

Summary
CRISPR‐Cas9 has proven to be highly valuable for genome editing in plants, including the model plant Physcomitrium patens. However, the fact that most of the editing events produced using the native Cas9 nuclease correspond to small insertions and deletions is a limitation.
CRISPR‐Cas9 base editors enable targeted mutation of single nucleotides in eukaryotic genomes and therefore overcome this limitation. Here, we report two programmable base‐editing systems to induce precise cytosine or adenine conversions in P. patens.
Using cytosine or adenine base editors, site‐specific single‐base mutations can be achieved with an efficiency up to 55%, without off‐target mutations. Using the APT gene as a reporter of editing, we could show that both base editors can be used in simplex or multiplex, allowing for the production of protein variants with multiple amino‐acid changes. Finally, we set up a co‐editing selection system, named selecting modification of APRT to report gene targeting (SMART), allowing up to 90% efficiency site‐specific base editing in P. patens.
These two base editors will facilitate gene functional analysis in P. patens, allowing for site‐specific editing of a given base through single sgRNA base editing or for in planta evolution of a given gene through the production of randomly mutagenised variants using multiple sgRNA base editing.
Keywords: adenine deaminase, APRT, base editing, Cas9, CRISPR, cytosine deaminase, Physcomitrella patens, Physcomitrium patens
Introduction
Modification of the sequence of a protein is a powerful approach to decipher the protein’s sequence–function relationship and understand its biological roles. In addition, production of different variants for a given gene and its corresponding protein also has practical applications and is a way to go beyond what evolution has shaped in terms of function for this protein. For a long time, intracellular protein engineering through substitution, insertion or deletion of nucleotides in its corresponding gene, was restricted to unicellular eukaryotes. One possible way to modify a gene in planta is the TILLING strategy, based on EMS‐induced mutations in the genome (Jacob et al., 2018). However, such in vivo mutagenesis is not targeted to a specific locus and, for this reason, needs first a thorough selection system to find the desired mutation in the target gene. In addition, TILLING will only very rarely allow the selection of simultaneous mutations in a given gene. Finally, such a strategy will necessitate the elimination of background mutations that could interfere with the functional analysis of the modified protein.
For a few years now, the CRISPR‐Cas9 system has been efficiently applied to induce targeted mutagenesis in plant genes. Guided by an sgRNA, the Cas9 nuclease produces a targeted DNA double‐stranded break (DSB) by site‐specific cleavage, which typically results in insertion or deletion mutations after error‐prone NHEJ DNA repair (Jiang & Doudna, 2017). In many cases these mutations correspond to loss of function due to out‐of‐frame mutations, premature terminations or aberrant splicing variants. By contrast, gene function analysis and the development of new traits of interest in crops need a more precise mutation system delivering more predictable mutations, including modification of one or more bases in a given gene. Such mutations that facilitate basic research can be achieved through different CRISPR‐derived tools, as recently reviewed (Manghwar et al., 2019; Veillet et al., 2020a), but also accelerate plant breeding (Zhang et al., 2019; Gaillochet et al., 2020). While modification of one or more bases via CRISPR‐mediated gene targeting constitutes an elegant strategy, its efficiency remains low in flowering plants (Huang & Puchta, 2019).
Recently, a strategy named Prime editing, has been developed in human cells (Anzalone et al., 2019) and then adapted to wheat and rice (Lin et al., 2020). Prime editing is based on the fusion of a Cas9 nickase to a reverse transcriptase that, guided by a Prime editing guide RNAs (pegRNAs), enables point mutations, insertions and deletions. This strategy is highly promising but still needs refinement to achieve editing that is efficient enough for routine use (Marzec & Hensel, 2020). As an alternative to gene targeting, and before the development of the promising Prime editing strategy, CRISPR‐mediated base editors (BEs) have been developed, to induce targeted base modification, first in human and murine cell lines (Komor et al., 2016) and then in rice (Li et al., 2017; Lu & Zhu, 2017). Like Prime editing, base editing is a gene editing strategy free of donor DNA and double‐stranded breaks. Two types of BEs exist, cytosine base editors (CBEs) and adenine base editors (ABEs) that correspond to a Cas9 nickase (nCas9) or a catalytically inactive Cas12a (dCas12a) (Komor et al., 2016; Li et al., 2018a) that is fused to either a cytosine‐ or an adenine‐deaminase domain, respectively. BEs generate targeted nucleotide substitutions on ssDNA in a small editing window that is accessible during the CRISPR‐mediated R‐loop formation. The action of the cytidine deaminase of the CBE complex on a cytosine can generate transition (C‐to‐T) and transversion (C‐to‐A and C‐to‐G) substitutions, however addition of a uracil DNA glycosylase inhibitor (UGI) to the CBE will permit the production mostly of transition (C‐to‐T) substitutions (Evanoff & Komor, 2019). ABEs almost exclusively result in A‐to‐G substitutions (Evanoff & Komor, 2019).
BEs using nCas9 fusions have been used in different model and crop plants including rice, wheat, maize, potato and tomato (Lu & Zhu, 2017; Shimatani et al., 2017; Zong et al., 2017; Li et al., 2018b; Bastet et al., 2019; Veillet et al., 2019a,b). These studies have shown that CBE or ABE activities can vary between plants, but also between target sites in term of editing efficiency of an effective deamination window and of occurrence of byproducts (insertions, deletions or unpredicted substitutions). In the model plant bryophyte Physcomitrium (Physcomitrella) patens, the use of CRISPR‐Cas9 or CRISPR‐Cas12a strategies has permitted efficient gene knock‐out (Nomura et al., 2016; Lopez‐Obando et al., 2016; Pu et al., 2019; Mallett et al., 2019) or gene knock‐in (Collonnier et al., 2017a; Yi & Goshima, 2020), but no base‐editing strategies have been reported so far.
Physcomitrium patens is a well recognised model to study evolutionary developmental biology questions, stem cell reprogramming, and the biology of nonvascular plants (Rensing et al., 2020). In order to expand the toolbox for gene function analysis in P. patens, we explored the possibility of editing specific bases of the genome through CBE and ABE in this model plant. For this purpose, we used the APT gene that we and other groups had previously used as a reporter of gene modification or modulation in P. patens (Trouiller et al., 2006; Holá et al., 2013; Orr et al., 2020). We demonstrate here for the first time that the CRISPR‐Cas9 deaminase systems CBE and ABE are very efficient tools for base editing, including multiplex editing, in P. patens. We characterised the respective efficiencies and deamination windows for CBE and ABE and demonstrated that they can be useful tools for gene function analysis. In addition, data gained from this study can be translated to drive directed in planta evolution of other targets. Finally, we propose here an original co‐editing selection strategy, named selecting modification of APRT to report gene targeting (SMART), based on the restoration of the APT gene function, for efficient and easy‐to‐screen base editing of any gene of interest in P. patens.
Materials and Methods
Molecular cloning
Guide RNA (sgRNA) sequences specific to the APT (Pp3c8_16590), Pp3c3_13220, Pp3c14_9040 and Pp3c17_3870 genes were chosen using the webtool CRISPOR 4.97 (Concordet & Haeussler, 2018). Expression cassettes sgRNA#5, sgRNA#7, sgRNA#21, sgRNA#23, sgRNA#24 and sgRNA#25, comprising the promoter of the P. patens U6 snRNA (Collonnier et al., 2017a), the 5′‐G‐N(19)‐3′ guide sequences targeting the APT gene and the tracrRNA scaffold were synthesised by Twist Bioscience (San Francisco, California, USA; Supporting Information Table S1). The sgRNA expression cassette sgRNAPp3c14, based on the same backbone but targeting the Pp3c14_9040 gene was synthesised by Twist Bioscience. sgRNA#5 and sgRNA#7 were subcloned into the pDONR207 vector by GatewayTM BP reaction (Invitrogen) to give psgRNA#5 and psgRNA#7. sgRNA#21, sgRNA#23, sgRNA#24, sgRNA#25 and sgRNAPp3c14 were cloned into the pDONR207‐neomycin resistance (NeoR) vector using a GatewayTM BP reaction (Invitrogen) to give psgRNA#21, psgRNA#23, psgRNA#24, psgRNA#25 and psgRNAPp3c14. pDONR207‐NeoR was obtained by cloning the 35S::neoR fragment (SmaI–ApaI, 1824 pb) from pBNRF (Schaefer et al., 2010) into pDONR207. The sgRNAPp3c3, sgRNAPp3c17 and sgRNArestor expression cassettes, targeting the Pp3c3_13220 gene, the Pp3c17_3870 gene and a mutated version of the APT gene respectively, were synthesised and cloned in the pTwist Amp vector by Twist Bioscience to give psgRNAPp3c3 spgRNAPp3c17and psgRNArestor. psgRNA#2 containing the expression cassette sgRNA#2 has been described previously (Collonnier et al., 2017a). All the expression cassettes used in this study are described in Table S1.
For the CBE system we used a CRISPR‐nCas9 cytosine deaminase consisting of a fusion of nCas9 (D10A) to the Petromyzon marinus cytosine deaminase (PmCDA1). The pnCas9‐CBE1 vector expressing this fusion enzyme is based on the pDicAID_nCas9‐PmCDA_NptII_Della vector (Shimatani et al., 2017), from which the sgRNA expression cassette targeting the tomato DELLA gene was dropped out (AanI digest). For The ABE system we used a CRISPR‐nCas9‐adenine deaminase consisting of a heterodimer of a wild‐type bacterial tRNA adenosine deaminase (TadA) and a mutated version (TadA*), fused to nCas9 (Gaudelli et al., 2017). For this purpose, using Invitrogen Platinum SuperFi DNA polymerase (Thermo Fisher Scientific), we PCR amplified the ABE7.10‐nCas9 gene fusion from pCMV‐ABE7.10 (Addgene plasmid #102919) with AttB1 and AttB2 flanking sequences using the ABE7.10‐AttB1 and ABE7.10‐AttB2 primers. The PCR fragment was cloned into pDONR207 using the GatewayTM BP reaction and then subcloned into pBS‐TPp‐B (Thévenin et al., 2012) using the GatewayTM LR reaction (Invitrogen) to give the pnCas9‐ABE1 plasmid, in which ABE7.10 is flanked by the rice Actin 1 promoter and CaMVter terminator. The pAct‐Cas9 plasmid used in this study has been described previously (Collonnier et al., 2017a). Sequences of the plasmids used in this study are listed in Table S2.
