

Abstract
Objective
The role of the survival of motor neuron (SMN) gene in amyotrophic lateral sclerosis (ALS) is unclear, with several conflicting reports. A decisive result on this topic is needed, given that treatment options are available now for SMN deficiency.
Methods
In this largest multicenter case control study to evaluate the effect of SMN1 and SMN2 copy numbers in ALS, we used whole genome sequencing data from Project MinE data freeze 2. SMN copy numbers of 6,375 patients with ALS and 2,412 controls were called from whole genome sequencing data, and the reliability of the calls was tested with multiplex ligation‐dependent probe amplification data.
Results
The copy number distribution of SMN1 and SMN2 between cases and controls did not show any statistical differences (binomial multivariate logistic regression SMN1 p = 0.54 and SMN2 p = 0.49). In addition, the copy number of SMN did not associate with patient survival (Royston‐Parmar; SMN1 p = 0.78 and SMN2 p = 0.23) or age at onset (Royston‐Parmar; SMN1 p = 0.75 and SMN2 p = 0.63).
Interpretation
In our well‐powered study, there was no association of SMN1 or SMN2 copy numbers with the risk of ALS or ALS disease severity. This suggests that changing SMN protein levels in the physiological range may not modify ALS disease course. This is an important finding in the light of emerging therapies targeted at SMN deficiencies. ANN NEUROL 2021;89:686–697
Amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA) are both motor neuron disorders leading to progressive muscle weakness and death of patients, mostly due to respiratory failure. 1 , 2
ALS is an adult‐onset disease with an estimated lifetime risk of 1 in 400. Approximately 50% of patients die within 3 to 5 years after onset, but there is a high level of variability between patients in age at onset and progression rate, with 5% of the patients surviving more than 10 years. 3 , 4 Patients are typically categorized into familial ALS representing 10 to 20% of patients and sporadic ALS. Genetically, ~ 66% of familial and ~ 10% sporadic patients are explained by a mutation in one of the ~ 25 genes that have been associated with ALS, with mutations in SOD1, TARDBP, FUS, and C9orf72 being the most common causes. 1 To date, several clinical and genetic factors have been put forward as disease modifiers, for example, sex, age at onset, site of onset, diagnostic delay, presence of frontotemporal dementia, C9orf72 repeat expansions and SNPs in or near UNC13A, and CAMTA1. 5 In addition, copy number (CN) variation in SMN, the gene that is deleted in childhood onset SMA, has been extensively studied as disease modifier with many conflicting reports. 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15
SMA is a monogenic disease usually caused by a homozygous loss of SMN1 and modulated by the number of SMN2 copies present. 16 In ~ 95% of the cases, both copies of SMN1 are missing or affected by a gene conversion, the remaining ~ 5% of the patients have a mutation on their remaining copy of SMN1. 17 The SMN genes are located on q13.2 of chromosome 5, a locus that underwent an inverted duplication of about 500 kb. 18 SMN2 is almost identical to SMN1, except for a few point mutations and small insertions and deletions, one of which is located at the splice junction of exon 7 and causes exon 7 skipping. 18 As a consequence, SMN2 mainly codes for a non‐fully functional SMN protein. 16 Recently, 3 treatment options became available that aim to increase full length functional SMN protein levels. The first one is a multi‐dose antisense oligonucleotide therapy named nusinersen, the second is the single dose gene replacement therapy onasemnogene abeparvovec, and the third option is experimental compound therapies, aiming to increase the expression of the SMN locus. 19 , 20 Nusinersen works through blocking the binding of heterogeneous nuclear ribonucleoproteins (hnRNPs) at a splicing silencer element, named ISS‐N1, leading to exon 7 inclusion and increases full length SMN protein levels. 2 , 19 Onasemnogene abeparvovec adds a functional copy of SMN1 to the genome using a self‐complementary adeno‐associated viral serotype 9, increasing the SMN protein levels. 20
These recent therapeutic breakthroughs in SMA could have large consequences for ALS as well, if a convincing role for SMN in ALS could be established.
Previous studies investigating the effect of the CNs of SMN on disease risk and progression of ALS have reported conflicting results. 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 According to some studies, loss of SMN1 is associated with ALS risk, 8 , 9 , 12 whereas others found that duplication of SMN1 associated with ALS risks. 6 , 7 , 11 , 12 , 13 Others did not find any association between ALS and the number of SMN1 copies. 10 , 14 , 15 Likewise for SMN2, some found a homozygous deletion to be protective, 8 whereas others found the deletion to be more frequent in ALS and reducing the survival time of these patients. 9 , 10 , 15 In addition, here, several other publications failed to find any association. 6 , 7 , 11 , 12 , 13 , 14
In this large study, we aimed to use the whole genome sequencing (WGS) data from Project MinE, which contains 6,375 patients with ALS and 2,412 controls to evaluate the effect of SMN CNs in the context of ALS risk and clinical phenotype. 21 This is the largest ALS cohort in which SMN genes have been analyzed in the hope to find a definitive answer.
Methods
Experimental Design
This WGS case–control study uses data freeze 2 from Project MinE, which contains a total of 9,600 whole genomes sequencing data from ALS cases and age‐matched and sex‐matched control samples. 21 Samples were mostly of people of European descent and collected from 17 centers across 13 countries: Belgium, Switzerland, Spain, France, United Kingdom, Ireland, Israel, Italy, The Netherlands, Portugal, Sweden, Turkey, and the United States.
Patients were diagnosed with ALS in their respective centers, mainly using the El‐Escorial criteria. Clinical information was collected from each center and centrally harmonized and passed though quality control. For patients who are still alive, a survival update was requested on a yearly basis. All participants signed an informed consent at their respective centers. This study was approved by the respective ethics committees of the participating centers.
Sequencing, Variant Calling, and Quality Control
The first batch of 2,250 cases and control samples were sequenced on the Illumina HiSeq 2000 platform. All remaining 7,350 cases and controls were sequenced on the Illumina HiSeq X platform. All samples were sequenced to ~ 35X coverage with 100 bp reads and ~ 25X coverage with 150 bp reads for the HiSeq 2000 and HiSeq X, respectively. Both sequencing sets used polymerase chain reaction (PCR)‐free library preparation. Samples were also genotyped on the Illumina 2.5 M array. Sequencing data were then aligned to GRCh37 using the iSAAC Aligner, and variants called using the iSAAC variant caller; both the aligner and caller are standard to Illumina's aligning and calling pipeline.
