

Abstract
Degeneration of the cartilage endplate (CEP) induces intervertebral disc degeneration (IVDD). Nucleus pulposus cell (NPC) apoptosis is also an important exacerbating factor in IVDD, but the cascade mechanism in IVDD is not clear. We investigated the apoptosis of NPCs and IVDD when stimulated by normal cartilage endplate stem cell (CESC)‐derived exosomes (N‐Exos) and degenerated CESC‐derived exosomes (D‐Exos) in vitro and in vivo. Tert‐butyl hydroperoxide (TBHP) was used to induce inflammation of CESCs. The bioinformatics differences between N‐Exos and D‐Exos were analyzed using mass spectrometry, heat map, and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis. NPC apoptosis was examined using TUNEL staining. The involvement of the AKT and autophagy signaling pathways was investigated using the signaling inhibitor LY294002. Magnetic resonance imaging, Western blotting, and immunofluorescence staining were used to evaluate the therapeutic effects of N‐Exos in rats with IVDD. TBHP effectively induced inflammation and the degeneration of CEP in rat. N‐Exos were more conducive to autophagy activation than D‐Exos. The apoptotic rate of NPCs decreased obviously after treatment with N‐Exos compared to D‐Exos. N‐Exos inhibited NPCs apoptosis and attenuated IVDD in rat via activation of the AKT and autophagy pathways. These results are the first findings to confirm that CEP delayed the progression of IVDD via exosomes. The therapeutic effects of N‐Exos on NPC apoptosis inhibition and the slowing of IVDD progression were more effective than D‐Exos due to activation of the PI3K/AKT/autophagy pathway, which explained the increase in the incidence of IVDD after inflammation of the CEP.
Keywords: apoptosis, autophagy, cartilage endplate stem cells, exosome, intervertebral disc degeneration
Graphical abstract of the mechanism that cartilage endplate (CEP) inflammation accelerated the progression of intervertebral disc degeneration (IVDD). Normal cartilage endplate stem cell (CESC)‐derived exosomes (N‐Exos) can more effectively inhibit nucleus pulposus cell (NPC) apoptosis than degenerated CEPC‐derived exosomes (D‐Exos) due to the anti‐apoptotic protein carried by exosomes decreasing after the CEP degeneration. Furthermore, N‐Exos also activate the PI3K/AKT signaling pathway in NPC more conducively compared with D‐Exos, enhancing autophagy, alleviating NPC apoptosis in vitro and ameliorating IVDD in vivo

Significance statement.
Degeneration of cartilage endplate (CEP) induces intervertebral disc degeneration (IVDD), and nucleus pulposus cells (NPCs) apoptosis is also an important factor in exacerbating IVDD. Tert‐butyl hydroperoxide (TBHP) could effectively induced inflammation and degeneration of CEP in rat. CESC‐derived exosomes (N‐Exos) were more conducive to activating autophagy than D‐Exos. N‐Exos inhibited NPCs apoptosis or attenuated IVDD in a rat tail by activating the AKT and autophagy signaling pathways. The therapeutic effects of N‐Exos on inhibiting NPCs apoptosis and slowing IVDD progression was more effective than D‐Exos by activating the PI3K/AKT/autophagy pathway.
1. INTRODUCTION
Intervertebral disc degeneration (IVDD) is a common cause of lower back pain that limits activity. 1 It is generally characterized by the upregulation of matrix metalloproteinase (MMP) and proinflammatory cytokine expression, a reduction in the number of functional nucleus pulposus cells (NPCs) and anatomical and morphological changes. 2 , 3 The mechanism of IVDD includes a reduction in the nutrient supply from cartilage endplates (CEPs) to the inner layer of the annulus fibrosus and NPCs 4 and weakened CEP‐mediated regulation of IVDD‐associated anabolism and catabolism, 5 which lead to NPC senescence and apoptosis.
The CEP is a hyaline cartilage located on the upper and lower sides of the intervertebral disc. Previous studies showed that many progenitor cells differentiated into osteoblasts, adipocytes, and chondrocytes in human CEP tissues. 6 , 7 These progenitor cells were defined as cartilage endplate stem cells (CESCs), which are important in maintaining the integrity of the structure and function of CEPs. 8 CESCs powerfully inhibit IVDD by promoting NPC regeneration and regulating the homeostasis of the intervertebral disc. 9 , 10 , 11 However, the detailed mechanism is not clear. IVDD is more likely to occur after inflammation and the degeneration of CEP, 12 , 13 but the mechanisms are not clear. Multiple experiments showed that tert‐butyl hydroperoxide (TBHP) effectively induced the degeneration of stem cells. 14 , 15 , 16 Therefore, we used TBHP to induce and simulate the degeneration and inflammation of CEP and CESCs in vivo and in vitro. The present study examined normal CESC‐derived exosomes (N‐Exos) and degenerated CESC‐derived exosomes (D‐Exos) to elucidate the mechanism underlying the increased incidence of IVDD under the condition of CEP inflammation compared to a noninflammatory state and the novel mechanism of CEP inhibition of IVDD.
Exosomes are extracellular vesicles with diameters of 30 to 150 nm. Exosomes transport membrane components, proteins, microRNAs, and mRNAs into the intracellular environment upon fusion with the cytoplasmic membrane 17 , 18 and exert therapeutic effects on liver ischemia‐reperfusion injury and degenerative diseases. 19 , 20 , 21 Research showed that IVDD may be repaired via the conversion of normal CESCs into NPCs or strengthening the nutrient supply of the intervertebral discs via CEP. 5 , 6 However, whether normal CESCs secrete exosomes and the role secreted exosomes play in IVDD are not clear. The present study primarily investigated whether exosomes secreted by CESCs (CESC‐Exos) had a therapeutic effect on IVDD and the specific regulatory mechanism of the therapeutic effect. Recent research showed that human umbilical cord MSC‐derived exosomes exerted antiapoptotic effects via activation of the autophagy‐related signaling pathway phosphatidylinositol 3‐kinase (PI3K)/AKT/mTOR. 22 Therefore, we hypothesized that CESC‐Exos would inhibit NPC apoptosis and ameliorate IVDD by activating intracellular autophagy pathways.
Autophagy is an essential intracellular catabolic process that contributes to intracellular quality control and maintains cell survival by degrading and recycling damaged components and toxic proteins and organelles under inflammatory, nutrient deprivation, and stress conditions. 23 , 24 , 25 Autophagy degrades apoptotic proteins to regulate the progression of various diseases, such as cancer, 26 neurodegenerative diseases, and osteoarthritis. 27 , 28 The enhancing of autophagy in IVDD via activation of the PI3K/p‐AKT signaling pathway significantly reduced the expression levels of SASP proteins, such as IL‐6, IL‐1β, and TNFα, and inhibited aging and apoptosis. 27 , 29 Moreover, exosomes derived from MSCs promote autophagy and inhibit apoptosis. 22 , 30 Therefore, we hypothesized that N‐Exos also inhibited NPC apoptosis via activation of the PI3K/AKT/autophagy signaling pathway.
The present study analyzed the differences in bioinformatics between N‐Exos and D‐Exos and investigated the inhibitory effect of N‐Exos on IVDD. We showed for the first time that normal CESCs secreted exosomes and inhibited NPC apoptosis in vitro and in vivo. Mechanistic experiments showed that N‐Exos effectively activated the PI3K/p‐AKT/autophagy signaling pathway and inhibited NPC apoptosis and IVDD compared to D‐Exos. Our study offers new insights into IVDD treatment strategies using N‐Exos as a therapeutic tool.
2. MATERIALS AND METHODS
2.1. Reagents and antibodies
The AKT inhibitor (LY294002) and antibodies against CD81 and CD9 were obtained from Beyotime (Shanghai, China). Antibodies against cleaved caspase3, Bax, Bcl‐2, Beclin‐1, IL‐6, IL‐1β, TNF‐α, GAPDH, p‐ERK1/2, p‐AKT, Tsg101, CD63, and Alix were purchased from Proteintech (Wuhan, China). Antibodies against AKT, ERK1/2, JNK/p‐JNK, and NFκB/p‐NFκB were obtained from Abcam (Cambridge, Massachusetts). Antibodies against LC3A/B were purchased from KleanAB (Shanghai, China). Antibodies against LC3B were purchased from Bioss (Beijing, China). Collagenase II was purchased from Sangon Biotech (Shanghai, China). TBHP and PKH67 were obtained from Sigma (St. Louis, Missouri). 1,1′‐Dioctadecyl‐3,3,3′,3′‐tetramethylindotricarbocyanine iodide (DIR) was obtained from Invitrogen (Carlsbad, California). MSC osteogenic differentiation medium, chondrogenic differentiation medium, and adipogenic differentiation medium were provided by Cyagen (Guangzhou, China), Cell‐Light EdU Apollo488 in vitro Kit was purchased from RIBOBIO (Guangzhou, China).
2.2. Statistics of patients with lumbar disc herniation
Clinical statistics on the recurrence rate of 243 patients with lumbar disc herniation with (163 cases; males: 70, females: 93; age: 56 [28‐76 years old]) or without (80 cases; males: 49, females: 31; age: 53 [32‐70 years old]) CEP degeneration after surgery were collected. We obtained written informed consent from patients or relatives prior to tissue collection. The Ethics Committee of the Xinqiao Hospital of Army Medical University approved the present study (AF/SC‐08/1.0).