Moss culture and transformation
P. patens wild‐type Gransden strain was propagated vegetatively as previously described (Cove et al., 2009). Plants were grown on PpNH4 medium (PpNO3 medium supplemented with 2.7 mM NH4‐tartrate) in growth chambers set at 60% humidity with 16 h of light (quantum irradiance of 80 μmol m−2 s−1) at 24°C and 8 h of dark at 22°C. Moss protoplast isolation and transfection were performed as previously described (Schaefer & Zryd, 1997). Protoplasts were transfected with a total of 20 µg of circular DNA divided as follow: 8 µg of the pAct‐Cas9, pDIC‐AID‐APTgRNA#21, pnCas9‐CBE1 or pnCas9‐ABE1 plasmids and a mix of 12 µg of sgRNA plasmids. Regenerating protoplasts were spread on cellophane disks on PpNH4 medium supplemented with 0.33 M mannitol for 1 wk. Plants on cellophane disks were then selected either on PpNH4 supplemented with 50 mg l−1 G418 (Duchefa) to select clones that were transiently transfected (Lopez‐Obando et al., 2016) or directly on PpNH4 supplemented with 10 μM 2‐FA (Fluorochem) to select clones that were mutated at the APT locus (Collonnier et al., 2017a). For the experiment consisting of the restoration of the APRT activity of the apt mutant ABEv#1, protoplasts isolated from this mutant were transfected with 10 µg of each of the pnCas9‐CBE1 and psgRNArestor plasmids. Regenerating protoplasts were spread on cellophane disks on PpNH4 medium supplemented with 0.33 M mannitol and 1.75 mM adenine (Sigma A8626) for 1 wk and then transferred onto PpNH4 supplemented with 1.75 mM adenine for 3 wk for selection of clones in which APRT function was restored.
PCR and sequence analysis of the edited plants
For PCR analysis, genomic DNA was extracted from 50 mg of fresh tissue as previously described (Lopez‐Obando et al., 2016). The quality of the DNA samples was controlled using primers targeting the P. patens RAD51‐1 gene, PpRAD51‐1#6 and PpRAD51‐1#7. Molecular analysis was based on Sanger sequencing (Genoscreen, Lille, France) of PCR fragments using primers surrounding the targeted locus. For edited plants obtained using a single sgRNA, PpAPT#25 and PpAPT#5 were used for APT, Pp3c3#1 and Pp3c3#2 for the Pp3c3_13220 locus, Pp3c14#1 and Pp3c14#2 for the Pp3c14_9040 locus, Pp3c17#1 and Pp3c17#2 for the Pp3c1783870 locus. For edited plants obtained using multiple sgRNAs, molecular analysis was carried out using primers PpAPT#8 and PpAPT#10 primers for the sgRNA#7 locus, primers PpAPT#60 and PpAPT#61 for the sgRNA#5 and primers PpAPT#25 and PpAPT#5 for sgRNA#2 and sgRNA#21 loci. PCR primers used in this study are listed in Table S3.
Structural analysis of mutations in P. patens APRT
Nine APRT templates were selected with Modeller 9.18 (Webb & Sali, 2017), based on sequence identity (>20%) from: Escherichia coli (PDB:2DY0), Saccharomyces cerevisiae (PDB:1G2Q), Giardia intestinalis (PDB:1L1Q), Thermoanaerobacter pseudethanolicus (PDB:4LZA), Rhodothermus marinus (PDB:4M0K), Yersinia pseudotuberculosis (PDB:4MB6) and Homo sapiens (PDB:4X45). Multiple alignment was used to develop high quality models and the best model was chosen using the discrete optimised protein energy (DOPE) method (Shen & Sali, 2006) and/or the GA341 method (John & Sali, 2003; Melo et al., 2009). Optimisation of the model was achieved using energy minimisation protocols available at Yasara software (Elmar et al., 2010). The visualisation of the multiple alignment with the secondary structure was made using the ESPript3 server (Robert & Gouet, 2014).
In vivo measurements of chlorophyll fluorescence
Here, 10‐d‐old plants grown on PpNO3 medium were probed for chlorophyll fluorescence with the modular version of the Dual PAM‐100 fluorometer (Walz). Plants were dark adapted for 40 min and then induction curve analyses were performed using 850 µmol of photons m−2 s−1 red actinic light for 8 min followed by 8 min of dark recovery. Chlorophyll fluorescence was measured in plants dark adapted for 30 min that were then exposed to actinic light of 800 μmol of photons m−2 s−1 for 8 min. Fm and Fm′ are the fluorescence values after exposure to saturating pulses (6000 μmol of photons m−2 s−1, duration 600 ms) in, respectively, dark adapted plants and plants exposed to actinic light. Non‐photochemical quenching (NPQ) was calculated as Fm/Fm′ − 1 (Klughammer & Schreiber, 1994).
Results
Efficient base editing in P. patens via RNA‐guided cytosine or adenine deaminases
The APRT enzyme is a member of the Type I phosporibosyltransferase family and is involved in the nucleotide salvage pathway, by which organisms, including plants (Ashihara et al., 2018), convert adenine to adenosine monophosphate (AMP). APRT enzyme, encoded by the APT gene, is also able to convert some adenine analogues into toxic compounds and this has been used for efficient selection of APRT‐deficient mutants in many organisms (Taylor et al., 1985). For this reason, we hypothesised that base modifications of the P. patens APT gene (Fig. 1a) could be easily monitored by selection on 2‐FA in order to quickly assess the efficiency of base editing (Fig. S1). To determine whether programmable BEs could catalyse site‐specific base editing in the genome of P. patens, we used both a CBE based on the cytosine deaminase from P. marinus (PmCDA1) and an ABE based on tRNA adenosine deaminase from E. coli (TadA) (Fig. 1b). We selected eight different sgRNAs targeting the APT reporter gene that contained cytosine or adenine residues in the predicted editing windows (Fig. 1a).
Fig. 1.
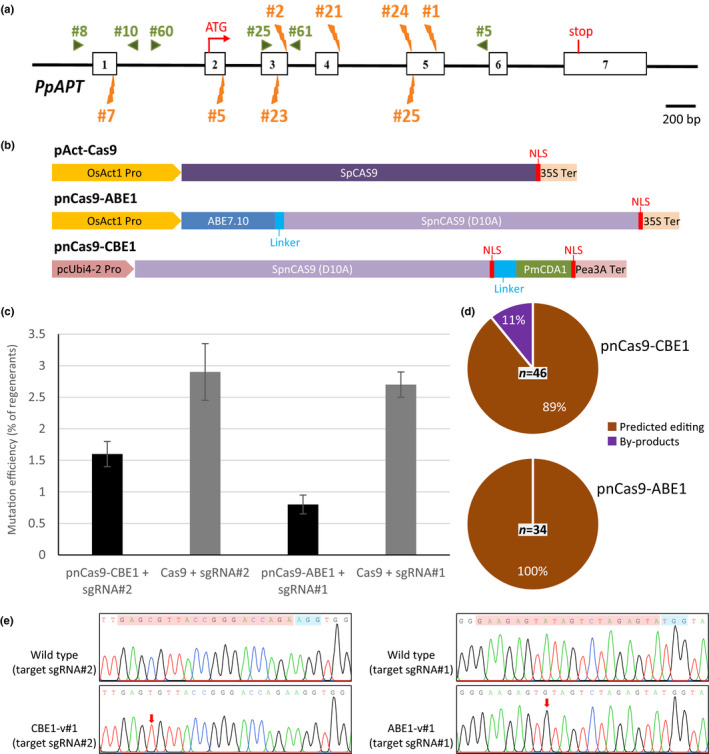
Relative efficiency of mutagenesis and the nature of editing by CBE and ABE in Physcomitrium patens. (a) Structure of the PpAPT gene and sgRNAs positions. Boxes in white represent the exons and black lines represent the introns. The eight sgRNAs positions are indicated in orange, at the top for sgRNAs that target forward strand, and at the bottom for sgRNA that target reverse strand. Green arrows represent the primers used for PCR and sequencing. (b) Schematic representation of the native Cas9 and the two base editors. Linker sequences are in light blue, NLS sequences are in red. (c) Mutation rates using pnCas9‐CBE1 or pnCas9‐ABE1 (in black) vs active Cas9 (in grey). (d) Nature of the base editing was characterised by sequence analysis of 2FA‐resistant plants. (e) Sequence chromatograms from wild‐type and CBE or ABE edited clones. Target sequence (in red) and PAM (in blue) are highlighted in the wild‐type (WT) sequence; red arrows point to the positions with edited base. Primers used for amplification and Sanger sequencing can be found in the Materials and Methods section. Number of analysed plants is indicated.
