typically manifests in the 2nd or 3rd decade of life [3, 4]. Disease severity is highly variable as roughly 20% of mutation carriers are asymptomatic [5] while 20% eventually require a wheelchair [2, 3]. In a rare, early-onset form of FSHD in which symptoms emerge by 10 years of age, severity is higher than the general FSHD population with most showing childhood facial weakness and more frequent wheelchair dependence [4, 6, 7].
As discussed below, the genetics underlying FSHD are extremely complex, and consensus on the etiology of the disease has only recently emerged, thus there is currently no effective treatment or cure. Furthermore, due to the wide range of disease severity, a single therapeutic approach may not be appropriate for all affected individuals, which complicates the development of therapeutics. Still, several attempts at FSHD drug development have been conducted beginning with initial untargeted therapeutics approaches, which provided the groundwork for the increasing success of emerging targeted therapies. In this review, we summarize the underlying genetics behind FSHD pathology, explore evolution of therapeutic approaches from untargeted drugs to new signaling and gene therapy approaches with translational potential, and discuss current gaps in clinical trial readiness.
FACIOSCAPULOHUMERAL MUSCULAR DYSTROPHY GENETICS AND PATHOLOGY
Genetics and DUX4
Genetically, FSHD is associated with a macrosatellite array consisting of tandem D4Z4 (see glossary) repeats at the distal end of chromosome region 4q35 [8] (Figure 1). This array is typically highly compacted, limiting transcription [9, 10] but in FSHD there is a ‘decompaction,’ resulting in a more open state [11, 12]. In FSHD1 (95% of cases), this occurs due to a contraction of the D4Z4 repeats to between 1 to 10 copies [13] while for FSHD2 (5% of cases), the region opens up due to mutations in epigenetic modifiers such as SMCHD1 and DNMT3B [14–16]. However, decompaction alone is not pathogenic, a permissive type A haplotype after the distal repeat is needed for disease manifestation. This 4qA allele, unlike the non-pathogenic 4qB, contains a polyadenylation sequence (PAS) that stabilizes the distal double homeobox protein 4 (DUX4) transcript [13]. Adding to this complexity, while fewer D4Z4 repeats tend to correlate with increased severity and earlier age of onset, this is not always the case [5, 6]. Explanations for this variability include somatic mosaicism in some individuals where the disease defining mutation is present only in a portion of cells, as well as FSHD2-associated mutations acting as modifiers of severity in FSHD1 [17, 18]. Nevertheless, consensus has emerged that the molecular pathology underlying FSHD is caused by DUX4 expression [2, 18].
Figure 1. The genetics and molecular mechanism for FSHD.
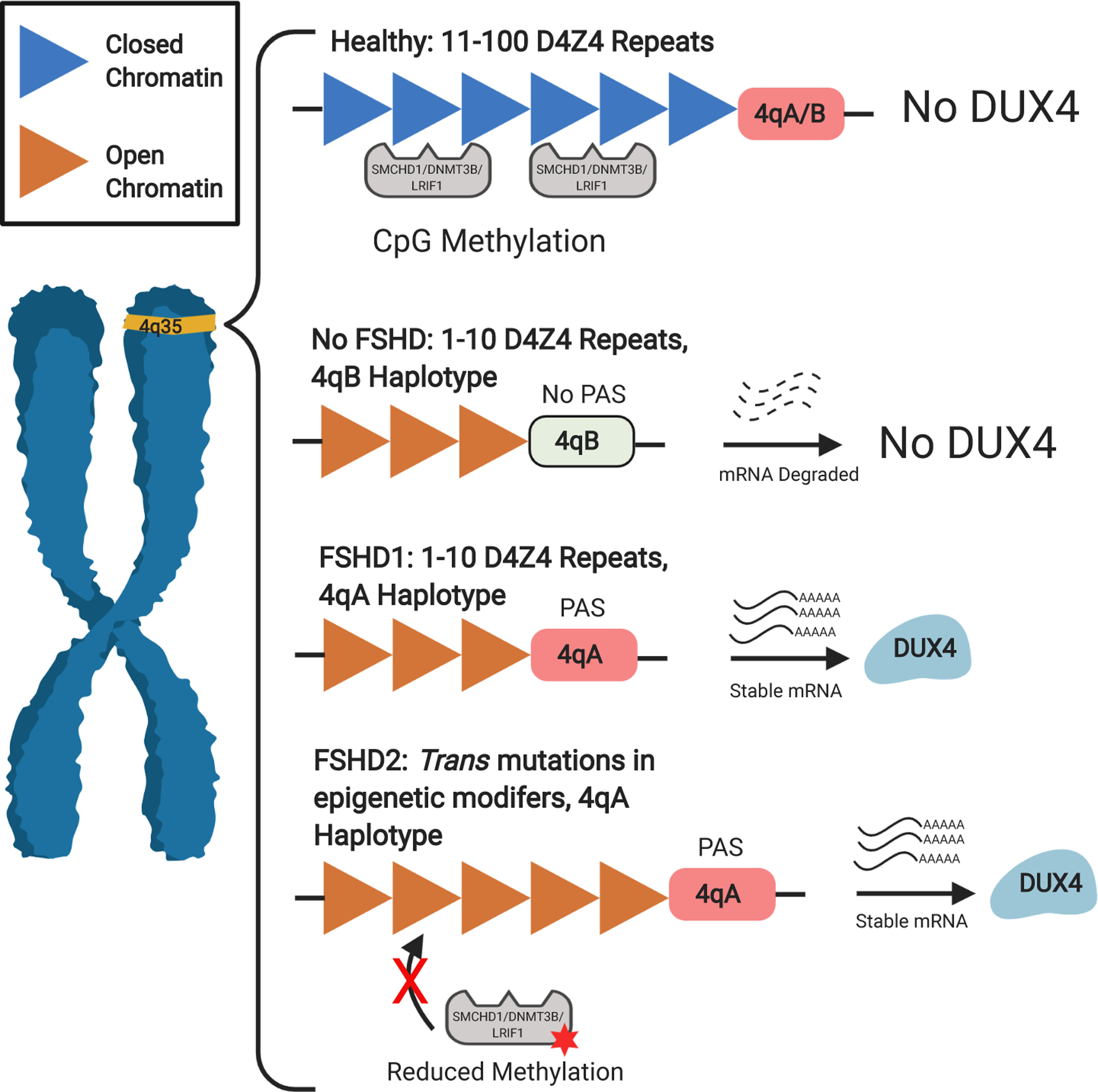
In a healthy individual, the subtelomeric region of chromosome 4q35 contains 11–100 highly compacted D4Z4 macro satellite repeats. FSHD occurs when two conditions in this region are met. First, there needs to be a relaxation of the chromatin structure, allowing expression of the DUX4 mRNA transcript. In FSHD1 (~95% of cases) contraction of the D4Z4 units (between 1–10 units) allows this change while in FSHD2 (~5% of cases) mutations in epigenetic modifiers such as SMCHD1, DNMT3B and LRIF1 results in hypomethylation to allow expression. The second condition is a permissive 4qA allele at the distal D4Z4 repeat. Unlike the non-permissive 4qB, 4qA contains a polyadenylation sequence (PAS) which adds a poly(A) tail to the DUX4 mRNA, stabilizing the transcript and allowing translation of the DUX4 transcription factor.
DUX4 and Disease Pathology
DUX4 is a transcription factor with an N-terminus containing two homeobox domains and a C-terminus containing a transcriptional activation domain [19]. The DUX protein family has a conserved function in mammals and DUX4 plays a role in the zygotic genome activation at the 4-cell stage in human embryos [20], after which it is epigenetically silenced in most tissues except the thymus and testes [21, 22]. Following the association of DUX4 and FSHD, elucidation of mechanisms associated with disease pathology began by exploring the impact of its overexpression, revealing several possible contributors. In relation to its role in embryonic development, DUX4 inappropriately activates stem cell and germline programs which may be incompatible with adult skeletal muscle, leading to dysfunction [23]. This program can also impact the immune response which is important given evidence of inflammation and oxidative damage in patients [24–26]. Most related to muscular atrophy, DUX4 can induce apoptosis either directly [27–29] or toxic build-up from inhibition of nonsense mediated RNA decay [30]. When paired with inhibition of myogenesis [31], regeneration is restricted. Despite elucidation of these pathological pathways, targeted therapies have only recently emerged as these discoveries occurred in the last decade. Most FSHD trials were untargeted which we describe in the next section.
ANABOLIC APPROACHES
Since consensus around DUX4 as the disease gene did not occur until fairly recently [13], early trials centered around ways to augment existing muscle in the hopes of improving function. As FSHD presents with progressive loss of muscle mass, anabolic therapeutics were considered attractive options for FSHD in the absence of a DUX4-specific therapy. In this section we describe the anabolic compounds that have been studied in FSHD patients.
