

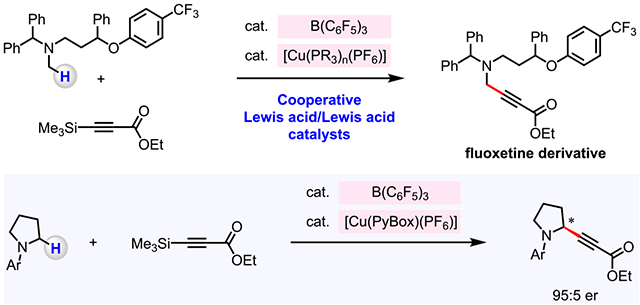
Abstract
An efficient catalytic method to convert an α-C–H bond of N-alkylamines into an α-C–alkynyl bond was developed. In the past, such transformations were carried out under oxidative conditions, and the enantioselective variants were confined to tetrahydroisoquinoline derivatives. Here, we disclose a method for union of N-alkylamines and trimethylsilyl alkynes, without the presence of an external oxidant, and promoted through cooperative actions of two Lewis acids, B(C6F5)3 and a Cu-based complex. A variety of propargylamines can be synthesized in high diastereo- and enantioselectivity. The utility of the approach is demonstrated by late-stage site-selective modification of bioactive amines. Kinetic investigations that shed light on various mechanistic nuances of the catalytic process are presented.
Graphical Abstract
1. INTRODUCTION
Propargylamines are prevalent in pharmaceuticals and are commonly used intermediates in synthesis of bioactive amines (Figure 1a).1 Enantiomerically enriched propargylamines have been prepared by addition of an alkynylmetal compound to an imine.2–4 An attractive alternative would entail the conversion of an α-amino C(sp3)–H bond into a α-C–alkynyl bond. One way to accomplish this would be through in situ generation of an iminium ion intermediate formed from the corresponding amine under oxidative conditions.5–6 An illustrative case is enantioselective Cu–PyBOX-catalyzed coupling of a benzylic α-amino C–H bond of N-phenyl tetrahydroisoquinoline 1a with ethynylbenzene 2a to afford propargylamine 3a (Figure 1b).6 Still, development of a precious transition metal- and oxidant-free catalytic C–H functionalization process represents a compelling research objective.5–7 Particularly noteworthy would be the direct conversion of α-C–H bonds contained in bioactive N-alkylamines into α-C–alkynyl bonds because these entities constitute over 50% of the top-selling drugs; the resulting derivatives of these pharmaceuticals possessing the alkyne unit can serve as modifiable intermediates for late-stage structural diversification that could lead to new leads and/or more effective therapeutics.8
Figure 1. Representative bioactive compounds containing N-propargylamines and the synthesis strategy to be pursued.
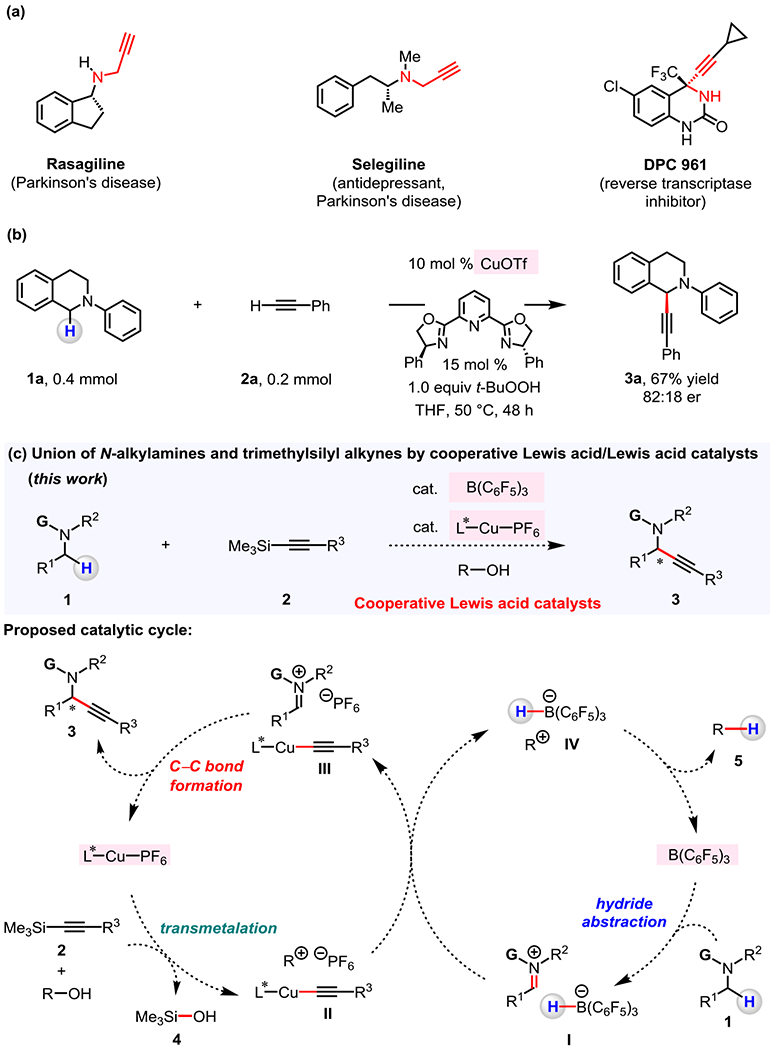
(a) Examples of pharmaceutical agents that contain a propargylamine unit. (b) Enantioselective organocopper-catalyzed transformation of an α-amino C–H bond of tetrahydroisoquinolines into C–alkyne bond under oxidative conditions. (c) Coupling of N-alkylamines with trimethylsilylacetylenes by cooperative Lewis acid/Lewis acid catalysis. A possible mechanism might involve enantioselective C–C bond formation between an iminium ion and a chiral alkynylcopper complex via reactive intermediates that are generated in situ by cooperative functions of a chiral and an achiral Lewis acid co-catalyst.
In contemplating ways to design a possible method for the reaction of an N-alkylamine 1 with trimethylsilylacetylene 2,9–10 which can be easily prepared, we envisioned utilizing the combination of two Lewis acid catalysts, an organoborane and a Cu-based complex, so that they might function cooperatively (Figure 1c).11–14 Specifically, we surmised that B(C6F5)3 might receive a hydride from an amine (1), generating a borohydride and an iminium ion (I).15–21 Subsequently, Cu-based catalyst might undergo transmetalation with alkynyl silane 2 with the aid of an alcohol additive (R–OH) to afford a LnCu–alkynyl complex (II) and trimethylsilanol 4.22 An ensuing C–C bond formation (III) between in situ generated LnCu–alkynyl complex and iminium ion would afford the desired propargylamine 3. Hydride transfer from borohydride to R–OH-derived cationic species (IV→ 5) would then regenerate B(C6F5)3, thereby closing the cycle. Here, we report the development of a cooperative Lewis acid/Lewis acid catalyst system for the transformation of α-amino C–H bonds of N-alkylamines into C–alkyne bonds and its utility in synthesis, including late-stage incorporation of alkynyl units into bioactive amines.
2. RESULTS AND DISCUSSION
2.1. Method Development
2.1.1. Identification of optimal conditions.
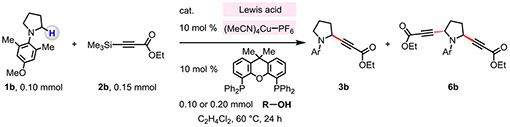
To begin, we set out to identify a suitable combination of catalysts (Table 1). We probed the ability of B(C6F5)3 and various Cu-based complexes to catalyze the reaction between 1b and 2b, generating α-alkynyl amines 3b and 6b. Treatment of 1b (0.10 mmol) and 2b (0.15 mmol) with B(C6F5)3, (MeCN)4CuPF6, Xantphos (10 mol % of each) afforded 3b in 7% yield (C2H4Cl2, 60 °C, 24 h; entry 1, Table 1).23 Use of an alcohol as an additive improved efficiency (entries 3–7), likely by accelerating the transmetalation between 2b and (MeCN)4CuPF6/Xantphos complex, releasing trimethylsilanol 4 as byproduct. Whereas the use of i-PrOH was ineffective (entry 2), addition of the more hindered t-BuOH resulted in the formation of 3b in 17% yield (entry 3). With Ph3COH as the hydroxyl source, a mixture of 3b (52% yield) and 6b (34% yield) was formed (entry 4) and Ph3C–H was obtained as a byproduct (i.e., 5, R = Ph3C; Figure 1c). When less Ph3COH was used (1.0 equiv.), 3b (83% yield) was formed more selectively (vs 6b in 15% yield; entry 5), and the desired product 3b was isolated in 90% yield when the reaction time was shortened to 12 h (vs 24 h; entry 6). The transformation was efficient with less B(C6F5)3 (5.0 mol %), affording 3b in 81% yield (entry 7). There was no transformation in the absence of B(C6F5)3 or when the less hindered BF3 or less Lewis acidic BPh3 were used (entries 8–10, Table 1).
Table 1.
 | |||||
|---|---|---|---|---|---|
| entry | Lewis acid (mol %) | R–OH (mmol) | yield (%) | ||
| 3b | 6b | ||||
| 1 | B(C6F5)3 | (10) | none | 7 | 0 |
| 2 | B(C6F5)3 | (10) | i-PrOH (0.20) | 0 | 0 |
| 3 | B(C6F5)3 | (10) | t-BuOH (0.20) | 17 | 0 |
| 4 | B(C6F5)3 | (10) | Ph3COH (0.20) | 52 | 34 |
| 5 | B(C6F5)3 | (10) | Ph3COH (0.10) | 83 | 15 |
| 6c | B(C6F5)3 | (10) | Ph3COH (0.10) | 90 | <5 |
| 7 | B(C6F5)3 | (5.0) | Ph3COH (0.10) | 81 | <5 |
| 8 | none | Ph3COH (0.10) | 0 | 0 | |
| 9 | BF3·OEt2 | (10) | Ph3COH (0.10) | 0 | 0 |
| 10 | BPh3 | (10) | Ph3COH (0.10) | 0 | 0 |
Conditions: Reactions were performed under N2 atmosphere. N-arylpyrrolidine (1b, 0.10 mmol), 3-(trimethylsilyl)propiolate (2b, 0.15 mmol), B-based Lewis acid, (MeCN)4CuPF6 (10 mol %), Xantphos (10 mol %), alcohol additive, C2H4Cl2 (0.4 mL), 60 °C, 24 h.
