

Abstract
Objectives
Early diagnosis of Alzheimer's disease (AD) is essential for early interventions. Symptoms of depression could represent a prodromal stage of AD. Very early mood alterations may help to stratify those at highest risk of late‐life AD. We aim to investigate associations between baseline/longitudinal scores for depression, presence of cognitive impairment and/or AD pathology at death.
Methods/Design
Between 1991 and 2015, participants from The University of Manchester Longitudinal Study of Cognition in Normal Healthy Old Age underwent 10 waves of assessment using the Geriatric Depression Scale (GDS). AD pathology at death was evaluated in 106 eligible cases. Analyses aimed to examine associations between GDS scores, cognitive status and AD pathology (as measured by Braak stage, Thal phase and CERAD).
Results
Baseline GDS scores were significantly higher for those cognitively impaired at death than those cognitively normal. Significantly higher baseline GDS scores were found for those with greater Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) scores than those with lower CERAD scores. Similarly, significantly higher baseline GDS scores were found for those with a greater Braak stage than those with lower tau burden. These correlations remained after controlling for age at death, education and APOE ε4, but were less robust. Mean longitudinal GDS scores associated with cognition but not pathology.
Conclusions
GDS scores collected approximately 20 years before death were associated with cognitive status and AD pathology at death. We postulate that early AD‐related pathological change produces raised GDS scores due to an overlapping neural basis with depression, and that this may be considered as an early diagnostic marker for AD.
Keywords: Alzheimer's disease, depression, early diagnosis, neuropathology, prodrome
1. INTRODUCTION
Depression is a frequent co‐morbidity in patients with dementia and it presents unique challenges for clinicians who assess older, often cognitively impaired, adults. 1 Large, population‐based studies have identified depression as a risk factor for dementia and, in particular, Alzheimer's disease (AD). 2 Recent epidemiological studies have replicated this finding.3, 4, 5, 6 However, critically, few studies have had neuropathological confirmation of AD diagnosis. 7
Key points.
Baseline depression scores correlate with cognition and Alzheimer pathology
Mean longitudinal depression scores associate only with cognition at death
Results suggest overlapping neural basis between Alzheimer's disease and depression
Depression affects about 50% of patients with AD, 8 and the cognitive performance of patients with both AD and depression declines at a greater rate than of patients without depression. 9 A history of depression has been shown to increase the risk of developing dementia. 10 Early‐onset depression has been associated with an increased risk of AD.11, 12 However, late‐onset depression has been strongly implicated in AD, suggesting that depression is a prodrome of AD.13, 14, 15 One study highlighted that differences in depressive symptoms between those who do and those who do not go on to develop AD can be seen a decade before dementia diagnosis. 16 However, it is noteworthy that other studies reported no depressive symptoms in the prodromal phase of AD. 17
The major pathological changes associated with AD are abnormal accumulation of extracellular beta‐amyloid (Aβ), which is thought to be an initial event of AD, and secondary intracellular tau in the form of neurofibrillary tangles. 18 It is thought that these pathological changes start many years before the onset of measurable cognitive decline. 19 Both Aβ and tau have also been implicated in depression. Pre‐clinical AD is associated with low plasma levels of Aβ42 when compared with levels of Aβ40 and a low Aβ42:Aβ40 ratio is associated with depression in AD patients. 20 This is exacerbated further in those individuals carrying the APOE ε4 allele. 21 Levels of total tau and phosphorylated tau have been helpful in distinguishing mild cognitive impairment from major depressive disorder (MDD) which can be clinically difficult due to overlap of symptoms. 22 In addition, a mutant R406W human tau mouse model exhibited changes in depression‐related behaviour which involved serotonergic neurons suggesting that tau may play a role in AD and depression. 23 Conversely, a PET imaging study found no difference in amyloid binding between depressed and non‐depressed individuals, 24 and other studies have also failed to associate amyloid burden with depression. 25
Although clinical studies have significantly contributed to the understanding of the relationship between depression and AD, they lack the benefit of neuropathological confirmation of the disease. There are few studies with pathological confirmation of AD and they lack consensus in the established literature. A recent review concluded that cognitive dysfunction in MDD and AD share a common origin relating to the hippocampus and the prefrontal cortex; specifically in the breakdown of connectivity between these regions. 26 It has previously been shown that AD patients with depression have a decreased number of neurons in the locus coeruleus compared to their non‐depressed counterparts. 27 In addition, those with depression at intermediate stages of AD pathology (which is typically limbic) had a worse cognitive outcome than those without depression. 28 Likewise, the presence of a past history of major depression seems to lead to an increase in Aβ plaques and tau tangles in AD patients when compared with AD patients without depression. 29 Conversely, two large cohort studies that had brain donation as an end point, concluded that depression is not associated with any dementia‐related pathology.30, 31
The Yesavage Geriatric Depression Scale (GDS) 32 is a widely used tool for assessing depressive symptoms in the elderly. Scores from the GDS have been shown to be reliable and relevant in those with AD.33, 34, 35 This study aims to establish whether GDS scores for depression, measured longitudinally and starting many years before death, associate with the degree of AD pathology found at death as measured by Braak stage, 36 Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) score, 37 and Thal phase. 38
2. MATERIALS AND METHODS
2.1. Participants and study design
Participants from The University of Manchester Longitudinal Study of Cognition in Normal Healthy Old Age (UMLCHA) 39 were approached in 2003 for consent to brain donation. From the originally recruited total of 6542 healthy individuals (aged between 42 and 92 years), 312 individuals consented to brain donation. So far, 134 donations have been collected. Information pertaining to the clinical and pathological profile of this cohort has been previously described.40, 41
Between 1991 and 2003, participants underwent four face‐to‐face assessments of depression using the long form of the GDS which contains 30 YES/NO questions whose answers lead to a score which can be used to assess level of depressive symptoms. The cut off points are: 0–9 = normal; 10–19 = mild depression and 20–30 = severe depression. Between 2004 and 2015, participants underwent a further six telephone assessments of depressive symptoms using the short form of the GDS which contains 15 YES/NO questions. The cut off points are 0–5 = normal; 6–10 = suggestive of depression and 11–15 = severe. Both GDS30 and GDS15 have been previously validated as accurate means of assessing depression 32 and scores on the GDS15 have been shown to correlate well with scores on GDS30. 42
In addition, these participants (n = 100; Telephone Instrument for Cognitive Status [TICSm] data missing for six individuals) underwent five waves of cognitive assessment using the modified TICSm which contains 13 questions testing orientation, concentration, immediate and delayed memory, naming, calculation, comprehension and reasoning. The TICSm test 43 has a maximum score of 39 and a score of 21 was used here as the cut‐off point, indicating cognitive impairment. 44 Cognitive status at death was ascertained using a combination of the last TICSm score, patient notes obtained via participants' general practitioner and cause of death as recorded on the death certificate.
