

Abstract
CC chemokine receptor 2 (CCR2) antagonists that disrupt CCR2/MCP‐1 interaction are expected to treat a variety of inflammatory and autoimmune diseases. The lack of CCR2 crystal structure limits the application of structure‐based drug design (SBDD) to this target. Although a few three‐dimensional theoretical models have been reported, their accuracy remains to be improved in terms of templates and modeling approaches. In this study, we developed a unique ligand‐steered strategy for CCR2 homology modeling. It starts with an initial model based on the X‐ray structure of the closest homolog so far, that is, CXCR4. Then, it uses Elastic Network Normal Mode Analysis (EN‐NMA) and flexible docking (FD) by AutoDock Vina software to generate ligand‐induced fit models. It selects optimal model(s) as well as scoring function(s) via extensive evaluation of model performance based on a unique benchmarking set constructed by our in‐house tool, that is, MUBD‐DecoyMaker. The model of 81_04 presents the optimal enrichment when combined with the scoring function of PMF04, and the proposed binding mode between CCR2 and Teijin lead by this model complies with the reported mutagenesis data. To highlight the advantage of our strategy, we compared it with the only reported ligand‐steered strategy for CCR2 homology modeling, that is, Discovery Studio/Ligand Minimization. Lastly, we performed prospective virtual screening based on 81_04 and CCR2 antagonist bioassay. The identification of two hit compounds, that is, E859‐1281 and MolPort‐007‐767‐945, validated the efficacy of our model and the ligand‐steered strategy.
Keywords: CCR2, flexible docking, ligand‐steered homology modeling, MUBD‐DecoyMaker, normal mode analysis, structure‐based virtual screening
The model of CCR2 in complex with Teijin lead, generated by a unique ligand‐steered strategy based on EN‐NMA and flexible docking with AutoDock Vina.
Abbreviations
- CCR2
CC chemokine receptor 2
- DS
Discovery Studio
- DS/Ligand Minimization
Ligand Minimization module in DS
- FD
flexible docking
- GPCRs
G protein‐coupled receptors
- HBAs
hydrogen bond acceptors
- HBDs
hydrogen bond donors
- LOO CV
leave‐one‐out cross‐validation
- LSHM
ligand‐steered homology modeling
- MCP‐1
monocyte chemoattractant protein‐1
- MD
molecular dynamics
- MUBD‐CCR2
maximal unbiased benchmarking set for CCR2
- MW
molecular weight
- NC
net charge
- NMA
normal mode analysis
- RBs
rotatable bonds
- SBDD
structure‐based drug design
- SBVS
structure‐based virtual screening
- Tc
Tanimoto coefficient
- β2AR
human β2 adrenergic receptor
1. INTRODUCTION
CC chemokine receptor 2 (CCR2), one subtype of chemokine receptors, belongs to the superfamily of rhodopsin‐like G protein‐coupled receptors (GPCRs; Covino et al., 2016). It is expressed on the cell surface of T cells, dendritic cells, and endothelial cells, but mainly on inflammatory monocytes associated with immune defense and inflammatory disorders (Fantuzzi et al., 2019; Serbina & Pamer, 2006; Zlotnik & Yoshie, 2012). Its main natural ligand (or chemokine), that is, monocyte chemoattractant protein‐1 (MCP‐1 or CCL2), mediates the migration of inflammatory monocytes to sites of inflammation following its binding to CCR2 (Carter, 2013). On the one side, excessive activation or overexpression of MCP‐1/CCR2 signaling has been implicated in a variety of diseases, including cancer progression and metastasis, neurological disorders (e.g. epilepsy), autoimmune disease (e.g. rheumatoid arthritis), obesity and insulin resistance, atherosclerosis, and pathogenesis of HIV‐1 infection (Bozzi & Caleo, 2016; Cerri et al., 2016; Covino et al., 2016; O'Connor et al., 2015; Zhang & Luo, 2019). On the other side, downregulation of MCP‐1/CCR2 signaling has been validated in vivo as an effective strategy to treat those diseases (Zhang et al., 2012; Zlotnik & Yoshie, 2012). Therefore, discovery of small‐molecule CCR2 antagonists is attractive for pharmaceutical industry and academia (Brown et al., 2016; Carter, 2013; Carter et al., 2015; Strunz et al., 2015; Vilums et al., 2015; Winters et al., 2014).
In modern drug discovery, structure‐based drug design (SBDD) is widely recognized as an effective technique that speeds up hit identification and helps lead optimization (Jazayeri et al., 2017). According to the specific aim, SBDD is classified into two methods, that is, structure‐based virtual screening (SBVS; Waszkowycz et al., 2011) and de novo drug design (Hartenfeller & Schneider, 2011; Rodrigues et al., 2015). Both methods require three‐dimensional structure(s) of the specific target in order to rationally design molecules of potential binding affinity or expected binding modes. For CCR2‐targeted drug design, however, the lack of available crystal structure(s) for this target hampered the wide application of SBDD.
Three‐dimensional theoretical models were regarded as an alternative to experimental structures. Since 2000, a few groups have attempted to construct CCR2 homology models for the aim of functional study or/and drug design. At the early stage, all the modeling was based on the structures of only resolved GPCR, that is, rhodopsin. Mirzadegan et al. reported the first model based on bacteria rhodopsin structure (Mirzadegan et al., 2000). Then, Shi et al. reported other two CCR2 receptor models that were built based on bovine rhodopsin (PDB Entry: 1F88) and refined by molecular mechanics and 1‐ns restrained molecular dynamics (MD) simulations (Shi et al., 2002). Berkhout et al. used bovine rhodopsin (PDB Entry: 1F88) as a template for CCR2 homology modeling. It was reported that the plausible binding modes of ligands from molecular docking against this model were completely consistent with site‐directed mutations they constructed (Berkhout et al., 2003). Nevertheless, not all unrefined CCR2 models based on rhodopsin were able to generate reasonable poses for diverse ligands. Kimura et al. noted this problem and attributed the failure to too small size of the constructed binding site. They thus proposed a balloon expansion approach to refine the active site. This approach optimized both backbone and side chains, and was able to make the binding site accommodate known ligands (Kimura et al., 2008). Afterward, two groups used human β2 adrenergic receptor (β2AR, PDB Entry: 2RH1) as the template due to its higher level of sequence identity to CCR2. Notably, they started to consider ligand‐induced arrangement in homology modeling. Kim et al. used Ligand Minimization module in Discovery Studio (DS) to construct the ligand‐induced binding site (Kim et al., 2011). Chavan et al. built a CCR2 receptor model, docked TAK779 into the binding site, and performed MD simulation to refine its bound state (Chavan et al., 2012). More recently, the crystal structure of a closer homolog to CCR2, that is, CXCR4 (PDB Entry: 3ODU), was resolved (Wu et al., 2010), which provided a better template for CCR2 homology modeling. And the modeling approach was consistently molecular docking plus MD simulations. Kothandan et al. firstly performed MD simulations to obtain a more stable apo‐CCR2 model and then docked multiple antagonists against this model (Kothandan et al., 2012). Shahlaei et al. adopted the same approach (Shahlaei et al., 2013). Singh et al. employed induced fit docking to explore the effect of side chain movement on performance of docking‐based VS right after the construction of an unrefined CCR2 model (Singh & Sobhia, 2013).
Ligand‐steered homology modeling (LSHM) is an effective approach for GPCR structure modeling and ligand docking. The resulting models have led to successful cases for GPCR‐targeted drug discovery (Cavasotto et al., 2008; Lin et al., 2012; Phatak et al., 2010). Also, it has shown advantages over other approaches in three‐round GPCR Dock competitions (Kufareva et al., 2011, 2014; Michino et al., 2009). This approach uses information about known ligands to refine the binding site by incorporating protein flexibility in homology modeling. In fact, DS/Ligand Minimization proposed by Kim et al. in the above approaches was a feasible strategy for LSHM. However, this strategy only optimized side chains of the binding site, and thus, the accuracy of the CCR2 model may be further improved. Long‐time MD simulations of a ligand‐bound state constructed by molecular docking are a better way to explore ligand‐induced fit effect. But it is well recognized as time‐consuming and computationally expensive. Normal mode analysis (NMA) methodology is also a good means to incorporate receptor flexibility for GPCR targets. Generation of multiple receptor conformations by NMA methodology is able to significantly improve the accuracy of receptor modeling (Dietzen et al., 2012; Rai et al., 2010; Rueda et al., 2009). It was encouraging and convincing that the blindly predicted A2A/ZM241385 complex models with the aid of NMA methodology ranked at the top of the model list in GPCR DOCK 2008 (Michino et al., 2009). To the best of our knowledge, NMA has never been applied to receptor conformation sampling and homology modeling for CCR2.
In this study, we attempted to develop a unique ligand‐steered strategy that employs NMA methodology and flexible docking (FD) to refine CCR2 models, for which CXCR4 crystal structure was used as a template. All the constructed models along with multiple scoring functions were evaluated for their screening power by retrospective small‐scale virtual screening. Specifically, the evaluation was based on a benchmarking set generated by our in‐house MUBD‐DecoyMaker (Xia et al., 2014), a tool that had effectively facilitated VS campaigns against multiple targets (Huang et al., 2016; Pei et al., 2015; Xia et al., 2015). Apart from the screening power, the binding mode proposed by the optimal model was explored. In addition, we constructed CCR2 models using the previously reported DS/Ligand Minimization strategy (Kim et al., 2011) and compared them with our selected models in terms of ligand enrichment. Eventually, we performed prospective VS against the optimal CCR2 structure and biological evaluation to explore the efficacy of the model and the modeling strategy.
2. MATERIALS AND METHODS
2.1. The general workflow for LSHM
LSHM is composed of three consecutive steps, (a) construction of crude model(s) from protein sequence, that is, model building; (b) construction of ligand‐induced fit protein model(s), that is, model optimization; and (c) evaluation of ligand‐induced fit models, that is, model selection. (cf. Figure 1) To be specific, step (i) is a common step shared by all homology modeling approaches. It builds an initial model of the target protein based on the three‐dimensional structure of a template protein and sequence alignment between template protein and target protein. Step (ii) is the most critical step for LSHM, as it varies among different homology modeling approaches and may significantly affect ligand enrichment of resulting models. Step (iii) is to select a protein model of optimal ligand enrichment from multiple ligand‐induced fit protein model(s) via benchmarking study. In this study, a novel approach using Elastic Network Normal Mode Analysis (EN‐NMA) plus FD was developed to optimize CCR2 crude models. As a reference for comparison, the reported protocol using Ligand Minimization in DS (DS/Ligand Minimization) was also constructed to optimize the initial CCR2 model (Kim et al., 2011).
