

Abstract
The stabilization of learned information into long-term memories requires new gene expression. CREB binding protein (CBP) is a coactivator of transcription that can be independently regulated in neurons. CBP functions both as a platform for recruiting other required components of the transcriptional machinery and as a histone acetyltransferase (HAT) that alters chromatin structure. To dissect the chromatin remodeling versus platform function of CBP or the developmental versus adult role of this gene, we generated transgenic mice that express CBP in which HAT activity is eliminated. Acquisition of new information and short-term memory is spared in these mice, while the stabilization of short-term memory into long-term memory is impaired. The behavioral phenotype is due to an acute requirement for CBP HAT activity in the adult as it is rescued by both suppression of transgene expression or by administration of the histone deacetylase inhibitor Trichostatin A (TSA) in adult animals.
Introduction
New information is stored as a transient short-term memory lasting minutes to hours, which can be stabilized into long-term memory (LTM) lasting years (McGaugh and Hertz, 1972; Squire, 1987). A variety of inhibitors of protein and RNA synthesis have been shown to effectively block long-term memory without altering short-term memory (Andrew, 1980; Davis and Squire, 1984). Environmental stimuli or high levels of neuronal activity are known to induce a variety of immediate-early genes such as c-fos, jun, and zif268 in many brain areas (Tischmeyer and Grimm, 1999; Worley et al., 1990). Recent genetic studies in mice suggest that zif268, creb, and c-fos may be involved in memory formation and consolidation (Bourtchuladze et al., 1994; Fleischmann et al., 2003; Jones et al., 2001; Josselyn et al., 2001; Kida et al., 2002; Pittenger et al., 2002). Thus, regulatory mechanisms directing transcription subsequent to learning-induced molecular changes in neurons play a pivotal role in the conversion of short- to long-term memory.
Regulation of gene expression requires not only activation of transcription factors but also recruitment of multifunctional coactivators that are independently regulated and utilized in a cell- and promoter-specific fashion to stimulate or repress transcription (Rosenfeld and Glass, 2001). Dynamic changes in the organization of chromatin control gene expression and histone acetylation is one mechanism for the local and global control of chromatin structure. Recent studies have shown that chromatin acetylation at a region of ongoing transcription is essential for high-level gene expression (Fischle et al., 2003; Neely and Workman, 2002). CBP and its homolog p300 are transcriptional coactivators (Chrivia et al., 1993; Eckner et al., 1994) that interact with multiple transcriptional regulators and facilitate the assembly of the basic transcriptional machinery (Janknecht, 2002). In addition to serving as molecular scaffolds, CBP and p300 each possess intrinsic histone acetyltransferase activities (HAT) (Ogryzko et al., 1996) that can be specifically and directly inhibited by phosphorylation or by association with viral proteins (Janknecht, 2002; Yuan et al., 2002). While CBP’s function as a platform to recruit other required coactivators appears to be indispensable, the requirement for HAT activity is transcription unit specific and may depend on the structure of chromatin at a specific locus (Korzus et al., 1998; Puri et al., 1997). Recently, studies have shown that CBP’s ability to function as a transcriptional coactivator is regulated by synaptic activity in neurons (Guan et al., 2002; Hardingham et al., 1999; Hu et al., 1999; Impey et al., 2002). Calcium influx via NMDA receptors or voltage-sensitive calcium channels induces CBP-dependent transactivation in cultured hippocampal and cortical neurons (Hardingham et al., 1999; Hu et al., 1999; Impey et al., 2002). CBP is also a direct target for calcium influx-induced CaM kinase IV in cultured neurons resulting in phosphorylation at S301, which is required for NMDA-induced transcription (Impey et al., 2002). Although the question of whether NMDA-dependent transcription requires CBP HAT activity has not been addressed, these findings suggest that in addition to the well-characterized function of transcription factors such as CREB in NMDA-induced gene expression, coactivators such as CBP are regulated by a separate pathway that plays a critical role in the activation of gene expression.
A role for CBP in higher cognitive function is suggested by the finding that Rubinstein-Taybi syndrome (RTS), a disorder in humans characterized by growth and psychomotor delay, abnormal gross anatomy, and severe mental retardation (Hennekam et al., 1990; Rubinstein and Taybi, 1963), is caused by heterozygous mutations at the CBP locus (Petrij et al., 1995). However, because of the complexity of developmental abnormalities and possible genetic compensation associated with this congenital disorder, it is difficult to establish a direct role for CBP in cognitive function in the adult brain.
While there has been extensive research into the molecular basis of CREB/CBP actions on gene expression and separate research on the role of CREB in memory, a fundamental question that has escaped resolution is whether the role of CBP as a platform protein or its chromatin remodeling histone acetyltransferase activity is the key component of its biological function. To test the function of CBP in the adult brain and specifically the role of CBP-mediated HAT activity, we generated transgenic mice carrying a dominant-negative CBP transgene that specifically blocks HAT activity. The mice were generated using the inducible tet system to allow for expression only in adult animals. We found that the mutant mice exhibit a long-term memory deficit while the encoding of new information and short-term memory are normal. This behavioral phenotype is reversible when we turn off transgene expression or when mutant animals are trained after administration of a histone deacetylase inhibitor. These results establish HAT activity as a critical component of long-term memory formation.
Results
Generation of CBP{HAT‒} Mutant Mice
On the basis of mutagenesis studies demonstrating that single amino acid substitutions in the acetyl coenzyme A (acetyl-CoA) binding domain of acetyltransferases result in loss of enzymatic activity (Korzus et al., 1998; Neuwald and Landsman, 1997), we have generated a CBP mutant (CBP{HAT‒}) harboring a substitution mutation of two conserved residues (Y1540/F1541 to A1540/A1541). This mutant has no intrinsic HAT activity (Figure 2A) but retains all protein-protein interaction domains.
Figure 2. CBP Acetyltransferase Activity Is Required for c-fos Gene Expression.
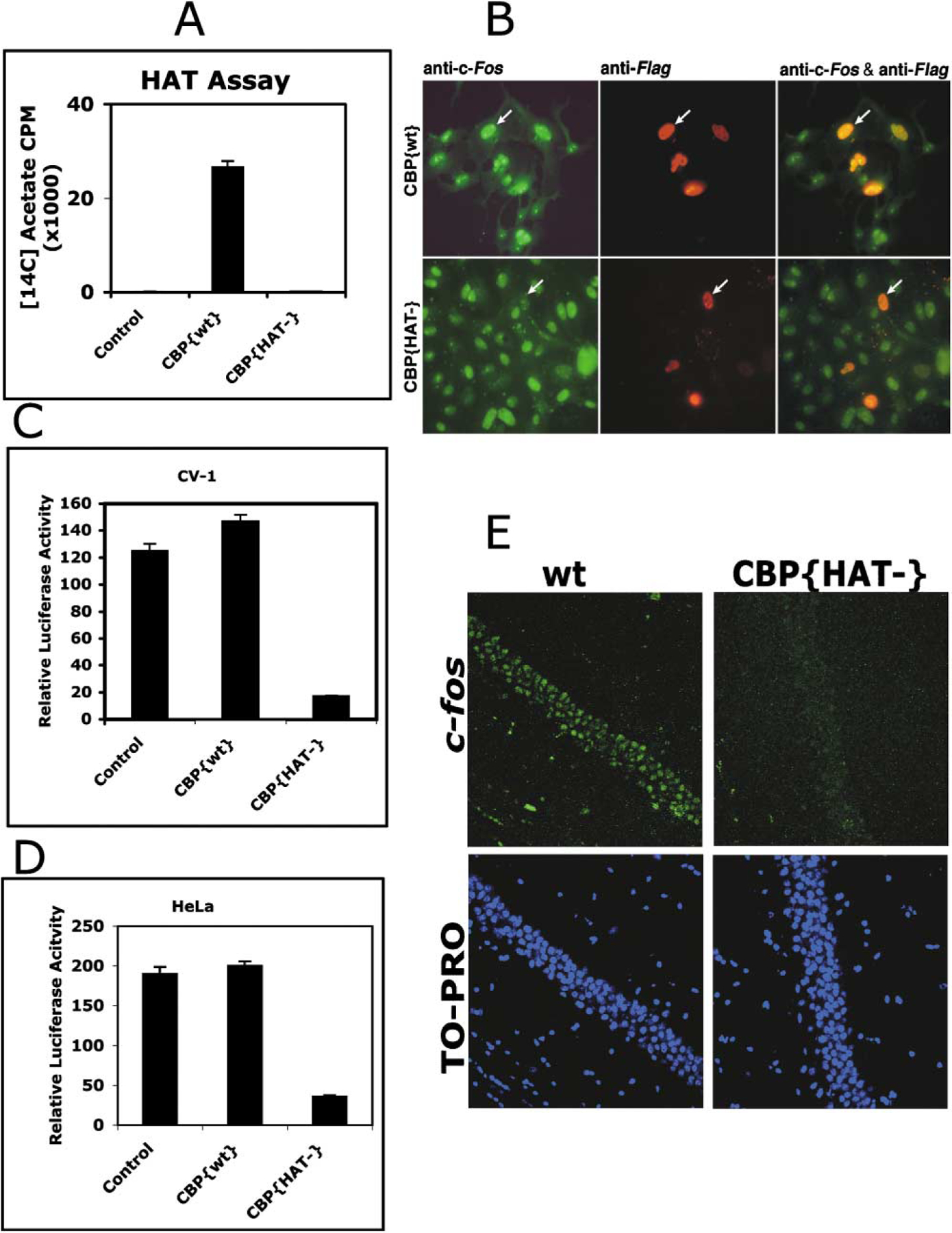
(A) Generation of substitution mutation in the mouse CBP (Y1540/F1541 to 1540/1541) abolished detectable acetylation of histones using[14C]acetyl-CoA as a substrate. Histone acetyltransferase activity was determined by liquid HAT assay (see Experimental Procedures) using purified CBP{wt} or CBP{HAT‒} proteins.
(B) CBP{HAT‒} expression blocks c-fos gene expression in vivo. CV-1 cells were transfected with Flag-tagged CBP wild-type or Flag-tagged CBP{HAT‒} expressing vectors. After 24 hr, cells were immunostained for c-fos protein (green) and for wild-type or mutant CBP proteins, both tagged with the Flag epitope (red). Left panels show immunostaining for endogenous c-fos protein expression. Middle panels show cells expressing transfected CBP{wt} or CBP{HAT‒}. Right panels show overlap.
