

Abstract
Key points
Ventrolateral thalamus (VL) integrates information from cerebellar nuclei and motor cortical layer VI.
Inputs from the cerebellar nuclei evoke large‐amplitude responses that depress upon repetitive stimulation while layer VI inputs from motor cortex induce small‐amplitude facilitating responses.
We report that the spiking of VL neurons can be determined by the thalamic membrane potential, the frequency of cerebellar inputs and the duration of pauses after cerebellar high frequency stimulation.
Inputs from motor cortical layer VI shift the VL membrane potential and modulate the VL spike output in response to cerebellar stimulation.
These results help us to decipher how the cerebellar output is integrated in VL and modulated by motor cortical input.
Abstract
Orchestrating complex movements requires well‐timed interaction of cerebellar, thalamic and cerebral structures, but the mechanisms underlying the integration of cerebro‐cerebellar information in motor thalamus remain largely unknown. Here we investigated how excitatory inputs from cerebellar nuclei (CN) and primary motor cortex layer VI (M1‐L6) neurons may regulate the activity of neurons in the mouse ventrolateral (VL) thalamus. Using dual‐optical stimulation of the CN and M1‐L6 axons and in vitro whole‐cell recordings of the responses in VL neurons, we studied the individual responses as well as the effects of combined CN and M1‐L6 stimulation. Whereas CN inputs evoked large‐amplitude responses that were depressed upon repetitive stimulation, M1‐L6 inputs elicited small‐amplitude responses that were facilitated upon repetitive stimulation. Moreover, pauses in CN stimuli could directly affect VL spiking probability, an effect that was modulated by VL membrane potential. When CN and M1‐L6 pathways were co‐activated, motor cortical afferents increased the thalamic spike output in response to cerebellar stimulation, indicating that CN and M1 synergistically, yet differentially, control the membrane potential and spiking pattern of VL neurons.
Keywords: cerebellum, cerebral cortex, dual‐optical stimulation, motor thalamus
Key points
Ventrolateral thalamus (VL) integrates information from cerebellar nuclei and motor cortical layer VI.
Inputs from the cerebellar nuclei evoke large‐amplitude responses that depress upon repetitive stimulation while layer VI inputs from motor cortex induce small‐amplitude facilitating responses.
We report that the spiking of VL neurons can be determined by the thalamic membrane potential, the frequency of cerebellar inputs and the duration of pauses after cerebellar high frequency stimulation.
Inputs from motor cortical layer VI shift the VL membrane potential and modulate the VL spike output in response to cerebellar stimulation.
These results help us to decipher how the cerebellar output is integrated in VL and modulated by motor cortical input.
Introduction
Successful movement requires an estimation of the sensory consequences of a motor plan and the integration of well‐timed error signals into ongoing sensorimotor processing (Ramnani, 2006; Brooks et al. 2015). This complex task requires the communication of multiple brain areas, such as the cerebellum, thalamus and motor cortex. The execution of acquired movements is mediated by cerebellar computation, in that genetic and functional lesions throughout various cortical and deeper cerebellar regions are known to disrupt execution of motor behaviour (Gao et al. 2018). Subsequently, the cerebellar output, embodied by projection neurons in the cerebellar nuclei (CN), is integrated in the ventrolateral nucleus (VL) of the thalamus with inhibitory input from reticular thalamus and cholinergic neuromodulatory input from mesencephalic nuclei and from there relayed to various layers of the motor cortex (Kha et al. 2000; Teune et al. 2000; Kuramoto et al. 2009; Proville et al. 2014; Svoboda & Li, 2017; Gornati et al. 2018). In addition, VL neurons receive excitatory input from cortical layer 6 neurons of the primary motor cortex (M1‐L6) (Yamawaki & Shepherd, 2015; Jeong et al. 2016). The interaction of subcortical and cortical inputs has been shown to determine thalamic output in the proprioceptive and visual system (Groh et al. 2008; Sherman & Guillery, 2011; Mease et al. 2014; Bickford et al. 2015). It is therefore of key importance to improve our understanding of cerebello‐cortical integration at the level of the motor thalamus. Here we focused on how M1‐L6 inputs modulate the responses in VL thalamus evoked by CN stimulation.
At rest, the baseline firing rates of CN range between 30 and 100 Hz (Hoebeek et al. 2010; Sarnaik & Raman, 2018), while VL and cortical L6 neurons fire at low frequencies between 5 and 20 Hz (Lamarre et al. 1971; Vitek et al. 1994; Beloozerova et al. 2003; Marlinski et al. 2012; Olsen et al. 2012; Proville et al. 2014). Once movement execution starts, neurons in the interposed nuclei evolve into phasic patterns including high‐frequency bursts of spiking (e.g. scratch movement in cats (Antziferova et al. 1980), locomotion in cats (Armstrong & Edgley, 1984) and mice (Sarnaik & Raman, 2018). It has been shown that stimulation of the cerebellar cortical crus I lobule results in well‐timed and rapid alterations in interposed CN activity patterns that precede an increase in spiking by VL thalamus neurons (Proville et al. 2014). These recent findings align with earlier published data about the thalamic and cortical responses evoked by cerebellar stimulation (Moruzzi, 1950; Sakata et al. 1966; Bava et al. 1967, 1986; Uno et al. 1970; Sasaki et al. 1972; Rispal‐Padel et al. 1973, 1987; Rispal‐Padel & Latreille, 1974; Shinoda, 1985; Jörntell & Ekerot, 1999). However, it remains an open question how CN‐VL synaptic transmission, which is subject to paired‐pulse depression (Uno et al. 1970; Sawyer et al. 1994; Gornati et al. 2018), and M1‐L6‐VL transmission interact in individual neurons and corroborate the VL spiking patterns that are characterized by burst‐pause and tonic spiking. In more detail, it is unclear how the pauses in the CN spiking, which are thought to decode the timing of specific sensory events (as reviewed by De Zeeuw et al. 2011), can affect VL spiking activity and how cortico‐thalamic modulation of VL membrane potential affects these supposed responses.
In the current study we investigated the interplay between CN and M1‐L6 inputs in VL neurons in an in vitro preparation, which allows us to pharmacologically and optogenetically manipulate the activities of these inputs. We recorded VL membrane potentials in whole‐cell patch‐clamp mode in combination with dual‐optical stimulation techniques to selectively stimulate CN and M1‐L6 axons. Our results show that pauses in CN spiking are a determinant of VL output and that M1‐L6 inputs modulate the cerebellar induced spiking in VL neurons, together constructing a low‐pass filter that can be fine‐tuned in a timing‐dependent manner.
Methods
Contact for reagent and resource sharing
Information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Freek E. Hoebeek (f.e.hoebeek@umcutrecht.nl).
Ethical approval
All experiments were performed in accordance with the European Communities Council Directive. All animal protocols were approved by the Dutch national experimental animal committee (DEC). For all experiments Tg( Ntsr1‐cre )GN220Gsat (Ntsr1‐Cre) transgenic mice were used, which in combination with Cre‐dependent AAV constructs allows expression in L6 neurons (Gong et al. 2007) as well as glutamatergic, thalamus‐projecting CN neurons (Houck & Person, 2015; Dumas et al. 2019). The colony was originally purchased from the MMRRC repository and maintained by backcrossing with C57Bl/6OlaHsd mice. The genotype was tested by PCR reaction using toe‐tissue gathered at postnatal days (P) 7–10. For physiology experiments Ntsr1‐Cre mice were injected with AAV‐particles (see below) at P21 and for anatomical experiments at P60–P120. All animals had ad libitum access to food and water and the total number of animals used for this study was 66. at the end of the experiment, the mice were anaesthetized with isoflurane and decapitated.
Surgical procedures and viral vectors
For surgery mice were anaesthetized with isoflurane (5% in 0.5 l/min O2 during the induction and 1.5% in 0.5 l/min O2 for maintainance). During the surgery the body temperature was maintained at 37°C and the depth of the anaesthesia was controled by monitoring the breathing frequency of the mouse. Following the systemic application of buprenorphine (i.p. injection; 50 μg/kg bodyweight) and local application of lidocaine (10%) on the dorsal skin of the head, a skin incision of ∼2 cm was made to expose dorsal skull bones and sagittal suture. Craniotomies of 0.5–2 mm were established above the planned injection sites. For injections to M1, 200 nl of adeno‐associated virus (AAV) was injected to each of the following stereotaxic coordinates relative to bregma and midline (x, y; in mm): (1) 1.5, 1; (2) 1.5, 1.25; (3) 1.5, 1.5 at −0.9 depth from the dura. For injections to the CN, 200 nl of AAV was injected 2 mm posterior to lambda, 2 mm lateral to the midline at a depth of −2 mm from the dura and on the contralateral side to M1 injections. For optical stimulation experiments, AAV2.9‐hSyn‐FLEX‐ChrimsonR‐tdTomato (provided by Professor Bryan Roth through the UNC vector core) and AAV2.9‐Ef1a‐FLEX‐ChR2(H134R)‐EYFP were injected into M1 and AAV2.9‐hsyn‐ChR2(H134R)‐EYFP was injected into CN (provided by Professor K. Deisseroth through the UNC vector core). Infected neurons express Channelrhodopsin2 (ChR2) or ChrimsonR molecules in their cell membrane, which form cation‐permeable ion channels that are activated by 470 nm light or 470–585 nm light, respectively (Klapoetke et al. 2014). ChR2 molecules also contain enhanced yellow‐fluorescent protein (EYFP) and ChrimsonR molecules the red‐fluorescent marker TdTomato. For all optical stimulation experiments, we injected the motor cortex that is ipsilateral to the recorded VL thalamus and the contralateral CN. Mice that showed ChR2 expression in vestibular nuclei neurons were excluded from analysis. For anterograde tracing of CN and M1‐L6 axons and identification of their terminals we injected 200 nl of AAV constructs (chimeric serotype 1 and 2) carrying CAG_Synaptophysin_eGFP or CAG_Synaptophysin_mOrange (kindly provided by Professor T. Kuner, Heidelberg University), respectively. For retrograde tracing of VL axons projecting to M1 we injected 1% cholera toxin subunit B (CTB).
Preparation of acute slices
Following 3—6 weeks of incubation time after the viral injection isoflurane‐anaesthetized mice were decapitated, their brains were quickly removed and placed into ice‐cold slicing medium containing (in mm): 93 NMDG, 93 HCl, 2.5 KCl, 1.2 NaHPO4, 30 NaHCO3, 25 glucose, 20 HEPES, 5 sodium ascorbate, 3 sodium pyruvate, 2 thiourea, 10 MgSO4, 0.5 CaCl2, 5 N‐acetyl‐l‐cysteine (osmolarity 310 ± 5; bubbled with 95% O2/5% CO2). Next, 250 μm thick coronal slices including the motor cortex (Fig. 3), cerebellum (Fig. 6) or thalamus (all other figures) were cut using a vibratome (VT1000S; Leica Biosystems, Nussloch, Germany). For the recovery, brain slices were incubated for 5 min in slicing medium at 34 ± 1°C and subsequently for ∼40 min in artifical cerebrospinal fluid (ACSF; containing in mm: 124 NaCl, 2.5 KCl, 1.25 Na2HPO4, 2 MgSO4, 2 CaCl2, 26 NaHCO3, and 20 d‐glucose, osmolarity 310 ± 5; bubbled with 95% O2/5% CO2) at 34 ± 1°C. After recovery brain slices were stored at room temperature for >30 min before the experiments started to extend the longivity of the recordings. For confirmation of the injection spots, motor cortices and hindbrain were conserved in 4% paraformaldehyde (PFA).