FIGURE 1.
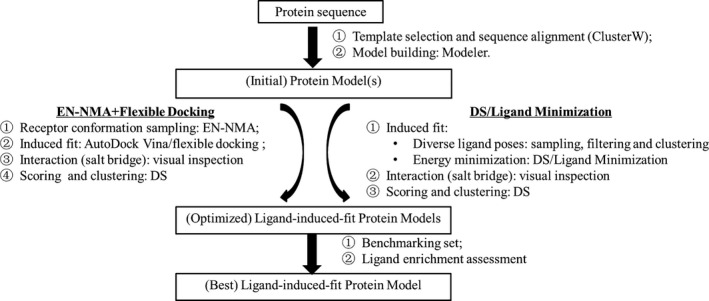
The LSHM flowcharts developed/used in this study. The strategy termed as EN‐NMA plus Flexible Docking was developed by our group, while the DS/Ligand Minimization strategy was proposed by others and used as a baseline in this study
2.2. Construction of initial models
2.2.1. Template selection and structure preparation
The X‐ray structure of protein‐ligand complex for CXCR4 (PDB ID: 3ODU) was used as a template protein for its high homolog to CCR2 (Wu et al., 2010). For protein preparation, the identical protein chain, water molecules, and T4 lysozyme fusion protein were removed from the X‐ray structure. The “Clean Protein” module of DS (version 2.5, Accelrys Software, Inc) was used to prepare the protein. It added hydrogens, modified chain termini, corrected nonstandard names, alternated conformations, repaired incomplete residues and atom order in amino acids, and protonated the whole protein at pH 7.0. The cognate ligand was kept in the binding site of the template protein.
2.2.2. Sequence alignment
ClustalW program (Thompson et al., 1994) implemented in DS was used for sequence alignment. Firstly, the full‐length sequences of all CCR subtypes (CCR1 ~ 11) were aligned to the sequence of target protein, that is, CXCR4, which locates conserved amino acids in the transmembrane domains of this chemokine receptor family. Secondly, CCR2 sequence (UniProt Entry: P61073) was aligned to the sequence corresponding to CXCR4 crystal structure. Lastly, the outcome of CCR2‐CXCR4 alignment was updated manually according to multiple sequence alignment (cf. Figure 1).
2.2.3. Building CCR2 initial model
The program of Modeler (version 9.4; Sali & Blundell, 1993) was used to build initial CCR2 models. The prepared structure of CXCR4 and the outcome of CCR2‐CXCR4 sequence alignment were the input of this program. The conserved disulfide bonds of Cys2/Cys247 and Cys83/Cys160 were constrained in model building. Besides, the cognate ligand, that is, IT1t, was copied from CXCR4 crystal structure to modeled CCR2 structures, which helped shape an initial binding site of CCR2. The number of models generated was set as 20. Among 20 models, three models, that is, MOD1‐3 with lower PDF Total Energy, were submitted for energy minimization with backbone constraints. CHARMm force field was added to the protein, and then, 500‐step minimization using the algorithm of conjugate gradient with was executed. Lastly, the stereochemical quality of the energy‐minimized CCR2 structures was checked by PROCHECK (http://nihserver.mbi.ucla.edu/SAVES/, accessed June 2016; Laskowski et al., 1993).
2.3. Construction of ligand‐induced fit protein models
2.3.1. Ligand selection and preparation
A CCR2 antagonist named Teijin lead (cf. Figure 2) was selected as a reference molecule to construct ligand‐induced fit protein models (Berkhout et al., 2003). This compound is a potent CCR2 antagonist, whose IC50 equals to 54 nM (Shiota, 2002). Mutagenesis study has indicated four residues, that is, Glu291, Thr292, Tyr120, and His121, may be involved in the interaction between this compound and CCR2 (Berkhout et al., 2003). These data are useful for the building and selection of ligand‐induced fit models. Tertiary amine in the compound is potentially charged at physiological pH. Therefore, “Prepare Ligands” module in DS was used to protonate the compound at pH of 7.3–7.5. Meanwhile, this module also generates the lowest‐energy conformer of the compound via Catalyst program. This conformer became the initial pose for docking.
FIGURE 2.
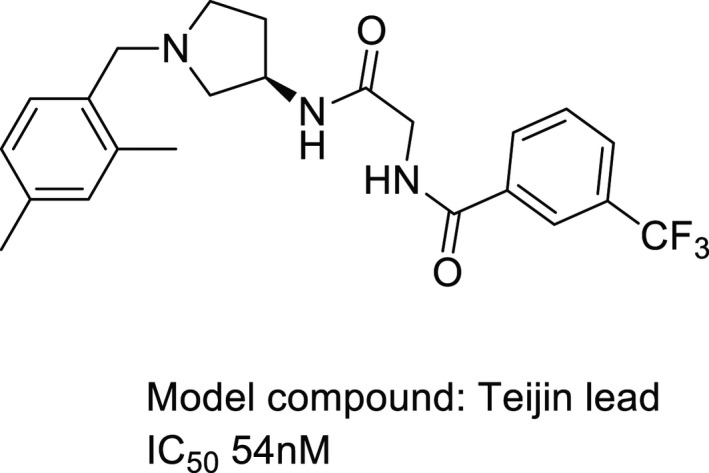
The chemical structure of a reference molecule used in the study
2.3.2. Strategy I for model optimization: EN‐NMA plus flexible docking
(a) Receptor conformation sampling. EN‐NMA MRC (http://abagyan.ucsd.edu/MRC, accessed June 2016) was used to generate Elastic Network Normal Mode‐guided multiple conformations based on one initial model, that is, MOD1. In this process, icmPocketFinder was used to automatically detect the largest pocket of the initial model. The residues within 15 Å from the center of the pocket were included in the NMA. The number of generated conformations before clustering was set as 100. Amplitude, that is, average atomic displacements, was set as 1 Å. All generated conformations were then clustered using 0.6 Å as a heavy‐atom RMSD cutoff. (b) Protein–ligand‐induced fit. The reference molecule was docked into binding sites of clustered conformations as well as the initial MOD1 (for reference) using FD module in AutoDock Vina software. Five reported important residues, that is, Tyr120, His121, Asp284, Glu291, and Thr292, were set as movable in order to incorporate their protein flexibility. The docking site was defined as a cubic box of 40 Å × 40 Å × 30 Å, centered on nitrogen atom of charged amine in the copied ligand from CXCR4 crystal structure. A maximum of twenty docking poses/binding modes were generated from FD. (c) Binding mode filtering by visual inspection. Salt bridge is critical for the interaction between aminergic GPCR and its ligand (Shi & Javitch, 2002). Therefore, those complexes that contained no salt bridge between the charged amine of the reference molecule and CCR2 were filtered. (d) Scoring and clustering. “Score Ligand Poses” module in DS was used to score protein‐ligand interaction for each remaining complex. As for scoring, the center of binding sphere was defined the same as that for FD while the radius was enlarged to 13 Å. The scoring functions were LigScore1, LigScore2, PLP1, PLP2, Jain, PMF, PMF04, LUDI1, LUDI2, and LUDI3. All complexes of Teijin lead bound to CCR2 were then ranked according to their respective scores from each scoring function, which generated ten ranks for each complex. The average value of ten ranks (i.e., 1, 2… n) was used as a consensus score to rank the complexes. Smaller consensus score (i.e., higher rank) is an indicator for lowest binding free energy between Teijin lead and CCR2, thus represents a more reliable complex (Kim et al., 2011; Wang & Wang, 2001). Meanwhile, the remaining complexes were clustered according to the following steps. Firstly, the reference molecule was excluded from each complex, after which all ligand‐induced fit CCR2 conformations were aligned to the initial model (i.e., MOD1) using the tool of “g_confrms” in GROMACS 3.3.1. Secondly, a matrix of heavy‐atom RMSDs was generated using the tool of “heavy‐atom RMSDs” in DS. Thirdly, the matrix was converted to a dendrogram as well as a heatmap by hierarchical clustering based on minimum distance in MATLAB (version 7.6.0.324). The outcome from clustering and consensus scoring was used as reference for model selection. According to the consensus score and cluster, ten complexes, that is, one complex of best consensus score in each cluster, were retained for further selection via benchmarking study.
2.3.3. Strategy II for model optimization: DS/ligand minimization
(a) Protein–ligand‐induced fit. In this strategy, all the three CCR2 initial models, that is, MOD1‐MOD3, were submitted for optimization. GOLD (version 3.0.1, Cambridge Crystallographic Data Center) was used to dock Teijin lead into the binding site of each CCR2 initial model, respectively. For molecular docking, two oxygen atoms in the carboxyl group of Glu291 were required to form hydrogen bonds with Teijin lead in docking simulation. The parameters were as follows: restraint constant equals to 10; minimum H‐bond geometry weight equals to 0.005, docking mode as default setting (precise docking), and scoring function as GoldScore. Multiple poses of Teijin lead were generated via molecular docking, from which those poses that contained salt bridge between charged amine and carboxyl group of Glu291 were selected. Subsequently, “Analyze Ligand Poses” module in DS was utilized to cluster the selected poses with a cutoff of 1.5 Å. The pose of highest GoldScore was kept in each cluster. The resulting complexes were submitted for energy minimization in order to relieve the restraints. To this end, “Ligand Minimization” module in DS was utilized and its parameters were set as follows: force field, CHARMm; algorithm, Smart Minimizer; Dielectric Constant, 4.0; flexible residues were set as the same as those in the cavity for EN‐NMA simulation. This type of energy minimization generated multiple ligand‐induced fit models. (b) Pose filtering. Via visual inspection, those complexes that contained no salt bridge were filtered. (c) Score protein‐ligand interaction and clustering. The same method with that in Strategy I was applied to this step. At first, “Score Ligand Poses” module in DS was used to predict binding affinity between Teijin lead and CCR2 for each ligand‐induced fit model. Then, consensus score was also calculated to rank those models. Lastly, models were selected according to both consensus scores and outcome of structure clustering.