2.2. Pathological examination
Standard blocks of frontal, cingulate, temporal (including superior and middle temporal gyrus), parietal and occipital cortex, hippocampus, amygdala, corpus striatum, thalamus, midbrain, brainstem and cerebellum were cut from the fixed tissue and processed into wax blocks. One section was stained with haematoxylin‐eosin and further sections (6 µm) were immunostained for Aβ (Cambridge Bioscience, clone 4G8, 1:3000), tau proteins phosphorylated at Ser202 and Thr205 (P‐tau; Innogenetics, clone AT8, 1:750), phosphorylated α‐synuclein (#1175–Gift from Dr Masato Hasegawa at Tokyo Metropolitan Institute of Medical Science, Japan; 1:1000) and TAR DNA‐binding protein 43 (TDP‐43; Proteintech, 1:1000). For antigen retrieval, sections were either immersed in 70% formic acid for 20 min (for Aβ only) or microwaved in 0.1 M citrate buffer, pH 6.0 (all other antibodies) prior to incubation with primary antibody. Staining was visualised using the VECTASTAIN Elite ABC kit (Vector Laboratories) which is an avidin/biotin based amplification system. For Aβ and P‐tau, horse anti‐mouse biotinylated secondary antibody was used. For TDP‐43 and phosphorylated α‐synuclein, goat anti‐rabbit biotinylated secondary antibody was used.
Neuropathological assessment was performed by two experienced neuropathologists (Daniela Montaldi & Federico Roncaroli) who used consensus criteria to establish the presence and staging of common neurodegenerative diseases.45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57 In addition, vascular pathology was assessed using the Vascular Cognitive Impairment Neuropathology Guidelines. 58 For the purpose of this study, we excluded all cases where the primary neuropathological diagnosis was not AD and also excluded AD cases where there was any secondary or concomitant pathology (other than cerebral amyloid angiopathy or small vessel disease). We included cases of Age‐Related Tau Astrogliopathy, Primary Ageing‐Related Tauopathy and Limbic‐predominant Age‐related TDP‐43 Encephalopathy owing to the fact that they are common ageing‐related pathologies. Of the 134 UMLCHA participants who had donated their brain, 106 were eligible. An overview of the pathology of eligible participants is available (Table S1).
2.3. Statistical analyses
The cohort was divided into pathology groups according to severity of AD pathology:
CERAD score: 0–A (low severity) versus B–C (high severity).
Thal phase: 0–3 (low severity) versus 4–5 (high severity).
Braak stage: 0–II (low severity) versus III–VI (high severity).
Differences between pathology groups in sex, cognitive impairment and presence of APOE ε4 allele(s) were analysed with the Chi‐squared test while those for age at death, and years of education were analysed with the T‐test.
Baseline GDS30 scores were not normally distributed. Therefore, differences between cognitive and pathology groups for baseline GDS30 scores were analysed with the Mann‐Whitney U‐Test. Further binary logistic regression analyses were used to determine whether the influence of sex, years of education and presence of APOE ε4 allele(s) affected the outcome.
To assess the association between longitudinal GDS scores and cognitive/pathology groups at death, we used binary logistic regression with mean depression score over the duration of the study as the independent variable of interest. For this analysis, both GDS15 and GDS30 measures were included, maximising the number of measures per patient. As these are on different scales, we standardised the scores by subtracting the mean and then dividing by the standard deviation of the sample. The mean was chosen as the measure of average due to the small number of depression scores over time per individual (mean number was 6). Covariates used in the model were sex, years of education and presence of APOE ε4 allele(s).
In all cases, a p value of <0.05 was considered significant.
3. RESULTS
3.1. Demographics
The demographics of the 106 eligible participants are shown in Table 1. There were no significant differences in sex, age at death or level of education between cognitive status groups, CERAD, Thal phase or Braak stage groups. As expected, a significantly greater proportion of cognitively impaired individuals were present in the high severity pathology groups for CERAD score (χ 2 = 27.2; p < 0.001), Thal phase (χ 2 = 6.1; p = 0.014) and Braak stage (χ 2 = 22.6; p < 0.001). Likewise, APOE ε4 allele(s) were more likely to be present in the high severity pathology groups for CERAD score (χ 2 = 5.7; p = 0.017), Thal phase (χ 2 = 10.1; p = 0.001) and Braak stage (χ 2 = 5.2; p = 0.023).
