

Abstract
Here we present the rational design and synthetic methodologies towards proteolysis‐targeting chimeras (PROTACs) for the recently‐emerged target leucine‐rich repeat kinase 2 (LRRK2). Two highly potent, selective, brain‐penetrating kinase inhibitors were selected, and their structure was appropriately modified to assemble a cereblon‐targeting PROTAC. Biological data show strong kinase inhibition and the ability of the synthesized compounds to enter the cells. However, data regarding the degradation of the target protein are inconclusive. The reasons for the inefficient degradation of the target are further discussed.
Keywords: cereblon, degradation, LRRK2, Parkinson's disease, PROTAC
Here, we present our efforts for developing LRRK2‐targeting PROTACs, starting from two highly‐potent LRRK2 inhibitors. Although the synthesized PROTACs showed cell permeability and interacted with the target, they failed to induce target degradation. This work highlights the challenges of PROTAC design for multidomain protein targets where the structure is not fully known and the dynamics are not fully understood.
Parkinson's disease (PD) is one of the most common neurodegenerative diseases and, although most PD cases are idiopathic and the etiology is largely unknown, both environmental and genetic factors are implicated. Leucine‐rich repeat kinase 2 (LRRK2) encoded by PARK8 is among the implicated genes for PD. LRRK2 mutations have been observed in a number of idiopathic late‐onset PD or Parkinson′s disease patients and are the most common cause of familial PD.[ 1 , 2 , 3 ] Importantly, recently it has been found that LRRK2 activity was enhanced in postmortem brain tissue from patients with idiopathic PD. [4] Therefore, LRRK2 is considered as an essential player in PD pathogenesis and targeting LRRK2 thus might be beneficial for PD in general.
LRRK2 is a large protein, consisting of 2527 amino acids and including multiple domains (Figure 1). Interestingly, it contains both a kinase and a GTPase domain. Regarding the mutations, they mostly occur in the GTPase and the kinase domain, leading to increased kinase activity and autophosphorylation. A significant number of disease‐associated LRRK2 mutations has been identified to date, among which five missense mutations (R1441C, R1441G, Y1699C, G2019S and I2020T) are linked to PD pathogenesis.[ 5 , 6 ] There have been extensive drug discovery efforts to develop inhibitors for LRRK2, focusing on ATP competitive active‐site kinase inhibitors in particular.
Figure 1.
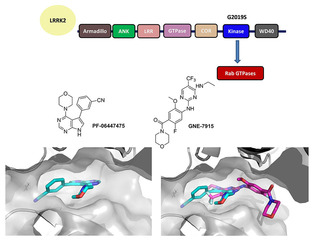
LRRK2, a PD target. Top: Functional LRRK2 moieties. Middle: 2D structures of two potent small molecules (PF‐06447475, GNE‐7915) inhibiting the LRKK2 kinase. Below left: the crystal structure of PF‐06447475 (cyan sticks) with MST3 (grey surface; PDB ID: 4 U8Z), below right overlap of a docking pose of GNE‐7915 (magenta sticks) over PF‐06447475 (cyan sticks) in MST3 active site (grey surface).
Starting in 2006, three patent reviews have been published, covering the numerous potent scaffolds against the target.[ 7 , 8 , 9 ] In 2018, the first clinical trial (NCT03710707) was announced by Denali Therapeutics, followed by a second clinical trial in 2019 (NCT04056689). [10] The structures of DNL201 and DNL151 are not yet disclosed.
Here we explore the possibility of degrading LRRK2 using a proteolysis targeting chimera (PROTAC) strategy, instead of inhibiting its activity. PROTACs in the last few years have shown tremendous opportunities for modulating challenging or traditionally considered “undruggable” targets. Despite the fact that kinase inhibitors for LRRK2 have yet failed to reach the market, the potential of degrading LRRK2, as an alternative aim, has not been thoroughly investigated. In our approach, the starting points for designing and synthesizing PROTACs were known kinase inhibitors. Due to the abundance of scaffolds in the literature, the following requirements were considered significant in the choice of the inhibitors: 1) high potency, preferably low nanomolar inhibitors; 2) high selectivity in the kinome; 3) penetration of the blood–brain barrier; 4) availability of structural data regarding the binding mode; 5) solvent exposed functional group to attach the linker without compromising the kinase binding; 6) number of synthetic steps; 7) cost and availability of starting materials.