2.4. Model selection via benchmarking study
2.4.1. Construction and validation of CCR2 benchmarking set
At first, all ligands for CCR2 were collected from CHEMBL19 (https://www.ebi.ac.uk/chembl/, accessed in Jun. 2014; Bento et al., 2014; Gaulton et al., 2012). Then, these ligands were cleaned according to the following criteria: (a) To ensure every collected ligand was indeed targeting the specific protein, confidence score of each ligand was required to be greater than 4 (cf. https://www.ebi.ac.uk/chembl/faq#faq24; Mysinger et al., 2012). Consequently, no compound was excluded for this criterion. (b) The reported IC50 value was required to be better than or equal to 1 µM. (c) Only chemical structure and activity class (i.e., active) of each ligand were kept, no matter how many IC50 values were reported for the ligand (e.g., IC50 values from different bioassay). Next, the cleaned ligands were curated using Pipeline Pilot (version 7.5, Accelrys Software, Inc), that is, components of “strip salts” and “standardize molecule,” as well as a customized property filter of “RBs > 20” or “MW ≥ 600” (Mysinger et al., 2012). After that, ligands were further protonated at pH ranging from 7.3 to 7.5.
Our in‐house program implemented in Pipeline Pilot (version 7.5, Accelrys Software, Inc.) and MATLAB (version 7.6.0.324), that is, MUBD‐DecoyMaker (Pei et al., 2015; Xia et al., 2014), was used to construct a maximal unbiased benchmarking set for CCR2, that is, MUBD‐CCR2. MUBD‐DecoyMaker includes three modules, that is, ligand processor, preliminary filter, and precise filter (cf. Figure 3). Ligand processor calculated pairwise similarity within the protonated ligands in terms of Tanimoto coefficient (Tc) based on MACCS fingerprints and excluded ligands whose pairwise similarity values are no less than 0.75. Besides, it calculated physicochemical properties of every remaining ligand, including AlogP, molecular weight (MW), hydrogen bond acceptors (HBAs), hydrogen bond donors (HBDs), rotatable bonds (RBs), and net charge (NC). Preliminary filter selected compounds as decoys from all‐purchasable subset of ZINC (http://zinc.docking.org/, accessed in Dec. 2012; Irwin & Shoichet, 2005; Irwin et al., 2012). To become a decoy, a compound must meet two criteria: (a) Its value of each physicochemical property is within the range defined by diverse ligands; (b) its topological similarity to every diverse ligand is between the minimum of pairwise similarity within ligands and 0.75. All selected decoys by preliminary filter constituted a potential decoy set, from which precise filter further selected 39 optimal decoys for each diverse ligand. The resulting decoy set and the diverse ligand set comprised MUBD‐CCR2.
FIGURE 3.
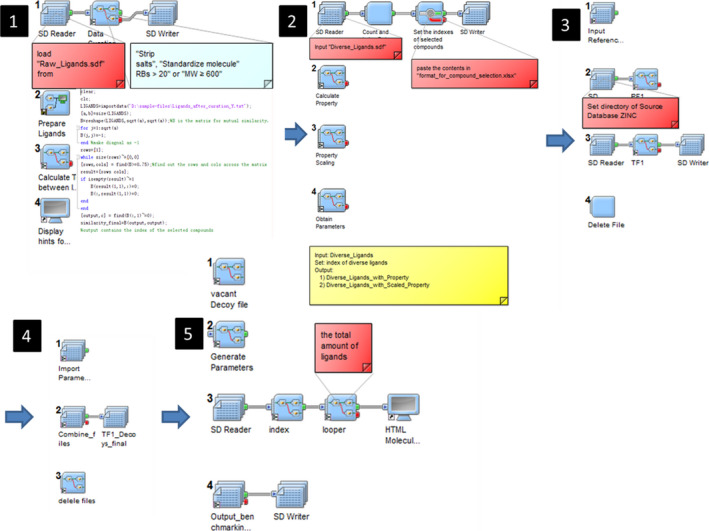
The GUI of MUBD‐DecoyMaker: Steps 1 and 2 for Ligand Processor; steps 3 and 4 for Preliminary Filter; step 5 for Precise Filter
MUBD‐CCR2 was extensively validated by retrospective VS using two nearest neighbor classifiers based on simp and MACCS sims in means of leave‐one‐out cross‐validation (LOO CV). For each nearest neighbor classifier, ROC curve of each cycle was generated and its AUC was calculated. These curves and the average value of AUCs were used to measure “artificial enrichment” and “analogue bias” in MUBD‐CCR2. Physicochemical property distribution curves were also used to show property matching between diverse ligands and decoys, which measured bias for “artificial enrichment” as well.
2.4.2. Enrichment assessment of CCR2 ligand‐induced fit models based on MUBD‐CCR2
Retrospective small‐scale VS was performed by docking diverse ligands and unbiased decoys in MUBD‐CCR2 against each ligand‐induced fit model. Prior to molecular docking, “Prepare Ligands” module in DS was used to generate lowest‐energy conformer as the starting docking pose of each compound. The program used for molecular docking was GOLD (version 3.0.1). In molecular docking, the oxygen atoms in carboxyl group of Glu291 were required to form hydrogen bond with compounds. Screening mode was 7–8 times speedup. Times for docking were set to 20, scoring function was ChemScore, and 10 docking poses were retained for each compound. After molecular docking, every scoring function in DS, that is, LigScore1, LigScore2, ‐PLP1, ‐PLP2, Jain, PMF, PMF04, LUDI1, LUDI2, and LUDI3, was applied to score binding affinity between each pose and CCR2 receptor, respectively. For each scoring function, the pose of best score for each compound was retained, which led to a ranked list of compounds. According to the list, ROC curve and its parameters, that is, ROCE1% and ROC AUC, were calculated to evaluate ligand enrichment of different pairs of models/scoring functions. ROCE1% is defined as the ratio of true positive rate to false‐positive rate, at 1% recovered known decoys (Jahn et al., 2009; Nicholls, 2008). Since early enrichment is more correlated to real‐world screening, ROCE1% was given priority for performance evaluation. Such an assessment aimed to select the optimal pair of model/scoring function for large‐scale VS from EN‐NMA plus Flexible Docking, DS/Ligand Minimization, and Modeler, respectively.
2.5. Structure‐based VS against the optimal CCR2 model
Three commercially available libraries, that is, TimTec Diversity Set (10,000), AnalytiCon NATx (5,000), and ChemDiv (100,000), were merged to constitute the source screening set. To increase the chances of hit identification, a focused library for CCR2 was built according to the following criteria. Firstly, only the compounds whose physicochemical properties were similar to the reported CCR2 antagonists were included, that is, AlogP, 0 ~ 6; MW, 400 ~ 600; HBDs, 1 ~ 5; HBAs, 1 ~ 6; RBs, 5 ~ 10, which reduced the library size to approximately 16K. Secondly, only the compounds predicted active by multiple support vector machine (SVM)‐based binary classifiers (Shiota et al., 2002) were included. As a result, the focused library used for molecular docking against the optimal CCR2 model was composed of 383 compounds. Prior to molecular docking, the compounds in the focused library were prepared by using the “Prepare Ligands” module in DS 2.5, including protonation at the pH range of 7.3–7.5, multiple isomer enumeration, and low‐energy conformer generation. For molecular docking, the parameters were set the same as those used in retrospective small‐scale VS. After molecular docking, all the poses of each compound were rescored by the best‐performing scoring function and the top‐ranking pose was kept for each compound. Eventually, the compounds for purchase and bioassay were selected out according to the predicted binding scores and modes.
2.6. CCR2 antagonist bioassay
The purchased compounds were submitted to EMD Millipore for biological evaluation services. To be specific, fluorescent imaging plate reader (FLIPR) assays were conducted to test the compounds for antagonist activities on CCR2b at the concentration of 100 μM.
2.6.1. Compound plate preparation
The compounds were prepared in specified solvent (i.e., DMSO, PBS, water) and ultimately prepared in EMD Millipore's GPCRProfiler® Assay Buffer to concentrations that were three‐fold higher than the final assay concentration, that is, 100 μM. Similarly, vehicle controls and positive control (reference agonist, MCP‐1) were prepared to ensure all assays were properly controlled. The GPCRProfiler® Assay Buffer was a modified Hanks Balanced Salt Solution (HBSS) where HBSS was supplemented to contain 20 mM HEPES and 2.5 mM Probenecid at pH 7.4.
2.6.2. Calcium flux assay
The compounds supplied were plated in duplicate for the concentration assayed. During the agonist assay, the concentration reflects accommodation for the dilution of compound during the Antagonist Assay. Reference agonist was prepared in a similar manner to serve as assay control and included at Emax (the concentration where the reference agonist elicited a maximal response).
The agonist assay was conducted on a FLIPRTETRA instrument where the test compounds, vehicle controls, and reference agonist were added to the assay plate after a fluorescence/luminescence baseline was established. The agonist assay was a total of 180 s and was used to assess each compound's ability to activate CCR2b. Upon completion of the 3‐min agonist assay, the assay plate was incubated at 25°C for a further 2 min. After the incubation period, the antagonist assay was initiated.
Using EC80 potency values determined during the agonist assay, all preincubated sample compound wells were challenged with EC80 concentration of reference agonist after establishment of a fluorescence/luminescence baseline. The antagonist assay was conducted using the same assay plate that was used for the agonist assay. The antagonist assay was conducted on a FLIPRTETRA instrument where vehicle controls and EC80 concentration of reference agonist were added to appropriate wells. The antagonist assay was a total of 180 s and was used to assess each compound's ability to inhibit CCR2b.
2.6.3. Data processing
All plates were subjected to appropriate baseline corrections. Once baseline corrections were processed, maximum fluorescence/luminescence values were exported and data were manipulated to calculate percentage activation and percentage inhibition. Data manipulation calculation is as follows: ((Max RLU) − (Baseline Avg.))/((Positive Avg.) − (Baseline Avg.)).
3. RESULTS AND DISCUSSION
3.1. Initial CCR2 models: sequence alignment and homology modeling
Kim et al. built CCR2 models using the X‐ray structure of β2AR (PDB Entry: 2RH1) as a template. Unlikely, we used X‐ray structure of CXCR4 bound to IT1t (PDB Entry: 3ODU) because CXCR4 is more homologous to CCR2. The sequence identity is 33.2%, and sequence similarity is 62.7% between CXCR4 and CCR2 (cf. Figure S1). Based on the outcome of sequence alignment, Modeler generated 20 CCR2 models according to default setting. As mentioned in the Method Section, three models of lower PDF total energy were submitted for energy minimization. Ramachandran plots generated by PROCHEK for these models are shown in Figure S2. It reflects stereochemistry of all amino acid residues except for Gly and Pro in a protein structure. As demonstrated by statistics from Ramachandran plots (cf. Table 1), generally three models are of equivalent stereochemical quality, as for all models over 99% residues are within allowed regions, that is, most favored region, additional allowed region and generally allowed region. To be specific, MOD1 is somewhat better than others. All of its residues are within allowed regions, and thus, this model was used as the initial model for optimization by EN‐NMA plus Flexible Docking. For both MOD2 and MOD3, 0.4% of residues are within disallowed region. For a fair comparison, we followed the earlier publication to retain all the three models for DS/Ligand Minimization (Kim et al., 2011). Figure S3 shows that the positions of Glu291 are different in three models, which may affect model performance in ligand enrichment. In this sense, the use of all three initial models would be a better choice.