TABLE 1.
Descriptive characteristics, stratified by AD neuropathological outcome and cognitive impairment at death, for eligible individuals from UMLCHA
| CI at death | CERAD score | Thal phase | Braak stage | Total cohort | |||||
|---|---|---|---|---|---|---|---|---|---|
| No | Yes | 0–A | B–C | 0–3 | 4–5 | 0–II | III–VI | ||
| N | 76 | 30 | 60 | 46 | 89 | 17 | 66 | 40 | 106 |
| Female | 54 (71.1%) | 22 (73.3%) | 40 (66.7%) | 36 (78.3%) | 64 (71.9%) | 12 (70.6%) | 47 (71.2%) | 29 (72.5%) | 76 (71.7%) |
| C.I at death | n/a | n/a | 5 (8.3%) | 25 (54.3%) | 21 (23.6%) | 9 (52.9%) | 8 (12.1%) | 22 (55.0%) | 30 (28.3%) |
| Age at death (mean ± SD) | 88.5 (±6.4) | 89.7 (±4.1) | 88.4 (±5.4) | 89.4 (±6.3) | 88.9 (±5.9) | 88.5 (±5.7) | 88.9 (±5.5) | 88.8 (±6.4) | 88.4 (±5.8) |
| Education in ISCED yrs (mean ± SD) | 15.7 (±3.5) | 15.6 (±3.8) | 15.8 (±3.6) | 15.4 (±3.6) | 15.8 (±3.5) | 14.8 (±4.1) | 15.6 (±3.6) | 15.7 (±3.5) | 15.7 (±3.6) |
| One or more ε4 alleles present | 18 (23.7%) | 11 (36.7%) | 11 (18.3%) | 18 (39.1%) | 19 (21.3%) | 10 (58.8%) | 13 (19.7%) | 16 (40.0%) | 29 (27.4%) |
Note: Bold indicates significant difference between inclusive cognitive/pathology groups.
Abbreviations: AD, Alzheimer's disease; CI, cognitive impairment; CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; UMLCHA, University of Manchester Longitudinal Study of Cognition in Normal Healthy Old Age.
3.2. Assessment of baseline GDS30 test scores and relationship to cognition and pathology at death.
Median baseline GDS30 score, mean age at baseline testing and mean number of years between test and death are shown in Table 2.
TABLE 2.
Data pertaining to baseline GDS scores, age at testing and time between death and baseline GDS testing, stratified by cognitive impairment and AD neuropathological outcome
| CI at death | CERAD score | Thal phase | Braak stage | Total cohort | |||||
|---|---|---|---|---|---|---|---|---|---|
| No | Yes | 0–A | B–C | 0–3 | 4–5 | 0–II | III–VI | ||
| Baseline | |||||||||
| Score (median/range) | 3.0 (25) | 7.0 (13) | 3.0 (25) | 6.5 (20) | 4.0 (25) | 5.0 (20) | 3.0 (25) | 6.0 (20) | 4.0 (25) |
| Age at testing (mean ± SD) | 68.4 (±5.5) | 68.4 (±4.9) | 68.2 (±5.2) | 68.6 (±5.6) | 68.0 (±5.3) | 70.2 (±5.5) | 68.1 (±5.2) | 68.8 (±5.6) | 68.4 (±5.3) |
| Years between test and death (mean ± SD) | 20.5 (±4.0) | 21.7 (±4.0) | 20.8 (±3.8) | 20.8 (±4.3) | 21.3 (±3.9) | 18.7 (±4.0) | 21.2 (±3.7) | 20.2 (±4.5) | 20.8 (±4.0) |
Note: Bold indicates significant difference between inclusive cognitive/pathology groups.
Abbreviations: AD, Alzheimer's disease; CI, cognitive impairment; CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; GDS, Geriatric Depression Scale.
The mean age at baseline GDS30 test was 68.4 (±5.3) years which was 20.8 (±4.0) years before death.
Those classified as having cognitive impairment at death scored higher on baseline GDS30 than their cognitively normal counterparts (p = 0.007). In addition, we found that baseline GDS30 scores were significantly higher in the severe AD pathology groups for CERAD and Braak stage when compared with the corresponding lower severity group (CERAD: p = 0.015; Braak stage; p = 0.018). However, this was not the case when examining Thal phase (p = 0.292; Figure 1).
FIGURE 1.
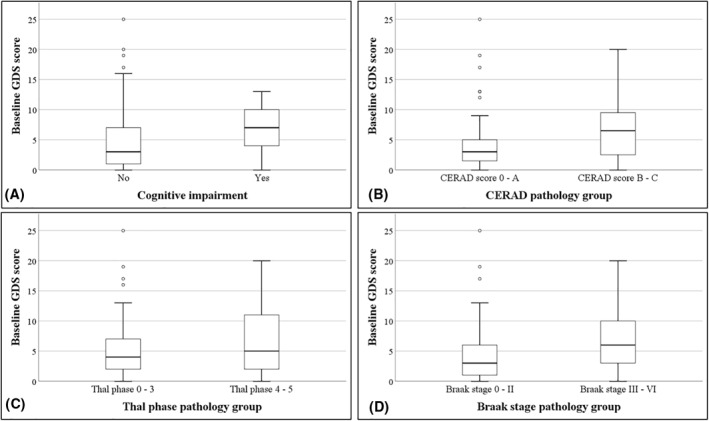
Boxplots comparing baseline Geriatric Depression Scale (GDS)30 scores between cognitive (Panel A) and Alzheimer's disease pathology groups (Panels B, C and D). The boxes represent the interquartile (IQ) range which contains the middle 50% of the records. The whiskers represent the highest and lowest values which are no greater than 1.5 times the IQ range. The line across the boxes indicates the median. Differences between cognitive and pathology groups for baseline GDS30 scores were analysed with the Mann–Whitney U‐Test
Regression analyses showed that after accounting for sex, level of education and presence of APOE ε4 allele(s), the association between baseline GDS30 scores and cognitive impairment (p = 0.104), CERAD scores (p = 0.094) and Braak stage (p = 0.060) were no longer significant (Table 3).