(C and D) Transiently expressed CBP{HAT‒} in CV-1 (C) or HeLa (D) cells efficiently reduced transcription of the c-fos promoter-driven luciferase.
(E) Physiological levels of c-fos expression is reduced in CBP{HAT‒}/Tg-ON. Fresh brain sections were prepared from wt and CBP{HAT‒}/Tg-ON animals expressing the transgene for 7 days. Rabbit anti-c-fos antibodies and Alexa488-goat anti-rabbit IgG were used to detect the physiological level of c-fos expression. Specimens were counterstained with selective nuclei fluorescent marker TO-PRO3 Iodine and analyzed using laser scanning confocal microscope Olympus BX61 (40× oil). Specimens are from animals exhibiting representative levels of c-fos expression for the CBP{HAT‒}/Tg-ON and wild-type littermate mice.
In order to target CBP{HAT‒} to temporal lobe in adult animals, we generated bitransgenic mutant mice referred to as CBP{HAT‒} mice (Figure 1A). The first transgene used the CamKII promoter to drive expression of the tetracycline transactivator (tTA) into forebrain neurons (Mayford et al., 1996). The second transgene carried CBP{HAT‒} (Figure 2A) driven by the tet-O-promoter. These two transgenic lines were crossed to obtain double transgenic mice expressing CBP{HAT‒} in forebrain neurons under doxycycline (Dox) control (Figure 1A). Histological analysis of CBP{HAT‒} mice revealed that CBP{HAT‒} expression is limited to the hippocampal formation (both CA1 neurons and dentate gyrus), caudate putamen, and at lower level in the neocortex (Figure 1B). Reverse transcription followed by PCR analysis performed on total RNA isolated from the entire hippocampus (Figure 1C, lines 1 and 2) or CA1 region (Figure 1C, lines 3 and 4) shows that expression of CBP{HAT‒} mRNA is tightly regulated by Dox and that the level of expression of wild-type CBP is not altered in CBP{HAT‒} mice. CBP wild-type and CBP{HAT‒} expression in CBP{HAT‒} mice was compared using reverse transcription followed by real-time PCR analysis as described in the Experimental Procedures. Analysis of total RNA isolated from whole hippocampus of CBP{HAT‒}/Tg-ON mice expressing the transgene for 7 days revealed presence of CBP{HAT‒} at 32.4% (SEM ± 1) of wild-type CBP. Considering that only a fraction of all cell types in the hippocampus express the transgene while CBP expression is ubiquitous, we estimate that the level of CBP{HAT‒} relative to CBPwt will be much higher in the individual expressing neurons.
Figure 1. Generation of CBP Mutant Mice.
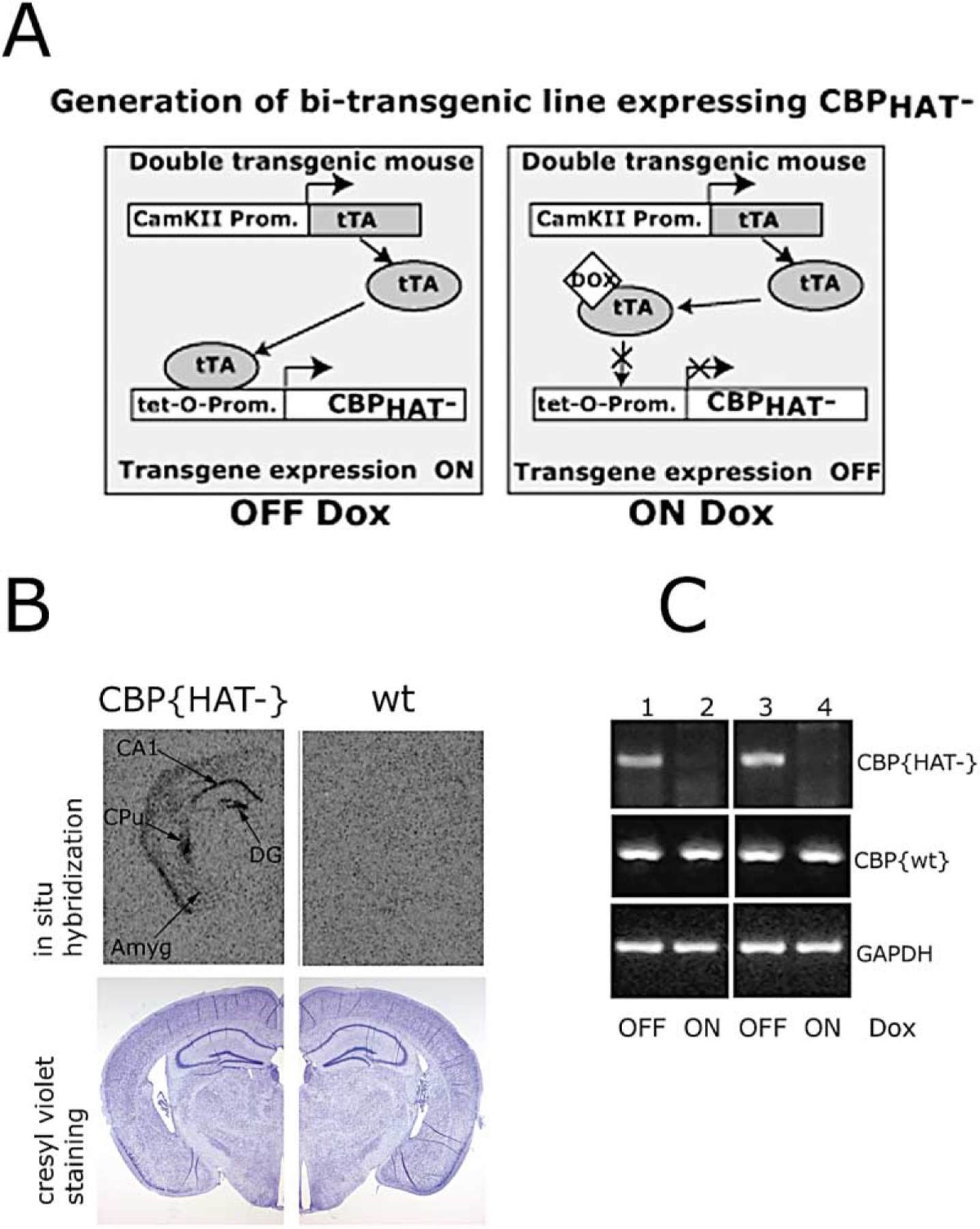
(A) Schematic of the generation of bitransgenic mice expressing CBP{HAT‒}.
(B) Coronal brain sections of CBP{HAT‒} mice and control wild-type (wt) littermates. Top: in situ analysis of CBP{HAT‒} transgene expression using 35S-labeled oligo probe. The transgene expression has been detected in CA1 and dentate gyrus fields of the hippocampus, caudate putamen, and at a low level throughout the neocortex. Bottom: coronal brain sections (10 µm) of CBP{HAT‒} and wt mice corresponding to top panels and stained with cresyl violet showed no alterations in gross neuroanatomy. Abbreviations: Amyg, amygdala; CA1, pyramidal cell field of hippocampus; DG, dentate gyrus; CPu, caudate putamen.
(C) RT-PCR analysis showed that the expression of CBP{HAT‒} transgene is tightly controlled by doxycycline. First strand cDNA was generated by reverse transcription followed by PCR analysis of CBP{HAT‒}, CBP{wt}, and GAPDH (internal control) expression. 50 ng of total RNA was used in reactions 1 and 2 with oligoes hybridizing with CBP{HAT‒} mRNA. For all other reactions, 10 ng of total RNA was used. 1, RNA from hippocampus, CBP{HAT‒} mice OFF-Dox for 4 days; 2, RNA from hippocampus, CBP{HAT‒} mice ON-Dox; 3, RNA from CA1, CBP{HAT‒} mice OFF-Dox for 7 days; 4, RNA from CA1, CBP{HAT‒} mice ON-Dox.
CBP{HAT‒} Blocked In Vivo c-fos Gene Expression
c-fos is one of the genes induced in the brain by environmentally driven neuronal activity (Tischmeyer and Grimm, 1999). CBP is a coactivator interacting with multiple transcription factor families and regulators of transcription; however, its intrinsic enzymatic activity is frequently not required for transcriptional activation even in the cases when the CBP protein is recruited to a target promoter (Janknecht, 2002; Korzus et al., 1998). For example, activation of retinoic acid receptor- or MyoD-directed transcription requires CBP/p300 platform function but not CBP/p300 acetyltransferase activity (Korzus et al., 1998; Puri et al., 1997). Although it has been demonstrated that CBP is recruited to the c-fos promoter in an NMDA-dependent fashion (Impey et al., 2002), the requirement for the acetyltransferase activity of CBP for c-fos transcription has not been addressed. We examined the effect of CBP{HAT‒} expression on c-fos-promoter-driven luciferase gene expression in CV-1 and HeLa cell as well as on endogenous c-fos gene expression in CV-1 cells (Figures 2B–2D). Overexpression of CBP{HAT‒} impaired c-fos induction, while simple overexpression of a wild-type CBP construct had no effect (Figures 2B–2D). The results demonstrate that the histone acetyltransferase activity of CBP is required for normal c-fos promoter activation (Figures 2B–2D).
We have also examined the levels of c-fos expression in the CA1 neurons in hippocampus of wild-type mice compared to CBP{HAT‒}/Tg-ON mice (Figure 2E). We found significantly decreased levels of c-fos expression in CBP{HAT‒} mice expressing the transgene for 7 days (Figure 2E). CBP{HAT‒}/Tg-On mice showed c-fos expression at 55% of wild-type littermates (wt, average integrated intensity = 3.41 × 106 units, n = 6, SEM ± 0.60 × 106; CBP{HAT‒}, average integrated intensity = 1.99 × 106 units, n = 6, SEM ± 0.43 × 106; t test for the average integrated intensity, p < 0.0087). These data indicate a deficiency in c-fos activation in the mutant animals and provide in vivo evidence that histone acetyltransferase activity of CBP is critical for transcriptional activation of c-fos gene expression in CA1 neurons.
Declarative Memory Was Impaired in CBP{HAT‒} Mice
Because CBP{HAT‒} mice showed expression of the CBP{HAT‒} transgene in hippocampus, we investigated the function of CBP acetyltransferase activity in two forms of declarative memory: recognition memory and spatial memory. All tested animals were raised with transgene expression suppressed. Mutant mice or wildtype littermates aged 11 weeks to 6 months were used for experiments after activation of the transgene in adult animals.