β2-Agonists
β2-adrenergic agonists (β2-agonists) were traditionally used in the treatment of bronchospasms, a sudden constriction of the muscles in the walls of the bronchioles which can lead to difficulty breathing [32]. It soon became apparent that β2-agonists had anabolic properties including the ability to increase muscle mass in animals and healthy human volunteers raising the possibility for β2-agonists to be used in the treatment of muscle wasting diseases [33, 34].
Promising results from a small open-label study of the β2-agonist albuterol in FSHD led to a large randomized placebo controlled trial of 90 patients [35, 36]. In this 1-year assessment, patients were given placebo, 8.0, or 16.0 mg albuterol twice/day while assessing muscle mass and strength. Overall global strength via maximal voluntary isometric contraction testing (MVICT) was not improved but increased grip strength was present in both treatment doses, and muscle mass increased at the higher dose, suggesting a minimal effect on functionality in FSHD patients. An alternate study examined the effect of strength training as exercise has been demonstrated to improve the effect of β-agonists on muscle [37]. While muscle mass was increased by albuterol as in previous studies, strength training did not enhance these results [38]. However, the dynamic strength of elbow flexors was improved, suggesting some benefit from the combination of albuterol and exercise. One explanation for the limited long-term effect of β-agonists in FSHD patients may be that β2-adrenoreceptors become desensitized after prolonged activation [39]. In order to assess this, a study was performed in FSHD patients that utilized a periodic albuterol regimen of 3 weeks on, one week off [40]. No effect was found between placebo and treatment; however, this could be explained by larger than expected variability between testing centers.
It is of interest that while the early studies of albuterol relied on its non-specific anabolic effects, a recent study demonstrated that β2-agonists inhibit DUX4 expression [41]. However, the study also showed that doses of albuterol needed to suppress DUX4 expression may interfere with muscle regeneration. Unlike albuterol, another more potent β2-agonist, clenbuterol, can suppress DUX4 expression at doses that do not interfere with myogenic markers making it a potential therapeutic candidate for FSHD [41]. Clenbuterol has been tested on FSHD patients but there were too few subjects to reliably interpret results [42]. Unfortunately, there is no unified assessment of clinical severity between these each of these trials to assess if degree of atrophy influences results. Future trials should therefore clearly assess severity with more recent generations of this compound class.
Creatine
Creatine is a naturally occurring non-protein amino acid mostly present in skeletal muscle which provides energy by converting adenosine diphosphate back to adenosine triphosphate [43]. Phosphocreatine stores may be depleted in dystrophic muscle [44] and supplementation has been considered as treatments for various myopathies [45, 46]. In a double-blind, placebo controlled trial of multiple muscular dystrophies including 12 with FSHD, 5g of creatine monohydrate per day was administered for 8 weeks [47]. Patient strength and function were evaluated by neurologists using the Medical Research Council (MRC) scale, Neuromuscular Symptoms Score (NSS) [48] and vital capacity (VC) measurements. Creatine supplementation resulted in minor improvements to MRC and NSS scoring, demonstrating promise as a method to improve muscle function. Functional benefits were found other trials including a mixed myopathy investigation that included 7 FSHD patients [49] and a case study [50]. However, being small uncontrolled trials and the case study using nutritional supplementation restrict interpretation. Nonetheless, creatine holds promise as a measure to restore minor muscle function in FSHD patients and the performance of additional trials would be beneficial. As of this writing, there is one ongoing clinical trial examining creatine in children with FSHD (iNCT02948244).
Myostatin Inhibitors
Myostatin, a member of the TGF-β family of growth factors is a potent negative regulator of muscle growth [51]. Myostatin’s ability to act as a strong check on muscle growth makes its inhibition a compelling option for the treatment of muscle wasting disorders [52]. A phase I/II mixed myopathy trial including 42 FSHD patients was conducted with MYO-029, an antibody that targets myostatin [53]. Muscle strength and function were assayed through manual muscle testing (MMT), quantitative muscle testing (QMT) and timed function tests. While MYO-029 was well tolerated, and an increase in muscle mass was found in some patients, no improvement to function was observed. However, the authors noted that the study was underpowered and larger studies are necessary. Recently, trials have been conducted using derivatives of follistatin, a naturally occurring protein inhibitor of myostatin [54]. Acceleron conducted a phase 2 clinical trial with ACE-083, a follistatin inhibitor that has been well tolerated in healthy individuals and improved muscle mass and function in mouse models of neuromuscular disease [55, 56] (iiNCT02927080). Unlike the systemic MYO-029 administration, ACE-083 was given as injections of a single muscle every 3 weeks for approximately 15 months while undergoing assessments of muscle strength, endurance and mass. Unfortunately, while treatment increased muscle mass, the trial was halted prematurely as it did not correspond to an improvement in muscle function (Box 1). As a DUX4-inducible mouse model of FSHD demonstrated improvements in muscle mass and function with adeno-associated virus (AAV)-follistatin injection [57] these results initially seem contradictory but can be explained by several factors. One explanation is the difference in modality of the therapeutic as well as its delivery mechanism. The clinical trial administered follistatin as a recombinant protein while the mouse study delivered the follistatin gene via an AAV, possibly creating differences in potency. The mouse also received injections in two muscles while only one was treated in the trial, suggesting that a more systemic means of administration to target more muscles is needed to produce a benefit in patients. Lastly, an alternate myostatin inhibitor compound may be needed. For example, a myostatin neutralizing antibody LY2495655 improved muscle function in frail older adults [58]. Ultimately, improvements in delivery and testing of muscle function are needed to determine the efficacy of myostatin inhibition as a therapy for FSHD patients.
Box 1. The Acceleron ACE-083 Study.
Acceleron’s clinical trial of ACE-083 on FSHD, initiated in 2016 was highly anticipated by the community. This was the first trial involving a myostatin inhibitor in FSHD since the conclusion of a MYO-029 trial in 2007. Initial results were extremely promising as increases in muscle mass were reported. Unfortunately, the trial was prematurely terminated as these changes in muscle mass did not lead to improvements in strength and function. While it is possible that continued treatment with ACE-083 would have revealed functional improvements, several other approaches including more sensitive measures, grouping by disease severity, or a systemic administration instead of targeting singular muscles might have detected earlier functional improvements. The FSHD field is now rapidly moving towards improving their outcome measures, closing the gap towards clinical trial readiness.
ANTI-INFLAMMATORY/OXIDATIVE APPROACHES
As more became known about FSHD, it became increasingly apparent that inflammation and oxidative stress are involved in the pathology. FSHD patients show signs of inflammation both in muscle biopsies and more modern techniques such as MRI [26, 59]. Muscle biopsies also demonstrated increased markers of oxidative damage [25]. Therefore, targeting the pathological insults present in affected muscle was considered as another treatment avenue in the absence of a targeted therapy as these insults may be causal to pathology. We next describe the anti-inflammatory/oxidative approaches tested.
Glucocorticoids
Glucocorticoids are a subset of corticosteroids that have potent anti-inflammatory properties [60]. They are routinely used in Duchenne muscular dystrophy to provide short term improvement in muscle strength and delay disease progression [61, 62]. In order to assess the efficacy of this compound class, early case studies tested the glucocorticoid prednisone on individuals with clinical diagnoses of FSHD, some of which had muscle biopsies with signs of inflammatory insult [63–65]. Results were mixed with subjects either improving for the duration of treatment, deteriorating after initial improvement, or having no response at all. Since these case reports produced inconsistent results and took place before genetic characterization of FSHD, a pilot study was conducted of 8 genetically confirmed FSHD patients that received 1.5mg/kg prednisone daily for 12 weeks [66]. MMT, MVICT showed no significant change and muscle mass decreased by an average of 1.17 kg. One explanation for the inconsistency between these trials are the year of occurrence. The case reports took place before genetic diagnoses of FSHD were available and it is possible the subjects were misdiagnosed as other myopathies can mimic the FSHD phenotype [67]. Another possibility is the level of inflammation as the authors of one case report proposed that this compound may be most effective in early stages of inflammation, but less useful in more advanced cases [63]. Even if there was a more consistent therapeutic benefit, side effects such as adrenal atrophy, osteoporosis and immune suppression preclude glucocorticoids from being an ideal therapy for long-term use [60].
Nutraceuticals
There is a growing trend towards the use of complementary and alternative medicine in concert with traditional medicine, especially in disorders for which there are currently no treatment or cure. One common approach is the use of nutraceuticals, oral dietary components naturally found in foods, containing properties with potential for medical or health benefit [68]. As more was discovered about FSHD pathology, it arose that nutraceuticals may prove beneficial. While a mechanistic approach attempting to restore D4Z4 methylation using folic acid and methionine failed [69], several studies demonstrated a potential role for oxidative stress in FSHD [24–26, 28], suggesting a benefit for anti-oxidant nutraceuticals. A pilot trial involved the antioxidants vitamin C, vitamin E, zinc gluconate, and selenomethionine, of which FSHD patients possess lower than average levels [25, 70]. Patients given these supplements were subjected to two-minute walking tests (2MWT), maximal voluntary contraction (MVCQ) and endurance time against a weight (TlimQ) of which only MVCQ and TlimQ were significantly higher than placebo. Interestingly, measures of oxidative stress such as lipid peroxide decreased compared to placebo, suggesting that the antioxidant properties of these supplements may have contributed to the functional improvement in patients. Another nutraceutical trial centered on flavonoids and omega-3 based compounds, molecules with anti-inflammatory and anti-oxidant effects. In this pilot trial, individuals with several different types of muscular dystrophy, including FSHD were given a daily mixture of such compounds, collectively referred to as FLAVOMEGA. Treatment significantly increased 6-min walk distance (6MWD) and measurements of knee extension. Unfortunately, as FSHD was grouped together with LGMD, it is uncertain if FSHD patients specifically have a benefit from this treatment. Additionally, the large number of compounds administered preclude identifying which supplement(s) produces the functional improvement.