TABLE 3.
Logistic regression analysis assessing the impact of sex, level of education and presence of APOE ε4 allele(s) on the significant outcomes in baseline GDS scores between cognitive impairment, CERAD score and Braak stage groups
| Cognitive impairment at death | CERAD score | Braak stage | |||||||
|---|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | p value | OR | 95% CI | p value | OR | 95% CI | p value | |
| Baseline GDS score | 1.08 | 0.98–1.18 | 0.104 | 1.08 | 0.99–1.17 | 0.094 | 1.09 | 1.00–1.18 | 0.060 |
| Sex | 1.39 | 0.44–4.36 | 0.575 | 1.51 | 0.56–4.02 | 0.413 | 0.96 | 0.35–2.63 | 0.941 |
| Education | 1.00 | 0.87–1.14 | 0.946 | 0.95 | 0.85–1.07 | 0.429 | 0.97 | 0.86–1.09 | 0.599 |
| Presence of APOE ε4 | 2.50 | 0.94–6.65 | 0.067 | 2.98 | 1.17–7.60 | 0.023 | 3.16 | 1.24–8.05 | 0.016 |
Note: Bold indicates significant difference.
Abbreviations: CI, confidence interval; CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; GDS, Geriatric Depression Scale; OR, odds ratio.
3.3. Assessment of longitudinal GDS test scores and relationship to cognition and pathology at death.
Binary logistic regression analyses were conducted using cognitive or pathology groups as the outcome measure and mean score from the GDS30 and GDS15 time points as the independent variable of interest. Covariates used in the model were sex, years of education and presence of APOE ε4 allele(s).
The mean longitudinal GDS score was significantly higher in cognitively impaired individuals when compared with cognitively normal individuals (p = 0.047) at death. However, there were no differences in mean longitudinal GDS score between CERAD score (p = 0.185), Thal phase (p = 0.783) or Braak stage (p = 0.195) groups (Table 4).
TABLE 4.
Logistic regression analysis assessing the impact of sex, level of education and presence of APOE ε4 allele(s) on the mean longitudinal GDS scores between cognitive impairment, CERAD score, Thal phase and Braak stage groups
| Outcome | OR | 95% CI | p value | AUROC (95% CI) |
|---|---|---|---|---|
| Cognitive impairment | 1.64 | 1.01–2.68 | 0.047 | 0.67 (0.56–0.78) |
| CERAD score | 1.06 | 0.97–1.16 | 0.185 | 0.67 (0.56–0.77) |
| Thal phase | 1.09 | 0.58–2.08 | 0.783 | 0.71 (0.54–0.87) |
| Braak stage | 1.36 | 0.85–2.18 | 0.195 | 0.67 (0.56–0.77) |
Note: Bold indicates significant difference
Abbreviations: AUROC, area under ROC; CI, confidence interval; CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; GDS, Geriatric Depression Scale; OR, odds ratio.
4. DISCUSSION
We have shown that scores from the baseline GDS30 test, undertaken approximately 20 years before death, associate with cognitive impairment and AD pathology at death. A general trend towards correlation remained after controlling for age at death, education level and presence of APOE ε4 allele(s). Although the difference in GDS30 score between the cognitive groups and also between the AD pathology groups is subtle and remains sub‐clinical for depression as measured by GDS, it nonetheless suggests that, if undertaken 20 years before death, the GDS is able to differentiate between, and could be predictive of, those who go on to develop AD pathology and associated cognitive impairment from those who will not.
Some studies have reported that cognitive test scores (usually of memory) are able to predict clinical dementia approximately 4–18 years before onset of symptoms.59, 60, 61, 62 Our findings show that a simple and widely used test for depression can also discriminate between those who become significantly cognitively impaired from those whose cognition remains relatively intact; and can do this many years before death.
A drawback to studies which are limited to clinical measures of dementia is the lack of neuropathological confirmation of disease type. Cognitive impairment is a ‘loose’ term encompassing many types of dementing illness. Here, instead, we ensured that our study group included only those with AD pathology and those with pathological findings that would be considered normal for the age of the subject. This gave us the opportunity to examine AD pathology alone as cases with possible concomitant pathology were excluded.
It is widely thought that the pathological processes underlying AD occur many years before any cognitive change that may lead to an individual seeking medical intervention. 63 Many studies have shown that cognitive performance scores, obtained 1–6 years before death, can predict ultimate levels of AD pathology.64, 65, 66 However, most recently our group has shown that these findings can be extrapolated to approximately 20 years before death. 67 The findings described in the present study, again, show that scores of cognitive performance, in this case GDS30 baseline scores, collected 20 years prior to death, associate with AD pathology at post‐mortem.