A visual-paired comparison (VPC) task has been successfully used to assess declarative memory formation in humans, monkeys, and rodents (Bachevalier, 1990; Ennaceur and Delacour, 1988; Fagan, 1970). This task assesses recognition memory by measuring a subject’s innate tendency to preferentially explore a novel object. It has been demonstrated that amnesic patients with damage to the temporal lobe performed poorly on the VPC task (McKee and Squire, 1993). Impaired performance on the VPC task has also been observed in studies involving monkeys with large medial temporal lobe lesions (Bachevalier, 1990) as well as monkeys and rodents with lesions restricted to the hippocampal formation (Clark et al., 2000; Zola et al., 2000).
Figure 3A shows performance on the VPC task of three independent groups of mice: CBP{HAT‒}/Tg-ON, CBP{HAT‒}/Tg-OFF, and wt mice (wt). Performance was measured at 30 min and again at a 24 hr delay to assess both short- and long-term memory. We found no difference in short-term memory measured at 30 min [ANOVA, F(2,33) = 0.23, p > 0.05]. However, at the 24 hr delay, ANOVA revealed a significant main effect of Group [F(2,33) = 6.72, p < 0.05], and post hoc tests showed that CBP{HAT‒}/Tg-ON mice exhibited a significant memory deficit (p < 0.05) when compared to the two control groups, wt and CBP{HAT‒}/Tg-OFF (Figure 3A). This demonstrates intact short-term memory but impaired long-term memory with expression of the dominant-negative CBP{HAT‒} transgene in adult animals.
Figure 3. Two Forms of Declarative Memory: Spatial and Recognition Memory Are Impaired in CBP{HAT‒} Mice.
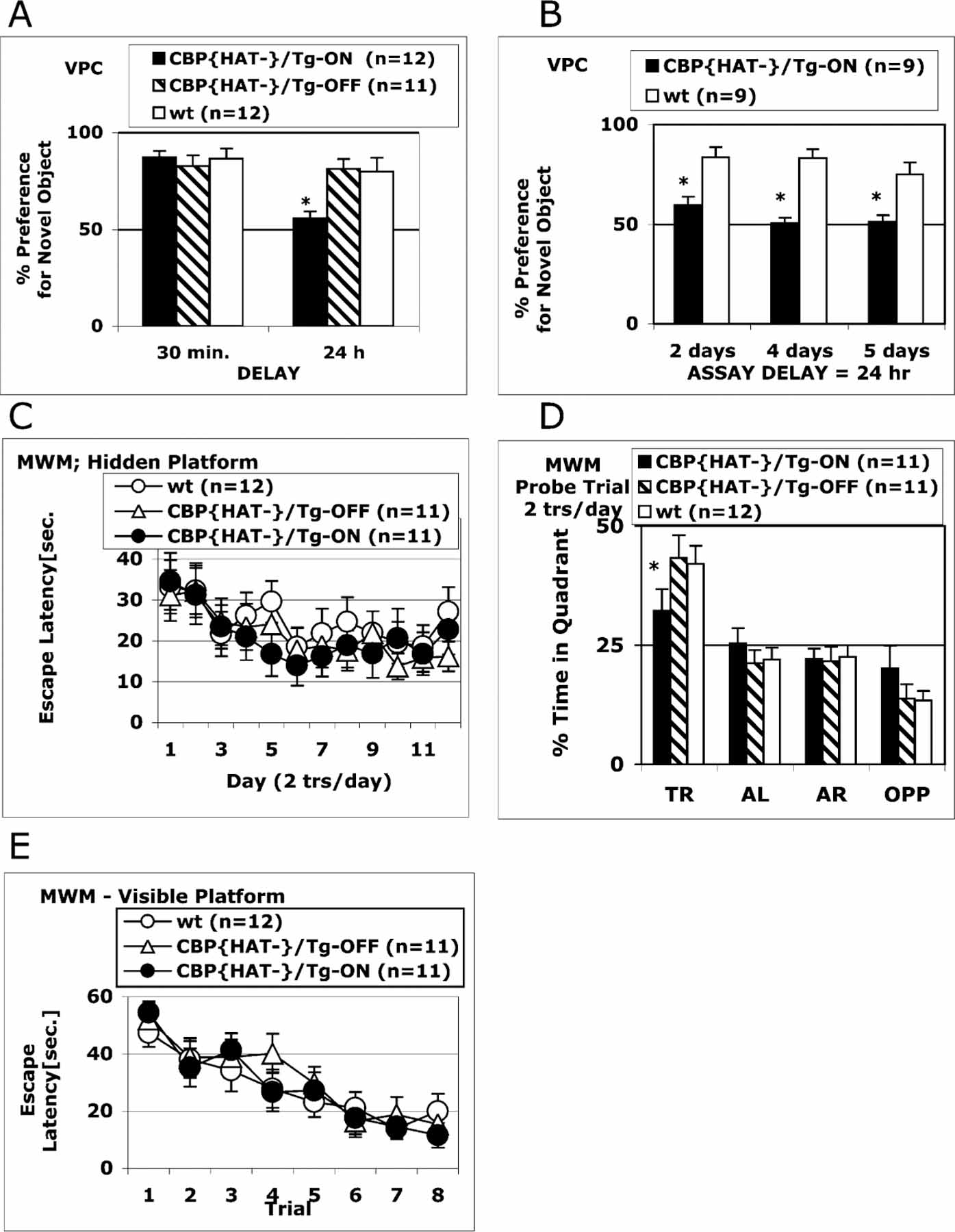
(A) CBP{HAT‒} mutant mice exhibit a severe impairment in recognition memory. Three groups were tested on the VPC task at 30 min and 24 hr delays: CBP{HAT‒}/Tg-ON (n = 12) that had expressed the transgene for 18 days before the assay, CBP{HAT‒}/Tg-OFF (n = 11) with blocked transgene expression, and wt (n = 13). CBP{HAT‒}/Tg-ON mice exhibited normal acquisition, short-term memory, and recall when tested on the VPC task at the 30 min delay. However, CBP{HAT‒}/Tg-ON mice were severely impaired on the VPC task at the 24 hr delay (*p < 0.05). We found no effect of Dox treatment in wild-type mice and therefore combined the data for wild-type ON/OFF Dox. t test for the % preference for novel object after 30 min delay: wt/OFF-Dox versus wt/ON-Dox, p > 0.85; wt/OFF-Dox, average = 86%, n = 7, SEM ± 0.8; wt/ON-Dox, average = 88%, n = 6, SEM ± 0.5. t test for the % preference for novel object after 24 hr delay: wt/OFF-Dox versus wt/ON-Dox, p > 0.89; wt/OFF-Dox, average = 79%, n = 7, SEM ± 0.8; wt/ON-Dox, average = 81%, n = 6, SEM ± 0.8.
(B) The performance of CBP{HAT‒}/Tg-ON and wt mice was tested on the VPC task at the 24 hr delay after CBP{HAT‒} transgene expression was activated for 2, 4, or 5 days before training. Expression of the CBP{HAT‒} transgene for 2 days before training was sufficient to block long-term memory formation assessed at the 24 hr delay. Both groups of animals, CBP{HAT‒}/Tg-ON and control wt littermates, received exactly the same Dox treatment before and during the experiment.
(C) Performance on the MWM task during training on Protocol 1 (see Experimental Procedures). CBP{HAT‒}/Tg-ON mice were trained together with two control groups: CBP{HAT‒}/Tg-OFF and wt mice for 12 days (2 trials/day, 1 hr ITI). No differences in escape latencies were found between CBP{HAT‒}/Tg-ON mice and two control groups.
(D) CBP{HAT‒}/Tg-ON mice show a deficiency in spatial memory on the MWM task. A probe trial performed 1 day after training was complete revealed a significant impairment in spatial localization in the CBP{HAT‒}/Tg-ON group compared to controls (t test for time in target quadrant: *CBP{HAT‒}/Tg-ON versus wt, p < 0.02; CBP{HAT‒}/Tg-ON versus CBP{HAT‒}/Tg-OFF, p < 0.05). We found no effect of Dox treatment in wild-type mice and therefore combined the data for wild-type ON/OFF Dox. t test for the time in target quadrant: wt/OFF-Dox versus wt/ ON-Dox, p > 0.9; wt/OFF-Dox: average = 43.2%, n = 7, SEM ± 4.8; wt/ON-Dox, average = 44.1%, n = 5, SEM ± 6.7.
(E) CBP{HAT‒}/Tg-ON mice showed normal acquisition on the visual platform version of the MWM task indicating normal vision, motivation, and swimming ability.
The design of the experiments in this figure (except B) includes two types of animals (wild-types and double transgenics) and two treatment groups (Dox or no Dox). We performed the behavioral analysis on these four groups. However, we found no effect of Dox treatment in wildtype mice and therefore combined the data for wild-type ON/OFF Dox.
Figure 3B shows results obtained from two groups of mice (CBP{HAT‒}/Tg-ON and wt control) tested on the VPC task at the 24 hr delay following 2, 4, or 5 days of CBP{HAT‒} expression before training. ANOVA revealed a significant main effect of Group (F = 39.1, p < 0.05), and post hoc tests showed severe memory impairment at the earliest time after initiation of transgene expression (p < 0.05) (Figure 3B).
We assessed spatial memory in CBP{HAT‒} mice using the hidden platform version of the Morris water maze (MWM) task (Morris et al., 1982). We tested performance on the MWM task in CBP{HAT‒}/Tg-ON compared to CBP{HAT‒}/Tg-OFF and wt littermates. Analysis of escape latencies during training on the hidden platform version of the MWM task showed no significant differences between groups (Figure 3C). However, compared to control groups, CBP{HAT‒}/Tg-ON mice exhibited a deficit in spatial localization on a probe trial performed after 12 days of training (2 trials per day) (Figure 3D; t test for time in target quadrant, p < 0.05). CBP{HAT‒}/Tg-ON mice showed normal performance on a visual platform version of the task (Figure 3E) indicating normal vision, swimming capabilities, and motivation to find the escape platform.
We also examined Pavlovian fear conditioning and found that CBP{HAT‒}/Tg-ON mice performed similar to controls in both the cued (CS, tone) and contextual (CS, context) versions of the task (Figure 4A). Both cued and contextual fear memories depend on the amygdala (Phillips and LeDoux, 1992), while contextual conditioning also requires hippocampal function. We did not observed CBP{HAT‒} transgene expression in amygdala (Figure 1B). CBP{HAT‒}/Tg-ON mice exhibit normal weight and performance on an open field activity assay that measures anxiety-related behavioral responses (Figures 4B–4E).