ATYR1940
Aminoacyl-tRNA synthetases (AARSs) are a family of enzymes involved in protein synthesis that also possess secondary biological activities including a group termed “physiocrines” with immune modulating properties [71]. Based on the association of inflammation with FSHD pathology, aTyr Pharma tested the therapeutic potential of the physiocrine ATYR1940 in FSHD. No peer reviewed results are available for the initial placebo-controlled and follow-up trials as of this writing (iiiNCT02239224, ivNCT02603562, vNCT02579239, viNCT02836418). However, promising improvements in MMT and quality of life scores have been reported (https://investors.atyrpharma.com/news-releases/news-release-details/atyr-pharma-presents-additional-data-resolaristm-phase-1b2-trial) [72].
CURRENT CLINICAL TRIALS
Overall, the anabolic and anti-inflammatory/oxidative trials demonstrated promising trends but were largely unsuccessful due to inconsistent results. This can partly be attributed to small sample sizes and a lack of pre-clinical testing in FSHD models but may also be due to their untargeted approach. Now that more is known about FSHD pathology, more targeted approaches are being developed and beginning to enter trials. We now summarize currently running trials.
Testosterone and rHGH
Testosterone, the primary male sex hormone has an anabolic, muscle-building function in healthy men [73]. It has therefore been used as a treatment for muscle wasting disorders including myotonic dystrophy and DMD [74, 75]. The peptide growth hormone has also been considered for muscular dystrophy treatment due to its anabolic effects [76, 77]. A single-center, open-label study is now being done to assess the safety and tolerability of combination therapy with testosterone and recombinant human growth hormone (rHGH) in FSHD patients (viiNCT03123913). 20 male FSHD patients will receive daily rHGH and intramuscular injections of testosterone every 2 weeks for 24 weeks followed by a 12-week washout. In addition to monitoring for adverse events, a preliminary assessment of muscle function will be performed through 6MWD, MMT, and quantitative muscle testing. Interpretation of results from this trial should be conservative until a larger, blinded, placebo-driven trial is conducted.
Losmapimod
One of the most anticipated compounds currently in trials is the p38 MAPK inhibitor losmapimod, the first compound to target FSHD mechanistically via inhibition of DUX4. One research group discovered the relevance of this compound and related pathway while screening for signaling involved in β2-agonist repression of DUX4 [78]. Since β2-agonist signaling activates p38 MAPK [79, 80], it was thought that p38 inhibitors would prevent β2-agonists from suppressing DUX4. Instead, it was found that targeting this pathway inhibited DUX4 expression independently of β2-agonists. Fulcrum additionally identified the potential of this compound [81] and has now launched a phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety and efficacy of losmapimod in FSHD (viiiNCT04003974). Subjects receive 15mg twice a day for 48 weeks during which the primary outcome is a change in baseline FSHD biomarkers to assess DUX4 activity in subsequent muscle biopsies. Other outcomes include assessing changes in muscle function through activities such as a reachable workspace test and strength through quantitative dynamometry. An open-label extension to assess long term effects is also underway for those that have completed the placebo-controlled trial (ixNCT04264442). In addition to being more targeted, losmapimod is an improvement over most untargeted trials by having supportive preclinical data and larger sample sizes, enhancing the chances of a conclusive result.
EMERGING APPROACHES TO FSHD THERAPEUTIC DEVELOPMENT
The FSHD field has now begun to mature due to improvements in understanding its pathophysiology, establishment of clinical outcome measures [82] and the development of DUX4 cellular and animal models [83, 84]. These advancements have accelerated preclinical development of targeted therapies, expanding the number of compounds that can be leveraged into clinical trials. In this next section we describe some of these emerging approaches.
Molecular Signaling Pathways
In recent years there has been much research into pathways that contribute to FSHD pathology. By modulating these pathways, it may be possible to halt disease progression without targeting DUX4 itself. Several signaling cascades have been revealed whose modulation affects DUX4 toxicity and may act as novel translational targets. These include MYC signaling [27], the hypoxia-inducible factor 1 (HIF1) pathway [85–87], Wnt/β-catenin signaling [85, 88] and hyaluronic acid (HA) signaling [89, 90]. Additional factors implicated in the pathology including RNA toxicity [27, 30, 91] and DUX4 interacting proteins such as the histone acetyltransferase (HAT) and transcriptional co-activator, p300 [92, 93], the multifunctional C1QBP protein and desmin [89, 90] have been identified. Interestingly, several of these factors have some degree of overlap with each other. HIF1α competitively binds β-catenin during hypoxia, increasing angiogenic genes such as VEGF [94]. p300 complexes with CBP as co-activators to allow transactivation of HIF1α [95] and MYC stabilization [96]. HA activates numerous signal transduction cascades upstream of p38MAPK and HIF1α such as phosphatidylinositol 3-kinase/Akt and Ras/ERK [95, 97]. Promisingly, the aforementioned losmapimod targets p38MAPK, which can regulate HIF1α signaling [98, 99]. This overlap suggests a convergence in the molecular signaling that contributes to FSHD pathophysiology and highlight the translational potential from their targeting.
RNA Therapeutics
Consensus on the underlying genetics behind FSHD allows for more precise molecular targeting. The most straightforward approach is to target the DUX4 transcript itself using RNA-based gene therapy. For example, antisense oligonucleotides (AOs) inhibit translation by binding to complementary mRNA sequences. Morpholinos, an AO which functions by ‘masking’ a sequence from translational machinery, have successfully been used to inhibit DUX4 by binding the PAS produced by the 4qA haplotype [100, 101]. FSHD myotubes had `25–50% DUX4 reduction and most promisingly, ~100% in patient xenografts [100]. Alternatively, morpholinos have successfully interfered with DUX4 splicing through exon binding [102]. However, as morpholinos have poor in vivo uptake, inhibition via exon targeting was tested with an alternate AO, LNA gapmers [103]. This AO reduced DUX4 more potently than morpholinos which suggests they could be a superior therapeutic. Another RNA-based approach is RNA interference (RNAi) where enzyme complexes are directed to degrade target mRNAs. In a proof-of-concept for the utility of RNAi in FSHD, mice that overexpressed DUX4 had reduced caspase levels and increased grip strength when given an AAV-miRNA against DUX4 [104]. In both approaches, there is a risk of toxicity and off-target silencing which need to be mitigated before use in the clinic. Underscoring this necessity, a toxicology screen in mice for two miRNAs targeting DUX4 demonstrated dose-dependent muscle toxicity in one but not the other [105].
Genome Editing
The emergence of CRISPR/Cas9 technology has created new possibilities for gene therapy in FSHD since with this tool, it enables precise editing of disease-causing mutations. One key approach is to correct mutations in epigenetic modifiers, which has been done successfully with SMCHD1 in FSHD myoblasts [106]. Specific targeting of the DUX4 gene itself is more challenging due to the highly repetitive nature of D4Z4 repeats with similar regions throughout the genome [107]. However, feasibility for this approach was demonstrated in a proof-of-concept where a fusion protein with a catalytically inactive Cas9 successfully silenced the DUX4 promotor and exon 1 [108]. Another approach would be to target the 4qA polyadenylation sequence, preventing stability of the transcript. These RNA and CRISPR-based approaches have many hurdles to overcome before widespread use (Box 2), but they are rapidly evolving and with time could be a powerful translational technology for FSHD.
Box 2. Challenges in Gene Therapy.
There are many challenges which need to be overcome before gene therapy becomes a common treatment for rare diseases. Current approaches involve the use of viral vectors, particularly adeno-associated viruses (AAV), which requires extremely large doses to treat adult patients. This requirement brings to light issues of safety, production-scaling limits and cost.
Immunogenicity is another concern with gene therapy as some viral vectors run the risk of a harmful or even deadly immune response. Even without deleterious effects, a large portion of the adult population possess pre-existing neutralizing antibodies to AAVs, rendering them ineffective [124]. Accordingly, individuals without resistance will gain immunity once exposed, preventing the opportunity for that vector to be used again. In order to get around these issues, several approaches including concurrent use of immunosuppressive drugs, injection into “immune-privileged” sites which elicit little to no immune response the use of non-viral delivery vectors such as lipids and nanoparticles, and an endopeptidase to cleave neutralizing antibodies [125] are being explored.