Yield values were determined by analysis of the 1H NMR spectra of unpurified mixtures with mesitylene as the internal standard.
Reaction mixture was allowed to stir for 12 h. See the Supporting Information for details.
2.1.2. Scope.
An assortment of cyclic and acyclic N-alkylanilines (1b–1g) may be used in reaction with 3-(trimethylsilyl)propiolate 2b to generate the corresponding propargylamines (3b–3g, Figure 2). With B(C6F5)3 and LnCu–Xantphos complex as catalysts, N-aryl pyrrolidines (1b, 1c), and N-aryl azepane (1d) were converted to 3b–3d in 77–90% yield. In a number of instances there was efficient hydride abstraction at the N-methyl site (cf. 1e–1j). 4-Methoxy-N,N,2,6-tetramethylaniline 1e reacted with 2b to afford 3e (90% yield) along with minimal amounts of the byproduct containing two propargyl amine moieties (<5%). With 1f and 1g, C–C bond formation occurred predominantly at the N-methyl site to furnish 3f (42% yield) and 3g (70% yield), respectively; there was <10% reaction at the α-amino C–H bonds of N-ethyl and N-benzyl groups. Tertiary amines 1h–1j, which lack the fused N-aryl group, readily underwent transformation to afford 3h–3j in 76%–97% yield. For synthesis of propargylamines 3h–3j the use of the less hindered and conformationally more flexible 1,2-bis(diphenylphosphino)ethane (dppe) ligand was optimal (vs <30% conv. with Xantphos).23 Furthermore, the more sizeable benzhydryl moiety was identified as a superior N-substituent as benzhydryl-substituted 3j was obtained in higher yield than benzyl-substituted 3i (97% vs 86% yield).
Figure 2. Incorporation of an alkyne unit into various N-alkylamines with 3-(triemthylsilyl)propiolate.
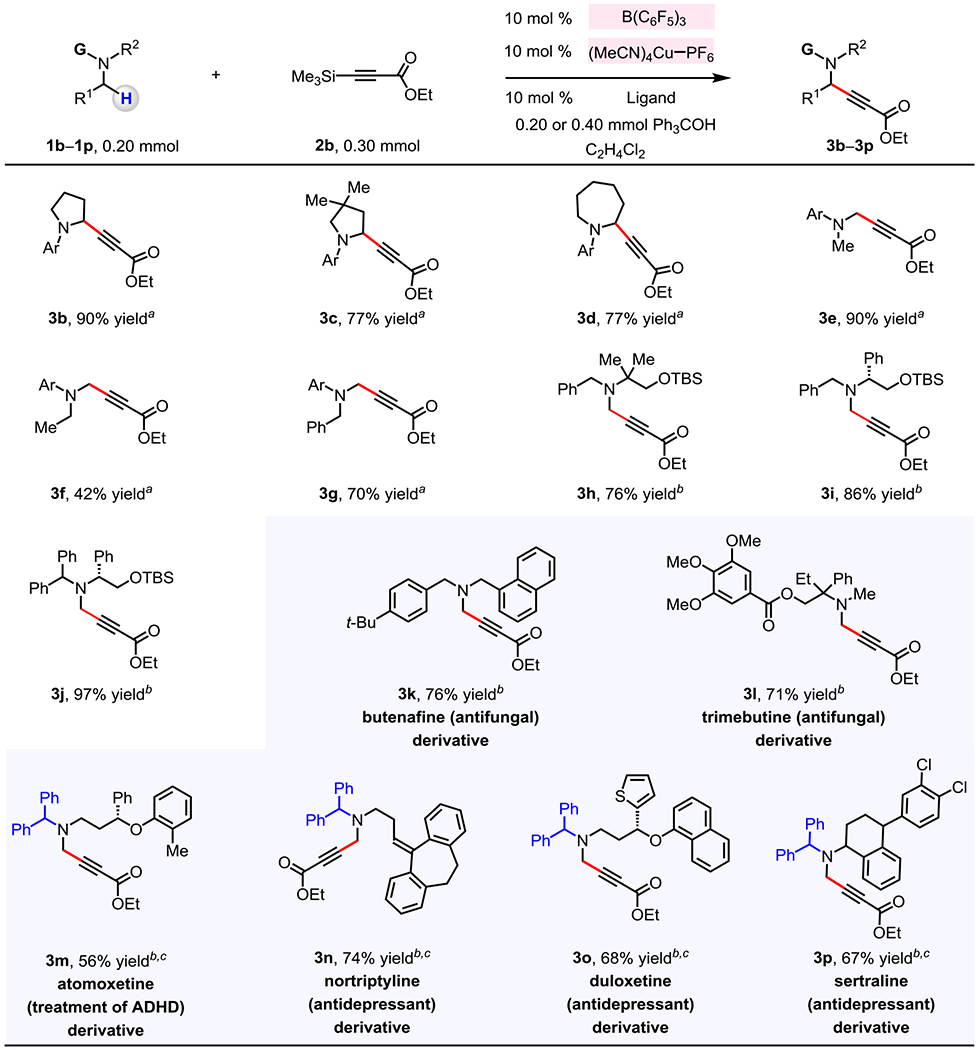
The values correspond to yields of isolated and purified products. a Conditions: N-alkylamine (1, 0.20 mmol), 3-(trimethylsilyl)propiolate (2b, 0.30 mmol), B(C6F5)3 (10 mol %), (MeCN)4CuPF6 (10 mol %), Xantphos (10 mol %), triphenylmethanol (0.20 mmol), C2H4Cl2 (0.4 mL), under N2 atmosphere, 60 °C, 12 h. b 1,2-Bis(diphenylphosphino)ethane (10 mol %) was used as a ligand, 0.40 mmol of triphenylmethanol was used, and the reaction mixture was allowed to stir at 80 °C for 24 h. c Blue color indicates protecting groups. See the Supporting Information for details.
The method is applicable to late-stage modification of N-containing bioactive molecules that possess an array of Lewis acid-sensitive functional groups (1k–1p; Figure 2). In addition to the N-alkylamine moieties of 1k–1p, an ester (1l), an ether (1l, 1m, 1o), a thienyl (1o) and an aryl chloride (1p) were tolerated, affording 3k–3p in 56–76% yield. The structures of antifungal compounds bearing a tertiary amine, such as butenafine 1k and trimebutine 1l, were readily altered (3k, 3l). For secondary amines, such as atomoxetine (used for treatment of ADHD), as well as antidepressants nortriptyline, duloxetine, and sertraline, incorporation of N-benzhydryl group was necessary for efficient generation of 3m–3p.
The catalytic protocol may involve the use of trimethylsilylacetylenes containing different alkynyl substituents (2b–2i, Figure 3). The reactions of fluoxetine derivative 1q with trimethylsilylacetylenyl esters (2b, 2c) and amide (2d) afforded 7b–7d in 76–82% yield. A series of phenyl-, para-(trifluoromethyl)phenyl-, para-chlorophenyl- or 3-thiophenyl-substituted trimethylsilylacetylenes were coupled with 1q to furnish 7e–7h in 74–82% yield. Whereas the transformation involving 1q and trimethylsilylacetylene (X = H) was inefficient (<10% yield),23 1,2-bis(trimethylsilyl)ethyne 2i proved to be a suitable reaction partner, affording 7i in 87% yield.
Figure 3. Reactions of N-Bzh fluoxetine with triemthylsilylacetylenes.
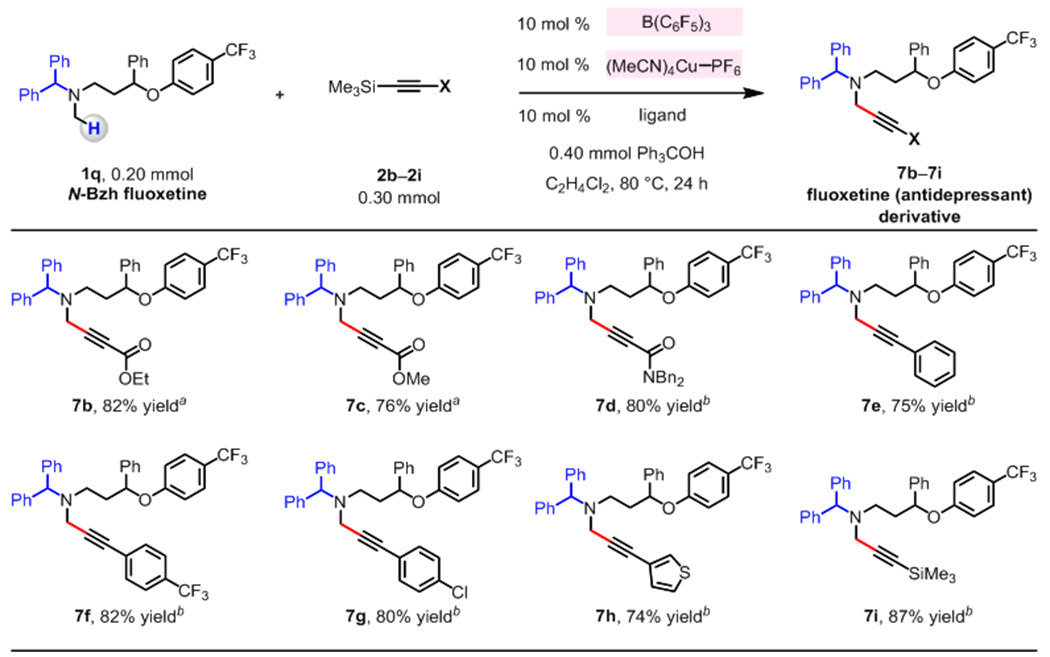
The values correspond to yields of isolated and purified products. The reaction conditions are identical to those in Figure 2, aside from the ligands used. Blue color indicates protecting groups. a 1,2-bis(diphenylphosphino)ethane was used as a ligand. b (S)-Ph–PyBOX was used as ligand. See the Supporting Information for details.