Based on those requirements, two scaffolds were selected: PF‐06447475, developed by Pfizer and GNE‐7915, developed by Genentech (Figure 1). In particular PF‐06447475, has an IC50 of 3 nM in the enzyme assay, 25 nM in the whole cell assay, is brain penetrant, highly selective in the kinome and does not show toxicity in rat models. The co‐crystal structure with MST3 kinase is reported (PDB ID: 4U8Z). [11] GNE‐7915 is also highly potent (K i 2 nM in the biochemical assay, IC50 9 nM in the cellular assay), brain penetrant and with high kinome selectivity (1 out of 187). [12] In this case, only a docking pose with JAK2 is published; however, the described extensive SAR is sufficient to guide the structural modifications for a PROTAC.
In particular, the available structural data for PF‐06447475 show that the oxygen of the morpholine participates in a hydrogen bond that is important for selectivity among the kinome, however, position 3 of the morpholine is solvent exposed and it could be used to attach a linker. On the contrary, for GNE‐7915, the morpholine moiety seems to be completely solvent exposed and the hypothesis is that it is not a strict requirement for binding. In order to choose which E3 ligase to target, the expression levels in different tissues were checked in the database of Protein Atlas. [13] A comparative analysis revealed that cereblon and mouse double minute 2 homolog (MDM2) are expressed in the same parts of the brain, whereas Von Hippel Lindau (VHL) is not. MDM2 is expressed in all tissues in high levels, so to address possible selectivity issues cereblon seemed a better choice to begin with. The original routes for resynthesizing PF‐06447475 and GNE‐7915 were followed and then small modifications were considered in order to improve the yields of intermediates and thus facilitate the synthesis of PROTACs (Scheme 1).
Scheme 1.
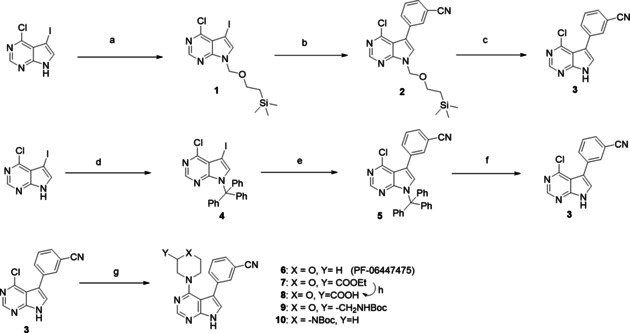
Synthetic routes to reach main intermediate (3) for PF‐06447475‐based PROTACs; original (top), modified (middle). Bottom: transformations to intermediates with functional groups. Reagents and conditions: a) NaH, SEM‐Cl, THF, 0 °C to RT, 3 h, 40 % yield; b) (3‐cyanophenyl)boronic acid, 1 % Pd(dppf)Cl2, K2CO3, DME/H2O, reflux 3 h, 30 % yield; c) TFA, RT, 24 h, 60 % yield; d) trityl chloride, CHCl3, Et3N, RT, 1 h, quantitative; e) (3‐cyanophenyl)boronic acid, 0.004 % Pd(dppf)Cl2, NaHCO3, toluene/EtOH, reflux 24 h, 60 % yield; f) TFA, CH2Cl2, RT, 24 h, 90 % yield; g) morpholine for (6) tert‐butanol, DIPEA, reflux 3 h, or ethyl morpholine‐2‐carboxylate for (7) or tert‐butyl N‐(morpholin‐2‐ylmethyl)carbamate for (9) or N‐Boc‐piperazine for (10), EtOH, DIPEA, MW, 150 °C, 1 h, yields 26–30 %; h) LiOH, THF/H2O, 65 % yield.