Per individual, gVCFs were merged using the Illumina gvcfgenotyper tool version 2018.10.15 (https://github.com/Illumina/gvcfgenotyper). Sites with a genotype quality (GQ) < 10 were set to missing and single nucleotide variations (SNVs) and indels with quality (QUAL) scores < 20 and < 30, respectively, were removed. Biological sex was inferred from the average coverage of chrY and chrX compared with the average coverage of the autosomal chromosomes.
Next sample‐level quality control was performed using the following filters at sample level: (1) more than 2% of the variant missing, (2) abnormal value of the autosomal homozygous genotype score calculated by Plink 1.9 |F| > 0.10, (3) aberrant transition/transversion ratio, defined as ±6 standard deviations (SDs), (4) aberrant number of variants, defined as ±6 SDs, (5) aberrant number of singletons, defined as ±6 SDs, (6) aberrant number of indels, defined as ±6 SDs, and (7) an inconsistency between the inferred and reported sex.
Variants in the merged vcf file were first decomposed and normalized using the corresponding commands in vt (version 2015.11.10) and then annotated by VEP (version 96), information from public databases dbNSFP (version 3.5a), dbscSNV (version 1.1), ExAC (version 0.3), gnomAD exome (version 2.1), ESP (ESP6500SI‐V2), 1,000 genomes (phase 3), dbsnp (version 151), and clinvar (version 20190513) were added using vcfanno (version 0.3.1). Annotated vcf files were loaded into gemini (version 0.30.1) and filtered for exonic variants, excluding synonymous variants not near splice sites, with an allele frequency of maximum 2%. Resulting variants in SOD1, FUS, and TARDBP were further inspected using the ACMG guidelines on the varsome platform. 22 Pathogenic, likely pathogenic, and variants of unknown significance with some evidence toward pathogenicity were retained for further analysis. C9ORF72 expansions were detected using Expansion Hunter (version 3.1.2) from Illumina. 23
SMN Calling
To call the CNs of SMN from WGS data we used the recently published SMNCopyNumberCaller (SMNCNC) tool from Illumina. 24 In brief, this method uses read depth information at the 8 loci in the SMN1 and SMN2 genes to determine the consensus CN of both genes (coordinates against hg19): site number 7 (intron 6, SMN1 chr5‐70246320‐G, SMN2 chr5‐69370895‐A), site number 8 (intron 6, SMN1 chr5‐70246793‐G, SMN2 chr5‐69371368‐A), site number 10 (intron 6, SMN1 chr5‐70247219‐G, SMN2 chr5‐69371799‐A), site number 11 (intron 6, SMN1 chr5‐70247290‐T, SMN2 chr5‐69371870‐C), site number 12 (intron 6, SMN1 chr5‐70247724‐G, SMN2 chr5‐69372304‐A), site number 13 (Splice site variant in exon 7, SMN1 chr5‐70247773‐C, SMN2 chr5‐69372353‐T), site number 14 (intron7, SMN1 chr5‐70247921‐A, SMN2 chr5‐69372501‐G), and site number 15 (intron7, SMN1 chr5‐70248036‐A, SMN2 chr5‐69372616‐G). Wet lab validation was performed using multiplexed ligation‐dependent probe amplification (MLPA) using standard protocols as described by Blauw et al. 7
Power Analysis
Power calculations were performed for the survival and onset analysis using the powerCT function of the R package powerSurvEpi (version 0.1.0), with a p value threshold of 0.05.
Statistical Analysis
To test the independence of the SMN CN distributions between cohorts we used a 2‐sided asymptotic generalized Pearson Chi‐Squared test (gχ2) provided by the chisq_test method of the R package coin (version 1.3–1), with SMN CN as ordinal and cohort as nominal. When comparing the SMN CN distribution of only two cohorts a 2‐sided asymptotic linear‐by‐linear (lbl) association test was used, implemented by the same chisq_test method.
For the corrected risk analysis, a 2‐sided binomial multivariate logistic regression was performed using the glm function from the R stats package. SMN CN was added to the model as a categorical variable with baseline a CN of 2, while correcting for the following terms: sex, sequencing technology, cohort, the first 20 HapMap projected principal components (PCs), and the mutation status of C9orf72, SOD1, FUS, and TARDBP.
Cox survival analysis was performed using the coxph function of the R package survival (version 3.1–8). Flexible survival regression using the Royston‐Parmar (RP) spline model was performed using the flexsurvspline function from the R package flexsurv (version 1.1.1). When assessing the effect of SMN CN on survival SMN CN was treated as a categorical variable with baseline a CN of 2. Survival analyses were corrected for age at onset, sex, site of onset, C9orf72 expansion status, sequencing technology, cohort, and the first 20 HapMap projected PCs. Onset analyses were corrected for sex, sequencing technology, cohort, and the first 20 HapMap projected PCs.
Meta‐analysis of the individual cohort was performed using the meta (version 4.13–0) and metafor (version 2.4–0) R packages. The same covariates were used as in the risk, survival, and onset analysis, but only the first 5 HapMap projected PCs were used.
Results
Validating SMN Calls from WGS Data
We used 2,412 controls and 6,375 cases that passed the QC metric and had sufficient clinical information (Table 1 and Table S1). For these samples, we ran SMNCNC to estimate the CN of SMN1 and SMN2. For 475 of our samples, we also had SMN CNs available from MLPA. We excluded samples where the MLPA results for exon 7 and exon 8 disagreed with each other. The concordance between the MLPA results of exon 7 and exon 8 was 89.3% for SMN1 and 97.9% for SMN2. Excluding discordant samples resulted in a set of 413 samples. This set was then further compared with the results of SMNCNC, with a concordance of 99.3% and 99.5% for SMN1 and SMN2, respectively, which is similar to previous findings (Figs 1 and 2). 24
TABLE 1.