2.1.3. Diastereo- and Enantio-selective Processes.
To develop a stereoselective version of the catalytic C–alkynyl bond forming process, we chose to use B(C6F5)3 in combination with an appropriate chiral organocopper complex, with N-arylpyrrolidine 1b and 3-(trimethylsilyl)propiolate 2b as model substrates. Accordingly, we performed systematic evaluation of catalyst systems comprised of (MeCN)4CuPF6 and a chiral ligand (Figure 4). The effectiveness of various bis-phosphine ligands (e.g., L1–L2) were evaluated in the presence of 10 mol % of B(C6F5)3.23 These transformations afforded 8b with minimal enantiomeric purity. We then explored the suitability of bis-oxazoline ligands (e.g., L3–L7), leading us to establish that with (S)-Ph–PyBOX (L3), 8b can be obtained in 84% yield and 82:18 er. Enantioselectivity improved when more sizeable 2,6-bis((S)-4-(m-tolyl)-4,5-dihydrooxazol-2-yl)pyridine (L4) and 2,6-bis((S)-4-(3,5-dimethylphenyl)-4,5-dihydrooxazol-2-yl)pyridine (L5) were used: 8b was isolated in 53% yield and 90:10 er, and 75% yield and 95:5 er, respectively. Neither efficiency nor enantioselectivity improved when L6 and L7 were used.
Figure 4. Evaluation of chiral ligands.
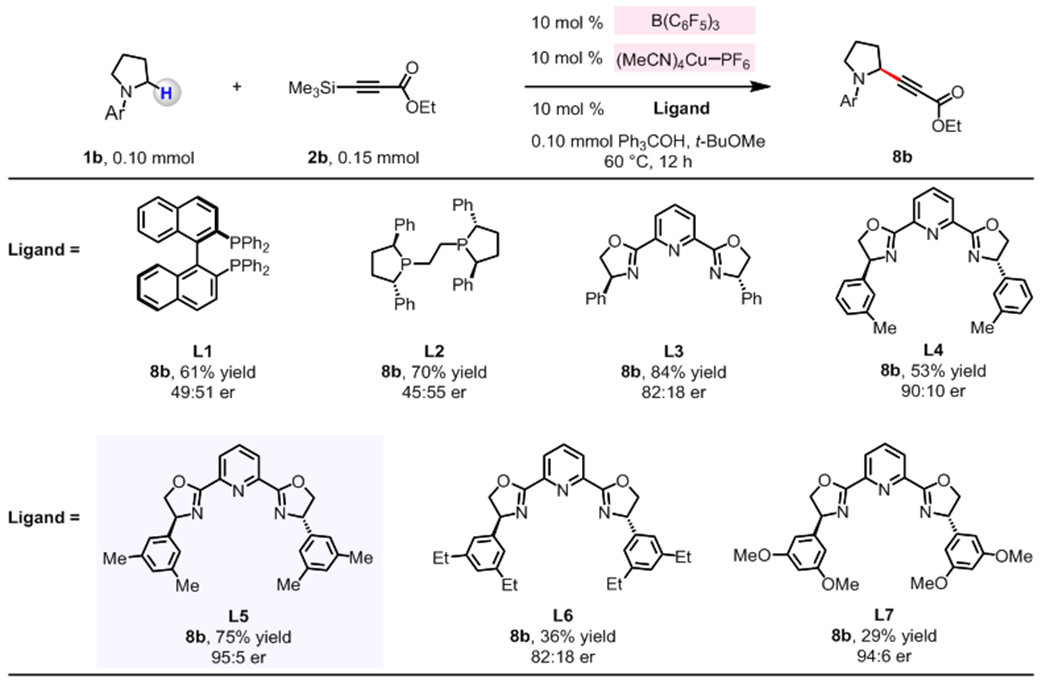
Yield values were determined by the 1H NMR analysis of unpurified reaction mixtures with mesitylene as the internal standard. Enantiomeric ratio (er) values were determined by the HPLC analysis of isolated and purified product. Conditions: N-arylpyrrolidine (1b, 0.10 mmol), 3-(trimethylsilyl)propiolate (2b, 0.15 mmol), B(C6F5)3 (10 mol %), (MeCN)4CuPF6 (10 mol %), ligand (10 mol %), triphenylmethanol (0.20 mmol), C2H4Cl2 (0.4 mL), under N2 atmosphere, 60 °C, 12 h. See the Supporting Information for details.
2.1.4. Stereoselective Synthesis and Functionalization of Propargylamines.
Reactions with an array of N-alkylamines were carried out in the presence of B(C6F5)3 and (MeCN)4CuPF6, L5 and 2b (Figure 5). N-Arylpyrrolidines ((S)-8b, 8c, 8d) as well as N-arylazepane (8e) bearing α-alkynyl group were thus synthesized in 64–75% yield and 83:17–95:5 er; there was minimal double-alkynyl byproduct formed (<5% yield). When rac-2-methyl-1-arylpyrrolidine was reacted with 2b, trans-8c was produced preferentially in 83:17 er. The reaction with 3,3-dimethyl-1-arylpyrrolidine and 2b furnished 8d as the sole regioisomer (vs the isomer formed through the formation of more sterically hindered iminium ion). An α-benzylic C–H bond of (E)-N,N-dibenzyl-4,4,4-trifluorobut-2-en-1-amine was functionalized to give propargylamine 8f in 45% yield and 84:16 er. Additionally, a range of enantiomerically enriched pyrrolidine substrates underwent transformation in the presence of B(C6F5)3 and (MeCN)4CuPF6/L5 to afford 8g–8i. With (S)-3-methyl- or (S)-3-phenyl-substituted pyrrolidine as substrate, reaction occurred at the less hindered α-amino C–H bond, affording 8g and 8h in 64% yield (11.8:1 trans:cis) and 68% yield (10.1:1 trans:cis), respectively. The union of a α-amino carbonyl compound19 and 2b resulted in the formation of 8i in 93% yield and 7.7:1 trans:cis ratio. The use of L5 was crucial in these latter processes, as 8g–8i were obtained in notably lower dr with an achiral ligand (e.g., Xantphos).23
Figure 5. Diastereo- and enantio-selective processes.
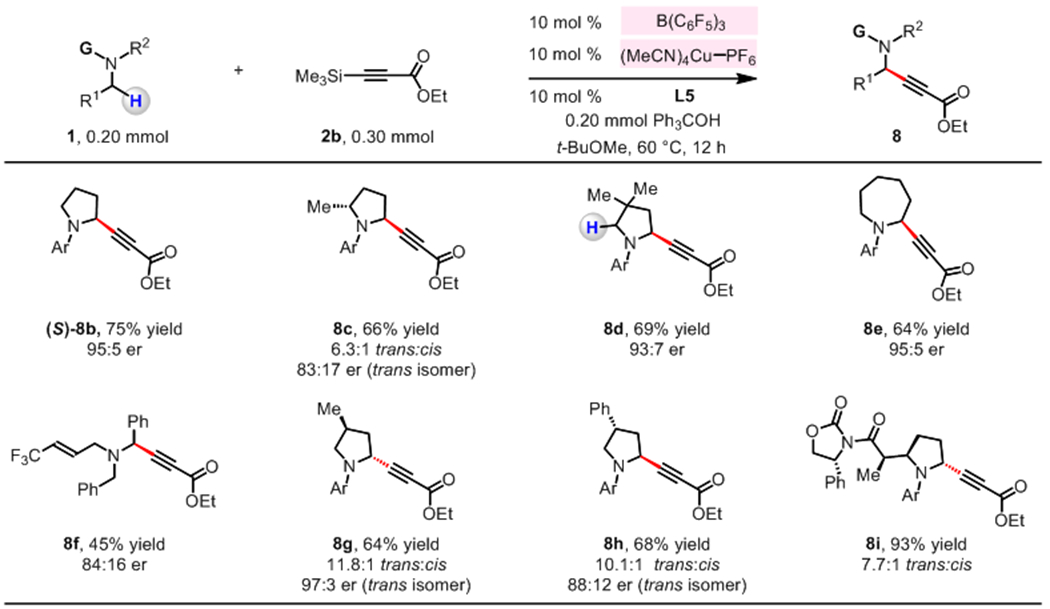
Cooperative functions of B(C6F5)3 and (MeCN)4CuPF6/L5 catalysts promote stereoselective conversion of N-alkylamines to the corresponding dialkyl propargylamines. Conditions: N-alkylamine (1, 0.20 mmol), 3-(trimethylsilyl)propiolate (2b, 0.30 mmol), B(C6F5)3 (10 mol %), (MeCN)4CuPF6 (10 mol %), L5 (10 mol %), triphenylmethanol (0.20 mmol), C2H4Cl2 (0.4 mL), under N2 atmosphere, 60 °C, 12 h. See the Supporting Information for details.
A benzhydryl group can be removed, as illustrated by the reaction of 1j and 2e with Et3SiH and trifluoroacetic acid, which afforded 9 in 64% yield (Figure 6a). The silyl moiety of fluoxetine derivative 7i (Figure 3) was excised by its treatment with (n-Bu)4NF, furnishing terminal alkyne 10 in >95% yield.23 Subjection of 10 with biotin-PEG3-azide to CuSO4/L-ascorbic acid and K2CO3 afforded heterocyclic derivative 11 in 70% yield (Figure 6b).24
Figure 6. Modification of propargyl amine products and scalability.