CRISPR-Cas9 technology is an enormous turning point in gene therapy, allowing for precise editing of the genome. However, one of the most major challenges to overcome with this tool is that of toxicity and off-target effects since it involves irreversibly altering one’s DNA. Changes in the wrong region could cause a cancerous mutation or dysfunction of a crucial protein, especially critical in germline cells where the mutation can be inherited. These are beginning to be overcome due to improvements in the technology. For example, variations of Cas9 have been developed which make a single strand break in DNA, requiring targeting of both strands in order to initiate editing of the region. This greatly reduces unwanted mutations as it is extremely unlikely for both targeting complexes to be off-target in the same region [126]. Altogether, as these issues are resolved, gene therapy will become more commonplace in the treatment of genetic disorders.
Overall, these preclinical developments (Figure 2) hold much promise over earlier trials for the future of FSHD therapeutics due to their targeted approach. However, it is important not to discount the trends from these earlier results suggesting an immune/oxidative component to FSHD pathology. Given the variable severity in FSHD patients, combining targeted and untargeted therapies may prove effective on the most affected individuals. Regardless of approach, effective clinical outcome measures are needed for effective testing which we describe in the next section.
Figure 2. Emerging DUX4-Targeting Therapies in FSHD.
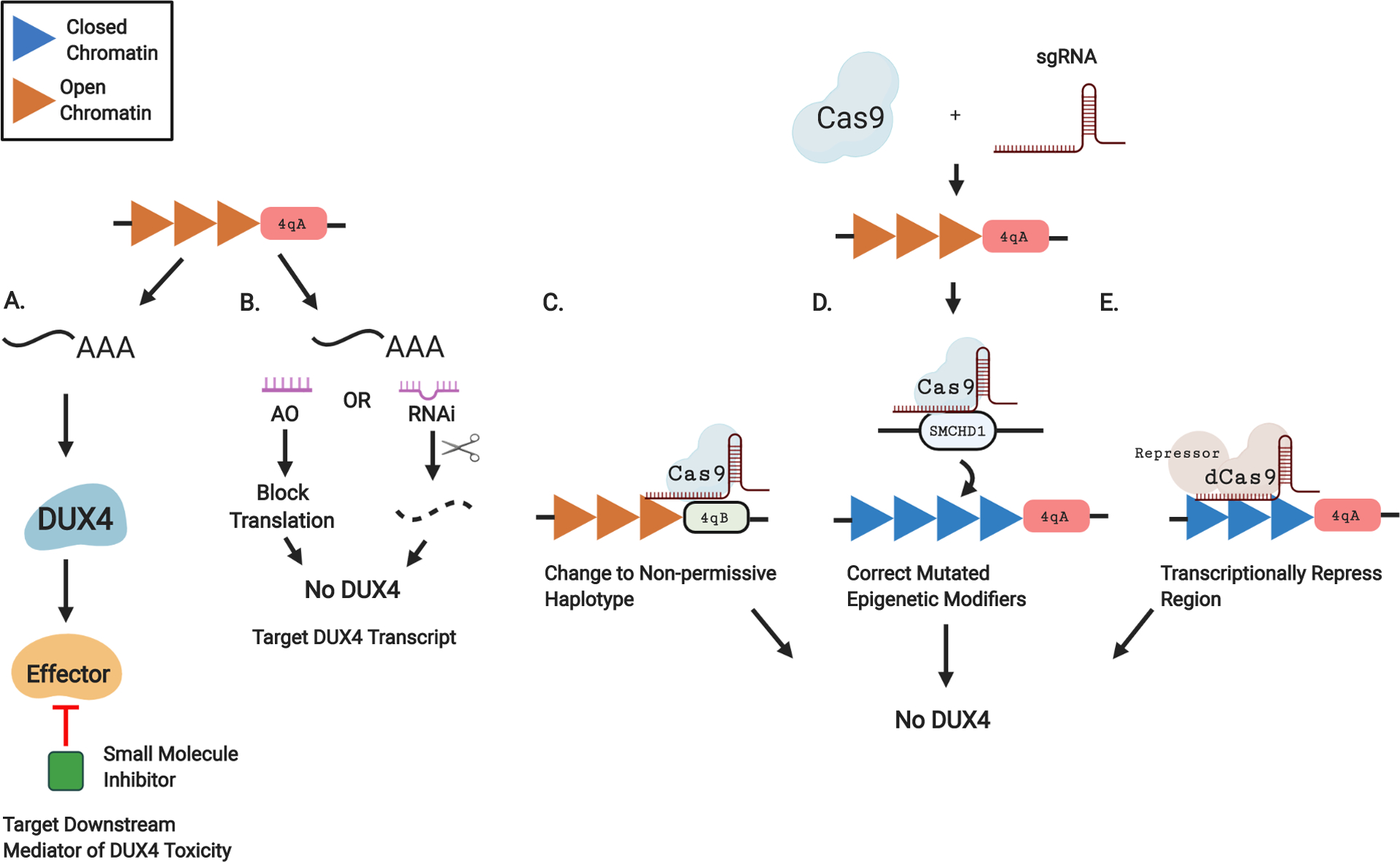
A. In lieu of directly targeting DUX4, the downstream effectors of DUX4 toxicity can be reduced using small molecule inhibitors. B. The DUX4 transcript can be targeted using RNA-based approaches. Antisense oligonucleotides (AO) can bind to a complimentary area of the transcript and block translation. Alternatively, RNA interference (RNAi) can direct the cleavage and degradation of the transcript. C-E. CRISPR-based approaches. CRISPR-Cas9 editing requires 2 components, a Cas9 endonuclease which cleaves DNA and a single guide RNA (sgRNA) to specify the target. C. The permissive 4qA haplotype at the end of the D4Z4 repeats can be changed to the non-permissive 4qB, eliminating the polyA sequence needed for stabilization of the DUX4 transcript. D. Mutations in epigenetic modifiers causative for FSHD2 such as SMCHD1 can be corrected to their functioning form. E. A catalytically inactive Cas9 that does not have endonuclease activity (dCas9) can be fused to a transcriptional repressor, condensing the region into a closed state which prevents DUX4 expression.
CLINICAL OUTCOME MEASURES
As new therapies begin to enter clinical trials it is important that the field is prepared with outcome measures to assess effectiveness. This has already been a challenge in other types of myopathies such as DMD where an incomplete understanding of a primary outcome measure led to inconclusive results and delays [109, 110]. Preventing the same mistakes will be particularly difficult in the case of FSHD as there is great variation in the degree of severity between patients and most significantly, the overall slow disease progression [1, 5, 6]. To address this problem, there needs to be consistency and standardization across clinical trials, which has yet to be achieved with the exception of MMT and quantitative myometry. Concurrent with standardization, it is necessary to develop measures which are sensitive to disease progression over the course of a year such as with MMT and MVICT in this natural history study [111]. More recently, a functional FSHD composite outcome measure (FSHD-COM) was developed that focused on patient-reported areas of functional impairment [82, 112]. Functional tasks in this measure include sit to stand time, time to pick a penny off the floor and time to put a coat on/off [82]. While this FSHD-COM holds promise due to its high test-retest reliability, sensitivity to disease progression has yet to be assessed and severely affected individuals may not be able to perform most tasks. These tasks are also snapshots in time, wearable technology should be considered as it allows for continuous/multiple points of measurement, providing more comprehensive functional assessments [113].
Biomarkers are another important tool for assessing disease progression. With the added benefit of being less subjective than functional tasks, a sensitive indicator of disease activity will be critical for clinical trials. Studies of serum biomarkers identified several protein classes in FSHD patients including protein folding, cell migration, and inflammation [114, 115]. However, those that also correlate with FSHD severity are associated with muscle breakdown such as creatine kinases, carbonic anhydrase and troponin. Alternatively, a molecular profile could be used rather than individual markers. A profile of transcriptionally downregulated targets of PAX7, a transcription factor competitively inhibited by DUX4, correlates with disease severity [116].
Imaging is another potential method to detect biomarkers of disease progression which has the advantage of being noninvasive and independent of fatigue. Muscle MRI can be used to visualize and quantitate fatty infiltration and structural changes in FSHD patients. Recently, a type of MRI tissue signal known as T2-STIR was found to correlate with DUX4 target gene expression and overall pathology [117]. A longitudinal one-year follow-up showed similar T2-STIR levels, however, biopsies from these imaged muscle revealed a pattern of DUX4-target, inflammatory and cell cycle genes that could distinguish levels of disease severity [118]. Another potential imaging tool for FSHD biomarkers is muscle ultrasound (MUS) which is much faster than MRI and can detect abnormalities in FSHD muscles that still appear normal on MRI [119]. A longitudinal study found that quantitative and qualitative MUS scores correlated with severity at baseline and increased over a year even as clinical scores remained stable, suggesting that this technique can detect subtle muscle changes before becoming symptomatic. With these and other imaging techniques, more longitudinal assessments are required in order to establish their utility.