Phenotype Information of the MinE Participants
| Project MinE | ALS (%) | Control (%) | ||
|---|---|---|---|---|
| 6375 | (72.55) | 2412 | (27.45) | |
| Sex | ||||
| Male | 3,815 | (59.84) | 1,272 | (52.74) |
| C9orf72 status | ||||
| Expanded | 377 | (5.91) | 7 | (0.29) |
| Onset | ||||
| Spinal | 4,226 | (66.29) | ||
| Bulbar | 1,734 | (27.2) | ||
| Generalized | 221 | (3.47) | ||
| Thoracic/respiratory | 112 | (1.76) | ||
| FTD | 5 | (0.08) | ||
ALS = amyotrophic lateral sclerosis; FTD = frontotemporal dementia.
FIGURE 1.
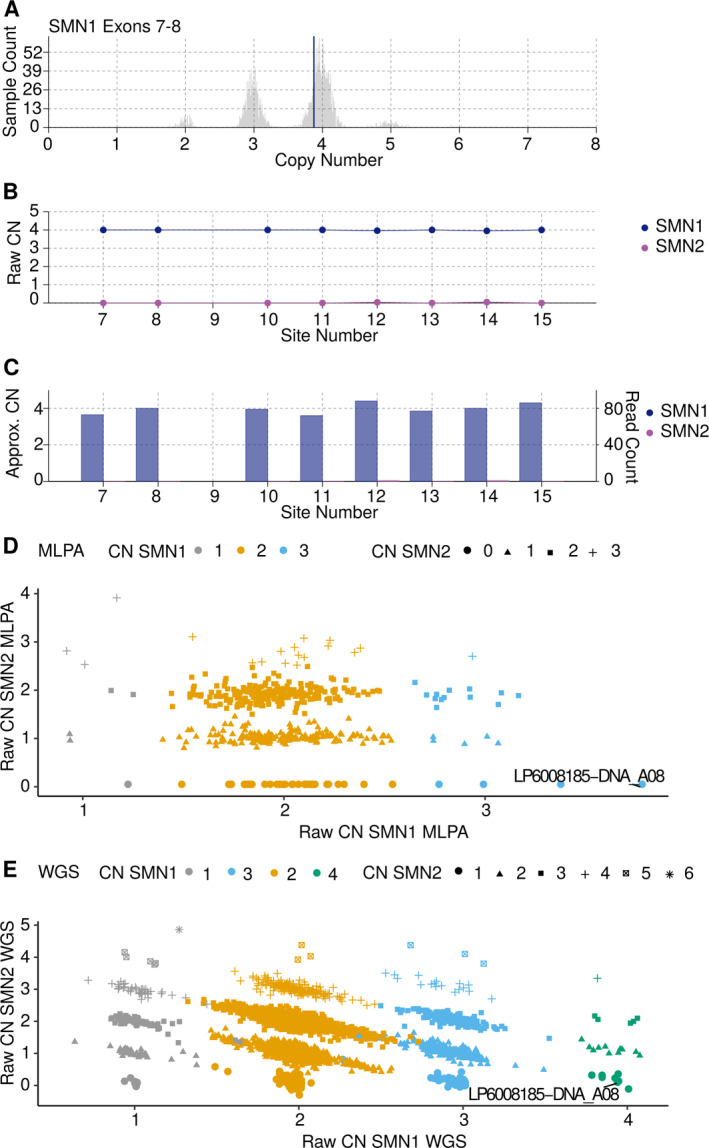
Raw data plots for discordant MLPA call. MPLA called 3 CNs for SMN1 in sample LP6008185‐DNA_A08, whereas SMNCNC called 4 CNs for SMN1. Closer inspection revealed SMNCNC to be correct. (A) Raw CN of full length SMN1 estimated by exon 7 and 8, histogram indicating the estimated CN of a large control cohort, vertical blue line indicates the discordant sample. (B) SMN1 and SMN2 raw CN values at the 8 loci used to determine the consensus CN. (C) SMN1 and SMN2 CNs normalized against coverage depth at 8 the loci. (D) Scatterplot of the raw CN calls by MLPA, on the x‐axis raw CN for SMN1, on the y‐axis raw CN for SMN2, discordant sample indicated with a line. (E) Scatter plot of the raw CN calls by SMNCNC, on the x‐axis raw CN for SMN1, and on the y‐axis raw CN for SMN2, discordant sample indicated with a line. CN = copy number; MLPA = multiplexed ligation‐dependent probe amplification; SMN = survival of motor neuron; SMNCNC = SMNCopyNumberCaller; WGS = whole genome sequencing.
FIGURE 2.
