

Abstract
With biomedical research transitioning into data-rich science, machine learning provides a powerful toolkit for extracting knowledge from large-scale biological datasets. The increasing availability of comprehensive kidney omics compendia (transcriptomics, proteomics, metabolomics, genome sequencing), as well as other data modalities such as electronic health records, digital nephropathology repositories, and radiology renal images, make machine learning approaches increasingly essential for analyzing human kidney datasets. Here, we discuss how machine learning approaches can be applied to the study of kidney disease, with a particular focus on how they can be used for understanding the relationship between genotype and phenotype.
Keywords: Machine Learning, Deep Learning, Genotype
Introduction
In this review, we provide an introduction to machine learning broadly targeted towards nephrologists and scientists studying the kidney, emphasizing applications of machine learning approaches to understand the relationship between genotype and phenotype. The recent generation of multidimensional, information-rich, kidney-specific datasets makes it increasingly fruitful to use machine learning approaches with the potential to yield biological and translational insight to address a variety of questions in kidney biology. Furthermore, the kidney is unusual in that it is an organ with a complex three-dimensional structure and a large number of distinct cell types that is routinely biopsied in living individuals. Urine produced directly from the diseased organ makes for an ideal source of non-invasive biomarkers. And, the recent explosion in the discovery of genetic causes underlying kidney disease etiology and progression have opened the way for discovery of novel biological mechanisms across many common and rare kidney diseases.1,2 These characteristics make kidney datasets well suited both for leveraging previously developed approaches to understand renal biology and for developing and refining machine learning strategies that can also be used for the study of other organs and organ systems.
We begin with a brief introduction to major concepts and techniques in machine learning. We then describe the increasing availability of high-dimensional data on renal disease that has facilitated the development and application of machine learning techniques, with an overview of some of the data modalities that can be integrated and investigated using machine learning approaches. We provide a summary of some of the major consortia that are collecting and assembling multidimensional kidney-related datasets at a large scale. We then describe a selection of machine learning approaches that can be applied to predict the relationship between patient genotype and phenotype. Finally, we discuss a selection of machine learning approaches that have been applied in the kidney domain to uncover this relationship between genetic variation and clinical disease presentation and progression.
This review is presented as part of a series of reviews in Kidney International on big science, artificial intelligence, and machine learning and focuses on introducing basic machine learning concepts, discussing how machine learning approaches can be broadly applied to kidney-disease related questions, and specifically describing how machine learning approaches may be leveraged to predict genotype from phenotype in the context of kidney disease. Several other recent reviews with related focuses may also be of interest to the reader.3–6
Introduction to Machine Learning
Machine Learning Approaches – Supervised and Unsupervised Methods.
Machine learning methods are computational approaches used to identify meaningful patterns in data. For example, suppose that we have genome-wide gene expression measurements from the blood of a large number of patients who have been treated with a particular drug. A typical problem that could be addressed with a machine learning technique would be to predict whether a new patient will be responsive or unresponsive to the drug based on his or her gene expression profile. There are two main subtypes of machine learning, supervised and unsupervised learning. In supervised learning, the algorithm takes a dataset of labeled examples (e.g. responder vs. non-responder) and the task is to predict labels in new (unseen) examples. In unsupervised learning, the task is to identify structure in the data without prior labels. A supervised approach to the problem might take as input the set of measured expression profiles across patients, with each patient labeled as responsive or unresponsive to the drug, train a model from these profiles, and then predict whether a new patient is responsive or unresponsive based on his or her expression profile (Figure 1a). An unsupervised approach might identify multiple clusters of patients based only on the expression profiles, without any information about the patient responses to the drug included (Figure 1b). These clusters could then be investigated for differential drug responsiveness, and a new patient could be assigned based on his or her own expression profile to the most similar cluster. There is also a class of machine learning methods known as semi-supervised approaches, which can be applied when the available data is only partially labeled, generally including a few labeled and many unlabeled examples.
Figure 1:
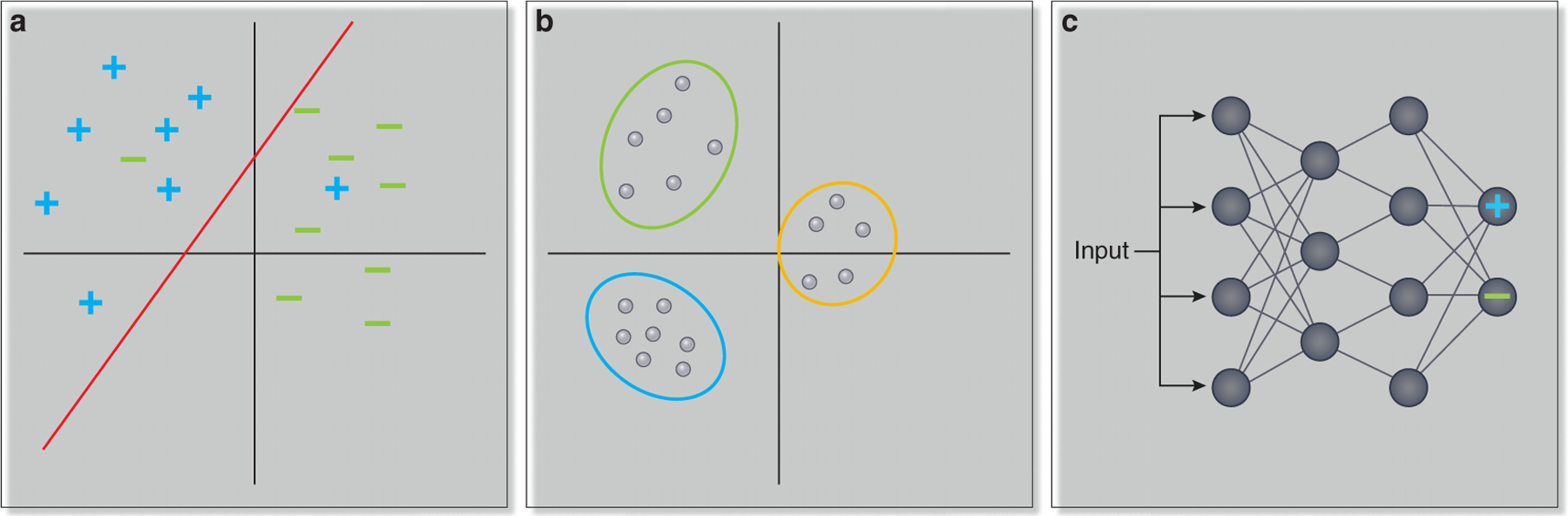
(a) In supervised learning approaches, a classifier is trained on labeled data and learns how to assign a label to a new (unseen) example. In this example, the classifier learns to distinguish between patients that are drug responders, denoted by + symbols, and drug non-responders, denoted by - symbols. (b) In unsupervised approaches, the task is to find patterns in unlabeled data. Here, patients are clustered based on similarity in gene expression profiles, and fall into three distinct groups. (c) Deep learning methods are approaches in which the input is transformed through multiple hidden layers. The output of each node in the deep learning network is a function of the inputs to that node. Here, input features might be the expression of individual genes, and the final output identifies whether the patient with the given expression profile is a drug responder (in which case the output node marked with a + will have a high value) or a drug non-responder (in which case the output node marked with a - will have a high value).