In order to overcome these barriers and hasten drug development for FSHD, a multi-center observational study has been established entitled clinical trial readiness to solve barriers to drug development in FSHD (ReSolve) [120]. The ReSolve study will further develop two clinical outcome assessments, the FSHD-COM [82] and a novel skeletal muscle biomarker called electrical impedance myography (EIM) [121–123]. ReSolve will also establish genetic and demographic correlations of disease severity to account for variability in disease progression, important for establishing a natural history baseline and create a large biorepository of serum, RNA and DNA samples from carefully characterized FSHD patients.
CONCLUDING REMARKS
In conclusion, therapeutic approaches for FSHD have evolved as the pathophysiology behind the disease has become elucidated (Box 3: Clinician’s Corner). Initial approaches were anabolic, attempting to improve muscle function in FSHD and other MDs by augmenting what remained. Other trials were anti-inflammatory and anti-oxidative in nature with minor success implying a role for these mechanisms in FSHD pathology. Results from these trials are unfortunately mixed and have not led to any routine treatments (Table 1). The Losmapimod trial is the first study to specifically target the mechanism underlying FSHD by addressing the toxic gene DUX4. Advances in FSHD research have revealed more potential translational avenues from targeting signaling pathways behind DUX4 toxicity to gene therapy which could be combined with nontargeted approaches. Critical to the success of future clinical trials in FSHD is the development of biomarkers for early proof of concept studies as well as validated clinical outcome measures for confirmatory phase 3 studies (see Outstanding Questions). The age of clinical trials in FSHD is here, and with proper preparation, the first effective treatment for FSHD can be just over the horizon.
Box 3. Clinician’s Corner.
Facioscapulohumeral muscular dystrophy (FSHD) is one of the most common myopathies with an estimated 1 in 8,000 individuals affected. Affected individuals suffer from gradual skeletal muscle weakness that typically starts in the face, shoulders and upper limbs before moving to other muscles. The age of onset and severity is highly variable, with most become symptomatic in young adulthood. In some patients, an early-onset form can lead to more pronounced symptoms and wheelchair-dependence.
The genetics behind FSHD is highly complex and has been traced to misexpression of the transcription factor DUX4, typically active only during development and toxic to muscle in adulthood. Despite advances in understanding the molecular pathology of FSHD, there is currently no treatment to halt or reverse muscle wasting.
In the absence of known disease mechanisms, early therapeutic approaches were untargeted, seeking to improve muscle function through anabolic and anti-inflammatory effects. Results were mixed and in some cases, myostatin inhibitors that improved muscle mass did not lead to functional changes. However, the first targeted clinical trial is now underway with Losmapimod, a p38MAPK inhibitor which reduces DUX4 expression.
More targeted approaches are now in the works with recent publications demonstrating that blocking downstream signaling of DUX4 could be therapeutic. Advances in gene therapy has reached the point where this technology could also be used to inhibit DUX4.
In order to prepare the FSHD field for drug development, natural history studies to establish the rate of disease progression as well as validate sensitive clinical outcome measures and biomarkers for future therapeutic trials. It is possible that a failure in outcome measure sensitivity led to the early termination of Acceleron’s myostatin inhibitor trial.
Table 1.
List of Compounds Tested in FSHD Subjects
| Effect | Class | Compound | Dosing | Effective Measures | Notes | Reference |
|---|---|---|---|---|---|---|
| Anabolic | β2-Adrenergic Agonist | Albuterol | placebo, 8, 16mg twice/day, 1 year | Grip strength, muscle mass | Only high dose showed mass improvement | [36] |
| 26 weeks strength training, then placebo, 8mg twice/day with training for 26 weeks | MVICT, muscle mass | Exercise did not enhance further | [38] | |||
| placebo, 16mg/day, 3 weeks on, 1 week off, 6 months | No benefit | Periodic dosing regimen, large variability between testing centers | [40] | |||
| Androgen and Growth Hormone | Testosterone and rHGH | 140mg testosterone every 2 weeks, 5μg/kg/day rHGH, 24 weeks | n/a | Ongoing | NCT03123913 | |
| Myostatin Inhibitor | ACE-083 | 150, 200, 250mg every 3 weeks, 12 weeks | n/a | Terminated as secondary muscle function unchanged | NCT02927080 | |
| MYO-029 | placebo, 1, 3, 10, 30mg/kg every 2 weeks, 6 months | Muscle mass | Study focused on safety, not functional efficacy | [53] | ||
| Non-Protein Amino Acid | Creatine | placebo, 5g/day, 8 weeks | MRC, NSS | 36 muscular dystrophy patients but 12 had FSHD | [47] | |
| placebo or creatine 100mg/kg/day, 3 months, 6 weeks washout, then reverse 3 months | n/a | Ongoing, testing in children with FSHD | NCT02948244 | |||
| Anti-inflammatory | Glucocorticoid | Prednisone | 1.5mg/kg/day, 12 weeks | No benefit | [66] | |
| Physiocrine | ATYR1940 | placebo or dose escalation 0.3, 1, 3mg/kg once a week, 12 weeks | n/a | Unpublished | NCT02239224, NCT02531217, NCT02603562, NCT02579239, NCT02836418 | |
| Anti-inflammatory/ oxidant | Nutraceutical | Flavomega | placebo, 161.4g/day, 24 weeks | 6MWD, isokinetic knee extension | FSHD analyzed as a group with LGMD | [127] |
| Vitamin C, vitamin E, zinc gluconate, and selenomethionine | placebo or 500 mg vitamin C, 400mg vitamin E, 25mg zinc gluconate and 200 μg selenomethionine/day, 17 weeks | MVCQ, TlimQ | [70] | |||
| DUX4 inhibition | p38MAPK inhibitor | Losmapimod | placebo, 15mg twice/day, 48 weeks | n/a | Ongoing | NCT04003974 |
| 15mg twice/day | n/a | Ongoing | NCT04264442 | |||
| Methylation | Nutraceutical | Folic acid and methionine | 5mg folic acid/day, 1g methionine thrice/day, 12 weeks | No increase | [69] |
Abbreviations
2MWT- Two-minute walk test; 6MWD- Six minute walk distance; MRC- Medical research council; MVCQ- Maximum voluntary contraction of quadriceps; MVICT- Maximum Isometric Voluntary Contraction Testing; NSS- Neurological symptom score; TlimQ- Endurance time limit of quadriceps
Supplementary Material
Highlights.
Initial approaches to treating FSHD were untargeted, generally seeking to improve muscle function through anabolic and anti-inflammatory effects which have had mixed results in clinical trials.
The first trial to target the pathological mechanism behind FSHD is underway with Losmapimod, a p38MAPK inhibitor that has been shown to reduce DUX4 levels.
Advances in molecular mechanisms of DUX4 toxicity and gene therapy localization to the FSHD locus provide novel targets for treatment.
Improvements in clinical outcome measures through natural history study characterization of the variable FSHD severity, standardization of methods across trials and development of measures sensitive to disease progression over a year will enhance testing of novel FSHD therapeutics.
FUNDING
JC is supported by The Chris Carrino Foundation for FSHD and MDA Development Grant (MDA631018). AD by the Wellstone Center for FSH Research grant (U54HD0060848) and Friends of FSH. ML by MDA Development Grant (MDA629095). AL is supported by the MDA Development Grant (MDA514330), FSHD Society (FSHS-22019-01), Wellstone Center for FSH Research grant (U54HD0060848) and Friends of FSH.
Glossary
- 2MWT/6MWD
Two-minute walk test/ Six-minute walk distance, a clinical outcome measure in which walking distance traveled over the indicated period is assessed.
- 4q35
Chromosomal region genetically linked to FSHD. ‘4’ pertains to chromosome 4, ‘q’ refers to the long arm of the chromosome, ‘35’ is the subtelomeric region on the chromosome arm.
- 4qA
Permissive allele associate with FSHD. Contains a polyadenylation sequence which allows stabilization of the DUX4 transcript located in the distal-most D4Z4 repeat unit.
- Antisense Oligonucleotides (AOs)
Synthetic oligonucleotide which bind to its complementary mRNA sequence, preventing translation through triggering mRNA degradation or by ‘masking’ a sequence from translational machinery
- Cas9
CRISPR associated protein 9, prokaryotic endonuclease that cleaves foreign DNA. In gene editing it and its variations are used to alter targeted regions of the genome.
- Clinical Outcome Measures
Change in the health of an individual, group of people, or population that is attributable to an intervention or series of interventions.
- CRISPR
Clustered regularly interspaced short palindromic repeats, family of DNA sequences used as part of prokaryotic immune system. Used in gene editing via targeting of similar sequences.
- D4Z4
Macrosatellite repeat unit in chromosome 4 which contains 11–100 copies in a healthy individual. Each unit contains a DUX4 open reading frame.
- DNMT3B
DNA methyltransferase 3 beta, epigenetic modifier which regulates gene expression through CpG methylation. In FSHD2, mutations in this gene results in hypomethylation of the D4Z4 region.