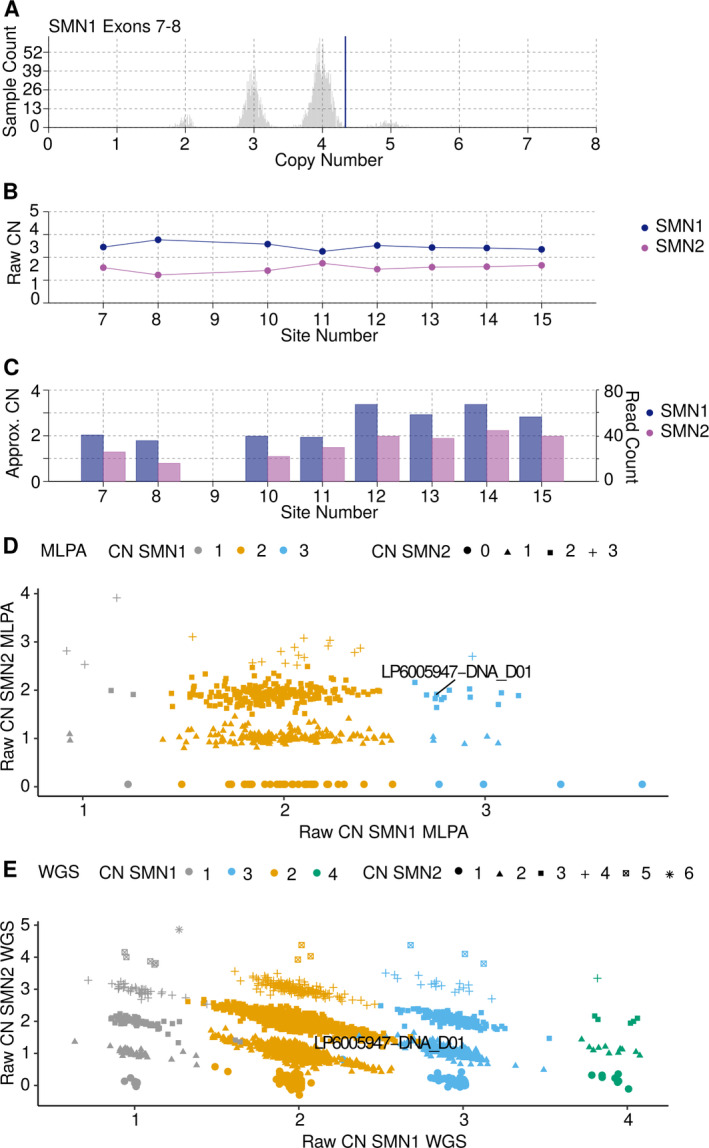
Raw data plots for discordant SMNCNC call. MPLA called 3 CNs for SMN1 in sample LP6005947‐DNA_D01, whereas SMNCNC called 2 CNs for SMN1. Closer inspection revealed MLPA to be correct. (A) Raw CN of full length SMN1 estimated by exon 7 and 8, histogram indicating the estimated CN of a large control cohort, vertical blue line indicates the discordant sample. (B) SMN1 and SMN2 raw CN values at the 8 loci used to determine the consensus CN. (C) SMN1 and SMN2 CN normalized against coverage depth at the 8 loci. (D) Scatterplot of the raw CN calls by MLPA, on the x‐axis raw CN for SMN1, and on the y‐axis raw CN for SMN2, discordant sample indicated with a line. (E) Scatter plot of the raw CN calls by SMNCNC, on the x‐axis raw CN for SMN1, and on the y‐axis raw CN for SMN2, discordant sample indicated with a line. CN = copy number; MLPA = multiplexed ligation‐dependent probe amplification; SMN = survival of motor neuron; SMNCNC = SMNCopyNumberCaller; WGS = whole genome sequencing.
SMN CNs as a Risk Factor for ALS
To investigate if our SMN CN estimates can be used in a risk factor analysis, we first compared the frequency of SMN copies in our controls to published literature (Fig 3A, B, and Table S2). 7 , 12 , 25 , 26 , 27 To assess the CN distributions across multiple groups we used a gχ2 test, whereas we used the lbl test when comparing the distribution between the 2 groups. Assessing the SMN1 and SMN2 CN distribution across the different control populations revealed geographic differences, as previously reported (SMN1; gχ2 p < 2.2 × 10‐16; SMN2 gχ2 p = 5.1 × 10‐7). 27 When comparing our SMN1 CN distribution to a Caucasian population of 2,175 individuals we did not find any difference (lbl p = 0.16; see Fig 3A), confirming that our control population is representative. 27 Within the control samples of Project MinE no difference in SMN1 or SMN2 CN frequency was observed between the cohorts (SMN1; gχ2 p = 0.45; SMN2; gχ2 p = 0.53; Fig 4 and Tables S3 and S4).
FIGURE 3.
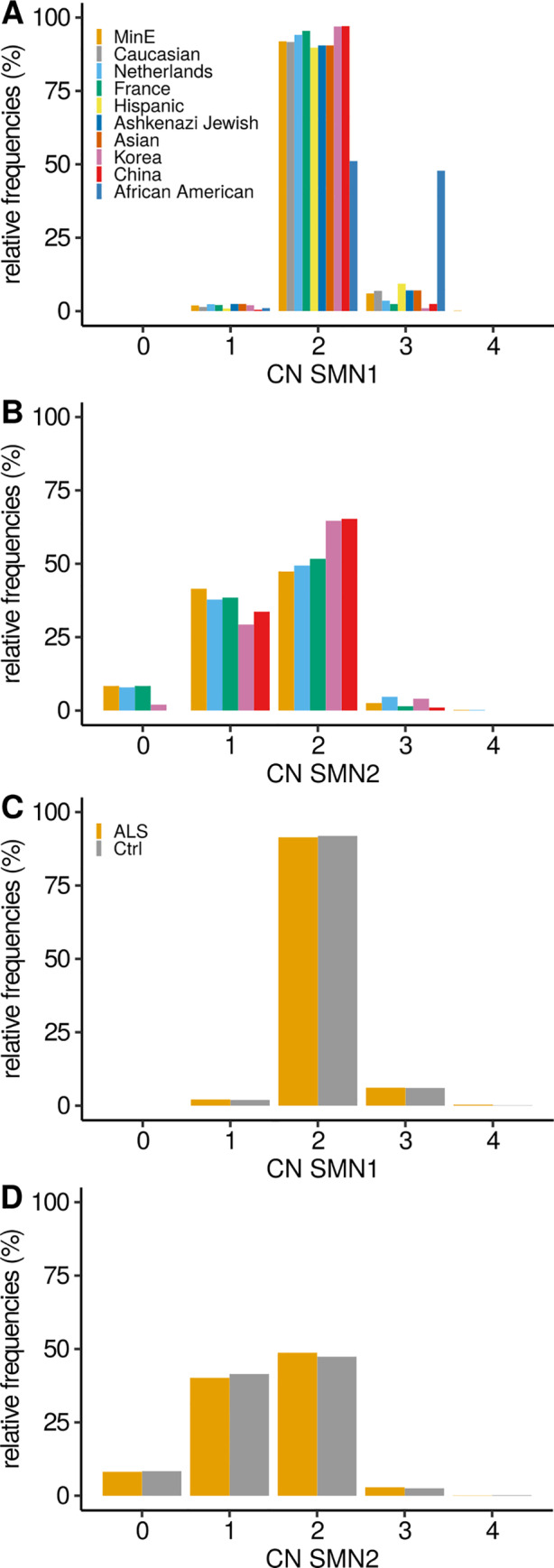
Frequency of the CNs of the SMN1 and 2 genes. Frequency of (A) SMN1 and (B) SMN2 copy number in control individuals of this study (MinE), Feng Y. et al. (White, Hispanic, Ashkenazi Jewish, Asian, and African American), Blauw HM. et al. (The Netherlands), Corcia P. et al. (France), Yoon S. et al. (Korea), and Fang P. et al. (China). Comparisons of the different cohort toward this study for SMN1 (lbl p value): White (0.16), Netherlands (3.2 × 10‐3), France (1.3 × 10‐3), Hispanic (3.4 × 10‐5), Ashkenazi Jewish (0.20), Asian (0.83), Korea (0.07), China (0.25), and African American (<1 × 10‐16); and SMN2: Netherlands (1.1 × 10‐2), France (0.63), Korea (2.3 × 10‐4), and China (7.6 × 10‐6). Frequency of (C) SMN1 and (D) SMN2 copy numbers in patients with ALS and control individuals in this study. ALS = amyotrophic lateral sclerosis; CN = copy number; lbl = linear‐by‐linear.
