

Abstract
Objective
To ascertain the role of platelet glycoprotein Ib α‐chain (GPIbα) plasma protein levels in cardiovascular, autoimmune, and autoinflammatory diseases and whether its effects are mediated by platelet count.
Methods
We performed a two‐sample Mendelian randomization (MR) study, using both a cis‐acting protein quantitative trait locus (cis‐pQTL) and trans‐pQTL near the GP1BA and BRAP genes as instruments. To assess if platelet count mediated the effect, we then performed a two‐step MR study. Putative associations (GPIbα/platelet count/disease) detected by MR analyses were subsequently assessed using multiple‐trait colocalization analyses.
Results
After correction for multiple testing (Bonferroni‐corrected threshold P ≤ 2 × 10−3), GPIbα, instrumented by either cis‐pQTL or trans‐pQTL, was causally implicated with an increased risk of oligoarticular and rheumatoid factor (RF)–negative polyarticular juvenile idiopathic arthritis (JIA). These effects of GPIbα appeared to be mediated by platelet count and were supported by strong evidence of colocalization (probability of all 3 traits sharing a common causal variant ≥0.80). GPIbα instrumented by cis‐pQTL did not appear to affect cardiovascular risk, although the GPIbα trans‐pQTL was associated with an increased risk of cardiovascular diseases and autoimmune diseases but a decreased risk of autoinflammatory diseases, suggesting that this trans‐acting instrument operates through other pathways.
Conclusion
The role of platelets in thrombosis is well‐established; however, our findings provide some novel genetic evidence that platelets may be causally implicated in the development of oligoarticular and RF‐negative polyarticular JIA, and indicate that GPIbα may serve as a putative therapeutic target for these JIA subtypes.
INTRODUCTION
Platelet glycoprotein Ib α‐chain (GPIbα) is a platelet surface membrane protein (1). It functions as a receptor for von Willebrand factor (vWF) and is implicated in atherothrombosis (2). Genetic evidence supports the assertion that GPIbα influences atherothrombosis via increased platelet counts (3). Given the potential of GPIbα as an antithrombotic target, its efficacy for the treatment of thrombotic thrombocytopenic purpura is currently being investigated in a phase II trial (3). Recent studies have also indicated a role for platelets in inflammation and immunity (4, 5), which may imply potential for repurposing GPIbα as a target for prevention/treatment of immune‐related disease. However, these putative associations have not been systematically evaluated.
Mendelian randomization (MR) studies utilize genetic variants, randomly allocated during conception, as instruments to infer causality and are less prone to confounding and reverse causation than observational studies (6). They are increasingly used to ascertain the health effects of potential therapeutic targets. Colocalization can further help to distinguish causal effects from confounding via linkage disequilibrium (LD) (7). Collectively, applying MR and colocalization to ‐omics data can provide a distinct strand of genetic validation for putative causal gene targets and thus improve the success rate of drug trials (8, 9).
To understand the effects of GPIbα on cardiovascular, autoimmune, and autoinflammatory diseases, and whether these are mediated by platelet count, we conducted a two‐step, two‐sample MR study. We subsequently performed multiple‐trait colocalization analyses (i.e., on GPIbα, platelet count, and a disease) to complement the evidence for causal associations detected in our MR study.
MATERIALS AND METHODS
Study design
MR relies on 3 core assumptions. First, the genetic variant is robustly associated with the exposure. Second, the genetic variant is independent of confounders of the exposure–outcome association. Third, the genetic variant is independent of the outcome except via the exposure (10).
In this study, we first performed a two‐sample MR study to assess the association of GPIbα with cardiovascular, autoimmune, and autoinflammatory disease risk (Figure 1A). To further assess whether platelets mediate the effects of GPIbα on disease, we subsequently performed a two‐step, two‐sample MR study. First, we assessed the association of GPIbα with platelet count. Second, we assessed the effect of platelet count on the disease outcome (Figure 1B).
Figure 1.
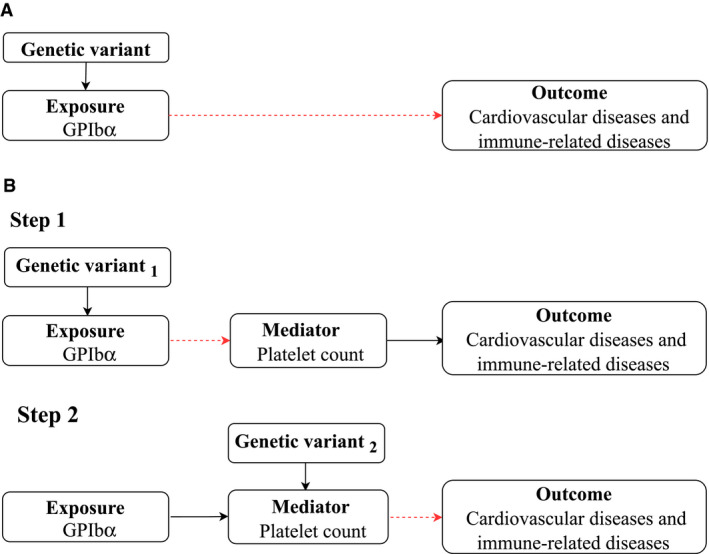
Schematic diagram of A, standard Mendelian randomization (MR) analysis of glycoprotein Ib α‐chain (GPIbα) and B, two‐step MR analysis of mediation by platelet count. Two‐step MR tests the association between a genetic variant and the exposure (GPIbα) postulated to influence the outcome (cardiovascular and immune‐related diseases) via an altered mediator (platelet count). Broken arrows indicate the causal pathway to be assessed. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.41561/abstract.