Machine learning purposes – Outcome Prediction, Subgroup Identification and Object Identification.
Two types of supervised learning problems commonly encountered in biological settings are classification and regression problems. In a classification problem, for example outcome prediction or subgroup identification, the goal is to assign each example to one of a set of distinct classes. Tasks might include classifying patients as drug responders or non-responders, as above, or distinguishing between patients who reach end-stage kidney disease and those who do not. A classification problem may be binary (assigning each example to one of two classes) or multiclass (assigning the example to one of more than two classes). In a regression problem, the task is to predict a continuous value (for example, to predict the level of a biomarker or estimated glomerular filtration rate given gene expression data). Finally, machine learning algorithms using imaging data are often presented with the problem of identifying an object in an image (for example, recognizing a glomerulus in a kidney biopsy).
Machine Learning Input and Process.
The input to a machine learning algorithm, a list of attributes for each example (e.g. patient) given to the algorithm, is called a feature set. In the example above, the features are the patient gene expression profiles, but a feature set could be any type of omics or clinical data. In the case of a supervised learning problem, the algorithm is also provided with a set of class labels, specifying to which category each example belongs. A classifier is the model which is developed by the machine learning algorithm to accomplish the specified task given the set of provided features. There are often one or more hyperparameters associated with the classifier. These are user-specified inputs that alter the behavior of the classifier. For example, for some algorithms which cluster data, the user must specify the desired number of clusters; in this case, the number of clusters is an input hyperparameter. There are also generally parameters associated with the classifier, which are classifier settings that are optimized automatically by the machine learning algorithm during the training of the model. For example, the variable coefficients in linear regression are parameters, because they are attributes of the model that are automatically optimized by the learning algorithm based on the input data.
Validation of the Classifier.
In order to be sure that a classifier will make accurate predictions on unseen data, it is critical to evaluate its performance on data that was not used for constructing the classifier. Often, input data is split into training, validation, and test sets. The training set is used to construct the classifier and learn parameters; the validation set is used to evaluate the classifier’s performance and fine-tune hyperparameters; and the test set, which is only examined at the final stage, is used to evaluate the performance of the classifier. Cross-validation is a popular approach for evaluating classifier performance. In k-fold cross-validation, the input data is split into k pieces (common choices of k are 5 and 10). The classifier is then trained on all but one of the pieces and validated on the final piece. This procedure is repeated until each piece has been left out once. We can evaluate the performance of a classifier based on the number of errors and correctly classified examples. For machine learning algorithms that produce ranked lists of predictions, we can produce a curve, such as a receiver operating characteristic curve or a precision-recall curve, that compares the order of recovery of correct and incorrect examples. Such curves can be used to compare the performance of different classifiers, since the best performing classifiers will assign a high rank to many correct but few incorrect predictions.
When constructing classifiers, care must be taken that the data used to train and evaluate the classifier is similar to the data to which the classifier will be applied. For example, if the drug response classifier above is trained based on adult patients, its performance may be poorer than expected on pediatric patients. It is also important in evaluating the performance of a classifier to ensure that the test set was not used to train parameters or hyperparameters; otherwise the performance of the classifier may be overestimated because the test data was not a truly independent sample. Ideally, just as with prediction models derived using non-machine learning methods, an independent data set is most helpful to assess the true prediction accuracy and validation of a model.
Deep Learning Methods.
One family of machine learning methods that is increasingly finding applications in biology is known as deep learning (reviewed in7). As biological datasets have grown in size, complexity, and number, deep learning methods have increasingly become suitable for addressing problems arising from these datasets. The problems deep learning approaches have been applied to include categorizing medical images, predicting patient outcome from electronic health records, and predicting drug response (reviewed in8). In deep learning, input features are subjected to multiple layers of transformations, in which the outputs of each layer are functions of subsets of the input to that layer (Figure 1c). Although deep learning approaches were first developed in the 1940’s, these approaches are generally most powerful when large numbers of training data points are available.9 Thus, algorithmic approaches similar to those used in early deep learning efforts have proven dramatically more successful as the size of available training datasets has increased. Another factor that has contributed to improvements in the performance of deep learning approaches is the availability of computational platforms that can support larger models.9
Machine Learning Limitations.
A number of challenges are common across many machine learning problems. Firstly, many biological applications of machine learning involve datasets in which each example is associated with many features. However, the presence of a high number of features tends to make problems harder, because few or no training examples may be associated with each combination of features. This difficulty is called the curse of dimensionality. To help address this problem, dimensionality reduction techniques can be applied to combine correlated features or select the most relevant subset of features. Another pitfall is that models with many parameters will sometimes overfit to the training data, meaning that they optimize for performance on the training data in a way that does not generalize to new examples. One approach to ameliorate overfitting is regularization, where additional constraints are added to the model parameters (for example, the model parameters may be constrained to take on smaller values or a subset of parameters may be set to zero to reduce the degrees of freedom of the model). Moreover, while many (and rapidly increasing numbers) of training examples are available in some biological domains, few examples are available in others, making validation datasets difficult to find. Labeled data, for some applications, can be particularly difficult to find. This problem is exacerbated by the presence of barriers to data sharing, which may be due to legal and ethical restrictions on sharing sensitive data, as well as a scientific culture that often does not promote rapid public release of datasets. Finally, machine learning methods vary in the interpretability of the resulting model. Understanding which features in the data drive the model’s predictions is often an important piece of the analysis process to allow the biomedical context to be linked to specific features.