First, in order to validate the CBE and ABE constructs, we compared their respective editing efficiencies to that of the native Cas9 (pAct‐Cas9). psgRNA#1 was transfected with pnCas9‐ABE1 (for ABE) or pAct‐Cas9 (for native Cas9) and psgRNA#2 was transfected with pnCas9‐CBE1 (for CBE) or pAct‐Cas9 (for native Cas9) respectively, in P. patens protoplasts. Regenerating protoplasts were transferred onto a medium containing 2‐FA in order to detect plants that had been mutated at the APT locus. The relative efficiencies of APT mutagenesis, estimated by dividing the number of 2‐FA‐resistant plants by the number of initially regenerating plants, were 0.8% for ABE using sgRNA#1 and 1.6% for CBE using sgRNA#2 (Table S4; Fig. 1c). Using the same two guides, these values were, respectively, 2.7 and 2.9% when mutagenesis was performed with the native Cas9 system (Fig. 1c). Analysis of the type of mutations obtained with the two different BE strategies showed that a majority of mutant plants obtained with the CBE strategy corresponded to precise base‐editing events (89%), and the remaining mutants corresponded to short insertions or deletions (hereafter called byproducts) (Fig. 1d,e). For the ABE strategy, 100% of the mutant plants corresponded to precise base editing (solely A‐to‐G substitution, Fig. 1d,e). This showed that CBE and ABE can be used to precisely modify cytosine or adenine bases in P. patens.
Multiplex base editing is efficient in P. patens
In order to evaluate the possibility of multiplex editing using the BE systems we co‐transfected P. patens protoplasts with plasmids psgRNA#2, psgRNA#5, psgRNA#7, psgRNA#21 (Fig. 1) and pnCas9‐CBE1 plasmids (for the CBE system), and with plasmids psgRNA#1, psgRNA#21, psgRNA#23 and pnCas9‐ABE1 plasmids (for the ABE system) (Fig. 1). As before, regenerating protoplasts were grown on medium containing 2‐FA, and plants that survived (apt mutants) were analysed. Sequence analysis of the apt mutants showed that, under these conditions, the relative efficiencies of mutagenesis (2.8% for CBE and 0.7% for ABE, Table S4) were comparable with those observed with the simplex strategy (1.6% for CBE and 0.8% for ABE, Table S4). For CBE, predictability of base editing was decreased, compared with the simplex strategy, as 47% (89% for the simplex) of the mutations corresponded to precise editing of a cytosine (Figs 2a, S2). Analysis of the mutations showed that the decrease observed was due to byproducts corresponding to small indels (Fig. S2), but also to the occurrence of deletions between the sgRNAs used for multiplexing. As expected, these deletions always involved sgRNAs that were in opposite orientation on the DNA (Figs 1a, S2), as nicking of both DNA strands by a pair of Cas9 nickases is known to lead to site‐specific DSBs and NHEJ, and by contrast nicks on the same DNA strand were predominantly repaired through the high‐fidelity base excision repair pathway (BER) (Ran et al., 2013). Analysis of the mutations generated by the CBE system at the different targets in a given plant showed that precise multiplex base editing could be achieved, as 40% of the precisely edited plants (C substitutions with no byproducts and no guide‐to‐guide deletions, n = 30) were modified at the four targeted loci (Fig. 2b,c). For ABE multiplexing, the predictability of base editing was comparable with the simplex strategy, as 98% (100% for the simplex) of the mutations corresponded with the precise editing of an adenine (Figs 2a, S2). In addition, 14% of the precisely edited plants (A substitutions with no byproduct) (n = 43) showed concomitant mutations at the three targeted loci (Fig. 2b,d). These data showed that, when a cell is subjected to the CBE or ABE systems with multiple sgRNAs, concomitant editing of the targeted loci is possible, making multiplexing a powerful tool in P. patens.
Fig. 2.
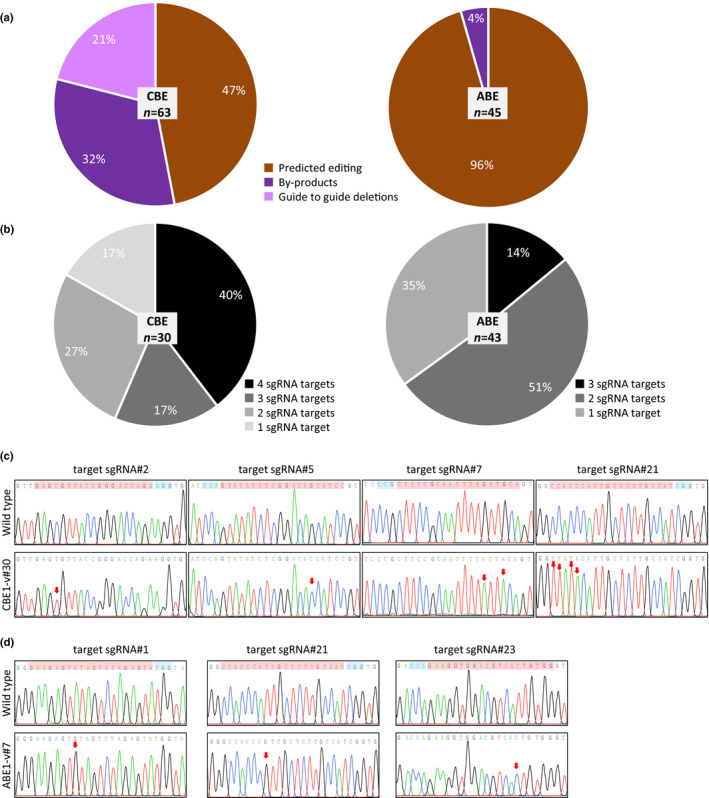
Predictability and efficiency of multiplex editing by CBE and ABE in Physcomitrium patens. (a) Frequencies of predicted base editing, byproducts and guide‐to‐guide deletions using pnCas9‐CBE1 with four sgRNAs or pnCas9‐ABE1 with three sgRNAs. (b) Percentage of plants where the APT gene has been modified precisely (no byproducts or guide‐to‐guide deletions) at multiple sgRNA targets concomitantly. Primers used for amplification and Sanger sequencing can be found in the Materials and Methods section. Number of analysed plants is indicated. (c, d) Sequence chromatograms from wild‐type and CBE or ABE edited clones. Target sequence (in red) and PAM (in blue) are highlighted in the WT sequence; red arrows point to the positions with edited base.
Characteristics of BE and nature of the substitutions
First, in order to check that selection of the edited plants on 2‐FA did not create a bias in the type of mutations that could be observed using BE, we aimed at analysing plants that were transfected with the CBE system without the a priori knowledge that editing would result in an alteration of APRT activity. For this purpose, protoplasts were transfected with the CBE system and sgRNA#2 or sgRNA#21. The regenerating protoplasts were transferred onto a medium containing the antibiotic G418 in order to select clones that expressed the transfected plasmids that contained a NeoR gene cassette (Table S5). We observed that 34.8% for sgRNA2 and 52.3% for sgRNA#21 of the G418 resistant transfected clones were mutated at the APT locus (Fig. 3). Of these mutated clones, 32.6% for sgRNA#2 and 20.9% for sgRNA#21 corresponded to precise base‐edited plants, the other mutants corresponded to deletions (Fig. 3). Interestingly, a small fraction of the G418‐resistant clones (2.2% for sgRNA#2 and 4.7% for sgRNA#21, Fig. 3) corresponded to chimeric clones composed of a mixture of wild‐type and mutated cells (Fig. S3). Such chimeric clones were not observed using direct 2‐FA selection. This suggests that, even if the BE system is preferentially active very early after protoplast transfection, it can still be active after multiple divisions of the originally transfected cell thanks to episomal replication of the vector (Muren et al., 2009). Even if illegitimate integration of a nonhomologous supercoiled plasmid is low in P. patens (Schaefer & Zryd, 1997), episomal persistence of the vector in the clones in the presence of G418 could, in theory, lead to stable integration in the genome. In order to check for the level of unexpected integration of the ABE or CBE vectors in the selected plants, we tested the sensitivity of 288 ABE‐ or CBE‐transfected clones to G418. We could observe that sensitivity to G418 was restored in most of the clones as only one clone out of 480 (0.21%) showed resistance to G418 (Fig. S4). All the edited clones presented in this study were G418 sensitive. Finally, in order to test whether the different mutations observed in the APT locus would have an effect on the activity of the APRT enzyme we sequenced the APT gene in the 132 G418‐resistant (transfected) clones obtained with sgRNA#2 or sgRNA#21 and placed them on a medium containing 2‐FA. Under these conditions, 100% of the clones containing a mutation (77 out of the 132 G418‐resistant clones) were resistant to 2‐FA (Table S6). This confirmed our hypothesis that the APRT enzyme is very constrained in terms of amino‐acid changes and is therefore very sensitive to mutations. Therefore 2‐FA screening is very effective in identifying any modification in the APT gene.
Fig. 3.
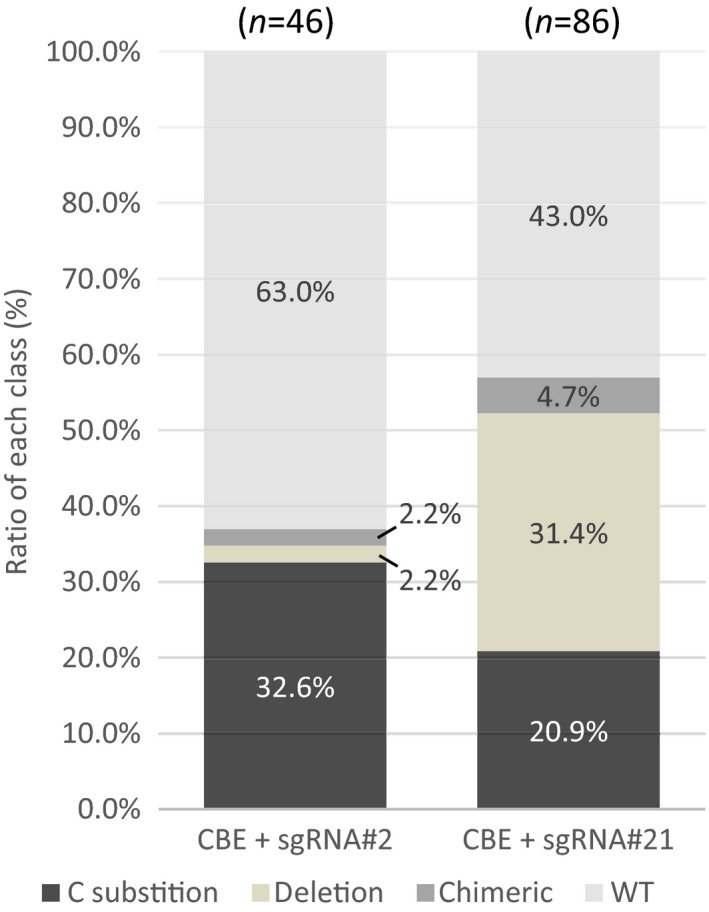
Efficiency of mutagenesis and nature of editing by CBE in Physcomitrium patens. Efficiency of mutagenesis using pnCas9‐CBE1 for sgRNA#2 and sgRNA#21 observed among transient transformants (G418 selection). Efficiency and nature of the mutagenesis were assessed by sequencing analysis using PpAPT#25/PpAPT#5 primers on G418‐resistant plants. Number of analysed plants is indicated.