Figure 4. Fear Conditioning, Anxiety-Related Response, Weight, and Locomotor Activity Are Normal in CBP{HAT‒} Mice.
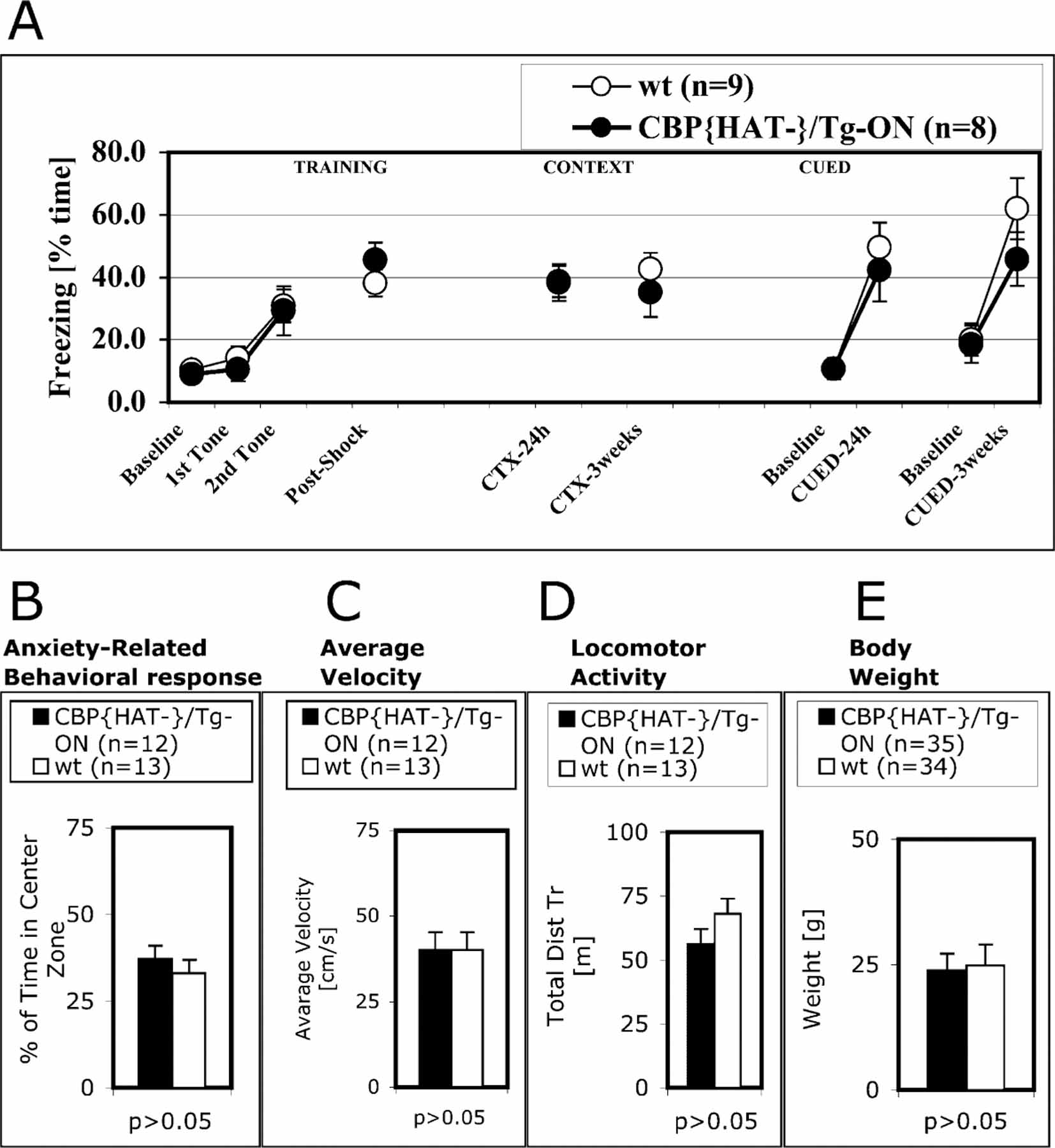
(A) CBP{HAT‒}/Tg-ON mice and control wt littermates showed the same freezing in tests of contextual and cued fear conditioning after 24 hr and 3 week delays.
(B–D) An open field activity test (see Experimental Procedures) showed that time in center (B), average velocity (C), and locomotor activity (D) were normal in CBP{HAT‒}/TgOn mice.
(E) Weights of CBP{HAT‒}/Tg-ON mice were the same as wt littermates.
Although we have observed expression of CBP{HAT‒} in hippocampus of the mutant mice, we did not observed a deficiency in contextual fear conditioning, while memory impairment was observed on the MWM and VPC tasks. The dissociation between spatial learning and contextual fear conditioning has been observed in several mutant mice harboring genetic mutations of genes such as BDNF, CaM Kinase Kinase β, or CREB targeted to forebrain including hippocampus (Gorski et al., 2003; Peters et al., 2003; Pittenger et al., 2002). The lack of deficit in contextual fear conditioning in mutant mice that have genetic mutations targeted to hippocampus and show deficits in spatial and recognition memory suggests that the genetic alteration can be compensated for, possibly at the molecular level. Our data support the idea that even if the different forms of memory involve the same brain region, the recruited nuclear mechanisms may differ.
Transgene Suppression Was Sufficient to Rescue the Memory Deficit in CBP{HAT‒} Mice
Mutation of CBP in RTS is known to produce a number of developmental phenotypes (Hennekam et al., 1990; Rubinstein and Taybi, 1963). While in the current study the transgene is induced only in adult animals to preclude developmental effects on the observed phenotypes, it is possible that transient disruption of CBP function in the adult brain leads to the observed phenotypes by inducing irreversible structural abnormalities rather that by interfering with the direct transcriptional signaling required for memory consolidation. To distinguish between these possibilities, we examined the effect on behavior of a prolonged exposure to CBP{HAT‒} transgene expression followed by suppression of expression prior to behavioral training (Figures 5A and 5B). We found that the behavioral phenotype was totally rescued following suppression of the CBP{HAT‒} trans-gene, indicating that the effect is due to an acute disruption of signaling rather than an irreversible alteration in neuronal function (Figures 5A and 5B).
Figure 5. Deficit in Recognition Memory in CBP{HAT‒} Mice Can Be Reversed by Elimination of CBP{HAT‒} Transgene Expression but not by Multiple Training Sessions.
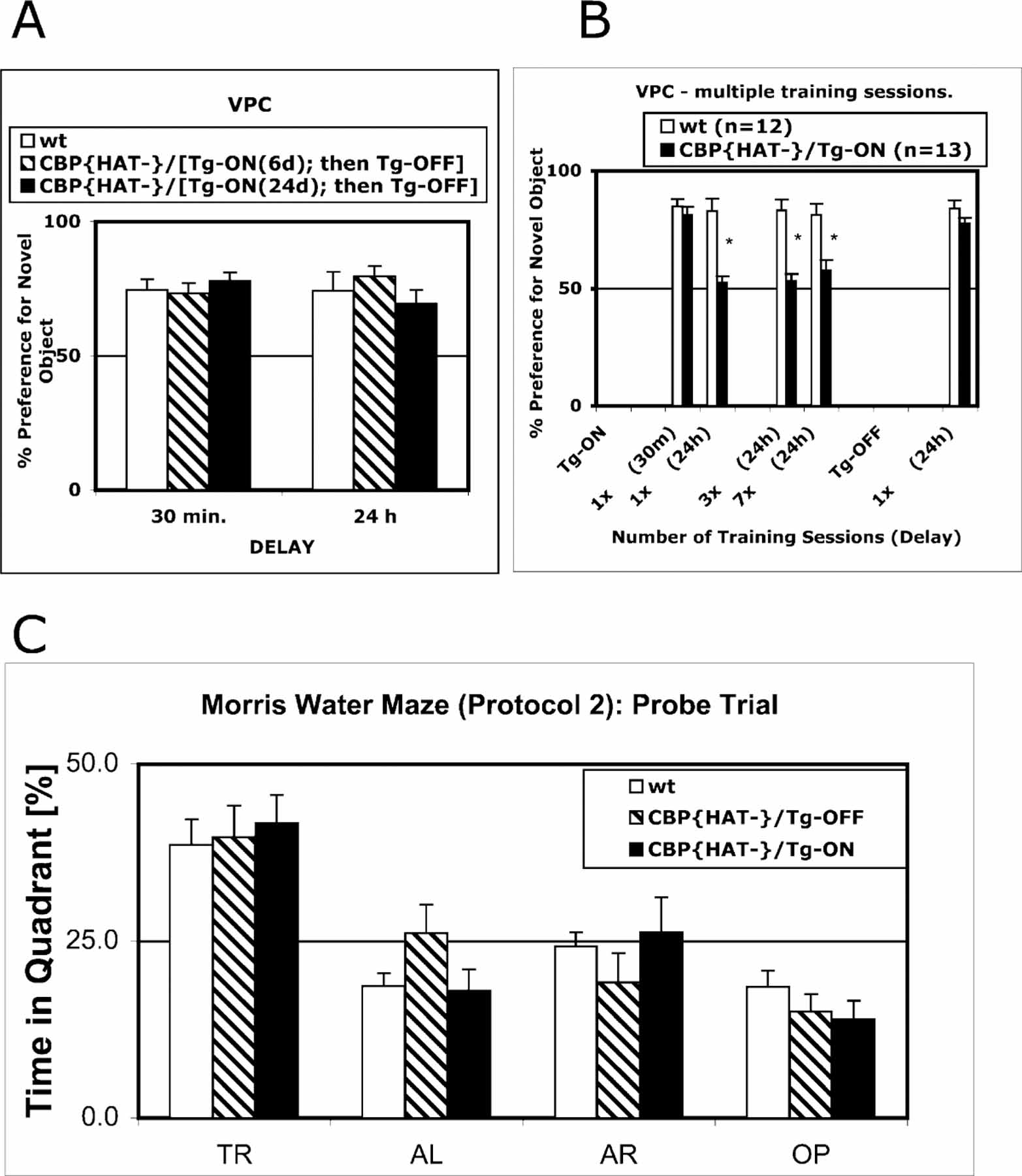
(A) Performance in CBP{HAT‒} mice was rescued when the expression of CBP{HAT‒} was suppressed. Three groups were used for the experiment: CBP{HAT‒}/Tg-ON(24d)-OFF (n = 12) that had expressed the transgene for 24 days, CBP{HAT‒}/Tg-ON(6d)-OFF that had expressed the transgene for 6 days (n = 11), and wt (n = 13). Following transgene expression for 6 or 24 days, we suppressed expression and tested the animals on the VPC task at 30 min and 24 hr delays using a new set of objects. CBP{HAT‒} mice with suppressed transgene expression exhibited normal performance on the VPC task at the 30 min delay [ANOVA, F(2,33) = 0.45, p > 0.05; CBP{HAT‒}/Tg-ON(24d)-OFF = 78.1 ± 3.0, CBP{HAT‒}/Tg-ON(6d)-OFF = 73.2 ± 3.9, wt = 74.5 ± 4.0] as well as at the 24 hr delay [ANOVA, F(2,33) = 0.75, p > 0.05; CBP{HAT‒}/Tg-ON(24d)-OFF = 69.5 ± 5.2, CBP{HAT‒}/Tg-ON(6d)-OFF = 79.6 ± 3.8, wt = 74.3 ± 7.1].