Increasing Availability of High-dimensional Genetic and Phenotypic Data for Machine Learning
Machine learning methods typically perform best when large datasets are available. As in other areas of biology, the availability of large datasets relevant to kidney disease is rapidly expanding. Researchers are increasingly generating large multimodal omics data compendia, including transcriptomic, proteomic, metabolomic, epigenomic, and exome or whole-genome sequencing datasets, now even linked to individual cells in single cell data or spatial context via profiling of tissue sections. As with the omics technologies, the study of high-dimensional datasets from the clinic is also increasingly feasible, including analyses of electronic medical records, high-intensity in-clinic monitoring data from intensive care unit and dialysis settings,10 patient-reported outcomes,11 and data from wearable technologies for capture of vital signs and physical activity.12 This phenotypic data matches the complexity and detail of the omics datasets. Developing approaches to efficiently analyze such datasets, as well as to organize, harmonize, and curate complex, complementary information across many sites and data types, represents a significant and crucial challenge. The use of ontologies, such as the Human Phenotype Ontology, aims to improve clinical data integration by use of standardized vocabulary and hierarchy to describe phenotypic information.13,14
The availability of genetic data has benefited from the rapid progress of nucleic acid sequencing technologies.15 The cost of generating the sequence of a human genome has decreased from ~$2.7 billion for the initial human genome project to less than $1000 using current approaches.16 Improvements in sequencing have led to the development and refinement of a broad spectrum of technologies, including whole-genome sequencing, whole-exome sequencing, bulk RNA sequencing, single-cell sequencing, single nucleus sequencing, identification of physical interactions between chromosome regions, and mapping of methylation sites and histone marks at bulk tissue and single-cell levels.
Evolving sequencing technologies have made it possible to sequence patients at a large scale to diagnose inherited kidney disease (reviewed in17). The earliest association of a specific genetic alteration in humans with kidney disease came in the mid-1980’s with the identification of a genetic marker of autosomal dominant polycystic kidney disease (reviewed in18). Since then, the number of genetic markers and alterations associated with kidney disease has rapidly expanded. For example, in 2010, two risk variants in the Apolioprotein L1 gene (APOL1) were associated with dramatically increased odds of focal segmental glomerulosclerosis and hypertension-associated nephropathy in people with sub-Saharan African ancestry.19,20 In recent years, genome-wide association studies have resulted in the discovery of many additional genomic regions contributing to kidney disease risk (reviewed in21).
Understanding the genetic causes of nephropathies has important clinical implications. A recent study found that exome sequencing of 3000 adult patients with chronic kidney disease led to a specific genetic diagnosis in approximately ten percent of cases.1 For many patients in this cohort, these genetic diagnoses had significant clinical utility, for example by clarifying underlying CKD etiology, suggesting referrals for non-renal disease manifestations or by guiding selection of the most appropriate therapeutic approach. Another recent study identified genetic causes of disease in approximately one third of a cohort of over a hundred pediatric renal transplant patients.22 These studies illustrate the power of a genetic diagnosis to guide clinical management of nephropathies.
However, integrating genetic findings with clinical care poses a significant challenge. As more pathogenic variants are defined, thousands of new genetic tests are increasingly moving into the clinic to inform patient diagnosis, prognosis, and care of diverse diseases.23 The penetrance, underlying biological mechanism, and clinical importance of many of the variants probed by these tests remain unclear, as they are frequently also seen in population level studies in individuals without the disease phenotype. Further work is needed to bridge the gaps between genotype, phenotype, clinical risk assessment, and treatment, and the population level sequencing projects currently ongoing will provide critical datasets for these efforts. Understanding how to utilize knowledge of risk posed by the APOL1 risk variants is a case in point. While the variants confer a significantly increased risk of nephropathy, most people carrying the risk variants will not develop kidney disease. Understanding how APOL1 risk genotypes affect donor risk and allograft survival, for example, and how this should affect transplant guidelines remains an area of active research.17,24 Moreover, as additional pathogenic genetic variants are discovered, reanalysis of sequence data using updated biological knowledge can often result in the discovery of additional causal variants. A recent study that re-analyzed the exomes of 40 individuals with suspected Mendelian disorders but no genetic diagnosis found causative variants in 4 (10%) of the cases based on literature support in work published since the time of the original analysis.25
Beyond sequencing data, a number of other technologies are also producing increasing quantities of phenotypic data which match the complexity and detail of the omics datasets. These datasets will require similar machine learning approaches for knowledge extraction. Imaging datasets, both comprised of images from radiology studies and of kidney biopsy tissue sections, provide important information on the physical characteristics and histological features of specific disease states. Electronic medical records, including high-intensity monitoring from intensive care units or dialysis settings, provide machine-readable clinical information on large patient cohorts. Patient reported outcomes can provide systematic information at a large scale on the subjective experience of individuals living with kidney disease.11 Wearable technologies can provide continuous monitoring of patient characteristics such as blood pressure, heart rhythm, blood glucose, and physical activity, and increasing use of such technologies can provide another important component for understanding renal disease characteristics and outcomes.12
Multi-layered high-dimensional datasets in kidney disease
Several large consortia are beginning to generate multimodal kidney disease datasets at previously unprecedented scales, including genetic and phenotypic data. By recruiting study participants across multiple sites and performing deep molecular and clinical profiling, these consortia allow integrated analyses of the characteristics predictive of disease subtypes, patient drug responses, and patient outcomes. The European Renal Biopsy cDNA Bank (ERCB) consortium is a multicenter European network that has generated microdissected transcriptomic profiles of patients with both common and rare kidney diseases, as well as clinical information on treatment regimens and disease progression over the last two decades.26,27 The NEPTUNE consortium is a multicenter effort dedicated to studying rare glomerular nephropathies, and has generated multilayered data sets including microdissected renal biopsy derived mRNA expression profiles, targeted urine and blood proteomic studies and genetic data, paired with digital pathology, patient reported outcomes, environmental exposures, and longitudinal clinical datasets.28 The CureGN consortium also recruits children and adults with glomerular disease for long term follow-up, collecting longitudinal clinical data, biospecimens, patient reported outcomes and digital kidney biopsy images.29 The Human Heredity and Health in Africa (H3 Africa) consortium is an effort to build African capacity to collect and analyze genomic data in order to understand factors contributing to chronic as well as infectious disease burden in Africa.30 Within the larger H3 Africa effort, the Kidney Disease Research Network focuses on kidney disease in sub-Saharan Africa, identifying clinical and genetic information from a large multinational patient cohort.31 The Kidney Precision Medicine Project (KPMP) aims to permit personalized treatment of patients with chronic kidney disease and acute kidney injury, generating deep genomic profiles (single-cell and microdissected transcriptomic, proteomic, metabolomic, and imaging) profiles of patient biopsies from patients with acute kidney injury and common forms of chronic kidney disease.32 The landmark CKD cohorts in adults (the Chronic Renal Insufficiency Cohort, CRIC33) and children (the Chronic Kidney Disease in Children Study, CKiD34) have rich phenotypic, genetic, and biosamples available. The Pima Indian35 cohort and the TRIDENT36 study both enroll patients with diabetes to assemble multi-layered datasets, including tissue transcriptomics. Together, these consortia and several others pursuing similar work represent extraordinarily rich data resources that can inform machinelearning based approaches to studying kidney disease biology.