FIGURE 4.
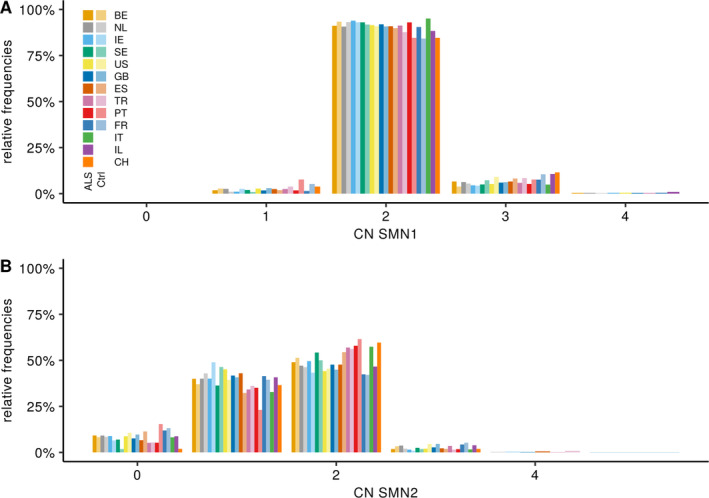
SMN frequencies by country. Frequency of SMN1 (A) and SMN2 (B) CN in ALS patients and control individuals in this study by cohort. Lighter colors indicate controls. Comparisons of the different ALS cohort toward the Turkish cohort for SMN1 (lbl p value): BE (0.41), NL (0.75), IE (0.86), SE (0.67), US (0.79), GB (0.55), ES (0.96), PT (0.90), FR (0.21), IT (0.83), IL (1.1 × 10‐2) and CH (0.42); and SMN2: BE (7.1 × 10‐5), NL (2.5e‐5), IE (1.6 × 10‐4), SE (0.19), US (0.4.0 × 10‐6), GB (9.9 × 10‐5), ES (5.9 × 10‐3), PT (0.73), FR (2.3 × 10‐4), IT (0.44), IL (0.05), and CH (0.81). ALS = amyotrophic lateral sclerosis; BE = Belgium; CH = Switzerland; CN = copy number; GB = United Kingdom; ES = Spain; FR = France; IE = Ireland; IL = Israel; IT = Italy; lbl = linear‐by‐linear; NL = The Netherlands; PT = Portugal; SE = Sweden; SMN = survival of motor neuron; TR = Turkey; US = United States.
When looking at SMN CN frequency in patients with ALS of published cohorts and our own we do observe a significant difference for SMN1 CN (gχ2 p = 7.3 × 10‐4) but not for SMN2 CN (gχ2 p = 0.10). Within Project MinE no differences between cohorts for SMN1 CN (gχ2 p = 0.52) were observed, whereas for SMN2 differences were observed (gχ2 p = 4.9 × 10‐4), in the Turkish cohort (see Fig 4B and Tables S3 and S4).
Comparing SMN1 frequency between cases and controls within Project MinE revealed no significant differences (lbl p = 0.54), similarly SMN2 also did not show any differences (lbl p = 0.23; see Fig 3C, D). Using a binomial logistic regression, we performed a corrected case control risk analysis for the SMN CN. Similarly, we did not observe any significant differences for the SMN CN status between cases and controls (binomial multivariate logistic regression; SMN1 p = 0.54 and SMN2 p = 0.49). Cohort analysis within Project MinE and meta‐analysis of the cohorts did not reveal any differences (see Fig 4). In addition, other CN categories, including duplications of SMN1 or SMN2 (lbl; SMN1 p = 0.55 and SMN2 p = 0.71; binomial multivariate logistic regression; SMN1 p = 0.73 and SMN2 p = 0.83) or deletions (lbl; SMN1 p = 0.69 and SMN2 p = 0.22; binomial multivariate logistic regression; SMN1 p = 0.98 and SMN2 p = 0.39) did not associate with ALS.
SMN CNs as a Modifier for ALS
As the CN of SMN has been proposed as a disease modifier, we also investigated the effect on age at onset and survival. 10 , 15 For this, we performed both a Cox regression and a RP analysis, 28 while correcting for covariates. This revealed no significant association with survival for SMN1 (Cox hazard ratio [HR] = 1.00, 95% confidence interval [CI] = 0.91–1.11, p = 0.89; RP HR = 0.98, 95% CI = 0.84–1.14, p = 0.78) or SMN2 (Cox HR = 1.01, 95% CI = 0.97–1.05, p = 0.67; RP HR = 1.04, 95% CI = 0.97–1.12, p = 0.23; Fig 5A, B). Meta analyzing the results in the individual cohorts with a random effect model revealed no significant associations for SMN1 (Cox HR = 1.00, 95% CI = 0.90–1.10, p = 0.96; RP HR = 0.95, 95% CI = 0.80–1.13, p = 0.59) or SMN2 (cox HR = 1.01, 95% CI = 0.95–1.07, p = 0.73; RP HR = 1.04, 95% CI = 0.95–1.13, p = 0.43). Power analysis showed that our cohort had an 80% power to detect a 25% change in expected hazard for SMN1 deletion (~ 9 months), 16% for SMN1 duplication (~ 6 months), 9% for SMN2 deletion (~ 3 months), and 22% for SMN2 duplications (~ 9 months).
FIGURE 5.
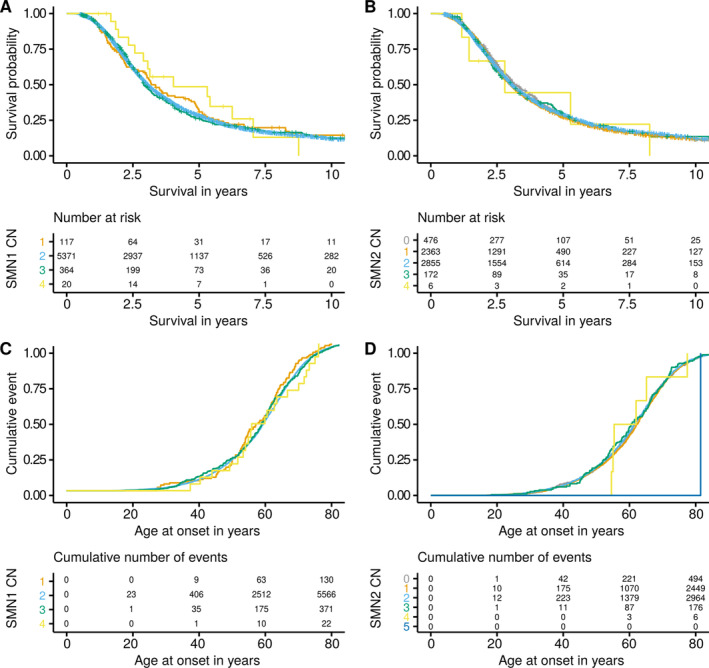
Survival and onset of patients, stratified by SMN CN. Kaplan Meier survival curves and risk table for (A) SMN1 and (B) SMN2 CNs. Kaplan Meier age at onset curves and cumulative events table for (C) SMN1 and (D) SMN2. CN = copy number; SMN = survival of motor neuron.
Similarly, we investigated the effect on onset, while correcting for covariates. Neither SMN1 nor SMN2 showed a significant association with onset at the level of the whole cohort (SMN1; Cox HR = 0.98, 95% CI = 0.90–1.06, p = 0.63, RP HR = 0.98, 95% CI = 0.85–1.13, p = 0.75; SMN2; Cox HR = 1.00, 95% CI = 0.97–1.04, p = 0.86, RP HR = 1.01, 95% CI = 0.95–1.08, p = 0.63) or by the cohort analysis (see Fig 5C, D). Likewise, meta analyzing using a random effect model revealed no significant associations for SMN1 (Cox HR = 0.97, 95% CI = 0.89–1.06, p = 0.49; RP HR = 0.94, 95% CI = 0.75–1.17, p = 0.57) or SMN2 (Cox HR = 1.00, 95% CI = 0.94–1.07, p = 0.85; RP HR = 1.01, 95% CI = 0.91–1.13, p = 0.83). Power analysis for the age at onset analysis showed we had an 80% power to detect a 21% change in expected hazard for SMN1 deletion, 14% for SMN1 duplication, 7% for SMN2 deletion, and 19% for SMN2 duplications.
Discussion
Our study, in a large cohort of patients with ALS for which SMN1 and SMN2 CNs have been assessed, shows that no association can be found between the CN of the SMN genes and ALS risk or disease severity. This is in line with some previously published studies, but inevitably also contradicts some previous findings in much smaller studies. 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 Given that our study is currently by far the largest available, includes samples spread out over a wide range of geographical areas, is well powered, and subgroup analyses did not reveal any associations, this study provides conclusive evidence that SMN genes do not have a role in ALS pathogenesis through SMN gene CN. Previous studies reported an increased risk of ALS up to 2‐fold to 5‐fold for carriers with only one copy of SMN1, which represent carrier status of SMA. 9 , 12 , 13 Our study clearly shows that SMA carriers, representing ~ 2% of the population, are not at an increased risk of developing ALS. The CN distribution in our control samples were consistent with an independent large control cohort, further validating our approach. 27
WGS data identify a multitude of genomic variations, including single point variants, small insertions, deletions, and structural variations. Additionally, an increasing amount of reliable bioinformatic tools become available for WGS data to assess complex genomic regions like, for example, repeat expansions or genomic duplications like the SMN region, allowing to extract new information from WGS data with unprecedented ease and speed, without the necessity of additional wet lab experiments. A recent study evaluating the reliability of wet lab testing of the C9orf72 repeat across multiple laboratories, showed a low concordance between different laboratories, highlighting the difficulties of wet lab testing and accompanying result interpretations. 29 It has recently been shown that C9orf72 calls made from WGS data were more reliable than their wet lab counterparts. 23 The power of WGS is that it offers a one‐stop solution for all the known genetically relevant ALS information, including genetic variation in complex regions.
Recently, SMA became a treatable disease currently with 3 treatment options. The first option is the use of chronic intrathecal antisense oligonucleotide therapy, that aims to convert SMN2 gene products that lack exon 7 into full length SMN1 protein. 19 The second option is a single dose viral gene replacement therapy that adds an extra copy of SMN1 into the patient's genome. 20 In addition, compound therapies are being developed to increase the expression of the SMN locus. These breakthroughs in SMA could be important for ALS as well. The SMN1 protein plays important roles in RNA metabolism, which has been implicated in ALS as well. However, the motor neuron death cascades seem to be distinct in both diseases. 30 , 31 Given our findings and the differences at genetic and molecular level that led to motor neuron death in ALS and SMA, current SMA treatment options may not be beneficial for patients with ALS.
Although our genetic findings show that SMN CN is not involved in ALS, it does not entirely rule out that changes in protein levels contribute to motor neuron degeneration in ALS. SMN protein levels are not solely determined by the CN state of the SMN genes, many processes influence protein level, including mRNA transcription, translation efficiency, post translational modification, and protein aggregation and sequestration. CN levels of SMN1 and SMN2 correlate with mRNA levels in blood, spinal cord, and cerebellum, as previously shown. 32 , 33 Protein levels do so to a lesser extent in blood and cerebellum, and not in the spinal cord. 32 , 34 Taken together these results show protein levels cannot be reliably predicted on SMN CN state alone. Furthermore, our findings do not rule out that overexpression of SMN to levels above the physiological range can have beneficial effects in protecting motor neurons in ALS. Indeed, previous work showed that motor neurons with the same genetic background display a wide heterogeneity of SMN expression and that mostly the motor neurons with a low level of SMN are vulnerable for motor neuron death. 35 Additionally, they showed that using compounds that increase SMN protein levels, through SMN1 expression promotion, but not SMN2, were beneficial for the survival of motor neurons, not only in SMA but also in an ALS context and in healthy controls. 35
This study has some limitations. Project MinE is not a population‐based study, but a multicenter study in which data and samples from different centers and countries are included. Samples are mostly from people of European descent. This could potentially limit the external validity to a wider, more diverse population and lead to population stratification. However, Project MinE explicitly aims to include balanced case control cohorts resulting in control samples not being significantly different from previously published data sets. 21 This, together with the large sample size and multivariate analyses, therefore add to the credibility of these results. Last, given the complexity of the SMN locus, we did not investigate the role of point mutations and small indel in the SMN genes in the context of ALS, as these mutations could also lead to a loss‐of‐function, like SMN1 gene deletions. In the light of our current results and the fact that they represent only 5% of SMN1 mutations, chances are small that these do play a role in ALS. 17
In summary, in our well‐powered study, using highly reliable and validated SMN calls, there was no association of SMN1 or SMN2 CNs with the risk of ALS or ALS disease severity. This suggests that changing SMN expression levels in the physiological range may not modify the progression of ALS. This is an important finding in the light of emerging therapies targeted at SMN deficiencies.
Author Contributions
M.M., P.V.D., and J.V. contributed to the conception and design of the study. All authors contributed to the acquisition and analysis of data. M.M., P.V.D., J.V., X.C., and M.A.E. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
X.C. and M.A.E. are employees of Illumina Inc.
Data Availability
Data can be accessed through EGAC00001000703. The authors declare that all the other data supporting the findings of this study are available within the article, its Supplementary Information files, or from the corresponding author upon reasonable request.
Supporting information
Table S1 Phenotype by country
Table S2 Frequency of SMN genes in different populations
Table S3 Frequency of the SMN1 gene in different countries within Project MinE
Table S4 Frequency of SMN2 in different countries within Project MinE
Acknowledgments
The authors thank the patients, their families, and healthy controls for their participation in this project. Samples were sequenced as part of Project MinE. Project MinE Belgium was supported by a grant from IWT (no. 140935), FWO‐Vlaanderen (G0B2819N), the ALS Liga België, the National Lottery of Belgium, and the KU Leuven Opening the Future Fund. P.V.D. holds a senior clinical investigatorship of FWO‐Vlaanderen and is supported by E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders, the ALS Liga België, and the KU Leuven funds “Een Hart voor ALS,”, “Laeversfonds voor ALS Onderzoek,” and the “Valéry Perrier Race against ALS Fund.”. Several authors of this publication are members of the European Reference Network for Rare Neuromuscular Diseases (ERN‐NMD). This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 772376 ‐ EScORIAL). This study was supported by the ALS Foundation Netherlands. The collaboration project is co‐funded by the PPP Allowance made available by Health ~ Holland, Top Sector Life Sciences & Health, to stimulate public‐private partnerships. This is in part an EU Joint Programme ‐ Neurodegenerative Disease Research (JPND) project. The project is supported through the following funding organizations under the aegis of JPND ‐ www.jpnd.eu (United Kingdom, Medical Research Council (MR/L501529/1; MR/R024804/1, the Netherlands, ZONMW, grant no. 733051071, BRAIN‐MEND), and through the Motor Neurone Disease Association. This study represents independent research part funded by the National Institute for Health Research (NIHR) Maudsley Biomedical Research Centre. Samples used in this research were in part obtained from the UK National DNA Bank for MND Research, funded by the MND Association and the Wellcome Trust. Funding was provided to J.E.L. by the NIH/NINDS (R01NS073873) and the ALS Association. V.S. receives or has received research supports from the Italian Ministry of Health (Grant RF‐201302355764), Fondazione Italiana di Ricerca per la SLA – AriSLA (Grant Exomefals and Novals), Fondazione Regionale per la Ricerca Biomedica Regione Lombardia (Project no. 2015–0023), and E‐RARE JTC (Project Repetomics). R.McL. is supported by Science Foundation Ireland (17/CDA/4737).
References
- 1. Van Damme P, Robberecht W, Van Den Bosch L. Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis Model Mech 2017;10:537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hagenacker T, Wurster CD, Günther R, et al. Nusinersen in adults with 5q spinal muscular atrophy: a non‐interventional, multicentre, observational cohort study. Lancet Neurol 2020;19:317–325. [DOI] [PubMed] [Google Scholar]
- 3. Pupillo E, Messina P, Logroscino G, et al. Long‐term survival in amyotrophic lateral sclerosis: a population‐based study. Ann Neurol 2014;75:287–297. [DOI] [PubMed] [Google Scholar]
- 4. Masrori P, Van Damme P. Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol 2020;27:1918–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fogh I, Lin K, Tiloca C, et al. Association of a locus in the CAMTA1 gene with survival in patients with sporadic amyotrophic lateral sclerosis. JAMA Neurol 2016;73:812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang X‐B, Cui N‐H, Gao J‐J, et al. SMN1 duplications contribute to sporadic amyotrophic lateral sclerosis susceptibility: evidence from a meta‐analysis. J Neurol Sci 2014;340:63–68. [DOI] [PubMed] [Google Scholar]
- 7. Blauw HM, Barnes CP, van Vught PWJ, et al. SMN1 gene duplications are associated with sporadic ALS. Neurology 2012;78:776–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Corcia P, Ingre C, Blasco H, et al. Homozygous SMN2 deletion is a protective factor in the Swedish ALS population. Eur J Hum Genet 2012;20:588–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Veldink JH, Kalmijn S, Van der Hout AH, et al. SMN genotypes producing less SMN protein increase susceptibility to and severity of sporadic ALS. Neurology 2005;65:820–825. [DOI] [PubMed] [Google Scholar]
- 10. Veldink JH, van den Berg LH, Cobben JM, et al. Homozygous deletion of the survival motor neuron 2 gene is a prognostic factor in sporadic ALS. Neurology 2001;56:749–752. [DOI] [PubMed] [Google Scholar]
- 11. Corcia P, Camu W, Praline J, et al. The importance of the SMN genes in the genetics of sporadic ALS. Amyotroph Lateral Scler 2009;10:436–440. [DOI] [PubMed] [Google Scholar]
- 12. Corcia P, Camu W, Halimi J‐M, et al. SMN1 gene, but not SMN2, is a risk factor for sporadic ALS. Neurology 2006;67:1147–1150. [DOI] [PubMed] [Google Scholar]
- 13. Corcia P, Mayeux‐Portas V, Khoris J, et al. Abnormal SMN1 gene copy number is a susceptibility factor for amyotrophic lateral sclerosis. Ann Neurol 2002;51:243–246. [DOI] [PubMed] [Google Scholar]
- 14. Gamez J, Barceló MJ, Muñoz X, et al. Survival and respiratory decline are not related to homozygous SMN2 deletions in ALS patients. Neurology 2002;59:1456–1460. [DOI] [PubMed] [Google Scholar]
- 15. Lee J‐B, Lee K‐A, Hong J‐M, et al. Homozygous SMN2 deletion is a major risk factor among twenty‐five Korean sporadic amyotrophic lateral sclerosis patients. Yonsei Med J 2012;53:53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harada Y, Sutomo R, Sadewa AH, et al. Correlation between SMN2 copy number and clinical phenotype of spinal muscular atrophy: three SMN2 copies fail to rescue some patients from the disease severity. J Neurol 2002;249:1211–1219. [DOI] [PubMed] [Google Scholar]
- 17. Sun Y, Grimmler M, Schwarzer V, et al. Molecular and functional analysis of intragenic SMN1 mutations in patients with spinal muscular atrophy. Hum Mutat 2005;25:64–71. [DOI] [PubMed] [Google Scholar]
- 18. Rochette CF, Gilbert N, Simard LR. SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens . Hum Genet 2001;108:255–266. [DOI] [PubMed] [Google Scholar]
- 19. Hua Y, Sahashi K, Hung G, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev 2010;24:1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mendell JR, Al‐Zaidy S, Shell R, et al. Single‐dose gene‐replacement therapy for spinal muscular atrophy. N Engl J Med 2017;377:1713–1722. [DOI] [PubMed] [Google Scholar]
- 21. Project MinE ALS Sequencing Consortium . Project MinE: study design and pilot analyses of a large‐scale whole‐genome sequencing study in amyotrophic lateral sclerosis. Eur J Hum Genet 2018;26:1537–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kopanos C, Tsiolkas V, Kouris A, et al. VarSome: the human genomic variant search engine. Bioinformatics 2019;35:1978–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dolzhenko E, Deshpande V, Schlesinger F, et al. ExpansionHunter: a sequence‐graph‐based tool to analyze variation in short tandem repeat regions. Bioinformatics 2019;35:4754–4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen X, BioResource N, Sanchis‐Juan A, et al. Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genet Med 2020;22:945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yoon S, Lee CH, Lee K‐A. Determination of SMN1 and SMN2 copy numbers in a Korean population using multiplex ligation‐dependent probe amplification. Korean J Lab Med 2010;30:93–96. [DOI] [PubMed] [Google Scholar]
- 26. Fang P, Li L, Zeng J, et al. Molecular characterization and copy number of SMN1, SMN2 and NAIP in Chinese patients with spinal muscular atrophy and unrelated healthy controls. BMC Musculoskelet Disord 2015;16:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feng Y, Ge X, Meng L, et al. The next generation of population‐based spinal muscular atrophy carrier screening: comprehensive pan‐ethnic SMN1 copy‐number and sequence variant analysis by massively parallel sequencing. Genet Med 2017;19:936–944. [DOI] [PubMed] [Google Scholar]
- 28. Royston P, Parmar MKB. Flexible parametric proportional‐hazards and proportional‐odds models for censored survival data, with application to prognostic modelling and estimation of treatment effects. Stat Med 2002;21:2175–2197. [DOI] [PubMed] [Google Scholar]
- 29. Akimoto C, Volk AE, van Blitterswijk M, et al. A blinded international study on the reliability of genetic testing for GGGGCC‐repeat expansions in C9orf72 reveals marked differences in results among 14 laboratories. J Med Genet 2014;51:419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bowerman M, Murray LM, Scamps F, et al. Pathogenic commonalities between spinal muscular atrophy and amyotrophic lateral sclerosis: converging roads to therapeutic development. Eur J Med Genet 2018;61:685–698. [DOI] [PubMed] [Google Scholar]
- 31. Simon CM, Dai Y, Van Alstyne M, et al. Converging mechanisms of p53 activation drive motor neuron degeneration in spinal muscular atrophy. Cell Rep 2017;21:3767–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramos DM, d’Ydewalle C, Gabbeta V, et al. Age‐dependent SMN expression in disease‐relevant tissue and implications for SMA treatment. J Clin Invest. 2019;129:4817–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Czech C, Tang W, Bugawan T, et al. Biomarker for spinal muscular atrophy: expression of SMN in peripheral blood of SMA patients and healthy controls. PLOS ONE. 2015;10:e0139950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sumner CJ, Kolb SJ, Harmison GG, et al. SMN mRNA and protein levels in peripheral blood. Neurology. 2006;66:1067–1073. [DOI] [PubMed] [Google Scholar]
- 35. Rodriguez‐Muela N, Litterman NK, Norabuena EM, et al. Single‐cell analysis of SMN reveals its broader role in neuromuscular disease. Cell Rep 2017;18:1484–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Phenotype by country
Table S2 Frequency of SMN genes in different populations
Table S3 Frequency of the SMN1 gene in different countries within Project MinE
Table S4 Frequency of SMN2 in different countries within Project MinE
Data Availability Statement
Data can be accessed through EGAC00001000703. The authors declare that all the other data supporting the findings of this study are available within the article, its Supplementary Information files, or from the corresponding author upon reasonable request.