Genetic instruments of GPIbα
A proteome genome‐wide association study (GWAS) was conducted in 3,301 healthy blood donors of European ancestry (3) randomly selected from the INTERVAL study (50% male) (11). The plasma protein concentrations were quantified by aptamer‐based multiplex protein assay (SOMAscan) (3). Genotyping was performed on an Affymetrix Axiom array and was imputed using a combined 1000 Genomes Phase 3‐UK10K reference panel (3). Genetic variants were excluded if they had a call rate of <99%, had a minor allele count of <8, deviated from Hardy‐Weinberg equilibrium (P < 5 × 10−6), or had an info score of <0.7 (3). The genetic associations were obtained in an additive genetic model adjusted for age, sex, duration between blood draw and processing, and the first 3 principal components of ancestry (3). Conditionally uncorrelated variants (with the lowest P value having LD r2 < 0.001) associated with GPIbα (P < 5 × 10−8) were selected as instruments.
Genetic instruments of platelet count
At the time of analyses, the largest hematologic GWAS that had been conducted included 173,480 participants of European ancestry (12). Participants were from the UK Biobank (n = 132,959; 48% male) and the INTERVAL studies (n = 40,521; 50% male) (11). Complete blood cell count was performed using a combination of fluorescence and impedance flow cytometry within 36 hours (12). Genotyping was undertaken using Affymetrix UK BiLEVE and UK Biobank Axiom arrays, and imputation was to a reference set combining the UK10K and Haplotype Reference Consortium reference panels (12). Genetic associations were obtained from a linear mixed model adjusted for the top 10 principal components of ancestry and recruitment center (12). Conditionally uncorrelated variants (with the lowest P value having LD r2 < 0.001) associated with platelet count (P < 8.31 × 10–9, a threshold for common, low frequency, and rare variants) (13) were selected as instruments. Since the genetic instruments for GPIbα were also strongly associated with platelet count, we undertook a sensitivity analysis which estimated the instrument‐specific effect of platelet count on the diseases of interest.
Genetic associations of selected outcomes
Outcomes included platelet count (per nl), 10 major cardiovascular diseases (coronary heart disease [CHD], myocardial infarction [MI], arterial embolism and thrombosis, deep venous thrombosis [DVT], phlebitis and thrombophlebitis, any stroke, any ischemic stroke, cardioembolic stroke, large artery stroke, and small vessel stroke), and 12 immune‐related diseases. Immune‐related diseases were classified (14) as autoimmune diseases (type 1 diabetes mellitus [type 1 DM], juvenile idiopathic arthritis [JIA; oligoarticular and rheumatoid factor [RF]–negative polyarticular subtypes], rheumatoid arthritis [RA], systemic lupus erythematosus, psoriasis, multiple sclerosis, primary sclerosing cholangitis, and primary biliary cirrhosis), autoinflammatory diseases (inflammatory bowel disease [IBD], Crohn's disease [CD], and ulcerative colitis [UC]), or atopic disease (eczema) (15). We obtained summary genetic associations (including estimates of regression coefficient, the corresponding standard error and P value, effect allele, other allele, and effect allele frequency) for each outcome from the largest publicly available GWAS at the time of analyses (16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27) (Supplementary Table 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41561/abstract).
MR analysis
To estimate the effect of exposure on outcome (βXY), we used the Wald estimate, i.e., the ratio of the genetic association with outcome (βGY) to the genetic association with exposure (βGX) (28) (Supplementary Figure 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41561/abstract). Wald estimates for multiple variants for the same exposure were combined using inverse variance–weighted (IVW) MR with multiplicative random effects (28), weighted median (29), and MR‐Egger (30) because these methods rely on different assumptions for valid causal inference. The MR‐Egger intercept with P < 0.05 indicates the presence of horizontal pleiotropy (30). Directionally consistent results from different methods increase confidence in the results of MR analyses. To orientate the direction of the effects of instruments, we applied Steiger filtering (31). Steiger filtering examines whether the variance explained between each variant–exposure () is larger than the variance explained between each variant–outcome effect (), and therefore whether the instrument primarily influences the outcome through the exposure (and not vice versa) (31). Two‐sided P values are reported throughout, with a Bonferroni correction for multiple testing threshold (P ≤ 2 × 10−3, given 22 disease traits were considered). Several of the traits examined in this study are likely to share clinical and underlying immunopathogenic features despite their distinct phenotypes; therefore, using this stringent correction provides a balance between reducing false positives and providing rigorous results.