2.3. Rat CEP and IVDD model and reagent treatment
Adult male SD rats (n = 59, 12 weeks old) were obtained from the Experimental Animal Center of the Army Military Medical University (ChongQing, China). Nine adult male rats were used to construct the CEP degenerated model. Ten adult male rats were used to construct the IVDD with or without CEP inflammation model. Forty adult male rats were used for the IVDD model and reagent treatment for the in vivo experiments. The injection method of the rat IVD site is shown in Figure S1. See the Supplemental Materials and Methods file for more details. The Animal Ethics Committee Army Medical University approved all studies (No. SYXK(yu)2017‐0002).
2.4. CESC and NPC isolation
Ten young rats (aged 2‐3 weeks) were euthanized via cervical dislocation after anesthetization to isolate and extract CESCs and NPCs. Six to eight segments of caudal vertebrae were obtained from the caudal root using a surgical tip blade. The fiber ring of the intervertebral disc was cut using a surgical blade to obtain normal rat NP tissues. The CEP tissue on the upper and lower sides of the NP tissues was removed. The NP tissues and CEP tissues were cut into 1‐mm3 pieces. Four milliliters of 0.2% type 2 collagenase was added, and the tissues were gently shaken and digested at 37°C for 1 to 2 hours and filtered through a 70‐μm filter. The rat NPCs and CESCs were collected, seeded in cell culture flasks and cultured in DMEM F12 medium containing 20% fetal bovine serum at 5% CO2 and 37°C.
2.5. Osteogenic, adipogenic, and chondrogenic differentiation
CESCs were cultured in 6‐well plates for osteogenic, adipogenic, and chondrogenic differentiation. See the Supplemental Materials and Methods file for more details.
2.6. Exosome isolation, characterization, and fluorescent staining
Third‐ to fifth‐generation CESCs in good condition were selected to extract exosomes. After the cells reached 80% confluence in T75 culture flasks, the culture supernatant was collected 3 days later to extract normal CESC‐derived exosomes, or the culture supernatants of CESCs treated with 100 μmoL/mL TBHP was collected 3 days later to extract degenerated CESC‐derived exosomes. The supernatant was centrifuged at 2000g for 10 minutes at 4°C to remove cell debris then centrifuged at 110 000g for 70 minutes at 4°C to isolate the exosomes. The pellet was resuspended in 1 mL of PBS, and a 0.22‐μm filter membrane was used for filter sterilization. The sample was centrifuged again at 110 000g for 70 minutes and washed once, and the exosome pellet was collected in 200 μL of PBS. Five microliters of PKH67 or DIR was added to 500 μL of diluent to form the reaction liquid, and 100 μL of exosomes was added to the reaction liquid. After 10 minutes, 5% BSA was added to stop the reaction, and exosomes labeled with different membrane dyes were obtained. The size and concentration of exosomes were measured using nanoparticle tracking analysis (NTA) (Wayen Biotechnologies, Shanghai). Exosome morphology was examined using electron microscopy, and the purity and characteristics were analyzed using Western blot (WB) based on the expression of exosome markers (Tsg101 and Alix).
2.7. Transmission electron microscopy (TEM)
The cells were separated via trypsin digestion and fixed with 2% glutaraldehyde at 4°C for 2 days. The samples were treated with 1% osmate for 30 minutes, and 50%, 70%, 80%, and 100% ethanol gradient dehydration was performed. After the samples were soaked in a 100% acetone/Epon 812 (Shell Chemical Co, Houston, Texas) solution, ultrathin sections (60 nm) were prepared, stained with 5% uranium acetate for 30 to 60 minutes and stained with lead citrate for 10 minutes. The samples were observed under a Tecnai‐10 transmission electron microscope (Philips, Amsterdam, The Netherlands).
2.8. Quantitative real‐time PCR
Total RNA was extracted from rat CESCs to synthesize cDNA according to the manufacturer's instructions. Reverse transcription and quantitative renal‐time PCR were performed using a PrimeScript RT reagent kit and SYBR Premix Ex Taq (Takara, Japan). All data are reported as means ± standard error of mean normalized to β‐actin. The primer sequences are listed in Table S1.
2.9. Western blotting
After the cells were lysed in RIPA buffer containing the protease inhibitor PMSF, the protein concentration was determined, and 4× Laemmli sample buffer (Catalog: 161074, Bio‐Rad) was added to the sample. Electrophoresis and semidry transfer electrophoresis were performed. After the membrane was sealed and the antibody was added, the membrane was incubated at 4°C with shaking overnight. Diluted second antibody was added and incubated for 1.5 hours. The membrane was covered with the ECL working solution (Millipore, St. Louis, Missouri) and placed in the imaging system (Bio‐Rad) to obtain images.
2.10. TUNEL staining
An apoptosis‐related TUNEL staining kit (Beyotime, Shanghai, China) was used to detect the level of apoptosis according to the protocol. The level of apoptosis was indicated by red staining in the nucleus. Different areas of the sample were randomly selected and captured under a fluorescence microscope (Olympus, Tokyo, Japan) to count the number of TUNEL‐positive cells.
2.11. Immunofluorescence and immunohistochemistry
When the NPCs reached 60% confluence, the NPCs were used for immunofluorescence staining. After dewaxing and hydration, tissue sections were used for immunohistochemical staining. Antigen repair, peroxidase elimination and serum blocking were performed according to the instructions. Specific primary antibodies were added and incubated at 4°C overnight. The NPC specimens were incubated with the fluorescently labeled secondary antibody at 37°C for 60 minutes. The tissue sections were also incubated with a special secondary antibody (KIT‐9707, MXB, China). Color development of tissue sections was performed using a DAB reagent. After the nuclei were stained with DAPI or hematoxylin (G1080, Solarbio, China), images of the specimens were captured using a microscope (Olympus) or laser confocal microscope (Lexia, Japan).
2.12. Flow cytometry to detect apoptosis and identify CESCs
The Annexin V‐APC/PI apoptosis detection kit (BD Biosciences, California) was used to detect and assess the rate of apoptosis via flow cytometry. The cells were digested and collected with 0.25% trypsin‐EDTA, washed with PBS, resuspended in 100 μL of binding buffer and stained with 5 μL of annexin V‐APC and 5 μL of 7‐AAD. After the cells were incubated for 30 minutes at room temperature, the apoptosis rate of the cells was detected in a flow cytometer (BD Biosciences). FlowJo software was used to analyze the collected data. To identify CESCs, antibodies against CD44 (103,005, BioLegend, San Diego, California), CD90 (202,503, BioLegend), and CD45 (103,107, BioLegend) were used. CESCs were collected within three generations and washed three times with PBS. After the cells were incubated at room temperature for 30 minutes and washed with PBS, the percentage of CESCs was detected and analyzed using flow cytometry (BD FACSCalibur).
2.13. Magnetic resonance imaging
One, 3, and 6 weeks after IVDD construction with or without the CEP inflammation model or 6 weeks after exosome injection or reagent treatment, the signal and structural changes in the disc were assessed using a 7.0‐T animal magnet (Bruker Pharmascan, Germany) based on the sagittal T2‐weighted images. To obtain clear imaging results, the parameters of the T2‐weighted sections were set as previously described. 31 The severity of IVDD was analyzed in a double‐blinded manner according to the Pfirrmann grading system.
2.14. 5‐Ethynyl‐29‐deoxyuridine (EdU) assay
The cells were treated with N‐Exos/D‐Exos and incubated for 2 hours with an EdU solution diluted with cell culture medium at a ratio of 1000:1. The cells were subjected to paraformaldehyde fixation. We performed Apollo staining using the Apollo staining reaction solution and DNA staining with Hoechst 33342 reaction solution. After the nuclei were stained with DAPI, images of the specimens were captured using a fluorescence microscope.
2.15. Data analysis
All data are presented as the means ± standard deviation of at least three independent experiments. Tukey's test for comparisons between the two groups was used to analyze and compare the results using GraphPad Prism 7.0 (GraphPad Software Inc, San Diego, California). P values <.05 were considered statistically significant.
3. RESULTS
3.1. Inflammation and degeneration of CEP accelerates IVDD in human and rat
The clinical data of IVDD patients revealed that the recurrence rate of patients with lumbar disc herniation with CEP inflammation after surgery was 11.66%, which was obviously higher than the 2.5% recurrence rate of IVDD patients without CEP inflammation (P = .03; Figure 1A). We speculated that the CEP played an important role in delaying IVDD. To verify this hypothesis, TBHP was used to construct an in vivo animal model of CEP inflammation and degeneration. PBS (50 μL) or TBHP (50 μL, 100 μmoL/mL) was injected into the CEP (Figure 1B). The results showed that the expression levels of inflammatory factors IL‐6 and TNF‐α in the normal control group and PBS group was significantly lower than the TBHP group (Figure 1C), which indicated that TBHP induced CEP Inflammation/degeneration in vivo. We studied whether CEP inflammation aggravated IVDD by constructing an IVDD with or without CEP inflammation model (Figure 1D). The MRI score based on the sagittal T2‐weighted images indicated that the severity of IVDD in the puncture + TBHP group was higher than the puncture group at 3 and 6 weeks (Figure 1E). The IL‐6 protein levels of CEP and NP in the puncture + TBHP groups were also increased significantly compared to the puncture groups at 3 and 6 weeks using immunohistochemical staining, and the nucleus pulposus became more narrowed in the puncture + TBHP group (Figure 1F). Therefore, TBHP may be used to induce and simulate the degeneration process of CEP and CESCs in vivo, which aggravated the progress of IVDD. Previous studies that demonstrated that the presence of CESCs in the CEP of humans and mice performed an important function in inhibiting IVDD 8 , 12 , 32 and the degeneration of CEP was accompanied by the occurrence and persistence of inflammation. We speculated that the CESCs played an important role in the inflammatory response of CEP degeneration.
FIGURE 1.
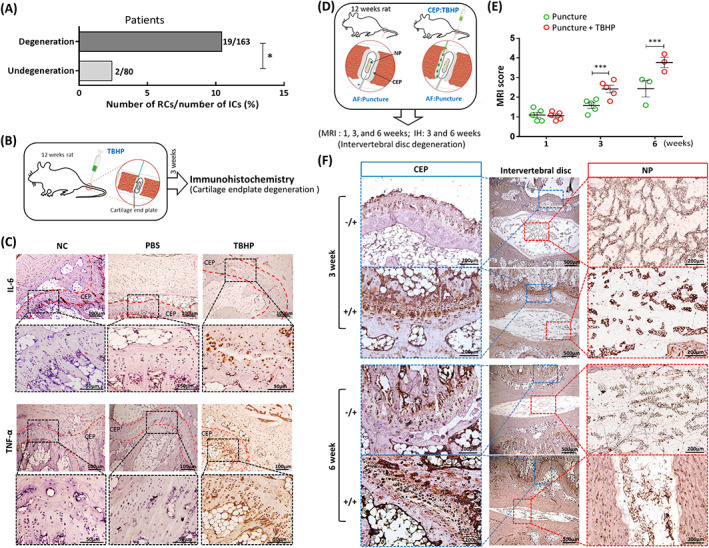
Degeneration of cartilage endplate (CEP) accelerates intervertebral disc degeneration (IVDD) in humans and rats. A, Clinical statistics on the recurrence rate of 243 patients with lumbar disc herniation with (163 cases) or without (80 cases) CEP degeneration after surgery. B, Tert‐butyl hydroperoxide (TBHP)‐induced model diagram of CEP degeneration in rat. C, Representative immunohistochemical staining of IL‐6 and TNF‐α in CEP under different treatments with microsyringe (NC, PBS [50 μL] or TBHP [50 μL,100 μmoL/mL]). D, The degeneration model diagram of puncture‐induced IVDD with or without TBHP‐induced CEP inflammation. AF:Puncture, puncture into annulus fibrosus; CEP:TBHP, TBHP was injected into CEP); IH, immunohistochemistry; MRI, magnetic resonance imaging; NP, nucleus pulposus. E, The IVDD of rats was evaluated using Pfirrmann grading according to the T2‐weighted images at 1, 3, and 6 weeks after the puncture or TBHP+puncture. F, Representative immunohistochemical staining of IL‐6 in CEP and NP 3 and 6 weeks after puncture or TBHP+puncture treatment (n = 5 per group; *P < .05). −/+, only puncture; +/+, TBHP and puncture; AF, annulus fibrosus; NC, normal control; ns: P > .05; *P < .05; **P < .01; ***P < .001
3.2. Identification of CESCs‐Exos and TBHP‐induced CESC degeneration
We first isolated CESCs from rat CEP tissues. We extracted CESCs from 2 to 3‐week‐old rats and identified CESCs by osteogenic, chondrogenic, and adipogenic differentiation (Figure 2A,B). The flow cytometry results showed that most CESCs positively expressed the stem cell surface markers CD90 and CD44 and did not express the differentiated cell surface marker CD45 (Figure 2C). After enrichment and extraction, the exosomes secreted by CESCs were identified by analyzing the morphology, size and marker proteins of the exosomes using TEM, NTA, and WB analyses, respectively (Figure 2D‐F). The results demonstrated that rat CESCs had functions similar to other types of stem cells that secreted exosomes, which were detected using marker proteins of exosomes, such as Alix, TSG101, CD9, CD63, and CD81. To verify whether TBHP induced the inflammation and degeneration of CESCs in vitro, different concentrations of TBHP or different induction durations were used to treat CESCs. The results of RT‐qPCR and WB indicated that gene expression levels of IL‐6, TNF‐α, IL‐1β, and MMP13 increased significantly with increasing TBHP concentrations and duration (Figure 2G,H). TBHP may be used in vitro to simulate the state of inflammation or degeneration of CESCs in vivo.
FIGURE 2.
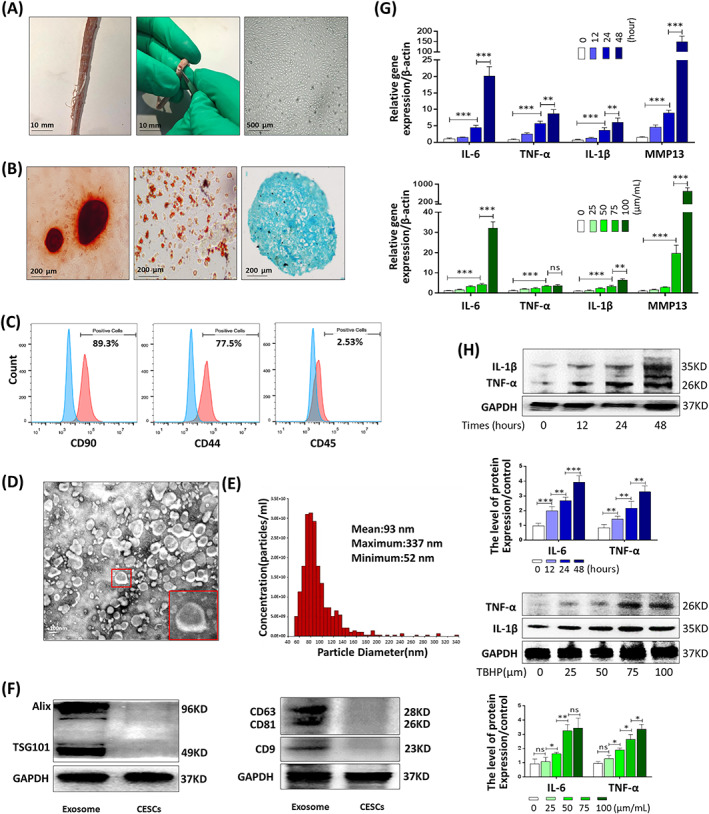
Identification of exosomes derived from rat cartilage end plate stem cells (CESCs‐Exos) and tert‐butyl hydroperoxide (TBHP)‐induced CESC degeneration. A, Horizontal views of rat cartilage endplate (CEP) and morphology of P3 CESCs at 100% confluence. B, After osteogenic induction for 14 days (left panel), adipogenic induction for 15 days (middle panel), and chondrogenic induction for 21 days (right panel), the ability of CESCs to differentiate into different cell lines was confirmed using Alizarin Red staining, Oil red O staining, and Alcian blue staining, respectively. C, Cell surface markers (CD90, CD44, and CD45) of CESCs was detected using flow cytometric analysis. The red curves represent the fluorescence intensity of CESCs stained with the corresponding antibodies. D, TEM images were used to identify the morphology of CESCs‐Exos. E, Nanoparticle trafficking analysis (NTA) was used to analyze the particle size distribution of CESCs‐Exos. F, Representative Western blots of Alix and TSG101 in CESCs‐Exos and CESCs. G, The gene expression of IL‐6, TNF‐α, IL‐1β, and MMP13 in the CESCs treated with different concentrations of TBHP (0, 25, 50, 75, or 100 μm) for 48 hours or TBHP (100 μm) for the indicated time points (0, 12, 24, or 48 hours). H, The Western blotting and quantitative protein levels of IL‐1β and TNF‐α in the CESCs as treated above. ns: P > .05; *P < .05; **P < .01; ***P < .001
3.3. N‐Exos were more conducive to activation of autophagy than D‐Exos
Because the incidence of IVDD increased after CEP inflammation or degeneration, we analyzed the differences in the regulation of cell signaling pathways and functions of exosomes secreted by normal CESCs or TBHP‐induced degenerated CESCs. Mass spectrometry and heat map analysis of the obtained N‐Exos and D‐Exos revealed a clear difference between the proteins carried by N‐Exos and D‐Exos (Figure 3A). KEGG enrichment analysis and gene ontology (GO) data analysis of the proteins contained in N‐Exos and D‐Exos, and the results showed that N‐Exos and D‐Exos regulated different cell biological functions, such as cell phagosome and leukocyte transendothelial migration (Figure 3B,C; Figure S2A,B). Furthermore, KEGG enrichment analysis of the top 50 quantitative differential proteins between N‐Exos and D‐Exos (Figure 3D) demonstrated that N‐Exos were more conducive to activation of autophagy than D‐Exos. These results suggested that N‐Exos suppressed IVDD via the regulation and activation of autophagy.
FIGURE 3.
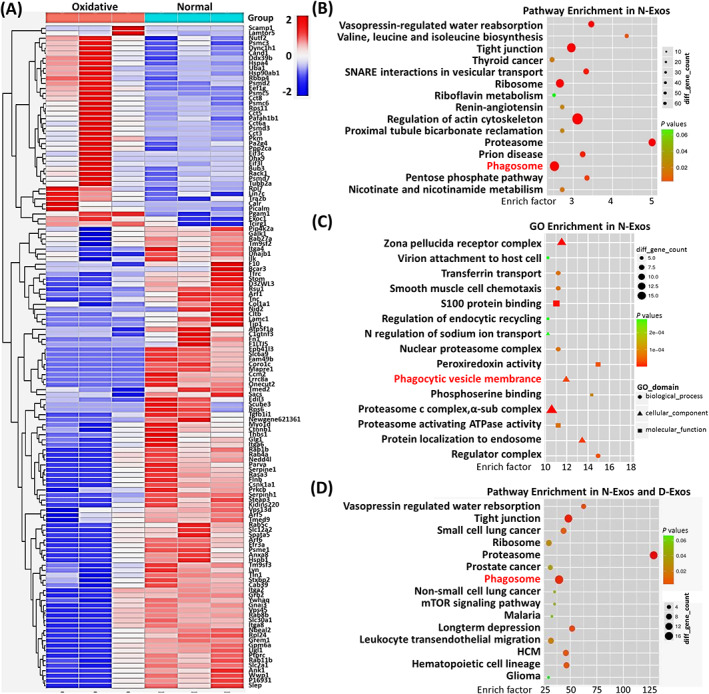
Bioinformatics analysis between normal cartilage end plate stem cell (CESC)‐derived exosomes (N‐Exos) and degenerated CESC‐derived exosomes (D‐Exos). A, Heat map analysis of differential proteins between N‐Exos and D‐Exos. B,C, KEGG enrichment analysis and gene ontology (GO) data analysis of proteins contained in N‐Exos. D, KEGG enrichment analysis of the top 50 quantitative differential proteins between N‐Exos and D‐Exos
3.4. N‐Exos more effectively attenuated rat NPC apoptosis than D‐Exos
Normal CEP and CESCs inhibit NPC apoptosis by providing nutrients, inhibiting the inflammatory response and differentiating into NPCs. 7 , 33 However, whether secreted exosomes inhibit NPC apoptosis is not clear. Mass spectrometry analysis and GO data analysis of the differential proteins between N‐Exos and D‐Exos revealed that many important proteins in the N‐Exos negatively regulated apoptosis (Figure 4A). We isolated, extracted, and cultured rat NPCs (Figure 4B). Rat NPCs were identified by a high expression of collagen II and low expression of collagen I (Figure 4C). N‐Exos and NPCs were cocultured, and we found that exosomes were taken up into the cytoplasm of NPCs (Figure 4D). We also verified that the expression level of the apoptotic protein Bax in the NPCs increased significantly with increased TBHP concentration (Figure 4E). Therefore, NPCs were treated with TBHP (100 μmoL/mL), TBHP (100 μmoL/mL) + D‐Exos (40 μg/mL), and TBHP (100 μmoL/mL) + N‐Exos (40 μg/mL) to verify the conclusion that the antiapoptotic effects of N‐Exos was more effective. TUNEL staining and flow cytometry analysis of apoptosis suggested that fewer NPCs underwent apoptosis in the presence of 40 μg/mL N‐Exos (Figure 4F,G). Representative WB and quantitative analysis of cleaved caspase3, Bax, and Bcl‐2 expression in NPCs also showed that N‐Exos had antiapoptotic effects, and the antiapoptotic effect was more obvious with increasing concentrations of exosomes (Figure 4H). Cell proliferation assay showed that N‐Exos promoted the proliferation of NPCs more than D‐Exos (Figure S3). We analyzed the concentration differences of other inflammatory/nutritional factors contained in the two exosomes and found that Igf2r and tgfb1i1 were more prevalent in N‐Exos than D‐Exos (Figure S4A,B).
FIGURE 4.
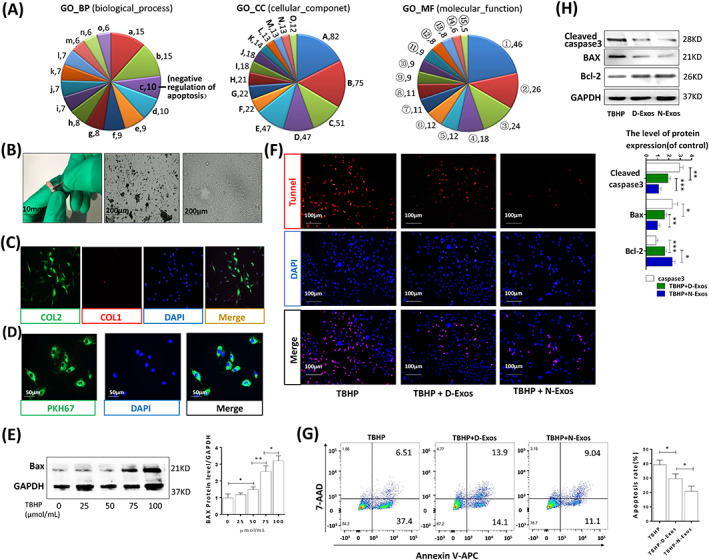
N‐Exos more effectively inhibited apoptosis compared to D‐Exos. A, Gene ontology (GO) analysis of all differential proteins carried in the normal cartilage end plate stem cell (CESC)‐derived exosomes (N‐Exos) and degenerated CESC‐derived exosomes (D‐Exos). a: small GTPase mediated signal transduction; b: protein transport; c: negative regulation of apoptotic process; d: response to drug; e: cell‐cell adhesion; f: cell adhesion; g: exocytosis; h: positive regulation of gene expression; i: cell‐matrix adhesion; j: protein folding; k: vesicle‐mediated transport; l: positive regulation of cell migration; m: substrate adhesion‐dependent cell spreading; n: protein heterooligomerization; o: positive regulation of angiogenesis. A: extracellular exosome; B: cytoplasm; C: membrane; D: plasma membrane; E: nucleus; F: Golgi apparatus; G: cytosol; H: focal adhesion; I: extracellular matrix; J: perinuclear region of cytoplasm; K: intracellular; L: cell‐cell adherens junction; M: protein complex; N: cell surface; O: centrosome. ①: protein binding, ②: poly(A) RNA binding, ③: ATP binding, ④: GTP binding, ⑤: cadherin binding involved in cell‐cell adhesion, ⑥: protein kinase binding, ⑦: protein domain specific binding, ⑧: identical protein binding, ⑨: integrin binding, ⑩: GTPase activity, ⑪: protein complex binding, ⑫: GDP binding, ⑬: unfolded protein binding, ⑭: ion channel binding, ⑮: glycoprotein binding. B, Morphological observations of rat NP tissue (left panel), P1 NPCs at 50% confluence (middle panel) and P3 NPCs at 100% confluence (middle panel). C, Double immunofluorescence staining of collagen II (green) and collagen I (red) in NPCs. D, Representative images of NPCs incubated with PBS or PKH67‐labeled N‐Exos for 24 hours. E, Western blotting and quantitative levels of apoptotic protein Bax in the NPCs treated with different concentrations of TBHP (0, 25, 50, 75, or 100 μmoL/mL) for 48 hours. F‐H, TUNEL staining, flow cytometry, representative Western blots, and quantification data of cleaved caspase3, Bax, and Bcl‐2 in NPCs treated with TBHP (100 μmoL/mL), TBHP (100 μmoL/mL) + D‐Exos (40 μg/mL), and TBHP (100 μmoL/mL) + N‐Exos (40 μg/mL). ns: P > .05; *P < .05; **P < .01; ***P < .001
3.5. N‐Exos regulated autophagic flux and inhibited rat NPC apoptosis
Exosomes attenuate diabetic nephropathy and D‐GaIN/LPS‐induced hepatocyte injury via the promotion of autophagic flux and apoptosis inhibition. 34 , 35 Therefore, we also examined whether exosomes inhibited NPCs apoptosis by promoting autophagic flux. Mass spectrometry and KEGG enrichment analysis of all differential proteins carried in the extracted N‐Exos and D‐Exos revealed that N‐Exos may also inhibit NPC apoptosis by activating autophagy (Figure 5A). The supernatants of TBHP‐treated or untreated CESCs with the same volume were collected after the same incubation time to extract N‐Exos and D‐Exos. We found that although D‐Exos contained 1.93 times the protein of N‐Exos, the average abundance of autophagy proteins and AKT pathway proteins in N‐Exos was more than three times D‐Exos. Overall, N‐Exos activated AKT and autophagy pathways more effectively than D‐Exos (Figure S5). We also detected that N‐Exos promoted the expression of collagen II and inhibited the release of inflammatory factors (such as IL‐6, IL‐1β) in NPCs, which may be caused by the differentiation of NPCs (Figure S6). We divided the NPCs into four groups for different treatments: NC, TBHP, TBHP+D‐Exos, and TBHP+N‐Exos. LC3‐B and cleaved caspase3 immunofluorescence double staining results showed that apoptotic protein expression was increased in cells treated with TBHP, but this increase in autophagosome fluorescence intensity and apoptotic protein expression were reversed in cells treated with TBHP+D‐Exos and TBHP+N‐Exos (Figure 5B). Although the number of autophagosomes increased in NPCs treated with TBHP+D‐Exos or TBHP+N‐Exos compared to the NC group or NPCs treated with TBHP, the number of autophagosomes was the largest in the TBHP+N‐Exos group as observed by TEM (Figure 5C). We also found that the LC3B/A ratio and levels of the autophagy‐associated protein Beclin‐1 were increased and the apoptosis‐related proteins cleaved caspase3 and Bax were reduced in NPCs treated with TBHP+D‐Exos or TBHP+N‐Exos, especially the TBHP+N‐Exos group, compared to the NPCs treated with TBHP (Figure 5D). These results suggested that N‐Exos inhibited NPC apoptosis by regulating autophagic flow, and N‐Exos was better than D‐Exos.
FIGURE 5.
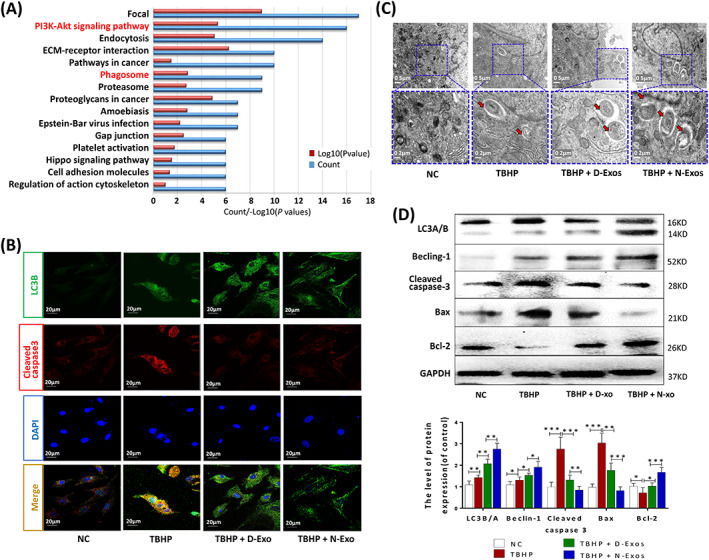
N‐Exos more effectively promoted nucleus pulposus cell (NPC) autophagy and apoptosis inhibition than D‐Exos. A, KEGG enrichment analysis of all differential proteins carried in the normal CESC‐derived exosomes (N‐Exos) and degenerated CESC‐derived exosomes (D‐Exos). B, Double immunofluorescence staining of LC3‐B (green) and cleaved caspase3 (red) in NPCs treated with NC (0 μg/mL), TBHP (100 μmoL/mL), TBHP (100 μmoL/mL) + D‐Exos (40 μg/mL), or TBHP (100 μmoL/mL) + N‐Exos (40 μg/mL). C, Autophagosomes (black arrow: autophagosome) were examined using TEM after NPCs were treated as described above. D, Representative Western blots and quantitative data of LC3A/B, Beclin‐1, cleaved caspase3, Bax, and Bcl‐2 expression in NPCs treated with NC, TBHP (100 μmoL/mL), TBHP (100 μmoL/mL) + D‐Exos (40 μg/mL), or TBHP (100 μmoL/mL) + N‐Exos (40 μg/mL). NC, normal control; ns: P > .05; *P < .05; **P < .01; ***P < .001
3.6. N‐Exos inhibit NPC apoptosis via activation of the PI3K/AKT/autophagy signaling pathway
MSC‐Exos effectively repaired critical size osteochondral defects via activation of AKT and ERK signaling in an immunocompetent rat model. 36 Based on the results of KEGG enrichment analysis (Figure 5A; Figure S5), we hypothesized that N‐Exos would activate autophagy via the PI3K/AKT pathway. We treated NPCs with NC, D‐Exos, and N‐Exos and found that the p‐AKT signaling pathway was activated when NPCs were treated N‐Exos. We examined other signaling pathways, such as p‐ERK1/2 and p‐JNK, but there were no significant changes (Figure 6A). We found that the apoptotic rate of NPCs and the apoptotic protein expression of cleaved caspase3 and Bax decreased significantly, and the ratio of the autophagy protein LC3B/A and the level of p‐AKT increased after activation of the p‐AKT signaling pathway. However, the AKT inhibitor LY294002 (20 μmoL/mL treatment for 3 days) inhibited these effects (Figure 6B,C). Immunofluorescence staining of p‐AKT and TUNEL staining also demonstrated that N‐Exos inhibited NPC apoptosis via activation of the PI3K/AKT/autophagy signaling pathway (Figure 6D).
FIGURE 6.
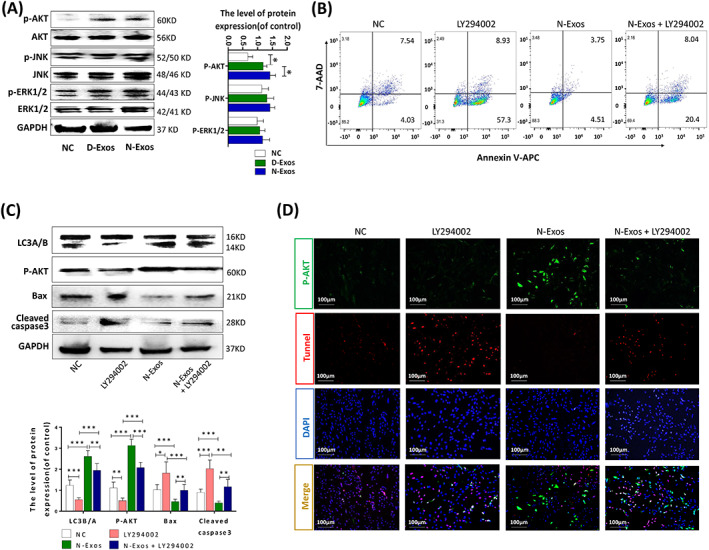
N‐Exos inhibited nucleus pulposus cells (NPCs) apoptosis by activating the PI3K/AKT/autophagy signaling pathway. A, Western blot analysis and quantitative data of p‐AKT, AKT, p‐JNK, JNK p‐ERK1/2, and ERK1/2 expression in NPCs treated with NC (0 μg/mL), D‐Exos (40 μg/mL), and N‐Exos (40 μg/mL). B, Flow cytometry was used to detect apoptosis of NPCs treated with NC (0 μg/mL), LY294002 (20 μmoL/mL), N‐Exo (40 μg/mL), and LY294002 (20 μmoL/mL) + N‐Exos (40 μg/mL). C,D, Representative Western blots and quantitative data of LC3A/B, p‐AKT, cleaved caspase3, and Bax expression and double immunofluorescence staining of p‐AKT (green) and TUNEL (red) in NPCs treated as described above. NC, normal control; ns: P > .05; *P < .05; **P < .01; ***P < .001
We investigated whether N‐Exos derived from CESCs promoted CESC differentiation. The results showed that the expression level of NPC biomarkers (such as SOX9 and collagen II) increased after the addition of N‐Exos to a CESC culture system (Figure S7). These suggested that N‐Exos promoted the differentiation of CSEC cells into NPCs. The mRNA levels of osteogenic and chondrogenic genes were also increased (Figure S8) in culture. N‐Exos may play more extensive roles in IVDD, and deeper research should be performed.
3.7. N‐Exos alleviated the progression of IVDD in rat
Exosome and reagent treatment in the IVD and the subsequent experimental steps are shown in Figure 7A. To confirm that exosomes can be injected into the IVD, we labeled exosomes with DIR and performed live imaging. We found that DIR‐labeled N‐Exos (DIR‐N‐Exos) had a signal in the IVD compared to unlabeled exosomes after 6 hours (Figure 7B). To further confirm that N‐Exos inhibited NPC apoptosis and ameliorated IVDD via activation of the AKT/autophagy pathway, we divided the rats into five groups: the NC group, puncture, puncture+LY294002 (20 μmol) group, puncture+N‐Exos (40 μg) group, and puncture+N‐Exos (40 μg) + LY204002 (20 μmol) group. MRI of the treatment site was performed 6 weeks later. The results suggested that puncture aggravated disc degeneration compared to the control group. The PI3K/AKT inhibitor LY294002 exacerbated the progression of IVDD, and exosomes alleviated disc degeneration, but LY294002 attenuated exosome‐mediated inhibition of IVDD progression (Figure 7C). We extracted NP tissue from the treatment site 3 weeks after surgery for WB. The results showed that the ratio of LC3B/A and the level of p‐AKT in the puncture+LY294002 group were significantly reduced compared to the control group, and the ratio of LC3B/A and the level of p‐AKT in the puncture+exosome group were significantly increased. The inhibitor LY294002 inhibited the exosome‐mediated promotion of NPC autophagy and activation of the AKT pathway (Figure 7D). The immunofluorescence staining results also showed that exosomes inhibited the expression of cleaved caspase3 and activated the AKT/autophagy pathway, but LY294002 attenuated these effects of N‐Exos (Figure 7E,F). We demonstrated the possible mechanism that CEP inflammation accelerated the progression of IVDD, that is, N‐Exos effectively inhibited NPCs apoptosis by activating autophagy via the PI3K/AKT signaling pathway compared to D‐Exos in vivo and in vitro (Figure 7G).
FIGURE 7.
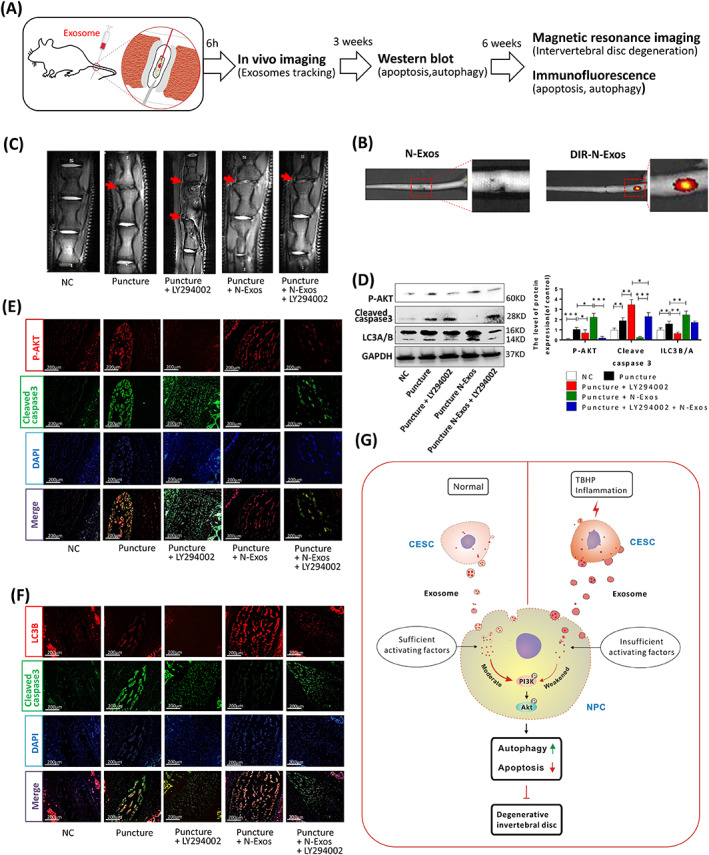
N‐Exos alleviated disc degeneration via activation of the PI3K/AKT/autophagy pathway in rat. A, Exosome and reagent treatment via microsyringe in the intervertebral disc degeneration (IVDD) and the subsequent experimental steps. B, in vivo imaging of rat IVD and the vertebral segments treated with unlabeled N‐Exos or DIR‐labeled N‐Exos (DIR‐N‐Exos). C, The representative images of MRI of rat intervertebral disc treated with NC, Puncture, Puncture+LY294002 (20 μmoL/mL), Puncture+ N‐Exos (40 μg/mL), and Puncture+ N‐Exos (40 μg/mL) + LY294002 (20 μmoL/mL). D, The Western blotting and quantification data of LC3A/B, p‐AKT, capase3, and Bax in the rat intervertebral disc 3 weeks after the above‐listed treatments. E,F, Representative double immunofluorescence of p‐AKT (red) and cleaved caspase3 (green) images and LC3B (red) and cleaved caspase3 (green) images of rat discs in each group (n = 5 per group; *P < .05). G, Graphical abstract of the mechanism of CEP inflammation acceleration of the progression of IVDD. Normal cartilage end plate stem cell (CESC)‐derived exosomes (N‐Exos) more effectively inhibit NPC apoptosis than degenerated CEPC‐derived exosomes (D‐Exos) due to the decrease in anti‐apoptotic proteins carried by exosomes after CEP degeneration. N‐Exos also better activated the PI3K/AKT signaling pathway in NPCs compared to D‐Exos, enhanced autophagy, alleviated NPC apoptosis in vitro, and ameliorated IVDD in vivo. NC, normal control; ns: P > .05; *P < .05; **P < .01; ***P < .001
4. DISCUSSION
Exosomes are extracellular vesicles that effectively deliver substances and are widely used in the treatment of various diseases by participating in intercellular communication. The present study first proposed that CESCs in the rat CEP located on the upper and lower sides of the IVD secreted exosomes. Normal CESC‐derived exosomes activated more autophagy than degenerated CESC‐derived exosomes. Although CESCs located near IVD are more difficult to obtain compared to mesenchymal stem cells (MSCs), CESCs better simulated the repair process of pathological changes in IVDD via activation of the AKT/autophagy pathway and inhibition of NPC apoptosis. Previous studies showed that the CEP played an important role in suppressing IVDD, 37 but whether this function was performed via exosomes derived from CESCs was not studied.
Previous research showed that CEP degeneration significantly accelerated the progress of IVDD via upregulation of the expression of inflammatory factors TNF‐α/IL‐1β in NPCs or promotion of NPC apoptosis. 38 , 39 The present study also found that the recurrence rate of patients with lumbar disc herniation with CEP inflammation increased significantly compared to patients without CEP after surgery. CESCs are the internal mediators of maintaining the integrity of CEP structure and physiological function. 7 We hypothesized that CEP inflammation exacerbated the progression of IVDD, primarily due to the weakened function of CESCs to inhibit IVDD. Because CESCs and MSCs have similar secretion characteristics of stem cells, CESCs also function as MSCs and secrete exosomes. Multiple experiments showed that TBHP effectively induced the degeneration of stem cells. 14 , 15 , 16 Therefore, we also used TBHP to induce and simulate the degeneration and inflammation of CEP and CESCs in vivo and in vitro. We found that TBHP effectively induced the inflammation of CEP and CESCs and exacerbated IVDD to simulate the degeneration process in vivo by checking the expression levels of inflammatory factors. MSC‐Exos inhibit NPC apoptosis and IVD via modulation of endoplasmic reticulum stress. 40 However, the mechanism of CESC‐derived exosomes in the inhibition of IVDD is not clear. We analyzed the differences between N‐Exos and D‐Exos in regulating cell signaling pathways and functions. The results showed N‐Exos were more conducive to autophagy activation than D‐Exos, which suggests that N‐Exos inhibit IVDD via activation of autophagy. This effect may explain the increased incidence of IVDD after inflammation of the CEP, that is, the degenerated CESC‐derived exosomes had a reduced ability to activate autophagy.
Recent studies confirmed that exosomes repaired tissue damage and inhibited tissue degeneration by inhibiting apoptosis and inflammation. 41 , 42 , 43 , 44 Based on the GO analysis of proteins carried in exosomes, we also found that N‐Exos may inhibit IVDD by inhibiting NPC apoptosis. We performed TUNEL staining, flow cytometry, and WB experiments to detect the effect of N‐Exos on apoptosis inhibition. The results confirmed that N‐Exos inhibited NPC apoptosis more effectively than D‐Exos. This result provided a new direction for treatments for IVDD. For example, it is possible to construct engineered exosomes that overexpressed a protein that inhibits IVDD to strengthen the therapeutic effect.
Exosomes are involved in the intercellular crosstalk and inter‐organelle communication 45 to enhance autophagy and inhibit cell apoptosis. 30 , 46 However, we did not know whether N‐Exos inhibited NPC apoptosis via autophagy activation. KEGG enrichment analysis, immunofluorescence staining, TEM analysis, and WB revealed that the effects of N‐Exos on autophagy activation to inhibit TBHP‐induced apoptosis was more effective than D‐Exos. MSC‐derived exosomes reduce myocardial ischemia reperfusion injury by inducing cardiac autophagy via the AMPK/mTOR or Akt/mTOR pathways. 47 However, the molecular mechanism of CESC exosomes entry in target cells was not clear. For the NPCs treated with NC, D‐Exos, and N‐Exos, the expression of p‐AKT was significantly increased in the N‐Exos group. AKT signaling was effectively activated in the presence of N‐Exos. Activation of the PI3K/AKT signaling pathway significantly reduced the levels of the apoptotic proteins cleaved caspase3 and Bax in NPCs, but the expression levels of the antiapoptotic protein Bcl‐2 were increased. These findings show that N‐Exos primarily activated autophagy in NPCs by enhancing the p‐AKT signaling pathway, which inhibited NPC apoptosis.
Various groups found that exosomes played important roles in protecting the heart and renal activity and inhibiting myocardial infarction in animal models via suppression of oxidative stress and apoptosis. 48 , 49 To clarify the function of exosome inhibition of rat IVDD via NPC apoptosis inhibition, we used a rat IVDD model and injected exosomes and the PI3K/AKT inhibitor LY294002 into the rat tail disc. Injection of DIR‐labeled N‐Exos into the IVD demonstrated that N‐Exos stayed in the IVD. Similar to LY294002 inhibition of autophagy in septic myocardial dysfunction, 50 we found that the injection of LY294002 increased the expression of LC3A/B, cleaved caspase3 and Bax and accelerated IVDD. However, after the injection of N‐Exos, the expression of LC3A/B, cleaved caspase3, and Bax was significantly reduced, and LY294002 effectively reversed the exosome‐mediated inhibition of IVDD. These results indicate that exosomes derived from CESCs enter NPCs to inhibit disc degeneration by enhancing autophagy via activation of the PI3K/AKT signaling pathway in vivo.
In summary, the present work demonstrated that there were many CESCs in the rat CEP that secreted exosomes. N‐Exos inhibited NPC apoptosis and weakened disc degeneration more effectively than D‐Exos by promoting autophagy via activation of the PI3K/AKT signaling pathway in vitro and in vivo. These results provide further support for the importance of exosomes derived from normal CESCs as therapeutic tools for IVDD prevention and treatment.
CONFLICT OF INTEREST
The authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
L.L., X.J., J.Q.: conception and design, conducting experiments, collection and/or assembly of data, data analysis and interpretation manuscript writing; H.S., Y.W.: provision of study material, data analysis; J.Z., Z.S., D.Y., C.L.: conducting experiments, animal modeling assistance; P.Z., M.L., Z.T., Y.Z.: revised the manuscript, administrative support and financial support.
Supporting information
Appendix S1. Supporting Information.
Figure S1 The injection method of the rat IVD site. Rats (about 250 g) were anesthetized with 2 mL 5% chloral hydrate. Incise the tail (the skin wound <1.0 cm in experiment. Here, the wound 4‐5 cm to show the details). 2. Find the IVD, microsyringe (50 μL, 32G) will to be used. 3. The microsyringe and tail form a 45° angle, and insert into cartilage endplate, the microsyringe close to the side of vertebrae. 4. After 1‐2 mm of needle insertion, the microsyringe and tail form vertical, and continue to insert 1‐2 mm, inject fluid. Stitch the wound and hemostasis.
{kind=link}
Figure S2 Bioinformatics Analysis between normal CESCs derived exosomes (N‐Exos) and degenerated CESCs derived exosomes (D‐Exos). (A,B) KEGG enrichment analysis and Gene Ontology (GO) data analysis of proteins contained in D‐Exos.
{kind=link}
Figure S3 Exosomes promote the proliferation of NPCs. Double immunofluorescence of EdU (green) and PKH26 (red) in NPCs treated with NC, D‐Exos (40ug/ml) or N‐Exos (40ug/ml).
{kind=link}
Figure S4 The expression of growth factors or anti‐inflammatory cytokines in N‐Exos and D‐Exos. (A) Heat map analysis and (B) LFQ intensity analysis of differential growth factors or anti‐inflammatory between N‐Exos and D‐Exos. Igf2r: Insulin‐like growth factor 2 receptor; Igfals: Insulin‐like growth factor‐binding protein complex acid labile subunit; Grb10: Growth factor receptor‐bound protein 10; Heg1: Heg homolog 1; Tgfb1i1: Transforming growth factor beta‐1‐induced transcript 1 protein; Ntrk1: High affinity nerve growth factor receptor; Igf2bp1: Insulin‐like growth factor 2 mRNA‐binding protein 1; Csf1: Macrophage colony‐stimulating factor 1; Pdgfra: Platelet‐derived growth factor receptor alpha; Epha2: Receptor protein‐tyrosine kinase; Pdgfrb: Platelet‐derived growth factor receptor beta; LFQ: label‐free quantification. ns: P > .05; * P < .05; ** P < .01; *** P < .001.
{kind=link}
Figure S5 Protein abundance analysis. (A) Relative protein quantification of N‐Exos and D‐Exos extracted from the supernatant of TBHP‐treated or untreated CESCs with the same volume and the same incubation time. (B,C) The average abundance of AKT pathway and autophagy proteins in N‐Exos and D‐Exos with the same protein quantification.
{kind=link}
Figure S6 N‐Exos treatment promotes NPCs extracellular matrix production and weakens IVDD. (A) The gene expression of Collagen II, Collagen I, IL‐6 and IL‐1β in the NPCs treated with different concentrations N‐Exos (0, 20 or 40 μg/mL). (B) The western blotting of Collagen II, Collagen I IL‐6 and IL‐1β in the NPCs as treated above. NC: Normal Control; ns: P > .05; * P < .05; ** P < .01; *** P < .001.
{kind=link}
Figure S7 N‐Exos promote the differentiation of CESCs into intervertebral disc cells. (A) The gene expression of Collagen II, Collagen I and Sox9 in the CESCs treated with different concentrations N‐Exos (0, 20 or 40 μg/mL). (B) The western blotting of Collagen II, Collagen I and Sox9 in the CESCs as treated above. (C) Double immunofluorescence of Collagen II (green) and PKH26 (red) in CESCs treated with different concentrations N‐Exos (0, 20 or 40 μg/mL). ns: P > .05; * P < .05; ** P < .01; *** P < .001.
{kind=link}
Figure S8 N‐Exos promote chondrogenic differentiation and osteogenic differentiation of CESCs. The gene expression of Acan, Runx2, BSP and adamts5 in the CESCs treated with different concentrations N‐Exos (0, 20 or 40 μg/mL). ns: P > .05; * P < .05; ** P < .01; *** P < .001.
{kind=link}
Table S1 Primers for quantitative RT‐PCR
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (Grant Number: 81874028), the Research Program of Foundation Science and Application Technology of Chongqing (Grant Number: cstc2018jcyjA1826), and Basic Medical College Foundation of Army Medical University (Grant Numbers: 2019JCZX10, 2019JCZX12).
Luo L, Jian X, Sun H, et al. Cartilage endplate stem cells inhibit intervertebral disc degeneration by releasing exosomes to nucleus pulposus cells to activate Akt/autophagy. Stem Cells. 2021;39:467–481. 10.1002/stem.3322
Funding information Basic Medical College Foundation of Army Medical University, Grant/Award Numbers: 2019JCZX10, 2019JCZX12; the National Natural Science Foundation of China, Grant/Award Number: 81874028; the Research Program of Foundation Science and Application Technology of Chongqing, Grant/Award Number: cstc2018jcyjA1826
Contributor Information
MingHan Liu, Email: liuminghan2008@hotmail.com.
Zhiqiang Tian, Email: tzhiq009@163.com.
Yue Zhou, Email: happyzhou@vip.163.com.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author.
REFERENCES
- 1. Andersson GB. Epidemiological features of chronic low‐back pain. Lancet. 1999;354:581‐585. [DOI] [PubMed] [Google Scholar]
- 2. Urban JP, Roberts S. Degeneration of the intervertebral disc. Arthritis Res Ther. 2003;5:120‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang F, Gao ZX, Cai F, et al. Formation, function, and exhaustion of notochordal cytoplasmic vacuoles within intervertebral disc: current understanding and speculation. Oncotarget. 2017;8:57800‐57812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wong J, Sampson SL, Bell‐Briones H, et al. Nutrient supply and nucleus pulposus cell function: effects of the transport properties of the cartilage endplate and potential implications for intradiscal biologic therapy. Osteoarthr Cartil. 2019;27:956‐964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li FC, Zhang N, Chen WS, Chen QX. Endplate degeneration may be the origination of the vacuum phenomenon in intervertebral discs. Med Hypotheses. 2010;75:169‐171. [DOI] [PubMed] [Google Scholar]
- 6. He Z, Jia M, Yu Y, Yuan C, Wang J. Roles of SDF‐1/CXCR4 axis in cartilage endplate stem cells mediated promotion of nucleus pulposus cells proliferation. Biochem Biophys Res Commun. 2018;506:94‐101. [DOI] [PubMed] [Google Scholar]
- 7. He Z, Pu L, Yuan C, Jia M, Wang J. Nutrition deficiency promotes apoptosis of cartilage endplate stem cells in a caspase‐independent manner partially through upregulating BNIP3. Acta Biochim Biophys Sin. 2017;49:25‐32. [DOI] [PubMed] [Google Scholar]
- 8. Liu LT, Huang B, Li CQ, Zhuang Y, Wang J, Zhou Y. Characteristics of stem cells derived from the degenerated human intervertebral disc cartilage endplate. PLoS One. 2011;6:e26285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang H, Zhou Y, Huang B, et al. Utilization of stem cells in alginate for nucleus pulposus tissue engineering. Tissue Eng Part A. 2014;20:908‐920. [DOI] [PubMed] [Google Scholar]
- 10. Chen S, Zhao L, Deng X, et al. Mesenchymal stem cells protect nucleus Pulposus cells from compression‐induced apoptosis by inhibiting the mitochondrial pathway. Stem Cells Int. 2017;2017:9843120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang W, Wang Y, Deng G, et al. Transplantation of hypoxic‐preconditioned bone mesenchymal stem cells retards intervertebral disc degeneration via enhancing implanted cell survival and migration in rats. Stem Cells Int. 2018;2018:7564159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zuo R, Wang Y, Li J, et al. Rapamycin induced autophagy inhibits inflammation‐mediated endplate degeneration by enhancing Nrf2/Keap1 signaling of cartilage endplate stem cells. Stem Cells. 2019;37:828‐840. [DOI] [PubMed] [Google Scholar]
- 13. Jiang C, Guo Q, Jin Y, et al. Inhibition of EZH2 ameliorates cartilage endplate degeneration and attenuates the progression of intervertebral disc degeneration via demethylation of Sox‐9. EBioMedicine. 2019;48:619‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ramos‐Ibeas P, Barandalla M, Colleoni S, Lazzari G. Pyruvate antioxidant roles in human fibroblasts and embryonic stem cells. Mol Cell Biochem. 2017;429:137‐150. [DOI] [PubMed] [Google Scholar]
- 15. Yao Y, Liang X, Shi Y, Lin Y, Yang J. Osthole delays Tert‐butyl hydroperoxide‐induced premature senescence in neural stem cells. Cell Reprogram. 2018;20:268‐274. [DOI] [PubMed] [Google Scholar]
- 16. Zhang KS, Chen LB, Huang XP, et al. Effects of astragali radix combined with angelicae sinensis radix on the proliferation of hematopoietic stem cells senescence model in mice. China J Chin Mater Med. 2017;42:4187‐4194. [DOI] [PubMed] [Google Scholar]
- 17. Montecalvo A, Larregina AT, Shufesky WJ, et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood. 2012;119:756‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nojima H, Freeman CM, Schuster RM, et al. Hepatocyte exosomes mediate liver repair and regeneration via sphingosine‐1‐phosphate. J Hepatol. 2016;64:60‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Phinney DG, Pittenger MF. Concise review: MSC‐derived exosomes for cell‐free therapy. Stem Cells. 2017;35:851‐858. [DOI] [PubMed] [Google Scholar]
- 20. Nong K, Wang W, Niu X, et al. Hepatoprotective effect of exosomes from human‐induced pluripotent stem cell‐derived mesenchymal stromal cells against hepatic ischemia‐reperfusion injury in rats. Cytotherapy. 2016;18:1548‐1559. [DOI] [PubMed] [Google Scholar]
- 21. Xia C, Zeng Z, Fang B, et al. Mesenchymal stem cell‐derived exosomes ameliorate intervertebral disc degeneration via anti‐oxidant and anti‐inflammatory effects. Free Radic Biol Med. 2019;143:1‐15. [DOI] [PubMed] [Google Scholar]
- 22. Liu H, Sun X, Gong X, et al. Human umbilical cord mesenchymal stem cells derived exosomes exert antiapoptosis effect via activating PI3K/Akt/mTOR pathway on H9C2 cells. J Cell Biochem. 2019;120:14455‐14464. [DOI] [PubMed] [Google Scholar]
- 23. Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728‐741. [DOI] [PubMed] [Google Scholar]
- 25. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weiner LM, Lotze MT. Tumor‐cell death, autophagy, and immunity. N Engl J Med. 2012;366:1156‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guo F, Liu X, Cai H, le W. Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathol. 2018;28:3‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luo P, Gao F, Niu D, et al. The role of autophagy in chondrocyte metabolism and osteoarthritis: a comprehensive research review. Biomed Res Int. 2019;2019:5171602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang S, Zhang F, Ma J, Ding W. Intervertebral disc ageing and degeneration: the antiapoptotic effect of oestrogen. Ageing Res Rev. 2020;57:100978. [DOI] [PubMed] [Google Scholar]
- 30. Wei F, Li M, Crawford R, et al. Exosome‐integrated titanium oxide nanotubes for targeted bone regeneration. Acta Biomater. 2019;86:480‐492. [DOI] [PubMed] [Google Scholar]
- 31. Pfirrmann CW, Metzdorf A, Zanetti M, et al. Magnetic resonance classification of lumbar intervertebral disc degeneration. Spine. 2001;26:1873‐1878. [DOI] [PubMed] [Google Scholar]
- 32. Liang L, Li X, Li D, et al. The characteristics of stem cells in human degenerative intervertebral disc. Medicine. 2017;96:e7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yuan C, Pu L, He Z, et al. BNIP3/Bcl‐2‐mediated apoptosis induced by cyclic tensile stretch in human cartilage endplate‐derived stem cells. Exp Ther Med. 2018;15:235‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao S, Liu Y, Pu Z. Bone marrow mesenchymal stem cell‐derived exosomes attenuate D‐GaIN/LPS‐induced hepatocyte apoptosis by activating autophagy in vitro. Drug Des Devel Ther. 2019;13:2887‐2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jin J, Shi Y, Gong J, et al. Exosome secreted from adipose‐derived stem cells attenuates diabetic nephropathy by promoting autophagy flux and inhibiting apoptosis in podocyte. Stem Cell Res Ther. 2019;10:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang S, Chuah SJ, Lai RC, Hui JHP, Lim SK, Toh WS. MSC exosomes mediate cartilage repair by enhancing proliferation, attenuating apoptosis and modulating immune reactivity. Biomaterials. 2018;156:16‐27. [DOI] [PubMed] [Google Scholar]
- 37. Huang YC, Urban JP, Luk KD. Intervertebral disc regeneration: do nutrients lead the way? Nat Rev Rheumatol. 2014;10:561‐566. [DOI] [PubMed] [Google Scholar]
- 38. Wang G, Huang K, Dong Y, et al. Lycorine suppresses endplate‐chondrocyte degeneration and prevents intervertebral disc degeneration by inhibiting NF‐kappaB signalling pathway. Cell Physiol Biochem. 2018;45:1252‐1269. [DOI] [PubMed] [Google Scholar]
- 39. Qiu X, Zhuang M, Lu Z, et al. RIPK1 suppresses apoptosis mediated by TNF and caspase‐3 in intervertebral discs. J Transl Med. 2019;17:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liao Z, Luo R, Li G, et al. Exosomes from mesenchymal stem cells modulate endoplasmic reticulum stress to protect against nucleus pulposus cell death and ameliorate intervertebral disc degeneration in vivo. Theranostics. 2019;9:4084‐4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Che Y, Shi X, Shi Y, et al. Exosomes derived from miR‐143‐overexpressing MSCs inhibit cell migration and invasion in human prostate cancer by downregulating TFF3. Mol Ther Nucleic Acids. 2019;18:232‐244. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42. Xu G, Zhang B, Ye J, et al. Exosomal miRNA‐139 in cancer‐associated fibroblasts inhibits gastric cancer progression by repressing MMP11 expression. Int J Biol Sci. 2019;15:2320‐2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fan C, Zhang E, Joshi J, Yang J, Zhang J, Zhu W. Utilization of human induced pluripotent stem cells for cardiac repair. Front Cell Dev Biol. 2020;8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Connor DE, Paulus JA, Dabestani PJ, et al. Therapeutic potential of exosomes in rotator cuff tendon healing. J Bone Miner Metab. 2019;37:759‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zappulli V, Friis KP, Fitzpatrick Z, Maguire CA, Breakefield XO. Extracellular vesicles and intercellular communication within the nervous system. J Clin Invest. 2016;126:1198‐1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gong XH, Liu H, Wang SJ, Liang SW, Wang GG. Exosomes derived from SDF1‐overexpressing mesenchymal stem cells inhibit ischemic myocardial cell apoptosis and promote cardiac endothelial microvascular regeneration in mice with myocardial infarction. J Cell Physiol. 2019;234:13878‐13893. [DOI] [PubMed] [Google Scholar]
- 47. Liu L, Jin X, Hu CF, Li R, Zhou Z’, Shen CX. Exosomes derived from mesenchymal stem cells rescue myocardial ischaemia/reperfusion injury by inducing cardiomyocyte autophagy via AMPK and Akt pathways. Cell Physiol Biochem. 2017;43:52‐68. [DOI] [PubMed] [Google Scholar]
- 48. Timmers L, Lim SK, Arslan F, et al. Reduction of myocardial infarct size by human mesenchymal stem cell conditioned medium. Stem Cell Res. 2007;1:129‐137. [DOI] [PubMed] [Google Scholar]
- 49. Zhou Y, Xu H, Xu W, et al. Exosomes released by human umbilical cord mesenchymal stem cells protect against cisplatin‐induced renal oxidative stress and apoptosis in vivo and in vitro. Stem Cell Res Ther. 2013;4:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yu T, Liu D, Gao M, et al. Dexmedetomidine prevents septic myocardial dysfunction in rats via activation of alpha7nAChR and PI3K/Akt‐ mediated autophagy. Biomed Pharmacother. 2019;120:109231. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information.
Figure S1 The injection method of the rat IVD site. Rats (about 250 g) were anesthetized with 2 mL 5% chloral hydrate. Incise the tail (the skin wound <1.0 cm in experiment. Here, the wound 4‐5 cm to show the details). 2. Find the IVD, microsyringe (50 μL, 32G) will to be used. 3. The microsyringe and tail form a 45° angle, and insert into cartilage endplate, the microsyringe close to the side of vertebrae. 4. After 1‐2 mm of needle insertion, the microsyringe and tail form vertical, and continue to insert 1‐2 mm, inject fluid. Stitch the wound and hemostasis.
Figure S2 Bioinformatics Analysis between normal CESCs derived exosomes (N‐Exos) and degenerated CESCs derived exosomes (D‐Exos). (A,B) KEGG enrichment analysis and Gene Ontology (GO) data analysis of proteins contained in D‐Exos.
Figure S3 Exosomes promote the proliferation of NPCs. Double immunofluorescence of EdU (green) and PKH26 (red) in NPCs treated with NC, D‐Exos (40ug/ml) or N‐Exos (40ug/ml).
Figure S4 The expression of growth factors or anti‐inflammatory cytokines in N‐Exos and D‐Exos. (A) Heat map analysis and (B) LFQ intensity analysis of differential growth factors or anti‐inflammatory between N‐Exos and D‐Exos. Igf2r: Insulin‐like growth factor 2 receptor; Igfals: Insulin‐like growth factor‐binding protein complex acid labile subunit; Grb10: Growth factor receptor‐bound protein 10; Heg1: Heg homolog 1; Tgfb1i1: Transforming growth factor beta‐1‐induced transcript 1 protein; Ntrk1: High affinity nerve growth factor receptor; Igf2bp1: Insulin‐like growth factor 2 mRNA‐binding protein 1; Csf1: Macrophage colony‐stimulating factor 1; Pdgfra: Platelet‐derived growth factor receptor alpha; Epha2: Receptor protein‐tyrosine kinase; Pdgfrb: Platelet‐derived growth factor receptor beta; LFQ: label‐free quantification. ns: P > .05; * P < .05; ** P < .01; *** P < .001.
Figure S5 Protein abundance analysis. (A) Relative protein quantification of N‐Exos and D‐Exos extracted from the supernatant of TBHP‐treated or untreated CESCs with the same volume and the same incubation time. (B,C) The average abundance of AKT pathway and autophagy proteins in N‐Exos and D‐Exos with the same protein quantification.
Figure S6 N‐Exos treatment promotes NPCs extracellular matrix production and weakens IVDD. (A) The gene expression of Collagen II, Collagen I, IL‐6 and IL‐1β in the NPCs treated with different concentrations N‐Exos (0, 20 or 40 μg/mL). (B) The western blotting of Collagen II, Collagen I IL‐6 and IL‐1β in the NPCs as treated above. NC: Normal Control; ns: P > .05; * P < .05; ** P < .01; *** P < .001.
Figure S7 N‐Exos promote the differentiation of CESCs into intervertebral disc cells. (A) The gene expression of Collagen II, Collagen I and Sox9 in the CESCs treated with different concentrations N‐Exos (0, 20 or 40 μg/mL). (B) The western blotting of Collagen II, Collagen I and Sox9 in the CESCs as treated above. (C) Double immunofluorescence of Collagen II (green) and PKH26 (red) in CESCs treated with different concentrations N‐Exos (0, 20 or 40 μg/mL). ns: P > .05; * P < .05; ** P < .01; *** P < .001.
Figure S8 N‐Exos promote chondrogenic differentiation and osteogenic differentiation of CESCs. The gene expression of Acan, Runx2, BSP and adamts5 in the CESCs treated with different concentrations N‐Exos (0, 20 or 40 μg/mL). ns: P > .05; * P < .05; ** P < .01; *** P < .001.
Table S1 Primers for quantitative RT‐PCR
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.