Figure 3. Fluctuations in membrane potential affect VL spike output after cerebellar stimulation.
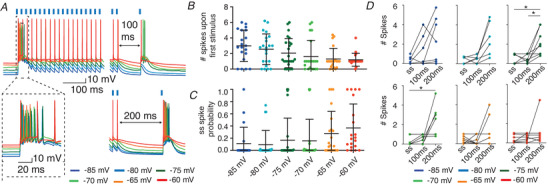
A, example showing VL responses to 50 Hz CN stimulus trains (1 ms 470 nm, in absence of ChrimsonR‐expression) and a subsequent 100 or 200 ms pause followed by a single CN stimulus. The increasing membrane potential (indicated by different colours) reduces the number of action potentials fired upon the first CN stimulus (B) and enhances the spike probability in the steady state (ss) (C). D, across membrane potentials, the average number of spikes increased after a pause of 100 and 200 ms compared to ss values. All error bars represent SD. * P < 0.05. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 6. Photosensitivity of ChR2‐EYFP‐expressing CN neurons.
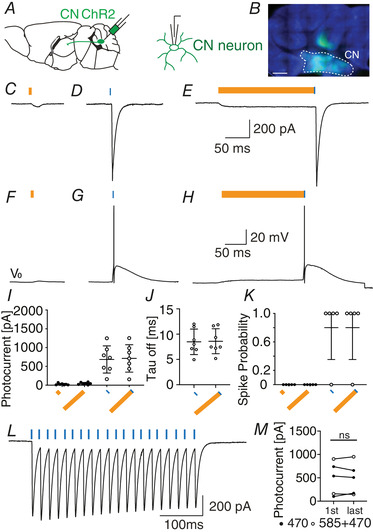
A, schematic illustration of injection strategy and patch‐clamp recording from ChR2‐EYFP‐expressing CN neurons. B, ChR2 expression in CN (scale bars: 500 μm). C–H, representative recordings of the photo‐response from ChR2‐expressing CN neurons after stimulation with 15 ms light at 585 nm (C), 1 ms light at 470 nm (D), and 200 ms light at 585 nm followed by 1 ms at 470 nm (E), when recorded in voltage clamp (VC) (C–E), and at resting potential (V 0 = −70 mV) in current clamp (F–H). I–K, quantification of the photocurrent (I) the decay (tau off) of the photocurrent (J), as well as the induced spike probabilities (K). L, representative recording of the photocurrent during stimulation with 50 Hz at 470 nm. M, the optical stimulation at 50 Hz reliably elicits inward currents, in that the peak current after first (1st) and last (last) stimulus in the train is stable. ‘ns’ indicates not significant. Error bars represent SD. [Color figure can be viewed at wileyonlinelibrary.com]
Electrophysiology and photostimulation
For all recordings, slices were bathed in 34 ± 1°C ACSF (bubbled with 95% O2/5% CO2) and supplemented with 100 μm picrotoxin to block for GABAergic inputs, e.g. evoked by neuronal activity in the adjacent reticular nucleus, which was present in the thalamic slices. Whole‐cell patch‐clamp recordings were performed using an EPC‐10 amplifier (HEKA Electronics, Lambrecht, Germany) for 20–60 min and digitized at 50 kHz. We recorded up to 3 cells per slice. Recordings were excluded if series or input resistances (RS and RI, respectively) varied by >25% over the course of the experiment or if RS exceeded a maximum of 25 MΩ. Voltage‐ and current‐clamp recordings were performed using borosilicate glass pipettes with a resistance of 3–6 MΩ when filled with K+‐based internal (in mm: 124 potassium gluconate, 9 KCl, 10 KOH, 4 NaCl, 10 HEPES, 28.5 sucrose, 4 Na2ATP, 0.4 Na3GTP (pH 7.25‐7.35; osmolarity 295 ± 5 mosmol l–1)). All recording pipettes were supplemented with 1 mg/ml Biocytin (Sigma‐Aldrich, St Louis, USA) to allow histological staining (see below). When necessary internal solution was supplemented with QX‐314 (10 mm, Sigma‐Aldrich), a blocker of voltage‐gated Na+‐channels, to prevent escape spikes in response to photocurrents in M1 and CN neurons. Current‐clamp recordings were corrected offline for the calculated liquid junction potential of −10.3 mV.
Dual‐optical stimulation was induced using a pE‐2 (CoolLED, Andover, UK) with LED wavelengths at 470 nm and 585 nm in combination with a dichroic mirror at 580 nm (filterset 15 without excitation filter, Carl Zeiss, Jena, Germany) and a 40× objective (Carl Zeiss). Light intensities were recorded by collecting photons across an area of 1 cm2 (PM400 Optical Power Meter, Thorlabs, Newton, USA) and the power was back calculated to the area of the focal spot to determine stimulation intensities. The photon flux was calculated by converting the irradiance via the following formula:
in which N is the number of photons, E is the irradiation (in W/m2), λ is the wavelength (in nm), h is the Max‐Planck constant and c is the speed of light. Full‐field dual‐optical stimulation with 585 nm was applied for 15 ms with an intensity of 1.66 mW/mm2 and a photonflux of ∼4.8 × 1024 photons/ms/m2, while stimulations at 470 nm were applied for 1 ms with intensities ranging from 0.99–7.65 mW/mm2 (maximally ∼18.08 × 1024 photons/ms/m2). The photostimulation resulted in maximally inducible response amplitudes from CN and M1‐L6 fibres. For the further refinement of this optical stimulation approach we restricted blue light to wavelengths above 460 nm (ET445/30x, Chroma Technology, Vermont, USA) and maximal intensities of 1.3 mW/mm2. To ensure that we recorded action potential driven neurotransmitter release, a portion of the CN and M1‐L6 stimulation experiments were concluded by bath application of 10 μm tetrodotoxin (TTX, Tocris, Bristol, UK), an antagonist of voltage‐gated sodium channels, which completely blocked the postsynaptic responses (data not shown).
Histology
For the histological reconstruction of the patched neurons, the brain slices were placed in 4% paraformaldehyde (PFA; in 0.1 m PB and pH 7.3) for 3–5 days. After rinsing the slices with 0.1 PB, they were placed in 10 mm sodium citrate at 80°C for 3 h and afterwards blocked for 2 h at RT (10% normal horse serum (NHS) and 0.5% Triton‐X100 in PBS). Biocytin‐filled neurons were visualized by overnight incubation with Streptavidin‐Cy5 conjugated antibody (1:400, Jackson Immunoresearch).
Image acquisition and analysis
Widefield images and confocal images were acquired on a LSM 700 microscope (Carl Zeiss) by using a 20×/0.30 NA and 63×/1.4NA objective, respectively. For the morphological reconstruction of the Synaptophysin‐mOrange and Synaptophysin‐GFP labelled synapses we used excitation wavelengths of 555 and 488 nm, respectively. For the morphological reconstruction of ChR2‐expressing fibers, ChrimsonR‐expressing fibres and biocytine‐filled, Cy5‐labelled cells we used excitation wavelengths of 488 nm, 555 nm and 639 nm, respectively.
Quantification and statistical analysis
All recording protocols (i.e. sweeps) were repeated 3–10 times and averaged for further analyses. Data analysis was performed using Clampfit software (HEKA Electronics) or custom written routines in IGOR Pro 6.21 (Wavemetrics, Lake Oswego, OR, USA). For trains of stimuli, the peak amplitude of each evoked postsynaptic current/potential (EPSC/EPSP) was detected relative to baseline. To normalize the EPSC amplitudes within the train, the amplitude of each EPSC was divided by the amplitude of the first EPSC. During train stimulation the current and voltage responses reached a plateau phase, which we defined as the ‘steady state’ response period. For calculating the average current amplitude as well as the average postsynaptic membrane depolarization, we calculated the responses during the last 100 ms of the train. To determine the average spike probability during the steady state of the train, we averaged the responses across the last 500 ms of the stimulus train. In Figs 2 and 4 we used a 2 s‐long stimulus train, while we used a 1 s‐long train in all other figures. To limit the impact of cerebellar response variability on the spike output, we excluded recordings in which the charge transferred from CN terminals was highly variable. We calculated the variability of the charge by the coefficient of variation (CV; the ratio of the standard deviation to the mean). VL recordings in which the CV of the CN‐evoked charge exceeded 0.2 were excluded from the analysis (n = 2). All data were tested for normal distribution with the Kolmogorov‐Smirnov test. For statistical comparisons Mann‐Whitney‐test, Wilcoxon matched‐pairs signed rank test or Friedman test with correction for missing values by pairwise exclusion were applied. For trend analysis the Cochran Armitrage test was applied. For statistical analyses GraphPad PRISM, SPSS and R software packages were used. In all datasets the n represents the number of recorded neurons. All datasets were corrected for multiple comparisons.
Figure 2. The effect of cerebellar stimulation on VL neurons.
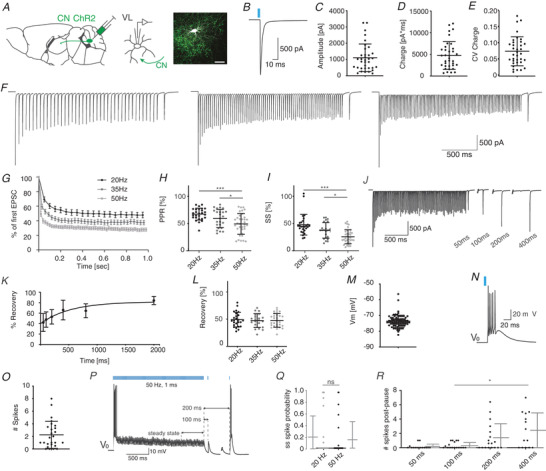
A, schematic representation of CN‐VL connectivity and experimental design in a parasagittal schematic. Immunofluorescent illustration of a patch‐clamped neuron embedded in ChR2‐expressing CN fibres taken from a coronal section (green, scale bar: 50 μm). B, example trace illustrating the optogenetically evoked response (1 ms, 470 nm) in a VL neuron. C–E, the amplitude (C), charge (D), and coefficient of variation (CV) of the charge (E) for CN‐evoked EPSCs. F, example traces depicting CN‐evoked responses to 20, 35 and 50 Hz stimulus trains of 2 s followed subsequently by a single 100 ms pause and a single CN stimulus. G, normalized steady state (ss) depression evoked by 20, 35 and 50 Hz stimulus train of 1 s. H and I, the paired pulse ratio (H, PPR) and ss current (I), evoked by train stimulation with 470 nm at 20, 35 and 50 Hz. J, the example trace depicts the time‐dependent recovery of the compound cerebellar event, which is quantified in K. L, the recovery of the CN‐evoked response in VL neurons after 100 ms pause was independent of frequency. M, the resting membrane potential (V m) of VL neurons is quantified. The thalamic burst response after cerebellar stimulation and at resting potential (V 0) is shown in the example trace in N, and quantified in O. P, example trace of VL responses to 50 Hz stimulation followed by 100 or 200 ms pauses at V 0. Q, spike probability during the steady state (ss), i.e. the last 500 ms of the stimulus train, at 20 Hz and 50 Hz. R, number of spikes evoked by single pulse CN stimulus after the extended pause following the stimulus train. All error bars represent standard deviation (SD) except for panel G, in which error bars represent standard error of the mean (SEM) for illustrative purposes. ‘ns’ indicates not significant and asterisks indicate significance level: * P < 0.05, ** P < 0.01, *** P < 0.005. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 4. M1‐L6 neurons modulate VL membrane potential.