Instrument strength
F statistics were calculated for each instrument of GPIbα as , where R2 indicates the proportion of exposure variability explained by the instrument, K indicates the number of instruments, and N indicates the sample size. R2 was calculated as 2EAF(1 – EAF)β2, where EAF is the effect allele frequency and β is the effect size of the effect allele. Higher F statistic values reflect a lower risk of weak instrument bias (32).
Multiple‐trait colocalization analysis
To differentiate whether any putative causal association detected by two‐step MR is driven by a common causal variant across multiple traits (i.e., GPIbα/platelet count/disease) or just confounded by LD, we subsequently performed multiple‐trait colocalization analyses at each locus (7). Under the assumption of a single causal variant within each region, the Bayesian statistical framework quantifies the posterior probability of association (PPA) for each of the possible hypotheses of colocalization (variant sharing) among the 3 traits (all hypotheses are listed in Supplementary Table 2, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41561/abstract). A lead variant‐centric approach was applied, where we extracted effect estimates and allele information for all variants within 1 megabase upstream and downstream of the cis‐acting protein quantitative trait locus (cis‐pQTL) and trans‐acting pQTL for each trait (GPIbα/platelet count/disease), respectively. To provide reliable evidence of colocalization, at least 50 variants (with minor allele frequency >1%), including the causal variant of interest, within the test region for all 3 traits were assessed (7). We assigned prior probabilities that a variant is equally associated with 1 trait (p1 = 1 × 10−4), 2 traits (p2 = 1 × 10−6), and 3 traits (p3 = 1 × 10−7), as recommended (7). The PPA for all 3 traits was ≥0.80, which was considered strong evidence of colocalization (7).
MR analyses were performed using the TwoSampleMR package, and multiple‐trait colocalization analyses were conducted using the moloc package. Results were visualized using the forestplot package in the R software platform (version 3.5.1; R Development Core Team).
RESULTS
Genetic instruments for GPIbα and instrument strength
Two conditionally uncorrelated (LD r2 < 0.001) pQTLs associated with GPIbα were used as instruments: cis‐pQTL (rs72835078 within the GP1BA gene) and trans‐pQTL (rs11065979 near the BRAP gene). The F statistic for cis‐pQTL was 48, with 1.4% of the variance in GPIbα explained by cis‐pQTL, and the F statistic for trans‐pQTL was 50, with 1.5% of the variance in GPIbα explained by trans‐pQTL (Supplementary Table 3, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41561/abstract).
Association of GPIbα with JIA
Using a Bonferroni‐corrected threshold of P ≤ 2 × 10−3 (equivalent to P ≤ 0.05 for a single test), the two‐sample MR analysis (Figure 1A) suggested that increased GPIbα level was positively associated with an increased risk of JIA. Higher GPIbα level instrumented by cis‐pQTL was associated with a higher risk of JIA, with an odds ratio (OR) of 2.45 (95% confidence interval [95% CI] 1.40–4.29) (P = 1.71 × 10−3) (Figure 2 and Supplementary Table 4, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41561/abstract). Higher GPIbα level instrumented by trans‐pQTL was also associated with a higher risk of JIA (OR 3.01 [95% CI 1.64–5.51], P = 3.66 × 10−4) (Figure 2 and Supplementary Table 5, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41561/abstract). When combining the estimates instrumented by both cis‐pQTL and trans‐pQTL, per unit increase in GPIbα level was associated with a 169% higher risk of JIA (OR 2.69 [95% CI 1.79–4.06], P = 2.33 × 10−6).
Figure 2.