Example machine learning approaches to predict effects of genetic variants on phenotype
Extracting biological meaning from the mountain of available data remains challenging. Approaches that can integrate multiple data types to extract biological signal are crucial for efficiently utilizing multimodal datasets. Many such approaches have been developed (reviewed in37–39). A selection of machine learning methods designed to predict phenotype from genotype data are summarized in Table 1. One example of an integrative approach that combines multiple data types to facilitate the interpretation of genotype data is the NetWAS algorithm, which re-prioritizes genes to identify likely causal genes from a genome-wide association (GWAS) study.40 NetWAS uses information from tissue-specific functional networks that quantify the probability that any two genes are functionally related in a specific tissue (e.g. kidney) by integrating the pairwise relationships between genes across thousands of experiments. The network connectivity patterns for genes of interest can be used as features for the NetWAS machine learning algorithm, using genes with marginally significant GWAS hits as positive examples and re-prioritizing all genes in the genome by likely association with the studied phenotype. This approach is especially useful when assessing multiple candidate genes with borderline significance because it uses the tissue specific network connectivity to improve the accuracy of disease-gene associations (see Supplemental Materials for use example).
Table 1:
Comparison of selected machine learning methods to predict phenotypic implications of genomic variants
| Method name | Summary | ML algorithm used | Tissue specific predictions? | Link |
|---|---|---|---|---|
| Basset | Predict DNA accessibility of sequence variants | Deep learning | Yes | github.com/davek44/Basset |
| CADD | Predict impact of coding or noncoding variant based on multiple annotation types | SVM (original version); logistic regression (updated version) | No | cadd.gs.washington.edu |
| ExPecto | Predict tissue-specific transcriptional impact of noncoding variants | Deep learning | Yes | https://hb.flatironinstitute.org/expecto/ |
| GWAVA | Identify which noncoding mutations are likely to be disease-causing | Random forest | No | www.sanger.ac.uk/sanger/StatGen_Gwava |
| LINSIGHT | Predict impact of noncoding variant by integrating evolutionary and genomic information | Generalized linear model; probabilistic model of sequence evolution | No (but applied to examine tissue-specific evolutionary properties of enhancers). | http://compgen.cshl.edu/~yihuang/LINSIGHT/ |
| NetWAS | Re-prioritize sub-significant GWAS hits based on functional network connectivity to identify disease-related genes | SVM | Yes | hb.flatironinstitute.org/netwas |
Given the rapidly increasing availability of sequence data and the biological and clinical importance of understanding the significance of genetic variants, approaches that can predict the significance of specific genetic alterations are critical. A large number of machine learning methods have been developed to predict the impact of coding and noncoding mutations on protein structure and function.41–46 One commonly applied approach to prioritize individual candidate variants (for example, GWAS hits or observed alterations in a patient’s genome) is the CADD framework, an integrative approach which uses a machine learning model to combine multiple classes of annotations into a single variant effect score.41,47 CADD includes both features that are specific to coding sequences and features that are informative for noncoding changes. Another approach for predicting the impact of noncoding mutations is the GWAVA algorithm, which integrates multiple classes of annotations using a random forest framework trained to distinguish between disease-causing and background noncoding mutations.48 The LINSIGHT algorithm, which integrates evolutionary information with functional genomics data, can also predict the effect of noncoding changes in the genome.49
More recently, a family of deep-learning based approaches have been developed to understand the impact of noncoding variants in the genome. The DeepSEA algorithm, which applies a deep learning-based method to predict the biological impact of sequence variants, is the first approach to predict the effect of a noncoding mutation based only on sequence input.50 DeepSEA assesses whether a genetic change is likely to alter the site’s transcription factor binding, chromatin mark, or chromatin accessibility profiles. For example, Zhou et al. applied DeepSEA to examine the transcriptional impact of noncoding mutations in individuals with Autism Spectrum Disorder.51 They found that probands had de novo noncoding changes predicted by the model to have significantly larger effects than noncoding changes in unaffected siblings. They also experimentally validated using cell-based assays the transcriptional effect predictions for prioritized variants.