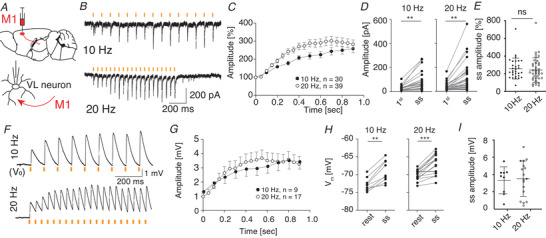
A, schematic representation of the optical stimulation approach by expressing ChrimsonR‐TdTomato (Chrimson) in M1‐L6 in Ntsr1‐Cre transgenic mice. B–E, optical stimulation with 15 ms, 585 nm light pulses selectively activated M1‐L6 fibres and result in an increased EPSC amplitude during the steady state (ss) of the stimulus trains of 10 and 20 Hz compared to the first stimulus (1st). E, the ss facilitation of M1‐L6 inputs to VL neurons is not different between 10 and 20 Hz stimulus trains. F–I, same as for B–E, but in current‐clamp condition: example traces (F) and average membrane depolarizations evoked by 10 and 20 Hz (G and H) M1‐L6 stimulus trains (15 ms, 585 nm). I, the ss facilitation of M1‐L6 inputs to VL neurons is not different between 10 and 20 Hz stimulus trains. V 0 indicates −73 mV for these example traces. ** P < 0.01 and *** P < 0.001. Error bars in C and G represent SEM for illustrative purposes and error bars in E and I represent SD. [Color figure can be viewed at wileyonlinelibrary.com]
Data and software availability
Data and software codes will be made available upon consent of the lead author (f.e.hoebeek@umcutrecht.nl).
Results
To confirm that in our preparation CN and M1‐L6 inputs converge on VL neurons that in turn project to the primary motor cortex, we injected adeno‐associated virus (AAV) expressing constructs that fluorescently label presynaptic terminals: Synaptophysin‐mOrange into CN and a mix of AAV expressing Synaptophysin‐GFP and cholera toxin‐B (CTB, retrograde tracer) into M1. We found that synapses originating from CN and M1‐L6 neurons indeed converged within close proximity on CTB labelled VL neurons, which innervate M1 (Fig. 1, n = 3).
Figure 1. Morphological evaluation of the cerebello‐thalamo‐cortical connectivity.
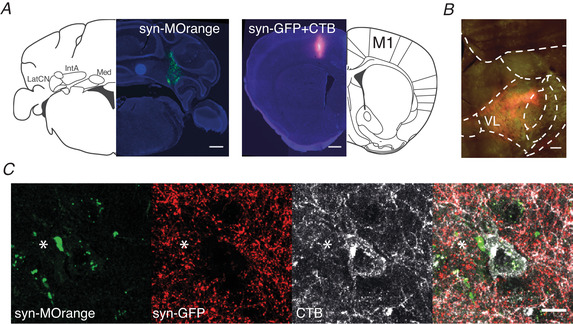
A, injection spots which represent synaptophysin‐mOrange expression in cerebellar nuclei and co‐labelling of synaptophysin‐GFP and cholera toxin subunit B (CTB) in primary motor cortex (M1) (scale bar: 500 μm). Synaptophysin‐mOrange is represented in green and synaptophysin‐GFP in red. B, the input from CN and M1 converges within the VL nucleus and overlaps with CTB labelling (scale bar: 500 μm). C, representative high‐magnification image of a cerebellar synapse (asterisk) and M1 synapses that converge on a CTB‐labelled VL neuron (white), which in turn projects back into the same area of M1 (n = 3, scale bar: 10 μm). Abbreviations: LatCN, lateral cerebellar nuclei; IntA, anterior interposed nucleus; Med, medial cerebellar nucleus; M1, primary motor cortex; VL, ventrolateral thalamic nucleus. [Color figure can be viewed at wileyonlinelibrary.com]
To identify the cell‐physiological mechanisms that enable VL neurons to integrate CN with M1‐L6 inputs we performed whole‐cell patch‐clamp recordings in acutely prepared coronal slices in Ntsr1‐Cre mice injected with AAV allowing the selective optical stimulation of CN axons (ChR2‐EYFP) and/or M1‐L6 axons (flex‐ChrimsonR‐TdTomato). We first evaluated the effect of optical CN stimulation on VL neurons in acute slices of Ntsr1‐Cre mice in which CN neurons express ChR2‐EYFP. For the 82 thalamic cells included in the study, the series resistance was 11.5 ± 2.6 MΩ and the membrane resistance was 186.2 ± 90.3 MΩ. Upon stimulation of ChR2‐expressing CN fibres with a single 1 ms light pulse at 470 nm (Fig. 2A, B), we recorded an EPSC of 1107 ± 849 pA carrying a charge of 4745 ± 3217 pA ms with a CV of 0.08 ± 0.04 (Fig. 2A –E, n = 35). The evoked current was independent of the incubation time, i.e. the time between the day of injection and the day of recording (linear regression: r 2 = 0.0045; n = 28). Next, we stimulated the cerebellar inputs with frequencies of 20, 35 and 50 Hz, which resulted in a decrease of EPSC amplitudes with paired pulse ratios of 67.3 ± 10.9% (n = 27), 59.3 ± 16.8% (n = 24) and 49.4 ± 19.1% (n = 35), respectively (Fig. 2F–H , Friedman test, 20 Hz vs. 50 Hz and 35 Hz vs. 50 Hz P values <0.001, Table 1) and steady state responses of 47.8 ± 19.0% (n = 27), 37.4 ± 14.4% (n = 24) and 25.6 ± 13.0% (n = 35) (Fig. 2F,G and I , Friedman test, 20 Hz vs. 50 Hz and 35 Hz vs. 50 Hz; P values <0.001, Table 1). To evaluate how the postsynaptic responses would recover from the depressed amplitude, we tested the impact of a pause length between the end of the stimulus train and the first subsequent CN stimulus. We tested pause lengths between 50 and 2000 ms. The recovery of the cerebellar EPSC amplitude evoked by 50 Hz stimulus train was best fitted by a single exponential fit (Fig. 2J–K , R 2 = 0.38, plateau = 81.9%, tau = 524.4 ms, Table 1). We also tested whether the recovery from a pause of 100 ms was dependent on the stimulus train frequency. The CN‐evoked response after 100 ms pauses were not significantly different between the recordings in which 20, 35 or 50 Hz stimulus trains were used (Fig. 2L , 20 Hz: 49.2 ± 13.5%, n = 25; 35 Hz: 47.4 ± 12.7%, n = 21; 50 Hz: 47.7 ± 12.6%, n = 28; Friedman test, P = 0.324, Table 1).
Table 1.
Statistical analysis for all data in Fig. 2
| Panel | Unit | Mean ± SD | Normal distribution | Pairing | Test applied | P value | n |
|---|---|---|---|---|---|---|---|
| C | Amplitude (pA) | 1107.0 ± 849.0 | No | NA | NA | NA | 35 |
| D | Charge (pa ms) | 4745.0 ± 3217.0 | Yes | NA | NA | NA | 35 |
| E | CV | 0.076 ± 0.045 | Yes | NA | NA | NA | 35 |
| H | Paired pulse ratio (PPR) (%) |
20 Hz: 67.3 ± 10.9 35 Hz: 59.3 ± 16.8 50 Hz: 49.4 ± 19.1 |
Yes Yes Yes |
No No No |
Friedman test, Bonferroni correction for multiple comparisons |
20 Hz vs. 35 Hz = 0.789 20 Hz vs. 50 Hz ≤0.001 35 Hz vs. 50 Hz ≤0.001 |
27 24 35 |
| I | Steady state (%) |
20 Hz: 47.8 ± 19.0 35 Hz: 37.8 ± 14.4 50 Hz: 25.6 ± 13.0 |
No Yes Yes |
No No No |
Friedman test, Bonferroni correction for multiple comparisons |
20 Hz vs. 35 Hz = 0.262 20 Hz vs. 50 Hz ≤0.001 35 Hz vs. 50 Hz ≤0.001 |
27 24 35 |
| K | Recovery (%) |
50 ms: 42.1 ± 17.1 100 ms: 47.7 ± 14.7 200 ms: 52.9 ± 13.8 400 ms: 65.9 ± 18.8 800ms: 64.5 ± 13.4 2000 ms: 84.2 ± 7.9 |
Yes Yes Yes Yes Yes Yes |
No No No No No No |
Single Exponential fit |
Y0: 39.7 Plateau: 81.9 Tau: 524.4 R 2: 0.38 |
12 11 12 12 6 5 |
| L | Recovery (%) |
20 Hz: 49.2 ± 13.5 35 Hz: 47.4 ± 12.7 50 Hz: 47.7 ± 12.6 |
Yes Yes Yes |
No No No |
Friedman test, Bonferroni correction for multiple comparisons |
0.324 |
25 21 28 |
| M | Resting potential (mV) | −73.6 ± 3.80 | No | NA | NA | NA | 82 |
| O | No. spikes | 2.24 ± 2.16 | No | NA | NA | NA | 26 |
| Q | Steady State spike probability |
20 Hz: 0.20 ± 0.38 50 Hz: 0.15 ± 0.32 |
No No |
Yes Yes |
Wilcoxon signed rank test |
0.0625 |
25 25 |
| R | No. spikes post‐pause (at resting potential) |
50 ms: 0.15 ± 0.35 100 ms: 0.28 ± 0.42 200 ms: 1.39 ± 1.96 400 ms: 2.43 ± 2.43 |
No No No Yes |
Yes Yes Yes Yes |
Friedman test, Bonferroni correction for multiple comparisons |
50 ms vs. 100 ms = 1.0 100 ms vs. 200 ms = 1.0 50 ms vs. 200 ms = 1.0 50 ms vs. 400 ms = 0.171 200 ms vs. 400 ms = 0.602 100 ms vs. 400 ms = 0.019 |
15 19 18 14 |
In current clamp, VL neurons showed a resting membrane potential of −73.4 ± 3.8 mV (n = 82, Fig. 2M , Table 1). Upon optical CN stimulation 13 VL neurons responded with an initial burst of 2.24 ± 2.16 action potentials, 9 VL neurons fired a single action potential or less and 4 did not fire an action potential in this setting (Fig. 2N and O). Following the initial response the spiking probability reached a steady state value of 0.20 ± 0.38 spikes per stimulus during the last 500 ms of the 20 Hz stimulus train and 0.15 ± 0.32 spikes per stimulus during the last 500 ms of the 50 Hz stimulus trains (Fig. 2P and Q; P = 0.0625, Wilcoxon signed‐rank (WCR) test, n = 25, Table 1). We investigated whether the spiking probability of VL neurons can be affected by implementing a pause after the CN stimulus train. We recorded the responses to a single CN stimulus after a pause of 50, 100, 200 or 400 ms and found that increasing the pause length increased the number of action potentials (Fig. 2R ; 0.15 ± 0.35, 0.28 ± 0.42, 1.39 ± 1.96 and 2.43 ± 2.43 respectively; Friedman test, 100 ms vs. 400 ms: P = 0.019, n = 19 and n = 14, respectively; Table 1). These data show that the spiking probability of VL neurons can be modulated by both the frequency during the CN stimulus train and by the duration of the pause between the end of train and the subsequent CN stimulus.