Based on the fact that a majority, if not all, of the mutations induced by CBE or ABE lead to a nonfunctional APRT, we decided to pool the different events of editing, obtained through selection on G418 or directly on 2‐FA, in order to analyse the nature of the CBE or ABE editing products. For CBE, the analysis of the nature of the cytosines modifications (n = 693 modified cytosines of 2005 analysed) showed that a majority corresponded to C‐to‐T substitutions (75.8%), but some C‐to‐G (18.6%) or C‐to‐A (5.8%) substitutions were also observed (Fig. 4a). Analysis of the positions of the cytosines that could be substituted in eight different sgRNAs targets showed that cytosines present in the 5‐bp editing window (positions −19 to −15 from the PAM) previously described (Nishida et al., 2016), are efficiently edited also in P. patens. However, a significant number of C substitutions (17%) could also be observed outside this editing window, including a cytosine in position −21 from the PAM that was outside the 20‐bp target sequence of sgRNA (Fig. 4c; Table S7). For target sequences presenting more than one C in the editing window, as is the case for sgRNA#21, at least two Cs and up to five Cs could be modified simultaneously (Fig. S3). In addition, important variation of the efficiency and nature of the substitutions could be observed for a cytosine in a given position from one targeted locus to another (Figs 4b, S5a, S5), confirming the influence of the environment of the cytosine on the efficiency and nature of its modification. Finally, we observed that the existence of multiple Cs in the editing window, as is the case for sgRNA#21, favoured the occurrence of deletion byproducts at the targeted locus, that in this case corresponded to 31.4% of the mutations (Fig. 3a). For ABE, analysis of the nature of the adenine modifications (n = 156 modified adenines) showed that, as observed in other organisms, all the modifications corresponded to A‐to‐G substitutions (Figs S5b, S7). It has been shown recently in animal cells that, in addition to converting adenine to guanine, ABEs could also convert cytosines that are in a narrow editing window (positions −16 to −14) and in a confined ‘TCN’ nucleic‐acid sequence context, into guanine or thymine (Kim et al., 2019). In order to check whether this unpredicted activity was present in P. patens, we designed two sgRNAs, sgRNA#24 and sgRNA#25 containing a ‘TCN’ sequence context with the C present in the narrow editing window (Fig. S7). Analysis of 75 plants that were modified by the ABE editor at the sgRNA#24 or #25 targets showed efficient substitution of the A in the editing window with no co‐substitution of the C in the ‘TCN’ sequence context (Fig. S7), suggesting that the phenomenon observed in animal cells could not be generalised. Concerning the positions of the adenines that could be substituted, analysis in five different sgRNAs targets showed that adenines present in the 4‐bp editing window (positions −17 to −14) previously reported (Eid et al., 2018), were efficiently edited also in P. patens. Interestingly, exclusive substitution of an A in position −13 could also be observed (sgRNA#1 and sgRNA#24, Figs S5b,S7). Finally, for target sequences presenting more than one A in the editing window, as was the case for sgRNA#23 (Fig. S5b), at least two As could be modified simultaneously.
Fig. 4.
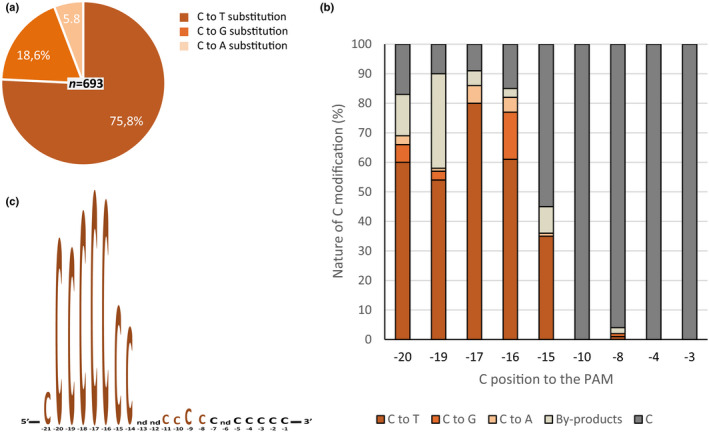
Type of cytosine substitutions and importance of the position of the cytosine. (a) Nature of cytosine editing using pnCas9‐CBE1 with the different sgRNAs used in this study was characterised (693 cytosine substitutions analysed). (b) Nature of cytosine editing using CBE with gRNA#21 for each cytosines present in this 20‐bp target (n = 100 Physcomitrium patens plants). (c) Frequency of substitution for cytosines at each position of the eight sgRNAs used in this study (2005 cytosines analysed, 693 substituted), only cytosines in positions −13, −12 and −6 were not present (for number of cytosines at each position see Supporting Information Table S7). Primers used for amplification and Sanger sequencing can be found in the Materials and Methods section.
Predicted potential off‐target sites are not affected by base‐editing activity in P. patens
The sgRNAs used in this study were designed to minimise potential off‐target cleavage in the P. patens genome (Phytozome 3.1) using the Crispor software package (Concordet & Haeussler, 2018). We focused our analysis on two sgRNAs (sgRNA#1 and sgRNA#2) for which no perfect 20‐bp matches were found, but potential off‐target sequences presenting three to six mismatches were identified: nine for sgRNA#1 and four for sgRNA#2 (Table S8). All these potential off‐target loci were amplified with surrounding primers and sequenced in 39 clones transformed with pnCas9‐ABE1 and psgRNA#1, and 39 clones transformed with pnCas9‐CBE1 and psgRNA#2 that were all previously identified as mutated at the APT locus. No mutation could be detected in the potential off‐target sequences for any of the tested clones, suggesting that Cas9‐dependent predicted off‐target activity may be low in P. patens.
BE allows the generation of multiple variants of the APRT enzyme
Altogether, use of CBE and ABE strategies with multiple sgRNAs allowed the selection of 38 plants containing different variants of the APRT enzyme and showing combinations of one to four amino‐acid modifications (Table S9; Fig. S8). To understand how CBE‐ and ABE‐induced substitutions were associated with APRT loss of function, we focused our analysis on apt mutants showing one amino‐acid change, and investigated the role of these single mutations on the APRT 3D structure. Because of the high level of identity of the P. patens APRT to the APRT proteins from different kingdoms (45% and 42% to E. coli and human APRT proteins, respectively) and the availability of several APRT 3D structures, we used homology modelling to build a P. patens APRT 3D model. All the sequences were aligned using Modeller 9.18 (Fig. S8). Based on multiple alignments of the APRT sequences, a model was created for the P. patens APRT (Fig. 5). The P. patens APRT monomer model is a single‐domain structure composed of eight α‐helices and nine β‐strands that can be divided further into the ‘hood’ (residues 1–39) responsible primarily for base recognition and the definition of substrate specificity, the ‘flexible loop’ (residues 98–115) and the core (residues 40–184) (Fig. S8) that corresponds to the conserved type I PRTase fold (Phillips et al., 1999; Shi et al., 2001).
Fig. 5.
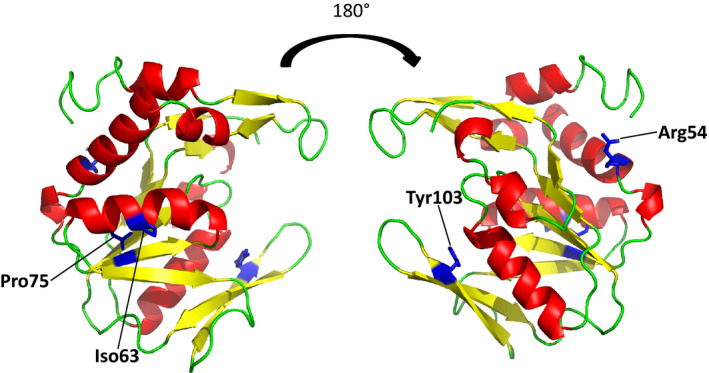
Single amino‐acid modifications by CBE or ABE in the Physcomitrium patens APRT. View of the Physcomitrium patens APRT 3D model. The APRT protein consists of eight α‐helices (in red) and nine β‐strands (in yellow) which can be divided further into the hood (residues 1–39), the flexible loop (residues 98–115; β5 and β6) and the core (residues 40–184). Amino acids that could be modified as single substitutions using CBE or ABE are indicated.