TABLE 1.
Statistics of the Ramachandran plots generated by PROCHECK for the three initial CCR2 homology models built by Modeler
| Initial model | Most favored region | Additional allowed region | Generally allowed region | Disallowed region | Subtotal (except Gly and Pro) | Total | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | n | % | |||
| MOD1 | 247 | 93.6 | 16 | 6.1 | 1 | 0.4 | 0 | 0 | 264 | 293 |
| MOD2 | 238 | 90.2 | 24 | 9.1 | 1 | 0.4 | 1 | 0.4 | 264 | 293 |
| MOD3 | 239 | 90.5 | 20 | 7.6 | 4 | 1.5 | 1 | 0.4 | 264 | 293 |
3.2. Rational selection of optimal ligand‐induced fit models
For strategy I that consists of EN‐NMA and Flexible Docking, EN‐NMA generated 100 receptor conformations based on the initial model, that is, MOD1. Flexible docking by AutoDock Vina generated ligand‐induced fit complexes, among which 46 complexes possessed a necessary salt bridge between the reference molecule and Glu291. These ligand‐induced fit models were clustered, which generated dendrogram and heatmap for model selection, that is, one model for each cluster (cf. Figure S4a). As mentioned in Method Section, consensus score is an essential indicator for model selection. The consensus scores of 46 complexes were shown in Table S1. According to that, we selected 10 ligand‐induced fit CCR2 models from different clusters, that is, 44_08, 65_04, 94_15, 71_20, 33_02, 09_11, 80_03, 20_04, 26_02, and 81_04. (cf. Table 2).
TABLE 2.
The consensus score (average rank) of each ligand‐induced fit model generated by EN‐NMA plus flexible docking
| ID | Ligscore1 | Ligscore2 | ‐PLP1 | ‐PLP2 | Jain | ‐PMF | ‐PMF04 | Ludi_1 | Ludi_2 | Ludi_3 | Consensus score |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 44_08 | 1 | 1 | 27 | 25 | 9 | 1 | 2 | 3 | 3 | 3 | 7.5 |
| 65_04 | 3 | 2 | 7 | 9 | 10 | 22 | 25 | 4 | 4 | 4 | 9 |
| 94_15 | 27 | 41 | 3 | 3 | 6 | 2 | 7 | 6 | 2 | 1 | 9.8 |
| 71_20 | 2 | 4 | 19 | 19 | 8 | 4 | 9 | 5 | 7 | 22 | 9.9 |
| 33_02 | 25 | 26 | 2 | 2 | 13 | 21 | 5 | 11 | 10 | 6 | 12.1 |
| 09_11 | 5 | 21 | 1 | 4 | 7 | 30 | 35 | 14 | 15 | 9 | 14.1 |
| 80_03 | 7 | 5 | 9 | 8 | 15 | 5 | 20 | 27 | 24 | 23 | 14.3 |
| 20_4 | 8 | 39 | 4 | 1 | 1 | 40 | 41 | 1 | 1 | 8 | 14.4 |
| 26_02 | 12 | 14 | 18 | 15 | 5 | 13 | 23 | 17 | 17 | 10 | 14.4 |
| 81_04 | 16 | 13 | 10 | 13 | 12 | 10 | 12 | 30 | 22 | 27 | 16.5 |
For strategy II that includes DS/Ligand Minimization, the reference molecule was docked into the binding site of each initial model, that is, MOD1‐MOD3 and submitted for energy minimization, which generated multiple ligand‐induced fit complexes. Similar to strategy I, we only kept those complexes that possessed salt bridge between the reference molecule and Glu291. As shown in Table 3, 12 ligand‐induced fit models met this criterion. Among them, six models were from MOD1, and three models were from MOD2, while other three models were from MOD3. By structure clustering, we observed all these models were in three clusters. To be specific, cluster 1 includes 1_1, 1_3, 1_4, 1_5, 1_6, and 1_7 from MOD1, and cluster 2 includes 2_1, 2_2, and 2_3 from MOD2. Likewise, cluster 3 includes 3_1, 3_2, and 3_3 from MOD3 (cf. Figure S4b). Each cluster was consistent with its individual source, which demonstrated trivial difference in structures within each cluster. This further indicated energy minimization was not capable of changing overall conformation in a significant way. The final ligand‐induced fit models we selected were 1_3, 2_3, and 3_2. (cf. Table 3), and they were of best consensus score in each cluster.
TABLE 3.
The consensus score (average rank) of each ligand‐induced fit model in each cluster
| Initial model | ID | Ligscore1 | Ligscore2 | ‐PLP1 | ‐PLP2 | Jain | ‐PMF | ‐PMF04 | Ludi_1 | Ludi_2 | Ludi_3 | Consensus score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MOD1 | 1_3 | 1 | 1 | 1 | 1 | 3 | 5 | 6 | 2 | 2 | 5 | 2.7 |
| 1_1 | 2 | 2 | 4 | 3 | 2 | 4 | 5 | 3 | 1 | 3 | 2.9 | |
| 1_5 | 3 | 4 | 2 | 2 | 6 | 2 | 2 | 4 | 3 | 1 | 2.9 | |
| 1_4 | 4 | 3 | 6 | 6 | 1 | 3 | 3 | 1 | 4 | 4 | 3.5 | |
| 1_7 | 6 | 6 | 3 | 4 | 5 | 1 | 1 | 6 | 6 | 2 | 4 | |
| 1_6 | 5 | 5 | 5 | 5 | 4 | 6 | 4 | 5 | 5 | 6 | 5 | |
| MOD2 | 2_3 | 3 | 1 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 2 | 1.5 |
| 2_2 | 2 | 2 | 2 | 2 | 1 | 1 | 2 | 2 | 2 | 1 | 1.7 | |
| 2_1 | 1 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 2.8 | |
| MOD3 | 3_2 | 1 | 2 | 1 | 2 | 2 | 1 | 1 | 2 | 1 | 1 | 1.4 |
| 3_1 | 2 | 1 | 2 | 1 | 1 | 2 | 2 | 1 | 2 | 2 | 1.6 | |
| 3_3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
3.3. The characteristics of MUBD‐CCR2
To build CCR2 ligand set, we initially collected 987 raw CCR2 antagonists (IC50 ≤ 1 µM). Then, they were carefully checked, curated, and prepared, which resulted in 891 protonated ligands (entities). Analogue excluding further reduced number of ligands to 60. Therefore, the final CCR2 ligand set includes 60 diverse and protonated ligands. Table S2 lists the CHEMBL IDs and the chemical structures of these ligands. As mentioned in the Method section, decoys were generated by our in‐house program named MUBD‐DecoyMaker. It generated 39 decoys for each ligand, and thus, a total of 2,340 unbiased decoys. Figure S5 shows most ROC curves of two nearest neighbor classifiers based on simp, and MACCS sims are close to the random distribution curve. Both values of mean(ROC AUCs) are 0.468 and 0.559, respectively, which are close to the value for random distribution, that is, 0.500. Besides, Figure S6 shows distribution curves for most physicochemical properties of ligands and decoys in MUBD‐CCR2 match well, for example, AlogP, RBs, and NC. All the above data indicate MUBD‐CCR2 is unbiased in terms of “artificial enrichment” and “analogue bias,” and thus, ligand enrichment assessment in this study based on MUBD‐CCR2 would be fair. Meanwhile, the data imply that the benchmarking set is quite challenging as the commonly used similarity search based on MACCS keys is insufficient to discriminate CCR2 antagonists from the presumed inactive compounds.
3.4. Ligand enrichment of ligand‐induced fit models
3.4.1. Models optimized by EN‐NMA plus FD
Among 110 pairs of models/scoring functions evaluated, the pairs of 81_04/PMF04 and 80_03/PMF04 show the maximum of ROCE1% values, that is, 14.29. (cf. Table 4) However, values of ROC AUC for 81_04/PMF04 (0.71) are greater than that for 80_03/PMF04 (0.67). (cf. Table 5) Meanwhile, Figure S7 clearly shows ROC curve for 81_04/PMF04 is above that for 80_03/PMF04. Therefore, 81_04/PMF04 performs better in terms of ligand enrichment than 80_03/PMF04. It is concluded that the best ligand‐induced fit model from EN‐NMA plus FD is 81_04 and the corresponding scoring function is PMF04. As shown in Tables 4 and 5, 81_04/PMF04 performs much better in terms of both early recognition and overall enrichment than MOD1/PMF04 (ROCE1%: 14.29 vs. 7.15; ROC AUC: 0.71 vs. 0.69) as well as the best pair for MOD1, that is, MOD1/PLP1 (ROCE1%: 14.29 vs. 8.93; ROC AUC: 0.71 vs. 0.59). These data demonstrate the use of strategy I, that is, EN‐NMA plus FD boosts ligand enrichment as expected.
TABLE 4.