To decipher how fluctuations in membrane potential affect responses of VL neurons to CN stimulus trains, we injected depolarizing currents while stimulating ChR2 expressing CN fibres at 50 Hz (Fig. 3A ). With increasingly depolarized membrane potentials, the number of action potentials fired upon the first CN‐stimulus decreased, but the spike probability following CN stimulation during the steady state increased (Fig. 3B and C; number of action potentials upon 1st stimulus: 3.00 ± 2.03 at −85 mV and 1.20 ± 0.82 at −60 mV; steady state: 0.11 ± 0.27 at −85 mV and 0.37 ± 0.39 at −60 mV, Cochran‐Armitrage test: P < 0.0001 and 0.03267; n = 22 and 20, respectively; Table 2). In a next step, we focus on thalamic cells in which cerebellar stimulation induces subthreshold spiking patterns and evaluate how 100 ms and 200 ms pauses as well as shifts in membrane potential affect the thalamic spike output. We found that pauses of 200 ms increase the thalamic spike output at resting and hyperpolarized potentials while depolarized potentials equalized that effect. In more detail, at potentials of −70 mV a pause of 200 ms significantly increased thalamic spiking when compared to responses during steady state (number of action potentials: 0.35 ± 0.49 at steady state; 0.62 ± 0.42 after 100 ms pause; 2.08 ± 1.52 after 200 ms pause, 200 ms vs. steady state: P = 0.0201, n = 9, Friedman test; Fig. 3D , Table 2). We found a similar effect at −75 mV, in that a pause of 200 ms results in significantly increased number of spikes compared to responses during steady state and after 100 ms pauses (number of action potentials: 0.49 ± 0.52 at steady state, 0.50 ± 0.0.44 after 100 ms pause and 2.18 ± 1.30 after 200 ms pause; steady state vs. 200 ms: P = 0.0121, 100 ms vs. 200 ms: P = 0.0179, n = 8; Friedman test, Fig. 3D , Table 2). Both at more hyperpolarized (−80 mV) and depolarized potentials (−65 mV and −60 mV) the effects of a pause in the 50 Hz CN stimulation train were not significant (all P values >0.456; Friedman test, Fig. 3D , Table 2).
Table 2.
Statistical analysis for all data in Fig. 3
| Panel | Unit | Mean ± SD | Normal distribution | Pairing | Test applied | P value | n |
|---|---|---|---|---|---|---|---|
| B | No. spikes |
−85 mV: 3.00 ± 2.03 −80 mV: 2.55 ± 1.97 −75 mV: 2.01 ± 1.87 −70 mV: 1.64 ± 2.07 −65 mV: 1.29 ± 1.37 −60 mV: 1.20 ± 0.82 |
Yes Yes No No No No |
No No No No No No |
Cochran Armitrage trend test | = 0.0001 |
20 22 25 21 21 20 |
| C | Steady state spike probability |
−85 mV: 0.11 ± 0.27 −80 mV: 0.09 ± 0.23 −75 mV: 0.17 ± 0.36 −70 mV: 0.16 ± 0.35 −65 mV: 0.28 ± 0.36 −60 mV: 0.37 ± 0.39 |
No No No No No No |
No No No No No No |
Cochran Armitrage trend test | = 0.03267 |
20 22 25 21 21 20 |
| D |
No. spikes |
−85 mV ss: 0.32 ± 0.39 −85 mV 100 ms: 1.23 ± 1.63 −85 mV 200 ms: 2.48 ± 2.35 −80 mV ss: 0.33 ± 0.37 −80 mV 100 ms: 0.45 ± 0.39 −80 mV 200 ms: 2.60 ± 1.73 −75 mV ss: 0.49 ± 0.52 −75 mV 100 ms: 0.50 ± 0.44 −75 mV 200 ms: 2.18 ± 1.30 −70 mV ss: 0.35 ± 0.49 −70 mV 100 ms: 0.62 ± 0.43 −70 mV 200 ms: 2.09 ± 1.52 −65 mV ss: 0.53 ± 0.43 −65 mV 100 ms: 0.49 ± 0.50 −65 mV 200 ms: 1.30 ± 1.34 −60 mV ss: 0.55 ± 0.43 −60 mV 100 ms: 0.67 ± 0.39 −60 mV 200 ms: 1.16 ± 1.25 |
No No No No No No No No No No No No No No No No No No |
Yes Yes Yes Yes Yes Yes Yes Yes Yes Yes Yes Yes Yes Yes Yes Yes Yes Yes |
Friedman test |
−85 mV: = 0.0505, ss vs. 100 ms: >0.9999, ss vs. 200 ms: = 0.0693, 100 ms vs. 200 ms: = 0.5443; −85 mV: = 0.0327, ss vs. 100 ms: >0.9999, ss vs. 200 ms: = 0.0911, 100ms vs. 200 ms: = 0.0911; −75 mV: = 0.0009, ss vs. 100 ms: >0.9999, ss vs. 200 ms: = 0.0121, 100 ms vs. 200 ms: = 0.0121; −70 mV: = 0.0017, ss vs. 100 ms: >07158, ss vs. 200 ms: = 0.0201, 100 ms vs. 200 ms: = 0.3765; −65 mV: = 0.4599, ss vs. 100 ms: >0.9999, ss vs. 200 ms: >0.9999, 100 ms vs. 200 ms: >0.9999; ‐60 mV: = 0.5303, ss vs. 100 ms: >0.9999, ss vs. 200 ms: >0.9999, 100 ms vs. 200 ms: >0.9999 |
7 7 7 6 6 6 8 8 8 9 9 9 9 9 9 9 9 9 |
In a next step, we tested the responses of VL neurons to optical stimulation of layer 6 pyramidal cells in the primary motor cortex (M1‐L6). We infected M1‐L6 neurons in Ntsr1‐Cre mice with flex‐ChrimsonR‐TdTomato and to further investigate the short‐term synaptic response patterns of M1‐L6 and CN inputs, we applied train stimuli (Fig. 4A ). After one second of 10 or 20 Hz stimulation at 585 nm the M1‐L6 EPSCs increased from 26.4 ± 19.7 pA to 75.4 ± 73.6 pA at 10 Hz and from 31.9 ± 31.7 pA to 99.8 ± 114.9 pA at 20 Hz (Fig. 4B –E; WCR test, P < 0.0001 for 10 Hz (n = 30) and 20 Hz (n = 39); Table 3), corresponding to 258.8 ± 118.8% and 242.8 ± 138.4% of initial amplitudes, respectively. We then evaluated the effect of M1‐L6 on the depolarization of the thalamic membrane potential (Fig. 4F ). On average, optical stimulation of M1‐L6 fibres with 585 nm pulses of 15 ms at 10 Hz resulted in a steady state depolarization of 3.3 ± 1.6 mV (Fig. 4G –I; range 0.5–5.5 mV), which shifted the average membrane potential from −72.8 ± 1.8 mV to −69.1 ± 3.1 mV (Fig. 4I ; WCR test, P = 0.0039, n = 9; Table 3). Stimulation at 20 Hz induced a depolarization of 3.5 ± 2.1 mV (Fig. 4G –I; ranging from 0.4 mV to 7.2 mV), resulting in a membrane potential shift from −72.5 ± 1.8 mV to −68.4 ± 3.5 mV (WCR test, P = 0.0005, n = 17; Table 3). To confirm the facilitating response pattern of layer 6 inputs from M1, we expressed AAV‐DIO‐ChR2‐EYFP in Ntsr1‐Cre mice. With this construct optical stimulation at 20 Hz also induced a facilitating postsynaptic responses in VL neurons, albeit with a stronger depolarizing effect (average steady state EPSP amplitude 10.9 ± 5.3 mV; ranging from 6.4 mV to 16.7 mV; shift in membrane potential from −69.3 ± 1.9 mV to −57.7 ± 3.9 mV, n = 3), confirming the modulatory role of L6 input on VL neurons.
Table 3.