For PF‐06447475, the main intermediate (3) was synthesized in three steps as shown in scheme 1. The original route[ 11 , 14 ] (top) led to intermediate (3) in only 8 % overall yield and required two column purifications. However, by simply changing the protecting group in the first step from (2‐chloromethoxyethyl)trimethylsilane (SEM‐Cl) to trityl‐chloride, [15] the yield became almost quantitative. Optimization of the Suzuki coupling also increased the yield significantly, and now the optimized route (middle route), led to intermediate (3) in 54 % yield over three steps, requiring only one column purification. After obtaining (3), the original inhibitor PF‐06447475 (6) was synthesized with a nucleophilic aromatic substitution with morpholine. In order to attach suitable linkers for the PROTACs, morpholines substituted on position 3 were used, bearing either an ester group or a Boc‐protected amine to obtain intermediates (7) and (9), respectively. Boc‐piperazine was also used in a similar way for intermediate (1). The nucleophilic aromatic substitution was performed under microwave irradiation instead of reflux.
For GNE‐7915 the original route [12] includes an amide coupling with morpholine and a nucleophilic aromatic substitution, with a starting material that also needs to be synthesized (Scheme 2, top). In GNE‐7915 the morpholine part is solvent exposed and is also the position where the linkers can be attached. For the benefit of the overall route it is best to introduce the morpholine at the end of the synthesis. We developed an alternative route, starting from the commercially available 4‐amino‐2‐fluoro‐5‐methoxybenzoic acid (Scheme 2, middle). The initial substitution of the fluorine with a methoxy group, was followed by an esterification and a reduction of the nitro group to obtain intermediate (14). The next step was a nucleophilic aromatic substitution using the bifunctional building block 2,4‐dichloro‐5‐(trifluoromethyl)pyrimidine. Interestingly, the substitution can be performed selectively by using zinc chloride as catalyst. [16] The obtained intermediate (15) undergoes a substitution on the second chloride (16), followed by a hydrolysis to give the carboxylic acid (17), which is the key intermediate in this synthesis. Overall, the yield is 32 % over 6 steps with only one column purification. The carboxylic acid (17) was used in an amide coupling reaction with morpholine to obtain GNE‐7915 (11) and also in amide couplings directly with cereblon (CRBN) building blocks or with a substituted morpholine to obtain the amine intermediate (18).
Scheme 2.

Synthetic routes to GNE‐7915: original (top), modified (middle) and intermediates for GNE‐7915‐based PROTACs (bottom). Reagents and conditions: a) MeOH, KOH, RT, 2 h, yield 92 %; b) EEDQ, EtOH, reflux 5 h, quantitative; c) stannous chloride, EtOH/H2O, reflux 4 h, quantitative; d) 2,4‐dichloro‐5‐(trifluoromethyl)pyrimidine, zinc chloride, diethyl ether, CH2Cl2, tert‐butanol, triethylamine, 0 °C to RT, 48 h, 40 % yield; e) ethanamine, triethylamine, THF, 0 °C to RT, 1 h, 88 % yield; f) LiOH, THF/H2O, 97 % yield; g) morpholine for (11) or tert‐butyl N‐(morpholin‐2‐ylmethyl)carbamate for (18), EEDQ, CHCl3, reflux 2 h, 40 % yield.
For the synthesis of cereblon building blocks two starting materials were used: the 4‐nitroisobenzofuran‐1,3‐dione and the 4‐fluoroisobenzofuran‐1,3‐dione (Scheme 3). The first synthetic step is the condensation of 4‐nitroisobenzofuran‐1,3‐dione with 3‐aminopiperidine‐2,6‐dione to obtain the nitro‐substituted imide (19), which is then reduced to the aniline group to obtain pomalidomide (20). Pomalidomide was then used in an anhydride opening reaction obtain the carboxylic acid (21). Pomalidomide was also used in an acylation reaction with 2‐chloroacetyl chloride to obtain the intermediate (22), which with a substitution reaction led to intermediate (23). In a similar way, the condensation of 4‐fluoroisobenzofuran‐1,3‐dione with 3‐aminopiperidine‐2,6‐dione was performed to obtain the fluoro‐substituted imide (24), which underwent a nucleophilic aromatic substitution with linear Boc‐protected diamines to obtain intermediates (25) and (26).