Early enrichment (i.e., ROCE1%) for different pairs of models/scoring functions
| Source | Model | ChemScore | Ludi_1 | Ludi_2 | Ludi_3 | Ligscore1 | Ligscore2 | ‐PLP1 | ‐PLP2 | Jain | ‐PMF | ‐PMF04 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EN‐NMA + FD | 81_04 | 0.00 | 1.79 | 1.79 | 0.00 | 3.57 | 1.79 | 0.00 | 1.79 | 3.57 | 3.57 | 14.29 |
| 94_15 | 0.00 | 1.79 | 1.79 | 1.79 | 3.57 | 5.36 | 1.79 | 1.79 | 3.57 | 1.79 | 7.15 | |
| 44_08 | 1.79 | 1.79 | 1.79 | 0.00 | 1.79 | 5.36 | 0.00 | 0.00 | 1.79 | 5.36 | 1.79 | |
| 33_02 | 0.00 | 3.57 | 3.57 | 0.00 | 0.00 | 0.00 | 0.00 | 1.79 | 1.79 | 3.57 | 3.57 | |
| 20_04 | 1.79 | 1.79 | 1.79 | 0.00 | 1.79 | 0.00 | 1.79 | 7.15 | 1.79 | 0.00 | 7.15 | |
| 09_11 | 1.67 | 0.00 | 0.00 | 1.67 | 0.00 | 0.00 | 1.67 | 3.33 | 1.67 | 5.00 | 5.00 | |
| 80_03 | 1.79 | 0.00 | 0.00 | 0.00 | 3.57 | 3.57 | 1.79 | 3.57 | 3.57 | 5.36 | 14.29 | |
| 71_20 | 1.79 | 0.00 | 0.00 | 1.79 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 3.57 | 3.57 | |
| 65_04 | 1.79 | 1.79 | 0.00 | 0.00 | 0.00 | 0.00 | 3.57 | 1.79 | 3.57 | 0.00 | 10.72 | |
| 26_02 | 1.79 | 0.00 | 0.00 | 0.00 | 3.57 | 5.36 | 0.00 | 1.79 | 1.79 | 0.00 | 3.57 | |
| DS/Ligand minimization | 1_3 | 1.79 | 0.00 | 0.00 | 0.00 | 8.93 | 3.57 | 1.79 | 0.00 | 3.57 | 1.79 | 1.79 |
| 2_3 | 1.79 | 0.00 | 1.79 | 0.00 | 1.79 | 1.79 | 1.79 | 1.79 | 1.79 | 0.00 | 7.15 | |
| 3_2 | 0.00 | 0.00 | 0.00 | 0.00 | 1.79 | 1.79 | 0.00 | 0.00 | 1.79 | 1.79 | 1.79 | |
| Modeler | MOD1 | 1.79 | 0.00 | 0.00 | 0.00 | 0.00 | 1.79 | 8.93 | 3.57 | 1.79 | 3.57 | 7.15 |
| MOD2 | 1.79 | 1.79 | 3.57 | 3.57 | 0.00 | 0.00 | 1.79 | 1.79 | 1.79 | 1.79 | 7.15 | |
| MOD3 | 1.79 | 3.57 | 5.36 | 1.79 | 1.79 | 0.00 | 3.57 | 8.93 | 3.57 | 1.79 | 10.72 |
TABLE 5.
Overall Enrichment (i.e., ROC AUC) for different pairs of models/scoring functions
| Source | Model | ChemScore | Ludi_1 | Ludi_2 | Ludi_3 | Ligscore1 | Ligscore2 | ‐PLP1 | ‐PLP2 | Jain | ‐PMF | ‐PMF04 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EN‐NMA + FD | 81_04 | 0.49 | 0.49 | 0.52 | 0.44 | 0.61 | 0.57 | 0.60 | 0.61 | 0.56 | 0.59 | 0.71 |
| 94_15 | 0.56 | 0.48 | 0.53 | 0.37 | 0.55 | 0.49 | 0.55 | 0.53 | 0.57 | 0.56 | 0.68 | |
| 44_08 | 0.53 | 0.44 | 0.47 | 0.33 | 0.58 | 0.56 | 0.55 | 0.57 | 0.58 | 0.53 | 0.63 | |
| 33_02 | 0.46 | 0.45 | 0.50 | 0.35 | 0.46 | 0.40 | 0.51 | 0.53 | 0.50 | 0.53 | 0.63 | |
| 20_04 | 0.46 | 0.44 | 0.48 | 0.35 | 0.55 | 0.52 | 0.60 | 0.58 | 0.54 | 0.59 | 0.67 | |
| 09_11 | 0.46 | 0.43 | 0.46 | 0.36 | 0.56 | 0.55 | 0.46 | 0.47 | 0.52 | 0.47 | 0.58 | |
| 80_03 | 0.52 | 0.45 | 0.46 | 0.42 | 0.57 | 0.56 | 0.56 | 0.55 | 0.51 | 0.53 | 0.67 | |
| 71_20 | 0.47 | 0.41 | 0.44 | 0.34 | 0.51 | 0.51 | 0.51 | 0.53 | 0.54 | 0.50 | 0.58 | |
| 65_04 | 0.50 | 0.47 | 0.51 | 0.40 | 0.52 | 0.49 | 0.55 | 0.57 | 0.63 | 0.61 | 0.74 | |
| 26_02 | 0.45 | 0.43 | 0.46 | 0.35 | 0.57 | 0.55 | 0.54 | 0.52 | 0.51 | 0.48 | 0.58 | |
| DS/Ligand Minimization | 1_3 | 0.47 | 0.49 | 0.53 | 0.37 | 0.65 | 0.63 | 0.52 | 0.52 | 0.57 | 0.53 | 0.66 |
| 2_3 | 0.51 | 0.47 | 0.49 | 0.41 | 0.54 | 0.52 | 0.57 | 0.56 | 0.55 | 0.59 | 0.69 | |
| 3_2 | 0.53 | 0.49 | 0.54 | 0.41 | 0.56 | 0.55 | 0.53 | 0.57 | 0.62 | 0.53 | 0.60 | |
| Modeler | MOD1 | 0.51 | 0.44 | 0.47 | 0.39 | 0.57 | 0.55 | 0.59 | 0.58 | 0.51 | 0.54 | 0.69 |
| MOD2 | 0.51 | 0.47 | 0.51 | 0.42 | 0.51 | 0.49 | 0.57 | 0.57 | 0.51 | 0.55 | 0.67 | |
| MOD3 | 0.53 | 0.49 | 0.53 | 0.41 | 0.50 | 0.49 | 0.55 | 0.60 | 0.57 | 0.55 | 0.66 |
3.4.2. Models optimized via DS/ligand minimization
In three optimized models from strategy II, model 1_3 performs the best when combined with Ligscore 1, with ROCE1% value of 8.93 and ROC AUC value of 0.65. For the model of 2_3, it shows the best performance when combined with PMF04. The values of ROCE1% and ROC AUC are 7.15 and 0.69, respectively. For the model of 3_2, its combinations with 11 scoring functions do not possess high ligand enrichment. Even for the pair of 3_2/PMF04, ROCE1% value is only 1.79 and ROC AUC is 0.60. Therefore, the best combination of models from DS/Ligand Minimization and 11 scoring functions is 1_3/Ligscore1.
To be noted, the best combination from DS/Ligand Minimization, that is, 1_3/Ligscore1, is worse than that from Modeler, that is, MOD3/PMF04 (ROCE1% = 10.72, ROC AUC = 0.66) in terms of ligand enrichment. We also compared the best combinations for each optimized model from DS/Ligand Minimization and its initial model. The result shows (a) the ligand enrichment of 1_3/Ligscore1 is equivalent to its initial model combined with PLP1 (ROCE1%: 8.93 vs. 8.93; AUC: 0.65 vs. 0.59); (b) ROCE1% value of 2_3/PMF04 is equal to that of MOD2/PMF04 (7.15) while its AUC value is slightly greater (0.69 vs. 0.67); (c) ROCE1% value of 3_2/PMF04 is 1.79, which is much less than that of MOD3/PMF04 (10.72). These data demonstrate the strategy that consists of DS/Ligand Minimization fails to generate ligand‐induced fit models of high enrichment as well as improve ligand enrichment of initial models. Structural clustering shows trivial difference between each initial model and its optimized model, which may explain their equivalent performance in terms of ligand enrichment (cf. Figure S8).
3.4.3. Comparison between EN‐NMA plus FD and DS/ligand minimization
From the above data (cf. Tables 4 and 5), we identified the best model/scoring function for each strategy, that is, 81_04/PMF04 from EN‐NMA plus FD, 1_3/Ligscore1 from DS/Ligand Minimization, and MOD3/PMF04 from Modeler. According to the respective ROC curve in Figure 4, it is obvious that 81_04/PMF04 is better than 1_3/Ligscore1. Specifically, the ROC enrichment of the 81‐04/PMF04 approach from our strategy was 5.36 times higher than the 1_3/Ligscore approach from DS/Minimization (14.29 vs. 8.93), when the false‐positive rate was 1% (cf. Table 4). The CCR2 antagonists retrieved by those two approaches at 1% recovered known decoys are listed in Table S3. As is shown, the 81_04/PMF04 approach can identify 8 CCR2 antagonists with eight Bemis‐Murcko scaffolds, whereas the 1_3/Ligscore1 approach only retrieved five actives with five Bemis‐Murcko scaffolds. Therefore, our unique strategy is better than the reported DS/Ligand Minimization.
FIGURE 4.
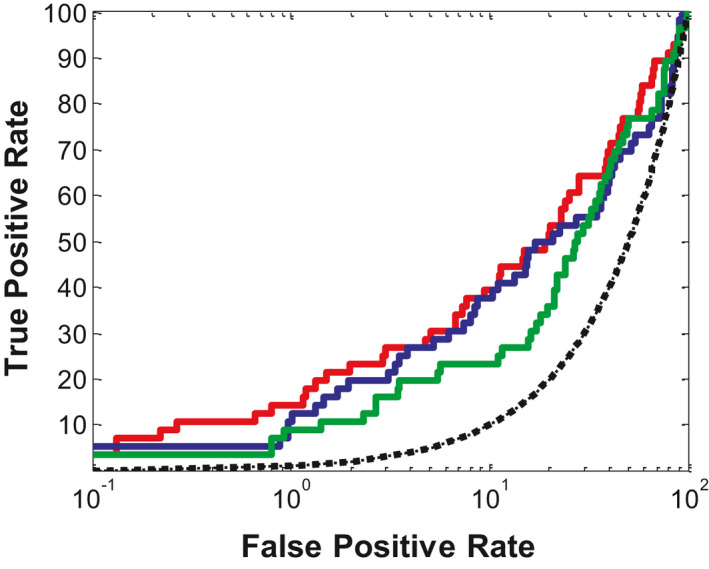
Ligand enrichments for three combinations of ligand‐induced fit model/scoring function. Color code: red, 81_04/PMF04; blue, MOD3/PMF04; green, 1_3/Ligscore1
3.5. The selected ligand‐induced fit model: binding mode and mutagenesis data
The model of 81_04 possesses high enrichment when combined with PMF04. Besides, its proposed binding mode with Teijin lead complies with the observations from mutagenesis study (Berkhout et al., 2003). It was predicted that the trifluoromethyl phenyl group of Teijin lead was located in a highly hydrophobic cavity composed of Tyr120, His121, and Tyr124. Consistently, the mutation from Tyr120 or His121 to an alanine did not significantly reduce the antagonistic activity of the Teijin lead, with only 5.4‐fold or 7.6‐fold change in binding IC50 (Kim et al., 2011). The charged amine group of pyrolidine forms salt bridge with Glu291. Such an interaction is plausible as the mutation from Glu291 to Gln291 led to complete loss of bioactivity. 2, 4‐Dimethylbenzyl group forms hydrophobic interactions with Trp128, Ala132, and Phe146 (cf. Figure 5). The binding mode acquired from the best model, that is, 81_04 may aid in the selection of potential hits in real‐world screening.