Neuroimaging and post‐mortem studies have attempted to elucidate the underlying pathophysiology of depression. Those with MDD show abnormal brain activity in frontal and temporal cortices, insula and cerebellum. 68 In addition, those with MDD have moderate volume reduction in the hippocampus and striatum 69 and a reduction of glia cell density in the prefrontal cortices and amygdalae.70, 71 However, the most striking findings are in the cingulate cortex where volume reduction has been shown early in MDD. 72 Although the present study does not include individuals with known MDD, it is highly pertinent that the areas affected in MDD overlap somewhat with those affected relatively early on in AD by Aβ and tau; specifically the hippocampus, temporal cortex and amygdala. Moreover, the presence of depressive symptoms, not severe enough to be considered MDD, could result from neurofibrillary damage to serotonergic neurones in areas considered to be the first affected in AD, such as the dorsal raphe and locus coeruleus.73, 74, 75 It has previously been shown that damage to serotonergic neurones can lead to depressive symptoms in animal models. 23 Thus, it would not be unreasonable to suggest that very early degenerative changes in these brain stem nuclei in humans could be responsible for generating mild depressive symptoms years before the onset of clinical AD. It is arguable therefore that the differences we report here in baseline GDS30 scores could reflect very early, subtle AD pathological changes, rather than the onset of a mild depressive disorder per se. This argument is bolstered by the fact that although the GDS30 baseline scores suggest mild depressive symptoms, they remain in the ‘normal’ range of the GDS protocol and do not indicate a diagnosis of depression.
Changes in neurogenesis could be another underlying mechanism explaining the very early differences in GDS scores between AD pathology groups. Both rodent models of AD and human AD post‐mortem tissue have shown decreased neurogenesis, especially within the ventral dentate gyrus of the hippocampus (and area of the brain implicated in both AD and MDD), while residual low‐level neurogenesis is likely to be still ongoing.76, 77 Interestingly, neurogenesis has also been implicated in the pathology of MDD and is argued to be one of the ways in which anti‐depressant medications exert their effect. 78 One hypothesis could therefore be that early AD pathology induces localised disruption of neurogenesis in the dentate gyrus. This would prevent new cell proliferation and network integration, thereby causing early depressive symptoms and later, mild cognitive impairment, and finally, dementia.
It is worthy of note that only the baseline scores for GDS30 differentiated between CERAD and Braak stage pathology groups. The mean longitudinal scores did not show the same differences, possibly due to the deaths of participants during the study, which diminished the number of cases with available data, shrinking the study group and reducing the statistical power. Moreover, in the elderly, as age increases so does frequency of depression which likely confounds the longitudinal GDS scores. It is also very possible that those with less severe pathology still undergo the very early pathological changes associated with AD but at a later time point and fail to progress to the later stages of the disease owing to death. These individuals may therefore score higher on GDS at later time points rendering them more comparable to their severe pathology counterparts on the longitudinal GDS.
Sample size is a limitation imposed on the UMLCHA and the recruitment method (self‐selection) suggests that the study samples may not be representative of the general population. Similarly, the geographical areas covered by UMLCHA (Greater Manchester and Newcastle) may not reflect society as a whole. The diagnosis of cognitive impairment was not confirmed by a diagnostic clinical interview. However, a consensus on cognitive impairment at death was reached by experts using a wide range of in‐life measures. In addition, a lack of follow‐up closer to death means that cognitive decline may have been missed. Binary categorisation of the pathology outcomes was an unfortunate, but necessary, limitation owing to an insufficient sample size to consider them in their more natural, ordinal form. Strengths of the study include the choice of GDS as a tool to measure depression, owing to its reliability and relevance in AD,33, 34, 35 the longitudinal aspect of data collection and the ability to confirm diagnosis and assess neuropathological findings post‐mortem.
In conclusion, GDS30 scores collected approximately 20 years before death were associated with cognitive status at death and AD pathology, but mean longitudinal GDS scores were only associated with cognition and not with pathology. We postulate that the very early pathological changes associated with AD lead to higher scores on the GDS30 due to the overlap between brain regions implicated in depression and those compromised in very early AD. We suggest that a test battery combining assessments of cognition and symptoms of depression may well identify those individuals who will go on to develop AD, many years before they would have otherwise been diagnosed using the usual clinical assessments. However, for this to be robust, factors such as age at death, education level and APOE genotype would have to be considered.
CONFLICTS OF INTEREST
The authors have declared that they have no conflict of interest to disclose.
AUTHOR CONTRIBUTIONS
All authors have read the manuscript and have agreed to be listed as authors. Andrew C Robinson devised and designed the study, performed data/statistical analysis and wrote the paper. Federico Roncaroli finalised neuropathological diagnosis and contributed to writing the manuscript. James Minshull assisted with writing the manuscript and performed immunohistochemistry. Calvin Heal performed data/statistical analysis and assisted with preparation of the manuscript. Daniela Montaldi assisted with preparation of the manuscript and advised on neuropsychology. Yvonne S Davidson performed immunohistochemistry and assisted with preparation of the manuscript. Antony Payton assisted with preparation of the manuscript. Michael A Horan helped to finalise clinical cognitive impairment diagnosis and provided clinical data. David MA Mann finalised neuropathological diagnosis and contributed to writing the manuscript. Neil Pendleton finalised clinical cognitive impairment diagnosis and assisted with preparation of the manuscript.
ETHICS COMMITTEE APPROVAL
The study was approved by Manchester Brain Bank Management Committee (REC reference 19/NE/0242). Under conditions agreed with the Research Ethics Committee, The Manchester Brain Bank can supply tissue or data to researchers, without requirement for researchers to apply individually to the REC for approval.