Statistical analysis for all data in Fig. 4
| Panel | Unit | Mean ± SD | Normal distribution | Pairing | Test applied | P value | n |
|---|---|---|---|---|---|---|---|
| D | Amplitude (pA) |
10 Hz 1st: 26.4 ± 19.7 10 Hz Steady state: 75.4 ± 73.6 |
No No |
Yes | Wilcoxon signed‐rank test | ≤0.0001 | 30 |
|
20 Hz 1st: 31.9 ± 31.7 20 Hz Steady state: 99.8 ± 114.9 |
No No |
Yes | Wilcoxon signed‐rank test | ≤0.0001 | 39 | ||
| E | Amplitude (%) |
10 Hz: 258.8 ± 118.8 20 Hz: 242.8 ± 138.4 |
No No |
No | Mann Whitney test | 0.3934 |
30 39 |
| H | V m (mV) |
10 Hz rest: −72.8 ± 1.8 10 Hz Steady state: −69.1 ± 3.1 |
Yes Yes |
Yes | Wilcoxon signed‐rank test | 0.0039 | 9 |
|
20 Hz rest: −72.5 ± 1.8 20 Hz Steady state: −68.4 ± 3.5 |
Yes Yes |
Yes | Wilcoxon signed‐rank test | 0.0005 | 17 | ||
| I | Amplitude (mV) |
10 Hz: 3.3 ± 1.6 20 Hz: 3.5 ± 2.1 |
Yes Yes |
No | Mann Whitney test | 0.7464 |
9 17 |
Furthermore, we aimed to prove that in our preparation both CN and M1‐L6 inputs converge onto single VL cells. Therefore we recorded VL responses following dual optical stimulation of AAV‐ChR2‐EYFP‐expressing CN fibres at 470 nm and AAV‐flex‐ChrimsonR‐TdTomato‐expressing M1‐L6 fibres at 585 nm in Ntsr1‐Cre mice (Klapoetke et al. 2014; Hooks et al. 2015). In this experimental setup, ChR2‐expressing CN axons are solely excited by 470 nm light pulses, but ChrimsonR‐TdTomato‐expressing M1‐L6 axons are sensitive to stimulation at 470 nm and 585 nm. To avoid potential cross‐talk we used prolonged stimulation at 585 nm to desensitize ChrimsonR‐TdTomato‐expressing motor cortical fibres to stimulation at 470 nm (Hooks et al. 2015). To ensure exclusive stimulation of M1‐L6 or CN fibres, we first evaluated photocurrents of ChrimsonR‐TdTomato‐expressing M1‐L6 neurons (Fig. 5A –D). For our optical stimulation approach we used pulses of 1 ms at 470 nm and 1, 15 and 200 ms at 585 nm, which corresponded to photonfluxes of 18.08 × 1024 photons/ms/m2 for 1 ms at 470 nm and 4.8 × 1024 to 960 × 1024 photons/ms/m2 for 1 to 200 ms at 585 nm, respectively (Fig. 5E ). We found that the photocurrents evoked by stimulation at 585 nm maximized at 15 ms (1 ms 585 nm: 251.5 ± 119.0 pA, 15 ms 585 nm: 938.8 ± 419.5 pA, 200 ms 585 nm: 1094.0 ± 302.0 pA; n = 8 for all groups; Fig. 5E and F; Table 4). In these recordings, we found that 1 ms stimulation at 470 nm evoked 636.5 ± 356.2 pA of current. We were able to confirm the efficacy of the desensitization protocol, in that after 200 ms pre‐stimulation at 585 nm, the additional current evoked by 1 ms stimulation at 470 nm is significantly decreased to 52.5 ± 35.0 pA (Fig. 5F , Kruskal‐Wallis test, P < 0.05, n = 8; Table 4). These maximal photocurrents induced action potential firing in ChrimsonR‐TdTomato‐positive M1‐L6 neurons (spiking probability at 1 ms 585 nm: 0.75 ± 0.46, n = 8; 15 ms 585 nm: 1.0 ± 0.0, n = 6; 200 ms 585 nm: 1.0 ± 0.0, n = 8; 1 ms 470 nm: 0.80 ± 0.45, n = 5), except when a 200 ms 585 nm pulse preceded the 1 ms 470 nm (0.0 ± 0.0, n = 8, Kruskal‐Wallis test, P < 0.05; Table 4). In addition, we desensitized the ChrimsonR‐TdTomato‐expressing M1‐L6 neurons to 470 nm light by applying a pre‐stimulation pulse for 200 ms at 585 nm, which saturates motor cortical photocurrents and prevents spike induction upon the subsequent 470 nm light pulse (Fig. 5F and G; Hooks et al. 2015; Klapoetke et al. 2014). This dual‐optical stimulation protocol prevents action potential firing in M1‐L6 neurons upon co‐stimulation with light at 585 nm and 470 nm. As a final control, we assessed the kinetics of the ChrimsonR photocurrent to determine the maximal stimulation frequency we can apply to thalamic slices (Fig. 2). Therefore, we measured the channel closing rate (‘tau off’) of ChrimsonR after 15 ms illumination at 585 nm, which at maximal stimulation intensity was 13.1 ± 1.2 ms, indicating that 20 Hz photostimulation is feasible.
Figure 5. Photosensitivity of ChrimsonR‐TdTomato‐expressing M1‐L6 neurons.
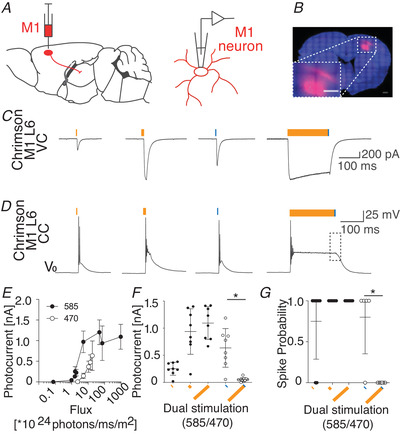
A, schematic illustration of ChrimsonR expression and patch‐clamp recording from ChrimsonR‐TdTomato‐expressing M1‐L6 neurons. B, layer 6 specific expression of ChrimsonR‐TdTomato in motor cortex of Ntsr1‐Cre mice. (inset: high‐magnification image of injection spot; scale bar: 200 μm). C and D, representative recordings of the photo‐response from ChrimsonR‐TdTomato‐expressing M1‐L6 neurons after stimulation with 1, 15 and 200 ms light at 585 nm and 1 ms at 470 nm, when recorded in voltage‐clamp (VC) (C), and at resting potential (V 0) in current clamp (CC) (D). To independently excite cerebellar synapses in thalamic slices (see Figs 6 and 7), the M1‐L6 neuron was desensitized to light at 470 nm by applying a 200 ms stimulation at 585 nm, following which a 1 ms stimulus at 470 nm failed to evoke an action potential in M1‐L6 neurons (square in right panel of D). V 0 indicates −71 mV for these example traces. E and F, pulses of 1, 15 and 200 ms at 585 and 1 ms at 470 nm with corresponding photon fluxes (E), resulted in photocurrents that maximized in response to 15 ms of 585 nm (F). The photocurrent in response to 1 ms stimulation at 470 nm is significantly decreased after 200 ms pre‐stimulation at 585 nm (F). G, these photocurrents induced AP firing after stimulation at 585 nm and 470 nm, but the dual‐optical stimulation protocol prevents AP firing in M1‐L6 neurons upon co‐stimulation by 585 nm and 470 nm. * P < 0.05. Error bars represent SD. [Color figure can be viewed at wileyonlinelibrary.com]
Table 4.
Statistical analysis of all data in Fig. 5
| Panel | Unit | Mean ± SD | Normal distribution | Test applied | P value | n |
|---|---|---|---|---|---|---|
| F | Photo‐current [nA] |
1 ms 585 nm: 251.5 ± 119.0 15 ms 585 nm: 938.8 ± 419.5 200 ms 585 nm: 1094 ± 302 1 ms 470 nm: 636.5 ± 356.2 585 nm + 470 nm: 52.5 ± 35.0 |
Yes Yes Yes Yes No |
Friedman test, Bonferroni correction for multiple comparisons |
1 ms 585 nm vs. 15 ms 585 nm: = 0.339 1 ms 585 nm vs. 200 ms 585 nm: = 0.133 1 ms 585 nm vs. 1 ms 470 nm: = 1.0 1 ms 585 nm vs. 585 nm + 470 nm: = 0.477 15 ms 585 nm vs. 200 ms 585 nm: = 1.0 15 ms 585 nm vs. 1 ms 470 nm: = 1.0 15 ms 585 nm vs. 585 nm ± 470 nm: ≤ 0.001 200 ms 585 nm vs. 1 ms 470 nm: = 1.0 200 ms 585 nm vs. 585 nm ± 470 nm ≤0.001 1 ms 470 nm vs. 585 nm ± 470 nm: = 0.003 |
8 8 8 8 10 |
| G | Spike probability |
1 ms 585 nm: 0.75 ± 0.46 15 ms 585 nm: 1.0 ± 0.0 200 ms 585 nm: 1.0 ± 0.0 1 ms 470 nm: 0.80 ± 0.45 585 nm + 470 nm: 0.0 ± 0.0 |
Yes NA NA NA NA |
Friedman test, Bonferroni correction for multiple comparisons |
1 ms 585 nm vs. 15 ms 585 nm: = 1.0 1 ms 585 nm vs. 200 ms 585 nm: = 1.0 1 ms 585 nm vs. 1 ms 470 nm: = 1.0 1 ms 585 nm vs. 585 nm + 470 nm: = 0.328 15 ms 585 nm vs. 200 ms 585 nm: = 1.0 15 ms 585 nm vs. 1 ms 470 nm: = 1.0 15 ms 585 nm vs. 585 nm ± 470 nm: = 0.09 200 ms 585 nm vs. 1 ms 470 nm: = 1.0 200 ms 585 nm vs. 585 nm + 470 nm: = 0.057 1 ms 470 nm vs. 585 nm ± 470 nm: = 0.012 |
8 6 8 5 8 |
To ensure that the wavelengths of our optical stimulation allow the selective stimulation of ChR2‐EYFP‐expressing CN axons by 470 nm but not by 585 nm, we set out to record photocurrent and spike probabilities in ChR2‐EYFP‐expressing CN neurons evoked by these wavelengths (Fig. 6A –H). Stimulation
pulses of 15 ms at 585 nm, 200 ms at 585 nm, 1 ms at 470 nm and the dual‐optical stimulation protocol (200 ms 585 nm followed by 1 ms 470 nm) induced a maximal photocurrent of 24.9 ± 23.2 pA, 42.8 ± 29.8 pA, 683.9 ± 361.5 pA and 712.0 ± 364.3 pA, respectively (n = 7 for all groups; Fig. 6I ; Table 5). The decay (‘tau off’) of the ChR2 photocurrent was 8.5 ± 2.5 ms for 1 ms at 470 nm and 8.6 ± 2.5 ms for 1 ms light at 470 nm preceded by 200 ms at 585 nm (n = 7 for all groups; Fig. 6J ; Table 5). As expected, the optical stimulation at 585 nm did not induce action potential firing (15 ms and 200 ms: 0% ± 0%), while stimulation with 1 ms at 470 nm as well as the dual stimulation protocol (200 ms 585 nm and 1 ms 470 nm) induced spiking in 80% ± 45% of the neurons (n = 5 for all groups; Fig. 6K ; Table 5). To investigate whether the closing kinetics of ChR2 and their dark‐state behaviour at the intended stimulus duration of 1 ms allows reliable stimulation with 50 Hz, we compared the relative change in photocurrent during a 50 Hz stimulus train of 1 ms light pulses at 470 nm with and without co‐stimulation at 585 nm (Fig. 6L and M). We recorded no significant difference between the photocurrents at the beginning and end of the stimulus train (1st stimulus: 487.4 ± 352.4 pA; last stimulus: 486.2 ± 339.1 pA; Fig. 6M ; WCR test, P > 0.99, n = 5; Table 5). These data show that stimulation at 585 nm does not induce spiking in CN neurons, while stimulation with short pulses of 1 ms at 470 nm induces strong photocurrents that do not depress at 50 Hz and reliably trigger action potentials.
Table 5.