FIGURE 5.
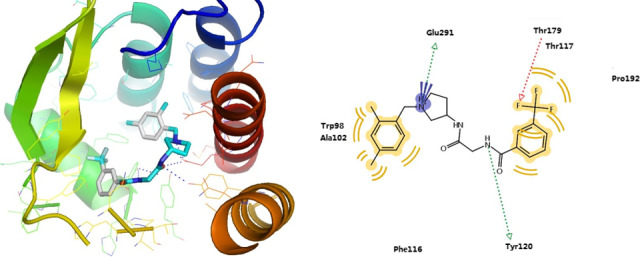
The best ligand‐induced fit model 81_04 generated by EN‐NMA plus FD and its binding mode with Teijin lead
3.6. The hit compound from virtual screening
We cherry‐picked four compounds, that is, E859‐1281, 0546‐0087, M077‐0425, and C561‐2092, from 15 compounds with the ‐PMF04 scores greater than 110 due to their similar binding mode to Teijin lead. Since these compounds were not annotated as active for CCR2 by PubChem, it was interesting to explore their inhibitory effects on CCR2. The outcome from calcium flux assay showed E859‐1281 was able to inhibit CCR2b at the concentration of 100 μM and the inhibition rate was 37.7 ± 2.0% (cf. Tables 6 and S4). By calculating pairwise similarity, that is, Tanimoto coefficient (Tc) based on FCFP_6 fingerprints of E859‐1281 to those reported CCR2 antagonists (IC50 ≤ 1 μM), we identified its nearest neighbor was CHEMBL1210670. The Tc value between E859‐1281 and CHEMBL1210670 was 0.182 (cf. Figure 6), which indicated its structure was diverse compared with the known CCR2 antagonists.
TABLE 6.
The three hit compounds as CCR2 antagonists from structure‐based virtual screening and substructure search
| Structure | Compound ID | MW | AlogP | No. of hydrogen bond donor | No. of hydrogen bond acceptors | CCR2 inhibition% at 100 μM |
|---|---|---|---|---|---|---|
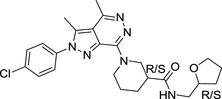
|
E859‐1281 (4 isomers) | 468.98 | 3.12 | 1 | 6 | 37.7 ± 2.0 |
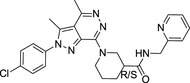
|
MolPort‐007‐767‐945 (2 isomers) | 475.97 | 3.41 | 1 | 6 | 33.2 ± 2.3 |
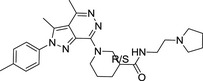
|
MolPort‐007‐768‐014 (2 isomers) | 461.60 | 3.19 | 1 | 6 | 16.6 ± 1.8 |
Their chemical structures, physicochemical properties, and biological activity are listed.
FIGURE 6.
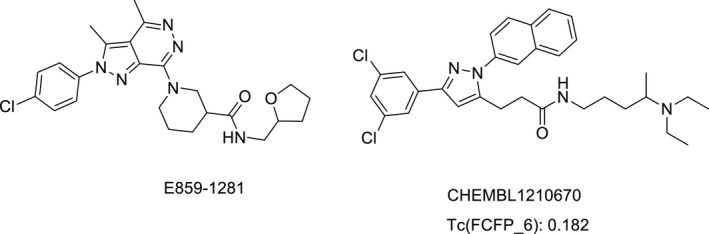
The hit compound E859‐1281 and its nearest neighbor, that is, the most similar CCR2 antagonist CHEMBL1210670
3.6.1. Analogues of E859‐1281 from substructure search
We performed substructure search to retrieve analogues of E859‐1281 from Molport database (https://www.molport.com, accessed in Jun. 2014). The substructures used are shown in Figure S9. By further molecular docking against the optimal model 81_04, we selected 10 analogues for bioassay. Among them, MolPort‐007‐767‐945 and MolPort‐007‐768‐014 showed 33.2 ± 2.3% and 16.6 ± 1.8% inhibition on CCR2 at the concentration of 100 μM, respectively (cf. Tables 6 and S3). By exploring the two‐dimension chemical structures of 10 bioassayed compounds, we identified the N‐3,4‐dimethyl‐2H‐pyrazolo[3,4‐d]pyridazin and 3‐carbonyl group substituted piperidine were shared by those active compounds. Besides, we noted the 3 carbon of piperidine in each active compound was chiral, and thus, the compounds we purchased and tested were composed of multiple isomers. To be specific, E859‐1281 contained four isomers, while both MolPort‐007‐767‐945 and MolPort‐007‐768‐014 included two isomers. To uncover the specific isomer that took effect, we docked all the isomers against the CCR2 model and explored their binding modes. Interestingly, only when the carbon atom of piperidine was R‐type, the binding modes of the shared core scaffold were the same for three hit compounds (cf. Figure 7), that is, the formation of salt bridge with Glu291 and hydrogen bond with Tyr120. The predicted binding modes also seemed to be consistent with the potency of these R‐type compounds. For instance, the terminal group of E859‐1281 interacted with Arg206 by hydrogen bond. Likewise, the compound of equivalent potency, that is, MolPort‐007‐767‐945 formed hydrogen bond with Thr117. By contrast, no hydrogen bond was observed for the weakest compound, MolPort‐007‐768‐014. Based on these observations, we proposed the R‐type isomers were the active components of the hit compounds and the core structure was the most important part to maintain its activity. Therefore, we anticipate the pure R‐type isomer of E859‐1281 and MolPort‐007‐767‐945 would possess higher potency.
FIGURE 7.
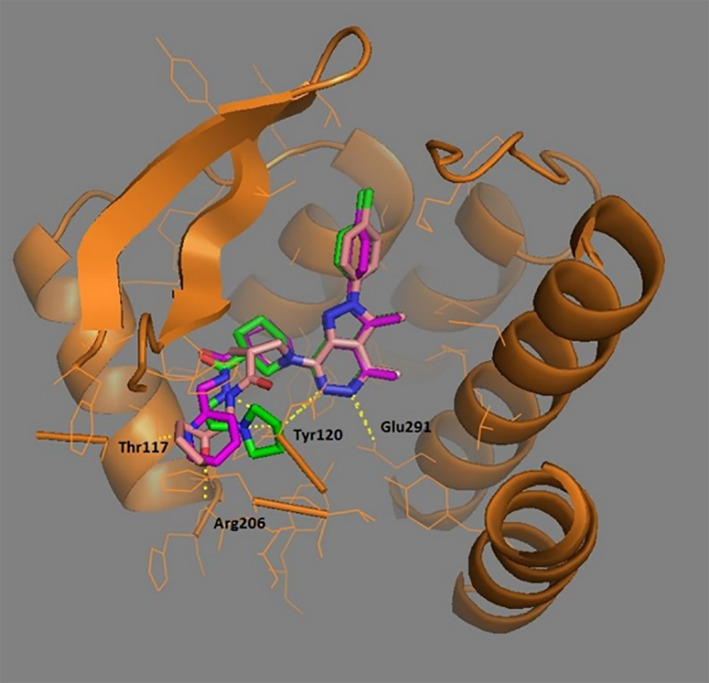
The proposed binding modes of three compounds. Color code: red, E859‐1281; magentas, MolPort‐007‐767‐945; green, MolPort‐007‐768‐014
4. CONCLUSION
In this study, we have developed a unique strategy for LSHM that consists of EN‐NMA and FD by AutoDock Vina. Using CXCR4 crystal structure as a template, we applied the proposed strategy to build multiple CCR2 homology models. Then, we used an in‐house program implemented in Pipeline Pilot, that is, MUBD‐DecoyMaker to construct a maximal unbiased benchmarking set for CCR2, that is, MUBD‐CCR2. Based on this dataset, we have performed retrospective small‐scale virtual screening to evaluate the performance of all the constructed models in enriching CCR2 antagonists. The model of 81_04 has been selected out as the best CCR2 model, as (a) it shows the highest enrichment when combined with the scoring function of PMF04, and (b) the proposed binding mode between CCR2 and Teijin lead by this model is consistent with the reported mutagenesis data. Nevertheless, it should be noted that the correlation coefficient between the ‐PMF04 scores and pIC50 values of the potent CCR2 antagonists in MUBD‐CCR2 (cf. Figure S10) was quite low, indicating the 81_04/PMF04 approach remains to be improved.
We also reproduced the reported LSHM protocol termed as DS/Ligand Minimization here, with the aim to uncover the advantage of our unique protocol. For a fair comparison, we used CXCR4 crystal structure as a template to build CCR2 homology models. Ligand enrichment of all generated CCR2 models has been evaluated. Among them, the model of 1_3 has been validated as the best when combined with Ligscore1. Nevertheless, ligand enrichment of 1_3/Ligscore1 is much worse than 81_04/PMF04. Surprisingly, it is even worse than the combination of an initial model, that is, MOD3 and PMF04. According to clustering analysis, optimized models by DS/Ligand Minimization are quite similar to initial models from Modeler. This may explain why DS/Ligand Minimization is not able to improve ligand enrichment. By contrast, the performance of 81_04/PMF04 is much better than its initial model, that is, MOD1 combined with PMF04 or PLP1. These data demonstrate our novel strategy using EN‐NMA plus FD is more capable of optimizing homology models than the reported one using DS/Ligand Minimization. In fact, we have found DS/Ligand Minimization is limited in model optimization, though it was reported to be effective.
Furthermore, we screened an in‐house focused library by molecular docking against 81_04 and scoring by PMF04. Cherry‐picking rendered four potential hits for CCR2 antagonist bioassay, of which E859‐1281 was confirmed as active. Interestingly, the DS/Ligand Minimization strategy with 1_3/Ligscore1 as a representative approach was not likely to identify E859‐1281 due to its low rank at the compound list (cf. Table S6). The substructure search and bioassay further identified an analogue; that is, MolPort‐007‐767‐945 was equivalent to E859‐1281 in terms of potency. We noticed E859‐1281 or MolPort‐007‐767‐945 was a mixture of multiple isomers, and thus predicted their R‐type isomer may be more potent than the mixture and proposed further isolation. This prospective SBVS campaign has demonstrated the efficacy of the constructed model and its modeling strategy.