(B) Multiple training sessions did not rescue the recognition memory deficit in CBP{HAT‒} mice. Two groups (wt [n = 12] and CBP{HAT‒}/ Tg-ON [n = 13]) were trained in multiple training sessions (40 min exposure to the same object every 24 hr) and tested on the VPC task at 24 hr delays except for the first test, which was performed at the 30 min delay (t test for performance, *p > 0.05).
(C) Performance on the hidden platform version of the MWM task was rescued by intense training (Protocol 2, see Experimental Procedures).
To assess the severity of the behavioral phenotype, we determined whether repeated training could rescue the memory deficits observed in the VPC and MWM tasks. We trained two groups, CBP{HAT‒}/Tg-ON mice and wt littermates, on the VPC task employing multiple training sessions (Figure 5B). Recognition memory was assessed after 1, 2, 3, and 7 training sessions with the same familiar object. Memory tests were performed at 24 hr delays with the exception of the first training session, which was followed by both a 30 min and 24 hr memory test. At the end of the experiment, we suppressed expression of the transgene and, after 2 weeks, we retested performance of animals on the VPC task using a different sets of objects. Statistical analysis of the results confirmed again that after one training session, CBP{HAT‒} mice performed the same as the wt group at the 30 min delay but showed a strong memory deficit when tested at the 24 hr delay (Figure 5B). Repeated training trials failed to rescue this deficit (Figure 5B). However, when the expression of CBP{HAT‒} in the same mice was suppressed and the mice were retrained, the memory deficit was no longer observed (Figure 5B, last two bars, p > 0.05).
In contrast to the deficit in recognition memory observed on the VPC task (Figure 5B), intense training rescued the spatial memory deficit in the MWM task in CBP{HAT‒}/Tg-ON mice (Figures 3D and 5C). The VPC task recruits an innate behavioral preference for novelty and does not involve an exogenous reinforcer such as a swim stress (water maze) or an electric shock (fear conditioning). It is possible that these stressful reinforcers activate alternative synaptic and nuclear mechanisms for the stabilization of long-term memories. This could explain why intense training on the MWM task can overcome the memory deficits (Figures 3D and 5C) and why hippocampus-dependent contextual fear conditioning is unaffected in the CBP{HAT‒} mice (Figure 4A).
Administration of a Deacetylase Inhibitor Rescued the Memory Deficit in CBP{HAT‒} Mice
Our results are consistent with a requirement for the intrinsic histone acetyltransferase activity of CBP in the transcriptional signaling required for long-term memory consolidation. If this interpretation is correct, we would expect that the phenotype could be reversed by a general increase in histone acetylation. To test this idea, we used Trichostatin A (TSA), a histone deacetylase inhibitor that has been successfully used to increase accumulation of acetylated histones in cell culture and in whole-mouse models without apparent toxicity (Nervi et al., 2001; Yoshida et al., 1990). Administration of TSA in mice increased the level of acetylated histone H3 in hippocampus (Figure 6A). To examine the effect of TSA on performance of CBP{HAT‒} mice, we injected TSA or vehicle into wt and CBP{HAT‒}/Tg-ON mice 2 hr before training on the VPC task (Figure 6B). We found no differences in performance between groups at the 30 min delay [ANOVA, F(3,32) = 0.61, p > 0.05; CBP{HAT‒}/TSA = 71.5 ± 3.7; wt/TSA ± 66.4 ± 2.5; CBP{HAT‒}/veh = 73.0 ± 3.2; wt/veh = 72.7 ± 6.6], indicating that TSA did not alter acquisition or recall under these experimental conditions. However, the same groups show significant differences in performance when tested at the 24 hr delay [ANOVA, F(3,32) = 3.51, p < 0.05; CBP{HAT‒}/TSA = 70.8 ± 6.1; wt/TSA = 75.7 ± 5.5; CBP{HAT‒}/veh = 54.1 ± 4.4; wt/veh = 76.9 ± 5.6]. Three-way ANOVA with repeated measure revealed a Genotype × Delay interaction [F(1,32) = 8.41, p < 0.05] and a Drug × Delay interaction [F(1,32) = 4.15, p < 0.05]. A post hoc test revealed that CBP{HAT‒}/veh was the only group showing a significant deficit in memory at the 24 hr delay when compared to each of the other groups. Levene’s test for homogeneity of variances demonstrated that groups did not differ (p > 0.05). These data demonstrate that administration of a histone deacetylase inhibitor rescues the memory deficit in CBP{HAT‒} mice. This reversal of the CBP{HAT‒} phenotype by TSA treatment was transient. When the VPC test was repeated on the same animals with different objects (Groups 1 and 2) 4 days following TSA treatment, the CBP{HAT‒} mice again showed a memory deficit at the 24 hr delay (Figure 6B, 2nd 24 hr; CBP{HAT‒} = 50.3 ± 2.2, wt = 75.5 ± 2.9, p < 0.05). This is consistent with the previously reported observation that administration of deacetylase inhibitors by a single i.p. injection causes a transient change in the level of histone acetylation (Butler et al., 2000). These results provide direct evidence that the memory impairment in CBP{HAT‒} mice results from a deficiency in acetyltransferase activity in forebrain neurons.
Figure 6. Administration of the Histone Deacetylase Inhibitor TSA Reverses the Recognition Memory Deficit in CBP{HAT‒} Mice.
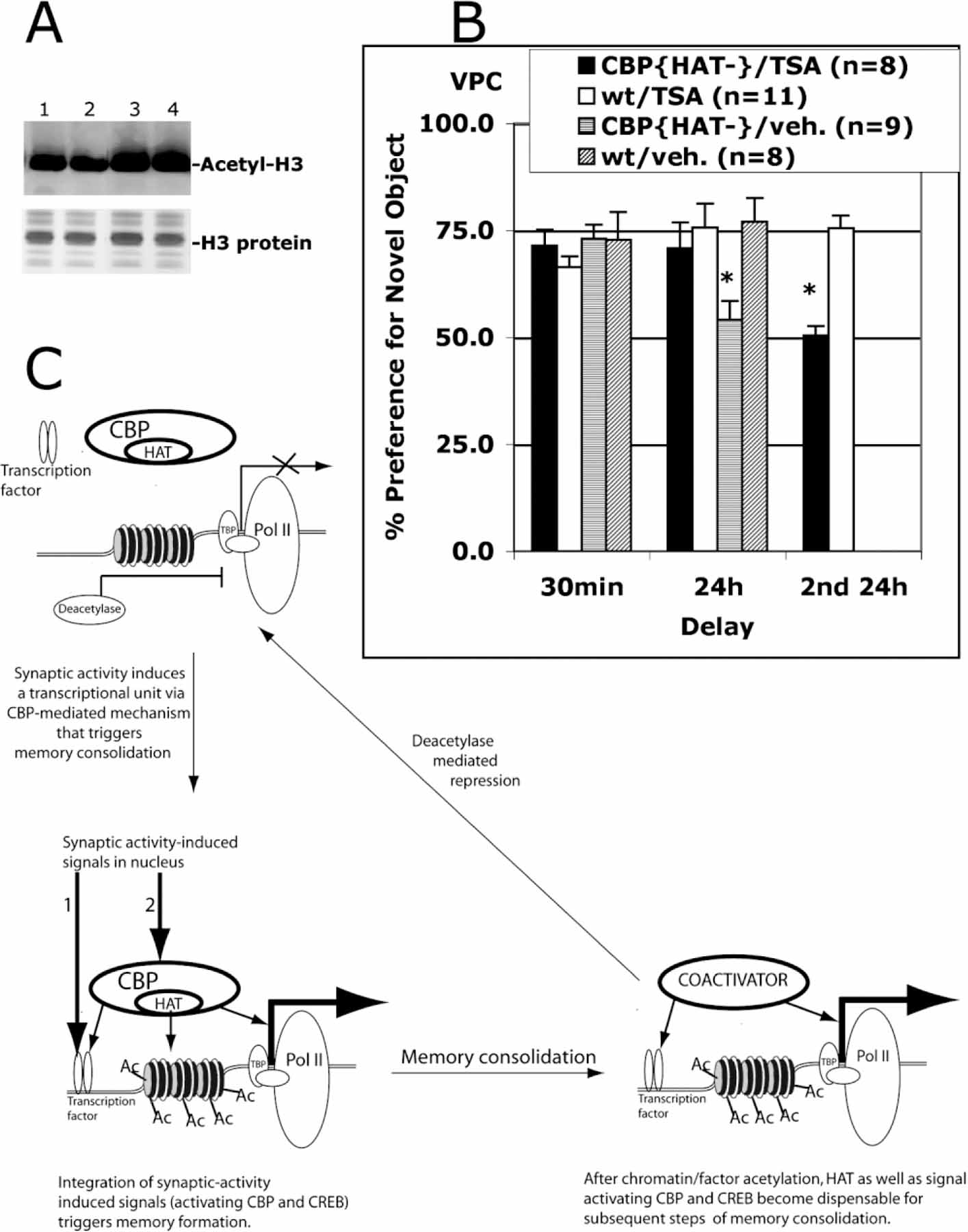
(A) I.p. TSA injection increases histone acetylation levels in the hippocampus. Nuclear extracts were prepared from hippocampi isolated from wt mice sacrificed 6 hr after i.p. injection of vehicle (lanes 1 and 2) or TSA (lanes 3 and 4). Acid-extracted histones were visualized with Coomassie blue staining. The level of histone H3 acetylation was assessed in Western blot assay using anti-acetylated H3 antibodies (Upstate).