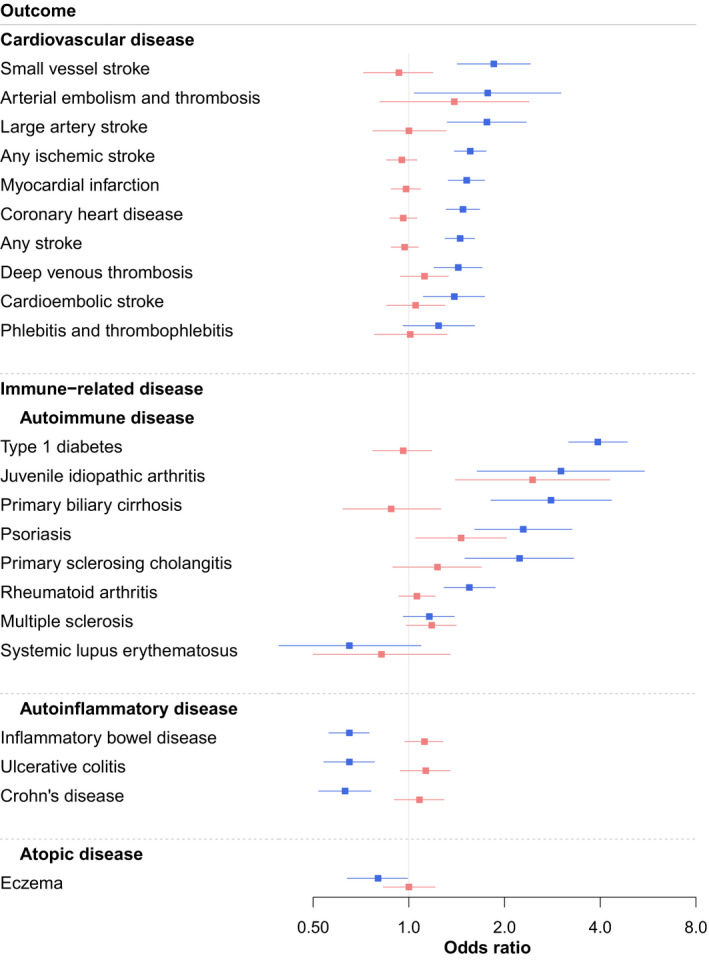
Mendelian randomization estimates for effect of glycoprotein Ib α‐chain on cardiovascular and immune‐related diseases. Values are the odds ratio (point estimate of effect) and 95% confidence interval. Red represents GPIbα instrumented by the cis‐acting protein quantitative trait locus (cis‐pQTL) within the GP1BA gene, and blue represents GPIbα instrumented by the trans‐pQTL near the BRAP gene.
There was little evidence of an association of GPIbα level instrumented by cis‐pQTL with cardiovascular diseases (Figure 2 and Supplementary Table 4). However, the GPIbα trans‐pQTL was associated with an increased risk of cardiovascular diseases (small vessel stroke, large artery stroke, any ischemic stroke, MI, CHD, any stroke, and DVT) and autoimmune diseases (type 1 DM, JIA, primary biliary cirrhosis, psoriasis, primary sclerosing cholangitis, and RA) but decreased risk of autoinflammatory diseases (IBD, UC, and CD) (Figure 2 and Supplementary Table 5). The Steiger filtering tests suggested that the instruments primarily influenced the outcome through the exposure (GPIbα).
The association of GPIbα with JIA is mediated by platelet count
Figure 1B demonstrates how two‐step MR estimates whether the effect of GPIbα on JIA is mediated by platelet count. In the first step, increased GPIbα level was associated with higher platelet count (β = 0.37 [95% CI 0.03–0.70]; P = 0.03), among which the effect instrumented by trans‐pQTL (β = 0.54 [95% CI 0.49–0.58]; P = 1.37 × 10−143) was larger than that instrumented by cis‐pQTL (β = 0.19 [95% CI 0.15–0.23]; P = 2.54 × 10−19) (Figure 3). In the second step, there were 135 conditionally uncorrelated variants (LD r2 < 0.001) associated with platelet count (P < 8.31 × 10−9) (Supplementary Table 6, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41561/abstract). Using the IVW method, genome‐wide genetically predicted platelet count was positively associated with the risk of JIA (OR 1.88 [95% CI 1.12–3.16], P = 0.02) (Figure 4). The weighted median and MR‐Egger methods provided consistent findings, with no evidence of horizontal pleiotropy (Supplementary Table 7, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41561/abstract). Sensitivity analyses restricted to each specific instrument for GPIbα also showed that platelet count increased the risk of JIA (Supplementary Table 7).
Figure 3.
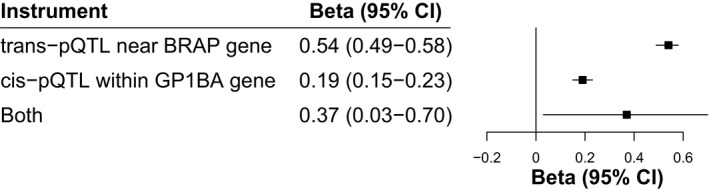
Mendelian randomization estimates for the effect of glycoprotein Ib α‐chain (GPIbα) on platelet count. Values are the beta (point estimate of effect) and 95% confidence interval (95% CI). pQTL = protein quantitative trait locus.
Figure 4.

Mendelian randomization (MR) estimates for effect of platelet count on juvenile idiopathic arthritis. Values are the odds ratio (OR; point estimates of effect) and 95% confidence interval (95% CI).