In fact, the strategy that comprises EN‐NMA plus FD is not limited to the CCR2 modeling, but also applicable for other GPCR targets. As long as a homologous protein that possesses certain sequence identity and crystal structures are available, then the protocol would be fairly good for structure prediction. We anticipate this new workflow for LSHM will be widely applied for GPCRs‐targeted drug discovery.
CONFLICT OF INTEREST
The authors declare no competing financial interest.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by District of Columbia Developmental Center for AIDS Research (P30AI087714), National Institutes of Health Administrative Supplements for U.S.‐China Biomedical Collaborative Research (5P30AI0877714‐02), the National Institute on Minority Health and Health Disparities of the National Institutes of Health under Award Number G12MD007597. We are also grateful to the China Scholarship Council (CSC) (201206010076) and National Natural Science foundation of China (NSFC 91213302, 81161120404).
Jin H, Xia J, Liu Z, Wang XS, Zhang L. A unique ligand‐steered strategy for CC chemokine receptor 2 homology modeling to facilitate structure‐based virtual screening. Chem Biol Drug Des. 2021;97:944–961. 10.1111/cbdd.13820
Contributor Information
Xiang Simon Wang, Email: x.simon.wang@gmail.com.
Liangren Zhang, Email: liangren@bjmu.edu.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Bento, A. P. , Gaulton, A. , Hersey, A. , Bellis, L. J. , Chambers, J. , Davies, M. , Krüger, F. A. , Light, Y. , Mak, L. , McGlinchey, S. , Nowotka, M. , Papadatos, G. , Santos, R. , & Overington, J. P. (2014). The ChEMBL bioactivity database: An update. Nucleic Acids Research, 42, D1083–D1090. 10.1093/nar/gkt1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkhout, T. A. , Blaney, F. E. , Bridges, A. M. , Cooper, D. G. , Forbes, I. T. , Gribble, A. D. , Groot, P. H. , Hardy, A. , Ife, R. J. , Kaur, R. , Moores, K. E. , Shillito, H. , & Willetts, J. , Witherington, J. (2003). CCR2: Characterization of the antagonist binding site from a combined receptor modeling/mutagenesis approach. Journal of Medicinal Chemistry, 46(19), 4070–4086. 10.1021/jm030862l [DOI] [PubMed] [Google Scholar]
- Bozzi, Y. , & Caleo, M. (2016). Epilepsy, seizures, and inflammation: Role of the C‐C motif ligand 2 chemokine. DNA Cell Biology, 35(6), 257–260. 10.1089/dna.2016.3345 [DOI] [PubMed] [Google Scholar]
- Brown, G. D. , Shi, Q. , Delucca, G. V. , Batt, D. G. , Galella, M. A. , Cvijic, M. E. , Liu, R.‐Q. , Qiu, F. , Zhao, Q. , Barrish, J. C. , & Carter, P. H. (2016). Discovery and synthesis of cyclohexenyl derivatives as modulators of CC chemokine receptor 2 activity. Bioorganic & Medicinal Chemistry Letters, 26(2), 662–666. 10.1016/j.bmcl.2015.11.051 [DOI] [PubMed] [Google Scholar]
- Carter, P. H. (2013). Progress in the discovery of CC chemokine receptor 2 antagonists, 2009–2012. Expert Opinion on Therapeutic Patents, 23(5), 549–568. 10.1517/13543776.2013.771168 [DOI] [PubMed] [Google Scholar]
- Carter, P. H. , Brown, G. D. , Cherney, R. J. , Batt, D. G. , Chen, J. , Clark, C. M. , Cvijic, M. E. , Duncia, J. V. , Ko, S. S. , Mandlekar, S. , Mo, R. , Nelson, D. J. , Pang, J. , Rose, A. V. , Santella, J. B. , Tebben, A. J. , Traeger, S. C. , Xu, S. , Zhao, Q. , & Barrish, J. C. (2015). Discovery of a potent and orally bioavailable dual antagonist of CC chemokine receptors 2 and 5. ACS Medicinal Chemistry Letters, 6(4), 439–444. 10.1021/ml500505q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavasotto, C. N. , Orry, A. J. , Murgolo, N. J. , Czarniecki, M. F. , Kocsi, S. A. , Hawes, B. E. , O’Neill, K. A. , Hine, H. , Burton, M. S. , Voigt, J. H. , Abagyan, R. A. , Bayne, M. L. , & Monsma, F. J. (2008). Discovery of novel chemotypes to a G‐protein‐coupled receptor through ligand‐steered homology modeling and structure‐based virtual screening. Journal of Medicinal Chemistry, 51(3), 581–588. 10.1021/jm070759m [DOI] [PubMed] [Google Scholar]
- Cerri, C. , Genovesi, S. , Allegra, M. , Pistillo, F. , Puntener, U. , Guglielmotti, A. , Perry, V. H. , Bozzi, Y. , & Caleo, M. (2016). The chemokine CCL2 mediates the seizure‐enhancing effects of systemic inflammation. Journal of Neuroscience, 36(13), 3777–3788. 10.1523/JNEUROSCI.0451-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavan, S. , Pawar, S. , Singh, R. , & Sobhia, M. F. (2012). Binding site characterization of G protein‐coupled receptor by alanine‐scanning mutagenesis using molecular dynamics and binding free energy approach: Application to C‐C chemokine receptor‐2 (CCR2). Molecular Diversity, 16(2), 401–413. 10.1007/s11030-012-9368-z [DOI] [PubMed] [Google Scholar]
- Covino, D. A. , Sabbatucci, M. , & Fantuzzi, L. (2016). The CCL2/CCR2 axis in the pathogenesis of HIV‐1 infection: A new cellular target for therapy. Current Drug Targets, 17(1), 76–110. 10.2174/138945011701151217110917 [DOI] [PubMed] [Google Scholar]
- Dietzen, M. , Zotenko, E. , Hildebrandt, A. , & Lengauer, T. (2012). On the applicability of elastic network normal modes in small‐molecule docking. Journal of Chemical Information and Modeling, 52(3), 844–856. 10.1021/ci2004847 [DOI] [PubMed] [Google Scholar]
- Fantuzzi, L. , Tagliamonte, M. , Gauzzi, M. C. , & Lopalco, L. (2019). Dual CCR5/CCR2 targeting: Opportunities for the cure of complex disorders. Cellular and Molecular Life Science, 76(24), 4869–4886. 10.1007/s00018-019-03255-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton, A. , Bellis, L. J. , Bento, A. P. , Chambers, J. , Davies, M. , Hersey, A. , Light, Y. , McGlinchey, S. , Michalovich, D. , & Al‐Lazikani, B. , & Overington, J. P. (2012). ChEMBL: A large‐scale bioactivity database for drug discovery. Nucleic Acids Research, 40, D1100–D1107. 10.1093/nar/gkr777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartenfeller, M. , & Schneider, G. (2011). De novo drug design. Methods in Molecular Biology, 672, 299–323. 10.1007/978-1-60761-839-3_12 [DOI] [PubMed] [Google Scholar]
- Huang, Y. X. , Zhao, J. , Song, Q. H. , Zheng, L. H. , Fan, C. , Liu, T. T. , Bao, Y. , Sun, L. , Zhang, L. , & Li, Y. X. (2016). Virtual screening and experimental validation of novel histone deacetylase inhibitors. BMC Pharmacology & Toxicology, 17, 32. 10.1186/s40360-016-0075-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin, J. J. , & Shoichet, B. K. (2005). ZINC‐ a free database of commercially available compounds for virtual screening. Journal of Chemical Information and Modeling, 45(1), 177–182. 10.1021/ci049714+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin, J. J. , Sterling, T. , Mysinger, M. M. , Bolstad, E. S. , & Coleman, R. G. (2012). ZINC: A free tool to discover chemistry for biology. Journal of Chemical Information and Modeling, 52(7), 1757–1768. 10.1021/ci3001277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn, A. , Hinselmann, G. , Fechner, N. , & Zell, A. (2009). Optimal assignment methods for ligand‐based virtual screening. Journal of Cheminformatics, 1, 14. 10.1186/1758-2946-1-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazayeri, A. , Andrews, S. P. , & Marshall, F. H. (2017). Structurally enabled discovery of adenosine A2A receptor antagonists. Chemical Reviews, 117(1), 21–37. 10.1021/acs.chemrev.6b00119 [DOI] [PubMed] [Google Scholar]
- Kim, J. H. , Lim, J. W. , Lee, S. W. , Kim, K. , & No, K. T. (2011). Ligand supported homology modeling and docking evaluation of CCR2: Docked pose selection by consensus scoring. Journal of Molecular Modeling, 17(10), 2707–2716. 10.1007/s00894-010-0943-x [DOI] [PubMed] [Google Scholar]
- Kimura, S. R. , Tebben, A. J. , & Langley, D. R. (2008). Expanding GPCR homology model binding sites via a balloon potential: A molecular dynamics refinement approach. Proteins‐Structure Function and Bioinformatics, 71(4), 1919–1929. 10.1002/prot.21906 [DOI] [PubMed] [Google Scholar]
- Kothandan, G. , Gadhe, C. G. , & Cho, S. J. (2012). Structural insights from binding poses of CCR2 and CCR5 with clinically important antagonists: A combined in silico study. PLoS One, 7(3), e32864. 10.1371/journal.pone.0032864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufareva, I. , Katritch, V. ,GPCR Dock 2008 participants , Stevens, R. C. , & Abagyan, R. (2014). Advances in GPCR modeling evaluated by the GPCR Dock 2013 assessment: Meeting new challenges. Structure, 22(8), 1120–1139. 10.1016/j.str.2014.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufareva, I. , Rueda, M. , Katritch, V. , Stevens, R. C. , GPCR Dock 2008 participants , Abagyan, R. (2011). Status of GPCR modeling and docking as reflected by community‐wide GPCR Dock 2010 assessment. Structure, 19(8), 1108–1125. 10.1016/j.str.2011.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, R. A. , Macarthur, M. W. , Moss, D. S. , & Thornton, J. M. (1993). PROCHECK—A program to check the stereochemical quality of protein structures. Journal of Applied Crystallography, 26, 283–291. 10.1107/S0021889892009944 [DOI] [Google Scholar]