(B) I.p. TSA administration before training rescued a recognition memory deficit in CBP{HAT‒} mice. Four groups of mice were tested: CBP{HAT‒}/TSA (Group 1), wt/TSA (Group 2), CBP{HAT‒}/veh (Group 3), and wt/veh (Group 4). TSA or DMSO alone (veh) was administered 2 hr before training on the VPC task as described in Experimental Procedures. Then, we measured performance of these four groups on the VPC task at 30 min and 24 hr delays. 4 days later, the recognition memory was assessed again (Groups 1 and 2) on the VPC task at the 24 hr delay using a new set of objects (2nd 24 hr). This assay provided a control showing that Group 1 has strong memory impairment if TSA was not administered.
(C) Model of acetylation-dependent memory consolidation. The proposed model takes into account five observations: (1) NMDAR-dependent neuronal activation is required for memory formation (Davis et al., 1992); (2) neuronal activity-dependent transcription is required for memory consolidation (Andrew, 1980; Davis and Squire, 1984); (3) NMDA-dependent transcription requires a signaling pathway to activate transcription factors such as CREB and a separate signaling pathway to activate CBP (Chawla et al., 1998; Impey et al., 2002); (4) behaviorally induced CREB phosphorylation is transient and does not correlate with peak Fos induction (Stanciu et al., 2001); and (5) CBP acetyltransferase activity is required for long-term memory consolidation but not for short-term memory (our data). This model suggests that CBP-mediated histone acetylation during transcriptional activation is a limiting step in the molecular mechanism controlling memory stabilization. Initial steps include induction of transient CREB phosphorylation, CBP activation, and CBP-mediated histone acetylation at a specific transcriptional unit in response to the initial learning event. Subsequently, prolonged elevated transcription required for memory consolidation could be maintained by CBP- and CREB phosphorylation-independent nuclear mechanisms even after signals to CREB and CBP are no longer present. This ongoing transcription would remain active until the competing deacetylase-dependent repression mechanism shut off transcription.
Discussion
Transcription factors including creb, c-fos, and zif268 have been implicated in long-term memory consolidation (Bourtchuladze et al., 1994; Fleischmann et al.,2003; Jones et al., 2001; Josselyn et al., 2001; Kida et al., 2002; Pittenger et al., 2002). However, transcriptional activation requires not only the induction of transcription factors but also the removal of repression imposed by promoter-specific repressors as well as by histone deacetylation-dependent chromatin silencing. This can be achieved by recruitment of specific coactivators of transcription such as CBP that are able to facilitate HAT-dependent nucleosomal remodeling and to promote the transition of silent to transcriptionally permissive chromatin (Fischle et al., 2003; Rosenfeld and Glass, 2001). Nucleosome acetylation mediated by coactivators of transcription such as CBP during gene activation may alter chromatin structure and mediate gene-specific removal of epigenetically controlled repression (Fischle et al., 2003; Jaenisch and Bird, 2003; Rosenfeld and Glass, 2001). This represents an attractive mechanism for the regulation of long-term transcriptional changes associated with long-lasting forms of neuronal and behavioral plasticity.
The acetyltransferase function of CBP has a strong impact on chromatin structure and the activity of transcription factors (Janknecht, 2002). Support for the idea that CBP may play a role in behavioral plasticity came from three findings. First, CREB-based transcriptional signaling has been implicated in memory formation in a variety of species (Bourtchuladze et al., 1994; Dash et al., 1990; Josselyn et al., 2001; Kida et al., 2002; Pittenger et al., 2002; Yin et al., 1994). Second, CBP can be recruited to CREB-dependent promoters in response to upstream signals (Chrivia et al., 1993). Third, it has been suggested, from studies in cultured hippocampal neurons, that NMDA receptor-induced transcription is mediated by activation of both transcription factors such as CREB and CBP independently (Hardingham et al., 1999; Hu et al., 1999; Impey et al., 2002). The current results link the CBP transcriptional coactivator to the molecular mechanisms underlying long-term memory formation. While it is possible that the behavioral phenotypes were the result of simple overexpression of the mutant CBP, the lack of effect of wild-type CBP overexpression on c-fos induction in culture and the rescue of the behavioral phenotype by histone deacetylase inhibitors strongly argue that there is a requirement for acetyltransferase activity in memory consolidation.
Prenatal lethality in CBP knockout mice demonstrates an essential role of this gene in embryogenesis (Yao et al., 1998). In humans, heterozygous mutations in CBP lead to RTS and cause a broad spectrum of developmental abnormalities and severe mental retardation (Hennekam et al., 1990; Rubinstein and Taybi, 1963). In addition, heterozygous mutations in the mouse resemble RTS phenotype and result in a variety developmental abnormalities including memory deficiency (Bourtchouladze et al., 2003; Oike et al., 1999a, 1999b; Tanaka et al., 1997, 2000). It is unclear to what extent the mental retardation associated with RTS results from a permanent abnormality in brain architecture or chronic neuronal dysfunction due to CBP deficiency. By altering CBP activity only in the adult, our data demonstrate that CBP acetyltransferase activity is an essential acute component of long-term memory consolidation in the adult independent of its role in development. It is possible that targeting CBP acetyltransferase deficiency, for example with deacetylase inhibitors, in RTS may restore some of the cognitive deficits associated with this syndrome. The observation that acetyltransferase activity plays a critical role in memory consolidation may open new avenues for the investigation and treatment of memory disorders as well as provide new opportunities for memory improvement.
How is the acetyltransferase activity of CBP involved in memory consolidation? There is a correlation between the level of histone acetylation and the rate of gene transcription (Pazin andKadonaga,1997). This is consistent with the finding that activation of transcription is assisted by specific recruitment of acetyltransferase activity to a promoter, which is critical to overcome the repressed, deacetylated state of chromatin. Histones are not the only targets for CBP acetyltransferase activity. For example, it has been demonstrated that in vivo acetylation of transcription factors such as p53, GATA-1, or Myo-D by CBP drastically changes their affinity for DNA and enhances transcription (Boyes et al., 1998; Gu and Roeder, 1997; Polesskaya et al., 2000). The finding that coactivators such as CBP are direct targets of cellular signaling pathways, which induce posttranslational modifications in CBP altering its HAT activity, ability to be recruited, or its transactivation activity, suggest an additional level of transcriptional regulation (Janknecht, 2002; Rosenfeld and Glass, 2001).
We suggest that CBP acetyltransferase activity is critical for activation of genes controlling memory consolidation (Figure 6C). It is well known that the transcription factor CREB is required for long-term memory consolidation (Bourtchuladze et al., 1994; Josselyn et al., 2001; Kida et al., 2002; Pittenger et al., 2002) and that CREB phosphorylation at S133 is necessary to recruit CBP and for subsequent transcriptional activation (Chrivia et al., 1993). However, behaviorally induced CREB phosphorylation is transient and does not correlate with peak c-fos induction (Stanciu et al., 2001). Moreover, S133 phosphorylation of CREB alone is not sufficient to induce transcription (Bading et al., 1993; Bito et al., 1996). NMDA-dependent phosphorylation of S301 on CBP has been also shown to be required for CBP-dependent gene expression (Impey et al., 2002). This suggests that a separate signaling pathway directly to CBP must be activated in neurons to allow normal CREB-mediated gene activation, as well as other non-CREB-dependent genes that use CBP as a coactivator. The current results suggest that this CBP pathway is also critical for memory consolidation by recruitment of functional CBP histone acetyltransferase activity and possible alteration of local chromatin structure. In addition to its chromatin remodeling function, it is possible that nuclear nonhistone substrates for CBP acetyltransferase activity might be critical during memory consolidation. This activation of CBP-mediated acetylation at specific gene targets could serve to alter the subsequent requirements for transcriptional activation of those genes in response to future cellular signals. This sort of the acetylation-mediated covalent modification would open up atemporal window in which cellular signals, which did not recruit an active acetyltransferase to the promoter, would nevertheless stimulate transcription as shown in Figure 6C. It is possible that after initial CBP-mediated integration of short-lasting neuronal signals leading to acetylation-dependent alteration of local chromatin structure, CBP HAT or even CBP platform function may become dispensable for subsequent steps. In fact, CBP-independent activation of CREB (or CREM, a CREB family member), which is mediated by other coactivators and bypasses the classical requirements for phosphorylation of CREB/CREM and interaction with CBP, has been described (Conkright et al., 2003; Fimia et al., 1999). This could allow for prolonged elevation of transcription in response to an initial learning event by maintaining transcription even after signals to CREB and CBP were no longer present.
Experimental Procedures
Generation of Mice
Mutations in CBP were introduced by site-directed mutagenesis using the Quick Change mutagenesis system according to the manufacturer’s instructions (Stratagene). Double-stranded oligonucleotides were designed such that the wild-type sequence corresponding to amino acids Y1540/F1541 (acetyl-CoA binding site) in cDNA encoding CBP isolated from a mouse pituitary library (Kamei et al., 1996) were substituted with alanines in order to generate a mutant of CBP lacking HAT activity, CBP{HAT‒}. Mutation was confirmed by sequence analysis. Mutant and wild forms of CBP were expressed in Baculovirus and tested for histone acetyltransferase activity (Korzus et al., 1998). CBP{HAT‒} was transfected into cells as previously described (Korzus et al., 1998), and the effect of CBP{HAT‒} on the c-fos gene expression was tested in tissue culture.
CBP{HAT‒} was subcloned into the EcoRV site of pMM400 (Mayford et al., 1996) to create ptet-O-CBP{HAT‒}. In ptet-O-CBP{HAT‒}, the CBP{HAT‒} gene is downstream of the tet-O promoter and flanked by artificial introns and a 3′ polyadenylation signal. The tetO-CBP{HAT‒} transgene was purified away from vector sequences and microinjected into B6/D2 F2 embryos. Founder animals were crossed to mice carrying the tetracycline transactivator (tTA) under the control of the CaMKII promoter (Mayford et al., 1996). Double transgenics males (carrying both the tetO-CBP{HAT‒} and CaMKII-tTA transgenes) were backcrossed to C57Bl/6 females to generate the animals used in the experiments. Animals were kept on 12/12 hr dark/light cycle and all experiments were performed during the light phase of the cycle. Experiments used mice 10 to 24 weeks of age for behavior studies. All mice were kept on a doxycycline diet during development and until they reached at least 10 weeks of age. To activate gene expression, doxycycline was removed from the diet 7 days before starting an experiment unless indicated otherwise.