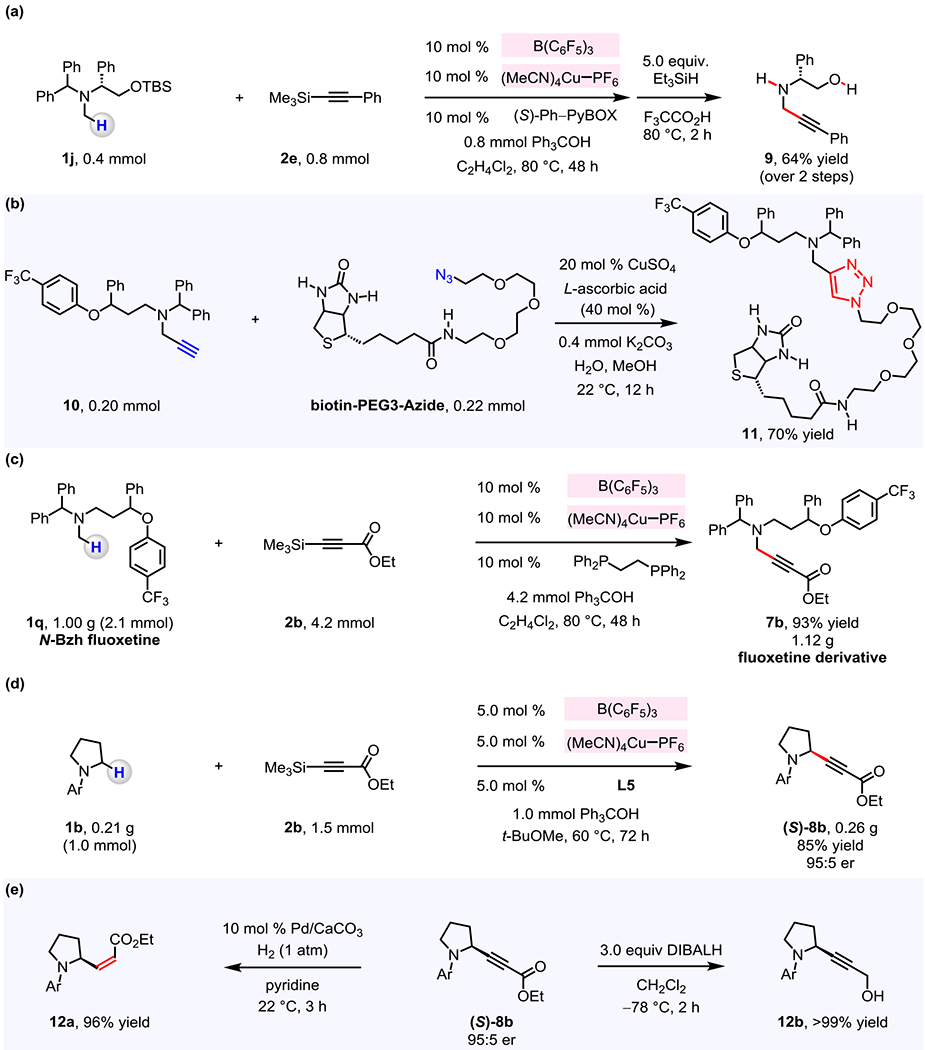
(a) Sequential conversion of C–H bond into C–alkynyl bond and removal of N-benzhydryl and O-TBS protecting groups can be achieved to afford propargylamine 9. (b) The fluoxetine derivative 10 can undergo organocopper-catalyzed Click reaction with biotin-PEG3-azide to give 11. (c) The method is amenable to gram-scale operations. (d) Enantioselective reactions may be carried out on 1.0 mmol scale. (e) The versatility of (S)-8b was demonstrated by its transformation to a Z-alkene 12a and a propargyl alcohol 12b. See the Supporting Information for details.
1.4. Scalability.
The catalytic method is scalable. For example, treatment of 1.0 g (2.1 mmol) of N-benzhydryl fluoxetine 1q and 2b with 10 mol % B(C6F5)3, 10 mol % (MeCN)4CuPF6/dppe, 2.0 equivalents of Ph3COH (C2H4Cl2, 48 h, 80 °C) afforded 7b in 93% yield (1.12 g; Figure 6c). Furthermore, enantioselective coupling of N-arylpyrrolidine 1b (0.21 g, 1.0 mmol) with 2b in the presence of 5.0 mol % B(C6F5)3, 5.0 mol % (MeCN)4CuPF6/L5, and 1.0 equivalent of Ph3COH (t-BuOMe, 72 h, 60 °C) gave (S)-8b in 85% yield (0.26 g) and 95:5 er (Figure 6d). Hydrogenation of (S)-8b delivered Z-alkene 12a in 96% yield and reduction of (S)-8b furnished propargyl alcohol 12b in >99% yield.
2.2. Mechanistic Investigations
We designed and performed studies aimed at shedding light on the mechanism of the catalytic process (a revised catalytic cycle, based on the investigations described below, is illustrated in Figure 7).
Figure 7. A catalytic cycle consistent with the results of mechanistic investigations.
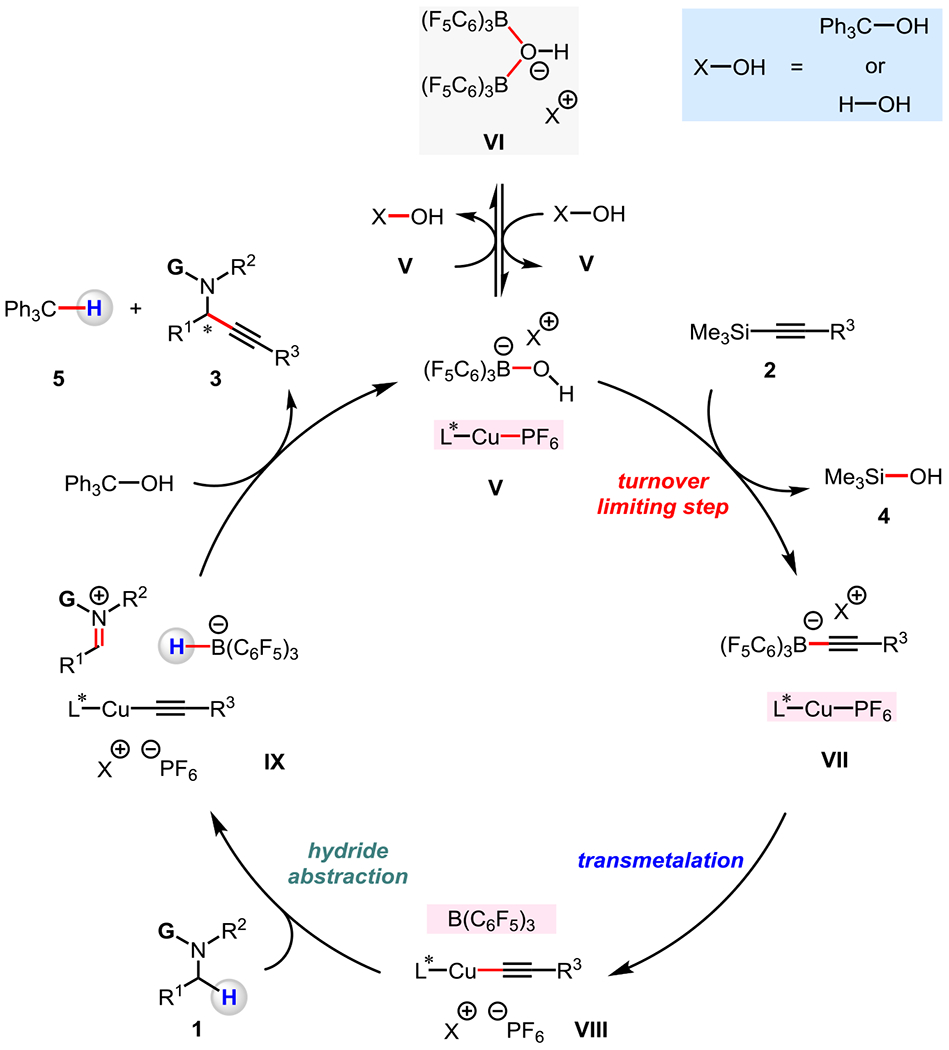
Kinetic and NMR studies indicate that the turnover-limiting step occurs prior to the (F5C6)3B-catalyzed hydride abstraction, and that the C–H bond cleavage step is irreversible.
2.2.1. Kinetic studies.
These investigations revealed that the rate of the reaction of 4-methoxy-N,N,2,6-tetramethylaniline 1e with ethyl 3-(trimethylsilyl)propiolate 2b is independent of the concentration of 1e, (MeCN)4CuPF6/Xantphos complex, and Ph3COH (Figure 8).23 However, there were 0.5-order dependence on the B(C6F5)3 concentration (Figure 8a),25 and 1.0-order dependence on the concentration of 2b (Figure 8b). These data imply that C–H bond cleavage through (F5C6)3B-catalyzed hydride abstraction (Figure 7, 1 → IX) occurs after the turnover-limiting step (energetic span).26,27 They further suggest that the transformation has a resting state that consists of two B(C6F5)3 units, such as an ionic complex containing a borate anion [(F5C6)3B(μ-OH)B(C6F5)3]– (VI, [X]+ = H+ and/or Ph3C+).28 The 11B NMR spectra acquired for the reaction mixture under the standard catalytic conditions (Figure 8) are in agreement with the formation of the borate anion VI.23, 28 In the presence of Ph3COH and/or H2O, two molecules of [(F5C6)3B–OH]–[X]+ (V) may be produced from VI.28 Ensuing reaction of V and trimethylsilylacetylene 2 to afford [(F5C6)3B–alkyne]– [X]+ (VII) is turnover-limiting. Treatment of preformed [(F5C6)3B–C≡C–CO2Et]−[H–NR3]+ (NR3 = 1e)29 with 100 mol % of (MeCN)4CuPF6/Xantphos complex was found to give propargylamine product 3e in 24% yield, thereby demonstrating the competency of intermediate VII in the alkyne incorporation process.23 Subsequent to the turn-over limiting step (V → VII), [(F5C6)3B–alkynyl]− [X]+ undergoes transmetalation with (MeCN)4CuPF6/Xantphos complex to afford a LnCu–alkynyl complex and B(C6F5)3, latter of which converts amine 1 into iminium ion through hydride abstraction (VII → VIII → IX). C–C bond formation between in situ generated LnCu–alkynyl complex and iminium ion would afford the desired propargylamine (IX → 3). The reaction between borohydride and Ph3COH would then produce Ph3C–H 5 and regenerate V, thereby closing the cycle.
Figure 8. Kinetic studies.