- MMT
Manual muscle testing, clinical outcome measure to assess muscle strength and function. Subject is instructed to flex the evaluated muscle or undergo a range of motion while evaluator applies resistance. Scored from 0 to 5.
- MVICT
Maximum Isometric Voluntary Contraction Testing, clinical outcome measure in which a subject is instructed to contract their indicated muscle while strapped to a transducer which measures the force of the contraction.
- NSS
Neurological symptom score, clinical outcome measure that focuses on 17 symptoms related to muscle weakness, sensory disturbances and autonomic symptoms. Evaluation is done by a clinician for each symptom and given a score of 1 if present and 0 if not.
- PAS
Polyadenylation signal, DUX4 mRNA derived from the distal-most unit of D4Z4 is spliced to a polyadenylation sequence in the 4qA haplotype which stabilizes the transcript.
- QMT
Quantitative muscle testing, clinical outcome measure in which muscle strength is assessed through a dynamometer.
- sgRNA
Single guide RNA, non-coding short RNA sequences which in gene editing bind the Cas9 enzyme and brings it to the complementary target DNA sequences.
- SMCHD1
Structural Maintenance of Chromosomes Hinge Domain containing 1, epigenetic modifier which represses genes by maintenance of CpG methylation. In FSHD2, the absence of SMCHD1 causes derepression of the D4Z4 region.
REFERENCES
- 1.Deenen JC, et al. , Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology, 2014. 83(12): p. 1056–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeSimone AM, et al. , Facioscapulohumeral Muscular Dystrophy. Compr Physiol, 2017. 7(4): p. 1229–1279. [DOI] [PubMed] [Google Scholar]
- 3.Hamel J and Tawil R, Facioscapulohumeral Muscular Dystrophy: Update on Pathogenesis and Future Treatments. Neurotherapeutics, 2018. 15(4): p. 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mah JK and Chen YW, A Pediatric Review of Facioscapulohumeral Muscular Dystrophy. J Pediatr Neurol, 2018. 16(4): p. 222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tonini MM, et al. , Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul Disord, 2004. 14(1): p. 33–8. [DOI] [PubMed] [Google Scholar]
- 6.Goselink RJM, et al. , A 22-year follow-up reveals a variable disease severity in early-onset facioscapulohumeral dystrophy. Eur J Paediatr Neurol, 2018. 22(5): p. 782–785. [DOI] [PubMed] [Google Scholar]
- 7.Klinge L, et al. , Severe phenotype in infantile facioscapulohumeral muscular dystrophy. Neuromuscul Disord, 2006. 16(9–10): p. 553–8. [DOI] [PubMed] [Google Scholar]
- 8.Wijmenga C, et al. , Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet, 1992. 2(1): p. 26–30. [DOI] [PubMed] [Google Scholar]
- 9.Gabriels J, et al. , Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene, 1999. 236(1): p. 25–32. [DOI] [PubMed] [Google Scholar]
- 10.Hewitt JE, et al. , Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystrophy. Hum Mol Genet, 1994. 3(8): p. 1287–95. [DOI] [PubMed] [Google Scholar]
- 11.Greco A, et al. , Consequences of epigenetic derepression in facioscapulohumeral muscular dystrophy. Clin Genet, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van der Maarel SM, et al. , Facioscapulohumeral muscular dystrophy: consequences of chromatin relaxation. Curr Opin Neurol, 2012. 25(5): p. 614–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lemmers RJ, et al. , A unifying genetic model for facioscapulohumeral muscular dystrophy. Science, 2010. 329(5999): p. 1650–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lemmers RJ, et al. , Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet, 2012. 44(12): p. 1370–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van den Boogaard ML, et al. , Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am J Hum Genet, 2016. 98(5): p. 1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamanaka K, et al. , Homozygous nonsense variant in LRIF1 associated with facioscapulohumeral muscular dystrophy. Neurology, 2020. 94(23): p. e2441–e2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lemmers RJ, et al. , Somatic mosaicism in FSHD often goes undetected. Ann Neurol, 2004. 55(6): p. 845–50. [DOI] [PubMed] [Google Scholar]
- 18.Himeda CL and Jones PL, The Genetics and Epigenetics of Facioscapulohumeral Muscular Dystrophy. Annu Rev Genomics Hum Genet, 2019. 20: p. 265–291. [DOI] [PubMed] [Google Scholar]
- 19.Mitsuhashi H, et al. , Functional domains of the FSHD-associated DUX4 protein. Biol Open, 2018. 7(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Iaco A, et al. , DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat Genet, 2017. 49(6): p. 941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hendrickson PG, et al. , Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat Genet, 2017. 49(6): p. 925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whiddon JL, et al. , Conservation and innovation in the DUX4-family gene network. Nat Genet, 2017. 49(6): p. 935–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geng LN, et al. , DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell, 2012. 22(1): p. 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sasaki-Honda M, et al. , A Patient-derived iPSC Model Revealed Oxidative Stress Increases Facioscapulohumeral Muscular Dystrophy-causative DUX4. Hum Mol Genet, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turki A, et al. , Functional muscle impairment in facioscapulohumeral muscular dystrophy is correlated with oxidative stress and mitochondrial dysfunction. Free Radic Biol Med, 2012. 53(5): p. 1068–79. [DOI] [PubMed] [Google Scholar]
- 26.Dahlqvist JR, et al. , Relationship between muscle inflammation and fat replacement assessed by MRI in facioscapulohumeral muscular dystrophy. J Neurol, 2019. 266(5): p. 1127–1135. [DOI] [PubMed] [Google Scholar]
- 27.Shadle SC, et al. , DUX4-induced dsRNA and MYC mRNA stabilization activate apoptotic pathways in human cell models of facioscapulohumeral dystrophy. PLoS Genet, 2017. 13(3): p. e1006658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rickard AM, Petek LM, and Miller DG, Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum Mol Genet, 2015. 24(20): p. 5901–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wallace LM, et al. , DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann Neurol, 2011. 69(3): p. 540–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng Q, et al. , A feedback loop between nonsense-mediated decay and the retrogene DUX4 in facioscapulohumeral muscular dystrophy. Elife, 2015. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knopp P, et al. , DUX4 induces a transcriptome more characteristic of a less-differentiated cell state and inhibits myogenesis. J Cell Sci, 2016. 129(20): p. 3816–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye X, et al. , Appropriate use of inhaled corticosteroid and long-acting beta(2)-adrenergic agonist combination therapy among asthma patients in a US commercially insured population. Curr Med Res Opin, 2009. 25(9): p. 2251–8. [DOI] [PubMed] [Google Scholar]
- 33.Ryall JG, et al. , Beta 2-agonist administration reverses muscle wasting and improves muscle function in aged rats. J Physiol, 2004. 555(Pt 1): p. 175–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lynch GS and Ryall JG, Role of beta-adrenoceptor signaling in skeletal muscle: implications for muscle wasting and disease. Physiol Rev, 2008. 88(2): p. 729–67. [DOI] [PubMed] [Google Scholar]
- 35.Kissel JT, et al. , Pilot trial of albuterol in facioscapulohumeral muscular dystrophy. FSH-DY Group. Neurology, 1998. 50(5): p. 1402–6. [DOI] [PubMed] [Google Scholar]
- 36.Kissel JT, et al. , Randomized, double-blind, placebo-controlled trial of albuterol in facioscapulohumeral dystrophy. Neurology, 2001. 57(8): p. 1434–40. [DOI] [PubMed] [Google Scholar]
- 37.Caruso JF, et al. , The effects of albuterol and isokinetic exercise on the quadriceps muscle group. Med Sci Sports Exerc, 1995. 27(11): p. 1471–6. [PubMed] [Google Scholar]
- 38.van der Kooi EL, et al. , Strength training and albuterol in facioscapulohumeral muscular dystrophy. Neurology, 2004. 63(4): p. 702–8. [DOI] [PubMed] [Google Scholar]
- 39.Newman-Tancredi A, et al. , Down-regulation of rat beta-adrenoceptors by clenbuterol or desipramine does not require chronic treatment: [3H] CGP-12177 binding reveals rapid (24 hour) modulation. Brain Res Bull, 1996. 41(2): p. 93–6. [PubMed] [Google Scholar]
- 40.Payan CA, et al. , Periodic salbutamol in facioscapulohumeral muscular dystrophy: a randomized controlled trial. Arch Phys Med Rehabil, 2009. 90(7): p. 1094–101. [DOI] [PubMed] [Google Scholar]
- 41.Campbell AE, et al. , BET bromodomain inhibitors and agonists of the beta-2 adrenergic receptor identified in screens for compounds that inhibit DUX4 expression in FSHD muscle cells. Skelet Muscle, 2017. 7(1): p. 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oya Y, Ogawa M, and Kawai M, [Therapeutic trial of beta 2-adrenergic agonist clenbuterol in muscular dystrophies]. Rinsho Shinkeigaku, 2001. 41(10): p. 698–700. [PubMed] [Google Scholar]
