

Abstract
Migraine patients often report (inter)ictal hypersensitivity to light, but the underlying mechanisms remain an enigma. Both hypo‐ and hyperresponsivity of the visual network have been reported, which may reflect either intra‐individual dynamics of the network or large inter‐individual variation in the measurement of human visual evoked potential data. Therefore, we studied visual system responsivity in freely behaving mice using combined epidural electroencephalography and intracortical multi‐unit activity to reduce variation in recordings and gain insight into visual cortex dynamics. For better clinical translation, we investigated transgenic mice that carry the human pathogenic R192Q missense mutation in the α1A subunit of voltage‐gated CaV2.1 Ca2+ channels leading to enhanced neurotransmission and familial hemiplegic migraine type 1 in patients. Visual evoked potentials were studied in response to visual stimulation paradigms with flashes of light. Following intensity‐dependent visual stimulation, FHM1 mutant mice displayed faster visual evoked potential responses, with lower initial amplitude, followed by less pronounced neuronal suppression compared to wild‐type mice. Similar to what was reported for migraine patients, frequency‐dependent stimulation in mutant mice revealed enhanced photic drive in the EEG beta‐gamma band. The frequency‐dependent increases in visual network responses in mutant mice may reflect the context‐dependent enhancement of visual cortex excitability, which could contribute to our understanding of sensory hypersensitivity in migraine.
Keywords: CaV2.1 calcium channels, electrophysiology, sensory systems, visual stimulation
Migraine patients often report hypersensitivity to light, but the underlying network mechanisms are unclear. We here studied visual network responsivity relevant to migraine by simultaneous recording of EEG and multi‐unit activity during visual stimulation paradigms with flashing light in freely behaving wild‐type and familial hemiplegic migraine type 1 (FHM1) R192Q mutant mice. Mutant mice showed intensity‐ and frequency‐dependent changes in visual evoked potentials that indicate context‐dependent visual network hyperexcitability relevant to migraine.

Abbreviations
- AUC
area‐under‐curve
- dB
decibel
- EEG
electroencephalography
- FHM1
familial hemiplegic migraine type 1
- ICC
intraclass correlation coefficient
- IO
input‐output paradigm
- ISI
inter‐stimulus interval
- LED
light‐emitting diode
- MUA
multi‐unit activity
- VEP
visual evoked potential
- WT
wild‐type
1. INTRODUCTION
Migraine is a common episodic brain disorder characterized by severe recurrent attacks of headache, associated with phono‐ and photophobia and other autonomic and neurological symptoms (Headache Classification Committee of the International Headache Society (IHS), 2018). Many patients report abnormal sensitivity or intolerance to light, not only during but also outside attacks, and show abnormal cortical activation in response to visual stimulation in imaging studies (Boulloche et al., 2010; Denuelle et al., 2011). Enhanced visual sensitivity before the onset of headache has been regarded as a sign that an attack has started (Schulte et al., 2015). The light sensitivity may result from cortical “hyper‐responsivity” (Coppola et al., 2007b) that is not restricted to the visual cortex as it was also reported for other brain structures implicated in migraine pathophysiology (Brennan & Pietrobon, 2018; De Tommaso et al., 2014; Ferrari et al., 2015; Noseda & Burstein, 2013).
It remains unresolved whether findings of altered visual responsivity in migraine patients translate to increased or decreased excitability of the visual cortex (Coppola et al., 2007b; Cosentino et al., 2014) as unaltered (Omland et al., 2013), reduced (Afra et al. 1998; Höffken et al., 2009) and enhanced (Lisicki et al., 2018; Magis et al., 2007) visual evoked potential (VEP) responses in between attacks have been reported. Apart from transient visual stimulation paradigms, visual processing in migraine has also been studied using steady‐state stimulation resulting in “photic driving” responses. Photic drive (also known as entrainment) is the frequency‐following EEG response of the visual cortex to various stimulation frequencies, resulting in a dominant EEG frequency (Herrmann, 2001). In migraineurs, an enhanced photic drive response between 10 and 20 Hz was observed that could reflect plasticity changes involving the visual cortex (Chorlton & Kane, 2000; De Tommaso et al., 2014; Nyrke et al., 1989). A shortened photic driving paradigm (“chirp” stimulation) showed enhanced responses in the beta band (18 to 26 Hz) in between migraine attacks (Gantenbein et al., 2014). Using this paradigm, we recently observed a similar enhanced photic drive response that was evident in the harmonics of the beta‐gamma band (22–32 Hz) in migraineurs, albeit not in between attacks but toward an impending attack (Perenboom et al., 2020). These observations support the view that enhanced visual network excitability contributes to attack initiation.
Contradictory findings of cortical hyper‐ or hyporesponsivity in migraine may be explained by the dynamics of the network and, even more likely, can be due to differences in stimulation procedures and readout parameters in clinical studies (Cosentino et al., 2014). Also, large inter‐individual variation may be caused by the low signal‐to‐noise ratio of scalp EEG in humans, which hampers the interpretation of human VEP findings. Performing VEP measurements in animals can circumvent most of the issues, as VEPs with an improved signal‐to‐noise ratio can be obtained (a) when intracortical or epidural electrodes are used, and (b) by controlling the influence of genetic background by inbred strains.
Visual stimulation by flashes of light has been widely used to elicit VEP responses in anesthetized (Ridder & Nusinowitz, 2006; Strain & Tedford, 1993), head‐fixed (Funayama et al., 2016), and freely behaving (Lopez et al., 2002; Mazzuchelli et al., 1995; Tomiyama et al., 2016) mice. To investigate changes in visual network responsivity, we here examined flash VEP responses in freely behaving mice. To better capture dynamical changes in visual system responsivity, as reported in migraineurs for stimulation at varying frequencies (Bjork et al., 2011; Gantenbein et al., 2014; Perenboom et al., 2020; Sand et al., 2008), both steady‐state responses and transitions between stimulation frequencies were investigated. We studied both wild‐type mice and FHM1 mutant mice that carry the R192Q missense mutation in the α1A subunit of neuronal CaV2.1 calcium channels (van den Maagdenberg et al., 2004), known to cause familial hemiplegic migraine type 1 (FHM1), a subtype of migraine with aura (Ferrari et al., 2015; Tolner et al., 2015). The mutant mice display a gain of CaV2.1 channel function with enhanced glutamatergic neurotransmission in the cortex (Tottene et al., 2009; Vecchia et al., 2014) and are considered a relevant model for studying mechanisms by which neuronal hyperexcitability contributes to migraine pathophysiology (Ferrari et al., 2015; Tolner et al., 2019). Our mouse model is also relevant given that triggers of attacks, including bright light, reported by FHM1 patients are similar to triggers reported by patients suffering from migraine with aura (Hansen et al., 2011). In addition, in line with clinical reports of photophobia symptoms in migraineurs during and sometimes also outside attacks (Mulleners et al., 2001; Perenboom et al., 2018), FHM1 mutant mice displayed behavioral signs of photophobia (Chanda et al., 2013) that may reflect enhanced visual system responsivity. Hence, insight into altered responses to visual stimulation in the transgenic migraine mice may help understand how visual system alterations are brought about in a migraine context.
2. MATERIALS AND METHODS
2.1. Animals
Male homozygous FHM1 R192Q knock‐in (“FHM1 mutant”) and wild‐type (“WT”) mice of 3–6 months were used. The mutant mice were generated by introducing the human pathogenic FHM1 R192Q missense mutation in the orthologous mouse Cacna1a gene using a gene‐targeting approach (van den Maagdenberg et al., 2004). Mice, backcrossed for 20 generations to C57BL/6J, were maintained on a normal 12:12 light‐dark cycle with water and food available ad libitum. All experiments were approved by the Animal Experiment Ethics Committee of Leiden University Medical Center and were carried out in accordance with ARRIVE guidelines and EU Directive 2010/63/EU for animal experiments. All efforts were made to minimize the suffering of the mice.
2.2. EEG recordings and visual stimulation in freely behaving mice
Under isoflurane anesthesia (1.5%, in oxygen‐enriched air), seven electrodes were stereotaxically implanted at the following coordinates (in mm relative to bregma): a pair of platinum (Pt) electrodes 3.5 posterior/2.0 lateral/0.8 ventral from dura (right visual cortex); a pair of Pt electrodes 1.5 anterior/1.5 lateral/0.8 ventral from dura (right motor cortex); a silver (Ag) ball‐tip electrode 3.5 posterior/2.0 lateral on the dura (left visual cortex); an Ag ball‐tip electrode and Ag‐AgCl ball‐tip electrode were placed above cerebellum to serve as reference and ground electrodes, respectively (Figure 1a‐top). Electrodes were attached to the skull using light‐activated bonding primer and dental cement (Kerr optibond/ premise flowable, DiaDent Europe, Almere, the Netherlands). Carprofen (5 mg/kg, s.c.) was administered for post‐operative pain relief.
Figure 1.