Statistical analysis of all data in Fig. 6
| Panel | Unit | Mean ± SD | Normal distribution | Test applied | P value | n |
|---|---|---|---|---|---|---|
| I | Photo‐current (nA) |
15 ms 585 nm: 24.9 ± 23.2 200 ms 585 nm: 42.8 ± 29.8 1ms 470 nm: 683.9 ± 361.5 585 nm + 470 nm: 712.0 ± 364.3 |
Yes Yes Yes Yes |
Friedman test, Dunn's correction for multiple comparisons |
15 ms 585 nm vs. 200 ms 585 nm: = 1.0 15 ms 585 nm vs. 1 ms 470 nm: = 0.011 15 ms 585 nm vs. 585 nm ± 470 nm: = 0.006 200 ms 585 nm vs. 1 ms 470 nm: = 0.078 200 ms 585 nm vs. 585 nm ± 470 nm = 0.043 1 ms 470 nm vs. 585 nm + 470 nm: = 1 |
7 7 7 7 |
| J | ‘tau off’ (ms) |
1 ms 470 nm: 8.5 ± 2.5 585 nm + 470 nm: 8.6 ± 2.5 |
Yes Yes |
Unpaired t test | 0.932 |
7 7 |
| K | Spike probability |
15 ms 585 nm: 0 ± 0 200 ms 585 nm: 0 ± 0 1 ms 470 nm: 0.8 ± 0.45 585 nm + 470 nm: 0.8 ± 0.45 |
No No No No |
Friedman test, Bonferroni correction for multiple comparisons |
15 ms 585 nm vs. 200 ms 585 nm: = 1.0 15 ms 585 nm vs. 1 ms 470 nm: = 0.3 15 ms 585 nm vs. 585 nm + 470 nm: = 0.3 200 ms 585 nm vs. 1 ms 470 nm: = 0.3 200 ms 585 nm vs. 585 nm + 470 nm: = 0.3 1 ms 470 nm vs. 585 nm + 470 nm: = 1 |
5 5 5 5 |
| M | Photo‐current (pA) |
first: 487.4 ± 352.4 last: 486.2 ± 339.1 |
Yes Yes |
Paired t test | 0.965 | 5 |
We next applied our dual optical stimulation protocol to independently activate ChrimsonR‐TdTomato‐expressing M1‐L6 fibres and ChR2‐EYFP‐expressing CN fibres in thalamic slices and evaluated the individual postsynaptic responses (Fig. 7A –E). For M1‐L6 stimulation we used a 20 Hz stimulus train of 15 ms pulses at 585 nm and found that the EPSC amplitudes increased from 18.6 ± 21.3 pA to a steady state value of 89.7 ± 130.0 pA. The subsequent single pulse stimulation of 1 ms at 470 nm of CN fibres (following a 200 ms pulse of 585 nm) evoked an EPSC of 688.4 ± 691.8 pA, which contrasts with M1‐L6 responses (WCR test CN vs. M1‐L6 facilitated, P < 0.0001, n = 28; Fig. 7C –E; Table 6). As we activate the full population of CN and M1‐L6 inputs synchronously, we analysed the charge and its CV to determine for variability of the multi‐synaptic inputs. The total charge transferred was 888 ± 1457 pA ms for M1‐L6 after the last 1 ms stimulus at 585 nm in the 1 s lasting train at 20 Hz and 3308 ± 3746 pA ms for the subsequent single pulse of 470 nm that selectively activated CN terminals (Fig. 7E ; WCR test CN vs. M1‐L6 facilitated, P < 0.0012, n = 28; Table 6). Despite this significant difference in total charge transferred, the variability was not different (CV M1‐L6 charge: 0.19 ± 0.21; CV CN charge: 0.16 ± 0.18; Fig. 7E ; WCR test, P = 0.6406, n = 28; Table 6).
Figure 7. Physiological convergence of CN and M1‐L6 in motor thalamus.
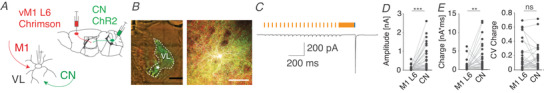
A, schematic representation of the dual‐optogenetic stimulation approach by expressing ChrimsonR‐TdTomato (Chrimson) in M1‐L6 and ChannelRhodopsin2‐EYFP (ChR2) in CN in Ntsr1‐Cre transgenic mice. B, fluorescent images of ChrimsonR‐TdTomato‐positive M1‐L6 fibres (red), ChR2‐EYFP‐positive CN fibres (green) and a biocytin‐filled VL neuron (white; scale bars: 100 μm). C, representative VL recording during 20 Hz M1‐L6 stimulation (15 ms 585 nm, orange) and a single CN stimulus (200 ms 585 nm and 1 ms 470 nm, blue). D and E, the amplitude (D), and the charge and the coefficient of variation (CV) of the charge evoked by M1‐L6 and CN stimuli (E). ‘ns’ indicates not significant, ** P < 0.01 and *** P < 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
Table 6.
Statistical analysis for all data in Fig. 7
| Panel | Unit | Mean ± SD | Normal distribution | Pairing | Test applied | P value | n |
|---|---|---|---|---|---|---|---|
| D | Amplitude (pA) |
CN: 688.4 ± 691.8 M1‐L6: 89.7 ± 130.0 |
No No |
Yes | Wilcoxon signed‐rank test | ≤0.0001 | 28 |
| E | Charge (pA ms) |
CN: 3308.0 ± 3746.0 M1‐L6: 888.5 ± 1457.0 |
No No |
Yes | Wilcoxon signed‐rank test | 0.0012 | 28 |
| E | CV |
CN: 0.16 ± 0.18 M1‐L6: 0.19 ± 0.21 |
No No |
Yes | Wilcoxon signed‐rank test | 0.6406 | 28 |
In order to investigate the convergence of postsynaptic responses evoked CN and M1‐L6 inputs in more detail, we adapted our optical stimulation protocol to allow co‐stimulation of CN and M1‐L6 inputs. Therefore we use the initial dual optical stimulation protocol to identify thalamic cells that are exclusively innervated by motor cortical or cerebellar fibres as well as thalamic cells that receive both inputs (Fig. 8A ). In a next step we want to further limit the responsiveness of Chrimson‐expressing motor cortical fibres to blue stimulation by limiting the light pulses to wavelengths up to 460 nm and minimizing the corresponding light intensity. Next, we use excitation with 20 Hz stimulus trains with 1 ms pulses of light at 460 nm as well as 15 ms pulses at 585 nm to show that motor cortical fibres are exclusively activated after stimulation at 585 nm. During the last 500 ms of the stimulus train, 20 Hz stimuli at 585 nm evoked significantly more current than at 460 nm, with the latter evoking practically no current (Fig. 8B and C; 20 Hz 585 nm: 68.0 ± 37.3 pA, 20 Hz 460 nm: 6.8 ± 4.7 pA, n = 8, WCR test, P = 0.0078, Table 7). The same amplitude difference was found for CN stimulation, in that at 460 nm 1 ms pulses evoked a significantly bigger response than 15 ms pulses at 585 nm, with the latter stimulus evoking practically no response (Fig. 8D and E; 20 Hz 585 nm: 6.6 ± 5.7 pA, 20 Hz 460 nm: 539.1 ± 468.4 pA, n = 11, WCR test, P = 0.0010, Table 7). We identified VL neurons of which both CN and M1‐L6 inputs responded to optical stimulation, i.e. neurons that showed a facilitating response to 585 nm train stimulation and a depressing response to 460 nm train stimulation. In these dually connected neurons we set out to pair the 460 nm and 585 nm stimuli (Fig. 8F ). We found that the postsynaptic currents induced by co‐stimulating motor cortical and cerebellar afferents resulted in a summation of the motor cortical and the cerebellar inputs at the soma. To quantify the summation of both compound responses, we quantified the accumulation of the total charge during the steady state of the stimulation (Fig. 8G ; M1‐L6: 28.6 ± 27.9 nA × ms, CN: 37.9 ± 31.5 nA ms, CN plus M1‐L6: 52.3 ± 42.1 nA ms, n = 13, Friedman test, P = 0.0004, Table 7). When motor cortical fibres were co‐activated with cerebellar stimulation, the steady state spike probability after pulses with 20 Hz was increased to 0.45 ± 0.47 spikes compared to 0.23 ± 0.39 spikes (Fig. 8 H‐J, n = 14, WCR test, P = 0.0078, Table 7) after activating cerebellar fibres exclusively. These data indicated that indeed the co‐activation of M1‐L6 modulates the spiking probability of VL neurons in response to CN stimulus trains.
Figure 8. Motor cortical afferents modulate cerebellar induced spike transfer in the VL motor thalamus.
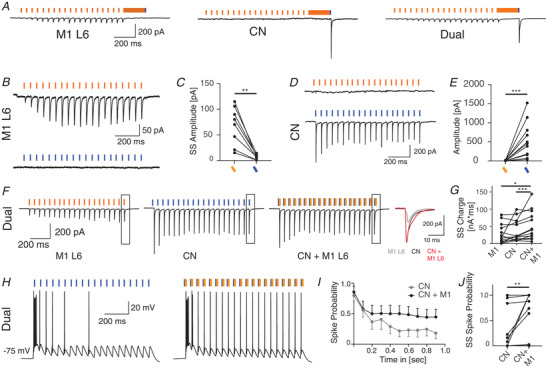
A, example traces illustrating the connectivity pattern of VL cells that are innervated by M1‐L6, CN or by both M1‐L6 and CN B, example trace of a VL cell that is exclusively innervated by ChrimsonR‐TdTomato‐expressing M1‐L6 fibres which are excited by 585 nm light, but not by 460 nm light C. D and E, example trace of a VL cell (D) that is exclusively innervated by ChR2‐EYFP‐expressing CN fibres, which are excited by 460 nm light, but not by 585 nm light (E). F, in VL cells that which are innervated by both ChrimsonR‐TdTomato‐expressing M1‐L6 fibres and ChR2‐EYFP‐expressing CN fibres, 585 nm light pulses evoked EPSCs with increasing amplitude and 460 nm light pulses evoked EPSCs with decreasing amplitude. The inset illustrates the accumulation of both currents in the charge of the compound responses. The total charge during the last 500 ms of the stimulus train is quantified in G. H, example traces illustrating that co‐stimulation of motor cortical afferents can shift the cerebellar induced spike output of VL cells. I and J, the spike probability is quantified in the average spike probability per stimulus (I), as well as in the average steady state (ss) spike probability per cell and stimulus condition (J). Error bars in I represent SEM for illustrative purposes. [Color figure can be viewed at wileyonlinelibrary.com]
Table 7.
Statistical analysis for all data in Fig. 8
| Panel | Unit | Mean ± SD | Normal distribution | Pairing | Test applied | P value | n |
|---|---|---|---|---|---|---|---|
| C | Amplitude (pA) |
585 nm: 68.0 ± 37.3 460 nm: 6.8 ± 4.7 |
No No |
Yes | Wilcoxon signed‐rank test | 0.0078 | 8 |
| E | Amplitude (pA) |
460 nm: 539.1 ± 468.4 585 nm: 6.6 ± 5.7 |
No No |
Yes | Wilcoxon signed‐rank test | 0.0010 | 11 |
| G | Steady state Charge (nA* ms) |
M1‐L6: 28.6 ± 27.9 CN: 37.9 ± 31.5 CN&M1‐L6: 52.3 ± 42.1 |
Yes No Yes |
Friedman test |
M1 vs. CN: 0.5094 M1 vs. CN±M1: 0.0324 CN vs. CN±M1: 0.0003 |
13 13 13 |
|
| J | Spike probability |
CN: 0.23 ± 0.39 CN&M1‐L6: 0.45 ± 0.47 |
No No |
Yes | Wilcoxon signed‐rank test | 0.0078 |
14 14 |
Discussion
Here we show that cortical projections from M1‐L6 evoke facilitating postsynaptic responses in VL neurons that modulate the membrane potential at the subthreshold level, whereas the cerebellar inputs from CN elicit responses sufficient to induce thalamic spiking. Our in vitro data reveal that attenuated responses to cerebellar stimulation in VL thalamic neurons are restored after a brief pause in the stimulus train. The modulation of the membrane potential of VL neurons by M1‐L6 in effect also controls the VL spiking, in that their probability to fire following repetitive CN stimulation was higher at depolarized levels. Accordingly, an increase in membrane potential depolarization dampened the differences between initial and steady state responses of VL neurons when CN input was paused for 100–200 ms. This synergistic modulation of both inputs enables the motor thalamus to operate as a low‐pass filter, in which a response to the high‐frequency cerebellar input can be adapted based on motor cortical feedback.
In the thalamus the transfer of subcortical inputs to the motor cortex is dictated by the state of the thalamic membrane potential (Jahnsen & Llinas, 1984a ; McCormick & Bal, 1997; Mease et al. 2014). Our findings show that repetitive stimulation of M1‐L6 fibres induces short‐term facilitation of postsynaptic responses in VL, which allows these modulatory inputs to depolarize the thalamic membrane potential, albeit below the threshold for action potential initiation. A previous report on synaptic connectivity between M1‐L6 and VL reported the responses to single pulse stimulation to be remarkably weak or absent compared to the impact of M1‐L6 neurons on other thalamic nuclei (Yamawaki & Shepherd, 2015). Still, the repetitive stimulation protocol in the current study revealed facilitating response patterns irrespective of whether the ChR2 or ChrimsonR construct was expressed.
The output of layer 6 neurons of the motor cortex probably affects the membrane potential of thalamic VL neurons by activating their t‐type calcium channels. Their activation in turn allows the thalamic cells to fire the characteristic low threshold calcium spike (LTS) and the burst of action potentials (Jahnsen & Llinas, 1984a , b ) that we typically found in response to CN stimulation (Fig. 3F ). The degree of t‐type channel de‐inactivation is time‐ and voltage‐dependent and determines the number of spikes transferred within a burst (Jahnsen & Llinas, 1984a ) and thereby the timing of thalamic spiking (Wolfart et al. 2005; Mease et al. 2014, 2017). In our data we found that the membrane potential level, which we modulated using somatic current injections or by selective activation of M1‐L6 axons, indeed modulated the number of action potentials that a single CN stimulus evoked in VL neurons, which is in accordance with the previous findings from the visual thalamo‐cortical system (McCormick & Von Krosigk, 1992).
In addition to the responses to single CN stimuli, we also investigated the responses of VL neurons to high‐frequency CN stimulus trains. In various in vitro and in vivo experimental settings it has been shown that CN neurons can fire continuously up to 100 Hz or higher (Antziferova et al. 1980; Armstrong & Edgley, 1984; Raman et al. 2000; Hoebeek et al. 2010; Ohmae et al. 2013; Ten Brinke et al. 2017). Our dual‐optogenetic patch‐clamp approach permitted us to synchronously activate the full population of cerebellar fibres at a maximum of 50 Hz (Klapoetke et al. 2014). Despite this limitations, we were able to investigate how the CN‐evoked responses in VL neurons altered after brief pauses in the stimulus train. We found that the responses rapidly recovered from the paired‐pulse depression and increased the number of action potentials evoked by CN stimuli. These data suggest that spike coding of thalamic neurons that project to motor cortex depends on the recovery of synaptic depression and de‐inactivation of t‐type calcium channels during a pause in high‐frequency CN spiking. Our findings in the motor system are in line with previous reports on the impact of conjunctive activation of subcortical and cortical driver inputs in sensory and visual thalamus (Hoogland et al. 1991; Groh et al. 2008, 2013; Bickford et al. 2015).
The low‐pass filter function within VL suggests that the paired‐pulse depression characteristic of the cerebello‐thalamic synapses transforms high‐frequency cerebellar spiking patterns into a low‐frequency thalamic spiking pattern that, upon pauses in cerebellar spiking, which release the synapses from paired‐pulse depression, shows a peak in spiking probability. In addition, our co‐stimulation experiments, in which we combined M1‐L6 with CN optical stimulation, revealed that synaptic transmission from M1‐L6 pyramidal cells onto VL neurons modulates the impact of the synaptic transmission from CN neurons and can increase the spiking probability evoked by individual CN stimuli during a stimulus train, restoring the effect of CN‐VL paired‐pulse depression (Fig. 8J ). Thereby our results indicate that the gain of the low‐pass filter can be modulated by M1‐L6 input. Future studies need to address the modulation of VL output by CN and M1‐L6 inputs during movement execution and behaviour. One aspect that will be of relevance is to investigate the potential role of feedforward inhibition: in the in vivo situation M1‐L6 neurons monosynaptically innervate excitatory neurons in VL as well as inhibitory neurons in the reticular thalamic nucleus (RTN), which in turn provide feed‐forward inhibition to thalamo‐cortical relay neurons (Yamawaki & Shepherd, 2015; Halassa & Acsády, 2016). The depressing short‐term release dynamics of RTN synapses in the thalamus shift the balance between excitation and inhibition induced by M1‐L6 towards depolarized membrane potentials (Mease et al. 2014; Crandall et al. 2015). In our current study we blocked inhibitory inputs from RTN to exclusively study the interaction of cerebellum and the feedback from motor cortex. Our data show that the depolarizing shift in membrane potential after M1‐L6 activation increases the spiking probability evoked by cerebellar stimulation, i.e. M1‐L6 input modulates the gain of cerebello‐thalamic transmission. Recent evidence indicates that the synchronicity of M1‐L6 inputs to thalamic neurons is important to determine the gain of spike transfer from VL to motor cortex (Wolfart et al. 2005; Mease et al. 2014). The combination of cerebellar spike timing, and the response amplitude in VL neurons as well as their membrane potential can modulate the spike transfer to motor cortex along a continuum, as has previously been shown for the sensory thalamo‐cortical processing (Whitmire et al. 2016).
Additional information
Competing interests
The authors declare no conflicts of interest.
Author contributions
C.B.S. performed all experimental work and analysis. C.B.S. and F.E.H. designed the experiments. F.E.H. and Z.G. provided technical support. F.E.H., Z.G. and C.I.D.Z. provided financial support. C.B.S. and F.E.H. wrote the original draft, Z.G. and C.I.D.Z. edited the manuscript. F.E.H. conceived and guided the project. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
C.B.S. and F.E.H. are supported by the Dutch organization for life sciences (NWO‐ALW; VIDI grant No. 016.121.346) and Medical Sciences (TOP‐GO No. 91210067). Z.G. is supported by the Dutch organization for life sciences (NWO‐ALM; VENI grant No. 863.14.001 and NWO‐CAS grant No. 012.200.14) and the Erasmus MC fellowship. C.I.D.Z. thanks the Dutch Organization for Medical Sciences (Zon‐MW; TOP‐GO No. 91210067), Life Sciences (ALW; No. 854.10.004), ERC‐adv (No. 294775), ERC‐POC (No. 768914) and LISTEN (No. 6) of the EU, as well as Medical NeuroDelta (No. 7) for support. In addition the authors are funded by the Dutch Organization for Medical Sciences (ZonMw), Life Sciences (ALW‐ENW‐Klein), the European Research Council (Advanced and Proof of Concept grants), the EU LISTEN Innovative Training Network programme, the Medical NeuroDelta programme, LSH‐NWO (Crossover, INTENSE), Albinism Vriendenfonds NIN, van Raamsdonk fonds, and the Trustfonds of Erasmus University, Rotterdam. None of the funding bodies had any input to the study design or outcome.
Supporting information
Statistical Summary Document
Acknowledgements
We thank Simona V. Gornati, Oscar H.J. Eelkman Rooda, Valentina Riguccini and Bas van Hoogstraten for fruitful scientific discussions and Professor Dr Thomas Kuner for providing Synaptophysin virus constructs. We thank Erika H. Goedknegt, Elize D. Haasdijk, Mandy Rutteman and Patrick Eikenboom for helping with immunohistochemical experiments.
Biography
Carmen B. Schäfer, now at the Erasmus Medical Center in Rotterdam, Department of Neuroscience, obtained a BSc in biology and an MSc with Major in neuroscience at the University of Heidelberg in Germany. During her Masters course she developed a strong interest in understanding the neurophysiological processes that shape brain activity on the cellular level. During her PhD research, she found it particularly fascinating that playing with light at different wavelengths allowed her to selectively control individual components of neuronal circuits. This allowed her to gather in depth insights on the technical challenges that need to be tackled for the successful use of optogenetics in neuroscience. For her future research, she aims to combine this knowledge with in vitro and in vivo recording approaches to understand how neuronal activity shapes behaviour.

Edited by: Richard Carson & Vatsala Thirumalai
This article was first published as a preprint. Schäfer CB, Gao Z, De Zeeuw CI, Hoebeek FE. 2020. Cerebello‐thalamic spike transfer via temporal coding and cortical adaptation. bioRxiv. https://doi.org/10.1101/2020.01.19.911610.
Data availability statement
Data and software codes will be made available upon consent of the lead author (f.e.hoebeek@umcutrecht.nl).