- Lin, X. , Huang, X. P. , Chen, G. , Whaley, R. , Peng, S. , Wang, Y. , Zhang, G. , Wang, S. X. , Wang, S. , Roth, B. L. , & Huang, N. (2012). Life beyond kinases: Structure‐based discovery of sorafenib as nanomolar antagonist of 5‐HT receptors. Journal of Medicinal Chemistry, 55(12), 5749–5759. 10.1021/jm300338m [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michino, M. , Abola, E. , GPCR Dock 2008 participants , Brooks, C. L. , Dixon, J. S. , Moult, J. , & Stevens, R. C. (2009). Community‐wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nature Reviews Drug Discovery, 8(6), 455–463. 10.1038/nrd2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzadegan, T. , Diehl, F. , Ebi, B. , Bhakta, S. , Polsky, I. , McCarley, D. , Mulkins, M. , Weatherhead, G. S. , Lapierre, J.‐M. , Dankwardt, J. , Morgans, D. , Wilhelm, R. , & Jarnagin, K. (2000). Identification of the binding site for a novel class of CCR2b chemokine receptor antagonists: Binding to a common chemokine receptor motif within the helical bundle. The Journal of Biological Chemistry, 275(33), 25562–25571. 10.1074/jbc.M000692200 [DOI] [PubMed] [Google Scholar]
- Mysinger, M. M. , Carchia, M. , Irwin, J. J. , & Shoichet, B. K. (2012). Directory of useful decoys, enhanced (DUD‐E): Better ligands and decoys for better benchmarking. Journal of Medicinal Chemistry, 55(14), 6582–6594. 10.1021/jm300687e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls, A. (2008). What do we know and when do we know it? Journal of Computer‐Aided Molecular Design, 22(3–4), 239–255. 10.1007/s10822-008-9170-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor, T. , Borsig, L. , & Heikenwalder, M. (2015). CCL2‐CCR2 signaling in disease pathogenesis. Endocrine Metabolic & Immune Disorders‐Drug Targets, 15(2), 105–118. 10.2174/1871530315666150316120920 [DOI] [PubMed] [Google Scholar]
- Pei, F. , Jin, H. W. , Zhou, X. , Xia, J. , Sun, L. D. , Liu, Z. M. , & Zhang, L. R. (2015). Enrichment assessment of multiple virtual screening strategies for toll‐like receptor 8 agonists based on a maximal unbiased benchmarking data set. Chemical Biology & Drug Design, 86(5), 1226–1241. 10.1111/cbdd.12590 [DOI] [PubMed] [Google Scholar]
- Phatak, S. S. , Gatica, E. A. , & Cavasotto, C. N. (2010). Ligand‐steered modeling and docking: A benchmarking study in class A G‐protein‐coupled receptors. Journal of Chemical Information and Modeling, 50(12), 2119–2128. 10.1021/ci100285f [DOI] [PubMed] [Google Scholar]
- Rai, B. K. , Tawa, G. J. , Katz, A. H. , & Humblet, C. (2010). Modeling G protein‐coupled receptors for structure‐based drug discovery using low‐frequency normal modes for refinement of homology models: Application to H3 antagonists. Proteins‐Structure Function and Bioinformatics, 78(2), 457–473. 10.1002/prot.22571 [DOI] [PubMed] [Google Scholar]
- Rodrigues, T. , Reker, D. , Welin, M. , Caldera, M. , Brunner, C. , Gabernet, G. , Schneider, P. , Walse, B. , & Schneider, G. (2015). De novo fragment design for drug discovery and chemical biology. Angewandte Chemie International Edition, 54(50), 15079–15083. 10.1002/anie.201508055 [DOI] [PubMed] [Google Scholar]
- Rueda, M. , Bottegoni, G. , & Abagyan, R. (2009). Consistent improvement of cross‐docking results using binding site ensembles generated with elastic network normal modes. Journal of Chemical Information and Modeling, 49(3), 716–725. 10.1021/ci8003732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali, A. , & Blundell, T. L. (1993). Comparative protein modelling by satisfaction of spatial restraints. Journal of Molecular Biology, 234(3), 779–815. 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- Serbina, N. V. , & Pamer, E. G. (2006). Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nature Immunology, 7(3), 311–317. 10.1038/ni1309 [DOI] [PubMed] [Google Scholar]
- Shahlaei, M. , Fassihi, A. , Papaleo, E. , & Pourfarzam, M. (2013). Molecular dynamics simulation of chemokine receptors in lipid bilayer: A case study on C‐C chemokine receptor type 2. Chemical Biology & Drug Design, 82(5), 534–545. 10.1111/cbdd.12179 [DOI] [PubMed] [Google Scholar]
- Shi, L. , & Javitch, J. A. (2002). The binding site of aminergic G protein‐coupled receptors: The transmembrane segments and second extracellular loop. Annual Review of Pharmacology & Toxicology, 42, 437–467. 10.1146/annurev.pharmtox.42.091101.144224 [DOI] [PubMed] [Google Scholar]
- Shi, X. F. , Liu, S. , Xiangyu, J. , Zhang, Y. , Huang, J. , Liu, S. , & Liu, C. Q. (2002). Structural analysis of human CCR2b and primate CCR2b by molecular modeling and molecular dynamics simulation. Journal of Molecular Modeling, 8(7), 217–222. 10.1007/s00894-002-0089-6 [DOI] [PubMed] [Google Scholar]
- Shiota, T. (2002). Cyclic amine derivatives and their use as drugs, WO PATENT 99/25686.
- Shiota, T. , Kataoka, K. , Imai, M. , Tsutsumi, T. , Sudoh, M. , Sogawa, R. , … Teig, S. (2002). Cyclic amine derivatives and their use as drugs. Official Gazette of the United States Patent and Trademark Office Patents, 1262(3), US 6451842. http://www.uspto.gov/web/menu/patdata.html [Google Scholar]
- Singh, R. , & Sobhia, M. E. (2013). Structure prediction and molecular dynamics simulations of a G‐protein coupled receptor: Human CCR2 receptor. Journal of Biomolecular Structure and Dynamics, 31(7), 694–715. 10.1080/07391102.2012.707460 [DOI] [PubMed] [Google Scholar]
- Strunz, A. K. , Zweemer, A. J. , Weiss, C. , Schepmann, D. , Junke, A. , Heitman, L. H. , Koch, M. , & Wunsch, B. (2015). Synthesis and biological evaluation of spirocyclic antagonists of CCR2 (chemokine CC receptor subtype 2). Bioorganic & Medicinal Chemistry, 23(14), 4034–4049. 10.1016/j.bmc.2015.02.019 [DOI] [PubMed] [Google Scholar]
- Thompson, J. D. , Higgins, D. G. , & Gibson, T. J. (1994). CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position‐specific gap penalties and weight matrix choice. Nucleic Acids Research, 22(22), 4673–4680. 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilums, M. , Zweemer, A. J. , Barmare, F. , van der Gracht, A. M. , Bleeker, D. C. , Yu, Z. , de Vries, H. , Gross, R. , Clemens, J. , Krenitsky, P. , Brussee, J. , Stamos, D. , Saunders, J. , Heitman, L. H. , & Adriaan, P. I. (2015). When structure–affinity relationships meet structure–kinetics relationships: 3‐((Inden‐1‐yl)amino)‐1‐isopropyl‐cyclopentane‐1‐carboxamides as CCR2 antagonists. European Journal of Medicinal Chemistry, 93, 121–134. 10.1016/j.ejmech.2015.01.063 [DOI] [PubMed] [Google Scholar]
- Wang, R. X. , & Wang, S. M. (2001). How does consensus scoring work for virtual library screening? An idealized computer experiment. Journal of Chemical Information and Modeling, 41(5), 1422–1426. 10.1021/ci010025x [DOI] [PubMed] [Google Scholar]
- Waszkowycz, B. , Clark, D. E. , & Gancia, E. (2011). Outstanding challenges in protein–ligand docking and structure‐based virtual screening. Wiley Interdisciplinary Reviews‐ Computational Molecular Science, 1(2), 229–259. 10.1002/wcms.18 [DOI] [Google Scholar]
- Winters, M. P. , Teleha, C. A. , Kang, F. A. , McComsey, D. , O'Neill, J. C. , Hou, C. , Kirchner, T. , Wang, P. , Johnson, D. , & Sui, Z. (2014). The discovery and SAR of cyclopenta[b]furans as inhibitors of CCR2. Bioorganic & Medicinal Chemistry Letters, 24(9), 2137–2140. 10.1016/j.bmcl.2014.03.036 [DOI] [PubMed] [Google Scholar]
- Wu, B. , Chien, E. Y. , Mol, C. D. , Fenalti, G. , Liu, W. , Katritch, V. , Abagyan, R. , Brooun, A. , Wells, P. , Bi, F. , Hamel, D. , Kuhn, P. , Handel, T. , Cherezov, V. , & Stevens, R. C. (2010). Structures of the CXCR4 chemokine GPCR with small‐molecule and cyclic peptide antagonists. Science, 330(6007), 1066–1071. 10.1126/science.1194396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, J. , Jin, H. , Liu, Z. , Zhang, L. , & Wang, X. S. (2014). An unbiased method to build benchmarking sets for ligand‐based virtual screening and its application to GPCRs. Journal of Chemical Information and Modeling, 54(5), 1433–1450. 10.1021/ci500062f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, J. , Tilahun, E. L. , Kebede, E. H. , Reid, T. E. , Zhang, L. R. , & Wang, X. S. (2015). Comparative modeling and benchmarking data sets for human histone deacetylases and sirtuin families. Journal of Chemical Information and Modeling, 55(2), 374–388. 10.1021/ci5005515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, K. , & Luo, J. (2019). Role of MCP‐1 and CCR2 in alcohol neurotoxicity. Pharmacological Research, 139, 360–366. 10.1016/j.phrs.2018.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. Q. , Hou, C. F. , Hufnagel, H. , Singer, M. , Opas, E. , McKenney, S. , Johnson, D. , & Sui, Z. (2012). Discovery of a 4‐azetidinyl‐1‐thiazoyl‐cyclohexane CCR2 antagonist as a development candidate. ACS Medicinal Chemistry Letters, 3(12), 1039–1044. 10.1021/ml300260s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotnik, A. , & Yoshie, O. (2012). The chemokine superfamily revisited. Immunity, 36(5), 705–716. 10.1016/j.immuni.2012.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.