The four mutated residues (single mutations) associated with APRT loss of function were reported on the APRT 3D model, where they were all in important structural or functional domains (Figs 5,S9). Arginine 54 (Arg54) at the end of the helix H3, isoleucine 63 (Ile63) in β‐strand S3, proline 75 (Pro75) in the helix H4, are all located in the core subdomain, and tyrosine 103 (Tyr103) is at the end of β‐strand S5. Arg54 is surrounded by residues implicated in intraside‐chain polar contact (Tyr55–Glu154–Gln58/Asp6–Tyr176) and the R54C mutation could impair the formation of this interaction, and therefore the correct folding of the protein. It is highly likely that the mutation at position 75 could disturb the helix H4 and lead to abnormal protein folding. Tyr103 is highly conserved in the eight APRT enzymes of the different kingdoms, and is thought to be involved in the conformation of the flexible loop that is proposed to close the active site during catalysis (Shi et al., 2002).
Efficient base editing of a gene of interest in P. patens using CBE and ABE
We have shown before that APT‐base‐edited plants could be isolated after selection on G418 of CBE‐transfected protoplasts. In order to test the portability of this BE strategy based on selection on G418, for genes for which the mutations cannot be positively selected we decided to target three genes located on three different chromosomes in the P. patens genome (Fig. S10) and encoding respectively, violaxanthin de‐epoxidase (VDE) protein (Pp3c3_13220), a C2H2 zinc finger transcription factor (Pp3c14_9040), and the tetratricopeptide‐repeat protein 39C (TTC39C) protein (Pp3c17_3870). Protoplasts were transfected with the pnCas9‐CBE1 plasmid and either of the sgRNAs expression vectors targeting the three genes. The regenerating protoplasts were transferred onto a medium containing the antibiotic G418 in order to select clones expressing the transfected plasmids. Sequence analysis of the targeted genes showed that 55% for sgRNAPp3c3, 23% for sgRNAPp3c14 and 51% for sgRNAPp3c17 of the transfected (G418 resistant) clones showed a cytosine edited at the targeted locus (Fig. 6a). As observed for the sgRNAs targeting the APT gene, the ratio of C‐to‐T, C‐to‐A and C‐to‐G varied from one sgRNA to the other, but with a majority of C‐to‐T substitutions (Figs 6b, S6). Analysis of the mutations induced at the three different loci showed that editing of the cytosines present in the editing window led to the production of different amino‐acid changes and premature stop codons for the three different proteins.
Fig. 6.
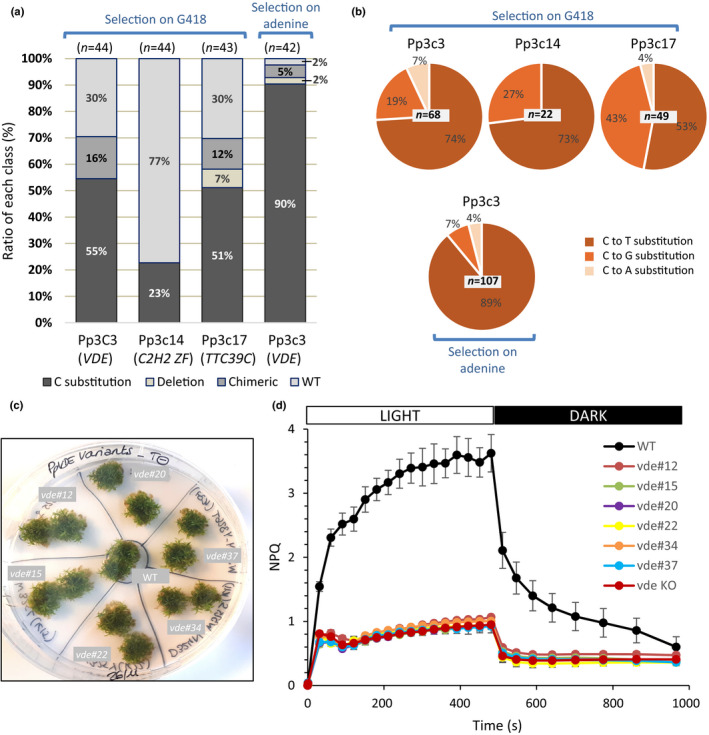
Efficiency and nature of editing by cytidine deaminase on Physcomitrium patens genes of interest and consequences on NPQ activity for the VDE (Pp3c3_13220) variants. (a) Efficiency of CBE editing of Pp3c3_13220 (VDE), Pp3c14_9040 (C2H2 ZF) and Pp3c17_3870 (TTC39C) via transfection of wild‐type (WT) protoplasts and selection of transient transformants on G418 and, for Pp3c3_13220 via transfection of ABEv#1 protoplasts and co‐selection on adenine. Editing efficiency was assessed by sequence analysis performed on G418‐resistant plants and also on adenine‐resistant plants for Pp3c3_13220. Primers used for amplification and Sanger sequencing can be found in the Materials and Methods section. Number of analysed plants is indicated. (b) Nature of editing for Pp3c3_13220, Pp3c17_3870 and Pp3c14_9040 loci was assessed on 68, 22 and 49 C substitutions for selection on G418, respectively, and 107 for selection on adenine for locus Pp3c3_13220. (c) Phenotype of 20‐d‐old wild‐type and VDE variant plants. (d) NPQ induction and relaxation kinetics of the wild‐type and VDE base‐edited variants. The wild‐type is compared with clones mutated for the VDE gene by cytosine base editing (Supporting Information Table S10). A null mutant of VDE (vde KO; Pinnola et al, 2013) was used as a control for VDE inactivation. Data are mean values of plants grown on three independent plates SD.
The genomic region targeted in the P. patens VDE gene has a key role in regulation of photosynthesis. When plants are exposed to excess light, VDE catalyses the conversion of the carotenoid violaxanthin into another one, zeaxanthin (Baroli et al., 2000). The VDE‐dependent zeaxanthin synthesis contributes to the dissipation of excess energy through a mechanism called NPQ. The assessment of NPQ levels, calculated from the measurement of chlorophyll a fluorescence, therefore allows inferring VDE activity in vivo (Pinnola et al., 2013). The availability of structural data on VDE allows the identification of key residues for its activity (Arnoux et al., 2009) such as amino acids involved in violaxanthin binding that have been shown to be essential for protein activity in Arabidopsis VDE thanks to in vitro assays (Saga et al., 2010) (Fig. S11). Base editing of the three cytosines present in this catalytic region led to the production of six variants. Variant vde#20 contains a premature stop codon leading to a truncated VDE, the other variants are modified on one or two of amino acids D177, W178 and Y179 (Table S10; Fig. S11). All vde‐edited plants showed an impaired NPQ response, with a phenotype similar to vde KO plants (Fig. 6c,d). These results confirmed on one side the essential in vivo role of the targeted amino acids in VDE activity in P. patens, and more importantly in this context, provided an in vivo demonstration of the specific alteration of protein activity using gene editing of selected amino acids (Fig. 6).
These data showed that selection of transfected clones on G418 can be used to detect base‐editing events in the three targeted genes of this study leading to efficiencies ranging from 23% to 55%. However, it must be noted that 12–16% of the edited clones were chimeric (Fig. 6a).
The SMART strategy for efficient base co‐editing of a gene of interest in P. patens
In order to increase the efficiency of selection for base‐edited clones and reduce the risk of chimerism we used the APT gene as a reporter of editing efficiency. Because the apt mutants show a developmental phenotype (decreased number of gametophores, see Fig. S8b) that could potentially interfere with phenotyping of a mutant in a given gene of interest, we could not use a strategy based on co‐editing of the wild‐type APT gene and the gene of interest. For this reason, we based our strategy on the reversion of an existing apt mutation toward the wild‐type allele of the APT gene. Co‐editing of this apt mutation and of a gene of interest should permit the selection of individuals mutated in the gene of interest but wild‐type for the rest of their genome. For this purpose, we took advantage of the hypersensitivity of the apt mutants, compared with the wild‐type, to the substrate of APRT, the adenine (Figs S1, S8b). We selected the apt mutant ABEv#1 obtained in this study (Fig. S8b) and tried to suppress the sensitivity to adenine in this mutant by reverting the initial mutation. In ABEv#1, the Y103C substitution that confers resistance to 2‐FA and hypersensitivity to adenine, can be restored to the wild‐type sequence via a C103Y substitution. We designed an sgRNA, sgRNArestor, that through a C‐to‐T base editing with the CBE system could cause this mutation. This reversion should lead to clones that are sensitive to 2‐FA but resistant to adenine (Figs 7,S12).
Fig. 7.
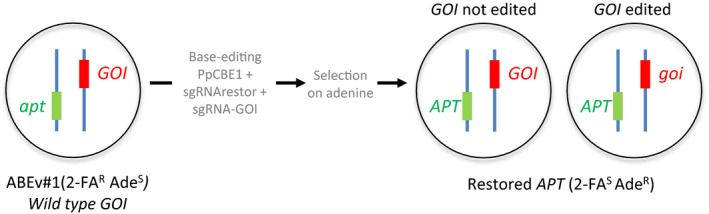
The SMART strategy for efficient base co‐editing of a gene of interest in Physcomitrium patens. Principle of selection of base‐edited events using adenine selection. The ABEv#1 mutant is co‐transfected with the CBE system and two different sgRNAs, sgRNArestor and a sgRNA targeting a gene of interest (GOI). Base editing at the apt locus with sgRNArestor can restore APRT function and confer resistance to adenine. Adenine‐resistant clones can be co‐edited or not at the GOI locus.