References
- Antziferova LI, Orlovsky GN & Pavlova GA (1980). Activity of neurons of cerebellar nuclei during fictitious scratch reflex in the cat I fastigial nucleus. Brain Res 200, 239–248. [DOI] [PubMed] [Google Scholar]
- Armstrong BYDM & Edgley SA (1984). Discharges of nucleus interpositus neurones during locomotion in the cat. J Physiol 351, 411–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bava A, Cicirata F, Giuffrida R, Licciardello S & Panto MR (1986). Electrophysiologic properties and nature of ventrolateral thalamic nucleus neurons reactive to converging inputs of paleo‐ and neocerebellar origin. Exp Neurol 91, 1–12. [DOI] [PubMed] [Google Scholar]
- Bava A, Manzoni T & Urbano A (1967). Effects of fastigial stimulation on thalamic neurones belonging to the diffuse projection system. Brain Res 4, 378–380. [DOI] [PubMed] [Google Scholar]
- Beloozerova IN, Sirota MG & Swadlow HA (2003). Activity of different classes of neurons of the motor cortex during locomotion. J Neurosci 23, 1087–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickford ME, Zhou N, Krahe TE, Govindaiah G & Guido W (2015). Retinal and tectal “driver‐like” inputs converge in the shell of the mouse dorsal lateral geniculate nucleus. J Neurosci 35, 10523–10534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Brinke MM, Heiney SA, Wang X, Proietti‐Onori M, Boele HJ, Bakermans J, Medina JF, Gao Z & De Zeeuw CI (2017). Dynamic modulation of activity in cerebellar nuclei neurons during pavlovian eyeblink conditioning in mice. Elife 6, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks JX, Carriot J & Cullen KE (2015). Learning to expect the unexpected : rapid updating in primate cerebellum during voluntary self‐motion. Nat Neurosci 18, 1310–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall SR, Cruikshank SJ & Connors BW (2015). A corticothalamic switch: controlling the thalamus with dynamic synapses. Neuron 86, 768–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Zeeuw CI, Hoebeek FE, Bosman LWJ, Schonewille M, Witter L & Koekkoek SK (2011). Spatiotemporal firing patterns in the cerebellum. Nat Rev Neurosci 12, 327–344. [DOI] [PubMed] [Google Scholar]
- Dumas DB, Gornati SV, Adolfs Y, Shimogori T, Pasterkamp RJ & Hoebeek FE (2019). Anatomical development of the cerebellothalamic tract in embryonic mice. bioRxiv DOI: 10.1101/731968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Davis C, Thomas AM, Economo MN, Abrego AM, Svoboda K, De Zeeuw CI & Li N (2018). A cortico‐cerebellar loop for motor planning. Nature 563, 113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Doughty M, Harbaugh CR, Cummins A, Hatten ME, Heintz N & Gerfen CR (2007). Targeting cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J Neurosci 27, 9817–9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gornati SV, Schäfer CB, Eelkman Rooda OHJ, Nigg AL, De Zeeuw CI & Hoebeek FE (2018). Differentiating cerebellar impact on thalamic nuclei. Cell Rep 23, 2690–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh A, Bokor H, Mease RA, Plattner VM, Hangya B, Stroh A, Deschenes M & Acsády L (2013). Convergence of cortical and sensory driver inputs on single thalamocortical cells. Cereb Cortex 24, 3167–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh A, de Kock CPJ, Wimmer VC, Sakmann B & Kuner T (2008). Driver or coincidence detector: modal switch of a corticothalamic giant synapse controlled by spontaneous activity and short‐term depression. J Neurosci 28, 9652–9663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM & Acsády L (2016). Thalamic inhibition : diverse sources, diverse scales. Trends Neurosci 39, 680–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoebeek FE, Witter L, Ruigrok TJH & De Zeeuw CI (2010). Differential olivo‐cerebellar cortical control of rebound activity in the cerebellar nuclei. Proc Natl Acad Sci U S A 107, 8410–8415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogland PV, Wouterlood FG, Welker E & Van der Loos H (1991). Ultrastructure of giant and small thalamic terminals of cortical origin: a study of the projections from the barrel cortex in mice using Phaseolus vulgaris leuco‐agglutinin (PHA‐L). Eperimental Brain Res 87, 159–172. [DOI] [PubMed] [Google Scholar]
- Hooks BM, Lin XJY, Guo C & Svoboda K (2015). Dual‐channel circuit mapping reveals sensorimotor convergence in the primary motor cortex. J Neurosci 35, 4418–4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houck BD & Person AL (2015). Cerebellar premotor output neurons collateralize to innervate the cerebellar cortex. J Comp Neurol 523, 2254–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnsen BYH & Llinas R (1984a). Electrophysiological properties of guinea‐pig thalamic neurones: an in‐vitro study. J Physiol 349, 205–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnsen BYH & Llinas R (1984b). Ionic basis for the electroresponsiveness and oscillatory properties of guinea‐pig thalamic neurones in vitro. J Physiol 349, 227–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong M, Kim Y, Kim J, Ferrante DD, Mitra PP, Osten P & Kim D (2016). Comparative three‐dimensional connectome map of motor cortical projections in the mouse brain. Sci Rep 6, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jörntell H & Ekerot CF (1999). Topographical organization of projections to cat motor cortex from nucleus interpositus anterior and forelimb skin. J Physiol 514, 551–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kha HT, Finkelstein DI, Pow DV, Lawrence AJ & Horne MK (2000). Study of projections from the entopeduncular nucleus to the thalamus of the rat. J Comp Neurol 426, 366–377. [PubMed] [Google Scholar]
- Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey‐Benson A, Cho YK, Morimoto TK, Chuong AS, Carpenter EJ, Tian Z, Wang J, Xie Y, Yan Z, Zhang Y, Chow BY, Surek B, Melkonian M, Jayaraman V, Constantine‐Paton M, Wong GK‐S & Boyden ES (2014). Independent optical excitation of distinct neural populations. Nat Methods 11, 338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramoto E, Furuta T, Nakamura KC, Unzai T, Hioki H & Kaneko T (2009). Two types of thalamocortical projections from the motor thalamic nuclei of the rat: a single neuron‐tracing study using viral vectors. Cereb Cortex 19, 2065–2077. [DOI] [PubMed] [Google Scholar]
- Lamarre Y, Filion M & Cordeau JP (1971). Neuronal discharges of the ventrolateral nucleus of the thalamus during sleep and wakefulness in the cat I. Spontaneous activity. Exp Brain Res 12, 480–498. [DOI] [PubMed] [Google Scholar]
- McCormick DA & Bal T (1997). Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci 20, 185–215. [DOI] [PubMed] [Google Scholar]
- Marlinski V, Nilaweera WU, Zelenin PV, Sirota MG & Beloozerova IN (2012). Signals from the ventrolateral thalamus to the motor cortex during locomotion. J Neurophysiol 107, 455–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA & Von Krosigk M (1992). Corticothalamic activation modulates thalamic firing through glutamate “metabotropic” receptors. Proc Natl Acad Sci U S A 89, 2774–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mease RA, Krieger P & Groh A (2014). Cortical control of adaptation and sensory relay mode in the thalamus. Proc Natl Acad Sci U S A 111, 6798–6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mease RA, Kuner T, Fairhall AL & Groh A (2017). Multiplexed spike coding and adaptation in the thalamus. Cell Rep 19, 1130–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moruzzi G (1950). Effects at different frequencies of cerebellar stimulation upon postural tonus and myotatic reflexes. Electroencephalogr Clin Neurophysiol 2, 463–469. [DOI] [PubMed] [Google Scholar]
- Ohmae S, Uematsu A & Tanaka M (2013). Temporally specific sensory signals for the detection of stimulus omission in the primate deep cerebellar nuclei. J Neurosci 33, 15432–15441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen SR, Bortone DS, Adesnik H & Scanziani M (2012). Gain control by layer six in cortical circuits of vision. Nature 483, 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proville RD, Spolidoro M, Guyon N, Dugué GP, Selimi F, Isope P, Popa D & Léna C (2014). Cerebellum involvement in cortical sensorimotor circuits for the control of voluntary movements. Nat Neurosci 17, 1233–1239. [DOI] [PubMed] [Google Scholar]
- Raman IM, Gustafson AE & Padgett D (2000). Ionic currents and spontaneous firing in neurons isolated from the cerebellar nuclei. J Neurosci 20, 9004–9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramnani N (2006). The primate cortico‐cerebellar system: anatomy and function. Nat Rev Neurosci 7, 511–522. [DOI] [PubMed] [Google Scholar]
- Rispal‐Padel L, Harnois C & Troiani D (1987). Converging cerebellofugal inputs to the thalamus ‐ I. Mapping of monosynaptic field potentials in the ventrolateral nucleus of the thalamus. Exp Brain Res 68, 47–58. [DOI] [PubMed] [Google Scholar]
- Rispal‐Padel L & Latreille J (1974). The organization of projections from the cerebellar nuclei to the contralateral motor cortex in the cat. Exp Brain Res 19, 36–60. [DOI] [PubMed] [Google Scholar]
- Rispal‐Padel L, Massion J & Grangetto A (1973). Relations between the ventrolateral thalamic nucleus and motor cortex and their possible role in the central organization of motor control. Brain Res 60, 1–20. [DOI] [PubMed] [Google Scholar]
- Sakata HS, Ishijma T & Toyoda Y (1966). Single unit stdies on ventrolateral ncles of the thalams in cat: its relation to the cerebellm, motor cortex and basal ganglia. Jpn J Physiol 16, 42–60. [DOI] [PubMed] [Google Scholar]
- Sarnaik R & Raman IM (2018). Control of voluntary and optogenetically perturbed locomotion by spike rate and timing of neurons of the mouse cerebellar nuclei. Elife 7, 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Kawaguchi S, Matsuda Y & Mizuno N (1972). Electrophysiological studies on cerebello‐cerebral projections in the cat. Exp Brain Res 16, 75–88. [DOI] [PubMed] [Google Scholar]
- Sawyer SF, Young SJ, Groves PM & Tepper JM (1994). Cerebellar‐responsive neurons in the thalamic ventroanterior‐ventrolateral complex of rats: in vivo electrophysiology. Neuroscience 63, 711–724. [DOI] [PubMed] [Google Scholar]
- Sherman SM & Guillery RW (2011). Distinct functions for direct and transthalamic corticocortical connections. J Neurophysiol 106, 1068–1077. [DOI] [PubMed] [Google Scholar]
- Shinoda Y (1985). Synaptic organization of the cerebello‐thalamo‐cerebral pathway in the cat. Projection of individual cerebellar nuclei to single pyramidal tract neurons in areas 4 and 6. Neurosci Res 2,133–156. [DOI] [PubMed] [Google Scholar]
- Svoboda K & Li N (2017). ScienceDirect Neural mechanisms of movement planning: motor cortex and beyond. Curr Opin Neurobiol 49, 33–41. [DOI] [PubMed] [Google Scholar]
- Teune TM, van der Burg J, van der Moer J, Voogd J & Ruigrok TJ (2000). Topography of cerebellar nuclear projections to the brain stem in the rat. Prog Brain Res 124, 141–172. [DOI] [PubMed] [Google Scholar]
- Uno M, Yoshida M & Hirota I (1970). The mode of cerebello‐thalamic relay transmission investigated with intracellular recording from cells of the ventrolateral nucleus of cat's thalamus. Exp Brain Res 10, 121–139. [DOI] [PubMed] [Google Scholar]
- Vitek JL, Ashe J, DeLong MR & Alexander GE (1994). Physiologic properties and somatotopic organization of the primate motor thalamus. J Neurophysiol 71, 1498–1513. [DOI] [PubMed] [Google Scholar]
- Whitmire CJ, Waiblinger C, Schwarz C, & Stanley GB (2016). Information Coding Through Adaptive Gating of Synchronized Thalamic Bursting. Cell Rep. 14, 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfart J, Debay D, Le Masson G, Destexhe A & Bal T (2005). Synaptic background activity controls spike transfer from thalamus to cortex. Nat Neurosci 8, 1760–1767. [DOI] [PubMed] [Google Scholar]
- Yamawaki N & Shepherd GMG (2015). Synaptic circuit organization of motor corticothalamic neurons. J Neurosci 35, 2293–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Statistical Summary Document
Data Availability Statement
Data and software codes will be made available upon consent of the lead author (f.e.hoebeek@umcutrecht.nl).
Data and software codes will be made available upon consent of the lead author (f.e.hoebeek@umcutrecht.nl).