Scheme 3.
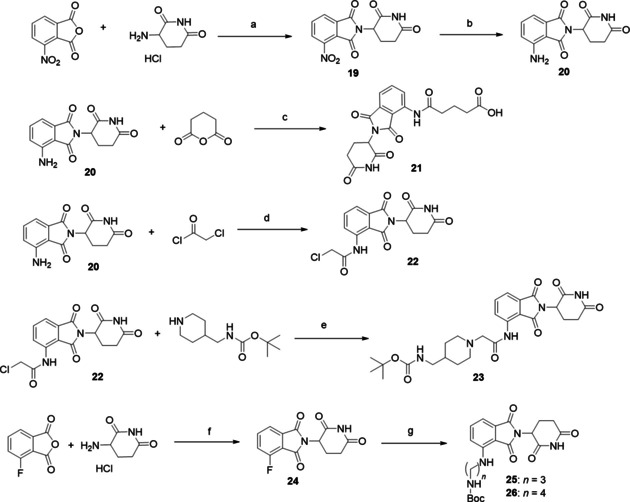
Synthetic routes for CRBN‐building blocks. Reagents and conditions: a) sodium acetate, acetic acid, overnight reflux, yield 90 %; b) H2, Pd/C, RT, 24 h, yield 92 %; c) potassium acetate, acetic acid, reflux 3 h, yield 32 %; d) THF, reflux, 30 min, yield 92 %; e) sodium iodide, potassium carbonate, THF, RT, yield 81 %; f) sodium acetate, acetic acid, overnight reflux, yield 85 %; g) N‐Boc‐propane‐1,3‐diamine or N‐Boc‐butane‐1,4‐diamine, DIPEA, DMF, overnight reflux, yield 60 %.
After synthesizing the appropriate kinase intermediates and the cereblon building blocks, the final PROTAC compounds were synthesized with amide coupling reactions. In all the cases of Boc‐protected intermediates, the deprotection was performed with HCl in dioxane and the obtained salts were used directly in the amide coupling without purification. An overview of structures and coupling yields is shown in Scheme 4.
Scheme 4.

Structures of PF‐06447475‐based‐CRBN‐PROTACs and GNE‐7915‐based‐CRBN‐PROTACs.
Then, the four PF‐06447475/CRBN‐based PROTACs and the three GNE‐7915/CRBN‐based PROTACs, as well as the original resynthesized inhibitors, were biologically evaluated. The original inhibitors were used as reference compounds. First, an in vitro kinase assay with the phosphorylation rate of a key LRRK2 autophosphorylation site (S1292‐LRRK2) as readout, revealed that the compounds were indeed potent kinase inhibitors (Figures S1 and S2 in the Supporting Information). Then, in order to test whether the PROTACs were cell‐permeable and able to bind to the kinase pocket of LRRK2, confocal microscopy was performed. It is known that incubation of LRRK2 with ATP competitive inhibitors results in relocalization of overexpressed GFP‐tagged LRRK2 onto microtubules.[ 17 , 18 , 19 , 20 ] HEK cells transfected with GFP‐tagged LRRK2 were incubated with different PROTACs along with the original kinase inhibitors and DMSO as controls. Formation of green filaments was observed after incubation with the original inhibitors and most of the PROTACs conferring the ability of the majority of the synthesized PROTACs to penetrate the cells and attach to the kinase pocket of LRRK2 (Figure 2A). We next determined the ability of different PROTACs to degrade LRRK2 in LRRK2 parental RAW 264.7 cells. For the initial screening, the cells were incubated with 2 concentrations (1 and 10 μM) of PROTACs for 24, 48 and 72 hours. Data from western blotting did not show any significant changes in the LRRK2 protein levels between the PROTAC treated cells and cells treated with the original kinase inhibitors, indicating that PROTACs were not able to cause LRRK2 degradation (Figures 2B and C). Furthermore, ubiquitination assays showed that one of the most potent and cell permeable PROTACs, PROTAC (33) (IC50=14.69±6.14 nM) was unable to increase the ubiquitin signal compared to the DMSO or original inhibitor compound (11) (IC50=17.3±6.76 nM) (Figure 2D).