Supporting information
Supplementary Material 1
ACKNOWLEDGEMENTS
Longitudinal Cognitive studies were funded by Medical Research Council, Economic and Social Research Council, The Wellcome Trust [Grant reference number 003889] and Unilever PLC. The work of Manchester Brain Bank is supported by Alzheimer's Research UK and Alzheimer's Society through the Brains For Dementia Research (BDR) Programme.
Neil Pendleton and David MA Mann contributed equally to the study.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
REFERENCES
- 1. Steffens DC. Late‐life depression and the prodromes of dementia. JAMA Psychiatry. 2017;74(7):673‐674. [DOI] [PubMed] [Google Scholar]
- 2. Kokmen E, Beard CM, Chandra V, Offord KP, Schoenberg BS, Ballard DJ. Clinical risk factors for Alzheimer's disease: a population‐based case‐control study. Neurology. 1991;41(9):1393‐1397. [DOI] [PubMed] [Google Scholar]
- 3. Steffens DC, Plassman BL, Helms MJ, Welsh‐Bohmer KA, Saunders AM, Breitner JC. A twin study of late‐onset depression and apolipoprotein E epsilon 4 as risk factors for Alzheimer's disease. Biol Psychiatry. 1997;41(8):851‐856. [DOI] [PubMed] [Google Scholar]
- 4. Barnes DE, Alexopoulos GS, Lopez OL, Williamson JD, Yaffe K. Depressive symptoms, vascular disease, and mild cognitive impairment: findings from the Cardiovascular Health Study. Arch Gen Psychiatry. 2006;63(3):273‐279. [DOI] [PubMed] [Google Scholar]
- 5. Dotson VM, Beydoun MA, Zonderman AV. Recurrent depressive symptoms and the incidence of dementia and mild cognitive impairment. Neurology. 2010;75(1):27‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Simões do Couto F, Lunet N, Ginó S, et al. Depression with melancholic features is associated with higher long‐term risk for dementia. J Affect Disord. 2016;202:220‐229. [DOI] [PubMed] [Google Scholar]
- 7. Wiels W, Baeken C, Engelborghs S. Depressive symptoms in the elderly‐an early symptom of dementia? A systematic review. Front Pharmacol. 2020;11:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Modrego PJ. Depression in Alzheimer's disease. Pathophysiology, diagnosis, and treatment. J Alzheimers Dis. 2010;21(4):1077‐1087. [DOI] [PubMed] [Google Scholar]
- 9. Bassuk SS, Berkman LF, Wypij D. Depressive symptomatology and incident cognitive decline in an elderly community sample. Arch Gen Psychiatry. 1998;55(12):1073‐1081. [DOI] [PubMed] [Google Scholar]
- 10. Ownby RL, Crocco E, Acevedo A, John V, Loewenstein D. Depression and risk for Alzheimer disease: systematic review, meta‐analysis, and metaregression analysis. Arch Gen Psychiatry. 2006;63(5):530‐538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Geerlings MI, den Heijer T, Koudstaal PJ, Hofman A, Breteler MM. History of depression, depressive symptoms, and medial temporal lobe atrophy and the risk of Alzheimer disease. Neurology. 2008;70(15):1258‐1264. [DOI] [PubMed] [Google Scholar]
- 12. Heser K, Tebarth F, Wiese B, et al. Age CoDe study group. Age of major depression onset, depressive symptoms, and risk for subsequent dementia: results of the German study on ageing, cognition, and dementia in primary care patients (AgeCoDe). Psychol Med. 2013;43(8):1597‐1610. [DOI] [PubMed] [Google Scholar]
- 13. Berger AK, Fratiglioni L, Forsell Y, Winblad B, Bäckman L. The occurrence of depressive symptoms in the preclinical phase of AD: a population‐based study. Neurology. 1999;53(9):1998‐2002. [DOI] [PubMed] [Google Scholar]
- 14. Chen P, Ganguli M, Mulsant BH, DeKosky ST. The temporal relationship between depressive symptoms and dementia: a community‐based prospective study. Arch Gen Psychiatry. 1999;56(3):261‐266. [DOI] [PubMed] [Google Scholar]
- 15. Yaffe K, Blackwell T, Gore R, Sands L, Reus V, Browner WS. Depressive symptoms and cognitive decline in nondemented elderly women: a prospective study. Arch Gen Psychiatry. 1999;56(5):425‐430. [DOI] [PubMed] [Google Scholar]
- 16. Singh‐Manoux A, Dugravot A, Fournier A, et al. Trajectories of depressive symptoms before diagnosis of dementia: a 28‐year follow‐up study. JAMA Psychiatry. 2017;74(7):712‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wilson RS, Arnold SE, Beck TL, Bienias JL, Bennett DA. Change in depressive symptoms during the prodromal phase of Alzheimer disease. Arch Gen Psychiatry. 2008;65(4):439‐445. [DOI] [PubMed] [Google Scholar]
- 18. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184‐185. [DOI] [PubMed] [Google Scholar]
- 19. Tan CC, Yu JT, Tan L. Biomarkers for preclinical Alzheimer's disease. J Alzheimers Dis. 2014;42(4):1051‐1069. [DOI] [PubMed] [Google Scholar]
- 20. Sun X, Steffens DC, Au R, et al. Amyloid‐associated depression: a prodromal depression of Alzheimer disease? Arch Gen Psychiatry. 2008;65(5):542‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qiu WQ, Zhu H, Dean M, et al. Amyloid‐associated depression and ApoE4 allele: longitudinal follow‐up for the development of Alzheimer's disease. Int J Geriatr Psychiatry. 2016;31(3):316‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schönknecht P, Pantel J, Kaiser E, Thomann P, Schröder J. Increased tau protein differentiates mild cognitive impairment from geriatric depression and predicts conversion to dementia. Neurosci Lett. 2007;416(1):39‐42. [DOI] [PubMed] [Google Scholar]
- 23. Egashira N, Iwasaki K, Takashima A, et al. Altered depression‐related behavior and neurochemical changes in serotonergic neurons in mutant R406W human tau transgenic mice. Brain Res. 2005;1059(1):7‐12. [DOI] [PubMed] [Google Scholar]