In Situ Hybridization and Immunohistology
Mouse brains were removed from animals that were perfused with 10% formalin. Brains were incubated overnight in 10% formalin and then in 20% sucrose. Brains were embedded in tissue-freezing medium and placed at 80°C. Sections were cut on a freezing cryotome at a thickness of 10 µm, mounted on glass slides, and hybridized with a poly-[α−35S]dA tailed DNA probe (5′-GCAGGATCC GCTTGGGCTGCAGTTGGA-3′) complementary to an untranslated region in the CBP{HAT‒} mRNA. To test the integrity of major brain structure, the gross neuroanatomy was analyzed on sections stained with cresyl violet. Immunostaining was performed on fresh brain sections (14 µm, Bregma: ‒2.20 mm to ‒2.40 mm) using rabbit anti-c-fos antibodies (Upstate, 1:20) and Alexa488-goat anti-rabbit IgG (Molecular Probes, 1:2000). TO-PRO3 Iodine (Molecular Probes, 1:1000) was used to counterstain nuclei. Immunostained tissue was analyzed on a semi-automatic laser scanning confocal microscope Olympus BX61 controlled by Fluoview500 software. Statistical analysis of c-fos expression in the CA1 field was performed using Metamorph software.
RNA Isolation and RT-PCR Detection of CBP{HAT‒} Transgene mRNA
The mouse brain was removed and immersed in ice-cold phosphate-buffered saline (PBS). After quick dissection, the hippocampus was cut into small pieces and total RNA was isolated (Qiaqen; RNeasy Kit). Alternatively, whole-brain was cut using microtome, dorsal CA1 field of hippocampus was dissected out, and total RNA was isolated using RNeasy kit (Qiagen). Reverse transcription followed by PCR was carried out with the Invitrogen RT-PCR kit using specific oligoes complementary to GAPDH, CBP, and CBP{HAT‒}. Reverse transcription followed by real-time PCR was carried out using M-MLV Reverse Transcriptase (Invitrogen) and DNA amplification kit from Roche. The real-time PCR was performed in multichannel Rotor-Gene 3000 thermocycler (Corbett Research) using specific oligoes complementary to CBP and CBP{HAT‒}. Plasmids containing DNA sequences encoding CBP or CBP{HAT‒} were used for standardization. Statistical analysis of ratios between CBP{HAT‒} and CBPwt was performed on multiple samples of total RNA isolated from whole hippocampus of CBP{HAT‒} mice using software provided from Corbett Research.
Visual-Paired Comparison Task
The visual-paired comparison (VPC) task has been adapted from a previously described protocol developed for rats (Clark et al., 2000; Ennaceur and Delacour, 1988). The task is divided into four phases: habituation, familiarization, delay, and test. (1) Mice were handled 4 times and were placed in the experimental room for 2 hr before the experiment. (2) During familiarization, two identical objects were placed in the home cage with the animals for 40 min. (3) Animals were tested at 30 min and 24 hr delays to examine short-term and long-term memory, respectively. (4) During the test phase, all animals were transferred to a temporary holding cage and 2 min later, both the familiar object (a replica of the original familiar object was used to avoid the use of odor cues) and a novel object were placed in the cage. Object exploration times (for both familiar and novel objects) were recorded for a 2 min test period. We employed computer-assisted scoring using software to measure and analyze performance on the VPC task that was developed by Dr. Raphael Bejar. Object exploration was scored when the animal’s head was oriented toward the object and vibrissae were moving. The objects varied in color, shape, and size and were balanced so that the same objects (replicas) were used for some animals as familiar and for others as novel in the same session. Also, the position of novel and familiar objects was randomized.
Fear Conditioning
Mice were trained in a standard fear conditioning chamber (Med Associates, Inc.). After a 3 min baseline period, two 20 s tones (2800 Hz, 75 dB) were played and a shock (0.5 mA, 1 s) was delivered during the final 1 s of the tone. 24 hr or 3 weeks later, mice were placed again in the training enclosure for 3 min and contextual freezing was scored. Alternatively, for the cued recall, mice were placed in a novel enclosure and after a 3 min baseline exposure, a series of three tones identical to that given in the training session were played. Freezing was scored and analyzed automatically by a video-based system, FreezeFrame (ActiMetrics).
Morris Water Maze Task
The Morris water maze (MWM) task was performed in a pool 48 inches in diameter and 20 inches deep with a 4 inch diameter platform. Animals were handled 4 times before training. During the visible platform version of the task, mice received 2 days of training (4 trials a day). Visual distal cues were removed and a flagged platform was placed in the target quadrant. Each trial was begun from one of four pseudo-randomly assigned start locations and lasted 60 s. Mice were placed at a start position along the wall of the tank and given 60 s to find the visibly flagged platform. If they did not find it in 60 s, they were led to the platform. Once on the platform, mice were given 40 s to rest before being returned to their home cage.
Protocol 1
During the hidden platform version of the MWM task, training lasted 12 days and consisted of 2 trials per day with approximately 60 min between trials. The platform always remained in the raised position, about 1 cm below the surface of the water. Mice were given 60 s to escape, after which they were led to the platform. Once on the platform, mice were allowed 40 s to rest before returning to their home cage. A probe trial test lasting 60 s was performed 1 day after training was complete.
Protocol 2
Protocol 2 employed intense training using the Atlantis platform. A training trial began with the mouse placed in a pseudo-random start position along the wall of the pool. With the exception of training day 1, the platform began in the lowered (inaccessible) position. In order to raise the platform, the mouse had to spend the requisite time in a 20 cm diameter area around the platform location. When the animal approached and stayed in the target zone (for 0, 1, 2, 3, or 3 s during 1st, 2nd, 3rd, 4th, and 5th day, respectively), the platform was raised and become available. If the mouse had not raised the platform after 60 s have elapsed, the platform was automatically raised. Mice were given a maximum of 90 s to find the raised platform, after which time the mouse was led to the platform. Once there, mice were given 40 s to rest before being returned to their home cage. Training was performed for 5 days: one block (4 trials per block with an ITI of 10 min) during the first day and two blocks a day during the next 4 days of training. A final probe trial was administered 1 day after training was complete. Performance on the MWM task was scored and analyzed automatically by a computer-operated video tracking system (ActiMetrics).
Open Field Activity
Anxiety-related behavioral responses and locomotor activity was scored and analyzed using an Open Field Activity Monitor (software, hardware, and manufacturer protocols from Med Associates, Inc.).
Histone Acetylation Assay and Trichostatin A Administration
Trichostatin A (TSA) form Biomol was solubilized in 100% dimethyl-sulfoxide (DMSO) at a concentration of 2 µg/µl. TSA or vehicle was administered by intraperitoneal injection (i.p.) of 1 µl per gram body weight. In order to analyze the accumulation of acetylated histone H3 in hippocampus, Western blot analysis was performed. 6 hr after injection, the mice were euthanized and hippocampus was isolated and snap-frozen in liquid nitrogen. Nuclear proteins from hippocampus were acid-extracted and separated onto a denaturing, 7%–12% acrylamide gel followed by electroblotting onto nitrocellulose. Acetylated histone H3 was detected using rabbit polyclonal anti-acetyl-H3 antibodies (Upstate) and the Amersham ECF Kit accordingly to the manufacturer’s protocol.
For behavioral experiments, TSA or DMSO was administered by i.p. injection (1 µl per gram body weight) 2 hr before training on the VPC task. Training on the VPC task was followed by memory tests at 30 min and 24 hr delays. 4 days later, mice that were treated with TSA were retested on the VPC task at the 24 hr delay using a new set of objects. This was the second 24 hr test.
Experimenter was blind to group membership through all behavioral experiments, and statistical analysis of data was performed using Microsoft Excel or Statistica.
Acknowledgments
This work was supported by the NIMH Research Career Award K01 MH01785 (to E.K.) and by grants from NIH (to M.M.). M.G.R. is an investigator of the HHMI. We thank Dr. J.L. McGaugh for helpful discussions. We also thank Kelly Harrington, Rett Bartolome, and Monica Roy for technical assistance.