Multiple‐trait colocalization analysis supports the causal association of GPIbα mediated by platelet count with JIA
The two‐step MR analyses suggested that GPIbα mediated by platelet count has an impact on JIA (P ≤ 2 × 10−3); this association was assessed using multiple‐trait colocalization analysis. The association (GPIbα instrumented by trans‐pQTL/platelet count/JIA) was supported by strong evidence of colocalization (PPA ≥ 0.80), indicating that the same causal variant affects 3 traits. The association of GPIbα instrumented by cis‐pQTL meditated by platelet count with JIA could not be assessed because the causal variant of interest was not available for the outcome (Supplementary Table 8, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41561/abstract).
DISCUSSION
In this MR study, we observed that GPIbα instrumented by cis‐pQTL and GPIbα instrumented by trans‐pQTL both increased the risk of oligoarticular and RF‐negative polyarticular JIA. We found no evidence of an association of GPIbα instrumented by cis‐pQTL with cardiovascular diseases. However, GPIbα instrumented by trans‐pQTL increased the risk of cardiovascular and autoimmune diseases but decreased the risk of autoinflammatory diseases, suggesting potential pleiotropic effects of this trans‐pQTL on multiple disease outcomes. Two‐step, two‐sample MR analysis showed that the effect of GPIbα on the increased risk of oligoarticular and RF‐negative polyarticular JIA was mediated by platelet count, which was supported by strong evidence of colocalization. Apart from the well‐established role of platelets in thrombosis (4), our findings provide novel evidence that platelets are causally implicated in oligoarticular and RF‐negative polyarticular JIA.
The GPIb–IX–V complex is a well‐characterized adhesion receptor for vWF and collagen, of which the subunit GPIbα is associated with an increased risk of ischemic cerebrovascular disease in genetic studies (33, 34). Murine data show that absence of GPIbα significantly reduces platelet count and down‐regulates atherosclerosis and inflammation (35), consistent with our findings. Our results also align with a large GWAS of 1 million participants of European ancestry which found that the lead variant at the BRAP gene (rs11065979) was positively associated with cardiometabolic and autoimmune diseases in overall and sex‐specific analyses (36). Activated platelets secrete a wide range of cytokines (e.g., interleukin‐6 [IL‐6] and IL‐1), neutrophil chemoattractant (e.g., IL‐8), growth factors, and potent vasoconstrictors (e.g., thromboxane) (4), which play an important role in amplifying inflammatory and thrombotic cascades in these conditions (37, 38). Consistent with our findings, in vivo, platelet‐derived cellular microparticles have been observed in synovial fluid from patients with inflammatory polyarthropathies (e.g., RA, JIA, and psoriatic arthritis) but not from patients with noninflammatory arthritis (osteoarthritis) (37). Furthermore, platelet indices were associated with increased disease activity and severity of JIA (oligoarticular, RF‐negative polyarticular, and systemic subtypes) and were highly labile, particularly in the acute phase (39).
Our findings are consistent with those of a growing number of studies that illustrate the close relationship between atherosclerotic and immune‐mediated disorders (40), leading to the exploration of the role of antiatherosclerotic agents in the autoimmune arena. The antiplatelet agent ticagrelor is under investigation in RA (Clinicaltrials.gov identifier: NCT02874092), and abciximab (a glycoprotein IIb/IIIa inhibitor) is used in children with Kawasaki disease (an inflammatory vasculitis that particularly affects the heart) (41). With regard to the role of platelets in JIA, JIA patients have been shown to have impaired vascular function and thus potentially increased cardiovascular risk (42). Existing therapies for JIA include nonselective nonsteroidal antiinflammatory drugs, which have been shown to antagonize platelet function, and escalation to biologic therapies including anti–tumor necrosis factor (e.g., infliximab, adalimumab, and golimumab), anti–IL‐6 (tocilizumab), and anti–IL‐1 (canakinumab and anakinra) (43). IL‐1 blockade with anakinra has limited efficacy in RA (44), and it has been postulated that this is, in part, due to difficulty in antagonizing platelet microparticle–derived IL‐1 (37). Conversely, IL‐1 blockade is highly effective in the treatment of systemic JIA (45), where very high platelet counts are common.
In our study, GPIbα was associated with an increased risk of both oligoarticular and RF‐negative polyarticular JIA, and this association was shown to be mediated by platelet count. Our findings imply a novel role for platelets in oligoarticular and RF‐negative polyarticular JIA, extending the pathogenic role of platelets in JIA to include disease causation. Therefore, GPIbα represents a potential new therapeutic strategy or a drug repurposing opportunity for these JIA subtypes, which is supported within the current literature. However, given multiple physiologic drivers and functions of platelets (46), such approaches need to be carefully explored to ensure therapeutic benefit. In addition, JIA consists of 7 subtypes (of which oligoarticular and polyarticular subtypes account for up to 90%) (47), and it is increasingly recognized that these comprise discrete clinical entities (48). Further work will be required to ascertain whether these findings are applicable to other JIA subtypes, in particular systemic JIA.