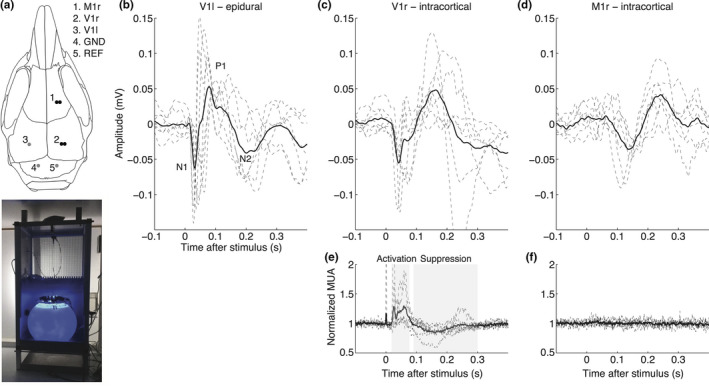
Approach and validation of single‐flash visual evoked potential measurements in freely behaving wild‐type mice. (a) Top: Electrode locations used for EEG (single epidural electrode in left V1; grey dot), or local field potential/neuronal multi‐unit activity (bipolar intracortical electrodes in right V1 and M1; black dots) recordings. Bottom: Home cage with light sphere (cf. van Diepen et al., 2013 for details) for housing a mouse during VEP recordings. (b–f) Individual (dashed lines) and group‐averaged (thick line) responses to 100 single flashes (presented at 1 Hz, 1 V) in visual and motor cortex. (b) EEG network responses recorded epidurally over the left visual cortex (n = 8) showing N1, P1, and N2 responses. (c) Local field potential (n = 6) and (e) baseline normalized multi‐unit activity (n = 6) recorded in the right visual cortex with activation between 20 and 80 ms, and transient suppression of activity between 90 and 300 ms. (d) local field potential and (f) normalized multi‐unit activity in the right motor cortex. Note the absence of time‐locked neuronal (multi‐unit) activity in relation to light stimulation in the motor cortex. MUA: multi‐unit activity; r = right; l = left; GND = ground; REF = reference
After a recovery period of 7 days, animals were placed in a shielded recording cage and connected to the recording hardware through a counterbalanced, low‐torque custom‐built electrical commutator. Epidural EEG and intracortical local field potential signals were pre‐amplified 3X and fed into separate amplifiers for EEG/local field potential and neuronal multi‐unit activity (MUA) recordings. EEG/local field potential signals were band‐pass filtered (0.05 to 500 Hz) and amplified 1,200X, and digitized (Power 1401, CED, Cambridge, UK) at a rate of 5,000 Hz. In addition, differential signals from paired intracortical Pt electrodes were used for MUA recordings by 36,000X amplification, band‐pass filtering (500 to 5,000 Hz), and digitizing at 25,000 Hz. For VEP measurements, the tethered mouse was placed inside a computer‐controlled custom‐built light‐emitting diode (LED) illuminated sphere in which it was able to move freely (van Diepen et al., 2013; Figure 1a‐bottom). The wavelength of blue light and irradiance was measured using a calibrated spectrometer (AvaSpec2048; Avantes, Apeldoorn, The Netherlands). The sphere was 30 cm in diameter, with the inside coated with high‐reflectance paint. On top of the sphere, around an opening for the swivel, monochromatic blue (wavelength: 469 nm) 1‐ms light flashes were presented at 1‐V stimulator output voltage, corresponding to a light intensity of ~2.2 log cd/m2, unless mentioned otherwise. A baffle prevented mice from looking directly at the LEDs. Water and food were provided inside the sphere during the experiment.
EEG/local field potential data were down‐sampled offline to 1,000 Hz. MUA was reduced in complexity by calculating the root mean square amplitude per 25 samples (1 ms), as the root mean square correlates to the spiking rate using template matching (Gummadavelli et al., 2015; Schridde et al., 2008), and next down‐sampled to 1,000 Hz. Stimulation sequence, data processing, and analysis using custom‐written scripts in MATLAB (version R2013b; The MathWorks) varied per paradigm. Resting periods in between different stimulation paradigms were at least 1 min for both WT and FHM1 mutant groups.
2.3. Single‐VEP paradigm
To assess whether light flashes evoked responses in the visual cortex, 100 flashes were presented at 1 Hz. Single‐VEP responses were high‐pass filtered at 1 Hz and low‐pass filtered at 35 Hz (4th order Butterworth, zero‐phase shift) and averaged between 100 ms prior and 500 ms after stimulation. N1 peak amplitude and latency were detected between 20 and 80 ms after stimulation, P1 peak amplitude and latency between the N1 latency and 120 ms, and N2 peak amplitude and latency between P1 latency and 250 ms. MUA activation was defined as the area‐under‐curve (AUC) between 20 and 80 ms after stimulation, the subsequent suppression phase was defined as AUC between 90 and 300 ms. EEG or MUA data per animal were excluded from further analyses in case of an absent response to the single‐VEP paradigm in the averaged traces.
2.4. Input–output paradigm
To assess the intensity dependency of VEP responses, 60 flashes of increasing light intensity between 0.01 and 0.1 V stimulator output (~0.4 to 1.1 log cd/m2) were presented at 2 Hz, and five flashes of increasing intensity between 0.2 and 1 V stimulator output (~1.4 to 2.2 log cd/m2) at 0.5 Hz (Figure 2a‐top). The paradigm was repeated 50 times with 20 s rest in‐between blocks. Input‐output (IO) curves were averaged over 50 repeats and N1 and P1 amplitude were determined as for single‐VEP analysis. P1‐N1 amplitudes between 0.01 and 0.1 V light intensity were fitted with the Naka‐Rushton equation (Ridder & Nusinowitz, 2006) providing V max, the maximum saturated amplitude, k, the semi‐saturation intensity, and n, the slope of the fitted line. P1‐N1 amplitudes between 0.2 and 1 V light intensity were fitted using least‐squares linear regression, providing slope and intercept (amplitude) parameters.
Figure 2.
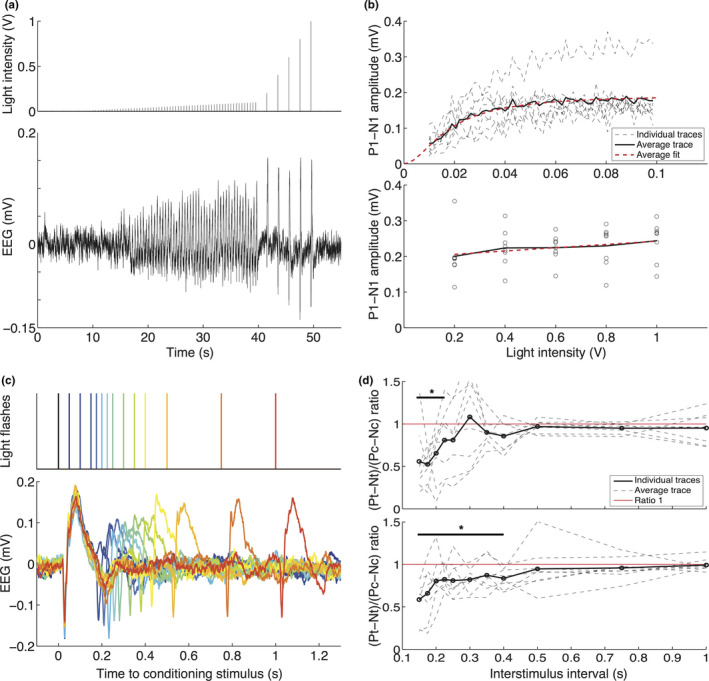
Visual evoked potential responses in freely behaving wild‐type mice show light intensity dependency with a plateau that is stable across animals, and show light‐intensity dependent recovery in a paired‐pulse paradigm after 225 or 500 ms. (a) Top: Light flash stimulation protocol used for generating input‐output curves, consisting of 60 flashes between 0.01 and 0.1 V at 2 Hz and five flashes between 0.2 and 1 V at 0.5 Hz. Bottom: Example EEG trace showing VEP responses to increasing stimulation intensity, illustrating increasing N1‐P1 peaks up to 0.1‐V stimulation intensity, reaching a plateau between 0.2‐ and 1‐V intensity. (b) Individual (dashed black line) and averaged (thick black line) P1‐N1 peak amplitudes for each light intensity. Top: For stimulation between 0.01‐ and 0.1‐V stimulation intensity; bottom: for stimulation between 0.2 and 1 V, with fitted Naka‐Rushton (top; dashed red line) and least‐squares linear regression lines (bottom). (c) Top: Paired light flashes were presented at 13 intervals after the conditioning stimulus (black), from blue to red: 50, 100, 150, 175, 200, 225, 250, 300, 350, 400, 500, 750, and 1,000 ms. Bottom: Example EEG traces per interval using the same color coding. (d) Top: For 0.1‐V stimulation voltage, individual (dashed black) and averaged (thick black line) ratio of P1‐N1 peak amplitudes in response to a test (Pt‐Nt) and conditioning stimulus (Pc‐Nc). Red line indicates a ratio of 1 at which the test stimulus amplitude equals that of the conditioning stimulus. Recovery of responses is present after 225 ms (difference not significant from 1). Bottom: Similar paired‐pulse response ratios shown for stimulation at 1 V, showing full recovery of the test response to that of the conditioning response at 500 ms
2.5. Paired‐pulse paradigm
To determine the recovery after evoked potentials, double light flashes at 13 inter‐stimulus intervals between the conditioning and the test stimulus (ISI; 1,000, 750, 500, 400, 350, 300, 250, 225, 200, 175, 150, 100, and 50 ms) were presented at 0.5 Hz and repeated 50 times per ISI (Figure 2c‐top). The paradigm was presented at two light intensities corresponding to 0.1 and 1 V stimulator output. Paired‐pulse responses were averaged over the 50 repeats per ISI, and P1‐N1 amplitude of the conditioning (Pc‐Nc) and test stimuli (Pt‐Nt) were determined. Recovery of the response amplitudes to the test stimuli was determined by calculating a paired‐pulse response curve. Test stimuli responses were normalized to responses to the conditioning stimulus, where a ratio of 1 indicates the return of the test response amplitude to the amplitude of the conditioning response. Possible habituation effects of the long duration of the paradigm on paired‐pulse responses were assessed by comparing the first 100 and the last 100 conditioning responses at the 1 V stimulator output.
2.6. Habituation VEP paradigm
To assess habituation to repeated light flashes, 600 flashes were presented at 3.1 Hz. Six consecutive blocks of 100 responses were filtered (see “Single‐VEP paradigm”) and averaged between 50 ms prior and 250 ms after stimulation. N1 and P1 peaks were extracted, and the ratio between the P1‐N1 amplitude of the 6th block and the P1‐N1 amplitude of the 1st block was calculated (cf. Omland et al., 2013). A ratio below 1 indicates habituation over 600 pulses, whereas a ratio above 1 indicates potentiation.