- 43.Kreider RB, et al. , International Society of Sports Nutrition position stand: safety and efficacy of creatine supplementation in exercise, sport, and medicine. Journal of the International Society of Sports Nutrition, 2017. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kemp GJ, et al. , Cellular energetics of dystrophic muscle. J Neurol Sci, 1993. 116(2): p. 201–6. [DOI] [PubMed] [Google Scholar]
- 45.Banerjee B, et al. , Effect of creatine monohydrate in improving cellular energetics and muscle strength in ambulatory Duchenne muscular dystrophy patients: a randomized, placebo-controlled 31P MRS study. Magn Reson Imaging, 2010. 28(5): p. 698–707. [DOI] [PubMed] [Google Scholar]
- 46.Kley RA, Tarnopolsky MA, and Vorgerd M, Creatine for treating muscle disorders. Cochrane Database Syst Rev, 2011(2): p. CD004760. [DOI] [PubMed] [Google Scholar]
- 47.Walter MC, et al. , Creatine monohydrate in muscular dystrophies: A double-blind, placebo-controlled clinical study. Neurology, 2000. 54(9): p. 1848–50. [DOI] [PubMed] [Google Scholar]
- 48.Molenaar DS, et al. , Impact of neurologic signs and symptoms on functional status in peripheral neuropathies. Neurology, 1999. 52(1): p. 151–6. [DOI] [PubMed] [Google Scholar]
- 49.Matsumura T, et al. , [A clinical trial of creatine monohydrate in muscular dystrophy patients]. Rinsho Shinkeigaku, 2004. 44(10): p. 661–6. [PubMed] [Google Scholar]
- 50.Pasotti S, et al. , An integrated approach in a case of facioscapulohumeral dystrophy. BMC Musculoskelet Disord, 2014. 15: p. 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee SJ, Regulation of muscle mass by myostatin. Annu Rev Cell Dev Biol, 2004. 20: p. 61–86. [DOI] [PubMed] [Google Scholar]
- 52.Smith RC and Lin BK, Myostatin inhibitors as therapies for muscle wasting associated with cancer and other disorders. Curr Opin Support Palliat Care, 2013. 7(4): p. 352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wagner KR, et al. , A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol, 2008. 63(5): p. 561–71. [DOI] [PubMed] [Google Scholar]
- 54.Sidis Y, et al. , Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology, 2006. 147(7): p. 3586–97. [DOI] [PubMed] [Google Scholar]
- 55.Glasser CE, et al. , Locally acting ACE-083 increases muscle volume in healthy volunteers. Muscle Nerve, 2018. 57(6): p. 921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pearsall RS, et al. , Follistatin-based ligand trap ACE-083 induces localized hypertrophy of skeletal muscle with functional improvement in models of neuromuscular disease. Sci Rep, 2019. 9(1): p. 11392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Giesige CR, et al. , AAV-mediated follistatin gene therapy improves functional outcomes in the TIC-DUX4 mouse model of FSHD. JCI Insight, 2018. 3(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Becker C, et al. , Myostatin antibody (LY2495655) in older weak fallers: a proof-of-concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol, 2015. 3(12): p. 948–57. [DOI] [PubMed] [Google Scholar]
- 59.Arahata K, et al. , Inflammatory response in facioscapulohumeral muscular dystrophy (FSHD): immunocytochemical and genetic analyses. Muscle Nerve Suppl, 1995(2): p. S56–66. [PubMed] [Google Scholar]
- 60.Becker DE, Basic and clinical pharmacology of glucocorticosteroids. Anesth Prog, 2013. 60(1): p. 25–31; quiz 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Angelini C, The role of corticosteroids in muscular dystrophy: a critical appraisal. Muscle Nerve, 2007. 36(4): p. 424–35. [DOI] [PubMed] [Google Scholar]
- 62.Matthews E, et al. , Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst Rev, 2016(5): p. CD003725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Munsat TL, et al. , Inflammatory myopathy with facioscapulohumeral distribution. Neurology, 1972. 22(4): p. 335–47. [DOI] [PubMed] [Google Scholar]
- 64.Bates D, Stevens JC, and Hudgson P, “Polymyositis” with involvement of facial and distal musculature. One form of the fascioscapulohumeral syndrome? J Neurol Sci, 1973. 19(1): p. 105–8. [DOI] [PubMed] [Google Scholar]
- 65.Wulff JD, Lin JT, and Kepes JJ, Inflammatory facioscapulohumeral muscular dystrophy and Coats syndrome. Ann Neurol, 1982. 12(4): p. 398–401. [DOI] [PubMed] [Google Scholar]
- 66.Tawil R, et al. , A pilot trial of prednisone in facioscapulohumeral muscular dystrophy. FSH-DY Group. Neurology, 1997. 48(1): p. 46–9. [DOI] [PubMed] [Google Scholar]
- 67.Sacconi S, et al. , Patients with a phenotype consistent with facioscapulohumeral muscular dystrophy display genetic and epigenetic heterogeneity. J Med Genet, 2012. 49(1): p. 41–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Souyoul SA, Saussy KP, and Lupo MP, Nutraceuticals: A Review. Dermatol Ther (Heidelb), 2018. 8(1): p. 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van der Kooi EL, et al. , No effect of folic acid and methionine supplementation on D4Z4 methylation in patients with facioscapulohumeral muscular dystrophy. Neuromuscul Disord, 2006. 16(11): p. 766–9. [DOI] [PubMed] [Google Scholar]
- 70.Passerieux E, et al. , Effects of vitamin C, vitamin E, zinc gluconate, and selenomethionine supplementation on muscle function and oxidative stress biomarkers in patients with facioscapulohumeral dystrophy: a double-blind randomized controlled clinical trial. Free Radic Biol Med, 2015. 81: p. 158–69. [DOI] [PubMed] [Google Scholar]
- 71.D JJ, et al. , Brugia malayi Asparaginyl-tRNA Synthetase Stimulates Endothelial Cell Proliferation, Vasodilation and Angiogenesis. PLoS One, 2016. 11(1): p. e0146132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Walker G, et al. , P.341 - Results of a Phase 1b/2 Study of ATYR1940 in adolescents and young adults with early onset facioscapulohumeral muscular dystrophy (FSHD) (ATYR1940-C-003). Neuromuscular Disorders, 2017. 27: p. S199. [Google Scholar]
- 73.Bhasin S, et al. , The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men. N Engl J Med, 1996. 335(1): p. 1–7. [DOI] [PubMed] [Google Scholar]
- 74.Griggs RC, et al. , Effect of testosterone on muscle protein synthesis in myotonic dystrophy. Ann Neurol, 1986. 20(5): p. 590–6. [DOI] [PubMed] [Google Scholar]
- 75.Wood CL, et al. , Testosterone Treatment of Pubertal Delay in Duchenne Muscular Dystrophy. Neuropediatrics, 2015. 46(6): p. 371–6. [DOI] [PubMed] [Google Scholar]
- 76.Nielsen S, et al. , The effect of long-term pharmacological antilipolysis on substrate metabolism in growth hormone (GH)-substituted GH-deficient adults. J Clin Endocrinol Metab, 2002. 87(7): p. 3274–8. [DOI] [PubMed] [Google Scholar]
- 77.Prahm KP, Feldt-Rasmussen U, and Vissing J, Human growth hormone stabilizes walking and improves strength in a patient with dominantly inherited calpainopathy. Neuromuscul Disord, 2017. 27(4): p. 358–362. [DOI] [PubMed] [Google Scholar]
- 78.Oliva J, et al. , Clinically Advanced p38 Inhibitors Suppress DUX4 Expression in Cellular and Animal Models of Facioscapulohumeral Muscular Dystrophy. J Pharmacol Exp Ther, 2019. 370(2): p. 219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McAlees JW and Sanders VM, Hematopoietic protein tyrosine phosphatase mediates beta2-adrenergic receptor-induced regulation of p38 mitogen-activated protein kinase in B lymphocytes. Mol Cell Biol, 2009. 29(3): p. 675–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moule SK and Denton RM, The activation of p38 MAPK by the beta-adrenergic agonist isoproterenol in rat epididymal fat cells. FEBS Lett, 1998. 439(3): p. 287–90. [DOI] [PubMed] [Google Scholar]
- 81.Rojas LA, et al. , P38alpha Regulates Expression of DUX4 in Facioscapulohumeral Muscular Dystrophy. J Pharmacol Exp Ther, 2020. [DOI] [PubMed] [Google Scholar]