The reaction of 1e and 2b to afford propargylamine 3e was found to be 0.5-order in B(C6F5)3 and 1.0-order in alkyne, suggesting that the resting state of B(C6F5)3 contains two B(C6F5)3 units and that the turnover-limiting involves the reaction of 2b with in situ generated [(F5C6)3B–OH]− to give [(F5C6)3B–alkynyl] − and TMS–OH. (a) Log(rate) vs Log[B(C6F5)3] plot is employed to determine the reaction order for B(C6F5)3. (b) Log(rate) vs Log[2b] plot is employed to determine the reaction order for 2b. See the Supporting Information for details.
2.2.2. Kinetic Isotope Effect Studies.
To shed light on the hydride abstraction step (Figure 7, 1 → IX), deuterium-labeled methylaniline 1g-d was prepared, and its reaction with 2b was studied (Figure 9). Based on the aforementioned rate studies (Figure 8), which suggested that C–H bond cleavage might not be turnover-limiting, the overall rate of the reaction should be unaffected for a reaction involving 1g-d, and, indeed, there was no significant kinetic isotope effect with independent rate measurements (Figure 9a).26 On the other hand, with competition rate measurements, there could be an observable KIE if (F5C6)3B-catalyzed C–H bond cleavage step were irreversible (Figure 9b), as these experiments measure a change in product distribution that results from a difference in the rate of an irreversible C–H bond cleavage event.26 That is, these experiments should provide a product ratio that reflects a primary KIE, despite the C–H bond cleavage not being turnover-limiting.26 In the event, independent rate measurements (Figure 9a) involving 1g and 1g-d was found to have kH/kD = 1.02 ± 0.02 (average of 2 measurements;).23 What is more, intermolecular competition rate measurements (Figure 9b) showed that 1g reacts 4.4 times faster than 1g-d (kH/kD = 4.4). These isotope effect experiments support the notion that the turnover-limiting step is before the (F5C6)3B-catalyzed hydride abstraction, and that C–H bond cleavage step is irreversible.
Figure 9. Kinetic isotope effect studies.
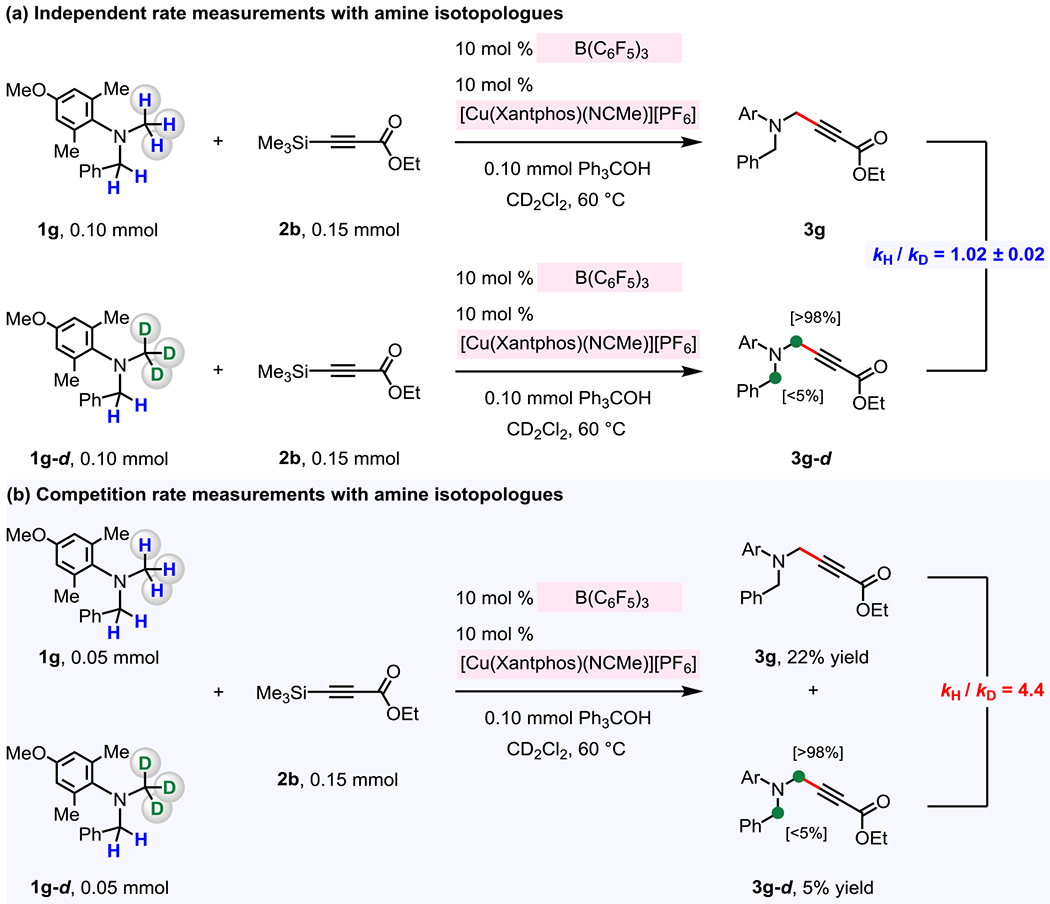
These studies indicate that hydride abstraction is not the turnover-limiting step, and yet the deuterium-labeling caused an amine to react 4.4 times slower in the competition rate measurement studies. See the Supporting Information for details.
2.2.3. Origin of Regioselectivity.
Next, we chose to investigate why an N-methyl C–H bond of an N-methyl-N-benzylamine moiety is preferentially activated (e.g., Figure 9b, 1g) while N-benzyl and N-benzhydryl groups remain intact (c.f., Figures 2, 3). We considered two possible scenarios. In one, B(C6F5)3 activation cannot convert an N-benzyl or an N-benzyhydryl group to the corresponding iminium intermediate ([ArMeN=CHPh]+ (e.g., Figure 10a, XI and XII), and in the other, the C-phenyl iminium intermediates are formed but are too hindered and/or not sufficiently electrophilic to react with a LnCu–alkynyl complex. To establish whether a C-phenyl iminium intermediate can be formed, we prepared 1g-d and subjected it to 10 mol % B(C6F5)3 at 60 °C for 16 hours (in C2H4Cl2; Figure 10a). The 1H NMR spectrum of purified 13g-d (>95% yield) indicated that 63% of benzylic C–H bonds were converted to C–D bonds, while 37% of N-methyl C–D bonds were transformed to C–H bonds. The finding that deuterium incorporation takes place at the benzylic site indicates that B(C6F5)3 is capable of generating a C-phenyl iminium ion (XI), which might occur through (F5C6)3B-catalyzed deuteride abstraction at the N–CD3 moiety of 1g-d to afford iminium ion X followed by isomerization to the lower energy intermediate XI.30 Subsequent reduction of C-phenyl iminium then furnishes a benzylic C–D bond (13g-d). Nonetheless, direct formation of C-phenyl iminium intermediate by (F5C6)3B-catalyzed benzylic C–H abstraction cannot be ruled out (1g-d → XII; Figure 10a).31
Figure 10. B(C6F5)3 promotes intramolecular and intermolecular H/D exchange.
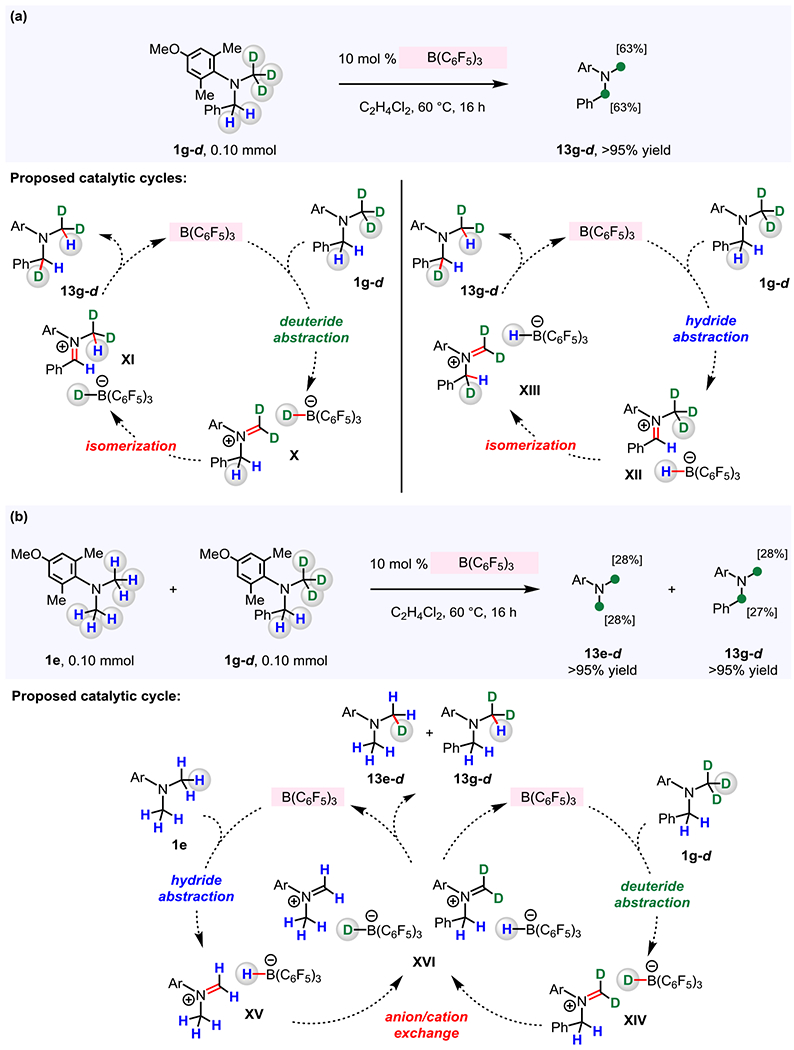
(a) (F5C6)3B-catalyzed H/D exchange occurs within N-benzyl-4-methoxy-2,6-dimethyl-N-(methyl-d3)aniline. (b) Intermolecular H/D exchange was shown to take place between 1e and 1g-d. See the Supporting Information for details.