The limitations of this study include, first, that ~8% of the participants from the INTERVAL study (3,300 of 40,521) overlapped between the proteome GWAS and the hematologic GWAS. Nonetheless, bias due to sample overlap is likely to be negligible in this study due to the presence of strong instruments (49). Second, exposures instrumented by a single variant precluded the use of pleiotropy‐robust MR methods, such as weighted median and MR‐Egger (29, 30), which require a large number of instruments. Therefore, to improve the reliability of causal inference, we used multiple‐trait colocalization to complement the MR findings, as recommended (50). Third, it is important to note that although multiple‐trait colocalization analysis provided strong evidence that GPIbα impacts disease via its effects on platelet count, other potential interpretations such as horizontal pleiotropy should be considered. Fourth, BRAP also associates with 3 other proteins (vascular cell adhesion molecule 1, β2‐microglobulin, and CXCL16), which may also play a role (3). Nevertheless, these proteins are also on the same biologic pathway as GPIbα (9). Fifth, we used platelet count as the mediator; however, platelet count alone may not represent a major or sole determinant of thrombosis and inflammation, and other platelet indices may also be important. Sixth, genetic contributions to complex traits are partitioned into effects from cis‐genes and trans‐genes (51). However, authoritative analysis conclusively assessing gene regulatory networks on complex traits is beyond the scope of this study. Seventh, summary statistics are subject to the quality control and covariable adjustments conducted by the original researchers of the GWAS based on the specific optimization requirements of their data sets; the use of summary statistics precluded re‐adjustment of data. Finally, this investigation was conducted using summary statistics obtained from participants of European ancestry, and therefore might not be generalizable to other ethnic populations (52). Replication of our findings in other ethnic populations will be helpful to improve the generalizability, and evaluate whether there are underlying ethnic differences in the pathogenesis of disease (53), once data become available.
Using two‐step MR and multiple‐trait colocalization approaches, we provide reliable genetic evidence that the genetic variants that regulate GPIbα proteomic pathways, with well‐characterized biology function on platelet count, have a causal etiologic role in oligoarticular and RF‐negative polyarticular JIA. Our findings highlight the active role of platelets in these JIA subtypes, and GPIbα as a putative therapeutic target for these JIA subtypes.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Luo had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Luo, Clarke, Ramanan, Schooling, Gaunt, Au Yeung, Zheng.
Acquisition of data
Luo, Thompson, Langefeld, Marion, Grom, Gaunt, Zheng.
Analysis and interpretation of data
Luo, Clarke, Ramanan, Schooling, Gaunt, Au Yeung, Zheng.
Supporting information
Fig S1
Table S1‐S8
Acknowledgments
This work would not have been possible without access to publicly available summary data. Summary data on coronary artery disease and myocardial infarction were contributed by CARDIoGRAMplusC4D investigators and downloaded from www.cardiogramplusc4d.org. Summary data on stroke and stroke subtypes were contributed by the MEGASTROKE investigators and downloaded from http://www.megastroke.org/. The MEGASTROKE project received funding from sources specified at http://megastroke.org/acknowledgements.html. Summary data on deep venous thrombosis were downloaded from the Neale Lab (http://www.nealelab.is/). Summary data on arterial embolism, and thrombosis and phlebitis and thrombophlebitis were downloaded from the Lee Lab (https://www.leelabsg.org/resources). Summary data on multiple sclerosis were contributed by the International Multiple Sclerosis Consortium and downloaded from http://imsgc.net/. Summary data on primary sclerosing cholangitis were contributed by the International Primary Sclerosing Cholangitis Study Group Consortium and downloaded from https://www.ipscsg.org/. Summary data on eczema were contributed by the EArly Genetics and Lifecourse Epidemiology Consortium and downloaded from http://copsac.com/. Summary data on inflammatory bowel disease, Crohn’s disease, and ulcerative colitis were contributed by investigators (26) and summary data on platelet count were contributed by investigators (12) and downloaded from the GWAS Catalog. Summary data on systemic lupus erythematosus (21), rheumatoid arthritis (20), juvenile idiopathic arthritis (19), type 1 diabetes mellitus (18), psoriasis (22), and primary biliary cirrhosis (25) were obtained from MR‐Base (http://www.mrbase.org/) and the IEU GWAS database (http://gwas.mrcieu.ac.uk).
The views expressed herein are those of the authors and not necessarily those of the NHS, the National Institute for Health Research, the Department of Health, or any other funder.