DeepSEA as well as other approaches can also be applied to predict the effects of variants in a tissue-specific manner.50,52,53 The Basset framework, which uses deep learning to predict the effect of noncoding changes on chromatin accessibility, was applied to predict DNA accessibility across 164 cell types.52 The DeepWAS framework uses the DeepSEA deep learning network architecture to predict the effect of genetic variants in specific cell types.54 Other approaches utilize tissue-informed deep-learning frameworks for tasks such as predicting DNA methylation or alternative splicing.55–57 Another example of an algorithm that can predict the tissue-specific transcriptional effects of a genetic variant is the ExPecto framework, which utilizes a deep learning model to predict tissue-specific transcriptional impact using only genetic sequence of interest as input (Figure 2).58 The ExPecto algorithm consists of three steps. First, a deep learning model is trained to predict chromatin marks, transcription factor binding sites, and DNA accessibility profiles across sliding windows of the input sequence. Next, these features are combined across sequence windows and transformed into a reduced feature set (smaller number of features derived by processing the predicted chromatin marks, transcription factor binding sites, and chromatin accessibility profiles). The reduced feature set is then used as input to a regularized linear model to predict tissue-specific gene expression (see Supplemental Materials for use example).
Figure 2:
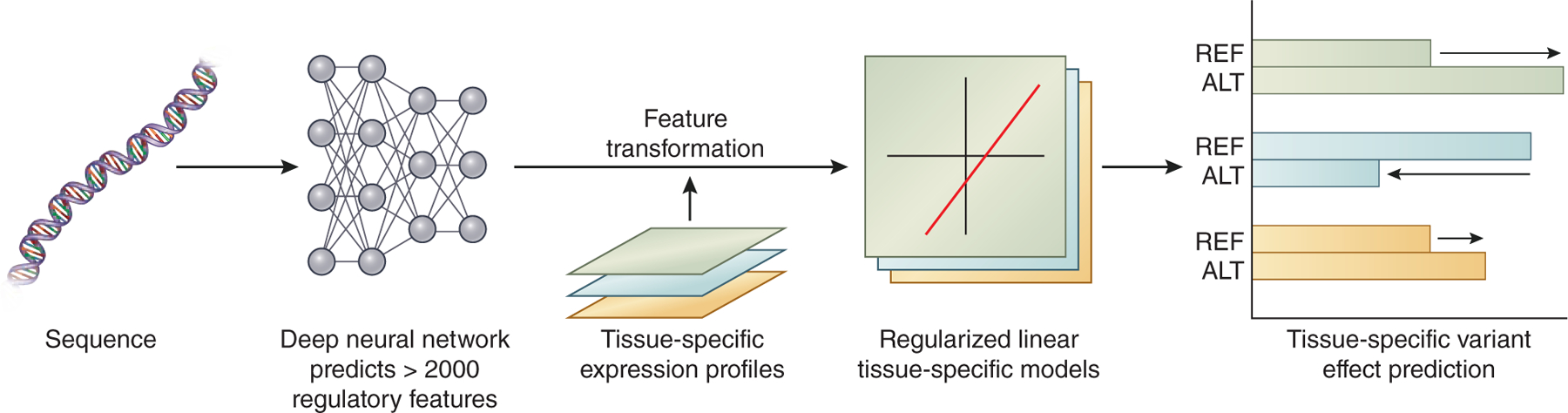
The ExPecto framework predicts tissue-specific variant effects given sequence data alone. To this end, it incorporates a deep neural network which predicts over 2000 regulatory features. After transforming and combining these regulatory features and integrating with tissue-specific expression profiles, the model generates regularized linear tissue-specific models, which can be used to predict the gene expression changes caused by individual sequence variants.
A variety of other approaches beyond machine learning can also help assess the significance of genetic sequence changes, including statistical methods to prioritize GWAS hits and methods examining the overlap between genetic variants and functional or conserved elements such as regulatory DNA sequence elements. For example, highly penetrant and pathogenic DNA changes are unlikely to be found frequently in large sets of sequence data from people unaffected by the condition of interest, so examining the frequency of a mutation in large genetic databases can help assess its pathogenicity.23,59,60 Large databases of variant frequencies, such as the gnomAD database, can facilitate such assessments.59,61
Examples of machine learning approaches in kidney disease using non-genomic data domains to predict phenotype
Many groups have applied machine learning algorithms in the renal domain, using a variety of high dimensional clinical and imaging datasets to improve disease classification, predict progression and inform therapeutic decisions. A few examples are discussed below.
Manually analyzing and annotating kidney images is time-consuming and must be performed by a specialist, so approaches that can automatically extract relevant data from images for focused review by radiologists are of great utility. Sharma and colleagues applied a deep learning-based method to segment computed tomography images from patients with polycystic kidney disease in order to automatically measure total kidney volume.62 Bukowy and colleagues used a convolutional neural net framework to identify glomeruli in rat renal tissue images.63 Park and colleagues trained a deep convolutional neural network to estimate glomerular filtration rate from single-photon emission computed tomography (SPECT)/computed tomography images.64 Hermsen and colleagues used a convolutional neural network approach to automatically identify ten structures from kidney transplant biopsy images.65 Machine learning algorithms have also been developed to categorize digital images of renal biopsy tissue. For example, a recent method applied a binary nearest-neighbors classifier to classify tissue based on whether or not it contained proliferative glomerular lesions.66 Another approach trained a convolutional neural network to identify glomeruli as either healthy or sclerotic in order to automatically assess the quality of potential donor kidneys.67 Ginley and colleagues developed a computational framework to segment and classify severity of diabetic nephropathy. 68 Finally, imaging data can also be used to predict clinical outcome. One study using convolutional neural networks to characterize trichrome-stained images from renal biopsy could outperform pathologist-estimated interstitial fibrosis in predicting 1-, 3- and 5-year survival.69
A number of studies have used machine learning methods to predict kidney transplant graft survival from clinical variables. A variety of methods, including Bayesian network models,70 logistic regression,71 ensemble methods,72,73 and decision tree-based approaches74 have been applied to this problem. These approaches predicted a variety of endpoints, such as graft survival after one year, graft survival after three years, or graft survival after ten years. A recent study also compared the efficacy of multiple machine learning methods at predicting the appropriate dose of tacrolimus for renal post-transplant patients based on clinical and genetic data.75
Several groups have investigated using machine learning approaches to predict disease onset or progression. Investigators have applied machine learning methods to patients’ electronic health records to predict which hospitalized patients are likely to be diagnosed with acute kidney injury and would benefit from early intervention.10,76–78 For example, a recent study used a deep learning approach to predict acute kidney injury onset in hospitalized patients using information from electronic health records.82 Another recent study applied a random forest regression-based approach to predict future eGFR in individuals with chronic kidney disease using electronic health record data collected in primary care clinics.79 Nadkarni and colleagues used supervised learning methods including a random forest classifier to predict rapid decline in kidney function from biomarkers combined with longitudinal clinical variables in individuals at high risk for kidney disease.80 Other studies have used urine biomarkers to predict acute kidney injury risk or to distinguish between subtypes of chronic kidney disease.81,82
Finally, machine learning algorithms can be used to inform treatment decisions. For example, mature AI algorithms are already used in clinical management to tailor erythropoietin dosing in chronic hemodialysis patients.83 These algorithms, integrated into the EHR, generate individualized erythropoietin dosing recommendations and result not only in lower overall dose levels, but also lower transfusion rates.