Based on this, protoplasts from the ABEv#1 mutant were co‐transfected with the CBE system and two different sgRNAs, sgRNArestor and sgRNAPp3c3 used previously to target the VDE gene. The regenerating protoplasts were transferred onto medium containing adenine in order to select clones of the ABEv#1 mutant that were edited and in which function of the APRT enzyme was restored. Sequence analysis of the APT gene in these clones (Table S11) showed that sgRNArestor can efficiently restore a wild‐type APRT activity by reverting the Y103C mutation to C103Y. This reversion was sufficient to restore a perfect wild‐type APRT sequence (38% of the clones), or it could be accompanied by the replacement of the glutamic acid in position 102 by an aspartic acid, a lysine or a glutamine. Interestingly, restoration of a wild‐type APRT activity could also be obtained by replacement of the cysteine in position 103 by a phenylalanine (C103F) accompanied or not by the replacement of the glutamic acid in position 102 by an aspartic acid. Sequence analysis of the VDE gene show that its concomitant editing with the APT gene was very efficient (Table S11; Fig. 6a). Using this strategy of selection on adenine for co‐editing of the gene of interest and of the APT reporter gene, the efficiency of base editing of the VDE gene reached 90%, compared with the 55% efficiency observed using selection on G418, and furthermore increased the number of VDE variants. In addition, the number of chimeric clones was decreased more than three‐fold (Fig. 6a), making this system of selection, that we named SMART, a very powerful tool for base editing of genes of interest in P. patens.
Discussion
A wide range of gene function analysis tools is available for the model plant P. patens, such as gene replacement through homologous recombination, gene knock‐down through RNA silencing or gene knock‐out through CRISPR‐Cas (Rensing et al., 2020). Here, we adapted the plant CBE and ABE systems to efficiently and specifically achieve targeted modification of single or multiple cytosines and adenines in P. patens. We showed that CBE and ABE strategies are efficient in P. patens and deciphered their different characteristics and outputs.
The results presented here show that, in P. patens, the PmCDA1‐based CBE strategy allows substitution of one or more cytosines in an editing window slightly larger than the one previously described (Nishida et al., 2016) including nucleotides −20 to −14 from the PAM. The majority of the modifications corresponded to C‐to‐T substitutions but, depending on the sgRNA used, the ratio of edited plants showing indel mutations at the targeted site could reach almost 50%. This is in line with what can be observed in rice, for which the different CBE strategies gave rise to 22% to 71% of plants showing indel mutations (Li et al., 2017; Ren et al., 2018). The observed byproducts occurred in the editing window, and were more frequent when the editing window contained more than one cytosine. These are likely to be due to the unfaithful BER of the uracil through nonhomologous end joining. As previously observed in other plants or animal cells, the TadA‐based ABE strategy allows efficient substitution of one or more adenines present in the predicted editing window by guanines with very few byproducts, which were observed only when multiple nearby sgRNAs were used at the same time. These byproducts are close to the predicted cutting site of the nickase, and are probably due to Cas9 nickase activity. For ABE, only very few byproducts could be found with more than 98% of the modifications corresponding to A‐to‐G substitutions. In animal cells, ABE has been shown to be able to convert cytosines that are in a confined ‘TCN’ nucleic‐acid sequence context into guanine or thymine (Lee et al., 2018; Kim et al., 2019). We could not detect this additional activity in P. patens, reinforcing the high level of precision of the ABE strategy in this organism.
Because BE strategies are based on dead or nickase Cas9, and as such do not produce double‐stranded breaks, a theoretical major advantage of BE over classical editing via Cas9 is the recovery of a more precise edited product with few or no off‐targets. Low levels of gRNA‐dependent off‐target DNA base‐editing activity for CBE or ABE editors have been shown in animals (for a review see Molla & Yang, 2019) and in plants such as rice or tomato (Shimatani et al., 2017; Hua et al., 2018). However, unbiased whole genome analyses (WGA) in rice, oilseed rape and mouse embryos (Jin et al., 2019; Zuo et al., 2019; Cheng et al., 2021), have shown that CBEs derived from the rat APOBEC1 deaminase can induce substantial genome‐wide Cas9‐independent off‐target mutations. Interestingly, this gRNA‐independent off‐target DNA base‐editing activity was not observed for the ABE editor in rice (Jin et al., 2019). Such unpredicted off‐target activity of CBEs was also demonstrated for alternate cytosine deaminase domains in E. coli, and motivated the production of engineered CBEs with low levels of Cas9‐independent off‐target activity (Doman et al., 2020). We have shown in this study that the PmCDA1‐based CBE and TadA‐based ABE editors used in this study have a low or null predicted gRNA‐dependent off‐target activity. Concerning a possible unpredicted off‐target activity of these BEs, it must be noticed that no such activity could be detected in rice for the ABE used in our study (Jin et al., 2019). For CBE, if the PmCDA1‐based CBE showed only low levels of Cas9‐independent deamination in E. coli compared with other cytosine deaminases, this activity was significantly higher in mammalian cells (Doman et al., 2020). Only an unbiased WGS analysis would address the possible unpredicted off‐target activity of these BEs in P. patens and permit a conclusion on the precision of these BEs in this plant.
Using selection on G418 for CBE‐ or ABE‐transfected protoplasts we could demonstrate efficient editing of genes of interest. This allowed us to produce multiple variants of the APRT enzyme. In humans, alterations of APRT activity can lead to kidney stone disease (nephrolithiasis), and more than 40 mutations have been described to date leading to this metabolic defect (Rumsby, 2016). Some of these mutations are present in domains that were affected in our P. patens APRT variants. For instance, the common D65V mutation found in British, Icelandic and Spanish patients affected by nephrolithiasis modifies APRT helix H4, as do our variants P75I, P75L and P75R. Mimicking in the P. patens the APRT mutations causing human nephrolithiasis could be potentially informative in terms of conservation between kingdoms of APRT functions. Taking into consideration the potential sgRNAs (using an NGG PAM) present in the APT gene, 40% of the APRT amino acids could theoretically be modified using a combination of the two ABE and CBE editors used in this study. Recently, we have shown that the SpCas9 variant, SpCas9‐NG, is active in P. patens for CRISPR‐mediated gene knock‐out applications (Veillet et al., 2020b). Because this variant recognises NGN PAMs, setting up a BE strategy based on a SpnCas9‐NG variant should theoretically permit the modification of 100% of the amino acids encoded by the APT gene, reinforcing the usefulness of these ABE and CBE strategies. The great potential of these BE strategies for functional analysis was confirmed for three other genes, including the VDE gene. The latter allowed us to demonstrate in vivo the essential role of the targeted amino acids in VDE activity in P. patens. Nevertheless, analysis of the edited clones obtained after selection on G418 showed that significant numbers of edited clones were chimeric. This should be taken into consideration when P. patens protoplasts clones were selected for transient transfection of BE editors using antibiotics. The possibility of the existence of such chimeric clones in other protoplast‐based BE editor transfection systems (e.g. potato or tomato) would probably deserve attention.
Finally, by using APT as a reporter gene, we propose the SMART approach to efficiently select base editing in target genes. Using the SMART selection system, based on co‐editing of a gene of interest and of a mutated version of the APT, and selection on the APRT substrate adenine, we could both diminish the proportion of chimeric clones and increase the efficiency of precise cytosine base editing significantly, reaching an efficiency of editing of 90%. This strategy is also possible for ABE starting from an apt mutant obtained via the CBE strategy. Furthermore, as multiplex base editing has been reported in dicot and monocot plants (Shimatani et al., 2017, 2018; Hua et al., 2018) and because the APRT function is a very conserved enzymatic function in all kingdoms, the SMART strategy presented here should be applicable to different flowering plants. It would potentially be an additional tool for transgene free editing in crops, already obtained using the ALS gene for example (Veillet et al., 2019b), but would present the advantage of not including a selection step on herbicides or antibiotics.
In conclusion, we demonstrated here that CBE and ABE editors can be very useful tools for in‐depth gene function analysis in P. patens. We provide information on the nature of the edited products, windows of editing, simplex versus multiplex systems and selection strategies, which should facilitate their use in this model plant. BE editors extend the already imposing tool box for precise genome editing in P. patens, such as gene replacement through homologous recombination (Schaefer, 2001), that could be made more efficient and precise using a CRISPR‐Cas9 strategy (Collonnier et al., 2017b), or via an elegant recent strategy also based on CRISPR‐Cas9 but using oligonucleotide templates (Yi & Goshima, 2020). In theory, all these strategies could benefit from the SMART co‐editing selection system described here. Finally, in addition to being an easy‐to‐implement alternative to these base modification strategies, the BE system described here makes possible the in vivo random mutagenesis of a given gene, a powerful new tool for gene function analysis in P. patens that should reinforce the status of P. patens as a powerful platform for functional analysis of plant genes.
Author contributions
FN and AG‐D designed the research; AG‐D performed the research with the help of AA, ZT, FC, FV, FB, PV‐M, JMC, TM and J‐LG; FN and AG‐D wrote the manuscript with contributions from all the authors.
Supporting information
Fig. S1 Schematic description of the APT reporter gene and APRT function.
Fig. S2 Examples of deletions observed during BE multiplexing.
Fig. S3 Examples of multiple cytosines editing or chimerism observed in some clones.
Fig. S4 G418 sensitivity of ABE and CBE single and multiplex edited clones after relaxing of the antibiotic selection pressure.
Fig. S5 Nature of editing using CBE or ABE for each cytosine or adenine present in the target locus.
Fig. S6 Nature of editing using CBE on genes of interest for each cytosine in the target locus.
Fig. S7 Sequence of two sgRNAs containing cytosines potentially target of ABE activity and nature of ABE editing using these sgRNAs.
Fig. S8 Alignment of APRT sequences from different species and phenotype of the apt P. patens mutants.
Fig. S9 View of the P. patens APRT 3D model with amino acids (in blue) that could be modified as single substitutions using CBE or ABE.
Fig. S10 Structure of the Pp3c3_13220, Pp3c14_9040 and Pp3c17_3870 targeted genes.
Fig. S11 Sequence alignment of VDE from Arabidopsis and Physcomitrella.
Fig. S12 Use of the APT gene as a marker of base‐editing efficiency.
Table S1 List of sgRNAs expression cassettes used in this study.
Table S2 Sequences of plasmids used in this study.
Table S3 List of PCR primers used in this study.
Table S4 Mutation rates of the CBE and ABE systems tested (2‐FA direct selection).
Table S5 Transfection efficiency of the CBE and ABE systems.