Supported by the NIHR Biomedical Research Centre at University Hospitals Bristol NHS Foundation Trust and the University of Bristol. Dr. Luo’s work was supported by the Bau Tsu Zung Bau Kwan Yeun Hing Research and Clinical Fellowship from the University of Hong Kong. Dr. Clarke’s work was supported by the Wellcome Trust GW4 Clinical Academic Training Programme (grant 211030/Z/18/Z0). Dr. Zheng’s work was supported by a Vice‐Chancellor Fellowship from the University of Bristol. Drs. Clarke, Gaunt, and Zheng’s work was supported by the MRC and the University of Bristol (grant MC_UU_00011/4). Drs. Gaunt and Zheng’s work was supported by a British Heart Foundation Accelerator award (grant AA/18/7/34219).
Drs. Schooling, Gaunt, Au Yeung, and Zheng contributed equally to this work.
Dr. Gaunt has received research support from GlaxoSmithKline and Biogen for previous work. No other disclosures relevant to this article were reported.
Contributor Information
Shan Luo, Email: luoshan@hku.hk.
Jie Zheng, Email: jie.zheng@bristol.ac.uk.
References
- 1. Luo SZ, Mo X, Afshar‐Kharghan V, Srinivasan S, Lopez JA, Li R. Glycoprotein Ibα forms disulfide bonds with 2 glycoprotein Ibβ subunits in the resting platelet. Blood 2007;109:603–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bergmeier W, Piffath CL, Goerge T, Cifuni SM, Ruggeri ZM, Ware J, et al. The role of platelet adhesion receptor GPIbα far exceeds that of its main ligand, von Willebrand factor, in arterial thrombosis. Proc Natl Acad Sci U S A 2006;103:16900–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, et al. Genomic atlas of the human plasma proteome. Nature 2018;558:73–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van der Meijden PE, Heemskerk JW. Platelet biology and functions: new concepts and clinical perspectives [review]. Nat Rev Cardiol 2019;16:166–79. [DOI] [PubMed] [Google Scholar]
- 5. Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res 2018;122:337–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Relton CL, Davey Smith G. Two‐step epigenetic Mendelian randomization: a strategy for establishing the causal role of epigenetic processes in pathways to disease. Int J Epidemiol 2012;41:161–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Giambartolomei C, Liu JA, Zhang W, Hauberg M, Shi H, Boocock J, et al. A Bayesian framework for multiple trait colocalization from summary association statistics. Bioinformatics 2018;34:2538–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nat Genet 2015;47:856–60. [DOI] [PubMed] [Google Scholar]
- 9. Zheng J, Haberland V, Baird D, Walker V, Haycock PC, Hurle MR, et al. Phenome‐wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat Genet 2020;52:1122–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lawlor DA, Harbord RM, Sterne JA, Timpson N, Smith GD. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med 2008;27:1133–63. [DOI] [PubMed] [Google Scholar]
- 11. Di Angelantonio E, Thompson SG, Kaptoge S, Moore C, Walker M, Armitage J, et al. Efficiency and safety of varying the frequency of whole blood donation (INTERVAL): a randomised trial of 45 000 donors. Lancet 2017;390:2360–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Astle WJ, Elding H, Jiang T, Allen D, Ruklisa D, Mann AL, et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 2016;167:1415–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. UK10K Consortium , Walter K, Min JL, Huang J, Crooks L, Memari Y, et al. The UK10K project identifies rare variants in health and disease. Nature 2015;526:82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Med 2006;3:e297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Doria A, Zen M, Bettio S, Gatto M, Bassi N, Nalotto L, et al. Autoinflammation and autoimmunity: bridging the divide [review]. Autoimmun Rev 2012;12:22–30. [DOI] [PubMed] [Google Scholar]
- 16. Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1,000 genomes‐based genome‐wide association meta‐analysis of coronary artery disease. Nat Genet 2015;47:1121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, et al. Multiancestry genome‐wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet 2018;50:524–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Onengut‐Gumuscu S, Chen WM, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet 2015;47:381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hinks A, Cobb J, Marion MC, Prahalad S, Sudman M, Bowes J, et al. Dense genotyping of immune‐related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nat Genet 2013;45:664–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014;506:376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S, et al. Association of systemic lupus erythematosus with C8orf13‐BLK and ITGAM‐ITGAX. N Engl J Med 2008;358:900–9. [DOI] [PubMed] [Google Scholar]