- 24. De Winter FL, Emsell L, Bouckaert F, et al. No association of lower hippocampal volume with Alzheimer's disease pathology in late‐life depression. Am J Psychiatry. 2017;174(3):237‐245. [DOI] [PubMed] [Google Scholar]
- 25. Chung JK, Plitman E, Nakajima S, et al. Cortical amyloid β deposition and current depressive symptoms in Alzheimer disease and mild cognitive impairment. J Geriatr Psychiatry Neurol. 2016;29(3):149‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sampath D, Sathyanesan M, Newton SS. Cognitive dysfunction in major depression and Alzheimer's disease is associated with hippocampal‐prefrontal cortex dysconnectivity. Neuropsychiatr Dis Treat. 2017;13:1509‐1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Förstl H, Burns A, Luthert P, Cairns N, Lantos P, Levy R. Clinical and neuropathological correlates of depression in Alzheimer's disease. Psychol Med. 1992;22(4):877‐884. [DOI] [PubMed] [Google Scholar]
- 28. Milwain EJ, Nagy Z. Depressive symptoms increase the likelihood of cognitive impairment in elderly people with subclinical Alzheimer pathology. Dement Geriatr Cogn Disord. 2005;19(1):46‐50. [DOI] [PubMed] [Google Scholar]
- 29. Rapp MA, Schnaider‐Beeri M, Grossman HT, et al. Increased hippocampal plaques and tangles in patients with Alzheimer disease with a lifetime history of major depression. Arch Gen Psychiatry. 2006;63(2):161‐167. [DOI] [PubMed] [Google Scholar]
- 30. Wilson RS, Capuano AW, Boyle PA, et al. Clinical‐pathologic study of depressive symptoms and cognitive decline in old age. Neurology. 2014;83(8):702‐709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wilson RS, Boyle PA, Capuano AW, et al. Late‐life depression is not associated with dementia‐related pathology. Neuropsychology. 2016;30(2):135‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yesavage JA, Brink TL, Rose TL, et al. Development and validation of a geriatric depression screening scale: a preliminary report. J Psychiatr Res. 1983;17(1):37‐49. [DOI] [PubMed] [Google Scholar]
- 33. Nyunt MS, Fones C, Niti M, Ng TP. Criterion‐based validity and reliability of the Geriatric Depression Screening Scale (GDS‐15) in a large validation sample of community‐living Asian older adults. Aging Ment Health. 2009;13(3):376‐382. [DOI] [PubMed] [Google Scholar]
- 34. Yamane Y, Sakai K, Maeda K. Dementia with Lewy bodies is associated with higher scores on the Geriatric Depression Scale than is Alzheimer's disease. Psychogeriatrics. 2011;11(3):157‐165. [DOI] [PubMed] [Google Scholar]
- 35. Conradsson M, Rosendahl E, Littbrand H, Gustafson Y, Olofsson B, Lövheim H. Usefulness of the Geriatric Depression Scale 15‐item version among very old people with and without cognitive impairment. Aging Ment Health. 2013;17(5):638‐645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82(4):239‐259. [DOI] [PubMed] [Google Scholar]
- 37. Mirra SS, Heyman A, McKeel D, et al. The consortium to establish a registry for Alzheimer's disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41(4):479‐486. [DOI] [PubMed] [Google Scholar]
- 38. Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791‐1800. [DOI] [PubMed] [Google Scholar]
- 39. Rabbitt PMA, McInnes L, Diggle P, et al. The University of Manchester longitudinal study of cognition in normal healthy old age, 1983 through 2003. Aging Neuropsychol C. 2004;11(2–3):245‐279. [Google Scholar]
- 40. Robinson AC, Davidson YS, Horan MA, Pendleton N, Mann DMA. Pathological correlates of cognitive impairment in the University of Manchester longitudinal study of cognition in normal healthy old age. J Alzheimers Dis. 2018;64(2):483‐496. [DOI] [PubMed] [Google Scholar]
- 41. Robinson AC, Chew‐Graham S, Davidson YS, et al. A comparative study of pathological outcomes in the University of Manchester longitudinal study of cognition in normal healthy old age and brains for dementia Research cohorts. J Alzheimers Dis. 2020;73(2):619‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sheikh JI, Yesavage JA. Geriatric Depression Scale (GDS): recent evidence and development of a shorter version. Clin Gerontol. 1986;5:165‐173. [Google Scholar]
- 43. Plassman BL, Welsh KA, Helms M, Brandt J, Page WF, Breitner JC. Intelligence and education as predictors of cognitive state in late life: a 50 year follow‐up. Neurology. 1995;45(8):1446‐1450. [DOI] [PubMed] [Google Scholar]
- 44. Evans O, Singleton N, Meltzer D, Stewart R, Prince M. The mental health of older people: Report based on the analysis of the ONS survey of psychiatric morbidity among adults in Great Britain. IBSN 0116216603. London, UK: Office of National Statistics; 2003. [Google Scholar]
- 45. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47(5):1113‐1124. [DOI] [PubMed] [Google Scholar]
- 47. McKeith IG, Dickson DW, Lowe J, et al. Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863‐1872. [DOI] [PubMed] [Google Scholar]
- 48. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology. 2017;89(1):88‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 2009;117(1):15‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD‐TDP pathology. Acta Neuropathol. 2011;122(1):111‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dickson DW, Bergeron C, Chin SS, et al. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002;61(11):935‐946. [DOI] [PubMed] [Google Scholar]