Table S6 Mutation rates of the CBE system after preselection on G418.
Table S7 Frequency of substitution for cytosines at each position of the eight sgRNAs used in this study.
Table S8 Sequences and positions of possible off‐target sites for sgRNA1 and sgRNA2.
Table S9 List of amino acids modified in the APT gene using the CBE or ABE strategy.
Table S10 Consequence of CBE editing in the different edited clones for the three genes of interest.
Table S11 Sequence analysis of the APT and Pp3c3_13220 locus in adenine‐resistant clones obtained after co‐transfection of the ABEv#1 mutant with the CBE system and the two sgRNAs, sgRNArestor and sgRNAPp3c3.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
We are grateful to Akihiko Kondo who provided the Target‐AID plasmid. We thank Mark Tepfer and Pierre‐François Perroud for their critical revision of the manuscript. The work, including study design, data collection, analysis and interpretation and manuscript writing, was supported by the French National Research Agency (ANR11‐BTBR‐0001‐GENIUS). The IJPB benefits from the support of the LabEx Saclay Plant Sciences‐SPS (ANR‐10‐LABX‐0040‐SPS). The authors declare no conflict of interest.
References
- Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A et al. 2019. Search‐and‐replace genome editing without double‐strand breaks or donor DNA. Nature 576: 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoux P, Morosinotto T, Saga G, Bassi R, Pignol D. 2009. A structural basis for the ph‐dependent xanthophyll cycle in Arabidopsis thaliana . The Plant Cell 21: 2036–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashihara H, Stasolla C, Fujimura T, Crozier A. 2018. Purine salvage in plants. Phytochemistry 147: 89–124. [DOI] [PubMed] [Google Scholar]
- Baroli I, Niyogi KK, Barber J, Heifetz P. 2000. Molecular genetics of xanthophyll‐dependent photoprotection in green algae and plants. Philosophical Transactions of the Royal Society B: Biological Sciences 355: 1385–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastet A, Zafirov D, Giovinazzo N, Guyon‐Debast A, Nogué F, Robaglia C, Gallois J‐L. 2019. Mimicking natural polymorphism in eIF4E by CRISPR‐Cas9 base editing is associated with resistance to potyviruses. Plant Biotechnology Journal 17: 1736–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Hao M, Ding B, Mei D, Wang W, Wang H, Zhou R, Liu J, Li C, Hu Q. 2021. Base editing with high efficiency in allotetraploid oilseed rape by A3A‐PBE system. Plant Biotechnology Journal 19: 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collonnier C, Epert A, Mara K, Maclot F, Guyon‐Debast A, Charlot F, White C, Schaefer DG, Nogué F. 2017a. CRISPR‐Cas9‐mediated efficient directed mutagenesis and RAD51‐dependent and RAD51‐independent gene targeting in the moss Physcomitrella patens . Plant Biotechnology Journal 15: 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collonnier C, Guyon‐Debast A, Maclot F, Mara K, Charlot F, Nogué F. 2017b. Towards mastering CRISPR‐induced gene knock‐in in plants: survey of key features and focus on the model Physcomitrella patens . Methods 121–122: 103–117. [DOI] [PubMed] [Google Scholar]
- Concordet J‐P, Haeussler M. 2018. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Research 46: W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cove DJ, Perroud P‐F, Charron AJ, McDaniel SF, Khandelwal A, Quatrano RS. 2009. Culturing the moss Physcomitrella patens. In: Emerging model organisms: a laboratory manual, Vol. 1. New York, NY, USA: Cold Spring Harbor Laboratory Press. [DOI] [PubMed] [Google Scholar]
- Doman JL, Raguram A, Newby GA, Liu DR. 2020. Evaluation and minimization of Cas9‐independent off‐target DNA editing by cytosine base editors. Nature Biotechnology 38: 620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid A, Alshareef S, Mahfouz MM. 2018. CRISPR base editors: genome editing without double‐stranded breaks. Biochemical Journal 475: 1955–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmar K, Keehyoung J, Jinwoo L, Jooyoung L, Srivatsan R, James T, Mike T, David B, Kevin K. 2010. Improving physical realism, stereochemistry and side‐chain accuracy in homology modeling: four approaches that performed well in CASP8 Elmar. Proteins 77: 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanoff M, Komor AC. 2019. Base Editors: Modular Tools for the Introduction of Point Mutations in Living Cells. Emerging topics in life sciences 3: 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillochet C, Develtere W, Jacobs TB. 2020. CRISPR screens in plants: approaches, guidelines, and future prospects. The Plant Cell tpc.00463.2020. doi: 10.1105/tpc.20.00463 [DOI] [PubMed] [Google Scholar]
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR. 2017. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551: 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holá M, Kozák J, Vágnerová R, Angelis KJ. 2013. Genotoxin induced mutagenesis in the model plant Physcomitrella patens . BioMed Research International 2013: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua K, Tao X, Yuan F, Wang D, Zhu JK. 2018. Precise A·T to G·C base editing in the rice genome. Molecular Plant 11: 627–630. [DOI] [PubMed] [Google Scholar]
- Huang TK, Puchta H. 2019. CRISPR/Cas‐mediated gene targeting in plants: finally a turn for the better for homologous recombination. Plant Cell Reports 38: 443–453. [DOI] [PubMed] [Google Scholar]
- Jacob P, Avni A, Bendahmane A. 2018. Translational research: exploring and creating genetic diversity. Trends in Plant Science 23: 42–52. [DOI] [PubMed] [Google Scholar]
- Jiang F, Doudna JA. 2017. CRISPR–Cas9 structures and mechanisms. Annual Review of Biophysics 46: 505–529. [DOI] [PubMed] [Google Scholar]
- Jin S, Zong Y, Gao Q, Zhu Z, Wang Y, Qin P, Liang C, Wang D, Qiu JL, Zhang F et al. 2019. Cytosine, but not adenine, base editors induce genome‐wide off‐target mutations in rice. Science 364: 292–295. [DOI] [PubMed] [Google Scholar]
- John B, Sali A. 2003. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Research 31: 3982–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Jeong YK, Hur JK, Kim JS, Bae S. 2019. Adenine base editors catalyze cytosine conversions in human cells. Nature Biotechnology 37: 1145–1148. [DOI] [PubMed] [Google Scholar]
- Klughammer C, Schreiber U. 1994. An improved method, using saturating light pulses, for the determination of photosystem I quantum yield via P700+‐absorbance changes at 830 nm. Planta 192: 261–268. [Google Scholar]
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. 2016. Programmable editing of a target base in genomic DNA without double‐stranded DNA cleavage. Nature 533: 420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Willi M, Miller SM, Kim S, Liu C, Liu DR, Hennighausen L. 2018. Targeting fidelity of adenine and cytosine base editors in mouse embryos. Nature Communications 9: 7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Sun Y, Du J, Zhao Y, Xia L. 2017. Generation of targeted point mutations in rice by a modified CRISPR/Cas9 system. Molecular Plant 10: 526–529. [DOI] [PubMed] [Google Scholar]
- Li X, Wang Y, Liu Y, Yang B, Wang X, Wei J, Lu Z, Zhang Y, Wu J, Huang X et al. 2018a. Base editing with a Cpf1‐cytidine deaminase fusion. Nature Biotechnology 36: 324–327. [DOI] [PubMed] [Google Scholar]
- Li ZY, Wang Y, Jin S, Zhang D, Song Q, Zhang R, Gao C, Li C, Zong Y et al. 2018b. Expanded base editing in rice and wheat using a Cas9‐adenosine deaminase fusion. Genome Biology 19: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Zong Y, Xue C, Wang S, Jin S, Zhu Z, Wang Y, Anzalone AV, Raguram A, Doman JL et al. 2020. Prime genome editing in rice and wheat. Nature Biotechnology 38: 582–585. [DOI] [PubMed] [Google Scholar]
- Lopez‐Obando M, Hoffmann B, Géry C, Guyon‐Debast A, Téoulé E, Rameau C, Bonhomme S, Nogué F. 2016. Simple and efficient targeting of multiple genes through CRISPR‐Cas9 in Physcomitrella patens . G3: Genes, Genomes, Genetics 6: 3647–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhu J‐K. 2017. Precise editing of a target base in the rice genome using a modified CRISPR/Cas9 system. Molecular Plant 10: 523–525. [DOI] [PubMed] [Google Scholar]
- Mallett DR, Chang M, Cheng X, Bezanilla M. 2019. Efficient and modular CRISPR‐Cas9 vector system for Physcomitrella patens . Plant Direct 3: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manghwar H, Lindsey K, Zhang X, Jin S. 2019. CRISPR/Cas system: recent advances and future prospects for genome editing. Trends in Plant Science 24: 1102–1125. [DOI] [PubMed] [Google Scholar]
- Marzec M, Hensel G. 2020. Prime editing: game changer for modifying plant genomes. Trends in Plant Science 25: 722–724. [DOI] [PubMed] [Google Scholar]
- Melo F, Sánchez R, Sali A. 2009. Statistical potentials for fold assessment. Protein Science 11: 430–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molla KA, Yang Y. 2019. CRISPR/Cas‐mediated base editing: technical considerations and practical applications. Trends in Biotechnology 37: 1121–1142. [DOI] [PubMed] [Google Scholar]
- Muren E, Nilsson A, Ulfstedt M, Johansson M, Ronne H. 2009. Rescue and characterization of episomally replicating DNA from the moss Physcomitrella. Proceedings of the National Academy of Sciences, USA 106: 19444–19449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida K, Arazoe T, Yachie N, Banno S, Kakimoto M, Tabata M, Mochizuki M, Miyabe A, Araki M, Hara KY et al. 2016. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 102: 553–563. [DOI] [PubMed] [Google Scholar]