- 22. Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet 2012;44:1341–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. International Multiple Sclerosis Genetics Consortium , Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, et al. Analysis of immune‐related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet 2013;45:1353–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ji SG, Juran BD, Mucha S, Folseraas T, Jostins L, Melum E, et al. Genome‐wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat Genet 2017;49:269–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cordell HJ, Han Y, Mells GF, Li Y, Hirschfield GM, Greene CS, et al. International genome‐wide meta‐analysis identifies new primary biliary cirrhosis risk loci and targetable pathogenic pathways. Nat Commun 2015;6:8019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. De Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, et al. Genome‐wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet 2017;49:256–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Paternoster L, Standl M, Waage J, Baurecht H, Hotze M, Strachan DP, et al. Multi‐ancestry genome‐wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet 2015;47:1449–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med 2016;35:1880–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bowden J, Smith GD, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 2016;40:304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bowden J, del Greco MF, Minelli C, Smith GD, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two‐sample Mendelian randomization analyses using MR‐Egger regression: the role of the I2 statistic. Int J Epidemiol 2016;45:1961–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hemani G, Tilling K, Smith GD. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet 2017;13:e1007081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Burgess S. Sample size and power calculations in Mendelian randomization with a single instrumental variable and a binary outcome. Int J Epidemiol 2014;43:922–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baker RI, Eikelboom J, Lofthouse E, Staples N, Afshar‐Kharghan V, Lopez JA, et al. Platelet glycoprotein Ibα Kozak polymorphism is associated with an increased risk of ischemic stroke. Blood 2001;98:36–40. [DOI] [PubMed] [Google Scholar]
- 34. Sonoda A, Murata M, Ito D, Tanahashi N, Ohta A, Tada Y, et al. Association between platelet glycoprotein Ibα genotype and ischemic cerebrovascular disease. Stroke 2000;31:493–7. [DOI] [PubMed] [Google Scholar]
- 35. Koltsova EK, Sundd P, Zarpellon A, Ouyang H, Mikulski Z, Zampolli A, et al. Genetic deletion of platelet glycoprotein Ibα but not its extracellular domain protects from atherosclerosis. Thromb Haemost 2014;112:1252–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Timmers PR, Mounier N, Lall K, Fischer K, Ning Z, Feng X, et al. Genomics of 1 million parent lifespans implicates novel pathways and common diseases and distinguishes survival chances. Elife 2019;8:e39856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, et al. Platelets amplify inflammation in arthritis via collagen‐dependent microparticle production. Science 2010;327:580–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Corken A, Russell S, Dent J, Post SR, Ware J. Platelet glycoprotein Ib‐IX as a regulator of systemic inflammation. Arterioscler Thromb Vasc Biol 2014;34:996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vakili M, Ziaee V, Moradinejad MH, Raeeskarami SR, Kompani F, Rahamooz T. Changes of platelet indices in juvenile idiopathic arthritis in acute phase and after two months treatment. Iran J Pediatr 2016;26:e5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sanjadi M, Sichanie ZR, Totonchi H, Karami J, Rezaei R, Aslani S. Atherosclerosis and autoimmunity: a growing relationship. Int J Rheum Dis 2018;21:908–21. [DOI] [PubMed] [Google Scholar]
- 41. Bachlava E, Loukopoulou S, Karanasios E, Chrousos G, Michos A. Management of coronary artery aneurysms using abciximab in children with Kawasaki disease. Int J Cardiol 2016;220:65–9. [DOI] [PubMed] [Google Scholar]
- 42. Vlahos AP, Theocharis P, Bechlioulis A, Naka KK, Vakalis K, Papamichael ND, et al. Changes in vascular function and structure in juvenile idiopathic arthritis. Arthritis Care Res (Hoboken) 2011;63:1736–44. [DOI] [PubMed] [Google Scholar]
- 43. Giancane G, Consolaro A, Lanni S, Davi S, Schiappapietra B, Ravelli A. Juvenile idiopathic arthritis: diagnosis and treatment. Rheumatol Ther 2016;3:187–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mertens M, Singh JA. Anakinra for rheumatoid arthritis. Cochrane Database Syst Rev 2009:CD005121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ruperto N, Brunner HI, Quartier P, Constantin T, Wulffraat N, Horneff G, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012;367:2396–406. [DOI] [PubMed] [Google Scholar]
- 46. Gremmel T, Frelinger AL III, Michelson AD. Platelet physiology. Semin Thromb Hemost 2016;42:191–204. [DOI] [PubMed] [Google Scholar]
- 47. Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet 2007;369:767–78. [DOI] [PubMed] [Google Scholar]
- 48. Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol 2004;31:390–2. [PubMed] [Google Scholar]
- 49. Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two‐sample Mendelian randomization. Genet Epidemiol 2016;40:597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wainberg M, Sinnott‐Armstrong N, Mancuso N, Barbeira AN, Knowles DA, Golan D, et al. Opportunities and challenges for transcriptome‐wide association studies. Nat Genet 2019;51:592–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu X, Li YI, Pritchard JK. Trans effects on gene expression can drive omnigenic inheritance. Cell 2019;177:1022–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Carlson CS, Matise TC, North KE, Haiman CA, Fesinmeyer MD, Buyske S, et al. Generalization and dilution of association results from European GWAS in populations of non‐European ancestry: the PAGE study. PLoS Biol 2013;11:e1001661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wojcik GL, Graff M, Nishimura KK, Tao R, Haessler J, Gignoux CR, et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature 2019;570:514–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Table S1‐S8