- 82.Eichinger K, et al. , Facioscapulohumeral muscular dystrophy functional composite outcome measure. Muscle Nerve, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lek A, et al. , Emerging preclinical animal models for FSHD. Trends Mol Med, 2015. 21(5): p. 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jagannathan S, et al. , Model systems of DUX4 expression recapitulate the transcriptional profile of FSHD cells. Hum Mol Genet, 2016. 25(20): p. 4419–4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Banerji CR, et al. , beta-Catenin is central to DUX4-driven network rewiring in facioscapulohumeral muscular dystrophy. J R Soc Interface, 2015. 12(102): p. 20140797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lek A, et al. , Applying genome-wide CRISPR-Cas9 screens for therapeutic discovery in facioscapulohumeral muscular dystrophy. Sci Transl Med, 2020. 12(536). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tsumagari K, et al. , Gene expression during normal and FSHD myogenesis. BMC Med Genomics, 2011. 4: p. 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Block GJ, et al. , Wnt/beta-catenin signaling suppresses DUX4 expression and prevents apoptosis of FSHD muscle cells. Hum Mol Genet, 2013. 22(23): p. 4661–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ansseau E, et al. , Homologous Transcription Factors DUX4 and DUX4c Associate with Cytoplasmic Proteins during Muscle Differentiation. PLoS One, 2016. 11(1): p. e0146893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.DeSimone AM, et al. , Identification of the hyaluronic acid pathway as a therapeutic target for facioscapulohumeral muscular dystrophy. Sci Adv, 2019. 5(12): p. eaaw7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shadle SC, et al. , DUX4-induced bidirectional HSATII satellite repeat transcripts form intranuclear double stranded RNA foci in human cell models of FSHD. Hum Mol Genet, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Choi SH, et al. , DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res, 2016. 44(11): p. 5161–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bosnakovski D, et al. , A novel P300 inhibitor reverses DUX4-mediated global histone H3 hyperacetylation, target gene expression, and cell death. Sci Adv, 2019. 5(9): p. eaaw7781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kaidi A, Williams AC, and Paraskeva C, Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat Cell Biol, 2007. 9(2): p. 210–7. [DOI] [PubMed] [Google Scholar]
- 95.Masoud GN and Li W, HIF-1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B, 2015. 5(5): p. 378–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Faiola F, et al. , Dual regulation of c-Myc by p300 via acetylation-dependent control of Myc protein turnover and coactivation of Myc-induced transcription. Mol Cell Biol, 2005. 25(23): p. 10220–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vigetti D, et al. , Hyaluronan: biosynthesis and signaling. Biochim Biophys Acta, 2014. 1840(8): p. 2452–9. [DOI] [PubMed] [Google Scholar]
- 98.Kwon SJ, Song JJ, and Lee YJ, Signal pathway of hypoxia-inducible factor-1alpha phosphorylation and its interaction with von Hippel-Lindau tumor suppressor protein during ischemia in MiaPaCa-2 pancreatic cancer cells. Clin Cancer Res, 2005. 11(21): p. 7607–13. [DOI] [PubMed] [Google Scholar]
- 99.Nys K, et al. , A p38(MAPK)/HIF-1 pathway initiated by UVB irradiation is required to induce Noxa and apoptosis of human keratinocytes. J Invest Dermatol, 2010. 130(9): p. 2269–76. [DOI] [PubMed] [Google Scholar]
- 100.Chen JC, et al. , Morpholino-mediated Knockdown of DUX4 Toward Facioscapulohumeral Muscular Dystrophy Therapeutics. Mol Ther, 2016. 24(8): p. 1405–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marsollier AC, et al. , Antisense targeting of 3’ end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: a new gene-silencing approach. Hum Mol Genet, 2016. 25(8): p. 1468–78. [DOI] [PubMed] [Google Scholar]
- 102.Ansseau E, et al. , Antisense Oligonucleotides Used to Target the DUX4 mRNA as Therapeutic Approaches in FaciosScapuloHumeral Muscular Dystrophy (FSHD). Genes (Basel), 2017. 8(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lim KRQ, et al. , Inhibition of DUX4 expression with antisense LNA gapmers as a therapy for facioscapulohumeral muscular dystrophy. Proc Natl Acad Sci U S A, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wallace LM, et al. , RNA interference inhibits DUX4-induced muscle toxicity in vivo: implications for a targeted FSHD therapy. Mol Ther, 2012. 20(7): p. 1417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wallace LM, et al. , Pre-clinical Safety and Off-Target Studies to Support Translation of AAV-Mediated RNAi Therapy for FSHD. Mol Ther Methods Clin Dev, 2018. 8: p. 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Goossens R, et al. , Intronic SMCHD1 variants in FSHD: testing the potential for CRISPR-Cas9 genome editing. J Med Genet, 2019. 56(12): p. 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Himeda CL, Jones TI, and Jones PL, Scalpel or Straitjacket: CRISPR/Cas9 Approaches for Muscular Dystrophies. Trends Pharmacol Sci, 2016. 37(4): p. 249–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Himeda CL, Jones TI, and Jones PL, CRISPR/dCas9-mediated Transcriptional Inhibition Ameliorates the Epigenetic Dysregulation at D4Z4 and Represses DUX4-fl in FSH Muscular Dystrophy. Mol Ther, 2016. 24(3): p. 527–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mendell JR, et al. , Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol, 2016. 79(2): p. 257–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Voit T, et al. , Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol, 2014. 13(10): p. 987–96. [DOI] [PubMed] [Google Scholar]
- 111.A prospective, quantitative study of the natural history of facioscapulohumeral muscular dystrophy (FSHD): implications for therapeutic trials. The FSH-DY Group. Neurology, 1997. 48(1): p. 38–46. [DOI] [PubMed] [Google Scholar]
- 112.Johnson NE, et al. , Patient-identified disease burden in facioscapulohumeral muscular dystrophy. Muscle Nerve, 2012. 46(6): p. 951–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ma CZ, et al. , Towards Wearable Comprehensive Capture and Analysis of Skeletal Muscle Activity during Human Locomotion. Sensors (Basel), 2019. 19(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Petek LM, et al. , A cross sectional study of two independent cohorts identifies serum biomarkers for facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul Disord, 2016. 26(7): p. 405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Statland J, et al. , Multiplex Screen of Serum Biomarkers in Facioscapulohumeral Muscular Dystrophy. J Neuromuscul Dis, 2014. 1(2): p. 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Banerji CRS and Zammit PS, PAX7 target gene repression is a superior FSHD biomarker than DUX4 target gene activation, associating with pathological severity and identifying FSHD at the single-cell level. Hum Mol Genet, 2019. 28(13): p. 2224–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang LH, et al. , MRI-informed muscle biopsies correlate MRI with pathology and DUX4 target gene expression in FSHD. Hum Mol Genet, 2019. 28(3): p. 476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wong CJ, et al. , Longitudinal measures of RNA expression and disease activity in FSHD muscle biopsies. Hum Mol Genet, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mul K, et al. , Quantitative muscle MRI and ultrasound for facioscapulohumeral muscular dystrophy: complementary imaging biomarkers. J Neurol, 2018. 265(11): p. 2646–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.LoRusso S, et al. , Clinical trial readiness to solve barriers to drug development in FSHD (ReSolve): protocol of a large, international, multi-center prospective study. BMC Neurol, 2019. 19(1): p. 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hamel J, et al. , Magnetic resonance imaging correlates with electrical impedance myography in facioscapulohumeral muscular dystrophy. Muscle Nerve, 2019. [DOI] [PubMed] [Google Scholar]
- 122.Mul K, et al. , Electrical impedance myography in facioscapulohumeral muscular dystrophy: A 1-year follow-up study. Muscle Nerve, 2018. 58(2): p. 213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Statland JM, et al. , Electrical impedance myography in facioscapulohumeral muscular dystrophy. Muscle Nerve, 2016. 54(4): p. 696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Boutin S, et al. , Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther, 2010. 21(6): p. 704–12. [DOI] [PubMed] [Google Scholar]
- 125.Leborgne C, et al. , IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat Med, 2020. [DOI] [PubMed] [Google Scholar]
- 126.Ran FA, et al. , Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell, 2013. 154(6): p. 1380–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sitzia C, et al. , Preliminary Evidences of Safety and Efficacy of Flavonoids- and Omega 3-Based Compound for Muscular Dystrophies Treatment: A Randomized Double-Blind Placebo Controlled Pilot Clinical Trial. Front Neurol, 2019. 10: p. 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
RESOURCES
- i-. NCT02948244. https://clinicaltrials.gov/ct2/show/NCT02948244.
- ii-. NCT02927080. https://clinicaltrials.gov/ct2/show/NCT02927080.
- iii-. NCT02239224. https://clinicaltrials.gov/ct2/show/NCT02239224.
- iv-. NCT02603562. https://clinicaltrials.gov/ct2/show/NCT02603562.
- v-. NCT02579239. https://clinicaltrials.gov/ct2/show/NCT02579239.
- vi-. NCT02836418. https://clinicaltrials.gov/ct2/show/NCT02836418.
- vii-. NCT03123913. https://clinicaltrials.gov/ct2/show/NCT03123913.
- viii-. NCT04003974. https://clinicaltrials.gov/ct2/show/NCT04003974.
- ix-. NCT04264442. https://clinicaltrials.gov/ct2/show/NCT04264442.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.