Together, these studies demonstrate the promise of applying machine learning approaches to understand kidney disease patient characteristics and outcomes. Machine learning methods will need to be developed that can integrate newly available data modalities to address pressing questions facing nephrologists, such as identifying the disease subtype, likely patient outcome, and optimal treatment plan given a patient’s clinical, genetic, and genomic profiles (Figure 3). As dramatically larger datasets and new data types are becoming available, applying machine learning frameworks to the areas of data-rich science in kidney disease will become increasingly feasible and provide a rich source of biological insight into renal disease.
Figure 3:
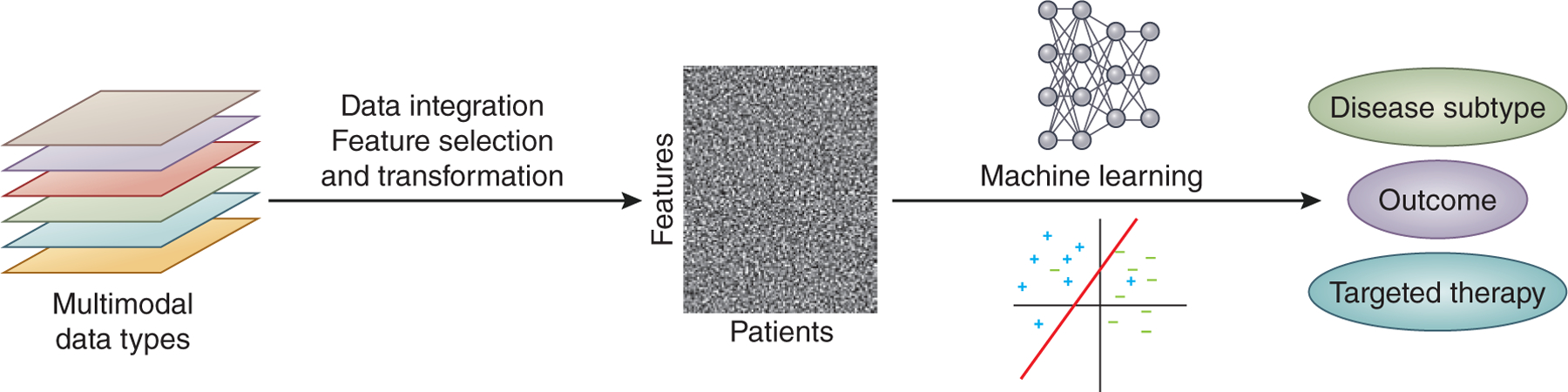
Developing methods to integrate multimodal data types and apply machine learning approaches to predict disease subtype, outcome, and patient-targeted therapies is one of the current challenges in kidney genomics.
Conclusions:
Linking the increasing quantity and diverse modalities of kidney disease data via powerful new machine learning frameworks will enable a wide range of questions in kidney disease to be addressed. An immediate application is the deployment of recently developed frameworks that can predict the effects of coding and noncoding genetic mutations from kidney disease GWAS hits and population level genetic databases. Machine learning approaches will need to be adapted to leverage rich multimodal datasets from large consortia in order to understand disease subtypes, biological mechanisms, patient outcomes, and optimal therapies. Understanding the methods, benefits and limitations of the various machine learning algorithms is critical to interpreting and applying the results to the clinical setting. Machine learning approaches can increase the analytical power in the rapid expansion of available multi-layered datasets. They can represent important tools for gaining insight into the molecular mechanisms underlying renal disease states and for improving the standard of clinical care, assisting but not replacing the scientific creativity of the kidney disease researcher.
Supplementary Material
Acknowledgements:
The authors thank Aaron K. Wong for feedback on the manuscript. LHM is supported by the National Institutes of Health (NIH)/National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (K08 DK115891). This work is supported by NIH/NIDDK grants U24DK100845, UGDK114907, and U2CDK114886 and NIH grant UH3TR002158 to OGT. OGT is a senior fellow of the Genetic Networks program of the Canadian Institute for Advanced Research (CIFAR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures:
All authors reported no financial interest related to the information contained in this manuscript.
Supplementary Material
Description of how to access and use the NetWAS and ExPecto resources, including website, description of input data and output interpretation.
Supplementary information is available at Kidney International’s website.