Figure 2.
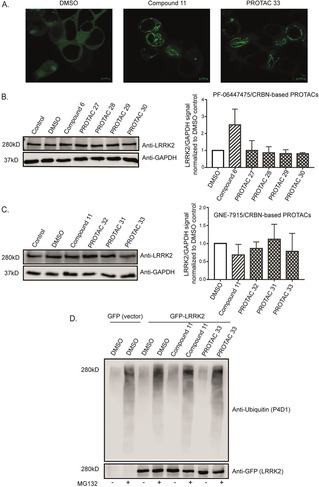
PROTACs are cell‐permeable yet not increasing LRRK2 degradation and ubiquitination. A) Confocal microscopy of GFP‐tagged LRRK2‐transfected HEK293 cells showing LRRK2 localization inside cells treated with DMSO, original kinase inhibitor and one of the PROTACs. The images show that the original kinase inhibitor and the PROTAC compound are cell‐permeable and able to induce LRRK2 localization to the microtubules depicted as green filaments. B) and C) Representative western blots of LRRK2 parental RAW 264.7 cells treated with 10 μM of different PROTACs together with the original kinase inhibitors and DMSO controls for 24 h and immunoblotted with LRRK2 and GAPDH as a loading control. The quantification of the blots (n=3) shows that different PROTACs are not affecting LRRK2 degradation compared to the original kinase inhibitor.. D) GFP‐tagged LRRK2 transfected HEK293 cells were treated with DMSO, original kinase inhibitor (11) and PROTAC (33) with or without MG132 for 24 h. An ubiquitin assay was performed on the collected cells. The samples were immunoblotted with anti‐ubiquitin (P4D1) for the ubiquitin signal and anti‐GFP for the LRRK2 signal. The blot shows that MG132 can enhance the ubiquitin signal and that neither the original kinase inhibitor (11) nor PROTAC (33) increase LRRK2 ubiquitination compared to the DMSO treated control.
Despite the fact that the synthesized PROTACs were able to enter the cells and bind to the target, there a few possible hypotheses explaining their inability to induce ubiquitination and degradation of LRRK2. It could be argued at this point that the number of synthesized PROTACs was limited and more variations on the linkers, including length, flexibility and attachment points, could be explored. However, as two different highly potent LRRK2 ligands failed to induce the degradation of the target, despite showing target engagement and cell permeability, there are also other possible explanations to consider. One plausible hypothesis, is that due to the observed re‐localization of LRRK2 to the microtubules and formation of stable filaments, LRRK2 might not be accessible to the E3 ligase component to form a ternary LRRK2‐PROTAC‐E3 ligase complex. Moreover, since the full‐length structure of LRRK2 is not known, the proximity of lysine residues suitable for ubiquitination degradation to the kinase site might not be optimal. To date, the full‐length structure of LRRK2 is not solved. Recently, a high resolution cryo‐EM structure of the catalytic half of LRRK2 including the RoC/GTPase, COR, kinase and WD40 domains was reported. [20] The structure revealed that the kinase and GTPase domains are in close proximity. Notably, in the absence of kinase inhibitors, the kinase was in an inactive/open conformation in the cryoEM structure, whereas data showed that the microtubule‐associated LRRK2 had the kinase domain in a closed and potentially active conformation. Moreover, kinase inhibitors also had an effect on the kinase domain conformation and in particular, inhibitors that promoted LRRK2‐microtubule binding favored the closed kinase conformation. The proposed model indicates the complexity of targeting LRRK2. Villa et al., [21] using cryoEM and integrative modeling, revealed the structure of LRRK2 in situ and showed that the GTPase domain is closer to the microtubule interface, in contrast to the kinase domain which is exposed to the cytoplasm.