References
- Andrew RJ (1980). The functional organization of phases of memory consolidation. In Advances in the Study of Behaviour, Hinde RA, Beer C, and Bunsel M, eds. (New York: Academic Press; ), pp. 337–367. [Google Scholar]
- Bachevalier J (1990). Ontogenetic development of habit and memory formation in primates. Ann. N Y Acad. Sci 608, 457–484. [DOI] [PubMed] [Google Scholar]
- Bading H, Ginty DD, and Greenberg ME (1993). Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260, 181–186. [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, and Tsien RW (1996). CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell 87, 1203–1214. [DOI] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, and Silva AJ (1994). Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79, 59–68. [DOI] [PubMed] [Google Scholar]
- Bourtchouladze R, Lidge R, Catapano R, Stanley J, Gossweiler S, Romashko D, Scott R, and Tully T (2003). A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc. Natl. Acad. Sci. USA 100, 10518–10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyes J, Byfield P, Nakatani Y, and Ogryzko V (1998). Regulation of activity of the transcription factor GATA-1 by acetylation. Nature 396, 594–598. [DOI] [PubMed] [Google Scholar]
- Butler LM, Agus DB, Scher HI, Higgins B, Rose A, Cordon-Cardo C, Thaler HT, Rifkind RA, Marks PA, and Richon VM (2000). Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res 60, 5165–5170. [PubMed] [Google Scholar]
- Chawla S, Hardingham GE, Quinn DR, and Bading H (1998). CBP: a signal-regulated transcriptional coactivator controlled by nuclear calcium and CaM kinase IV. Science 281, 1505–1509. [DOI] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, and Goodman RH (1993). Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 365, 855–859. [DOI] [PubMed] [Google Scholar]
- Clark RE, Zola SM, and Squire LR (2000). Impaired recognition memory in rats after damage to the hippocampus. J. Neurosci 20, 8853–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, and Montminy M (2003). TORCs: transducers of regulated CREB activity. Mol. Cell 12, 413–423. [DOI] [PubMed] [Google Scholar]
- Dash PK, Hochner B, and Kandel ER (1990). Injection of the cAMP-responsive element into the nucleus of Aplysia sensory neurons blocks long-term facilitation. Nature 345, 718–721. [DOI] [PubMed] [Google Scholar]
- Davis HP, and Squire LR (1984). Protein synthesis and memory: a review. Psychol. Bull 96, 518–559. [PubMed] [Google Scholar]
- Davis S, Butcher SP, and Morris RG (1992). The NMDA receptor antagonist D-2-amino-5-phosphonopentanoate (D-AP5) impairs spatial learning and LTP in vivo at intracerebral concentrations comparable to those that block LTP in vitro. J. Neurosci 12, 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB, and Livingston DM (1994). Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev 8, 869–884. [DOI] [PubMed] [Google Scholar]
- Ennaceur A, and Delacour J (1988). A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav. Brain Res 31, 47–59. [DOI] [PubMed] [Google Scholar]
- Fagan JF 3rd. (1970). Memory in the infant. J. Exp. Child Psychol 9, 217–226. [DOI] [PubMed] [Google Scholar]
- Fimia GM, De Cesare D, and Sassone-Corsi P (1999). CBP-independent activation of CREM and CREB by the LIM-only protein ACT. Nature 398, 165–169. [DOI] [PubMed] [Google Scholar]
- Fischle W, Wang Y, and Allis CD (2003). Histone and chromatin cross-talk. Curr. Opin. Cell Biol 15, 172–183. [DOI] [PubMed] [Google Scholar]
- Fleischmann A, Hvalby O, Jensen V, Strekalova T, Zacher C, Layer LE, Kvello A, Reschke M, Spanagel R, Sprengel R, et al. (2003). Impaired long-term memory and NR2A-type NMDA receptor-dependent synaptic plasticity in mice lacking c-Fos in the CNS. J. Neurosci 23, 9116–9122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Balogh SA, Wehner JM, and Jones KR (2003). Learning deficits in forebrain-restricted brain-derived neurotrophic factor mutant mice. Neuroscience 121, 341–354. [DOI] [PubMed] [Google Scholar]
- Gu W, and Roeder RG (1997). Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595–606. [DOI] [PubMed] [Google Scholar]
- Guan Z, Giustetto M, Lomvardas S, Kim JH, Miniaci MC, Schwartz JH, Thanos D, and Kandel ER (2002). Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell 111, 483–493. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Chawla S, Cruzalegui FH, and Bading H (1999). Control of recruitment and transcription-activating function of CBP determines gene regulation by NMDA receptors and L-type calcium channels. Neuron 22, 789–798. [DOI] [PubMed] [Google Scholar]
- Hennekam RC, Stevens CA, and Van de Kamp JJ (1990). Etiology and recurrence risk in Rubinstein-Taybi syndrome. Am. J. Med. Genet. Suppl 6, 56–64. [DOI] [PubMed] [Google Scholar]
- Hu SC, Chrivia J, and Ghosh A (1999). Regulation of CBP-mediated transcription by neuronal calcium signaling. Neuron 22, 799–808. [DOI] [PubMed] [Google Scholar]
- Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, and Goodman RH (2002). Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron 34, 235–244. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, and Bird A (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet 33 (Suppl), 245–254. [DOI] [PubMed] [Google Scholar]
- Janknecht R (2002). The versatile functions of the transcriptional coactivators p300 and CBP and their roles in disease. Histol. Histopathol 17, 657–668. [DOI] [PubMed] [Google Scholar]
- Jones MW, Errington ML, French PJ, Fine A, Bliss TV, Garel S, Charnay P, Bozon B, Laroche S, and Davis S (2001). A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat. Neurosci 4, 289–296. [DOI] [PubMed] [Google Scholar]
- Josselyn SA, Shi C, Carlezon WA Jr., Neve RL, Nestler EJ, and Davis M (2001). Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J. Neurosci 21, 2404–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK, and Rosenfeld MG (1996). A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell 85, 403–414. [DOI] [PubMed] [Google Scholar]
- Kida S, Josselyn SA, de Ortiz SP, Kogan JH, Chevere I, Masushige S, and Silva AJ (2002). CREB required for the stability of new and reactivated fear memories. Nat. Neurosci 5, 348–355. [DOI] [PubMed] [Google Scholar]
- Korzus E, Torchia J, Rose DW, Xu L, Kurokawa R, McInerney EM, Mullen TM, Glass CK, and Rosenfeld MG (1998). Transcription factor-specific requirements for coactivators and their acetyltransferase functions. Science 279, 703–707. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, and Kandel ER (1996). Control of memory formation through regulated expression of a CaMKII transgene. Science 274, 1678–1683. [DOI] [PubMed] [Google Scholar]
- McGaugh JL, and Hertz MJ (1972). Memory Consolidation (San Francisco: Albion; ). [Google Scholar]
- McKee RD, and Squire LR (1993). On the development of declarative memory. J. Exp. Psychol. Learn. Mem. Cogn 19, 397–404. [DOI] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, and O’Keefe J (1982). Place navigation impaired in rats with hippocampal lesions. Nature 297, 681–683. [DOI] [PubMed] [Google Scholar]
- Neely KE, and Workman JL (2002). Histone acetylation and chromatin remodeling: which comes first? Mol. Genet. Metab 76, 1–5. [DOI] [PubMed] [Google Scholar]
- Nervi C, Borello U, Fazi F, Buffa V, Pelicci PG, and Cossu G (2001). Inhibition of histone deacetylase activity by trichostatin A modulates gene expression during mouse embryogenesis without apparent toxicity. Cancer Res 61, 1247–1249. [PubMed] [Google Scholar]
- Neuwald AF, and Landsman D (1997). GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem. Sci 22, 154–155. [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, and Nakatani Y (1996). The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87, 953–959. [DOI] [PubMed] [Google Scholar]
- Oike Y, Hata A, Mamiya T, Kaname T, Noda Y, Suzuki M, Yasue H, Nabeshima T, Araki K, and Yamamura K (1999a). Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism. Hum. Mol. Genet 8, 387–396. [DOI] [PubMed] [Google Scholar]
- Oike Y, Takakura N, Hata A, Kaname T, Akizuki M, Yamaguchi Y, Yasue H, Araki K, Yamamura K, and Suda T (1999b). Mice homozygous for a truncated form of CREB-binding protein exhibit defects in hematopoiesis and vasculo-angiogenesis. Blood 93, 2771–2779. [PubMed] [Google Scholar]
- Pazin MJ, and Kadonaga JT (1997). What’s up and down with histone deacetylation and transcription? Cell 89, 325–328. [DOI] [PubMed] [Google Scholar]
- Peters M, Mizuno K, Ris L, Angelo M, Godaux E, and Giese KP (2003). Loss of Ca2+/calmodulin kinase kinase beta affects the formation of some, but not all, types of hippocampus-dependent long-term memory. J. Neurosci 23, 9752–9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ, et al. (1995). Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 376, 348–351. [DOI] [PubMed] [Google Scholar]
- Phillips RG, and LeDoux JE (1992). Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav. Neurosci 106, 274–285. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Huang YY, Paletzki RF, Bourtchouladze R, Scanlin H, Vronskaya S, and Kandel ER (2002). Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron 34, 447–462. [DOI] [PubMed] [Google Scholar]
- Polesskaya A, Duquet A, Naguibneva I, Weise C, Vervisch A, Bengal E, Hucho F, Robin P, and Harel-Bellan A (2000). CREB-binding protein/p300 activates MyoD by acetylation. J. Biol. Chem 275, 34359–34364. [DOI] [PubMed] [Google Scholar]
- Puri PL, Sartorelli V, Yang XJ, Hamamori Y, Ogryzko VV, Howard BH, Kedes L, Wang JY, Graessmann A, Nakatani Y, and Levrero M (1997). Differential roles of p300 and PCAF acetyl-transferases in muscle differentiation. Mol. Cell 1, 35–45. [DOI] [PubMed] [Google Scholar]
- Rosenfeld MG, and Glass CK (2001). Coregulator codes of transcriptional regulation by nuclear receptors. J. Biol. Chem 276, 36865–36868. [DOI] [PubMed] [Google Scholar]
- Rubinstein JH, and Taybi H (1963). Broad thumbs and toes and facial abnormalities. Am. J. Dis. Child 105, 588–608. [DOI] [PubMed] [Google Scholar]
- Squire LR (1987). Memory and Brain (New York: Oxford; ). [Google Scholar]
- Stanciu M, Radulovic J, and Spiess J (2001). Phosphorylated cAMP response element binding protein in the mouse brain after fear conditioning: relationship to Fos production. Brain Res. Mol. Brain Res 94, 15–24. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Naruse I, Maekawa T, Masuya H, Shiroishi T, and Ishii S (1997). Abnormal skeletal patterning in embryos lacking a single Cbp allele: a partial similarity with Rubinstein-Taybi syndrome. Proc. Natl. Acad. Sci. USA 94, 10215–10220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Naruse I, Hongo T, Xu M, Nakahata T, Maekawa T, and Ishii S (2000). Extensive brain hemorrhage and embryonic lethality in a mouse null mutant of CREB-binding protein. Mech. Dev 95, 133–145. [DOI] [PubMed] [Google Scholar]
- Tischmeyer W, and Grimm R (1999). Activation of immediate early genes and memory formation. Cell. Mol. Life Sci 55, 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley PF, Cole AJ, Murphy TH, Christy BA, Nakabeppu Y, and Baraban JM (1990). Synaptic regulation of immediate-early genes in brain. Cold Spring Harb. Symp. Quant. Biol 55, 213–223. [DOI] [PubMed] [Google Scholar]
- Yao TP, Oh SP, Fuchs M, Zhou ND, Ch’ng LE, Newsome D, Bronson RT, Li E, Livingston DM, and Eckner R (1998). Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 93, 361–372. [DOI] [PubMed] [Google Scholar]
- Yin JC, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, and Tully T (1994). Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila. Cell 79, 49–58. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Kijima M, Akita M, and Beppu T (1990). Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem 265, 17174–17179. [PubMed] [Google Scholar]
- Yuan LW, Soh JW, and Weinstein IB (2002). Inhibition of histone acetyltransferase function of p300 by PKCdelta. Biochim. Biophys. Acta 1592, 205–211. [DOI] [PubMed] [Google Scholar]
- Zola SM, Squire LR, Teng E, Stefanacci L, Buffalo EA, and Clark RE (2000). Impaired recognition memory in monkeys after damage limited to the hippocampal region. J. Neurosci 20, 451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]