References:
- 1.Groopman EE, Marasa M, Cameron-Christie S et al. Diagnostic utility of exome sequencing for kidney disease. N. Engl. J. Med 2019; 380: 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Connaughton DM, Hildebrandt F. Personalized medicine in chronic kidney disease by detection of monogenic mutations. Nephrol. Dial. Transplant 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torres R, Olson E. AI: what have you done for us lately? J. Am. Soc. Nephrol 2018; 29: 2031–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Susztak K, Böttinger EP. Diabetic nephropathy: a frontier for personalized medicine. J. Am. Soc. Nephrol 2006; 17: 361–367. [DOI] [PubMed] [Google Scholar]
- 5.Wu H, Humphreys BD. The promise of single-cell RNA sequencing for kidney disease investigation. Kidney Int. 2017; 92: 1334–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saez-Rodriguez J, Rinschen MM, Floege J et al. Big science and big data in nephrology. Kidney Int. 2019; 95: 1326–1337. [DOI] [PubMed] [Google Scholar]
- 7.LeCun Y, Bengio Y, Hinton G. Deep learning. Nature 2015; 521: 436–444. [DOI] [PubMed] [Google Scholar]
- 8.Ching T, Himmelstein DS, Beaulieu-Jones BK et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 2018; 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goodfellow I, Bengio Y, Courville A. Deep Learning (adaptive Computation And Machine Learning). The Mit Press; 2016:775. [Google Scholar]
- 10.Koyner JL, Carey KA, Edelson DP et al. The development of a machine learning inpatient acute kidney injury prediction model. Crit. Care Med. 2018; 46: 1070–1077. [DOI] [PubMed] [Google Scholar]
- 11.Tang E, Bansal A, Novak M et al. Patient-Reported Outcomes in Patients with Chronic Kidney Disease and Kidney Transplant-Part 1. Front Med (Lausanne) 2017; 4: 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wieringa FP, Broers NJH, Kooman JP et al. Wearable sensors: can they benefit patients with chronic kidney disease? Expert Rev. Med. Devices 2017; 14: 505–519. [DOI] [PubMed] [Google Scholar]
- 13.Köhler S, Øien NC, Buske OJ et al. Encoding Clinical Data with the Human Phenotype Ontology for Computational Differential Diagnostics. Curr. Protoc. Hum. Genet 2019; 103: e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Groza T, Köhler S, Moldenhauer D et al. The human phenotype ontology: semantic unification of common and rare disease. Am. J. Hum. Genet 2015; 97: 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet 2016; 17: 333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The Cost of Sequencing a Human Genome - National Human Genome Research Institute (NHGRI).
- 17.Groopman EE, Rasouly HM, Gharavi AG. Genomic medicine for kidney disease. Nat. Rev. Nephrol 2018; 14: 83–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Devuyst O, Knoers NVAM, Remuzzi G et al. Rare inherited kidney diseases: challenges, opportunities, and perspectives. Lancet 2014; 383: 1844–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freedman BI, Kopp JB, Langefeld CD et al. The apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. J. Am. Soc. Nephrol 2010; 21: 1422–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Genovese G, Friedman DJ, Ross MD et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010; 329: 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wuttke M, Köttgen A. Insights into kidney diseases from genome-wide association studies. Nat. Rev. Nephrol 2016; 12: 549–562. [DOI] [PubMed] [Google Scholar]
- 22.Mann N, Braun DA, Amann K et al. Whole-Exome Sequencing Enables a Precision Medicine Approach for Kidney Transplant Recipients. J. Am. Soc. Nephrol 2019; 30: 201–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diao JA, Kohane IS, Manrai AK. Biomedical informatics and machine learning for clinical genomics. Hum. Mol. Genet 2018; 27: R29–R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newell KA, Formica RN, Gill JS et al. Integrating APOL1 gene variants into renal transplantation: considerations arising from the american society of transplantation expert conference. Am. J. Transplant 2017; 17: 901–911. [DOI] [PubMed] [Google Scholar]
- 25.Wenger AM, Guturu H, Bernstein JA et al. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet. Med 2017; 19: 209–214. [DOI] [PubMed] [Google Scholar]
- 26.Schmid H, Cohen CD, Henger A et al. Gene expression analysis in renal biopsies. Nephrology Dialysis Transplantation 2004; 19: 1347–1351. [DOI] [PubMed] [Google Scholar]
- 27.Ju W, Eichinger F, Bitzer M et al. Renal gene and protein expression signatures for prediction of kidney disease progression. Am. J. Pathol 2009; 174: 2073–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gadegbeku CA, Gipson DS, Holzman LB et al. Design of the Nephrotic Syndrome Study Network (NEPTUNE) to evaluate primary glomerular nephropathy by a multidisciplinary approach. Kidney Int. 2013; 83: 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mariani LH, Bomback AS, Canetta PA et al. Curegn study rationale, design, and methods: establishing a large prospective observational study of glomerular disease. Am. J. Kidney Dis 2019; 73: 218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.H3Africa Consortium, Rotimi C, Abayomi A et al. Research capacity. Enabling the genomic revolution in Africa. Science 2014; 344: 1346–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Osafo C, Raji YR, Burke D et al. Human Heredity and Health (H3) in Africa Kidney Disease Research Network: A Focus on Methods in Sub-Saharan Africa. Clin. J. Am. Soc. Nephrol 2015; 10: 2279–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Norton JM, Ketchum CJ, Narva AS et al. Complementary Initiatives from the NIDDK to Advance Kidney Health. Clin. J. Am. Soc. Nephrol 2017; 12: 1544–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feldman HI, Appel LJ, Chertow GM et al. The chronic renal insufficiency cohort (CRIC) study: design and methods. J. Am. Soc. Nephrol 2003; 14: S148–53. [DOI] [PubMed] [Google Scholar]
- 34.Warady BA, Abraham AG, Schwartz GJ et al. Predictors of rapid progression of glomerular and nonglomerular kidney disease in children and adolescents: the chronic kidney disease in children (ckid) cohort. Am. J. Kidney Dis 2015; 65: 878–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pavkov ME, Knowler WC, Hanson RL et al. Predictive power of sequential measures of albuminuria for progression to ESRD or death in Pima Indians with type 2 diabetes. Am. J. Kidney Dis 2008; 51: 759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Transformative Research in Diabetic Nephropathy - Full Text View - ClinicalTrials.gov.