Another aspect to be taken into account, is the properties of LRRK2 inhibitors. Although numerous scaffolds have been reported, in most of the cases, there is lack of structural data and differences in selectivity. This seems to be affecting also the degradation potential of LRRK2 inhibitors. At the time of manuscript submission, a patent highlight indicated the degradation of LRRK2 and compared two types of inhibitors: the aminopyrimidine analogs and the indazole analogs.[ 22 , 23 ] In agreement with our observations, the aminopyrimidine analogs failed to degrade the target. On the other hand, the indazole analogs seemed to be able to reduce the levels of LRRK2. The data taken together with our results, show that the selection of LRRK2 inhibitors is crucial for the development of degraders. Additionally, very recently, a high‐throughput screen resulted in the discovery of a small molecule, which showed remarkable selectivity for G2019S‐LRRK2, the most common LRRK2 pathogenic mutation. [24] This could be an interesting starting point for future studies of selective G2019S‐LRRK2 degraders.
To date, although PROTACs have been successful in challenging targets, the task of developing degraders is not trivial. Especially in the case of LRRK2, which remains an elusive drug target, a better understanding of its structure and conformational changes, as reported very recently, is necessary in order to further explore the possibility of degrading it and additionally to understand the reasons for the observed differences when highly potent LRRK2 inhibitors were modified into potential degraders. Overall, in this work, we aim to underline the challenges in degrading a target, for which the full structure is not yet known. We believe that future work will enable the rational development of a successful LRRK2‐targeting PROTAC, following a more complete picture of LRRK2 structure and dynamics.
Experimental Section
Experimental details. General procedures, characterization data (1H NMR, 13C NMR), and biological screening (kinase assays, cell culture, microscopy, western blotting, ubiquitin assay) data are given in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research has been supported (to A.D.) through ITN “Accelerated Early stage drug dIScovery” (AEGIS, grant agreement no. 675555). Moreover, funding was received from the National Institutes of Health (2R01 GM097082‐05), the European Lead Factory (IMI; grant agreement no. 115489), the Qatar National Research Foundation (NPRP6‐065‐3‐012). and, COFUNDs ALERT (grant agreement no. 665250) and Prominent (grant agreement no. 754425) and KWF Kankerbestrijding grant (grant agreement no. 10504). A.M.D. is the recipient of a Stichting Parkinson Fonds (SPF) grant and a Rosalind Franklin Fellowship co‐funded by the European Union and the University of Groningen. A.K. is holder of a TUBITAK 2232 scholarship and additional funding was received from The Michael J. Fox Foundation for Parkinson's Research.
M. Konstantinidou, A. Oun, P. Pathak, B. Zhang, Z. Wang, F. ter Brake, A. M. Dolga, A. Kortholt, A. Dömling, ChemMedChem 2021, 16, 959.
Contributor Information
Dr. Markella Konstantinidou, http://www.drugdesign.nl
Dr. Alexander Dömling, Email: a.s.s.domling@rug.nl.