2.7. Frequency‐chirp paradigm
To assess frequency‐dependent entrainment, “chirp” stimulation consisting of four flashes per frequency between 10 and 40 Hz with 1‐Hz increments (Figure 5a‐top; cf. Gantenbein et al., 2014) was repeated 25 times with 15 s rest in‐between blocks. Chirp responses between 2 s prior and 8 s after the start of stimulation were subjected to Morlet wavelet analyses between 5 and 125 Hz, in 1‐Hz frequency steps. Wavelet scales increased logarithmically between 3 and 10 cycles (from lowest to highest frequency). The averaged response power over all repetitions was baseline‐corrected by calculating the decibel (dB) change in power relative to the mean power between 1.6 and 0.1 s prior to the start of stimulation. For each stimulation frequency, the total response power was calculated as average power between 5 and 125 Hz in the time window between the four flashes in the particular frequency plus 50 ms (cf. Gantenbein et al., 2014). In the same time windows, we also extracted the response power at driving frequencies (EEG responses between 10 and 40 Hz) and 2nd and 3rd harmonic frequencies (responses between 20 and 80 Hz and between 30 and 120 Hz, respectively) by averaging the time‐frequency response power at the frequencies between −1 and +1 Hz of each particular stimulation frequency (i.e., for driving frequencies), the stimulation frequency times two (for 2nd harmonic frequencies) or times three (3rd harmonic frequencies) (cf. Perenboom et al., 2020). Next, the mean of the total response power and the mean response power at driving frequencies and 2nd and 3rd harmonic frequencies were calculated within three frequency bands: 10–15 Hz (alpha band), 16–30 Hz (beta band), and 31–40 Hz (gamma band).
Figure 5.
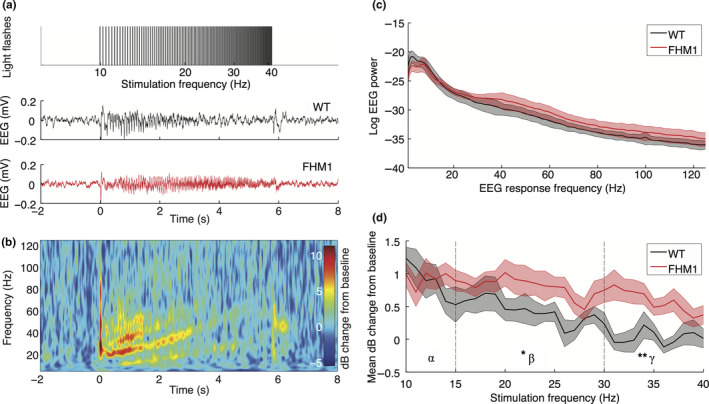
Chirp‐stimulation‐induced “photic drive” is more pronounced in the EEG beta‐gamma bands of the visual cortex in familial hemiplegic migraine type 1 (FHM1) mutant compared to wild‐type (WT) mice. (a) Top: Stimulation at increasing frequencies between 10 and 40 Hz in 1‐Hz steps, with four light flashes per frequency, is used to generate a chirp stimulation of ~6 s. Example traces of the averaged EEG response to the chirp stimulation paradigm of a WT (middle) and a mutant (bottom) animal. (b) Time‐frequency domain representation of averaged and baseline‐corrected EEG responses of the WT example trace. Baseline correction was performed over the averaged trials by calculating the log10 decibel (dB) change with respect to EEG activity 160 to 10 ms prior to stimulation onset. Note the presence of higher‐order responses, especially the second harmonic (20 to 80 Hz), up to halfway the chirp stimulation at 3 s. (c) Power spectral density of the non‐baseline‐normalized EEG response power, showing similar EEG spectra for WT and mutant mice. (d) Averaged EEG response power between 5 and 125 Hz for each stimulation frequency is enhanced in mutant mice in the gamma band (30–40 Hz) compared to WT mice but not in the alpha (10–15 Hz) and beta (15–30 Hz) bands (significance indicated: **p = .028). EEG response power at the driving frequencies in the beta band is enhanced for mutant mice in the beta band (*p = .028), whereas response power at driving and 2nd harmonic frequencies is increased in the gamma band (driving: p = .021; 2nd harmonics: p = .038; see text for details)
2.8. Frequency‐shift paradigm
To investigate transitions between two stimulation frequencies, stimulation consisting of two blocks of flashes at 8 Hz (24 flashes) and 14 Hz (42 flashes) was repeated 50 times without rest (Figure 6a‐top). Frequency‐shift transition responses were subjected to Morlet wavelet analyses between 3 and 45 Hz in 0.5‐Hz frequency steps. Wavelet scales increased logarithmically between 3 and 10 (from lowest to highest frequency). Time‐frequency power per response was averaged over all repetitions, and the mean frequency of the response was calculated for each time point. For each stimulation frequency the averaged power in the EEG and MUA signals was determined for a −0.5‐ to +0.5‐Hz window around stimulation frequencies and 2nd and 3rd harmonic frequencies (i.e., 7.5–8.5 Hz, 15.5–16.5 Hz, and 23.5–24.5 Hz for 8‐Hz stimulation; 13.5–14.5 Hz, 27.5–28.5 Hz, and 41.5 to 42.5 Hz for 14‐Hz stimulation). In addition, the average EEG power over time was normalized for each stimulation frequency (8 Hz, resp. 14 Hz) to the EEG power during stimulation at the other frequency (14 Hz, resp. 8 Hz). As an outcome, the multiplicative effect of the photic drive on EEG power for both stimulation frequencies is presented (Figure 6b). The responsivity of the visual cortex to each stimulation frequency was calculated as a difference in normalized power during stimulation at that frequency (average of 1 to 2 s after stimulation frequency onset) compared to stimulation at the other frequency (1 to 2 s after frequency switch).
Figure 6.
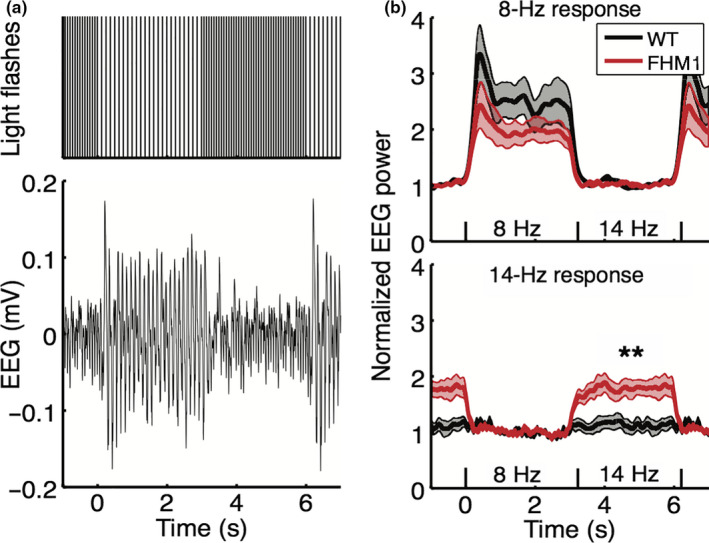
Stronger response to 14‐Hz stimulation in the frequency‐shifted paradigm in familial hemiplegic migraine type 1 (FHM1) mutant compared to wild‐type (WT) mice. (a) Top: Frequency‐shifted stimulation paradigm consisting of alternating blocks of 24 flashes at 8 Hz (duration: 3 s) and blocks of 42 flashes at 14 Hz (duration: 3 s). Bottom: Example trace of the averaged EEG response to the frequency‐shifted stimulation paradigm. (b) Top: Normalized EEG power at 8 Hz in response to the frequency‐shifted paradigm shows a similar response and rest pattern in WT and mutant mice. Bottom: Normalized EEG power at 14 Hz shows increased response to 14 Hz stimulation in mutant mice compared to WT (significance indicated: **p = .005)
2.9. Statistical analysis
Non‐parametric two‐sided Wilcoxon rank‐sum tests were used to determine whether differences between two or multiple groups were significant. Test‐retest reproducibility of IO curve readouts was compared using intraclass correlation coefficients. Intensity‐specific effects of IO responses on peak amplitude and latency were tested using two‐way Analysis of Variance (ANOVA) using intensity (0.01 to 1.0 V) and group (WT and mutant) as factors. Paired‐pulse response recovery was tested using a two‐tailed one‐sample t‐test per group using the ratio of conditioning and test stimulus amplitude versus a ratio of 1. The presence of chirp responses (mean dB change from 0) was tested as indicated for paired‐pulse recovery. Multiple comparisons were corrected for with false discovery rate (FDR) using the Benjamini‐Hochberg procedure. All statistical tests parameters were tested with GraphPad Prism 7 software (GraphPad Software, La Jolla, CA). P‐values < .05 were considered significant.
3. RESULTS
3.1. Visual evoked potentials induced by blue light specifically activate the visual cortex in freely behaving wild‐type mice
Visual evoked potentials (VEPs) in response to single‐flash blue light pulses, presented at a suprathreshold intensity at 1 Hz, were clearly recognizable in the EEG recorded with an epidural electrode over the visual cortex of all (n = 8) freely behaving wild‐type (WT) mice (Figure 1b). Averaged N1 amplitude and latency were −0.10 ± 0.03 mV at 45 ± 15 ms; P1 amplitude was 0.09 ± 0.04 mV at latency 80 ± 26 ms (in line with the literature, Lopez et al., 2002), whereas N2 amplitude and latency were −0.05 ± 0.04 mV at 197 ± 40 ms. Combining neuronal MUA and local field potential data from intracortical electrodes in the visual and motor cortex (n = 6 animals; Figure 1c‐f) revealed that blue light stimulation specifically activated the visual cortex (Figure 1c,e) and not the motor cortex, as no time‐locked multi‐unit neuronal activity was observed in the motor cortex (Figure 1f). The time‐locked local field potential activity in the motor cortex (Figure 1d) was later than the light‐evoked activity in the visual cortex and likely the result of volume conduction. Visual cortex neuronal activity showed an activation phase between 20 and 80 ms (AUC: 14.6 ± 10.1 mVms followed by a suppression below baseline between 90 and 300 ms (AUC: −17.3 ± 5.6 mVms). For subsequent VEP analyses, we used the less invasive epidurally recorded EEG signals from the primary visual cortex to assess overall visual network activity changes in response to light without confounding effects of varying electrode depth. Moreover, recording epidural VEP responses may allow for a more direct comparison with human studies that use scalp EEG. Additional visual cortex measures of intracortically recorded neuronal MUA were used to provide information on cortical neuronal network changes underlying VEP features.
3.2. Visual cortex responses in wild‐type mice are intensity‐dependent and show paired‐pulse suppression
The intensity‐dependence of VEP responses and time to recover to baseline following stimulation were tested in WT mice by assessing IO and paired‐pulse responses, respectively. IO responses were clearly visible in the averaged EEG data in response to light intensities ranging between 0.01 and 1V (Figure 2a‐bottom) and comparable between animals (Figure 2b). Naka‐Rushton fitting of the first 60 pulses (Figure 2b‐top; between 0.01 and 0.1 V) indicated a V max of 0.19 ± 0.07 mV, a slope n of 2.0 ± 0.3 mV/V, and a semi‐saturation intensity k of 0.02 ± 0.008 mV. Linear regression over the 5 pulses with highest intensity (Figure 2b‐bottom; between 0.2 and 1 V) showed a V max of 0.20 ± 0.07 mV and slope of 0.05 ± 0.07 mV/V. The amplitude parameters of IO curves were reproducible when comparing two measurements separated by ~3 hr; test‐retest reliability was good for amplitude (Naka‐Rushton amplitude: ICC of 0.81; linear amplitude: ICC 0.80) but poor to medium for slope (Naka‐Rushton slope: ICC 0.53 and linear slope: ICC 0.37), and poor to medium for semi‐saturation intensity (Naka‐Rushton semi‐saturation intensity: ICC 0.53). In subsequent IO curve analyses, therefore, only amplitude parameters were investigated.
Recovery from flash light stimulation at low (0.1 V) and high (1 V) intensity in WT mice was tested using a paired‐pulse paradigm with interstimulus intervals between 50 and 1,000 ms (Figure 2c‐bottom). Test responses at intervals 50 and 100 ms overlapped with the response to the conditioning pulse and were omitted from further analyses. The total duration of the paired‐pulse paradigm did not affect the responses, as the ratio of the conditioning responses of the last 100 pulses to the first 100 pulses was neither reduced nor enhanced (P1‐N1 amplitude ratio: 0.96 ± 0.34), indicating that habituation to repeated stimulation did not occur. Therefore, the averaged response per interval was calculated over all stimulation blocks. For a low stimulation intensity of 0.1 V, paired‐pulse suppression was observed for intervals of 225 ms and shorter, with the response amplitude to the test pulse recovering to that of the conditioning amplitude (i.e., reaching a ratio of 1) at intervals longer than 225 ms (Figure 2d‐top). At the high‐intensity stimulation of 1 V, the test response amplitude showed later recovery, i.e., after 500 ms (Figure 2d‐bottom). When using a VEP habituation paradigm consisting of six blocks of 100 repeated single‐VEP stimuli at 3.1 Hz, also no habituation of P1‐N1 responses was observed (block 6 to block 1 ratio: 0.83 ± 0.30), in line with the absence of habituation to conditioning stimuli observed in the paired‐pulse paradigm.
3.3. Familial hemiplegic migraine type 1 mutant mice show aberrant intensity‐dependent visual responses to single‐pulse stimulation
To assess migraine‐relevant network changes in the visual cortex, we next compared responses to the above‐described paradigms between FHM1 mutant and WT mice (n = 8 per genotype) (Figure 3a). Averaged single‐VEP N1 peak responses to suprathreshold stimulation at 1 Hz in mutant compared to WT mice were reduced in amplitude (WT vs. FHM1: −0.10 ± 0.03 mV vs. −0.06 ± 0.08 mV; p = .04; Figure 3b) and faster (45 ± 15 ms vs. 28 ± 5 ms; p = .01; Figure 3d), but did not differ for P1 peak amplitude (0.09 ± 0.04 mV vs. 0.08 ± 0.06 mV; p = .88; Figure 3c) and latency (80 ± 26 ms vs. 75 ± 15 ms; p = .70; Figure 3e) nor for N2 peak amplitude (−0.05 ± 0.04 mV vs. −0.05 ± 0.04 mV; p = .88; Figure 3g) and latency (197 ± 40 ms vs. 184 ± 53 ms; p = .49; Figure 3h). Neuronal activity in the visual cortex of FHM1 mutant and WT mice (example MUA traces in Figure 3f) showed similar initial activation between 20 and 80 ms (AUC: 14.6 ± 10.1 mVms vs. 13.1 ± 11.2 mVms, p = .81; Figure 3i) but less neuronal suppression between 90 and 300 ms (AUC: −17.3 ± 5.6 mVms vs. −5.0 ± 5.0 mVms; p = .01; Figure 3j) in mutant mice. These data indicate a faster recovery of visual cortex activity following flash light stimulation in mutant mice. Paired‐pulse conditioning and test responses, including their ratio and effect of the duration of the paired‐pulse paradigm with respect to the observed suppression and recovery, did not differ between mutant and WT mice. In mutant mice, responses to the test pulse showed recovery to the conditioning amplitude for low (0.1 V) and high intensity (1 V) stimulation after about 225 and 500 ms, respectively (Figure 4a,b), similar to WT mice. The repeated conditioning stimulations in the paired‐pulse paradigm did not result in the habituation of the VEP responses in FHM1 mice (P1‐N1 amplitude ratio WT vs. FHM1: 0.96 ± 0.34 vs. 0.98 ± 0.55; p = .96). Also for the stimulation paradigm of six blocks of 100 pulses at 3.1 Hz, in FHM1 mice no habituation was observed, similar as was observed for WT mice (block 6 to block 1 ratio, WT vs. FHM1: 0.83 ± 0.30 vs. 1.53 ± 1.25; p = .23).
Figure 3.
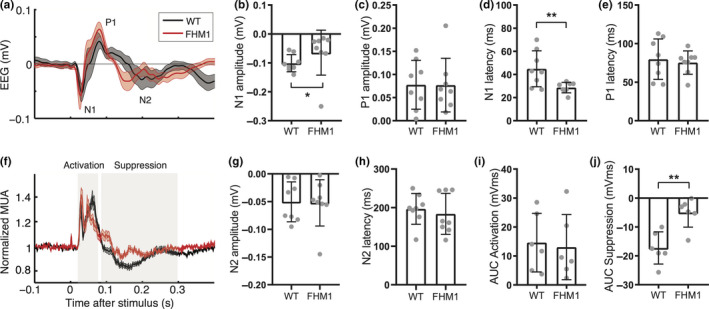
Single‐flash VEP responses differ between familial hemiplegic migraine type 1 (FHM1) mutant and wild‐type (WT) mice. (a) Average traces (mean with the shaded standard error of the mean) of epidural recordings over the visual cortex in WT (black) and FHM1 (red) mice. (b–e) Individual and mean amplitude and latency for N1 and P1 peaks (n = 8 mice per group). (b) N1 amplitude is smaller in FHM1 (significance indicated: *p = .04). (c) P1 amplitude is similar. (d) N1 latency is shorter in FHM1 (**p = .01). (e) P1 latency is similar. (f) Average traces of intracortical neuronal MUA recordings over the visual cortex of WT (black) and FHM1 (red) mice. (g, h) Individual and mean amplitude and latency for N2 peak (n = 8 mice per group). (g) N2 amplitude is similar. (h) N2 latency is similar. (i, j) Individual and mean area‐under‐curve (AUC) for two phases of the neuronal response to visual stimulation (n = 6 mice per group). (i) AUC for initial activation (between 20 and 80 ms) is similar between groups. (j) AUC for suppression (between 90 and 300 ms) is smaller in FHM1 mice (**p = .01). Error bars in B‐E and G‐J show standard deviation
Figure 4.
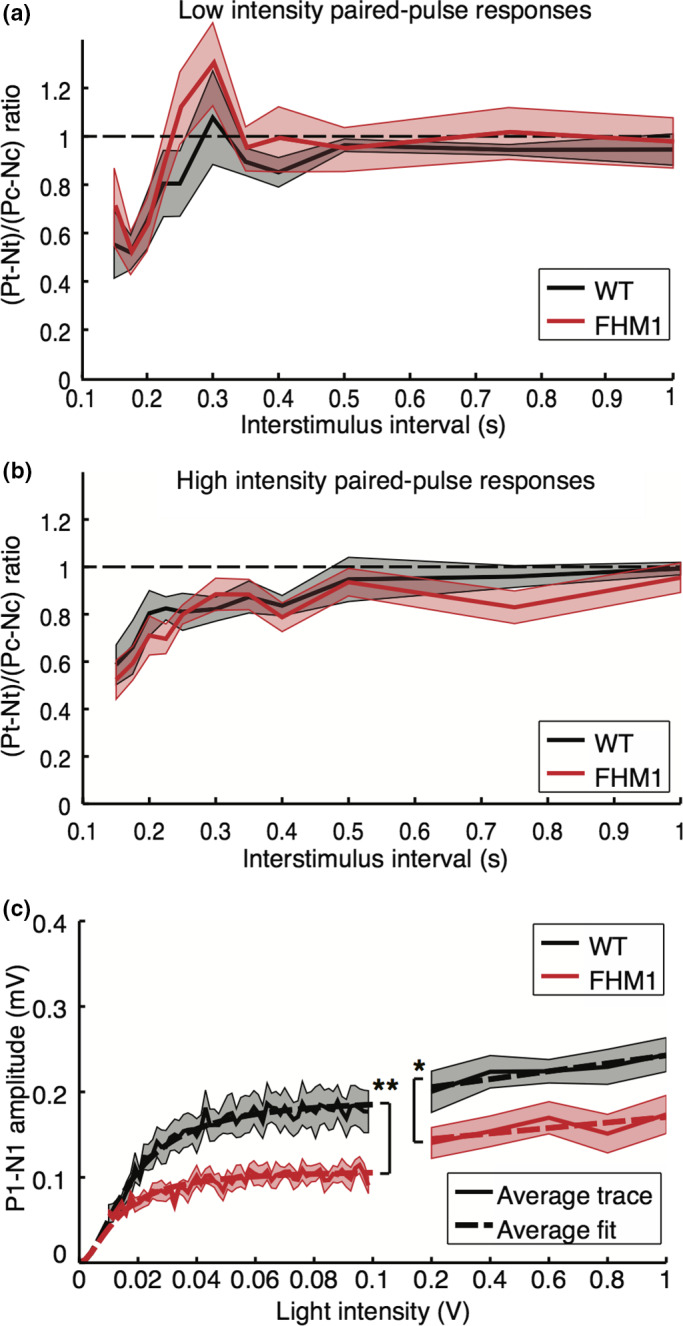
Decreased visual evoked potential input‐output (IO) responses, but similar paired‐pulse responses, in familial hemiplegic migraine type 1 (FHM1) mutant compared to wild‐type (WT) mice. (a) Averaged mutant (red; n = 8) and WT mice (black; n = 8) responses to paired‐pulse stimulation at low intensity (0.1 V) reveal recovery in both genotypes after 225 ms. Dashed line indicates recovery to baseline, at a ratio of 1. (b) Paired‐pulse responses at high intensity (1 V) show recovery at 500 ms, for both mutant and WT mice. (c) IO curves show lower VEP amplitude in mutant compared to WT mice in response to stimulation between 0.01‐ and 0.1‐V (significance indicated: **p = .005) and between 0.2‐ and 0.1‐V (*p = .028) stimulation. Dashed lines indicate averaged Naka‐Rushton fit for lower stimulation intensities and averaged least‐squares fit for higher intensities. All plots: mean and standard error of the mean (patched)
With respect to IO responses, the maximum P1‐N1 amplitude was reduced in FHM1 mutant compared to WT mice for low (0.19 ± 0.07 mV vs. 0.11 ± 0.03 mV; p = .0002; Figure 4c;), as well as higher stimulation intensities (0.20 ± 0.07 mV vs. 0.14 ± 0.05 mV; p = .03; Figure 4c). Also, N1 peaks were of smaller amplitude and had a shorter latency in mutant mice, as was also the case for the P1 peaks (all p < .001 for group effects between WT and mutants). Intensity‐specificity of the effect was observed for N1 peak amplitude, and P1 peak amplitude and latency (intensity effect: all p < .001). Only for N1 amplitude, an interaction effect was present between group and intensity (p = .003), with post hoc tests showing differences between WT and mutants for stimulation intensities of 0.02 V and higher (all p < .01 with post hoc FDR correction). Such interaction effects per intensity level were not observed for N1 latency nor for P1 peak amplitude and latency (interaction group and intensity: all p > .88). Mutant mice, compared to WT mice, showed similar local visual cortex neuronal activity levels during the N1 peak during activation (not shown; p = .27), albeit with less suppression afterward (p < .001). The intensity‐dependence of the MUA data was similar for mutants compared to WT mice (interaction effects: p > .99). Our findings reveal that FHM1 mutant mice displayed lower VEP amplitude and shorter latency, with reduced neuronal suppression, following single‐flash stimulation over a range of stimulation intensities.
3.4. Familial hemiplegic migraine type 1 mutant mice show enhanced beta‐gamma band power during chirp stimulation
To assess changes in the frequency dependency of VEP responses in FHM1 mutant mice, photic driving of visual cortex responses was tested using chirp stimulation (Figure 5a). Validation of this paradigm in WT mice revealed that the averaged EEG response showed frequency‐following between 10 and 25 Hz (Figure 5a‐middle), indicating a photic drive phenomenon. Higher‐order responses at multiples of the stimulation frequencies were also present (Figure 5b). The mutant mice showed a photic drive in response to chirp stimulation between 10 and 40 Hz (Figure 5a‐bottom). Non‐baseline‐corrected EEG response power did not differ between mutant and WT mice (Figure 5c), indicating that differences in chirp responses are not due to altered EEG spectra. Mutant mice showed an increased overall EEG power during stimulation in the 31–40 Hz gamma band (Figure 5d; p = .028) but not in the 10–15 Hz alpha (p = .80) or 16–30 Hz beta (p = .10) bands, compared to WT mice. Analysis of the separate EEG response power at the driving and harmonic frequencies revealed an increased response power in mutant mice in the gamma band for driving (p = .021) and 2nd harmonic (p = .038) frequencies, but not for 3rd harmonic frequencies (p = .16). In addition, EEG response power was enhanced in the beta frequency range for the driving (p = .028), but not 2nd harmonic (p = .083) or 3rd harmonic (p = .57) frequencies. Local visual cortex neuronal MUA did not display a clear driving or harmonic response above 15 Hz; for the alpha band MUA response, no group difference was observed (p = .10). Enhanced photic drive in the EEG beta and gamma band in response to chirp stimulation in mutant mice is in line with findings in migraine patients (Gantenbein et al., 2014), and suggestive of hyperresponsivity of the visual system.
3.5. Familial hemiplegic migraine type 1 mutant mice show enhanced VEP amplitude during 14‐Hz stimulation in a frequency‐shift paradigm
We next assessed the visual system dynamics in FHM1 mutant and WT mice using a novel frequency‐shift paradigm with alternating frequencies of 8 and 14 Hz (i.e., below and in the alpha band range). The frequency‐shifted transition between 8 and 14 Hz in WT mice (Figure 6a‐bottom) was parameterized by two amplitude shifts in the normalized EEG‐following response (drop in 8‐Hz power upon transition from 8 to 14 Hz: 1.50 ± 0.85; drop in 14‐Hz power upon transition from 14 to 8 Hz: 0.16 ± 0.31. Mutant mice showed an enhanced response power to 14‐Hz stimulation compared to WT mice (drop in 14‐Hz power: 0.77 ± 0.44; p = .005; Figure 6b‐bottom), while the response to 8 Hz was similar between genotypes (drop in 8‐Hz power: 0.91 ± 0.55; p = .19; Figure 6b‐top). Amplitude drops in the normalized visual cortex MUA were not different between genotypes (8 Hz: p = .94; 14 Hz: p = .13; data not shown).
4. DISCUSSION
Here we investigated visual system responsivity to existing and novel flash‐VEP paradigms in freely behaving mice to gain insight into mechanisms underlying visual sensitivity, particularly in the context of migraine. VEP responses were first assessed in WT mice, which showed time‐locked neuronal activation—as indicated by local multi‐unit activity (MUA) responses—in the visual cortex and intensity‐dependence. Compared to WT animals, FHM1 mutant mice carrying the R192Q missense mutation in the α1A subunit of CaV2.1 channels displayed: (a) shorter latency of the VEP N1, as well as lower VEP N1 amplitude followed by less pronounced neuronal suppression, in response to single‐flash stimulation over a range of light intensities, (b) enhanced EEG photic drive for the beta (15–30 Hz) and gamma (31–40 Hz) frequency bands in response to visual “chirp” stimulation, and (c) enhanced power in the EEG response to 14‐Hz stimulation. Together these findings indicate a frequency‐dependent enhancement of visual system responsivity in FHM1 mutant mice. The findings are in line with observations that functional effects of the FHM1‐related gain of CaV2.1 channel function across cortical (and other) regions may be context‐dependent, due to dynamic disturbances in the balance between neuronal excitation and inhibition (Vecchia & Pietrobon, 2012; Tolner et al., 2019) .
By combining EEG and local field potential with intracortical neuronal MUA recordings, we could obtain direct information on visual cortex neuronal activity, thus complementing standard VEP approaches using only EEG or local field potentials. We could thereby distinguish between local (based on MUA responses) and global (based on EEG responses) cortical network interactions in response to the different visual stimulation paradigms. A previous study in anesthetized mice indicated that VEPs largely reflect visual cortex activity and thus can be used as the measure of visual cortex responsivity to light (Ridder & Nusinowitz, 2006). The time‐locked MUA confined to the visual cortex during local field responses in our study demonstrates the specificity of the flash stimulations to activate the visual cortex, in accordance with MUA data from anesthetized mice (Land et al., 2013). While we used blue light flashes, the shape, and characteristics of our single‐pulse flash VEPs in WT mice had similar intensity‐dependent peak amplitudes and latencies as reported studies in freely behaving mice in which white light flashes were used (Mazzuchelli et al., 1995; Lopez et al., 2002; Tomiyama et al., 2016). Blue light flashes have been used earlier for VEP studies in anesthetized rats (Hudetz et al., 2009; Noseda et al., 2016) and mice (Ridder & Nusinowitz, 2006), as well as in freely behaving mice in which effects of light‐dark shifts on neuronal activity in the suprachiasmatic nucleus were investigated (van Diepen et al., 2013). Paired‐pulse VEP responses showed intensity‐dependent suppression, at low‐intensity stimulation for intervals up to 225 ms (i.e., above 4.4 Hz) and at high‐intensity stimulation up to 500 ms (i.e., 2 Hz). Differences with paired‐pulse VEP data from anesthetized mice, for which suppression occurred for intervals up to 1,000 ms (i.e., 1 Hz; Strain & Tedford, 1993) are likely due to slowing down of visual evoked potential components by anesthesia (Tomiyama et al., 2016).
In migraine patients, lack of habituation to visual pattern stimulation is an often (e.g., de Tomasso et al., 2014), but not always (e.g., Omland et al., 2016), reported feature distinguishing patients from controls. A paired‐pulse paradigm in migraine patients showed a lack of paired‐pulse suppression in the 80–130 ms interval range (Höffken et al., 2009), but longer intervals, as we presented in mice, were not studied. In our mouse experiments, we observed paired‐pulse suppression for both the WT and FHM1 mutant groups for intervals between 75 and 150 ms, as well as for longer time intervals. Both lack of habituation and lack of paired‐pulse suppression have been attributed to cortical hyperexcitability or “hyper‐responsitivity” (Coppola et al., 2007b; Höffken et al., 2009), but to our knowledge, a possible mechanistic link between the two experimental observations has not been studied directly. Contradictory findings supporting hyper‐ or hyporesponsivity across patient studies might be due to various factors: (a) in which phase of the attack the patient is when being investigated, (b) features of the stimulation paradigm such as the (spatial) frequency, and (c) differences in readout parameters (Cosentino et al., 2014; Omland et al., 2016). In our study, the clinically used habituation paradigm (consisting of 600 flashes at 3.1 Hz) did not result in habituation of the P1‐N1 component in WT or FHM1 mutant mice; neither did the longer paired‐pulse paradigm involving 650 paired flashes at 0.5 Hz. In awake restrained rats, using five blocks of 50 repeated single‐pulse light flashes at 1 Hz, for N1 and P1 components no habituation but potentiation was observed, that was influenced by dark‐ or light‐adaptation; habituation was evident though for the later P2 component (Bartus & Ferris, 1974). In anesthetized mice, local post‐synaptic potentials in the visual cortex showed rapid habituation to 4 Hz light flashes after the first of 10 pulses, which where stable for later pulses (Nestvogel et al., 2020). To allow comparison to the clinical studies, we averaged over blocks of 100 pulses without investigating possible short‐term habituation changes to single light flashes. To further study habituation to visual stimuli in freely behaving (mutant) mice, it will be useful to test other paradigms including effects of prior dark‐ or light adaptation and investigate both short‐term and longer term changes.
To better capture dynamic changes in cortical excitability two frequency‐dependent visual stimulation paradigms (i.e., chirp and frequency‐shift) were used, for the first time in mice. Visual chirp stimulation has been used to discriminate between migraine patients and controls (Gantenbein et al., 2014; Perenboom et al., 2020). We showed the applicability of this paradigm to freely behaving mice by the presence of EEG‐following and higher harmonic responses for stimulations between 10 and 25 Hz. The frequency‐shift paradigm around the alpha band (with a shift between 8 and 14 Hz) revealed the entrainment of the lower (8 Hz) but not the higher (14 Hz) frequency in WT mice. Lower responsivity to 14‐Hz stimulation in WT mice was also evident in the chirp response, which showed a dip around 14–15 Hz. This frequency‐dependency might relate to the “critical flicker frequency,” i.e., the maximum stimulation frequency at which EEG‐following responses can be measured (Kowacs et al., 2005). For mice, this maximum was estimated between 7 and 9 Hz for flash VEPs (Ridder & Nusinowitz, 2006) and around 12 Hz for pattern VEPs (Porciatti et al., 1999). Visual frequency‐following responses up to 15 Hz have been related to thalamo‐cortical interactions (Reinhold et al., 2015). The EEG‐following response above 14–15 Hz, i.e., up to 25 Hz with even higher harmonics, in our experiments, likely reflects global (including thalamic interactions) rather than local cortical network activity (Herrmann, 2001). This is further supported by the observation that local visual cortex neuronal activity did not follow chirp stimulation above ~15 Hz.
The shorter N1 latency observed for single‐VEP and input‐output paradigms in FHM1 mutant mice suggests hyperresponsivity of the visual system in mutants, likely as the result of genetically enhanced neuronal glutamatergic transmission. Earlier studies showed effects on enhancing the excitability of the R192Q mutation in cortical neuronal cultures (Vecchia et al., 2014), sensorimotor cortex brain slices in vitro (Tottene et al., 2009), and hippocampus in anesthetized mice in vivo (Dilekoz et al., 2015). The reduced suppression of neuronal activity following single‐VEP N1 responses suggests faster recovery of visual cortex activity following stimulation. Faster recovery of neuronal activity following stimulus‐related synaptic depression was also observed in brainstem slices of FHM1 mutant mice, which was hypothesized to be linked with enhanced presynaptic residual calcium levels (Inchauspe et al., 2012).
VEP P1‐N1 amplitude responses were highly repeatable in both WT and mutant mice, as shown by input‐output curve retests, whereas, in humans, VEP features can show profound temporal fluctuations (Labecki et al., 2019). The reduction of N1 VEP amplitude in mutant mice was accompanied by levels of local neuronal MUA that did not differ from that in WT animals. While VEPs are local field potentials reflecting activity within a brain region spanning at least hundreds of micrometers, the underlying MUA reflects extracellular spike activity of groups of neurons directly surrounding the tip of the electrode, dominated by activity from large (pyramidal) excitatory neurons (Buzsáki et al., 2012; Land et al., 2013; Super & Roelfsema, 2005). This suggests that the reduced VEP N1 response in mutant mice is likely caused by stronger recruitment of inhibitory neurons, also of more distantly located neurons. Enhanced inhibitory recruitment was previously implicated for the somatosensory cortex in brain slices of FHM1 mutant mice (Vecchia et al., 2014). The apparent conflicting observation of reduced neuronal suppression following N1 suggests that inhibitory networks contributing to this suppression phase are distinct from those impacting the initial N1 response.
For the VEP N2 component, amplitude and latency did not differ between FHM1 and WT mice. In migraine patients, using pattern‐VEP stimulation, N2 amplitude was reported to be enhanced (Fong et al., 2020) and latency prolonged (Oelkers et al., 1999), which may explain aversive responses of migraineurs to specific patterns and frequencies of light (Fong et al., 2020; Oelkers et al., 1999). Since early and late N2 components are proposed to reflect distinct visual system responsivity to contour and luminescence (Diener et al., 1989; Fong et al., 2020; Oelkers et al., 1999), respectively, pattern stimulation may reveal whether similar N2 changes exist for FHM1 mice.
Given the complexity of the neuronal networks involved in sensory‐evoked responses, it is not surprising that the effects of mutated CaV2.1 channels on VEP responses are not identical for the different VEP features and paradigms. For instance, possible brain region‐specific effects of the R192Q mutation may also explain the absence of a difference between FHM1 and WT mice for evoked response features following somatosensory stimulation (Khennouf et al., 2016). Hence, network‐specific changes in excitability may contribute to variable reports on hypo‐ versus hyperresponsivity in patient studies with different experimental designs (Cosentino et al., 2014). Regardless, extrapolation of findings from mouse studies to the human situation needs to be performed with great caution, not only because of species differences, but also because findings from hemiplegic migraine may not extend to the common forms of migraine.
We observed a clear effect of the FHM1 mutation on frequency‐following responses using visual chirp stimulation, whereby mutant mice were able to follow the chirp frequency stimulation up to 40 Hz, compared to a maximum of 25 Hz in WT mice. The increased EEG response power in the beta‐band (15–30 Hz) and lower gamma‐band (30–40 Hz) following chirp stimulation is in line with an enhanced beta‐gamma band response (18–26 Hz) reported interictally in migraine patients (Gantenbein et al., 2014). For patients, enhanced gamma‐power reported for pattern VEP responses (Coppola et al., 2007a) was proposed to reflect dysfunctional thalamocortical connectivity. CaV2.1 Ca2+ channels were shown to play a key role in thalamocortical gamma‐oscillatory activity in mice, as evidenced by in vitro and in vivo experiments in mice lacking CaV2.1 channels, for which EEG data showed strongly reduced gamma‐band power (Llinás et al., 2007). This suggests that the enhanced beta‐gamma power for FHM1 mutant mice in the chirp experiments may reflect enhanced thalamocortical excitability. A role of network interactions outside the visual cortex is reinforced by the absence of local neuronal activity above 15 Hz during chirp stimulation, while EEG photic drive remained present. Mutant mice also showed stronger responses to 14‐Hz stimulation in the frequency‐shift paradigm (with shifts between 8 and 14 Hz). Together, these findings indicate more pronounced frequency‐following features in response to light flashes in FHM1 mutant compared to WT mice, which may reflect visual system hyperexcitability in the mutant mice.
Transgenic FHM1 mutant mice can be used to unravel mechanisms underlying migraine susceptibility that are difficult to study in humans, whereby we consider VEPs a powerful translational tool to assess migraine‐related changes in visual network responsivity. The paradigm‐specific alterations in visual network responsivity we observed in the present study indicate frequency‐dependent enhancement of visual system excitability in FHM1 mutant mice. This may help understand how sensory hypersensitivity is brought about in migraine patients. The possibility to use VEPs in longitudinal studies, in freely behaving animals, thereby yields novel opportunities for translational studies on mechanisms and effects of attack‐modulatory triggers or migraine drugs.
CONFLICT OF INTEREST
The authors have no conflicts of interest.
AUTHOR CONTRIBUTIONS
MJLP, AMJMvdM, and EAT conceptualized and designed the study. MJLP and EAT analyzed and interpreted the data. MS collected the data. MJLP, AMJMvdM, and EAT wrote the manuscript with inputs from all other authors.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1111/ejn.15041.
ACKNOWLEDGMENTS
This work was supported by the Dutch Organization for Scientific Research NWO (Spinoza 2009 to MDF; VIDI 91711319 to GMT); the European Union "Euroheadpain" grant (grant number 602633 to MDF and AMJMvdM), “Brainpath” (grant number 612360 to AMJMvdM and EAT); LUMC Fellowship to EAT; Marie Curie Career Integration Grant to EAT; Medical Delta program “Medical NeuroDelta: Ambulant Neuromonitoring for Prevention and Treatment of Brain Disease” (to AMJMvdM). The authors thank Dr. Johanna Meijer, Sander van Berloo, Jan Jansen, and Cedric de Wijs for the development and fabrication of the recording cages and light spheres, Dr. Thijs Houben for setting up initial experiments, and Dr. Alexander Heimel for helpful discussions.
Perenboom MJL, Schenke M, Ferrari MD, Terwindt GM, van den Maagdenberg AMJM, Tolner EA. Responsivity to light in familial hemiplegic migraine type 1 mutant mice reveals frequency‐dependent enhancement of visual network excitability. Eur J Neurosci.2021;53:1672–1686. 10.1111/ejn.15041
Edited by Vidita Vaidya
DATA AVAILABILITY STATEMENT
The raw data that support the findings of this study are available from the corresponding author (EAT), upon request.
REFERENCES
- Áfra, J. , Cecchini, A. P. , De Pasqua, V. , Albert, A. , & Schoenen, J. (1998). Visual evoked potentials during long periods of pattern‐reversal stimulation in migraine. Brain, 121, 233–241. 10.1093/brain/121.2.233 [DOI] [PubMed] [Google Scholar]
- Bartus, R. T. , & Ferris, S. H. (1974). Neural correlates of habituation and dark adaptation in the visual cortex of the rat. Physiol. Psychol., 2, 55–59. [Google Scholar]
- Bjørk, M. , Hagen, K. , Stovner, L. , & Sand, T. (2011). Photic EEG‐driving responses related to ictal phases and trigger sensitivity in migraine: A longitudinal, controlled study. Cephalalgia, 31, 444–455. 10.1177/0333102410385582 [DOI] [PubMed] [Google Scholar]
- Boulloche, N. , Denuelle, M. , Payoux, P. , Fabre, N. , Trotter, Y. , & Géraud, G. (2010). Photophobia in migraine: An interictal PET study of cortical hyperexcitability and its modulation by pain. Journal of Neurology, Neurosurgery and Psychiatry, 81, 978–984. [DOI] [PubMed] [Google Scholar]
- Brennan, K. C. , & Pietrobon, D. (2018). Perspective a systems neuroscience approach to migraine. Neuron, 97, 1004–1021. 10.1016/j.neuron.2018.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki, G. , Anastassiou, C. A. , & Koch, C. (2012). The origin of extracellular fields and currents – EEG, ECoG, LFP and spikes electric current contributions from all active cellular processes within a volume of brain tissue superimpose at a given location in the extracellular medium and generate a potent. Nature Reviews Neuroscience, 13, 407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda, M. L. , Tuttle, A. H. , Baran, I. , Atlin, C. , Guindi, D. , Hathaway, G. , Israelian, N. , Levenstadt, J. , Low, D. , Macrae, L. , O’Shea, L. , Silver, A. , Zendegui, E. , Mariette Lenselink, A. , Spijker, S. , Ferrari, M. D. , Van Den Maagdenberg, A. M. J. M. , & Mogil, J. S. (2013). Behavioral evidence for photophobia and stress‐related ipsilateral head pain in transgenic Cacna1a mutant mice. Pain, 154, 1254–1262. 10.1016/j.pain.2013.03.038 [DOI] [PubMed] [Google Scholar]
- Chorlton, P. , & Kane, N. (2000). Investigation of the cerebral response to flicker stimulation in patients with headache. Clinical EEG and Neuroscience, 31, 83–87. [DOI] [PubMed] [Google Scholar]
- Coppola, G. , Ambrosini, A. , Di Clemente, L. , Magis, D. , Fumal, A. , Gérard, P. , Pierelli, F. , & Schoenen, J. (2007). Interictal abnormalities of gamma band activity in visual evoked responses in migraine: An indication of thalamocortical dysrhythmia? Cephalalgia, 27, 1360–1367. 10.1111/j.1468-2982.2007.01466.x [DOI] [PubMed] [Google Scholar]
- Coppola, G. , Pierelli, F. , & Schoenen, J. (2007). Is the cerebral cortex hyperexcitable or hyperresponsive in migraine? Cephalalgia, 27, 1429–1439. 10.1111/j.1468-2982.2007.01500.x [DOI] [PubMed] [Google Scholar]
- Cosentino, G. , Fierro, B. , & Brighina, F. (2014). From different neurophysiological methods to conflicting pathophysiological views in migraine: A critical review of literature. Clinical Neurophysiology, 125, 1721–1730. 10.1016/j.clinph.2014.05.005 [DOI] [PubMed] [Google Scholar]
- de Tommaso, M. , Ambrosini, A. , Brighina, F. , Coppola, G. , Perrotta, A. , Pierelli, F. , Sandrini, G. , Valeriani, M. , Marinazzo, D. , Stramaglia, S. , & Schoenen, J. (2014). Altered processing of sensory stimuli in patients with migraine. Nature Reviews. Neurology, 10, 144–155. [DOI] [PubMed] [Google Scholar]
- Denuelle, M. , Boulloche, N. , Payoux, P. , Fabre, N. , Trotter, Y. , & Géraud, G. (2011). A PET study of photophobia during spontaneous migraine attacks. Neurology, 76, 213–218. 10.1212/WNL.0b013e3182074a57 [DOI] [PubMed] [Google Scholar]
- Diener, H.‐C. , Scholz, E. , Dichgans, J. , Gerber, W.‐D. , Jäck, A. , Bille, A. , & Niederberger, U. (1989). Central effects of drugs used in migraine prophylaxis evaluated by visual evoked potentials. Annals of Neurology, 25, 125–130. [DOI] [PubMed] [Google Scholar]
- Dilekoz, E. , Houben, T. , Eikermann‐Haerter, K. , Balkaya, M. , Lenselink, A. M. , Whalen, M. J. , Spijker, S. , Ferrari, M. D. , van den Maagdenberg, A. M. J. M. , & Ayata, C. (2015). Migraine mutations impair hippocampal learning despite enhanced long‐term potentiation. Journal of Neuroscience, 35, 3397–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari, M. D. , Klever, R. R. , Terwindt, G. M. , Ayata, C. , & van den Maagdenberg, A. M. J. M. (2015). Migraine pathophysiology: Lessons from mouse models and human genetics. The Lancet Neurology, 14, 65–80. [DOI] [PubMed] [Google Scholar]
- Fong, C. Y. , Law, W. H. C. , Braithwaite, J. , & Mazaheri, A. (2020). Differences in early and late pattern‐onset visual‐evoked potentials between self‐ reported migraineurs and controls. NeuroImage Clinical, 25, 102122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funayama, K. , Hagura, N. , Ban, H. , & Ikegaya, Y. (2016). Functional organization of flash‐induced V1 offline reactivation. Journal of Neuroscience, 36, 11727–11738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantenbein, A. R. , Sandor, P. S. , Goadsby, P. J. , & Kaube, H. (2014). Chirp stimulation: H‐response short and dynamic. Cephalalgia, 34, 554–558. 10.1177/0333102413517777 [DOI] [PubMed] [Google Scholar]
- Gummadavelli, A. , Motelow, J. E. , Smith, N. , Zhan, Q. , Schiff, N. D. , & Blumenfeld, H. (2015). Thalamic stimulation to improve level of consciousness after seizures: Evaluation of electrophysiology and behavior. Epilepsia, 56, 114–124. 10.1111/epi.12872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, J. M. , Hauge, A. W. , Ashina, M. , & Olesen, J. (2011). Trigger factors for familial hemiplegic migraine. Cephalalgia, 31, 1274–1281. 10.1177/0333102411415878 [DOI] [PubMed] [Google Scholar]
- Headache Classification Committee of the International Headache Society (IHS) . (2018). The International Classification of Headache Disorders, 3rd edition. Cephalalgia, 38, 1–211. [DOI] [PubMed] [Google Scholar]
- Herrmann, C. S. (2001). Human EEG responses to 1–100 Hz flicker: Resonance phenomena in visual cortex and their potential correlation to cognitive phenomena. Experimental Brain Research, 137, 346–353. 10.1007/s002210100682 [DOI] [PubMed] [Google Scholar]
- Höffken, O. , Stude, P. , Lenz, M. , Bach, M. , Dinse, H. R. , & Tegenthoff, M. (2009). Visual paired‐pulse stimulation reveals enhanced visual cortex excitability in migraineurs. European Journal of Neuroscience, 30, 714–720. [DOI] [PubMed] [Google Scholar]
- Hudetz, A. G. , Vizuete, J. A. , & Imas, O. A. (2009). Desflurane selectively suppresses long‐latency cortical neuronal response to flash in the rat. Anesthesiology, 111, 231–239. 10.1097/ALN.0b013e3181ab671e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inchauspe, C. G. , Urbano, F. J. , Di Guilmi, M. N. , Ferrari, M. D. , van den Maagdenberg, A. M. J. M. , Forsythe, I. D. , & Uchitel, O. D. (2012). Presynaptic CaV2.1 calcium channels carrying familial hemiplegic migraine mutation R192Q allow faster recovery from synaptic depression in mouse calyx of Held. Journal of Neurophysiology, 108, 2967–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khennouf, L. , Gesslein, B. , Lind, B. L. , van den Maagdenberg, A. M. J. M. , & Lauritzen, M. (2016). Activity‐dependent calcium, oxygen, and vascular responses in a mouse model of familial hemiplegic migraine type 1. Annals of Neurology, 80, 219–232. [DOI] [PubMed] [Google Scholar]
- Kowacs, P. A. , Piovesan, E. J. , Werneck, L. C. , Fameli, H. , Zani, A. C. , & Da Silva, H. P. (2005). Critical flicker frequency in migraine. A controlled study in patients without prophylactic therapy. Cephalalgia, 25, 339–343. 10.1111/j.1468-2982.2004.00861.x [DOI] [PubMed] [Google Scholar]
- Labecki, M. , Nowicka, M. M. , & Suffczynski, P. (2019). Temporal modulation of steady‐state visual evoked potentials. International Journal of Neural Systems, 29, 1850050. [DOI] [PubMed] [Google Scholar]
- Land, R. , Engler, G. , Kral, A. , & Engel, A. K. (2013). Response properties of local field potentials and multiunit activity in the mouse visual cortex. Neuroscience, 254, 141–151. 10.1016/j.neuroscience.2013.08.065 [DOI] [PubMed] [Google Scholar]
- Lisicki, M. , D’Ostilio, K. , Coppola, G. , Scholtes, F. , Maertens de Noordhout, A. , Parisi, V. , Schoenen, J. , & Magis, D. (2018). Evidence of an increased neuronal activation‐to‐resting glucose uptake ratio in the visual cortex of migraine patients: A study comparing 18FDG‐PET and visual evoked potentials. J. Headache Pain, 19(19), 49. 10.1186/s10194-018-0877-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás, R. R. , Choi, S. , Urbano, F. J. , & Shin, H. S. (2007). γ‐Band deficiency and abnormal thalamocortical activity in P/Q‐type channel mutant mice. Proceedings of the National Academy of Sciences of the United States of America, 104, 17819–17824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez, L. , Brusa, A. , Fadda, A. , Loizzo, S. , Martinangeli, A. , Sannita, W. G. , & Loizzo, A. (2002). Modulation of flash stimulation intensity and frequency: Effects on visual evoked potentials and oscillatory potentials recorded in awake, freely moving mice. Behavioral Brain Research, 131, 105–114. 10.1016/S0166-4328(01)00351-5 [DOI] [PubMed] [Google Scholar]
- Magis, D. , Ambrosini, A. , Bendtsen, L. , Ertas, M. , Kaube, H. , & Schoenen, J. (2007). Evaluation and proposal for optimalization of neurophysiological tests in migraine: Part 1–electrophysiological tests. Cephalalgia, 27, 1323–1338. 10.1111/j.1468-2982.2007.01440.x [DOI] [PubMed] [Google Scholar]
- Mazzucchelli, A. , Conte, S. , D’Olimpio, F. , Ferlazzo, F. , Loizzo, A. , Palazzesi, S. , & Renzi, P. (1995). Ultradian rhythms in the N1–P2 amplitude of the visual evoked potential in two inbred strains of mice: DBA/2J and C57BL/6. Behavioral Brain Research, 67, 81–84. 10.1016/0166-4328(94)00107-Q [DOI] [PubMed] [Google Scholar]
- Mulleners, W. M. , Aurora, S. K. , Chronicle, E. P. , Stewart, R. , Gopal, S. , & Koehler, P. J. (2001). Self‐reported photophobic symptoms in migraineurs and controls are reliable and predict diagnostic category accurately. Headache, 41, 31–39. 10.1046/j.1526-4610.2001.111006031.x [DOI] [PubMed] [Google Scholar]
- Nestvogel, D. B. , Merino, R. M. , Leon‐Pinzon, C. , Schottdorf, M. , Lee, C. K. , Imig, C. , Brose, N. , & Rhee, J. S. (2020). The synaptic vesicle priming protein CAPS‐1 shapes the adaptation of sensory evoked responses in mouse visual cortex. Cell Reports, 30, 3261–3269.e4. 10.1016/j.celrep.2020.02.045 [DOI] [PubMed] [Google Scholar]
- Noseda, R. , Bernstein, C. A. , Nir, R. , Lee, A. J. , Fulton, A. B. , Bertisch, S. M. , Hovaguimian, A. , Cestari, D. M. , Saavedra‐Walker, R. , Borsook, D. , Doran, B. L. , Buettner, C. , & Burstein, R. (2016). Migraine photophobia originating in cone‐driven retinal pathways. Brain, 139, 1971–1986. 10.1093/brain/aww119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noseda, R. , & Burstein, R. (2013). Migraine pathophysiology: Anatomy of the trigeminovascular pathway and associated neurological symptoms, cortical spreading depression, sensitization, and modulation of pain. Pain, 154, S44–S53. 10.1016/j.pain.2013.07.021 [DOI] [PubMed] [Google Scholar]
- Nyrke, T. , Kangasniemi, P. , & Lang, A. H. (1989). Difference of steady‐state visual evoked potentials in classic and common migraine. Electroencephalography and Clinical Neurophysiology, 73, 285–294. [DOI] [PubMed] [Google Scholar]
- Oelkers, R. , Grosser, K. , Lang, E. , Geisslinger, G. , Kobal, G. , Brune, K. , & Lötsch, J. (1999). Visual evoked potentials in migraine patients: Alterations depend on pattern spatial frequency. Brain, 122, 1147–1155. 10.1093/brain/122.6.1147 [DOI] [PubMed] [Google Scholar]
- Omland, P. M. , Nilsen, K. B. , Uglem, M. , Gravdahl, G. , Linde, M. , Hagen, K. , & Sand, T. (2013). Visual evoked potentials in interictal migraine: No confirmation of abnormal habituation. Headache, 53, 1071–1086. 10.1111/head.12006 [DOI] [PubMed] [Google Scholar]
- Omland, P. M. , Uglem, M. , Hagen, K. , Linde, M. , Tronvik, E. , & Sand, T. (2016). Visual evoked potentials in migraine: Is the “neurophysiological hallmark” concept still valid? Clinical Neurophysiology, 127, 810–816. 10.1016/j.clinph.2014.12.035 [DOI] [PubMed] [Google Scholar]
- Perenboom, M. J. L. , Zamanipoor Najafabadi, A. H. , Zielman, R. , Carpay, J. A. , & Ferrari, M. D. (2018). Quantifying visual allodynia across migraine subtypes: The Leiden Visual Sensitivity Scale. Pain, 159, 2375–2382. 10.1097/j.pain.0000000000001343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perenboom, M. J. L. , van de Ruit, M. , Zielman, R. , van den Maagdenberg, A. M. J. M. , Ferrari, M. D. , Carpay, J. A. , & Tolner, E. A. (2020). Enhanced pre‐ictal cortical responsivity in migraine patients assessed by visual chirp stimulation. Cephalalgia, 40, 913–923. 10.1177/0333102420912725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porciatti, V. , Pizzorusso, T. , & Maffei, L. (1999). The visual physiology of the wild type mouse determined with pattern VEPs. Vision Research, 39, 3071–3081. [DOI] [PubMed] [Google Scholar]
- Reinhold, K. , Lien, A. D. , & Scanziani, M. (2015). Distinct recurrent versus afferent dynamics in cortical visual processing. Nature Neuroscience, 18, 1789–1797. [DOI] [PubMed] [Google Scholar]
- Ridder, W. H. , & Nusinowitz, S. (2006). The visual evoked potential in the mouse–origins and response characteristics. Vision Research, 46, 902–913. [DOI] [PubMed] [Google Scholar]
- Sand, T. , Zhitniy, N. , White, L. R. , & Stovner, L. J. (2008). Visual evoked potential latency, amplitude and habituation in migraine: A longitudinal study. Clinical Neurophysiology, 119, 1020–1027. 10.1016/j.clinph.2008.01.009 [DOI] [PubMed] [Google Scholar]
- Schridde, U. , Khubchandani, M. , Motelow, J. E. , Sanganahalli, B. G. , Hyder, F. , & Blumenfeld, H. (2008). Negative BOLD with large increases in neuronal activity. Cerebral Cortex, 18, 1814–1827. 10.1093/cercor/bhm208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte, L. H. , Jürgens, T. P. , & May, A. (2015). Photo‐, osmo‐ and phonophobia in the premonitory phase of migraine: Mistaking symptoms for triggers? Journal of Headache and Pain, 16, 1–5. 10.1186/s10194-015-0495-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strain, G. M. , & Tedford, B. L. (1993). Flash and pattern reversal visual evoked potentials in C57BL/6J and B6CBAF1/J mice. Brain Research Bulletin, 32, 57–63. [DOI] [PubMed] [Google Scholar]
- Super, H. , & Roelfsema, P. R. (2005). Chronic multiunit recordings in behaving animals: Advantages and limitations. Progress in Brain Research, 147, 263–282. [DOI] [PubMed] [Google Scholar]
- Tolner, E. A. , Chen, S. P. , & Eikermann‐Haerter, K. (2019). Current understanding of cortical structure and function in migraine. Cephalalgia, 39, 1683–1699. 10.1177/0333102419840643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolner, E. A. , Houben, T. , Terwindt, G. M. , De Vries, B. , Ferrari, M. D. , & Van Den Maagdenberg, A. M. J. M. (2015). From migraine genes to mechanisms. Pain, 156, S64–S74. 10.1097/01.j.pain.0000460346.00213.16 [DOI] [PubMed] [Google Scholar]
- Tomiyama, Y. , Fujita, K. , Nishiguchi, K. M. , Tokashiki, N. , Daigaku, R. , Tabata, K. , Sugano, E. , Tomita, H. , & Nakazawa, T. (2016). Measurement of electroretinograms and visually evoked potentials in awake moving mice. PLoS ONE, 11, 1–12. 10.1371/journal.pone.0156927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tottene, A. , Conti, R. , Fabbro, A. , Vecchia, D. , Shapovalova, M. , Santello, M. , van den Maagdenberg, A. M. J. M. , Ferrari, M. D. , & Pietrobon, D. (2009). Enhanced excitatory transmission at cortical synapses as the basis for Facilitated spreading depression in CaV2.1 knockin migraine mice. Neuron, 61, 762–773. 10.1016/j.neuron.2009.01.027 [DOI] [PubMed] [Google Scholar]
- Van Den Maagdenberg, A. M. J. M. , Pietrobon, D. , Pizzorusso, T. , Kaja, S. , Broos, L. A. M. , Cesetti, T. , Van De Ven, R. C. G. , Tottene, A. , Van Der Kaa, J. , Plomp, J. J. , Frants, R. R. , & Ferrari, M. D. (2004). A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron, 41, 701–710. 10.1016/S0896-6273(04)00085-6 [DOI] [PubMed] [Google Scholar]
- van Diepen, H. C. , Ramkisoensing, A. , Peirson, S. N. , Foster, R. G. , & Meijer, J. H. (2013). Irradiance encoding in the suprachiasmatic nuclei by rod and cone photoreceptors. The FASEB Journal, 27, 4204–4212. [DOI] [PubMed] [Google Scholar]
- Vecchia, D. , & Pietrobon, D. (2012). Migraine: A disorder of brain excitatory–inhibitory balance? Trends in Neurosciences, 35, 507–520. [DOI] [PubMed] [Google Scholar]
- Vecchia, D. , Tottene, A. , van den Maagdenberg, A. M. J. M. , & Pietrobon, D. (2014). Mechanism underlying unaltered cortical inhibitory synaptic transmission in contrast with enhanced excitatory transmission in CaV2.1 knockin migraine mice. Neurobiology of Diseases, 69, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data that support the findings of this study are available from the corresponding author (EAT), upon request.