- 52. Hauw JJ, Daniel SE, Dickson D, et al. Preliminary NINDS neuropathologic criteria for Steele‐Richardson‐Olszewski syndrome (progressive supranuclear palsy). Neurology. 1994;44(11):2015‐2019. [DOI] [PubMed] [Google Scholar]
- 53. Trojanowski JQ, Revesz T. Neuropathology Working Group on MSA. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol. 2007;33(6):615‐620. [DOI] [PubMed] [Google Scholar]
- 54. Braak H, Braak E. Argyrophilic grain disease: frequency of occurrence in different age categories and neuropathological diagnostic criteria. J Neural Transm. 1998;105(8‐9):801‐819. [DOI] [PubMed] [Google Scholar]
- 55. Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age‐related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128(6):755‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kovacs GG, Ferrer I, Grinberg LT, et al. Aging‐related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol. 2016;131(1):87‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain. 2019;142(6):1503‐1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Skrobot OA, Attems J, Esiri M, et al. Vascular cognitive impairment neuropathology guidelines (VCING): the contribution of cerebrovascular pathology to cognitive impairment. Brain. 2016;139(11):2957‐2969. [DOI] [PubMed] [Google Scholar]
- 59. Aretouli E, Tsilidis KK, Brandt J. Four‐year outcome of mild cognitive impairment: the contribution of executive dysfunction. Neuropsychology. 2013;27(1):95‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Amieva H, Jacqmin‐Gadda H, Orgogozo JM, et al. The 9 year cognitive decline before dementia of the Alzheimer type: a prospective population‐based study. Brain. 2005;128(Pt 5):1093‐1101. [DOI] [PubMed] [Google Scholar]
- 61. Amieva H, Le Goff M, Millet X, et al. Prodromal Alzheimer's disease: successive emergence of the clinical symptoms. Ann Neurol. 2008;64(5):492‐498. [DOI] [PubMed] [Google Scholar]
- 62. Rajan KB, Wilson RS, Weuve J, Barnes LL, Evans DA. Cognitive impairment 18 years before clinical diagnosis of Alzheimer disease dementia. Neurology. 2015;85(10):898‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vickers JC, Mitew S, Woodhouse A, et al. Defining the earliest pathological changes of Alzheimer's disease. Curr Alzheimer Res. 2016;13(3):281‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Powell MR, Smith GE, Knopman DS, et al. Cognitive measures predict pathologic Alzheimer disease. Arch Neurol. 2006;63(6):865‐868. [DOI] [PubMed] [Google Scholar]
- 65. Carlson JO, Gatz M, Pedersen NL, et al. Antemortem prediction of Braak stage. J Neuropathol Exp Neurol. 2015;74(11):1061‐1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cholerton B, Larson EB, Baker LD, et al. Neuropathologic correlates of cognition in a population‐based sample. J Alzheimers Dis. 2013;36(4):699‐709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Robinson AC, McNamee R, Davidson YS, et al. Scores obtained from a simple cognitive test of visuospatial episodic memory performed decades before death are associated with the ultimate presence of Alzheimer disease pathology. Dement Geriatr Cogn Disord. 2018;45(1–2):79‐90. [DOI] [PubMed] [Google Scholar]
- 68. Fitzgerald PB, Laird AR, Maller J. A meta‐analytic study of changes in brain activation in depression. Hum Brain Mapp. 2008;29(6):683‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Koolschijn PC, van Haren NE, Lensvelt‐Mulders GJ. Brain volume abnormalities in major depressive disorder: a meta‐analysis of magnetic resonance imaging studies. Hum Brain Mapp. 2009;30(11):3719‐3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rajkowska G, Miguel‐Hidalgo JJ, Wei J. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45(9):1085‐1098. [DOI] [PubMed] [Google Scholar]
- 71. Hercher C, Turecki G, Mechawar N. Through the looking glass: examining neuroanatomical evidence for cellular alterations in major depression. J Psychiatr Res. 2009;43(11):947‐961. [DOI] [PubMed] [Google Scholar]
- 72. Price JL, Drevets WC. Neurocircuitry of mood disorders. Neuropsychopharmacology. 2010;35(1):192‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Braak H, Del Tredici K. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol. 2011;121(2):171‐181. [DOI] [PubMed] [Google Scholar]
- 74. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70(11):960‐969. [DOI] [PubMed] [Google Scholar]
- 75. Davidson YS, Robinson A, Prasher VP, Mann DMA. The age of onset and evolution of Braak tangle stage and Thal amyloid pathology of Alzheimer's disease in individuals with Down syndrome. Acta Neuropathol Commun. 2018;6(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Li B, Yamamori H, Tatebayashi Y, et al. Failure of neuronal maturation in Alzheimer disease dentate gyrus. J Neuropathol Exp Neurol. 2008;67(1):78‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Anacker C, Luna VM, Stevens GS, et al. Hippocampal neurogenesis confers stress resilience by inhibiting the ventral dentate gyrus. Nature. 2018;559(7712):98‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hanson ND, Owens MJ, Nemeroff CB. Depression, antidepressants, and neurogenesis: a critical reappraisal. Neuropsychopharmacology. 2011;36(13):2589‐2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material 1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.