Alternatively, intermolecular H/D exchange between two 1g-d molecules, promoted by B(C6F5)3, might generate 13g-d. That is, iminium/borohydride complexes X and XII (Figure 10a) could exchange their anionic and cationic components, after which hydride or deuteride iminium reduction might produce 13g-d. To probe whether H/D exchange is intermolecular, we treated a mixture of 1e and 1g-d in the presence of 10 mol % B(C6F5)3 (Figure 10b). Analysis of the corresponding 1H NMR spectrum and HRMS data of the resulting products 13e-d and 13g-d (both were obtained in >95% yield) revealed that 28% of N-methyl C–H bonds in 1e was converted to C–D bonds and 27% of N-benzylic C–H bonds in 1g-d was transformed to C–D bonds. This intermolecular H/D exchange reaction might proceed through formation of iminium complexes XIV and XV, generated by (F5C6)3B-catalyzed hydride or deuteride abstraction from 1e and 1g-d, respectively. The iminium/borohydride complexes XIV and XV could then exchange their anionic and cationic components followed by hydride or deuteride reduction of XVI to furnish 13e-d and 13g-d. These results (Figure 10) imply that in the absence of (MeCN)4CuPF6/Xantphos and 2b, the iminium/borohydride complexes are sufficiently long-lived to undergo isomerization and/or intermolecular anion/cation exchange.
To determine whether the H/D exchange reaction is possible under the standard reaction conditions, we performed the reaction involving 0.10 mmol 1g-d, 0.15 mmol of 3-(trimethylsilyl)propiolate 2b and 0.10 mmol Ph3COH (Figure 11a); this allowed us to isolate propargylamine product 3g-d in 31% yield. However, <5% of the benzylic C–H bonds in 3g-d were converted to C–D bonds, whereas the propargylic position of 3g-d retained >98% of C–D bonds from 1g-d. Additionally, there was no detectable H/D exchange in the recovered 1g-d (0.067 mmol of 1g-d was isolated); namely, there was no H/D exchange under the catalytic conditions (vs H/D exchange processes shown in Figure 10). The above findings indicate that the in situ generated [ArBnN=CD2]+[D–B(C6F5)3]– is short-lived and rapidly consumed by its reaction with LnCu–alkynyl complex and Ph3COH to afford propargylamine 3g-d and Ph3C–D 5-d (1 → IX → 3, Figure 7); thus, it neither undergoes intra- and/or intermolecular H/D exchange (vs the pathway in Figure 10) nor does borodeuteride reduction generate B(C6F5)3 and 1g-d.
Figure 11. Structure Determination of Products.
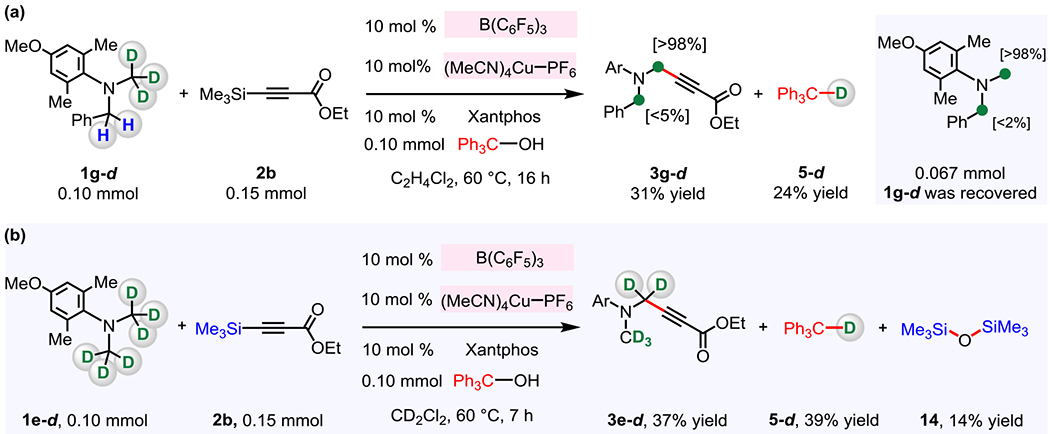
(a) Under the standard reaction conditions for installation of the alkyne unit, no intra- and/or intermolecular H/D exchange was observed between N–CD3 and N–CH2Ph groups, indicating that in situ generated iminium salt is rapidly consumed through C–alkynyl bond forming reaction vs H/D scrambling. (b) Spectroscopic studies (1H and 2H NMR) involving pure compounds revealed that Ph3C–D and Me3Si–O–SiMe3 are the stable byproducts. See the Supporting Information for details.
We examined the structure of byproducts to determine the fate of deuteride from amines 1g-d and 1e-d as well as trimethylsilyl group from 2b (Figure 11a–b). Based on our proposed mechanism (Figure 7), the expected byproducts generated by the reaction between 1g-d and 2b (to give 3g-d in 31% yield; Figure 11a) would be Ph3C–D (5-d) and Me3Si–OH (4). Although the formation of Ph3C–D (5-d) could be confirmed by 2H NMR spectroscopy of the unpurified mixture (24% yield), we were unable to detect any Me3Si–OH (4). To confirm that 4 is formed, we reacted 1e-d and 2b, which led to the formation of 3e-d (together with other byproducts; Figure 11b). Spectroscopic analysis indicated that when the latter mixture was heated at 60 °C for 7 hours, 3e-d (37% yield), Ph3C–D (5-d, 39% yield) and Me3SiO–SiMe3 (14, 14% yield) are generated; Me3Si–O–SiMe3 is likely produced by (F5C6)3B-catalyzed condensation between two molecules of Me3Si–OH (4) to afford 14 and H2O.32
3. CONCLUSIONS
In summary, we have developed an efficient and diastereo- and enantio-selective method for activation of α-amino C–H bonds to generate propargyl amines. We find that by using a blend of B(C6F5)3 and an organocopper complex, it is possible to generate an iminium from an N-alkylamine and a LnCu–alkynyl complex from an alkynylsilane. The catalyst system tolerates a wide variety of Lewis acid-sensitive functional groups and is therefore applicable to late-stage transformation of a complex (and bioactive) trialkyl amine molecule to its derived propargylamine. Mechanistic investigations indicate that the turnover-limiting step occurs prior to (F5C6)3B-catalyzed C–H abstraction, and that (F5C6)3B-catalyzed C–H abstraction is an irreversible process under the reaction conditions for alkyne incorporation. The principles outlined here demonstrate that proper combination of an achiral organoborane and a chiral organometallic catalyst can be used for chemo- and enantioselective C–H bond activation, providing a rational framework for further development of processes involving the late-stage stereoselective α-functionalization of bioactive amines. Studies aimed at achieving these objectives are currently underway.
Supplementary Material
ACKNOWLEDGEMENTS
Financial support was provided by the NIH (GM-128695), the Sloan Foundation, and Boston College. J.Z.C. and A. Y. were supported as John LaMattina Graduate Fellows in Chemical Synthesis. We thank Professor Amir H. Hoveyda for helpful discussions, and Professor Shih-Yuan Liu for assistance with mechanistic investigations. We are grateful to Professor Abhishek Chatterjee for a loan of biotin-PEG3-azide. Dr. Juan del Pozo del Valle, Dr. Yuebiao Zhou, Dr. Ryan Morrison, Ms. Katherine Grasso, and Ms. Eleni Kisty (Boston College) are also thanked for their assistance on this project. We are grateful to Dr. Bo Li (Boston College) for the X-ray crystallographic analysis.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.(a) Birks J; Flicker L Selegiline for Alzheimer’s disease. Cochrane Database of Systematic Reviews; John Wiley & Sons, Ltd: Chichester, UK, 2003,; Vol. 1, p CD00044210.1002/14651858.CD000442. [DOI] [PubMed] [Google Scholar]; Boulton AA; Davis BA; Durden DA; Dyck LE; Juorio AV; Li X-M; Paterson IA; Yu PH Aliphatic propargylamines: new antiapoptotic drugs. Drug Dev. Res 1997, 42, 150–156. [DOI] [PubMed] [Google Scholar]; (b) Arshadi S; Vessally E; Edjlali L; Hosseinzadeh-Khanmiri R; Ghorbani-Kalhor R N-Propargylamines: versatile building blocks in the construction of thiazole cores. Beilstein J. Org. Chem 2017, 13, 625–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Zani L; Bolm C Direct addition of alkynes to imines and related C=N electrophiles: A convenient access to propargylamines. Chem. Commun. 2006, 41, 4263–4275. [DOI] [PubMed] [Google Scholar]; (b) Trost BM; Weiss AH The enantioselective addition of alkyne nucleophiles to carbonyl groups. Adv. Synth. Catal. 2009, 351, 963–983. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lauder K; Toscani A; Scalacci N; Castagnolo D Synthesis and reactivity of propargylamines in organic chemistry. Chem. Rev 2017, 117, 14091–14200. [DOI] [PubMed] [Google Scholar]
- 3.(a) Wei C; Li C-J Enantioselective direct-addition of terminal alkynes to imines catalyzed by copper(I)pybox complex in water and in toluene. J. Am. Chem. Soc 2002, 124, 5638–5639. [DOI] [PubMed] [Google Scholar]; (b) Gommermann N; Koradin C; Polborn K; Knochel P Enantioselective, copper(I)-catalyzed three-component reaction for the preparation of propargylamines. Angew. Chem., Int. Ed 2003, 42, 5763–5766. [DOI] [PubMed] [Google Scholar]; (c) Knöpfel TF; Aschwanden P; Ichikawa T; Watanabe T; Carreira EM Readily available biaryl P,N ligands for asymmetric catalysis. Angew. Chem., Int. Ed 2004, 43, 5971–5973. [DOI] [PubMed] [Google Scholar]; (d) Bisai A; Singh VK Enantioselective one-pot three-component synthesis of propargylamines. Org. Lett 2006, 8, 2405–2408. [DOI] [PubMed] [Google Scholar]; (e) Hatano M; Asai T; Ishihara K Enantioselective alkynylation to aldimines catalyzed by chiral 2,2’-di(2-aminoaryloxy)-1,1’-binaphthyl-copper(I) complexes. Tetrahedron Lett. 2008, 49, 379–382. [Google Scholar]; (f) Yin L; Otsuka Y; Takada H; Mouri S; Yazaki R; Kumagai N; Shibasaki M Direct catalytic asymmetric alkynylation of ketoimines. Org. Lett 2013, 15, 698–701. [DOI] [PubMed] [Google Scholar]; (g) Takada H; Kumagai N; Shibasaki M Stereoselective total synthesis of KAE609 via direct catalytic asymmetric alkynylation to ketimine. Org. Lett 2015, 17, 4762–4765. [DOI] [PubMed] [Google Scholar]
- 4.Koradin C; Polborn K; Knochel P Enantioselective synthesis of propargylamines by copper-catalyzed addition of alkynes to enamines. Angew. Chem., Int. Ed 2002, 41, 2535–2538. [DOI] [PubMed] [Google Scholar]
- 5.For selected examples of α-C(sp3)–H alkynylation of N-alkylamines under oxidative conditions, see:; (a) Li Z; Li C-J CuBr-catalyzed efficient alkynylation of sp3 C–H bonds adjacent to a nitrogen atom. J. Am. Chem. Soc 2004, 126, 11810–11811. [DOI] [PubMed] [Google Scholar]; (b) Li Z; MacLeod PD; Li C-J Studies on Cu-catalyzed asymmetric alkynylation of tetrahydroisoquinoline derivatives. Tetrahedron: Asymmetry 2006, 17, 590–597. [Google Scholar]; (c) Lin W; Cao T; Fan W; Han Y; Kuang J; Luo H; Miao B; Tang X; Yu Q; Yuan W; Zhang J; Zhu C; Ma S Enantioselective double manipulation of tetrahydroisoquinolines with terminal alkynes and aldehydes under copper(I) catalysis. Angew. Chem., Int. Ed 2014, 53, 277–281. [DOI] [PubMed] [Google Scholar]; (d) Zhao C; Seidel D Enantioselective A3 reactions of secondary amines with a Cu(I)/acid-thiourea catalyst combination. J. Am. Chem. Soc 2015, 137, 4650–4653. [DOI] [PubMed] [Google Scholar]; (e) Sun S; Li C; Floreancig PE; Lou H; Liu L Highly enantioselective catalytic cross-dehydrogenative coupling of N-carbamoyl tetrahydroisoquinolines and terminal alkynes. Org. Lett 2015, 17, 1684–1687. [DOI] [PubMed] [Google Scholar]; (f) Xie Z; Liu X; Liu L Copper-catalyzed aerobic enantioselective cross-dehydrogenative coupling of N-aryl glycine esters with terminal alkynes. Org. Lett 2016, 18, 2982–2985. [DOI] [PubMed] [Google Scholar]
- 6.Li Z; Li C-J Catalytic enantioselective alkynylation of prochiral sp3 C–H bonds adjacent to a nitrogen atom. Org. Lett 2004, 6, 4997–4999. [DOI] [PubMed] [Google Scholar]
- 7.For selected examples of β- or γ-selective amino C–H functionalization chemistry, see:; (a) Shabashov D; Daugulis O Auxiliary-assisted palladium-catalyzed arylation and alkylation of sp2 and sp3 carbon–hydrogen bonds. J. Am. Chem. Soc 2010, 132, 3965–3972. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) He J; Li S; Deng Y; Fu H; Laforteza BN; Spangler JE; Homs A; Yu J-Q Ligand-Controlled C(sp3)–H Arylation and Olefination in Synthesis of Unnatural Chiral α–Amino Acids. Science 2014, 343, 1216–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Topczewski JJ; Cabrera PJ; Saper NI; Sanford MS Palladium-catalysed transannular C–H functionalization of alicyclic amines. Nature, 2016, 531, 220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Willcox D; Chappell BGN; Hogg KF; Calleja J; Smalley AP; Gaunt MJ A general catalytic β-C–H carbonylation of aliphatic amines to β-lactams. Science 2016, 354, 851–857. [DOI] [PubMed] [Google Scholar]; (e) Wang H; Tong H-R; He G; Chen G An enantioselective bidentate auxiliary directed palladium-catalyzed benzylic C–H arylation of amines using a BINOL phosphate ligand. Angew. Chem., Int. Ed 2016, 55, 15387–15391. [DOI] [PubMed] [Google Scholar]; (f) Zhang J ;Park S, Chang S Catalytic access to bridged sila-N-heterocycles from piperidines via cascade sp3 and sp2 C–Si bond formation. J. Am. Chem. Soc 2018, 140, 13209–13213. [DOI] [PubMed] [Google Scholar]
- 8.McGrath NA; Brichacek M; Njardarson JT A graphical journey of innovative organic architectures that have improved our lives. J. Chem. Educ 2010, 53, 1348–1349. [Google Scholar]
- 9.(a) Gevorgyan C; Liu J-X, Rubin M; Benson S; Yamamoto Y A novel reduction of alcohols and ethers with a HSiEt3/catalytic B(C6F5)3 system. Tetrahedron Lett. 1999, 40, 8919–8922. [Google Scholar]; (b) Parks DJ; Blackwell JM; Piers WE Studies on the mechanism of B(C6F5)3-catalyzed hydrosilation of carbonyl functions. J. Org. Chem 2000, 65, 3090–3098. [DOI] [PubMed] [Google Scholar]; (c) Oestreich M; Hermeke J; Mohr J A unified survey of Si–H and H–H bond activation catalysed by electron-deficient boranes. Chem. Soc. Rev 2015, 44, 2202–2220. [DOI] [PubMed] [Google Scholar]; (d) Siegel JS Silylium ions: from controversial beginnings to useful catalysts. Nat. Rev. Chem 2020, 4, 4–5. [Google Scholar]; (e) Walker JCL; Klare HFT; Oestreich M Cationic silicon Lewis acids in catalysis. Nat. Rev. Chem 2020, 4, 54–62. [Google Scholar]
- 10.(a) Rendler S; Oestreich M Conclusive evidence for an S(N)2-Si mechanism in the B(C6F5)3-catalyzed hydrosilylation of carbonyl compounds: implications for the related hydrogenation. Angew. Chem., Int. Ed 2008, 47, 5997–6000. [DOI] [PubMed] [Google Scholar]; (b) Sakata K; Fujimoto H Quantum Chemical Study of B(C6F5)3-Catalyzed Hydrosilylation of Carbonyl Group. J. Org. Chem 2013, 78, 12505–12512. [DOI] [PubMed] [Google Scholar]; (c) Houghton AY; Hurmalainen J; Mansikkamäki A; Piers WE; Tuononen HM Direct observation of a borane-silane complex involved in frustrated Lewis-pair-mediated hydrosilylations. Nat. Chem 2014, 6, 983–988. [DOI] [PubMed] [Google Scholar]; (d) Fallon T; Oestreich M A Constellation of Deuterium-Labeled Silanes as a Simple Mechanistic Probe not Requiring Absolute Configuration Determination. Angew. Chem., Int. Ed 2015, 54, 12488–12491. [DOI] [PubMed] [Google Scholar]
- 11.(a) Frustrated Lewis Pairs I: Uncovering and Understanding; Stephan DW; Erker G Eds.; Springer: Berlin, 2013; Vol. 332. [Google Scholar]; (b) Frustrated Lewis Pairs II: Expanding the Scope; Erker G; Stephan DW Eds.; Springer: Berlin, 2013; Vol. 334. [Google Scholar]; (c) Ashley AE; O’Hare D FLP-mediated activations and reductions of CO2 and CO. Top. Curr. Chem. 2013, 334, 191–218. [DOI] [PubMed] [Google Scholar]; (d) Feng X; Du H Metal-free asymmetric hydrogenation and hydrosilylation catalyzed by frustrated Lewis pairs. Tetrahedron Lett. 2014, 55, 6959–6964. [Google Scholar]; (e) Stephan DW; Erker G Frustrated Lewis pair chemistry: Development and perspectives. Angew. Chem., Int. Ed. 2015, 54, 6400–6441. [DOI] [PubMed] [Google Scholar]; (f) Stephan DW Frustrated Lewis pairs. J. Am. Chem. Soc 2015, 137, 10018–10032. [DOI] [PubMed] [Google Scholar]; (g) Stephan DW The broadening reach of frustrated Lewis pair chemistry. Science 2016, 354, aaf7229. [DOI] [PubMed] [Google Scholar]
- 12.(a) Ishihara K; Yamamoto H Arylboron Compounds as Acid Catalysts in Organic Synthetic Transformations. Eur. J. Org. Chem 1999, 527–538. [Google Scholar]; (b) Gevorgyan C; Liu J-X, Rubin M; Benson S; Yamamoto Y A novel reduction of alcohols and ethers with a HSiEt3/catalytic B(C6F5)3 system. Tetrahedron Lett. 1999, 40, 8919–8922. [Google Scholar]; (c) Blackwell JM; Foster KL; Beck VH; Piers WE B(C6F5)3-catalyzed silation of alcohols: A mild, general method for synthesis of silyl ethers. J. Org. Chem 1999, 64, 4887–4892. [DOI] [PubMed] [Google Scholar]; (d) Adduci LL; Bender TA; Dabrowski JA; Gagné MR Chemoselective conversion of biologically sourced polyols into chiral synthons. Nat. Chem 2015, 7, 576–581. [DOI] [PubMed] [Google Scholar]
- 13.For selected reviews on enantioselective cooperative catalysis, see:; (a) Yamamoto H; Futatsugi K “Designer acids”: combined acid catalysis for asymmetric synthesis. Angew. Chem., Int. Ed 2005, 44, 1924–1942. [DOI] [PubMed] [Google Scholar]; (b) Kobayashi S; Mori Y; Fossey JS; Salter MM Catalytic enantioselective formation of C–C bonds by addition to imines and hydrazones: a ten-year update. Chem. Rev 2011, 111, 2626–2704. [DOI] [PubMed] [Google Scholar]; (c) Shibasaki M; Kumagai N in Cooperative Catalysis: Designing Efficient Catalysts for Synthesis, Peters R, Eds.; Wiley-VCH: New York, 2015; Chapter 1. [Google Scholar]; (d) Romiti F; del Pozo J; Paioti PHS; Gonsales SA; Li X; Hartrampf FWW; Hoveyda AH Different Strategies for Designing Dual-Catalytic Enantioselective Processes: From Fully Cooperative to Non-cooperative Systems. J. Am. Chem. Soc 2019, 141, 17952–17961. [DOI] [PubMed] [Google Scholar]