- 37.Ritchie MD, Holzinger ER, Li R et al. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet 2015; 16: 85–97. [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Wu F-X, Ngom A. A review on machine learning principles for multi-view biological data integration. Brief. Bioinformatics 2018; 19: 325–340. [DOI] [PubMed] [Google Scholar]
- 39.Yao V, Wong AK, Troyanskaya OG. Enabling Precision Medicine through Integrative Network Models. J. Mol. Biol 2018; 430: 2913–2923. [DOI] [PubMed] [Google Scholar]
- 40.Greene CS, Krishnan A, Wong AK et al. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet 2015; 47: 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kircher M, Witten DM, Jain P et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 2014; 46: 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015; 31: 761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shihab HA, Rogers MF, Gough J et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 2015; 31: 1536–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet 2013; Chapter 7: Unit7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saunders CT, Baker D. Evaluation of structural and evolutionary contributions to deleterious mutation prediction. J. Mol. Biol 2002; 322: 891–901. [DOI] [PubMed] [Google Scholar]
- 46.Fariselli P, Martelli PL, Savojardo C et al. INPS: predicting the impact of non-synonymous variations on protein stability from sequence. Bioinformatics 2015; 31: 2816–2821. [DOI] [PubMed] [Google Scholar]
- 47.Rentzsch P, Witten D, Cooper GM et al. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019; 47: D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ritchie GRS, Dunham I, Zeggini E et al. Functional annotation of noncoding sequence variants. Nat. Methods 2014; 11: 294–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang Y-F, Gulko B, Siepel A. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat. Genet 2017; 49: 618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 2015; 12: 931–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou J, Park C, Theesfeld C et al. Whole-genome deep learning analysis reveals causal role of noncoding mutations in autism. BioRxiv 2018. [Google Scholar]
- 52.Kelley DR, Snoek J, Rinn JL. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 2016; 26: 990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kelley DR, Reshef YA, Bileschi M et al. Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res. 2018; 28: 739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eraslan G, Arloth J, Martins J et al. DeepWAS: Directly integrating regulatory information into GWAS using deep learning supports master regulator MEF2C as risk factor for major depressive disorder. BioRxiv 2016. [Google Scholar]
- 55.Angermueller C, Lee HJ, Reik W et al. DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol. 2017; 18: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leung MKK, Xiong HY, Lee LJ et al. Deep learning of the tissue-regulated splicing code. Bioinformatics 2014; 30: i121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019; 176: 535–548.e24. [DOI] [PubMed] [Google Scholar]
- 58.Zhou J, Theesfeld CL, Yao K et al. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet 2018; 50: 1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lek M, Karczewski KJ, Minikel EV et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Minikel EV, Vallabh SM, Lek M et al. Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med 2016; 8: 322ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karczewski KJ, Francioli LC, Tiao G et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv 2019. [Google Scholar]
- 62.Sharma K, Rupprecht C, Caroli A et al. Automatic Segmentation of Kidneys using Deep Learning for Total Kidney Volume Quantification in Autosomal Dominant Polycystic Kidney Disease. Sci. Rep 2017; 7: 2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bukowy JD, Dayton A, Cloutier D et al. Region-Based Convolutional Neural Nets for Localization of Glomeruli in Trichrome-Stained Whole Kidney Sections. J. Am. Soc. Nephrol 2018; 29: 2081–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park J, Bae S, Seo S et al. Measurement of Glomerular Filtration Rate using Quantitative SPECT/CT and Deep-learning-based Kidney Segmentation. Sci. Rep 2019; 9: 4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hermsen M, de Bel T, den Boer M et al. Deep Learning-Based Histopathologic Assessment of Kidney Tissue. J. Am. Soc. Nephrol 2019; 30: 1968–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barros GO, Navarro B, Duarte A et al. PathoSpotter-K: A computational tool for the automatic identification of glomerular lesions in histological images of kidneys. Sci. Rep 2017; 7: 46769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marsh JN, Matlock MK, Kudose S et al. Deep learning global glomerulosclerosis in transplant kidney frozen sections. IEEE Trans. Med. Imaging 2018; 37: 2718–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ginley B, Lutnick B, Jen K-Y et al. Computational segmentation and classification of diabetic glomerulosclerosis. J. Am. Soc. Nephrol 2019; 30: 1953–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kolachalama VB, Singh P, Lin CQ et al. Association of pathological fibrosis with renal survival using deep neural networks. Kidney Int. Rep 2018; 3: 464–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brown TS, Elster EA, Stevens K et al. Bayesian modeling of pretransplant variables accurately predicts kidney graft survival. Am. J. Nephrol 2012; 36: 561–569. [DOI] [PubMed] [Google Scholar]
- 71.Goldfarb-Rumyantzev AS, Scandling JD, Pappas L et al. Prediction of 3-yr cadaveric graft survival based on pre-transplant variables in a large national dataset. Clin. Transplant 2003; 17: 485–497. [DOI] [PubMed] [Google Scholar]
- 72.Yoo KD, Noh J, Lee H et al. A machine learning approach using survival statistics to predict graft survival in kidney transplant recipients: A multicenter cohort study. Sci. Rep 2017; 7: 8904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mark E, Goldsman D, Gurbaxani B et al. Using machine learning and an ensemble of methods to predict kidney transplant survival. PLoS ONE 2019; 14: e0209068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Greco R, Papalia T, Lofaro D et al. Decisional trees in renal transplant follow-up. Transplant. Proc 2010; 42: 1134–1136. [DOI] [PubMed] [Google Scholar]
- 75.Tang J, Liu R, Zhang Y-L et al. Application of Machine-Learning Models to Predict Tacrolimus Stable Dose in Renal Transplant Recipients. Sci. Rep 2017; 7: 42192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kate RJ, Perez RM, Mazumdar D et al. Prediction and detection models for acute kidney injury in hospitalized older adults. BMC Med. Inform. Decis. Mak 2016; 16: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Davis SE, Lasko TA, Chen G et al. Calibration drift in regression and machine learning models for acute kidney injury. J. Am. Med. Inform. Assoc 2017; 24: 1052–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tomašev N, Glorot X, Rae JW et al. A clinically applicable approach to continuous prediction of future acute kidney injury. Nature 2019; 572: 116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhao J, Gu S, McDermaid A. Predicting outcomes of chronic kidney disease from EMR data based on Random Forest Regression. Math. Biosci 2019; 310: 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nadkarni GN, Fleming F, McCullough JR et al. Prediction of rapid kidney function decline using machine learning combining blood biomarkers and electronic health record data. BioRxiv 2019. [Google Scholar]
- 81.Kashani K, Al-Khafaji A, Ardiles T et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit. Care 2013; 17: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fernando BNTW, Alli-Shaik A, Hemage RKD et al. Pilot study of renal urinary biomarkers for diagnosis of CKD of uncertain etiology. Kidney Int. Rep 2019; 4: 1401–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brier ME, Gaweda AE. Artificial intelligence for optimal anemia management in end-stage renal disease. Kidney Int. 2016; 90: 259–261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.