- Nomura T, Sakurai T, Osakabe Y, Osakabe K, Sakakibara H. 2016. Efficient and heritable targeted mutagenesis in mosses using the CRISPR/Cas9 system. Plant and Cell Physiology 57: 2600–2610. [DOI] [PubMed] [Google Scholar]
- Orr RG, Foley SJ, Sherman C, Abreu I, Galotto G, Liu B, González‐Guerrero M, Vidali L. 2020. Robust survival‐based RNA interference of gene families using in tandem silencing of adenine phosphoribosyltransferase. Plant Physiology 184: 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CL, Ullman B, Brennan RG, Hill CP. 1999. Crystal structures of adenine phosphoribosyltransferase from Leishmania donovani . EMBO Journal 18: 3533–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinnola A, Dall’Osto L, Gerotto C, Morosinotto T, Bassi R, Alboresi A. 2013. Zeaxanthin binds to light‐harvesting complex stress‐related protein to enhance nonphotochemical quenching in Physcomitrella patens . The Plant Cell 25: 3519–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu X, Liu L, Li P, Huo H, Dong X, Xie K, Yang H, Liu L. 2019. A CRISPR/LbCas12a‐based method for highly efficient multiplex gene editing in Physcomitrella patens . The Plant Journal 100: 863–872. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin C‐Y, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et al. 2013. Double nicking by RNA‐guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154: 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Yan F, Kuang Y, Li N, Zhang D, Zhou X, Lin H, Zhou H. 2018. Improved base editor for efficiently inducing genetic variations in rice with CRISPR/Cas9‐guided hyperactive hAID mutant. Molecular Plant 11: 623–626. [DOI] [PubMed] [Google Scholar]
- Rensing SA, Goffinet B, Meyberg R, Wu SZ, Bezanilla M. 2020. The moss physcomitrium (Physcomitrella) patens: A model organism for non‐seed plants. The Plant Cell 32: 1361–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Research 42: 320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumsby G. 2016. Genetic defects underlying renal stone disease. International Journal of Surgery 36: 590–595. [DOI] [PubMed] [Google Scholar]
- Saga G, Giorgetti A, Fufezan C, Giacometti GM, Bassi R, Morosinotto T. 2010. Mutation analysis of violaxanthin de‐epoxidase identifies substrate‐binding sites and residues involved in catalysis. Journal of Biological Chemistry 285: 23763–23770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer D. 2001. Gene targeting in Physcomitrella patens . Current Opinion in Plant Biology 4: 143–150. [DOI] [PubMed] [Google Scholar]
- Schaefer DG, Delacote F, Charlot F, Vrielynck N, Guyon‐Debast A, Le Guin S, Neuhaus JM, Doutriaux MP, Nogué F. 2010. RAD51 loss of function abolishes gene targeting and de‐represses illegitimate integration in the moss Physcomitrella patens . DNA Repair 9: 526–533. [DOI] [PubMed] [Google Scholar]
- Schaefer DG, Zryd J‐P. 1997. Efficient gene targeting in the moss Physcomitrella patens . The Plant Journal 11: 1195–1206. [DOI] [PubMed] [Google Scholar]
- Shen M, Sali A. 2006. Statistical potential for assessment and prediction of protein structures. Protein Science 15: 2507–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Sarver AE, Wang CC, Tanaka KSE, Almo SC, Schramm VL. 2002. Closed site complexes of adenine phosphoribosyltransferase from Giardia lamblia reveal a mechanism of ribosyl migration. Journal of Biological Chemistry 277: 39981–39988. [DOI] [PubMed] [Google Scholar]
- Shi W, Tanaka KSE, Crother TR, Taylor MW, Almo SC, Schramm VL. 2001. Structural analysis of adenine phosphoribosyltransferase from Saccharomyces cerevisiae . Biochemistry 40: 10800–10809. [DOI] [PubMed] [Google Scholar]
- Shimatani Z, Fujikura U, Ishii H, Matsui Y, Suzuki M, Ueke Y, Taoka KI, Terada R, Nishida K, Kondo A. 2018. Inheritance of co‐edited genes by CRISPR‐based targeted nucleotide substitutions in rice. Plant Physiology and Biochemistry 131: 78–83. [DOI] [PubMed] [Google Scholar]
- Shimatani Z, Kashojiya S, Takayama M, Terada R, Arazoe T, Ishii H, Teramura H, Yamamoto T, Komatsu H, Miura K et al. 2017. Targeted base editing in rice and tomato using a CRISPR‐Cas9 cytidine deaminase fusion. Nature Biotechnology 35: 441–443. [DOI] [PubMed] [Google Scholar]
- Taylor MW, Simon AE, Kothari RM. 1985. The APRT system. In Gottesman MM, ed. Molecular cell genetics. New York, NY, USA: John Wiley & Sons: 311–332. [Google Scholar]
- Thévenin J, Dubos C, Xu W, Le Gourrierec J, Kelemen Z, Charlot F, Nogué F, Lepiniec L, Dubreucq B. 2012. A new system for fast and quantitative analysis of heterologous gene expression in plants. New Phytologist 193: 504–512. [DOI] [PubMed] [Google Scholar]
- Trouiller B, Schaefer DG, Charlot F, Nogué F. 2006. MSH2 is essential for the preservation of genome integrity and prevents homeologous recombination in the moss Physcomitrella patens . Nucleic Acids Research 34: 232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veillet F, Chauvin L, Kermarrec MP, Sevestre F, Merrer M, Terret Z, Szydlowski N, Devaux P, Gallois JL, Chauvin JE. 2019a. The Solanum tuberosum GBSSI gene: a target for assessing gene and base editing in tetraploid potato. Plant Cell Reports 38: 1065–1080. [DOI] [PubMed] [Google Scholar]
- Veillet F, Durand M, Kroj T, Cesari S, Gallois J‐L. 2020a. Precision breeding made real with CRISPR: illustration through genetic resistance to pathogens. Plant Communications 1: 100102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veillet F, Perrot L, Chauvin L, Kermarrec M‐P, Guyon‐Debast A, Chauvin J‐E, Nogué F, Mazier M. 2019b. Transgene‐free genome editing in tomato and potato plants using Agrobacterium‐mediated delivery of a CRISPR/Cas9 cytidine base editor. International Journal of Molecular Sciences 20: 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veillet F, Perrot L, Guyon‐Debast A, Kermarrec M, Chauvin L, Chauvin J, Gallois J, Mazier M, Nogué F. 2020b. Expanding the CRISPR toolbox in P. patens using SpCas9‐NG variant and application for gene and base editing in solanaceae crops. International Journal of Molecular Sciences 21: 1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B, Sali A. 2017. Protein structure modeling with MODELLER. Methods in Molecular Biology 1654: 39–54. [DOI] [PubMed] [Google Scholar]
- Yi P, Goshima G. 2020. Transient cotransformation of CRISPR/Cas9 and oligonucleotide templates enables efficient editing of target loci in Physcomitrella patens . Plant Biotechnology Journal 18: 599–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Malzahn AA, Sretenovic S, Qi Y. 2019. The emerging and uncultivated potential of CRISPR technology in plant science. Nature Plants 5: 778–794. [DOI] [PubMed] [Google Scholar]
- Zong Y, Wang Y, Li C, Zhang R, Chen K, Ran Y, Qiu JL, Wang D, Gao C. 2017. Precise base editing in rice, wheat and maize with a Cas9‐cytidine deaminase fusion. Nature Biotechnology 35: 438–440. [DOI] [PubMed] [Google Scholar]
- Zuo E, Sun Y, Wei W, Yuan T, Ying W, Sun H, Yuan L, Steinmetz LM, Li Y, Yang H. 2019. Cytosine base editor generates substantial off‐target single‐nucleotide variants in mouse embryos. Science 364: 289–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Schematic description of the APT reporter gene and APRT function.
Fig. S2 Examples of deletions observed during BE multiplexing.
Fig. S3 Examples of multiple cytosines editing or chimerism observed in some clones.
Fig. S4 G418 sensitivity of ABE and CBE single and multiplex edited clones after relaxing of the antibiotic selection pressure.
Fig. S5 Nature of editing using CBE or ABE for each cytosine or adenine present in the target locus.
Fig. S6 Nature of editing using CBE on genes of interest for each cytosine in the target locus.
Fig. S7 Sequence of two sgRNAs containing cytosines potentially target of ABE activity and nature of ABE editing using these sgRNAs.
Fig. S8 Alignment of APRT sequences from different species and phenotype of the apt P. patens mutants.
Fig. S9 View of the P. patens APRT 3D model with amino acids (in blue) that could be modified as single substitutions using CBE or ABE.
Fig. S10 Structure of the Pp3c3_13220, Pp3c14_9040 and Pp3c17_3870 targeted genes.
Fig. S11 Sequence alignment of VDE from Arabidopsis and Physcomitrella.
Fig. S12 Use of the APT gene as a marker of base‐editing efficiency.
Table S1 List of sgRNAs expression cassettes used in this study.
Table S2 Sequences of plasmids used in this study.
Table S3 List of PCR primers used in this study.
Table S4 Mutation rates of the CBE and ABE systems tested (2‐FA direct selection).
Table S5 Transfection efficiency of the CBE and ABE systems.
Table S6 Mutation rates of the CBE system after preselection on G418.
Table S7 Frequency of substitution for cytosines at each position of the eight sgRNAs used in this study.
Table S8 Sequences and positions of possible off‐target sites for sgRNA1 and sgRNA2.
Table S9 List of amino acids modified in the APT gene using the CBE or ABE strategy.
Table S10 Consequence of CBE editing in the different edited clones for the three genes of interest.
Table S11 Sequence analysis of the APT and Pp3c3_13220 locus in adenine‐resistant clones obtained after co‐transfection of the ABEv#1 mutant with the CBE system and the two sgRNAs, sgRNArestor and sgRNAPp3c3.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.