- 14.For selected reviews on enantioselective non-covalent catalysis, see; (a) Ooi T; Maruoka K Recent advances in asymmetric phase-transfer catalysis. Angew. Chem., Int. Ed 2007, 46, 4222–4266. [DOI] [PubMed] [Google Scholar]; (b) Zhang Z; Schreiner PR (Thio)urea organocatalysis—What can be learnt from anion recognition? Chem. Soc. Rev 2009, 38, 1187–1198. [DOI] [PubMed] [Google Scholar]; (c) Brak K; Jacobsen EN Asymmetric ion-pairing catalysis. Angew. Chem., Int. Ed 2013, 52, 534–561. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Neel AJ; Hilton MJ; Sigman MS; Toste FD Exploiting non-covalent π interactions for catalyst design. Nature 2017, 543, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Millot N; Santini CC; Fenet B; Basset JM Formation and characterization of zwitterionic stereoisomers from the reaction of B(C6F5)3 and NEt2Ph: (E)- and (Z)-[EtPhN+=CHCH2–B−(C6F5)3]. Eur. J. Inorg. Chem 2002, 2002, 3328–3335. [Google Scholar]; (b) Dureen MA; Brown CC; Stephan DW Addition of enamines or pyrroles and B(C6F5)3 “Frustrated Lewis pairs” to alkynes. Organometallics 2010, 29, 6422–6432. [Google Scholar]
- 16.Focante F; Mercandelli P; Sironi A; Resconi L Complexes of tris(pentafluorophenyl)boron with nitrogen-containing compounds: Synthesis, reactivity and metallocene activation. Coord. Chem. Rev 2006, 250, 170–188. [Google Scholar]
- 17.(a) Maier AFG; Tussing S; Schneider T; Flörke U; Qu Z-W; Grimme S; Paradies J Frustrated Lewis pair catalyzed dehydrogenative oxidation of indolines and other heterocycles. Angew. Chem., Int. Ed 2016, 55, 12219–12223. [DOI] [PubMed] [Google Scholar]; (b) Kojima M; Kanai M Tris(pentafluorophenyl)borane-catalyzed acceptorless dehydrogenation of N-heterocycles. Angew. Chem., Int. Ed 2016, 55, 12224–12227. [DOI] [PubMed] [Google Scholar]
- 18.(a) Farrell JM; Heiden ZM; Stephan DW Metal-free transfer hydrogenation catalysis by B(C6F5)3. Organometallics 2011, 30, 4497–4500. [Google Scholar]; (b) Schwendemann S; Fröhlich R; Kehr G; Erker G Intramolecular frustrated N/B Lewis pairs by enamine hydroboration. Chem. Sci 2011, 2, 1842–1849. [Google Scholar]; (c) Chen J; Chen EX-Y Reactivity of amine/E(C6F5)3 (E = B, Al) Lewis pairs toward linear and cyclic acrylic monomers: hydrogenation vs. polymerization. Molecules 2015, 20, 9575–9590. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen G-Q; Kehr G; Daniliuc CG; Bursch M; Grimme S; Erker G Intermolecular redox-neutral amine C–H functionalization induced by the strong boron Lewis acid B(C6F5)3 in the frustrated Lewis pair regime. Chem. Eur. J. 2017, 23, 4723–4729. [DOI] [PubMed] [Google Scholar]
- 19.Shang M; Chan JZ; Cao M; Chang Y; Wang Q; Cook B; Torker S; Wasa M C–H Functionalization of amines via alkene-derived nucleophiles through cooperative action of chiral and achiral Lewis acid catalysts: Applications in enantioselective synthesis. J. Am. Chem. Soc 2018, 140, 10593–10601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Maier AFG; Tussing S; Zhu H; Wicker G; Tzvetkova P; Flörke U; Daniliuc CG; Grimme S; Paradies J Borane-catalyzed synthesis of quinolines bearing tetrasubstituted stereocenters by hydride abstraction-induced electrocyclization. Chem. Eur. J 2018, 24, 16287–16291; [DOI] [PubMed] [Google Scholar]; (b) Li R; Chen Y; Jiang K; Wang F; Lu C; Nie J; Chen Z; Yang G; Chen Y-C; Zhao Y; Ma C B(C6F5)3-Catalyzed redox-neutral β-alkylation of tertiary amines using p-quinone methides via borrowing hydrogen. Chem. Commun, 2019, 55, 1217–1220 [DOI] [PubMed] [Google Scholar]; (c) Tian J-J; Zeng N-N; Liu N; Tu X-S; Wang X-C Intramolecular cyclizations of vinyl-substituted N,N-dialkyl arylamines enabled by borane-assisted hydride transfer. ACS Catalysis 2019, 9, 295–300. [Google Scholar]; (d) Basak S; Alvarez-Montoya A; Winfrey L; Melen RL; Morrill LC; Pulis APB (C6F5)3-Catalyzed Direct C3 Alkylation of Indoles and Oxindoles. ACS Catal. 2020, 10, 4835–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chatterjee I; Qu Z-W; Grimme S; Oestreich M B(C6F5)3-catalyzed transfer of dihydrogen from one unsaturated hydrocarbon to another. Angew. Chem., Int. Ed 2015, 54, 12158–12162. [DOI] [PubMed] [Google Scholar]; (b) Keess S; Simonneau A; Oestreich M Direct and transfer hydrosilylation reactions catalyzed by fully or partially fluorinated triarylboranes: A systematic study. Organometallics 2015, 34, 790–799. [Google Scholar]
- 22.(a) Herron JR; Ball ZT Synthesis and reactivity of functionalized arylcopper compounds by transmetalation of organosilanes. J. Am. Chem. Soc 2008, 130, 16486–16487. [DOI] [PubMed] [Google Scholar]; (b) Lee K.-s.; Hoveyda AH Monodentate non-C2-symmetric chiral N-heterocyclic carbene complexes for enantioselective synthesis. Cu-catalyzed conjugate additions of aryl- and alkenylsilylfluorides to cyclic enones. J. Org. Chem 2009, 74, 4455–4462. [DOI] [PubMed] [Google Scholar]; (c) Gurung SK; Thapa S; Vangala AS; Giri R Copper-catalyzed Hiyama coupling of (hetero)aryltriethoxysilanes with (hetero)aryl iodides. Org. Lett 2013, 15, 5378–5381. [DOI] [PubMed] [Google Scholar]; (d) Denmark SE; Tymonko SA Cross-Coupling of Alkynylsilanols with Aryl Halides Promoted by Potassium Trimethylsilanolate. J. Org. Chem 2003, 68, 9151–9154. [DOI] [PubMed] [Google Scholar]
- 23.For further details, see the Supporting Information.
- 24.(a) Rostovtsev VV; Green LG; Fokin VV; Sharpless KB A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem., Int. Ed 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]; (b) Kolb HC; Sharpless KB The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 24, 1128–1137. [DOI] [PubMed] [Google Scholar]
- 25.Burés J A Simple Graphical Method to Determine the Order in Catalyst. Angew. Chem., Int. Ed. 2016, 55,2028–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Simmons EM; Hartwig JF On the Interpretation of Deuterium Kinetic Isotope Effects in C–H Bond Functionalizations by Transition-Metal Complexes. Angew. Chem., Int. Ed. 2012, 51, 3066–3072. [DOI] [PubMed] [Google Scholar]; (b) Blackmond DG Kinetic Profiling of Catalytic Organic Reactions as a Mechanistic Tool. J. Am. Chem. Soc 2015, 137, 10852–10866. [DOI] [PubMed] [Google Scholar]
- 27.(a) Kozuch S; Martin JML The Rate-Determining Step is Dead. Long Live the Rate-Determining State! ChemPhysChem 2011, 12, 1413–1418. [DOI] [PubMed] [Google Scholar]; (b) Kozuch S; Shaik S How to Conceptualize Catalytic Cycles? The Energetic Span Model. Acc. Chem. Res 2011, 44, 101–110. [DOI] [PubMed] [Google Scholar]
- 28.(a) Strasǎk T; Sykora J; Lamac M; Kubista J; Horaćek M; Gyepes R; Pinkas J Reactivity of a Titanocene Pendant Si–H Group toward Alcohols. Unexpected Formation of Siloxanes from the Reaction of Hydrosilanes and Ph3COH Catalyzed by B(C6F5)3. Organometallics 2013, 32, 4122–4129. [Google Scholar]; (b) Di Saverio A; Focante F; Camurati I; Resconi L; Beringhelli T; D’Alfonso G; Donghi D; Maggioni D; Mercandelli P; Sironi A Oxygen-Bridged Borate Anions from Tris(pentafluorophenyl)borane: Synthesis, NMR Characterization, and Reactivity. Inorg. Chem 2005, 44, 5030–5041. [DOI] [PubMed] [Google Scholar]
- 29.Dureen MA; Stephan DW Terminal Alkyne Activation by Frustrated and Classical Lewis Acid/Phosphine Pairs. J. Am. Chem. Soc 2009, 131, 8396–8397. [DOI] [PubMed] [Google Scholar]
- 30.Ma L; Paul A; Breugst M; Seidel D Redox-neutral aromatization of cyclic amines: Mechanistic insights and harnessing of reactive intermediates for amine α- and β-C–H functionalization. Chem. Eur. J 2016, 22, 18179–18189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farrell JM; Heiden ZM; Stephan DW Metal-free transfer hydrogenation catalysis by B(C6F5)3. Organometallics 2011, 30, 4497–4500. [Google Scholar]
- 32.Satoh Y; Igarashi M; Sato K; Shimada S Highly selective synthesis of hydrosiloxanes by Au-catalyzed dehydrogenative cross-coupling reactions of silanols with hydrosilanes. ACS Catal. 2017, 7, 1836–1840. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.