References
- 1. Gilks W. P., Abou-Sleiman P. M., Gandhi S., Jain S., Singleton A., Lees A. J., Shaw K., Bhatia K. P., Bonifiati V., Quinn N. P., Lynch J., Healy D. G., Holton J. L., Revesz T., Wood N. W., Lancet 2005, 365, 415–416. [DOI] [PubMed] [Google Scholar]
- 2. Lesage S., Janin S., Lohmann E., Leutenegger A. L., Leclere L., Viallet F., Pollak P., Durif F., Thobois S., Layet V., Vidailhet M., Agid Y., Dürr A., Brice A., Arch. Neurol. 2007, 64, 425–430. [DOI] [PubMed] [Google Scholar]
- 3. Bardien S., Lesage S., Brice A., Carr J., Parkinsonism Relat. Disord. 2011, 17, 501–508. [DOI] [PubMed] [Google Scholar]
- 4. Di Maio R., Hoffman E. K., Rocha E. M., Keeney M. T., Sanders L. H., De Miranda B. R., Zharikov A., Van Laar A., Stepan A. F., Lanz T. A., Kofler J. K., Burton E. A., Alessi D. R., Hastings T. G., Greenamyre J. T., Sci. Transl. Med. 2018, 10,eaar5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Giasson B. I., Van Deerlin V. M., Neurosignals 2008, 16, 99–105. [DOI] [PubMed] [Google Scholar]
- 6.D. J. Moore, Parkinsonism Relat. Disord. 2008, 14 Suppl. 2, S92-S98. [DOI] [PubMed]
- 7. Deng X., Choi H. G., Buhrlage S. J., Gray N. S., Expert Opin. Ther. Pat. 2012, 22, 1415–1426. [DOI] [PubMed] [Google Scholar]
- 8. Kethiri R. R., Bakthavatchalam R., Expert Opin. Ther. Pat. 2014, 24, 745–757. [DOI] [PubMed] [Google Scholar]
- 9. Galatsis P., Expert Opin. Ther. Pat. 2017, 27, 667–676. [DOI] [PubMed] [Google Scholar]
- 10. https://clinicaltrials.gov/.
- 11. Henderson J. L., Kormos B. L., Hayward M. M., Coffman K. J., Jasti J., Kurumbail R. G., Wager T. T., Verhoest P. R., Noell G. S., Chen Y., Needle E., Berger Z., Steyn S. J., Houle C., Hirst W. D., Galatsis P., J. Med. Chem. 2015, 58, 419–432. [DOI] [PubMed] [Google Scholar]
- 12. Estrada A. A., Liu X., Baker-Glenn C., Beresford A., Burdick D. J., Chambers M., Chan B. K., Chen H., Ding X., Di Pasquale A. G., et al., J. Med. Chem. 2012, 55, 9416–9433. [DOI] [PubMed] [Google Scholar]
- 13. https://www.proteinatlas.org/.
- 14.P. Galatsis (Pfizer), US 2017002000 A1, 2017.
- 15.Y L. Chen (Novartis), WO 2010015637 A1, 2010.
- 16.J. C. Kath (Pfizer), WO 2005023780 A1,2005.
- 17. Dzamko N., Deak M., Hentati F., Reith A. D., Prescott A. R., Alessi D. R., Nichols R. J., Biochem. J. 2010, 430,405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deng X., Dzamko N., Prescott A., Davies P., Liu Q., Yang Q., Lee J. D., Patricelli M. P., Nomanbhoy T. K., Alessi D. R., Gray N. S., Nat. Chem. Biol. 2011, 7,203–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ramírez M. B., Ordóñez A. J., Fdez E., Madero-Pérez J., Gonnelli A., Drouyer M., Chartier-Harlin M. C., Taymans J. M., Bubacco L., Greggio E., Hilfiker S., Hum. Mol. Genet. 2017, 26,2747–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deniston C. K., Salogiannis J., Mathea S., Snead D. M., Lahiri I., Matyszewski M., Donosa O., Watanabe R., Böhning J., Shiau A. K., Knapp S., Villa E., Reck-Peterson S. L., Leschziner A. E., Nature 2020, 588, 344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Watanabe R., Buschauer R. L., Böhning J., Audagnotto M., Lasker K., Tsan-Wen L., Boassa D., Taylor S., Villa E., Cell 2020, 182, 1508–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kargbo R. B., ACS Med. Chem. Lett. 2020, 11, 2070–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.N. S. Gray, J. Hatcher (Dana-Farber Cancer Institute), WO 2020/081682 A, 2020.
- 24. Garofalo A. W., Bright J., De Lombaert S., Toda A. M. A., Zobel K., Andreotti D., Beato C., Bernardi S., Budassi F., Caberlotto L., et al., J. Med. Chem. 2020, 63, 14821–14839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary